

Abstract
6-Deoxyerythronolide B (6dEB) is the macrocyclic aglycone precursor of the antibiotic natural product erythromycin. Heterologous production of 6dEB in Escherichia coli was accomplished, in part, by designed over-expression of a native prpE gene (encoding a propionyl-CoA synthetase) and heterologous pcc genes (encoding a propionyl-CoA carboxylase) to supply the needed propionyl-CoA and (2S)-methylmalonyl-CoA biosynthetic substrates. Separate E. coli metabolism includes three enzymes, Sbm (a methylmalonyl-CoA mutase), YgfG (a methylmalonyl-CoA decarboxylase), and YgfH (a propionyl-CoA:succinate CoA transferase), also involved in propionyl-CoA and methylmalonyl-CoA metabolism. In this study, the sbm, ygfG, and ygfH genes were individually deleted and over-expressed to investigate their effect on heterologous 6dEB production. Our results indicate that the deletion and over-expression of sbm did not influence 6dEB production; ygfG over-expression reduced 6dEB production by fourfold while ygfH deletion increased 6dEB titers from 65 to 129 mg/L in shake flask experiments. It was also found that native E. coli metabolism could support 6dEB biosynthesis in the absence of exogenous propionate and the substrate provision pcc genes. Lastly, the effect of the ygfH deletion was tested in batch bioreactor cultures in which 6dEB titers improved from 206 to 527 mg/L.
Keywords: heterologous, polyketide, propionyl-CoA, methylmalonyl-CoA, metabolic engineering
Introduction
The heterologous biosynthesis of natural products has received increasing interest since the approach offers access to structurally complex and medicinally relevant small molecules. Natural product heterologous production requires successful introduction and expression of foreign genes encoding a new metabolic pathway. However, once accomplished, improving heterologous titers remains a challenge since it is often unknown how native and heterologous metabolism will interact. Many strategies have been adopted to improve natural product titers, including codon optimization (Strohl et al., 1991), auto-inducer utilization (Wang et al., 2007), promoter enhancement (Wenzel and Müller, 2005), and gene copy number control (Zhang et al., 2009). Because these efforts primarily focus upon the reconstitution of the heterologous pathway, results may be compromised by a metabolic bottleneck resulting from native metabolism limiting the availability of required substrates or cofactors. It is therefore essential to characterize native host metabolism in order to identify and remove undesired pathways and/or enhance those favorably supporting heterologous biosynthesis.
Many microorganisms have been employed for heterologous natural product production (Zhang et al., 2008). Among them, E. coli is perhaps the most popular and certainly one of the easiest to genetically manipulate. More importantly, the well-known genetic makeup of E. coli provides adequate information to identify native metabolic pathways related to heterologous natural product biosynthesis. The information and tools available for E. coli helped facilitate the production of the erythromycin macrocyclic core, a polyketide termed 6-deoxyerythronolide B (6dEB). E. coli 6dEB production was established by introducing genes encoding the deoxyerythronolide B synthase (DEBS) from Saccharopolyspora erythraea and a phosphopantetheinyl transferase gene (sfp) from B. subtilis to posttranslationally modify the DEBS complex (Pfeifer et al., 2001). To provide elevated levels of the required substrates propionyl-CoA and (2S)-methylmalonyl-CoA, a hybrid native-heterologous metabolic pathway was engineered composed of an E. coli propionyl-CoA synthetase (encoded by the prpE gene) and a propionyl-CoA carboxylase (PCC, encoded by Streptomyces coelicolor pccB and accA genes). The newly implemented metabolism included both chromosomal- and plasmid-borne gene expression under the control of inducible T7 promoters. A working knowledge of E. coli metabolism was also used to design heterologous 6dEB production. Except for prpE, E. coli’s propionate catabolism genes within the prp operon were disrupted during the chromosomal introduction of the sfp gene. This step was a deliberate effort to eliminate undesired propionyl-CoA consumption as a result of metabolism encoded by the prp operon. Despite this design, propionate to 6dEB conversion was typically only between 5% and 15%, implying the activity of native metabolism detracting from desired 6dEB biosynthesis (Lau et al., 2004; Pfeifer et al., 2002).
As a way to better characterize the impact of the heterologously introduced genes required for 6dEB biosynthesis, we previously analyzed the Sfp, PCC, and DEBS components through gene dosage modulation (Zhang et al., 2009). However, to date, similar efforts have not been made to probe the impact native E. coli metabolism has upon 6dEB production. By taking advantage of static E. coli metabolic maps, three enzymes (Sbm, YgfG, and YgfH) were identified as being capable of connecting native E. coli metabolism to heterologous 6dEB production, specifically through the propionyl-CoA and methylmalonyl-CoA substrates (Fig. 1; Haller et al., 2000; Froese et al., 2009). Based upon this relationship, we modified levels of Sbm, YgfG, and YgfH by either deleting or over-expressing the corresponding genes in an attempt to confirm the predicted impact on 6dEB biosynthesis. In addition, 6dEB biosynthesis was also observed and quantified in the absence of either heterologous PCC or exogenous propionate. In doing so, this study makes efforts to more thoroughly explore, characterize, and engineer native E. coli metabolism to support the production of a complex polyketide natural product.
Figure 1.
