

Abstract
Genome-wide association studies (GWAS) have markedly advanced our understanding of the genetics of Parkinson’s disease (PD), but they currently do not account for the full heritability of PD. In many cases it is difficult to unambiguously identify a specific gene within each locus because GWAS does not provide functional information on the identified candidate loci. Here we present an integrative approach that combines transcriptome-wide association study (TWAS) with high-throughput neuronal dysfunction analyses in Drosophila to discover and validate candidate PD genes. We identified 160 candidate genes whose misexpression is associated with PD risk via TWAS. Candidates were validated using orthogonal in silico methods and found to be functionally related to PD-associated pathways (i.e. endolysosome). We then mimicked these TWAS-predicted transcriptomic alterations in a Drosophila PD model and discovered that 50 candidates can modulate α-Synuclein(α-Syn)-induced neurodegeneration, allowing us to nominate new genes in previously known PD loci. We also uncovered additional novel PD candidate genes within GWAS suggestive loci (e.g. TTC19, ADORA2B, LZTS3, NRBP1, HN1L), which are also supported by clinical and functional evidence. These findings deepen our understanding of PD, and support applying our integrative approach to other complex trait disorders.
Introduction
Genome-wide association studies (GWAS) have discovered dozens of genetic susceptibility loci associated with late-onset Parkinson disease (PD) and have revealed potentially causal genes (1,2). Despite remarkable advances in identifying genetic factors contributing to PD, it is estimated that current PD GWAS data only account for 16–36% of PD heritability (2). Thus, revealing the missing heritability would require much larger GWAS meta-analysis and approaches to reveal the contributions of rare variants (2,3). An alternative strategy is to use orthogonal methods that would reveal causal genes among suggestive GWAS signals and thus circumvent the lack of power in current GWAS datasets. An additional limitation of GWAS data is that it does not reveal the mechanisms through which variants impart disease risk (4–6). One way to minimize this gap is to integrate GWAS risk loci and quantitative trait loci to delineate causal genes and potential mechanisms by which disease risk is mediated.
GWAS risk variants often locate in regulatory regions and can ultimately influence the trait by affecting gene expression (7,8). Several publicly available resources have facilitated the study of gene expression across multiple human tissues, for instance the Genotype-Tissue Expression (GTEx) project, which provides genotypes and gene expression data (9). Computational approaches have been developed to integrate GWAS variant data with expression quantitative trait loci (eQTLs) to probe the link from single-nucleotide polymorphism (SNP) to gene expression to complex traits. Previous studies have deployed such methodologies to ascribe putative risk-associated genes to GWAS loci for PD, leading to the identification of many novel susceptibility genes for PD (10–13). However, in most cases, we do not know whether these genes modulate disease outcome due to the lack of systematic experimental validation.
In this study, we present an integrative approach that combines the computational analyses of genes increasing risk for PD and neuronal dysfunction assays in vivo. We used a transcriptome-wide association study (TWAS) method to prioritize genes whose misexpression correlates with PD and leveraged clinical data to further characterize them. Using high-throughput functional assays that assess neuronal dysfunction both quantitatively and longitudinally, we nominated 86 candidates for experimental validation in a well-established PD Drosophila model (14,15). These assays revealed that manipulating 50 candidates can ameliorate or aggravate neuronal dysfunction caused by human α-Synuclein(α-Syn). Remarkably, we found 27 candidates had congruent in silico (TWAS) and in vivo effects on PD. Interestingly, some of these candidates map to GWAS suggestive loci, while others were not the leading gene (defined as the closest gene to the corresponding significant variant) under a recognized PD GWAS locus. Together, these results show that this multilayered approach is a powerful method to nominate novel PD risk genes and systematically validate them in vivo, and suggest that it can be applied successfully to other GWAS datasets in which significant signals only partially account for the genetic heritability.
Results
Overview
Our multilayered approach first nominated 160 potential PD risk-associated genes through multi-tissue TWAS (Fig. 1A). To assess the robustness of those computationally predicted candidate PD genes, we used complementary methods including colocalization analysis and fine-mapping of TWAS association to corroborate their association to increased PD. We then investigated their potential to inform biomarkers for PD risk and connection to PD-related clinical traits, which further support many of our candidate genes’ relevance to the disease (Fig. 1B). Finally, using a high-throughput platform, we tested our candidate PD genes in a well-established α-Syn Drosophila model and found 50 out of 160 can modulate neurodegeneration in vivo (Fig. 1C).
Figure 1.

Integrative approach combining TWAS and in vivo functional assays to identify PD genes. (A) TWAS was used to identify potential PD risk-associated genes using multi-tissue eQTL weights as predictive models followed by orthogonal in silico validation (Focus, Coloc, etc.). (B) Further assessment of candidate genes associated to PD pathology using blood transcriptome and clinical data from PD patients and controls. (C) Experimental validation of PD candidate genes using high-throughput neuronal dysfunction assay platform in Drosophila PD model, complemented with longitudinal differentially expressed gene analysis and PPI network integration.
