

Abstract
Inner ear malformations are an important cause of sensorineural hearing loss ranging from severe to profound and they also contribute to Cerebrospinal fluid leakage and recurrent meningitis. The most severe is the complete labyrinthine aplasia (Michel Deformity), then there is cochlear aplasia, cochlear hypoplasia, incomplete partition of the cochlea, enlarged vestibular acqueduct and cochlear aperture abnormality.
Introduction
Cerebrospinal fluid leak of temporal bone origin is due to abnormal communication between the subarachnoid space and air spaces of temporal bone. The condition of tympanic membrane and Eustachian tube decides the clinical presentation of the patient. If the tympanic membrane is intact patient presents with rhinorrhea (leak from nose) and condition labelled as otorhinorrhea. If the tympanic membrane is perforated, patient presents as CSF otorrhea.
In children with congenital sensorineural hearing loss about 20% of them are reported to have cochlear malformations (Jackler et al.) [1]. There are many classifications, which exist for the same, including those by Phelps etal [2] and Senarroglu etal [3] and the most recent SMS Classifcation [4].
Case Report
A 9-month-old female child presented to Pediatrics Outpatient department of SMS Medical College, Jaipur, India, a tertiary referral center, with complaint of discharge from nose since 4 months. Perinatal history was unremarkable. Nasal discharge was attributed to a fall from bed at 5 months of age as discharge was noticed from left side nasal cavity after 10 days of fall.
The discharge was watery in consistency, non-foul smelling, non-blood tinged and did not get relieved on medications. Later the child developed high-grade fever and suffered from two episodes of convulsions, which were diagnosed to be due to pyogenic meningitis.
Nasal fluid analysis revealed B2 transferrin in nasal discharge to be very high and therefore diagnosis of CSF rhinorrhea was made. The patient was then referred to Department of Otorhinolaryngology for further evaluation.
Brainstem evoked response audiometry showed bilateral profound hearing loss and Otoacoustic emissions were “refer” in both ears.
MRI Brain showed ill-defined area of restricted diffusion involving right high fronto-pareital region. Multiple foci of restricted diffusion also seen in left frontal lobe, right temporal lobe, right basal ganglia and left occipital lobe in periventricular region probably representing acute infarcts.
For CSF rhinorrhea, MR cisternography was done which showed no obvious CSF leak from anterior cranial fossa into the nasal cavities.(Fig. 1) Right inner ear was normal however left side showed IP1 malformation (Sennaroglu and Bajin) [3]/SMS type IIIa cochleovestibular malformation (Grover et al.). [4] Hyperintensities were also seen in left Eustachian tube, nasopharynx, around palate and left nasal cavity, which was suggestive of CSF leak from left ear due to cochlear malformation into nasopharynx and nasal cavity through Eustachian tube (Fig. 2).
Fig. 1.

T2 weighted MRI showing fluid in middle ear and Eustachian tube with malformed inner ear
Fig. 2.
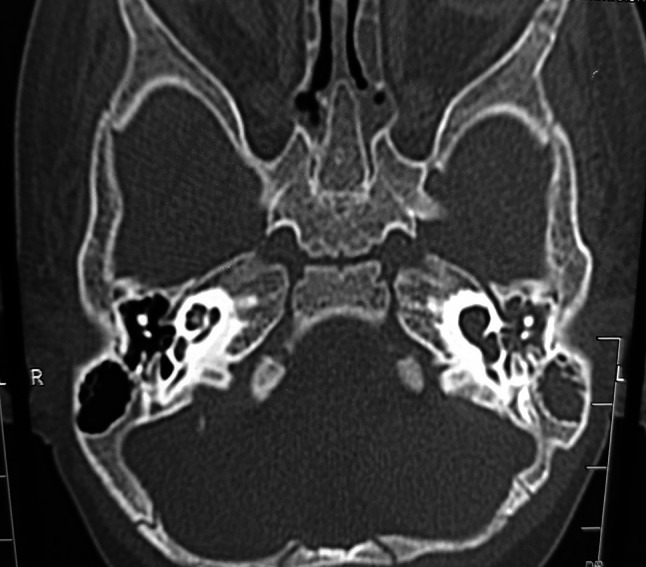
Left inner ear Sennaroglu IP-1 defect/SMS typeIIIb cochleovestibular malformation soft tissue density in left middle ear
High-resolution temporal bone CT scan was done which confirmed the inner ear findings of MRI. Considering the examination and radiology we assumed that a previous episode of meningitis was due to the left ear congenital malformation that led to CSF leak.