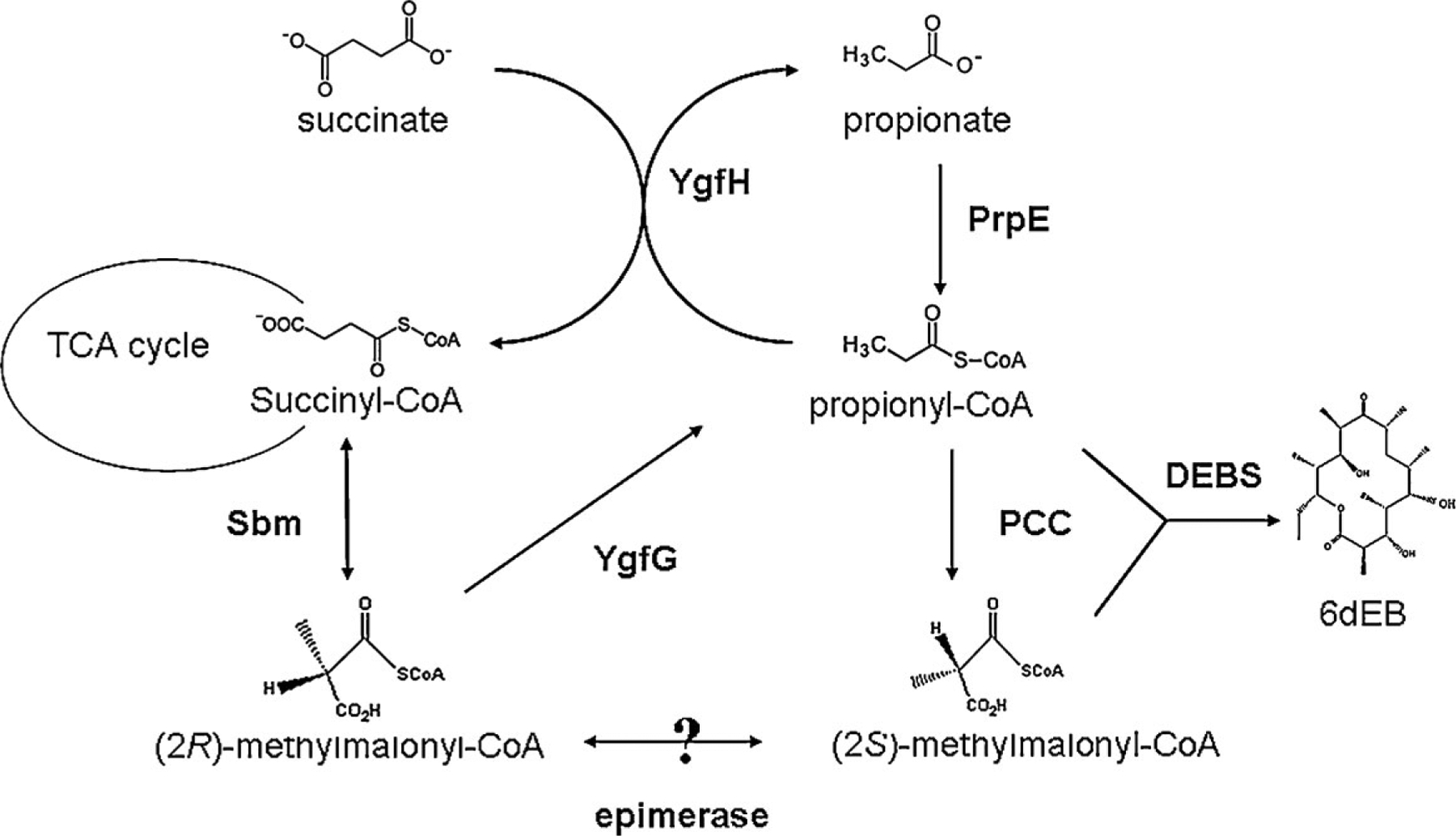
Metabolism connecting Sbm, YgfG, and YgfH activity to 6dEB heterologous production in E. coli. The map has been constructed from previous studies (Dayem et al., 2002; Haller et al., 2000; Marsden et al., 1994; Pfeifer et al., 2001).
Materials and Methods
Reagents and Chemicals
The reagents and chemicals used in this study were purchased from Fisher Scientific (Pittsburgh, PA) or Sigma (St. Louis, MO). All restriction enzymes and the Phusion High-Fidelity PCR Master Mix were purchased from New England Biolabs (Ipswich, MA). PCR primers were synthesized by Operon (Huntsville, AL).
Strains and Plasmid Construction
Restriction enzyme digestions, SDS–PAGE, and other standard molecular biology techniques were performed as described by Sambrook et al. (1989). E. coli BAP1, a derivative of BL21(DE3) (Studier and Moffatt, 1986), was constructed previously (Pfeifer et al., 2001). BAP1 sbm and ygfG deletions were conducted by directly disrupting the target genes by kanamycin marker insertion using λ-Red recombination (Datsenko and Wanner, 2000) followed by excision of the kanamycin resistance cassette through recombination of flanking flippase recognition targets mediated by the flippase recombination enzyme harbored by pCP20 (Cherepanov and Wackernagel, 1995). Inserting the kanamycin cassette to the ygfH location was accomplished through P1 transduction from E. coli ECK2916 (Baba et al., 2006) to BAP1. The kanamycin cassette was removed in the same way as described for the sbm and ygfG deletions.
For gene over-expression, sbm was PCR amplified using primers 5′-GCCATATGATGTCTAACGTGCAGGAGT-3′ and 5′-GCGAATTCTTAATCATGATGCTGGCTT-3′. The NdeI/EcoRI digested fragment was then inserted into pACYCDuet-1 digested with the same restriction enzymes. The ygfG gene was PCR amplified using primers 5′-GCCTGCAGATGTCTTATCAGTATGT-3′ and 5′-GCCTGCAGTTAATGACCAACGAAATT-3′ and the PCR product was digested by PstI and inserted into the PstI site of pACYCDuet-1. The ygfH gene was PCR amplified using primers 5′-GCCCATATGGAAACTCAGTGGACAAG-3′ and 5′-GCCCTCGAGTTAACCCAGCATCGAGCCG-3′. It has been reported that the 6x-hisitine-tagged YgfH lacked in vitro catalytic activity (Haller et al., 2000). Therefore, the NdeI/XhoI digested ygfH gene fragment was inserted into the second MCS of pACYCDuet-1 which does not produce a 6x-histidine-tag product. The sbm, ygfG, and ygfH insertions into pACYCDuet-1 led to plasmids pHZ1, pHZ2, and pHZ3, respectively. These constructs were verified by Sanger sequencing at the Tufts University Core Facility. Table I summarizes the E. coli strains and plasmids utilized in this study.
Table I.
Plasmid and E. coli strains used in this study.
