

Abstract
Enzymes that use a [4Fe-4S]1+ cluster plus S-adenosyl-l-methionine (SAM) to initiate radical reactions (radical SAM) form the largest enzyme superfamily, with over half a million members across the tree of life. This review summarizes recent work revealing the radical SAM reaction pathway, which ultimately liberates the 5´-deoxyadenosyl (5´-dAdo•) radical to perform extremely diverse, highly regio- and stereo-specific, transformations. Most surprising was the discovery of an organometallic intermediate Ω exhibiting an Fe-C5´-adenosyl bond. Ω liberates 5´-dAdo• through homolysis of the Fe-C5´ bond, in analogy to Co-C5´ bond homolysis in B12, previously viewed as biology’s paradigmatic radical generator. The 5´-dAdo• has been trapped and characterized in radical SAM enzymes via a recently-discovered photoreactivity of the [4Fe-4S]+/SAM complex, and has been confirmed as a catalytically active intermediate in enzyme catalysis. The regioselective SAM S-C bond cleavage to produce 5´-dAdo• originates in the Jahn-Teller effect. The simplicity of SAM as a radical precursor, and the exquisite control of 5´-dAdo• reactivity in radical SAM enzymes, may be why radical SAM enzymes pervade the tree of life, while B12 enzymes are only a few.
Keywords: radical, S-adenosylmethionine, radical SAM, adenosylcobalamin, B12, mechanism, deoxyadenosyl radical, EPR
Graphical Abstract
Recent advances in understanding the radical S-adenosyl-l-methionine (SAM) reaction pathway are summarized. Regioselective reductive cleavage of SAM yields an organometallic intermediate Ω exhibiting an Fe-C5´-adenosyl bond. Ω undergoes Fe-C bond homolysis to liberate the 5´-deoxyadenosyl radical, which is shown to be a true intermediate that initiates extremely diverse, highly regio- and stereo-specific, radical-based transformations.
Introduction to adenosylcobalamin and SAM as Radical Reservoirs
The idea that radicals could play essential roles in biology was controversial for many years. In fact, at one time much of the discussion of radicals as they relate to biology focused on the damaging effects of radicals and their putative roles in cancer, aging, and other serious disease states.[1] In the 1980s this began to change, with the increasing recognition that organic radicals, while highly reactive and potential dangerous in a ‘free’ state, have unique and powerful reactivity that can be exploited by biology if the radicals can be appropriately constrained.[2, 3] These developments paralleled similar recognitions being made at the time in synthetic organic chemistry, where it became clear that appropriate steric and stereoelectronic constraints could lead to stereoselective, regioselective, and chemoselective radical reactions.[4] Since this time, radicals have become recognized as prominent players in biochemical reactions, both as key reaction intermediates and as catalytically essential protein cofactors.[3, 5–10] Perhaps the most prominent examples are in the cases of the ribonucleotide reductases, which were first shown to harbor ‘free radicals’ by Sjöberg, Reichard, Gräslund, and Ehrenberg in the 1970s.[11–14] These remarkable enzymes are found throughout nature, catalyzing one of the most central reactions in all of biology: the conversion of RNA precursors into DNA precursors. Although this fundamentally important reaction is initiated by different metallocofactors in different organisms, the mechanisms are unified by the involvement of radical chemistry in the fundamental chemical steps, as so elegantly elaborated by JoAnne Stubbe and coworkers over the course of several decades.[15–19]
Among the earliest cofactors implicated in biological radical reactions was adenosylcobalamin (AdoCbl or coenzyme B12) which was structurally characterized by Lenhert and Hodgkin in 1961 to reveal a direct bond between the 5´-C of a 5´-deoxyadenosyl group to the cobalt of cobalamin.[20] This seminal structure of the first known organometallic cofactor in biology led to key biochemical studies by Perry Frey and Robert Abeles implicating the 5´-deoxyadenosyl moiety as directly involved in enzymatically-catalyzed hydrogen atom transfer reactions.[21–23] Subsequent mechanistic studies using both biochemical and chemical systems provided new insights into this remarkable cofactor, supporting a mechanism in which reversible cobalt-carbon bond homolysis generates a 5´-deoxyadenosyl (5´-dAdo•) radical to mediate radical rearrangement reactions via H-atom abstraction.[24–28] Noteworthy, despite the importance of the AdoCbl cofactor, there are fewer than 20 enzymes known to use AdoCbl to initiate radical reactions.
S-adenosyl-l-methionine (SAM) is an important biological methyl donor,[29] playing key roles in methylation of DNA and RNA, proteins, and a variety of small molecules.[30] An alternative function for SAM was suggested in 1970, when Barker and coworkers showed that SAM stimulates the reaction catalyzed by lysine 2,3-aminomutase (LAM), a reaction with no known involvement in methylation.[31] An important clue to the role of SAM in LAM catalysis was revealed by Marcia Moss and Perry Frey, who provided evidence for the involvement of the 5´-deoxyadenosyl moiety of SAM in hydrogen atom transfer, suggesting that SAM was playing a role similar to that of AdoCbl in radical rearrangements.[32]
SAM is a much simpler, purely organic cofactor compared to AdoCbl, and has a 5´-deoxyadenosyl C-S bond to the sulfonium sulfur of SAM, rather than the critical 5Ć-Co organometallic bond linking the adenosyl moiety to cobalamin (Fig. 1).[33] Despite these significant differences, a growing body of evidence in the 1980s and 1990s supported a role for the adenosyl moiety of SAM in mediating radical reactions, not only in LAM, but in the pyruvate formate-lyase activating enzyme (PFL-AE),[34–36] the anaerobic ribonucleotide reductase,[37, 38] spore photoproduct lyase,[39] and biotin synthase.[40, 41] The involvement of an iron-sulfur cluster in all of these SAM-dependent enzymes provided another striking similarity among them,[42] further distinguishing them from adenosylcobalamin enzymes.