TWAS integrate PD GWAS and gene expression data across multiple tissues
Since a limitation of GWAS is that it only pinpoints risk loci without identifying genes, we used TWAS to integrate GWAS and gene expression to link gene expression alterations with increased PD risk. We performed TWAS using the PD GWAS summary statistics (without 23andMe) from Nalls et al. (33 674 cases (18 618 proxy cases from UK Biobank) and 449 056 controls) and GTEx multi-tissue expression reference weights (see Data availability) to identify cis-regulated genes that associate with PD risk (see Materials and Methods). TWAS integrate the information in a reference panel, where genotype and gene expression from the same individuals are available, and the PD GWAS summary statistics to impute the gene expression onto GWAS cohorts and model the association between genes and the trait of interest (see Materials and Methods). We used the FUSION software to fit predictive models and compute TWAS association statistics (16). Using GTEx multi-tissue reference, 235 583 tissue-specific models were used for TWAS prediction. We observed nearly 20–30% improvement in prediction accuracy from Elastic net (ENET) and least absolute shrinkage and selection operator (LASSO) in comparison to other predictive models (Supplementary Material, Fig. S1). We found that the average prediction accuracy (normalized R2) across all predictive models is 56%, which is consistent with previous results and indicate a large amount of cis-regulated expression can be accounted for by these predictive models (10,16,17).
To identify candidate genes with significant transcriptome-wide PD associations, we performed multiple hypothesis testing using the Bonferroni correction and selected the nominal TWAS P-value significance at 5 × 10−5 to reduce the false positive rate in various tissues with distinct sizes. Based on these TWAS models, we found 160 genes whose imputed expression, across multiple tissues, significantly correlated with increased PD risk (Fig. 2A; Supplementary Material, Table S1). Among these genes 118 are protein coding and include known PD genes such as SNCA, LRRK2, CTSB, CRHR1, DYRK1A and TMEM175. In addition to known PD genes, this analysis also revealed other candidate genes (including NSF, ARL17B, NUPL2, P2RY12, ASH1L, PRSS53, STX4, BCKDK, FMNL1) whose expression associated with PD but are not the closest gene to the corresponding significant variant (see pathway analysis in Discussion). Furthermore, as we adopted the suggestive GWAS threshold, our TWAS analysis highlighted genes with only suggestive evidence of GWAS association. These genes include TTC19, FAHD1, ADORA2B, PNLIPRP3 and HN1L. These results are consistent with previous findings that unearthed disease causal genes with suggestive significant GWAS associations (18–20).
Figure 2.

Combination of TWAS with orthogonal gene fine mapping approaches reveals a set of consistent PD candidate genes. (A) Manhattan plot showing the TWAS PD hits. Chart indicates the genomic position of each gene on the X-axis versus − log10(P-value) on the Y axis. The 160 genes shown as triangles have both a TWAS and GWAS P-value of <5e-5 (dotted line). Empty circles represent genes with a TWAS P < 5e-5 but a GWAS P > 5e-5 (not considered as TWAS candidates in this study). Gene names are only displayed for the most significant ones in each locus. (B) Bar plot displaying 108 TWAS candidate genes that are independent following joint conditional testing. Each bar shows the –log(P) of the probability of conditioning GWAS association on the corresponding gene. Inset shows an example plot of the PRSS36 locus, where PD GWAS signals before conditioning (gray) and after removing the effect of PRSS36 expression (black) are shown. This analysis shows that the association is explained by PRSS36 (with asterisk), and RP11-388 M20.9 (with triangle) that is marginally TWAS significant. (C) Ternary plot showing the results of the colocalization test (Coloc) for the TWAS PD candidates. A total of 81 TWAS candidate genes whose GWAS signals colocalized with cis-eQTL signals are shown in black. Gray dots represent lack of colocalization. The standard criteria of PP3 ± PP4 ≥ 0.8 and PP4/PP3 ≥ 2 were selected as colocalized variants on a candidate gene (black dot) (briefly: PP0, no causal variant, PP1, causal variant for PD GWAS only, PP2, causal variant for eQTL only, PP3, two distinct causal variants in two different genes, PP4- one common causal variant). (D) Upset plot summarizing all 160 TWAS candidate genes supported by three complimentary in silico methods (Joint/conditional test, Coloc, FOCUS). The number of candidates whose imputation models were brain-derived is also indicated. A total of 37 genes are supported by all three orthogonal in silico methods.