| Description | Reference or source | |
|---|---|---|
| BAP1 | F−ompT hsdSB (rB-mB-) gal dcm (DE3) ΔprpRBCD::T7prom–sfp,T7prom–prpE | Pfeifer et al. (2001) |
| TB1 | BAP1 Δsbm | This study |
| TB2 | BAP1 ΔygfG | This study |
| TB3 | BAP1 ΔygfH | This study |
| pYW7317 | pET28 (Kanr) derivative, T7prom–DEBS1–T7term | Zhang et al. (2009) |
| pBP130 | pET21c (Carbr) derivative, T7prom–DEBS2–DEBS3–T7term | Pfeifer et al. (2001) |
| pBP144 | pET28 (Kanr) derivative, T7prom–pccB–rbs–accA1–T7prom–DEBS1–T7term | Pfeifer et al. (2001) |
| pHZ1 | pACYCDuet–1 (Cmr) derivative, T7prom–sbm–T7term | This study |
| pHZ2 | pACYCDuet–1 (Cmr) derivative, T7prom–ygfG–T7term | This study |
| pHZ3 | pACYCDuet–1 (Cmr) derivative, T7prom–ygfH–T7term | This study |
6dEB Production and Analysis
HPLC coupled to an evaporative light scattering detector (ELSD) was utilized to measure the 6dEB concentration in cultures of E. coli strains harboring the pcc genes (carried by pBP144), since these strains consistently produced easily detectable (>5 mg/L) concentrations of 6dEB. Shake flask cultures of E. coli BAP1 harboring pBP130, pBP144, and pHZ1, pHZ2, or pHZ3 were grown in 15mL production medium containing 100 mg/L carbenicillin, 50 mg/L kanamycin, 34 mg/L chloramphenicol, 100 μM IPTG, and 20 mM propionate at 22°C and 250 rpm for 5 days. One liter of production medium contained 5 g yeast extract, 10 g tryptone, 15 g glycerol, 10 g sodium chloride, 3 mL 50% (v/v) Antifoam B, 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesuffonic acid (HEPES) buffer, and was adjusted to pH 7.6 by NaOH before use. Since hydroxocobalamin is required for Sbm activity (Haller et al., 2000; Kellermeyer et al., 1964), the medium for BAP1 (pBP130, pBP144, pHZ1) also included 10 μM hydroxocobalamin (Dayem et al., 2002). After 5 days, 1 mL aliquots of the cultures were centrifuged for 10 min at 10,000 rpm and the supernatants were analyzed by an Agilent (Palo Alto, CA) 1100 series HPLC coupled with an Alltech (Deerfield, IL) 800 series ELSD. The cell pellet collected after centrifugation was used for gene expression SDS–PAGE analysis. Cell density was assessed after the 5-day culturing period by OD600 measurements. All experiments were repeated at least three separate times.
Due to the detection limitations of the HPLC–ELSD system, mass spectrometry was used for the measurement of 6dEB production levels in the absence of the pcc genes or propionate (Wang and Pfeifer, 2008; Zhang et al., 2009). BAP1, TB1, TB2, and TB3 containing pBP130 and pYW7317 (together carrying the DEBS1, 2, and 3 genes) were grown in 3 mL LB medium containing 100 mg/L carbenicillin, 50 mg/L kanamycin, 100 μM IPTG, and 20 mM propionate at 22°C and 250 rpm for 3 days. After culturing, 6dEB was extracted with 1.5 mL ethyl acetate and dried. The extract was dissolved in 50 μL methanol for analysis. To prepare a suitable calibration curve, the culture procedure was repeated using BAP1 without any plasmids and 6dEB standards added at different concentrations at the end of the culture period prior to ethyl acetate extraction. The standard samples were then used to prepare the calibration curve that correlated 6dEB MS peak intensity and 6dEB concentration. Erythromycin was used as an internal standard during the MS analysis to account for internal measurement drift of the instrument; erythromycin was added to the final methanol solution at a concentration of 5 mg/L. The calibration curve was generated for every sample set and experiments were repeated four separate times using a Thermo Electron Corporation (Waltham, MA) LTQ XL Linear Ion Trap Mass Spectrometer. For each experiment, there were at least three replicate samples for each E. coli strain.