Figure 1.

Adenosylcobalamin, or coenzyme B12 (left) and the SAM-[4Fe-4S] cluster complex at the active site of Radical SAM enzymes (right).
A key turning point came in 2001, when the SAM-dependent iron-sulfur enzymes were identified through a bioinformatics study as members of a new enzyme superfamily that was named radical SAM.[43] The Radical SAM enzyme superfamily now is known to be the largest in nature, with hundreds of thousands of members spanning all kingdoms of life.[44] Although only a small fraction of superfamily members have been characterized, already more than 100 distinct reactions have been identified for radical SAM enzymes.[44]
A central motivation of research in our labs has been the question, how does an enzyme-bound iron-sulfur cluster together with SAM recapitulate the remarkable biochemical reactions mediated by AdoCbl, in which a 5´-dAdo• radical generated by reversible Co-C bond homolysis initiates chemistry via H-atom abstraction? The remarkable and surprising findings that provide answers to this question are the focus of this article.
SAM-Cluster Interaction and Implications for Mechanism
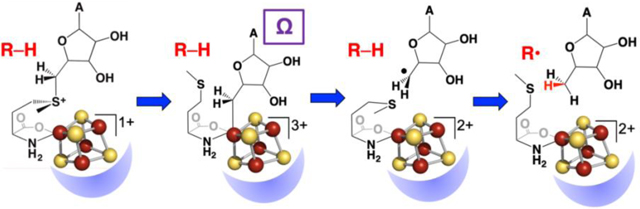
Radical SAM enzymes bind a [4Fe-4S] cluster at a three-cysteine motif that is most commonly CX3CX2C; the three cysteines bind three of the four irons of the [4Fe-4S] cluster, providing a site-differentiated cluster in which one iron is not coordinated by cysteine.[45, 46] The unique iron of the site-differentiated [4Fe-4S] cluster coordinates the amino and carboxylate groups of SAM (Fig. 1), a binding interaction first demonstrated using electron-nuclear double resonance (ENDOR) spectroscopy[47] and subsequently observed in a large number of X-ray crystal structures of radical SAM enzymes.[48, 49] The coordination of SAM to the [4Fe-4S] cluster places the sulfonium of SAM sufficiently close to the cluster to provide orbital overlap.[50, 51] The catalytically active state of the enzyme was shown to be the SAM-bound reduced [4Fe-4S]1+ cluster, in which this cluster provides the electron required for the reductive cleavage of SAM to generate a 5´-deoxyadenosyl radical (Fig. 2).[52] The orbital overlap between the sulfonium and cluster was therefore thought to provide a pathway for inner-sphere electron transfer during reductive cleavage.[50, 51] In at least one radical SAM enzyme, the SAM bridges the [4Fe-4S] cluster to a monovalent cation that is important for activity.[53]
Figure 2. A mechanism for radical initiation in Radical SAM enzymes.

Inner-sphere electron transfer from the reduced [4Fe-4S] cluster to the sulfonium of SAM leads to reductive cleavage of the S-C5´ bond of SAM (left). The resulting 5´-dAdo• radical abstracts an H-atom from substrate R-H (center) to yield a substrate radical (right).
Substrates bind in the active site pocket of radical SAM enzymes close to the SAM-bound [4Fe-4S] cluster, with the site of H-atom abstraction generally within 4 – 5 Å of the 5Ć of SAM. This is seen for example in the X-ray crystal structure of pyruvate formate-lyase activating enzyme (PFL-AE), where a peptide mimicking the pyruvate formate-lyase G734 site of H-atom abstraction is bound such that the G734 Cα is only 3.9 Å from the 5Ć of SAM.[54] Other examples include the structures of lysine bound in the active site of LAM,[55] peptide substrate bound to an anaerobic sulfatase maturating enzyme,[56] peptide substrate bound to SuiB,[57] among others. The proximity of the substrate to the 5Ć of SAM bound to the [4Fe-4S] cluster supports a simple mechanism involving reductive SAM cleavage followed by direct H-atom abstraction from substrate by the SAM-derived 5´-deoxyadenosyl radical, Fig. 2.