Orthogonal computational methods further corroborate TWAS candidate genes expressed in relevant brain regions
To improve the robustness of the 160 TWAS-predicted PD candidate genes, we used three orthogonal computational approaches to generate corroborating evidence for their relevance to PD. Since TWAS can identify multiple disease-associated genes within the same locus, we sought to identify which ones are conditionally independent. Applying joint and conditional tests within their corresponding 1 Mb regions, we found that 108 genes out of 160 were significantly associated with PD even when analyzed jointly with genes in their corresponding loci (Fig. 2B; Supplementary Material, Fig. S2 and Table S2). Next, we further assessed if both PD GWAS signals and eQTL signals were driven by the same causal variants. We used the FUSION tool to compute the probability of colocalization between the PD association signal at these TWAS loci and eQTLs. We found that the GWAS and eQTL signals colocalized for 81 genes of the 160 TWAS candidates (Fig. 2C; Supplementary Material, Fig. S3 and Table S1) (16,21). Lastly, to prioritize putatively causal genes based on TWAS gene-trait association signals, we applied FOCUS (fine-mapping of causal gene sets) to compute the refined TWAS statistics by taking into account the correlation of linkage disequilibrium (LD) and the SNP weights used in prediction into account (see Materials and Methods)(22). We computed the posterior inclusion probability (PIP) for genes at each TWAS region by taking cumulative sums of PIP until 90%. This generated a list of 56 genes from 160 TWAS-prioritized candidates, many of which fine-mapped at a handful of shared GWAS loci (Supplementary Material, Table S3) due to highly local patterns of LD. A total of 37 TWAS candidates were significant across all three methods. Interestingly, the expression of 35 of those 37 candidates was imputed via TWAS models from brain-derived tissues which showed significant association with PD (using LD score regression for specifically expressed genes, LDSC-SEG) (Fig. 2D; Supplementary Material, Fig. S4) (23). Therefore, the overrepresentation of these high confidence genes in predictive models from brain-derived tissues implies a strong connection to PD.
Candidate genes are altered in PD transcriptome and correlated with PD traits
TWAS implies that the change in expression of the 160 candidates is correlated with PD; thus, we checked if their expression were altered in PD patient transcriptome. We evaluated their expression in whole-blood transcriptome of the Parkinson’s Progression Marker Initiative (PPMI) cohort (we used blood data because there is no adequate available data from PD brain) and found that 35 PD candidate genes nominated by our approach were significantly dysregulated (adjusted P value < 0.05; see Materials and Methods). This suggests that these 35 genes may potentially constitute peripheral blood biomarkers to inform PD risk (Fig. 3A; Supplementary Material, Table S5) (24).
Figure 3.

Integration with transcriptomic and clinical data from PD patients reveals TWAS candidates linked to PD pathology. (A) Volcano plot showing genes dysregulated in PD blood transcriptome from PPMI (adjusted P-value cutoff at 0.05—black). There are 35 TWAS candidates that were significantly dysregulated in patients (names indicted in the chart). (B) Result of applying a linear mixed model to correlate genes expression with clinical traits (see Materials and Methods). Among the 35 TWAS genes dysregulated in patients, 18 of them shown correspond to genes whose differences in expression are correlated with 5% or more of the summed variance of the indicated clinical traits among defined PD cases and controls.
To further establish the link of our TWAS candidate genes to PD pathology, we measured the association of their expression levels in brains of 93 individuals with PD traits studied in the ROSMAP (Religious Order Study and Memory and Aging Project) cohort meeting pathological criteria for a PD diagnosis. A total of 72 controls were chosen as participants lacking a pathological diagnosis of PD or Alzheimer’s disease (see Materials and Methods; Supplementary Material, Table S6) (25,26). The association analysis was performed using a linear mixed model (R package VariancePartition), which computes the proportion of variance explained by gene expression association with five clinical traits (tremor, rigidity, gait, bradykinesia, motor) related to PD (17,27). Using this model, we found that 55 of the TWAS genes explained around 5–20% of total variation in five PD-associated traits in those selected samples (individuals with or without pathological diagnosis of PD) after accounting for biological and technical covariates (Supplementary Material, Fig. S5). Interestingly, 17 of these 55 candidates were also dysregulated in PD blood transcriptome (Fig. 3B), further strengthening their association to PD risk as these potential biomarkers might also link to clinical traits of the disease.
Neuronal dysfunction assays confirm the potential of TWAS candidate genes to modify neurodegeneration in vivo
As detailed above, 118 TWAS protein coding candidates are correlated with increased risk in PD, and we also note that many of them are dysregulated in PD blood transcriptome or their misexpression can be associated with PD-related motor defects. To further validate the potential role played by the TWAS candidates in PD-associated neurodegeneration, we assessed whether these candidate genes could modulate α-Syn induced neurodegeneration in vivo. We used a well-established automated assay that quantitatively assesses neuronal dysfunction using behavioral outputs based on the Drosophila negative geotaxis response (28–30). For this study, we used a well-characterized PD Drosophila model that expresses human wild-type α-Syn in all neurons (see Materials and Methods) (14,15). This PD model manifests late onset, progressive behavioral impairments, thereby allowing assessment of neuronal dysfunction over time as the animals age. In this model, we tested loss of function and/or overexpression alleles if available for the Drosophila homologs of the TWAS candidates (specific genotypes used for assays are summarized in Supplementary Material, Table S7).