For bioreactor experiments, 50 mL overnight flask cultures of either BAP1 (pBP130/pBP144) or TB3 (pBP130/pBP144) were used to seed a 3 L bioreactor (New Brunswick Scientific, BioFlo110, Edison, NJ) containing 1 L bioreactor-production medium (with the same composition as the previously introduced shake flask medium except that glycerol concentration was 45 g/L). Bioreactor conditions were maintained as follows: air flow rate, 3 L/min; pH value, 7.6; temperature, 22°C; and agitation, 500 rpm. Samples were withdrawn every 12 hr for OD600 measurements and 6dEB concentration analysis by HPLC–ELSD.
Results and Discussion
Deletion and Over-Expression of the sbm, ygfG, and ygfH Genes
The sbm, ygfG, and ygfH genes are found within the same operon. Individual deletions of the sbm, ygfG, and ygfH genes were conducted by λ-Red recombination or P1 transduction and the resulting strains were confirmed by PCR verification (data not shown). Over-expression of these genes together with the genes encoding for the PCC and DEBS enzymes were tested by SDS–PAGE analysis. As shown in Figure 2, the Sbm, YgfG, YgfH protein products were clearly visible after IPTG induction. PCC and DEBS were also expressed in E. coli as indicated by the arrows in Figure 2. Together, the gene deletion and over-expression experiments confirmed efforts to generate strains ranging in the levels of native enzymes potentially influencing the heterologous 6dEB pathway.
Figure 2.
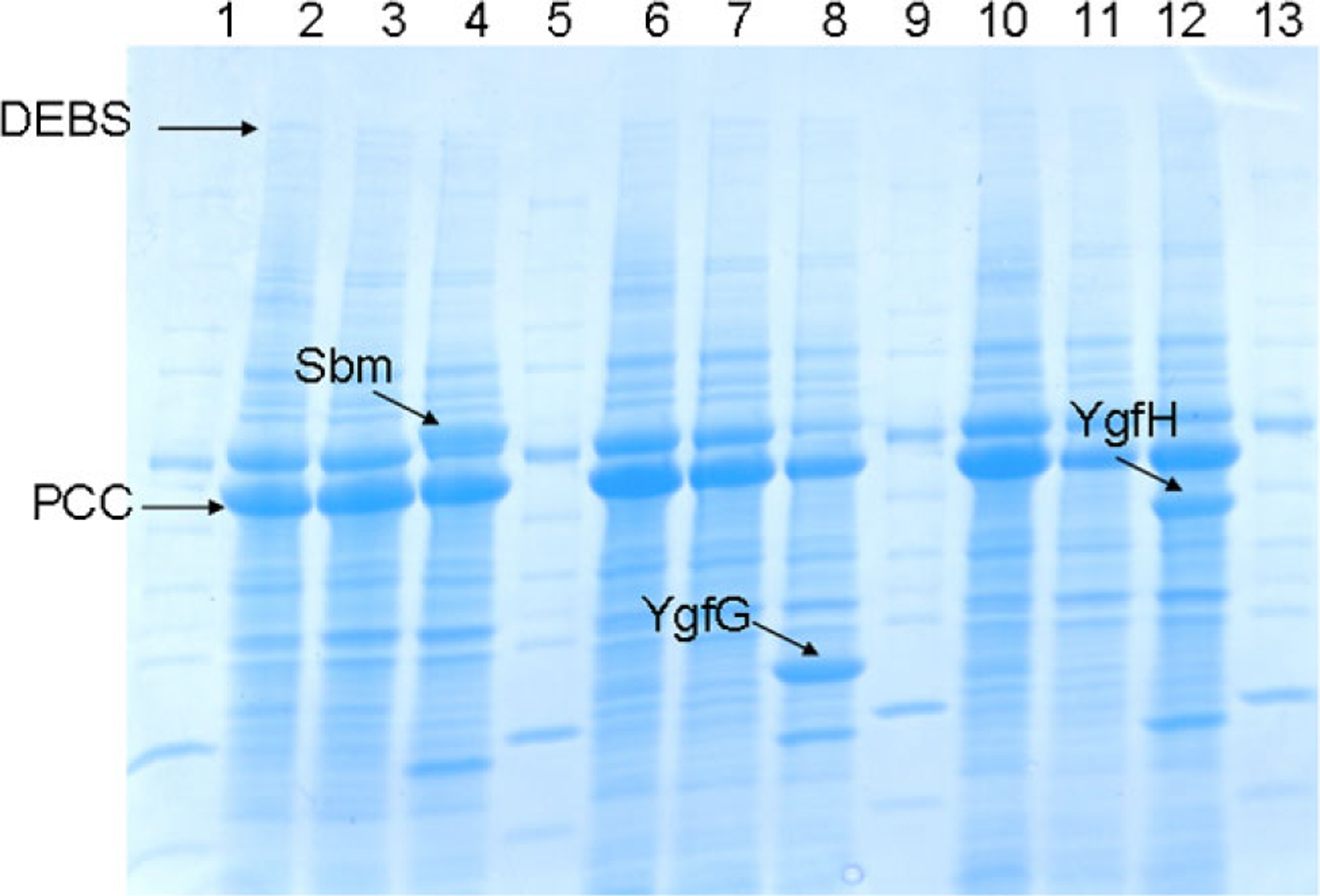
SDS–PAGE analysis of PCC, DEBS, sbm, ygfG, and ygfH over-expression. Lane 1: marker; lane 2: BAP1 (pBP130/pBP144); lane 3: TB1 (pBP130/pBP144); lane 4: BAP1 (pBP130/pBP144/pHZ1); lane 5: maker; lane 6: BAP1 (pBP130/pBP144); lane 7: TB2 (pBP130/pBP144); lane 8: BAP1 (pBP130/144/pHZ2); lane 9: marker; lane 10: BAP1 (pBP130/pBP144); lane 11: TB3 (pBP130/pBP144); lane 12: BAP1 (pBP130/144/ pHZ3); lane 13: marker.
sbm, ygfG, and ygfH Gene Deletion and Over-Expression and the Influence Upon 6dEB Production in the Presence of the pcc Genes and Propionate
Propionyl-CoA and (2S)-methylmalonyl-CoA are the two required substrates for production of 6dEB. These substrates link the heterologous 6dEB biosynthetic pathway and E. coli native metabolism involving the Sbm, YgfG, and YgfH enzymes, and it was unknown what effect the native pathway would have on heterologous 6dEB production. Therefore, the deletion and over-expression of the sbm, ygfG, and ygfH genes was performed to investigate the impact on 6dEB production. The effect was first characterized under conditions meant to maximize 6dEB levels, namely, when both the S. coelicolor pcc genes and exogenous propionate were utilized. The 6dEB production levels of different E. coli strains are compared in Figure 3. To take into account the effect of strain-associated cell growth, specific 6dEB production levels are also indicated in Figure 3. The results show that heterologous 6dEB biosynthesis is unaffected by either sbm deletion or over-expression. The deletion of ygfG also did not significantly impact 6dEB production levels as compared with the control BAP1 harboring pBP130/pBP144 (the fourth column in Fig. 3). However, the over-expression of ygfG led to a 4-fold decrease in 6dEB production. The deletion of ygfH increased 6dEB production levels by twofold (from 65 to 129 mg/L); whereas, ygfH over-expression did not have a significant effect on production. Specific 6dEB production levels showed the same trends as described above.
Figure 3.
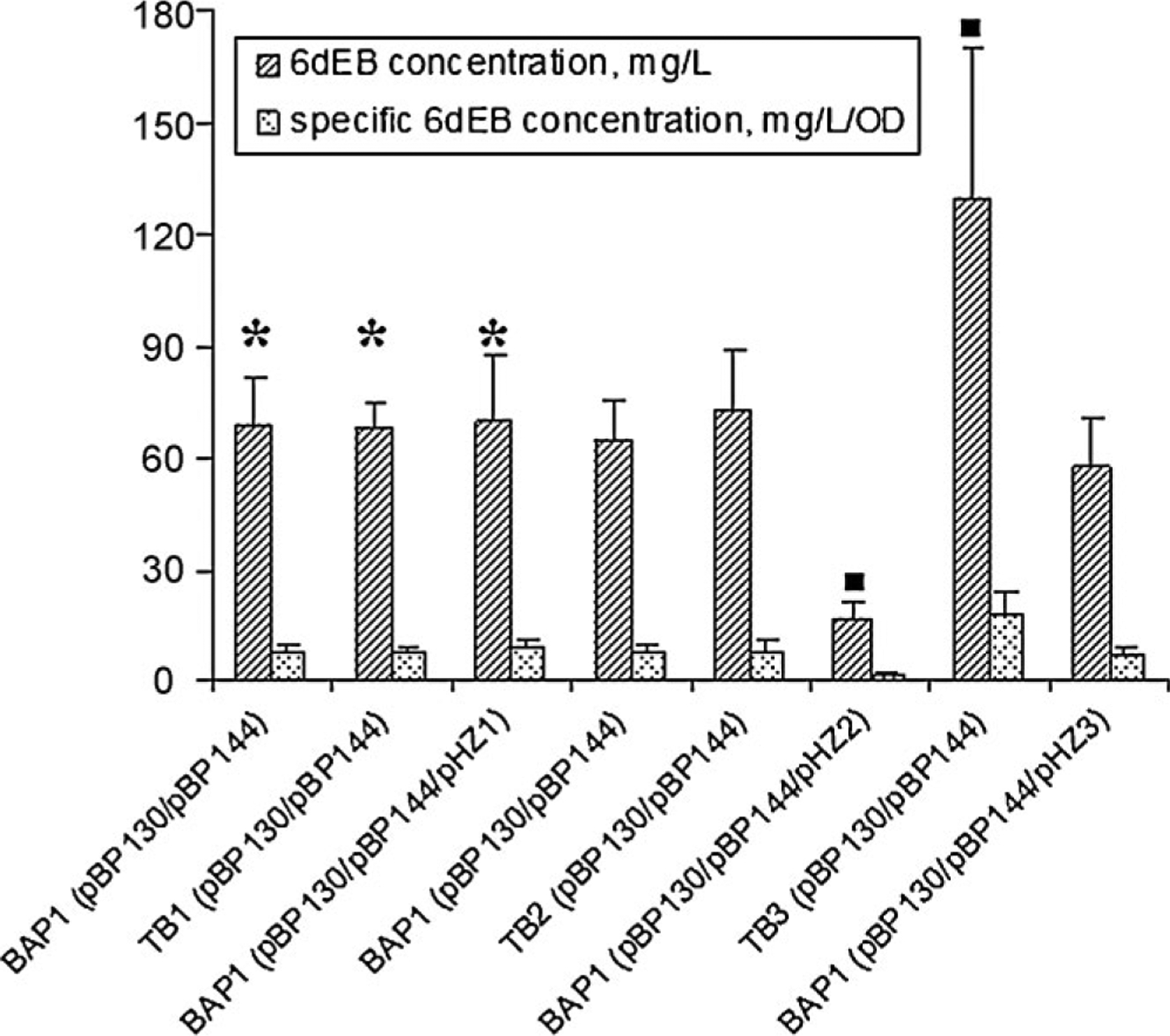
6dEB production comparisons in E. coli strains with sbm, ygfG, and ygfH genes deleted or over-expressed. Symbol *denotes that hydroxocobalamin was added Symbol ■ denotes that 6dEB production levels are significantly different (95% confidence) from the control BAP1 (pBP130/pBP144).