Further evidence for the close proximity of substrate to the [4Fe-4S] cluster-bound SAM in the active sites of radical SAM enzymes was found through use of the SAM analog 3´,4´-anhydroadenosylmethionine (anSAM). This functional SAM analog has a double bond between the 3´ and 4´ carbons that provides allylic stabilization of the anhydroadenosyl radical (anAdo•), a 5´-deoxyadenosyl radical analog, formed upon reductive S-C5´ bond cleavage (Fig. 3).[58] The use of anSAM in the LAM reaction led to direct observation of the anAdo• radical by electron paramagnetic resonance (EPR) spectroscopy.[58, 59] Subsequent studies of the anAdo• radical trapped in the LAM active site during turnover revealed the remarkable constraints placed on the positioning of the reactive radical species, and the extremely limited movement of the anAdo• radical required for H-atom abstraction from substrate.[60] These results provided the most direct evidence at the time that SAM, like adenosylcobalamin, is involved in enzymatic radical reactions by generation of a 5´-deoxyadenosyl radical species.[60] Further, they revealed that radical SAM enzymes can exert more constraints on the positioning of the reactive 5´-dAdo• radical than B12 enzymes, thereby providing much tighter control of the radical. This suggests that it is not solely the simplicity in biosynthesis of the active-site cluster + SAM components of the radical SAM enzymes as compared to B12, but primarily the higher precision with which radical SAM enzymes can guide 5´-dAdo• to its substrate target, that has led evolution to utilize radical SAM enzymes so widely in biology, while only a handful of B12 enzymes are found.[60]
Figure 3. Products of reductive cleavage of SAM and of anSAM.

Reductive cleavage of SAM leads to the primary carbon radical 5´-dAdo• (top), while reductive cleavage of the SAM analog anSAM leads to the allylically stabilized anAdo• radical (bottom).
Trapping of Radical Intermediates Reveals New Parallels to B12 Enzymes
Rapid freeze-quench (RFQ) trapping techniques were used to probe the reaction of PFL-AE with SAM and substrate PFL for radical intermediates. These revealed a previously unrecognized paramagnetic species that formed maximally at approximately 500 ms and then decayed with longer reaction times as the product PFL glycyl radical formed.[61]. The EPR spectrum of this species has g-values and lineshape consistent with its assignment to a [4Fe-4S]3+ cluster-based species. The nature of this intermediate was revealed via ENDOR spectroscopy with the use of isotopically-labeled SAM and 57Fe-labeled PFL-AE. The results revealed the remarkable finding that this intermediate, which was named omega (Ω), was an organometallic species in which the adenosyl moiety derived from SAM is directly bound to the unique iron of the [4Fe-4S] cluster through a 5Ć-Fe bond (Fig. 4).[61] This organometallic Ω intermediate was subsequently observed for a number of other radical SAM enzymes that carry out a wide array of differing reactions, implying that it plays a central role during radical initiation throughout the superfamily.[62]
Figure 4. The organometallic intermediate Ω.

The organometallic intermediate Ω (upper left) was first discovered in PFL-AE, and the presence of the iron-carbon bond was identified by 13C-ENDOR (lower left). The Ω intermediate has since been identified for a wide range of radical SAM enzymes.
A particularly striking feature of Ω is its structural analogy to adenosylcobalamin, both being organometallic species in which an adenosyl group is directly bound to a transition metal through a 5Ć-metal bond. This structural similarity implies mechanistic parallels: in both cases homolytic metal-carbon bond cleavage liberates the 5´-dAdo• radical. As a functional difference, the Co-C bond in adenosylcobalamin is stable at ambient temperature, and its homolysis requires significant activation, whereas the reactive Fe-C5´ bond of Ω homolyzes in the frozen solid at temperatures of ~ 170 K and above.
Direct Observation of the 5´-dAdo• Radical and Implications for Regioselectivity
Half a century of efforts to trap and characterize the 5´-dAdo• radical intermediate, driven by its role in both radical SAM and adenosylcobalamin enzymes, had proved fruitless. Although photolysis of adenosylcobalamin was demonstrated to induce homolysis of the Co-C bond, this produces a cob(II)alamin – 5´-dAdo• coupled radical pair rather than a “free” 5´-dAdo• radical.[63–65] The similarity of Ω to AdoCbl led us to explore the photochemical properties of both Ω and the precursor [4Fe-4S]+/SAM complex in the PFL-AE active site. This led to our discovery that the [4Fe-4S]+/SAM complex in radical SAM enzymes undergoes novel photochemistry, reminiscent of known photocatalytic reactions of sulfonium salts used in organic synthesis.[66, 67] Under cryogenic conditions, exposure of the PFL-AE [4Fe-4S]+/SAM complex to blue laser light (absorbed by the cluster) resulted in photoinduced electron transfer from the [4Fe-4S]+ cluster to SAM, which led to regioselective homolysis of the S-C5´ bond to generate a 5´-dAdo• radical cryo-trapped in the PFL-AE active site.[66] As this 5´-dAdo• is trapped adjacent to the diamagnetic [4Fe-4S]2+ cluster, it could be fully characterized for the first time through use of isotopic labelling coupled with EPR spectroscopy and computational studies (Fig. 5).[66] In a different approach to accessing the 5´-dAdo• radical in a radical SAM enzyme, Britt and coworkers incubated the HydG [4Fe-4S]+/SAM complex with a substrate analog lacking an abstractable H-atom, revealing a 5´-dAdo• radical species similar to that generated using photolysis.[68]
Figure 5. Blue light-induced reductive cleavage of SAM bound to the [4Fe-4S]+ cluster of PFL-AE.

Blue light induces the reductive cleavage of SAM bound to the [4Fe-4S]+ cluster of PFL-AE, forming 5´-dAdo• cryotrapped in the active site. The 5´-dAdo• has been fully characterized through use of SAM isotopologs and EPR spectroscopy (right).