In total, 85 genes had Drosophila homologs and available strains. We found that modulating the expression levels of 50 of these genes could ameliorate or aggravate α-Syn-induced neuronal dysfunction (Fig. 4A; Supplementary Material, Fig. S6). Our results showed that 31 genes ameliorated the α-Syn-induced dysfunction (29 when knocked down and 2 when overexpressed) while 19 enhanced the α-Syn-induced dysfunction when knocked down. Among these genes, the knockdown of Drosophila homologs of BCKDK, ELOVL7, FMNL1, PRSS36, MCCC1 and STX1B ameliorated the neuronal dysfunction in α-Syn animals, while knockdown of Drosophila homologs of IDUA, NSF, MAPT, TTC19 and LZTS3 exacerbated neuronal dysfunction in these models. Therefore, these gene perturbation data indicate that these genes may play roles in mechanisms underlying α-Syn-induced PD pathogenesis. It is noteworthy that our candidate genes have a significant enrichment of PD modifiers (hit rate: 58.8%; ~P < 0.00001, Fisher), thus strongly supporting the relevance of these targets in PD pathology. As expected, these genes include previously identified PD risk genes, but importantly they also include other genes not previously associated to PD and highlight their potential to modulate α-Syn-induced pathology. Note that PD is a complex condition that may be linked to genetic risk factors unrelated to α-Syn. However, we used a α-Syn model, which may lead to an overrepresentation of PD modifiers that modulate α-Syn-induced neurodegeneration while overlooking other risk factors.
Figure 4.

High-throughput behavioral assay reveals TWAS candidates that can modify α-Syn-induced neuronal dysfunction in vivo. (A) The average worsening or amelioration (%) of neuronal dysfunction measured as the loss in climbing speed of Drosophila expressing human α-Syn in the nervous system together with the indicated allele in the fruit fly homolog of the gene shown. Control animals expressing α-Syn and a non-targeting RNAi in the nervous system (elav-GAL4) are used as the reference. Error bars indicate standard deviation. All effects were statistically significant (P < 0.05) using ANOVA applied to linear mixed effect model with spline regressions (see Materials and Methods). (B) A total of 27 TWAS candidates were concordant between in silico (TWAS) and in vivo (Drosophila) effects. We considered as concordant those cases in which we observed a worsening of α-Syn-induced neurodegeneration when mimicking TWAS prediction (red) and cases where we observed amelioration of α-Syn-induced neurodegeneration when opposing TWAS prediction (green). (C) Circular plot for differentially expressed genes measured in a longitudinal RNA-seq experiment using brains of UAS-alpha-synuclein flies and controls. Plot shows 13 concordant TWAS candidate genes whose fly homologs were differentially expressed in the PD Drosophila model relative to controls are shown on the top panel, and the bottom panel shows the six time points (days) when samples were taken for the longitudinal measurement of transcriptome profile. A green bar means the gene is upregulated in PD flies relative to controls, whereas a red bar means the gene is downregulated. Curves connecting a gene and a time point indicate a gene is dysregulated at that time point.
We note that for genes identified through TWAS prediction and modeling, the direction of effect estimate infers the covariance between imputed gene expression and the GWAS trait (16,31). As such, a positive effect suggests that increased expression of the gene may increase PD risk, while a negative effect indicates that decreased expression of the gene may increase PD risk. Remarkably, the behavioral in vivo perturbation assays revealed 27 candidates where the effects were consistent with the TWAS prediction, such that the direction of gene expression predicted to increase PD risk in humans coincided with a similar perturbation modulating α-Syn-induced neuronal dysfunction in flies (Fig. 4B; Table 1; Supplementary Material, Table S8). Strikingly, opposing the TWAS predicted pathogenic effect of 14 of these 27 genes (HN1L, P2RY12, ASH1L, MCCC1, LRRK2, ADORA2B, PRSS53, BCKDK, NUPL2, SCARB2, ELOVL7, FMNL1, STX1B, STX4) conferred neuroprotection in vivo, underscoring their therapeutic potential.