Neither the deletion nor over-expression of sbm had any observable effect on 6dEB production. This result suggests that there is no metabolic flux from (2S)-methylmalonyl-CoA to succinyl-CoA (or vice versa) that significantly influences the substrate availability for 6dEB production. It has previously been confirmed that the native E. coli methylmalonyl-CoA mutase could catalyze the (2R)-methylmalonyl-CoA to succinyl-CoA conversion (Dayem et al., 2002). Whereas, 6dEB biosynthesis requires the (2S)-methylmalonyl-CoA isomer (Marsden et al., 1994). Therefore, the Sbm enzyme’s lack of influence upon 6dEB production implies the absence of a suitable (2R)/(2S)-methylmalonyl-CoA epimerase. This possibility is supported by the absence of a clear epimerase homolog encoded within the E. coli genome by BLAST sequence analysis (data not shown). YgfG natively decarboxylates (2R)-methylmalonyl-CoA which normally would not influence 6dEB production. However, the reduction in 6dEB levels with ygfG over-expression illustrates a connection between YgfG activity and 6dEB biosynthesis, supporting a revised metabolic model that includes the YgfG-catalyzed decarboxylation of (2S)-methylmalonyl-CoA to propionyl-CoA. This impact is only apparent at elevated YgfG levels since the ΔygfG mutant had no influence upon 6dEB production. The twofold improvement of 6dEB titer through the deletion of ygfH provides further evidence that the propionyl-CoA:succinate CoA transferase encoded by ygfH produces succinyl-CoA from propionyl-CoA, reducing substrate for 6dEB production under these conditions. Over-expression of ygfH showed no statistically significant reduction in 6dEB, indicating that basal YgfH levels and those resulting from over-expression have a similar impact upon heterologous 6dEB biosynthesis.
6dEB Production in the Presence of Propionate But Without the pcc Genes
Although exogenous propionate and the S. coelicolor propionyl-CoA carboxylase (PCC) were both required for high-level 6dEB production, in the course of this study, it was also observed that E. coli metabolism can support heterologous 6dEB biosynthesis in the absence of exogenously fed propionate or expression of the S. coelicolor propionyl-CoA carboxylase genes.
Without the pcc genes, 6dEB production levels were greatly reduced (from 65 to 0.04 mg/L). The absence of the pcc genes eliminates the heterologously introduced conversion of propionyl-CoA to (2S)-methylmalonyl-CoA (Fig. 1). However, 6dEB could still be produced implying that native E. coli metabolism was capable of supplying (2S)-methylmalonyl-CoA. To confirm whether this provision of (2S)-methylmalonyl-CoA is relevant to the pathway encompassing Sbm, YgfG, and YgfH, the corresponding BAP1 deletion mutants were tested for 6dEB production in the absence of the pcc genes. As shown in Figure 4, the deletions of sbm, ygfG, and ygfH did not change the 6dEB production levels. The result suggests the availability of alternative native pathways (unrelated to Sbm, YgfG, and YgfH) capable of providing (2S)-methylmalonyl-CoA for 6dEB biosynthesis.
Figure 4.
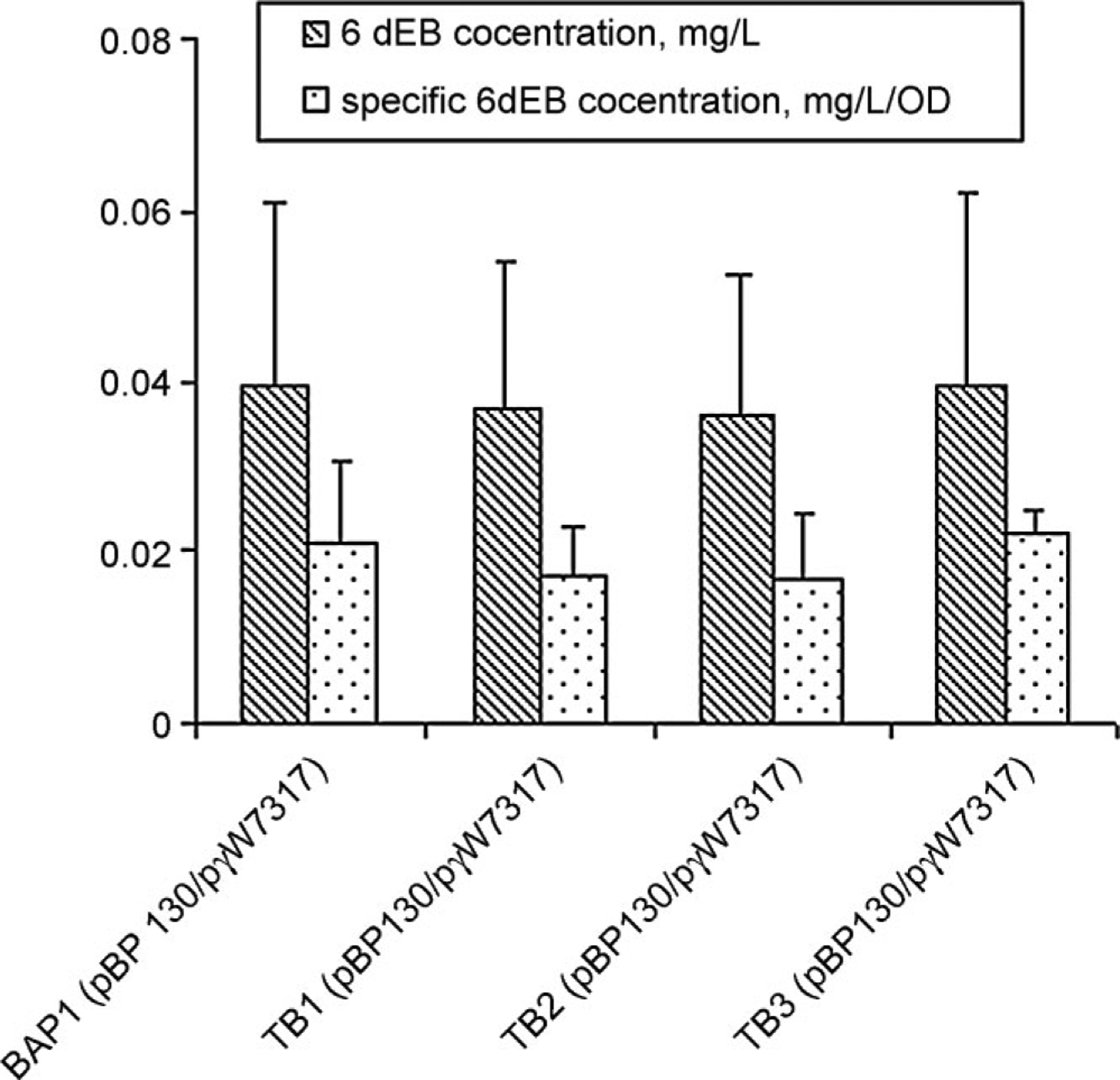
6dEB production levels in BAP1 and BAP1 mutant strains in the absence of the S. coelicolor pcc genes.