Surprisingly, when HydG [4Fe-4S]+/SAM was subjected to cryogenic photolysis, quantitative formation of a •CH3 radical resulting from S-CH3 bond homolysis was observed, rather than a 5´-dAdo• radical; this change in regioselectivity of S-C bond cleavage was unexpected and resulted in the first example of S-CH3 bond homolysis in a radical SAM enzyme.[69] This raised the questions: why does the S-C bond cleavage regioselectivity change with the enzyme under photolytic conditions, and more generally, what is the basis of reductive cleavage regioselectivity in radical SAM enzymes.
To address these questions, photolysis experiments were carried out on other radical SAM enzymes in the SAM-bound [4Fe-4S]+ state in the absence of substrate; all underwent photoinduced reductive cleavage of SAM, with some enzymes forming the 5´-dAdo• radical due to S-C5´ homolysis, while others formed a •CH3 radical due to S-CH3 homolysis (Fig. 6).[70, 71] The different regioselectivities under photolysis were distinct from the observations under enzyme catalytic conditions, where all the radical SAM enzymes selectively cleave the S-C5´ bond. Seeking a structure-reactivity correlation, analysis of the SAM conformations in radical SAM crystal structures available for the photolyzed enzymes revealed that while the overall SAM binding conformation was the same, the SAM ribose-ring ‘pucker’ correlated with the photolytic regioselectivity: enzymes where we observed S-C5´ homolysis with 5´-dAdo• formation had a 3’-endo ribose conformation, while those undergoing S-CH3 homolysis to form •CH3 had a 2’-endo conformation (Fig. 6).[70]
Figure 6. Photoinduced electron transfer of SAM bound to [4Fe-4S]+ clusters.

Photoinduced electron transfer of SAM bound to [4Fe-4S]+ clusters in a range of radical SAM enzymes results in cleavage of either the S-C5´ bond to generate 5´-dAdo•, or the S-CH3 bond to generate •CH3, with the radicals cryotrapped in the active site (left). The regioselectivity of photoinduced S-C bond cleavage correlates with the SAM ribose ring pucker (upper right) and is explained by the Jahn-Teller effect on the one-electron reduced sulfonium radical (right).
How does SAM ribose ring pucker impact regioselectivity in photoinduced reductive cleavage of SAM? In the process of carrying out computational analysis of the photoinduced reductive cleavage process, we recognized that the one-electron reduced R3S0 sulfonium center is Jahn-Teller active and thus undergoes a distortion, i.e. S-C bond lengthening, leading to S-C bond cleavage (Fig. 6). Ultimately, the experimental and computational results revealed that the origin of regioselectivity in photolysis is the Jahn-Teller effect coupled to active-site interactions that favor one or the other ribose conformations in the different enzymes. The differing interactions in these conformations localize the JT distortion in a particular S-C bond, resulting in regioselective elongation and cleavage of this favored S-C bond.[70] With this foundation, it was then proposed that the regioselective S-C5´ bond cleavage during actual radical SAM catalysis occurs because the thermally induced conformational changes that enable ET from the [4Fe-4S]+ cluster to sulfonium generate dominant additional forces that specifically select S−C5′ for cleavage.[70]
5´-dAdo• as a Catalytically Competent Intermediate Observable During Catalysis
The ability to trap and characterize the 5´-dAdo• radical for the first time via cryogenic photolysis was highly significant, but the radical was generated under conditions that were not catalytic, since substrate was not present. Thus while these studies provided the first opportunity to fully characterize this central radical species implicated in both B12 and radical SAM enzymes, they left unresolved the question of whether the 5´-dAdo• is generated as a true intermediate in these reactions, or is better described as a transition state species. Arguments in fact had been made, that the 5´-dAdo• radical is too high in energy, and thus too reactive, to be formed as a discrete species during catalysis.[49, 60, 72, 73]
Recent efforts to probe sequential radical intermediates during catalysis by PFL-AE have used a modified peptide substrate in which the target glycyl residue is replaced by a dehydroalanyl residue (Fig. 7).[74] By replacing the abstractable hydrogen on the glycine Cα with a double bond to Cβ, the peptide was expected to change the catalytic reaction from H-atom abstraction to adenosylation. Such altered reactivity had in fact been observed previously by Knappe and coworkers using a similar modified peptide.[75] The dehydroalanine peptide (Dha-pep) was shown to undergo reaction with PFL-AE and SAM under reducing conditions, generating an adenosylated peptide product that was characterized by mass spectrometry.
Figure 7. Radical intermediates observed during an adenosylation reaction catalyzed by PFL-AE on a dehydroalanine-containing substrate peptide.

The organometallic intermediate Ω is observed by rapid freeze-quench EPR at early times. Longer quench times from 10 to 18 s allows observation of both the catalytically competent 5´-dAdo• intermediate, and the adenosylated peptide radical intermediate Ado-Dha-pep•.