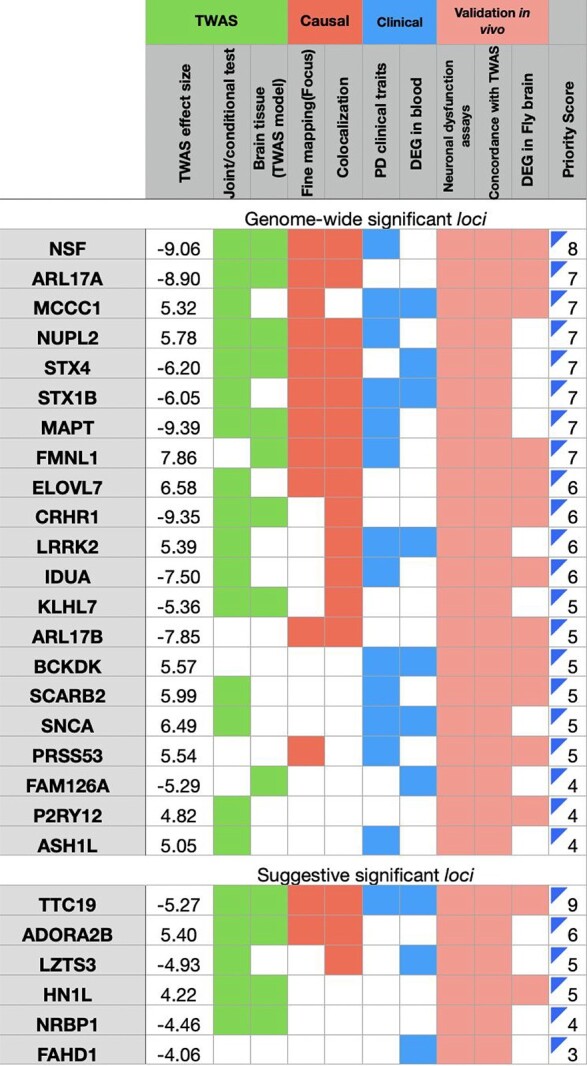
Table 1.
Concordant PD candidate genes with multi-layered evidence. The columns indicate (1) TWAS Z-score; (2) joint and conditional test of TWAS signals; (3) expression imputed via models from PD-relevant brain region/s; (4) fine-mapping of TWAS association in a locus; (5) colocalization of a GWAS signal and an eQTL signal; (6) correlation with PD motor deficits; (7) dysregulated in PPMI blood transcriptome; (8) modifier of α-syn induced neuronal dysfunction in vivo; (9) concordance between in silico and in vivo (Drosophila) tests; (10) DEGs in brains of α-Syn Drosophila model. Top panel built using the GWAS significant loci and bottom panel indicates the suggestive genome-wide loci. Each criterion is assigned an equal weight of 1, with the priority score summing over of all categories. Colored fields indicate that the gene meets the criteria
|
To deepen our mechanistic understanding of those concordant TWAS candidates, we evaluated their transcriptome profiles with time-series transcriptional data from the Drosophila PD model. We leveraged longitudinal RNA-seq on the Drosophila PD model and identified transcripts differentially expressed at each time point (see Materials and Methods). Among the 27 TWAS candidate genes with concordant in silico (TWAS) and in vivo effects on PD, we found that 13 of them were differentially expressed genes in PD models relative to controls across one or multiple time points (Fig. 4C; Supplementary Material, Table S9). Among them, four genes (BCKDK, ELOVL7, P2RY12, PRSS53), whose overexpression were predicted to increase PD via TWAS, showed elevated expression levels in α-Syn flies, while three genes (ARL17A, ARL17B, NSF), whose downregulation was correlated with PD risk, had reduced expression levels. Here we highlight BCKDK, a mitochondrial kinase regulating amino acid catabolic pathways whose mutations can cause autism and epilepsy as previously described (32). We showed that knockdown of BCKDK conferred neuroprotection in vivo as we reversed the pathogenic effect predicted by TWAS (Fig. 4A; Supplementary Material, Fig. S6). BCKDK expression was elevated in both PD Drosophila and human (blood) transcriptomes, suggesting that its pathogenic response can also be found in humans. These results might pinpoint distinct molecular alterations that could help to design mechanistic investigations or plan effective therapeutic interventions based on findings from past and ongoing studies.