After proper assessment of the child and an informed consent of the patient, CSF leak was repaired by postaural route. After elevating the tympanomeatal flap, CSF gusher was encountered. Leak was identified from oval window. Stapes suprastructure was removed. Vestibule was plugged with fat harvested from thigh. Tissue glue was used to seal the defect completely.
Discussion
There have been few classifications for inner ear malformations, however the most accepted one is that given by Sennaroglu [3]. Lately, a more practical and simplified classification has been given by Grover etal [4].
The most common labyrinthine abnormality, which is associated with CSF leak of temporal bone origin (Tyagi et al. 2005). [5] In our case there was bilateral profound hearing loss but malformation was unilateral.
Meningitis is more commonly seen in IP-1 patients or SMS Type IIIa/b4as it has been associated with defective stapes footplate and CSF fills the cochlea (Sennaroglu L, 2016). [6] In our patient also an episode of pyogenic meningitis was encountered and CSF was filled in left cochlea.
According to Sennaroglu and Bajin, 2017 patients who are diagnosed as IP-1 and with episodes of recurrent meningitis and fluid filling the middle ear cleft, their middle ear should be explored with special emphasis on stapes footplate. [3] In our patient the tympanic membrane was intact and there was a defect in oval window, so our patient presented only with rhinorrhea.
Our patient was identified with bilateral profound sensorineural hearing loss on BERA. Such patients are candidates for cochlear implant.
In our case CSF leak was repaired by postaural route. CSF gusher was encountered. Leak was identified from oval window. Stapes suprastructure was removed. Vestibule was plugged with fat harvested from thigh. Tissue glue was used to seal the defect completely.
Kontorinis et al. (2012) reported that among patients with IP, those with IP-1 are more at risk of intraoperative CSF gusher, due to a large bony defect in the lamina cribrosa. [7] The classification by Grover et al. differentiates between these two types as Type IIIa (intact lamina cribrosa) and Type IIIb (deficient lamina cribrosa).
Conclusion
Congenital CSF otorrhoea/otorhinorrhoea is a rare condition and cochlear malformation should always be ruled out in these patients. Deficient lamina cribrosa with absent modiolus and/or septations is commonly seen. These patients need to be operated on emergency basis and also planned for cochlear implantation as early as possible.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97(suppl 40):2–14. doi: 10.1002/lary.5540971301. [DOI] [PubMed] [Google Scholar]
- 2.Phelps PD, King A, Michaels L. Cochlear dysplasia and meningitis. Am J Otol. 1994;15:551–557. [PubMed] [Google Scholar]
- 3.Sennaroglu L, Bajin MD. Classification and current management of inner ear ealformations. Balk Med J. 2017;34:397–411. doi: 10.4274/balkanmedj.2017.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grover M, Sharma S, Preetam C, et al. J LaryngolOtol. 2019;133:368–375. doi: 10.1017/S0022215119000884. [DOI] [PubMed] [Google Scholar]
- 5.Tyagi I, Syal R, Goyal A. Cerebrospinal fluid otorhinor- rhoea due to inner-ear malformations: clinical presentation and new perspectives in management. J Laryngol Otol. 2005;119(9):714–718. doi: 10.1258/0022215054797934. [DOI] [PubMed] [Google Scholar]
- 6.Sennaroglu L. Histopathology of inner ear malformations: do wehave enough evidence to explain pathophysiology? Cochlear Implant Int. 2016;17:3–20. doi: 10.1179/1754762815Y.0000000016. [DOI] [PubMed] [Google Scholar]
- 7.Kontorinis G, Goetz F, Giourgas A, et al. Radiological diag- nosis of incomplete partition type I versus type II: significance for cochlear implantation. Eurradiol. 2012;22:525–532. doi: 10.1007/s00330-011-2301-5. [DOI] [PubMed] [Google Scholar]