Investigation of the reaction of reduced PFL-AE, SAM, and Dha-pep using RFQ trapping revealed the formation of Ω at early times (500 ms – 2 s).[74] Freeze-quenching the reaction at slightly longer times (8 – 10 s) revealed the formation of the adenosylated peptide radical (Ado-Dha-pep•) intermediate, which was identified and characterized by isotopic labeling of both SAM and peptide combined with EPR spectroscopy (Fig. 7).[74] Furthermore, thermal annealing of the Ω intermediate obtained by RFQ results in conversion to the Ado-Dha-pep• radical, demonstrating that Ω is a catalytically competent intermediate. The EPR signal for Ado-Dha-pep• was best observed at 70 K; when temperature was varied, an additional EPR-active species was observed at lower temperatures.[74] This additional species could be observed in isolation after spectral subtraction of the Ado-Dha-pep• radical signal; EPR analysis and isotopic labeling revealed the additional signal to arise from the 5´-dAdo• radical.[74] To evaluate whether this high-energy primary 5´-dAdo• radical was an on-pathway radical intermediate, its ability to convert to the capto-datively stabilized tertiary Ado-Dha-pep• product radical [76] by adenosylation of Dha-pep was evaluated both by thermal annealing and by carrying out time-dependent freeze-quench between 8 and 18 s. Both methods confirmed this observed 5´-dAdo• radical converts to Ado-Dha-pep•, demonstrating its catalytic competence as a true intermediate in this enzyme-catalyzed adenosylation reaction (Fig. 7).[74] This is the first demonstration of the 5´-dAdo• radical caught in action as an observable intermediate during a radical SAM reaction.
Conclusions
This review has summarized the mechanism for generation of the 5´-dAdo• radical in Radical SAM enzymes, Nature’s overwhelming choice as the catalyst for radical reactions across the tree of life. The mechanism incorporates three distinct and sequential intermediates prior to substrate transformation. First, reductive cleavage of SAM generates the organometallic intermediate Ω, with its Fe-C5´ bond. Next, cleavage of the Fe-C5´ bond of Ω liberates ‘free’ 5´-dAdo•. Finally, the 5´-dAdo• radical generates a substrate-based radical, generally by regio- and stereoselective H-atom abstraction from substrate; this radical then proceeds in the specific transformation characteristic of the enzyme. Alternatively, 5´-dAdo• can add to a double bond in an adenosylation reaction, as it does for Dha-pep, forming the Ado-Dha-pep• radical (Fig. 7). The simplicity of SAM as a radical precursor, as compared to the more elaborate coenzyme B12, with its large corrinoid ring, and the higher precision with which radical SAM enzymes can guide 5´-dAdo• to its substrate target, may well be why evolution utilizes radical SAM enzymes so widely, while only a handful of B12 enzymes are found.
Acknowledgements
The work described herein has been funded by the U.S. National Institutes of Health (GM131889 to J.B.B. and GM111097 to B.M.H.) and the U.S. Department of Energy (DE-SC0005404 to J.B.B.).
Abbreviations:
- SAM
S-adenosylmethionine
- AdoCbl
adenosylcobalamin
- 5´-dAdo
5´-deoxyadenosyl radical
- PFL-AE
pyruvate formate-lyase activating enzyme
- PFL
pyruvate formate-lyase
- LAM
lysine 2,3-aminomutase
- anSAM
3’,4’-anhydro-S-adenosylmethionine
- RFQ
rapid freeze-quench
- EPR
electron paramagnetic resonance
- ENDOR
electron-nuclear double resonance
References
- 1.Pryor WA (1978) The formation of free radicals and the consequences of their reactions in vivo, Photochem Photobiol. 28, 787–797. [DOI] [PubMed] [Google Scholar]
- 2.Williams RJP (1985) The necessary and the desirable production of radicals in biology, Phil Trans R Soc Lond B. 311, 593–603. [DOI] [PubMed] [Google Scholar]
- 3.Stubbe J.(1988) Radicals in Biological Catalysis, Biochemistry. 27, 3893–3900. [DOI] [PubMed] [Google Scholar]
- 4.Giese B.(1986) Radicals in organic synthesis: Formation of carbon-carbon bonds, Pergamom, Elmsford, NY. [Google Scholar]
- 5.Stubbe J.(1989) Protein radical involvement in biological catalysis?, Annu Rev Biochem. 58, 257–285. [DOI] [PubMed] [Google Scholar]
- 6.Frey PA (1990) Importance of organic radicals in enzymatic cleavage of unactivated C-H bonds, Chem Rev. 90, 1343–1357. [Google Scholar]
- 7.Frey PA (2001) Radical Mechanisms of Enzymatic Catalysis, Annu Rev Biochem. 70, 121–148. [DOI] [PubMed] [Google Scholar]
- 8.Stubbe J.& van der Donk W.(1998) Protein Radicals in Enzyme Catalysis, Chemical reviews. 96, 705–762. [DOI] [PubMed] [Google Scholar]
- 9.Sivaraja M, Goodin DB, Smith M.& Hoffman BM (1989) Identification by ENDOR of Trp191 as the free-radical site in cytochrome C peroxidase compounds ES, Science. 245, 738–740. [DOI] [PubMed] [Google Scholar]