Discussion
As the size of GWAS studies for complex polygenic diseases continues to expand, our ability to discover novel risk-associated variants and genes has greatly exceeded our ability to interpret and validate their biological functions. Several previous studies also conducted TWAS to identify new associations to PD within known PD loci, and some genes highlighted by these studies overlapped with our TWAS candidates (e.g. ZSWIM7, LRRC37A2, CD38, NUPL2, etc.) at known PD loci (10–13,33). However, they do not provide experimental validation to investigate candidates’ potential to modulate PD pathology, and they focus mostly on significant loci, which could lead to overlooking potential targets in suggestive loci. We present an integrative approach that combines TWAS and neuronal dysfunction assays in a PD Drosophila model to interrogate whether PD candidates could modulate α-Syn-induced neuropathology. In total, we have identified 69 PD candidate genes in known GWAS loci (some of which were not previously designated as the closest to the corresponding variant) and 27 in GWAS suggestive loci, many of which were further supported by orthogonal in silico methods and/or had clinical relevance in human (Supplementary Material, Tables S10 and S11). To gauge the functional connectivity between these TWAS candidates and assess which biological pathways they are involved in, we performed network analysis using the String-DB protein–protein interaction (PPI) network (34). We found that many of the candidates (15 novel candidates in known loci and 9 in suggestive loci) were connected in the String network (P < 1e-16) and coalesced around endolysosomal and lysosome-related pathways known to be relevant for PD (Fig. 5) (35–37).
Figure 5.
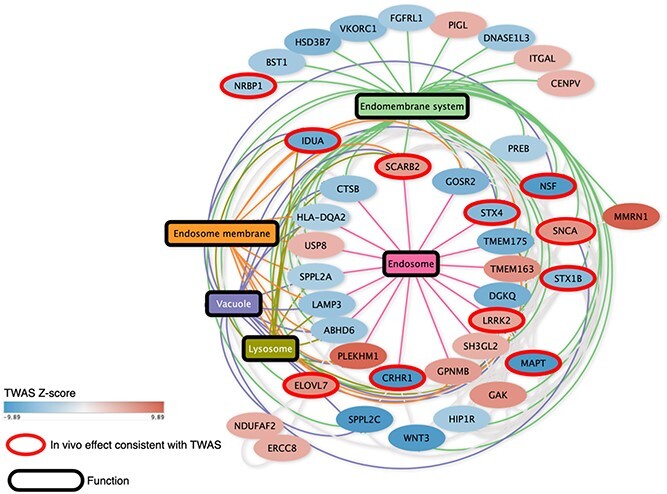
PPI network reveals that the candidates identified by our integrative analysis are enriched in the lysosomal and endolysosomal pathways. Candidates that showed a consistent effect between the TWAS prediction and the in vivo modifier effect in Drosophila are shown with a red outline in the network. Five significantly enriched pathways were all closely related to endolysosomal biology. Pathways are labeled as rectangular nodes with colored edges connecting them to the nodes (candidate genes). The filling of the nodes represents their TWAS score as a gradient.
Note that some well-known PD susceptibility genes were not identified (e.g. GBA, PARK2, PINK1, etc.) using our integrative approach. One example is GBA that was identified by previous PD GWAS but is not TWAS significant. The reason is that the GWAS significant variant for GBA is rs76763715 (Nalls et al., GWAS), which results in a coding variant that leads to the N370S substitution in the GBA protein. This likely results in a partial loss of function in the GBA protein independent of its level of expression. Other well-known PD susceptibility genes (e.g. PARK2, PARK7, FBXO7, PINK1, etc.) were also not nominated in this study since they were identified using familial forms of PD and do not contain functional common genetic variants that can be identified via GWAS and/or TWAS.
As we altered the expression level of TWAS candidates in vivo, we discovered that 50 candidates could ameliorate or aggravate α-Syn-induced neuronal dysfunction. Importantly, we found 27 candidates (including 13 novel genes in known PD GWAS loci and 6 in suggestive loci) were concordant between TWAS-predicted effect and in vivo validation (Table 1; Supplementary Material, Table S10), and 10 of them involve in the endolysosomal pathway (Fig. 5). This convergence in directions of effect to PD between computational and experimental methods emphasizes the potential of our integrative approach to discover disease causal genes. Remarkably, we also found 14 concordant candidates had therapeutic potential since they could generate neuroprotection in vivo when opposing the TWAS predicted pathogenic effect. These findings imply that our integrative approach could identify genes that modulate the risk of developing PD and that our strategy could be applied to a wide range of complex traits for which GWAS data are available.
Materials and Methods
PD GWAS summary statistics
We performed TWAS using PD GWAS summary statistics from Nalls et al. (2). The summary statistics include 15 056 PD cases, 18 618 UK Biobank proxy cases and 449 055 controls. Cohort information and quality control were reported by Nalls et al. (2).
Transcriptome-wide association studies
TWAS is a method that leverages eQTL cohorts with expression and genotype data to compute gene–trait associations for GWAS datasets. We performed a GTEx multi-tissue TWAS using the FUSION package (http://gusevlab.org/projects/fusion/) with pre-computed gene expression weights (GTEx v7 multi-tissue expression) and GWAS summary statistics to estimate the association of genes to PD (2,16). The detailed steps implemented in FUSION are as follows: first, it estimates the heritability of gene expression attributable to cis-SNPs within 1 Mb of gene TSS using GCTA software (38). Only features with heritability of Bonferroni-corrected P < 0.05 were retained for further analysis. Second, expression predictive weights were computed by five different models: best linear unbiased predictor, Bayesian sparse linear mixed model, LASSO, Elastic Net and top SNPs. Cross-validation for each of the desired models was performed, and the model with the largest cross-validation R2 was chosen for imputing gene expression for the GWAS. Lastly, the imputed expression–trait association was estimated as TWAS statistics.