The observation of 6dEB production in the absence of the PCC enzyme (and the similar situation in which production is observed without propionate supplementation) is of interest because it is unknown to what extent E. coli native metabolism may be able to support heterologous 6dEB biosynthesis. The S. coelicolor PCC is responsible for the production of (2S)-methylmalonyl-CoA in the engineered strain of E. coli used to generate 6dEB. It was found that an E. coli BAP1 strain harboring DEBS but no PCC complex could still produce low levels of 6dEB in the presence of propionate. The significant decrease in production level is consistent with previous results that indicate the relative importance of the heterologous PCC in the E. coli 6dEB production scheme (Zhang et al., 2009).
The Sbm methylmalonyl-CoA mutase is involved in methylmalonyl-CoA metabolism. However, 6dEB production was unchanged after deletion of the sbm, suggesting that the required (2S)-methylmalonyl-CoA was not produced through the activity of Sbm. The data then support other yet-unknown native E. coli pathways capable of directing the biosynthesis of the (2S)-methylmalonyl-CoA substrate for 6dEB production. These pathways, however, are dependent upon exogenous propionate since no 6dEB was produced in the absence of both the pcc genes and propionate (Fig. 5). In other organisms, it has been observed that native acetyl-CoA carboxylase enzymes are also capable of accepting propionyl-CoA as a substrate for methylmalonyl-CoA formation (Rodriguez and Gramajo, 1999). However, it is unknown whether the E. coli acetyl-CoA carboxylase has such activity. In the same study demonstrating the succinyl-CoA to (2R)-methylmalonyl-CoA activity of the native E. coli methylmalonyl-CoA mutase, co-expression of a P. shermanii methylmalonyl-CoA mutase and epimerase in E. coli allowed (2S)-methylmalonyl formation and production of 6dEB at approximately 1 mg/L (Dayem et al., 2002). As mentioned, there is no evidence for methylmalonyl-CoA epimerase activity in E. coli. This then helps explain the lack of impact upon 6dEB levels caused by deleting the sbm gene in the absence of the pcc genes since E. coli Sbm is only known to accept or produce (2R)-methylmalonyl-CoA.
Figure 5.
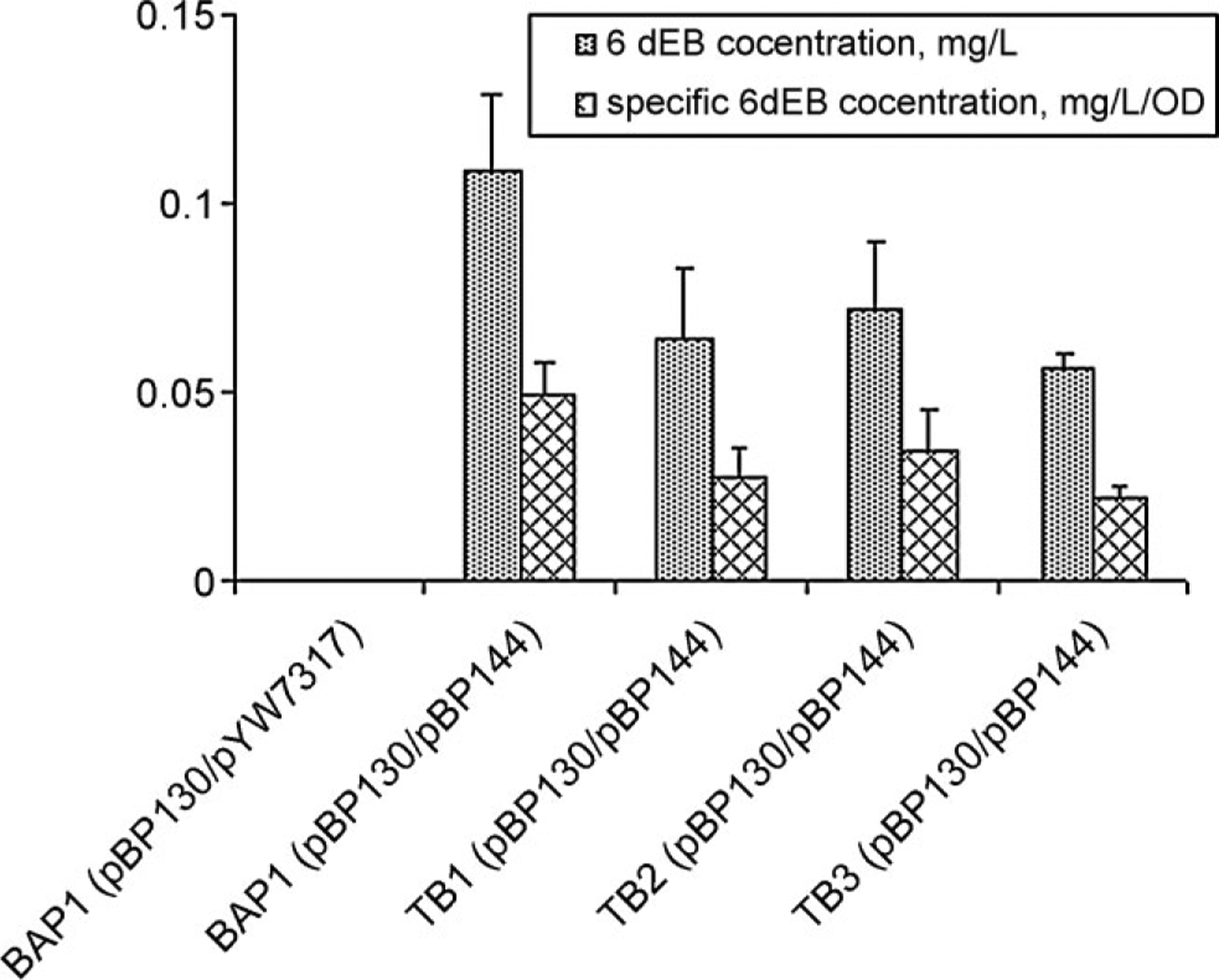
6dEB production levels in BAP1 and BAP1 mutant strains in the absence of propionate.