- 10.Barry BA & Babcock GT (1987) Tyrosine radicals are involved in the photosynthetic oxygen-evolving system, Proc Natl Acad Sci USA. 84, 7099–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sjöberg B-M, Reichard P, Gräslund A.& Ehrenberg A.(1977) Nature of the free radical in ribonucleotide reductase from Escherichia coli, J Biol Chem. 252, 536–541. [PubMed] [Google Scholar]
- 12.Sjöberg B-M, Reichard P, Gräslund A.& Ehrenberg A.(1978) The tyrosine free radical in ribonucleotide reductase from Escherichia coli, J Biol Chem. 253, 6863–6865. [PubMed] [Google Scholar]
- 13.Ehrenberg A.& Reichard P.(1972) Electron spin resonance of the iron-containing protein B2 from ribonucleotide reductase, J Biol Chem. 247, 3485–3488. [PubMed] [Google Scholar]
- 14.Reichard P.& Ehrenberg A.(1983) Ribonucleotide reductase - A radical enzyme, Science. 221, 514–519. [DOI] [PubMed] [Google Scholar]
- 15.Stubbe J.(1990) Ribonucleotide Reductases: Amazing and Confusing, Journal of Biological Chemistry. 265, 5329–5332. [PubMed] [Google Scholar]
- 16.Booker S, Broderick J.& Stubbe J.(1993) Ribonucleotide reductases: radical enzymes with suicidal tendencies, Biochemical Society transactions. 21, 727–730. [DOI] [PubMed] [Google Scholar]
- 17.Stubbe J.& van der Donk WA (1995) Ribonucleotide reductases: radical enzymes with suicidal tendencies, Chemistry & biology. 2, 793–801. [DOI] [PubMed] [Google Scholar]
- 18.Licht S, Gerfen GJ & Stubbe J.(1996) Thiyl radicals in ribonucleotide reductases, Science. 271. [DOI] [PubMed] [Google Scholar]
- 19.Stubbe J.& Nocera DG (2021) Radicals in biology: Your life is in their hands, J Am Chem Soc. 143, 13463–13472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenhert PG & Hodgkin DC (1961) Structure of the 5,6-dimethylbenzimidazolylcobamide coenzyme, Nature. 192, 937–938. [DOI] [PubMed] [Google Scholar]
- 21.Frey PA & Abeles RH (1966) The role of the B12 coenzyme in the conversion of 1,2-propanediol to propionaldehyde, J Biol Chem. 241, 2732–2733. [PubMed] [Google Scholar]
- 22.Abeles RH (1971) Dehydrations requiring vitamin B12 coenzyme, The Enzymes. 5, 481–497. [Google Scholar]
- 23.Barker HA (1972) Coenzyme B12-dependent mutases causing carbon chain rearrangements, The Enzymes. 6, 509–537. [Google Scholar]
- 24.Halpern J.(1985) Mechanisms of Coenzyme B12-Dependent Rearrangements, Science. 227, 869–875. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee R.(2003) Radical carbon skeleton rearrangements: catalysis by coenzyme B12-dependent mutases, ChemRev. 103, 2083–94. [DOI] [PubMed] [Google Scholar]
- 26.Brown KL (2005) Chemistry and Enzymology of Vitamin B12, Chem Rev. 105, 2075–2149. [DOI] [PubMed] [Google Scholar]
- 27.Fischer H.(2001) The persistent radical effect: A principle for selective radical reactions and living radical polymerizations, Chem Rev. 101, 3581–3610. [DOI] [PubMed] [Google Scholar]
- 28.Fischer H.(1986) Unusual selectivities of radical reactions by internal suppression of fast modes, J Am Chem Soc. 108, 3925–3927. [Google Scholar]
- 29.Cantoni GL (1953) S-Adenosylmethionine - A new intermediate formed enzymatically from L-methionine and adenosinetriphosphate, J Biol Chem. 204, 403–416. [PubMed] [Google Scholar]
- 30.Markham GD (2002) S-Adenosylmethionine in Nature Encyclopedia of Life Sciences, Nature Publishing Group, London. [Google Scholar]
- 31.Chirpich TP, Zappia V, Costilow RN & Barker HA (1970) Lysine 2,3-aminomutase. Purification and properties of a pyridoxal phosphate and S-adenosylmethionine activated enzyme, J Biol Chem. 245, 1778–1789. [PubMed] [Google Scholar]
- 32.Moss M.& Frey PA (1987) The role of S-adenosylmethionine in the lysine 2,3-aminomutase reaction, J Biol Chem. 262, 14859–14862. [PubMed] [Google Scholar]
- 33.Frey PA (1993) Lysine 2,3-aminomutase: is adenosylmethionine a poor man’s adenosylcobalamin?, FASEB J. 7, 662–670. [DOI] [PubMed] [Google Scholar]
- 34.Knappe J.& Schmitt T.(1976) A novel reaction of S-adenosyl-L-methionine correlated with the activation of pyruvate formate-lyase, Bioch Biophys Res Commun. 71, 1110–1117. [DOI] [PubMed] [Google Scholar]
- 35.Knappe J, Neugebauer FA, Blaschkowski HP & Gänzler M.(1984) Post-translational activation introduces a free radical into pyruvate formate-lyase, Proc Natl Acad Sci USA. 81, 1332–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frey M, Rothe M, Wagner AFV & Knappe J.(1994) Adenosylmethionine-dependent synthesis of the glycyl radical in pyruvate formate-lyase by abstraction of the glycine C-2 pro-S hydrogen atom, J Biol Chem. 269, 12432–12437. [PubMed] [Google Scholar]
- 37.Eliasson R, Fontecave M, Jörnvall H, Krook M, Pontis E.& Reichard P.(1990) The anaerobic ribonucleotide triphosphate reductase from Escherichia coli requires S-adenosylmethionine as a cofactor, Proc Natl Acad Sci USA. 87, 3314–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mulliez E, Fontecave M, Gaillard J.& Reichard P.(1993) An iron-sulfur cluster and a free radical in the active anaerobic ribonucleotide reductase of Escherichia coli, J Biol Chem. 268, 2296–2299. [PubMed] [Google Scholar]