Additional FUSION parameters include running colocalization analysis by COLOC (−coloc_P 0.05). For each locus, it analyzed all SNPs within 500 Mb of the SNP identified as the top eQTL in TWAS and ran coloc.abf using the default parameters and priors. We applied the following criteria to call a signal causal: PP3 + PP4 ≥ 0.8 and PP4/PP3 ≥ 2 (21).
Joint and conditional analyses
Joint and conditional analysis of TWAS was performed using a method previously described to determine how much GWAS signal remains after the expression association from TWAS is removed (6,16). The analysis is based on the TWAS statistics and a gene correlation matrix to evaluate the joint or conditional model, where the correlation matrix can be estimated via predicting the expression for each TWAS gene and Pearson correlations across genes were computed. The joint and conditional test was run on all TWAS hit regions to assess the independence of these associations within their respective 1 Mb regions. We used FUSION tool to perform the analysis and generate the regional scatterplots.
FOCUS fine-mapping
To further prioritize candidate susceptibility genes predicted by TWAS, we performed Bayesian fine-mapping using FOCUS (22). FOCUS models the correlation structure induced by LD and overlapping eQTL weights across predictive models to compute posterior inclusion probabilities for a gene. Using the FOCUS software, we computed 90% higher confidence gene sets by taking genes with largest posterior probability until 90% density was explained.
Identifying disease-relevant tissues via LDSC-SEG
We used LD score regression for specifically expressed genes (LDSC-SEG) to compute enrichment of tissues in PD (23), which takes gene expression data and GWAS summary statistics as input datasets. It uses stratified LD score regression to partition heritability from GWAS summary statistics to specifically expressed genes to identify disease-relevant tissues. We considered a tissue as PD relevant when there was enrichment of SNP heritability (P < 0.05) near genes expressed in that given tissue.
Integration of TWAS candidates with PD blood transcriptome
To assess the potential dysregulation of TWAS candidate genes in PD patients, we used the publicly available PD transcriptome dataset where whole-blood RNA data were collected from 1570 clinically phenotyped individuals (including both cases and controls). The transcriptome analysis and quality control have been described in ref (24). We obtained the transcriptome analysis results, available at https://doi.org/10.1038/s43587-021-00088-6, and determined TWAS candidate genes that were significantly dysregulated (adjusted P value < 0.05).
Association of gene expression with PD-associated traits
To test for association with PD, we limited the comparison to cases with neuropathological evidence for PD (e.g. Lewy bodies in Nigra, limbic system or neocortex) to a control group without pathological diagnosis of PD. We used the ROSMAP study to select 93 individuals with pathological diagnosis of PD ranging from moderate to severe, and presence of Lewy bodies as DLBDX is greater than 0 as PD cases (39,40). We also selected 72 individuals with no pathological diagnosis of PD and no presence of Lewy bodies as controls. We excluded individuals with AD pathology indicated with clinical and pathological evidence (detailed subject characteristics described in Supplementary Material, Table S6) (17,41). The association analysis with five PD traits and TWAS candidate genes was performed using a linear model, adjusting for experimental batch, RIN, sex, age at death and postmortem interval. We used variancePartition to compute the proportion of variance explained by candidate genes association with motor and gait, tremor, bradykinesia and rigidity of extremities (27).
Drosophila models and strains tested
The Drosophila lines carrying UAS-α-Syn have been previously characterized (14,15). For pan-neuronal expression in Drosophila, we used the elav-GAL4C155 driver obtained from the Bloomington Drosophila Stock Center (BDSC). The alleles tested as candidate modifiers of the α-Syn-induced neurodegeneration, which target the Drosophila homologs of TWAS candidates, were obtained from the BDSC and Vienna Drosophila Resource Center (Supplementary Material, Table S7). We used DIOPT (DRSC Integrative Ortholog Prediction Tool) to determine the corresponding Drosophila homologs (42). Specifically, we tested all fly homologs with DIOPT scores equal or greater than 10. In those cases where the scores are lower than 10, we only tested the best score.