As shown in Figure 1, the E. coli methylmalonyl-CoA decarboxylase encoded by ygfG removes the carboxyl group of methylmalonyl-CoA to make propionyl-CoA. However, 6dEB production was unchanged after ygfG deletion, consistent with what was observed in the presence of both the pcc genes and propionate. The E. coli YgfH is responsible for the CoA moiety transfer between propionyl-CoA and succinyl-CoA. Without PCC, the ygfH deletion did not impact 6dEB production, suggesting that this enzyme primarily affects propionyl-CoA levels which appear to be non-limiting.
6dEB Production in the Presence of the pcc Genes But Without Propionate
Propionate is the substrate required for the engineered production of propionyl-CoA and (2S)-methylmalonyl-CoA. In the absence of exogenous propionate, 6dEB production was greatly reduced. The 6dEB production level of different E. coli strains was compared in Figure 5. When neither propionate nor pcc genes were present (using strain BAP1 harboring pBP130 and pYW7317), no 6dEB production was detected. In the presence of pcc genes but no propionate, E. coli BAP1 containing the DEBS genes produced 0.11 mg/L 6dEB. When this condition was tested with the strains containing the sbm, ygfG, and ygfH deletions, a statistically significant decrease in final 6dEB production was observed for each strain. These findings indicate that without exogenous propionate, E. coli native metabolism is able to synthesize propionyl-CoA which is later converted to (2S)-methylmalonyl-CoA (facilitated by the PCC enzyme) for 6dEB production. Furthermore, the observation of propionyl-CoA biosynthesis without exogenously fed propionate may be partly attributed to the Sbm, YgfG, and YgfH pathway since removal of the encoding genes decreased the heterologous 6dEB production levels.
Exogenous propionate was believed to be required for heterologous 6dEB production in E. coli. It was found in this study that E. coli supports low-level 6dEB biosynthesis without the addition of exogenous propionate. The results indicate that E. coli can synthesize propionyl-CoA from native metabolic pathways. Data in Figure 5 support a metabolic model where succinyl-CoA can be converted to (2R)-methylmalonyl-CoA by Sbm and then to propionyl-CoA by YgfG, since the deletion of sbm and ygfG led to a decrease in 6dEB production. Similar results were observed previously where an over-expressed E. coli Sbm-YgfG pathway was used to produce propionyl-CoA in Salmonella enterica serovar Typhimurium (Aldor et al., 2002). In the current study, we hypothesize propionyl-CoA is now limiting and that the ability of Sbm and YgfG to provide propionyl-CoA gains importance during 6dEB formation because of no exogenously derived propionyl-CoA. The deletion of ygfH caused a similar decrease in 6dEB levels, which suggests that propionyl-CoA may be directly produced from succinyl-CoA by YgfH, in contrast to the model presented in Figure 1.
Bioreactor Production of 6dEB in the Absence of ygfH and Presence of the pcc Genes and Propionate
To confirm the effect of the ygfH deletion, bioreactor production cultures were used to compare BAP1 (pBP130/pBP144) and TB3 (pBP130/pBP144). As shown in Figure 6a, the growth curves of these two strains were similar. The production of 6dEB was observed after 1.5 day for BAP1 (pBP130/pBP144) and 1 day for TB3 (pBP130/pBP144). Final 6dEB titers reached approximately 206 mg/L in BAP1 (pBP130/pBP144) after 5.5 days; whereas, TB3 (pBP130/pBP144) produced approximately 527 mg/L 6dEB after the same time. In this scaled-up comparison, the 6dEB production level was increased by approximately 2.5-fold, slightly beyond the difference observed when using shake flask cultures. The scaled and well-controlled environment provided by the bioreactor allowed significantly improved 6dEB levels and helped to emphasize the positive impact provided by the ygfH deletion. This study shows how straightforward metabolic engineering techniques, coupled to modest process scale-up, can be used to significantly increase titers in complex compound biosynthesis even without extension to fed-batch bioreactor configurations previously used to boost titers to >1 g/L (Lau et al., 2004; Tsuruta et al., 2009).
Figure 6.