- 39.Rebeil R, Sun Y, Chooback L, Pedraza-Reyes M, Kinsland C, Begley TP & Nicholson WL (1998) Spore Photoproduct Lyase from Bacillus subtilis spores is a novel iron-sulfur DNA repair enzyme which shares features with proteins such as class III anaerobic ribonucleotide reductases and pyruvate formate-lyases, J Bacteriol. 180, 4879–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guianvarc’h D, Florentin D, Bui BTS, Nunzi F.& Marquet A.(1997) Biotin synthase, a new member of the family of enzymes which uses S-adenosylmethionine as a source of deoxyadenosyl radical, Biochem Biophys Res Commun. 236, 402–406. [DOI] [PubMed] [Google Scholar]
- 41.Escalettes F, Florentin D, Bui BTS, Lesage D.& Marquet A.(1999) Biotin synthase mechanism: Evidence for hydrogen transfer from the substrate into deoxyadenosine, J Am Chem Soc. 121, 3571–3578. [Google Scholar]
- 42.Cheek J.& Broderick JB (2001) Adenosylmethionine-dependent iron-sulfur enzymes: versatile clusters in a radical new role, J Biol Inorg Chem. 6, 209–226. [DOI] [PubMed] [Google Scholar]
- 43.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF & Miller NE (2001) Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods, Nucleic Acids Res. 29, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oberg N, Precord TW, Mitchell DA & Gerlt JA (2022) RadicalSAM.org: A resource to interpret sequence-function space and discover new radical SAM chemistry, ACS Bio Med Chem Au. 2, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frey PA, Hegeman AD & Ruzicka FJ (2008) The radical SAM superfamily, Crit Rev Biochem Mol Biol. 43, 63–88. [DOI] [PubMed] [Google Scholar]
- 46.Broderick JB, Duffus BR, Duschene KS & Shepard EM (2014) Radical S-Adenosylmethionine Enzymes, Chem Rev. 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walsby CJ, Ortillo D, Broderick WE, Broderick JB & Hoffman BM (2002) An anchoring role for FeS Clusters: Chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme, J Am Chem Soc. 124, 11270–11271. [DOI] [PubMed] [Google Scholar]
- 48.Vey JL & Drennan CL (2011) Structural Insights into Radical Generation by the Radical SAM Superfamily, Chem Rev. 111, 2487–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicolet Y.(2020) Structure-function relationships of radical of radical SAM enzymes, Nat Catal. 3, 337–2020. [Google Scholar]
- 50.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB & Hoffman BM (2002) Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active [4Fe-4S]+ cluster of pyruvate formate-lyase activating enzyme, J Am Chem Soc. 124, 3143–3151. [DOI] [PubMed] [Google Scholar]
- 51.Dey A, Peng Y, Broderick WE, Hedman B, Hodgson KO, Broderick JB & Solomon EI (2011) S K-edge XAS and DFT Calculations on SAM Dependent Pyruvate Formate-Lyase Activating Enzyme: Nature of Interaction between the Fe4S4 Cluster and SAM and its Role in Reactivity, J Am Chem Soc. 133, 18656–18662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henshaw TF, Cheek J.& Broderick JB (2000) The [4Fe-4S]+ cluster of pyruvate formate-lyase activating enzyme generates the glycyl radical on pyruvate formate-lyase: EPR-detected single turnover, J Am Chem Soc. 122, 8331–8332. [Google Scholar]
- 53.Shisler KA, Hutcheson RU, Horitani M, Duschene KS, Crain AV, Byer AS, Shepard EM, Rasmussen A, Yang J, Broderick WE, Vey JL, Drennan CL, Hoffman BM & Broderick JB (2017) Monovalent cation activation of the radical SAM enzyme pyruvate formate-lyase activating enzyme, J Am Chem Soc. 139, 11803–11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vey JL, Yang J, Li M, Broderick WE, Broderick JB & Drennan CL (2008) Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme, Proc Natl Acad Sci USA. 105, 16137–16141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lepore BW, Ruzicka FJ, Frey PA & Ringe D.(2005) The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale, Proc Natl Acad Sci USA. 102, 13819–13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ & Drennan CL (2013) X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification, Proc Natl Acad Sci USA. 110, 8519–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis KM, Schramma KR, Hansen WA, Bacik JP, Khare SD, Seyedsayamdost MR & Ando N.(2017) Structures of the peptide-modifying radical SAM enzyme SuiB elucidate the basis of substrate recognition, Proc Natl Acad Sci U S A. 114, 10420–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Magnusson OT, Reed GH & Frey PA (1999) Spectroscopic Evidence for the participation of an allylic analogue of the 5’-deoxyadenosyl radical in the reaction of lysine 2,3-aminomutase, J Am Chem Soc. 121, 9764–9765. [Google Scholar]
- 59.Magnusson OT, Reed GH & Frey PA (2001) Characterization of an Allylic Analogue of the 5’-Deoxyadenosyl Radical: An Intermediate in the Reaction of Lysine 2,3-Aminomutase, Biochemistry. 40, 7773–7782. [DOI] [PubMed] [Google Scholar]
- 60.Horitani M, Byer AS, Shisler KA, Chandra T, Broderick JB & Hoffman BM (2015) Why Nature Uses Radical SAM Enzymes so Widely: Electron Nuclear Double Resonance Studies of Lysine 2,3-Aminomutase Show the 5’-dAdo• “Free Radical” is Never Free, J Am Chem Soc. 137, 7111–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horitani M, Shisler KA, Broderick WE, Hutcheson RU, Duschene KS, Marts AR, Hoffman BM & Broderick JB (2016) Radical SAM catalysis via an organometallic intermediate with an Fe-[5’-C]-deoxyadenosyl bond, Science. 352, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Byer AS, Yang H, McDaniel EC, Kathiresan V, impano S, Pagnier A, Watts H, Denler C, Vagstad AL, Piel J, Duschene KS, Shepard EM, Shields TP, Scott LG, Lilla EA, Yokoyama K, Broderick WE, Hoffman BM & Broderick JB (2018) Paradigm shift for radical S-adenosyl-L-methionine reactions: The organometallic intermediate Ω is central to catalysis, J Am Chem Soc. 140, 8634–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robertson WD & Warncke K.(2009) Photolysis of adenosylcobalamin and radical pair recombination in ethanolamine ammonia-lyase probed on the micro- to millisecond time scale by using time-resolved optical absorption spectroscopy, Biochemistry. 48, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robertson WD, Wang M.& Warncke K.(2011) Characterization of protein contributions to cobalt-carbon bond cleavage catalysis in adenosylcobalamin-dependent ethanolamine ammonia-lyase by using photolysis in the ternary complex, J Am Chem Soc. 133, 6968–6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rury AS, Wiley TE & Sension R.(2015) Energy cascades, escited state dynamics, and photochemistry in cob(III)alamins and ferric porphyrins, Acc Chem Res. 48, 860–867. [DOI] [PubMed] [Google Scholar]
- 66.Yang H, McDaniel EC, Impano S, Byer AS, Jodts RJ, Yokoyama K, Broderick WE, Broderick JB & Hoffman BM (2019) The elusive 5’-deoxyadenosyl radical: Captured and Characterized by Electron Paramagnetic Resonance and Electron Nuclear Double Resonance Spectroscopies, J Am Chem Soc. 141, 12139–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Péter Á, Perry GJP & Procter DJ (2020) Radical C-C bond formation using sulfonium salts and light, Adv Synth Catal. 362, 2135–2142. [Google Scholar]
- 68.Sayler RI, Stich TA, Joshi S, Cooper N, Shaw JT, Begley TP, Tantillo DJ & Britt RD (2019) Trapping and electron paramagnetic resonance characterization of the 5’-dAdo• radical in a radical S-adenosylmethionine enzyme reaction with a non-native substrate, ACS Cent Sci. 5, 1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang H, Impano S, Shepard EM, James CD, Broderick WE, Broderick JB & Hoffman BM (2019) Photoinduced electron transfer in a radical SAM enzyme generates an S-adenosylmethionine derived methyl radical, J Am Chem Soc. 141, 16117–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Impano S, Yang H, Jodts RJ, Pagnier A, Swimley R, McDaniel EC, Shepard EM, Broderick WE, Broderick JB & Hoffman BM (2021) Active-Site Controlled, Jahn-Teller Enabled Regioselectivity in Reductive S-C Bond Cleavage of S-Adenosylmethionine (SAM) in Radical-SAM Enzymes, J Am Chem Soc. 143, 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Impano S, Yang H, Shepard EM, Swimley R, Pagnier A, Broderick WE, Hoffman BM & Broderick JB (2021) S-Adenosyl-L-ethionine is a catalytically competent analog of S-adenosyl-l-methionine (SAM) in the radical SAM enzyme HydG, Angew Chem Int Ed. 60, 4666–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frey PA & Magnusson OT (2003) S-Adenosylmethionine: A wolf in sheep’s clothing, or a rich man’s adenosylcobalamin?, Chem Rev. 103, 2129–2148. [DOI] [PubMed] [Google Scholar]
- 73.Rohac R, Amara P, Benjdia A, Martin L, Ruffié P, Favier A, Berteau O, Mouesca J-M, Fontecilla-Camps JC & Nicolet Y.(2016) Carbon-sulfur bond-forming reaction catalysed by the radical SAM enzyme HydE, Nat Chem. 8, 491–500. [DOI] [PubMed] [Google Scholar]
- 74.Lundahl MN, Sarksian R, Yang H, Jodts RJ, Pagnier A, Smith DF, Mosquera MA, van der Donk WA, Hoffman BM, Broderick WE & Broderick JB (2022) Mechanism of radical S-adenosyl-L-methionine adenosylation: Radical intermediates and the catalytic competence of the 5’-deoxyadenosyl radical, J Am Chem Soc. 144, 5087–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wagner AFV, Demand J, Schilling G, Pils T.& Knappe J.(1999) A dehydroalanyl residue can capture the 5’-dAdo radical generated from SAM by PFL-AE, Biochem Biophys Res Commun. 254, 306–310. [DOI] [PubMed] [Google Scholar]
- 76.Zipse H.(2006) Radical stability - A theoretical perspective, Top Curr Chem. 263, 163–189. [Google Scholar]