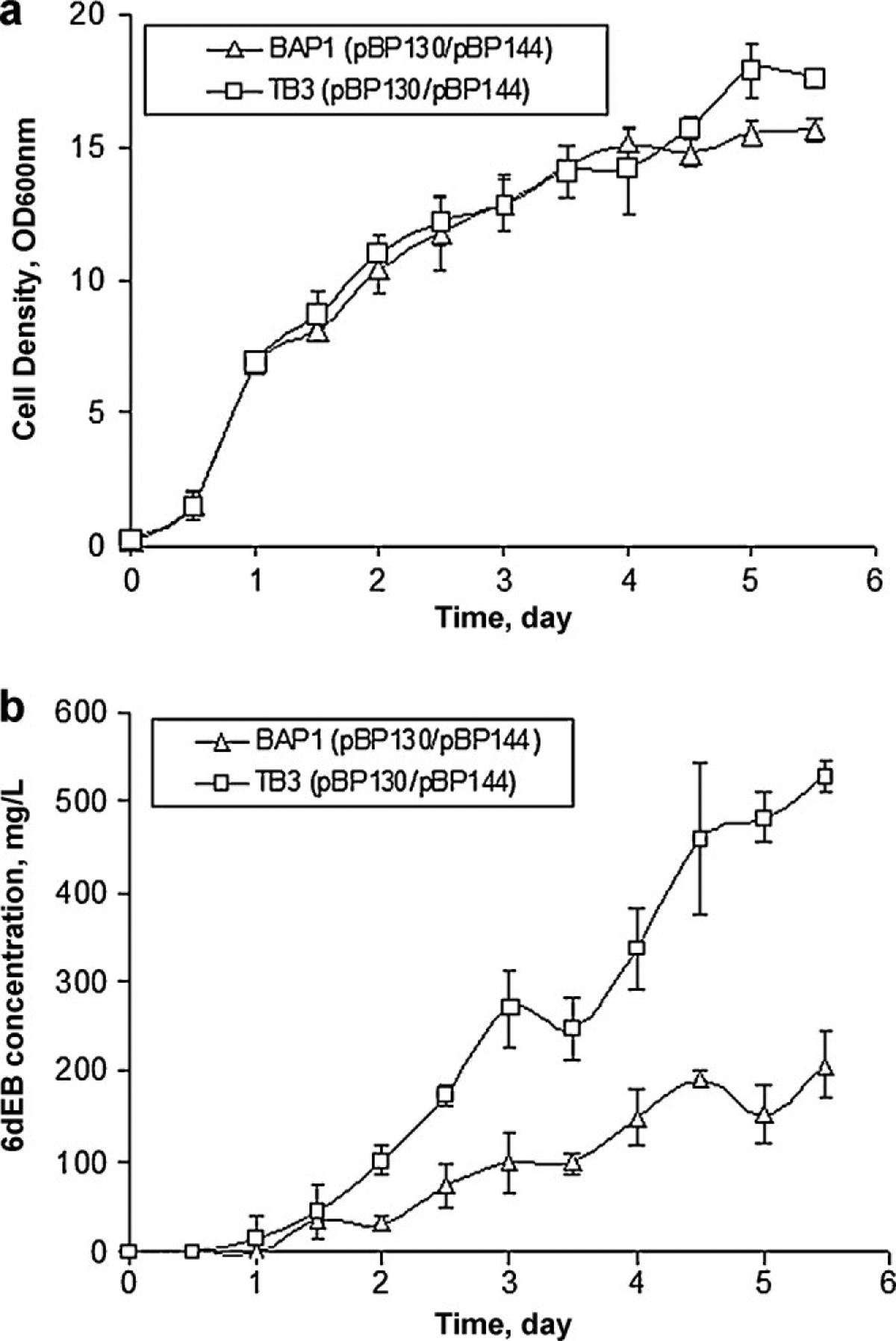
E. coli batch bioreactor comparison. a: Growth curves of BAP1 (pBP130/pBP144) and TB3 (pBP130/pBP144). b: 6dEB production levels of BAP1 (pBP130/pBP144) and TB3 (pBP130/pBP144).
Acknowledgments
The authors acknowledge support from NSF and NIH (0712019 & AI74224). The authors are also grateful to the National Institute of Genetics, Japan for providing E. coli strains used in this study.
References
- Aldor IS, Kim SW, Prather KL, Keasling JD. 2002. Metabolic engineering of a novel propionate-independent pathway for the production of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) in recombinant Salmonella enterica serovar typhimurium. Appl Environ Microbiol 68:3848– 3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko K, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol 2:0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–9–14. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayem LC, Carney JR, Santi DV. 2002. Metabolic engineering of a methylmalonyl-CoA mutase-epimerase pathway for complex polyketide biosynthesis in Escherichia coli. Biochemistry 41:5193–5201. [DOI] [PubMed] [Google Scholar]
- Froese DS, Dobson CM, White AP, Wu X, Padovani D, Banerjee R, Haller T, Gerlt JA, Surette MG, Gravel RA. 2009. Sleeping beauty mutase (sbm) is expressed and interacts with ygfd in Escherichia coli. Microbiol Res 164:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller T, Buckel T, Rétey J, Gerlt JA. 2000. Discovering new enzymes and metabolic pathways: Conversion of succinate to propionate by Escherichia coli. Biochemistry 39:4622–4629. [DOI] [PubMed] [Google Scholar]
- Kellermeyer RW, Allen SHG, Stjernholm R, Wood HG. 1964. Methylmalonyl isomerase. IV. Purification and properties of the enzyme from propionibacteria. J Biol Chem 239:2562–2569. [PubMed] [Google Scholar]
- Lau J, Tran C, Licari P, Galazzo J. 2004. Development of a high cell-density fed-batch bioprocess for the heterologous production of 6-deoxyerythronolide B in Escherichia coli. J Biotechnol 110:95–103. [DOI] [PubMed] [Google Scholar]
- Marsden AF, Caffrey P, Aparicio JF, Loughran MS, Staunton J, Leadlay PF. 1994. Stereospecific acyl transfers on the erythromycin-producing polyketide synthase. Science 263:378–380. [DOI] [PubMed] [Google Scholar]
- Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. 2001. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291:1790–1792. [DOI] [PubMed] [Google Scholar]
- Pfeifer BA, Hu Z, Licari P, Khosla C. 2002. Process and metabolic strategies for improved production of Escherichia coli-derived 6-deoxyerythronolide B. Appl Environ Microbiol 68:3287–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez E, Gramajo H. 1999. Genetic and biochemical characterization of the a and b components of a propionyl-CoA carboxylase complex of Streptomyces coelicolor A3(2). Microbiology 145:3109–3119. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular Cloning : A laboratory manual. New York : Cold Spring Harbor Laboratory. [Google Scholar]
- Strohl WR, Bartel PL, Li Y, Connors NC, Woodman RH. 1991. Expression of polyketide biosynthesis and regulatory genes in heterologous streptomycetes. J Ind Microbiol 7:163–174. [DOI] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189: 113–130. [DOI] [PubMed] [Google Scholar]
- Tsuruta H, Paddon CJ, Eng D, Lenihan JR, Horning T, Anthony LC, Regentin R, Keasling JD, Renninger NS, Newman JD. 2009. High-level production of amorpha-4,11-diene, a precursor of the antimalarial agent artemisinin, in Escherichia coli. PLoS ONE 4(2):e4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Pfeifer BA. 2008. 6-Deoxyerythronolide B production through chromosomal localization of the deoxyerythronolide B synthase genes in E. coli. Metab Eng 10:33–38. [DOI] [PubMed] [Google Scholar]
- Wang Y, Boghigian BA, Pfeifer BA. 2007. Improving heterologous polyketide production in Escherichia coli by overexpression of an S-adenosylmethionine synthetase gene. Appl Microbiol Biotechnol 77:367– 373. [DOI] [PubMed] [Google Scholar]
- Wenzel SC, Müller R. 2005. Recent developments towards the heterologous expression of complex bacterial natural product biosynthetic pathways. Curr Opin Biotechnol 16:594–606. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wang Y, Pfeifer BA. 2008. Bacterial hosts for natural product production. Mol Pharm 5:212–225. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wang Y, Boghigian BA, Pfeifer BA. 2009. Probing the heterologous metabolism supporting 6-deoxyerythronolide B biosynthesis in Escherichia coli. Microb Biotechnol 2:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]