Drosophila behavioral assay
We used a custom-made automated robot to quantitatively access neuronal dysfunction in Drosophila using quantitative behavioral readouts, which are based on the Drosophila startle-induced negative geotaxis response (28–30). To model the motor readouts as a function of age, we collected 10 age-matched virgin females per replica per genotype, and four replicates were collected per genotype. Flies are collected at day 1 and transferred into a new vial with 300 μl of media daily until the end of the experiment. Using this automated system, flies are taped to the bottom of a plastic vial and recorded for a period of 7.5 s in which climbing speed for each fly is analyzed using custom software. In total, three trials are performed per replicate for each day shown, and four replicates per genotype are used. We used a linear mixed model analysis of the variance to analyze each one of four replicates to obtain statistical significance across genotypes.
Specifically, we analyzed variations in regression between genotypes and time (additive effect, represented by a shift in the curve) or the interaction between genotype and time (interactive effect, represented by a change in the slope of the curve). Using a stringent threshold for statistical significance (alpha = 0.001), we calculated the expected statistical power of each of our models to detect differences. We reported P-values from pairwise post-hoc tests to determine whether all possible pairs of genotype curves are different in both models. Differences between positive controls and experimental perturbations were significant when P < 0.001. Holm’s procedure was used to adjust P-values for multiplicity. Code for this analysis is available upon request from the Botas Laboratory. Graphing and statistical analyses were performed in R.
We estimated the enrichment of PD modifiers among TWAS candidate genes based on a large α-Syn Drosophila screening of the druggable genome as previously described (20% PD modifier discovery rate among druggable genome) (15).
Drosophila RNA-seq
All flies were raised in identical conditions, and in parallel to the behavioral assay. Three replicates per genotype/time point were generated. Each replicate was made of 100 heads collected from flash frozen age-matched virgin females. mRNA was extracted using TRIzol (#15596026, Invitrogen) followed by DNAse treatment. A minimum of 500 ng of total DNase-treated RNA was used per replicate. Samples were prepared using mRNA TruSeq Stranded library preparation followed by Novaseq processing to generate 60 M paired reads per replicate (Illumina).
PPI Network analysis
String DB release v11.5 was used to compute the connections between candidate nodes using medium stringency. Markov Clustering Algorithm followed by pathway enrichment of each cluster was used to identify the most significantly enriched biological functions (fdr < 0.05). Cytoscape 3.9.1 was used to render the network, color it and arrange it for publication.
Supplementary Material
Acknowledgements
We thank Dr Rami AI-Quran, Jingyuan Hu, Dr Kwanghyuk Lee, Dr Olivier Lichtarge and members of the Botas laboratory at Baylor College of Medicine for insightful discussions and suggestions on this manuscript.
Conflict of Interest statement. The authors declare that no competing interests exist.
Contributor Information
Jiayang Li, Program in Quantitative and Computational Biosciences, Baylor College of Medicine, Houston, TX, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA.
Bismark Kojo Amoh, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Emma McCormick, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Akash Tarkunde, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Katy Fan Zhu, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Alma Perez, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Megan Mair, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Justin Moore, Program in Quantitative and Computational Biosciences, Baylor College of Medicine, Houston, TX, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA.
Joshua M Shulman, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA; Department of Neuroscience, Baylor College of Medicine, Houston, TX, USA; Center for Alzheimer’s and Neurodegenerative Diseases, Baylor College of Medicine, Houston, TX, USA.
Ismael Al-Ramahi, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA; Center for Alzheimer’s and Neurodegenerative Diseases, Baylor College of Medicine, Houston, TX, USA.
Juan Botas, Program in Quantitative and Computational Biosciences, Baylor College of Medicine, Houston, TX, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA; Center for Alzheimer’s and Neurodegenerative Diseases, Baylor College of Medicine, Houston, TX, USA.
Funding
Huffington Foundation, a gift from Terry and Bob Lindsay and the National Institutes of Health (R01 AG057339) to J.B.
Data availability
The reduced GWAS summary statistic of PD was shared by Nalls and colleagues (https://bit.ly/2ofzGrk) (2). Pre-computed predictive models for GTEx v7 multi-tissue expression are available to download from TWAS/FUSION datahub (http://gusevlab.org/projects/fusion/) (16). Single-tissue cis-QTL data are available from the GTEx portal (8). The PD blood transcriptomic dataset is available from Craig et al. (24). The ROS/MAP mapped RNA-seq data that support the findings of this study are available from the AMP-AD Knowledge Portal (https://www.synapse.org/!Synapse:syn2580853) upon authentication.
References
- 1. Chang, D., Nalls, M.A., Hallgrímsdóttir, I.B., Hunkapiller, J., Van Der Brug, M., Cai, F., Kerchner, G.A., Ayalon, G., Bingol, B., Sheng, M.et al. (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat. Genet., 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nalls, M.A., Blauwendraat, C., Vallerga, C.L., Heilbron, K., Bandres-Ciga, S., Chang, D., Tan, M., Kia, D.A., Noyce, A.J., Xue, A.et al. (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol., 18, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ohnmacht, J., May, P., Sinkkonen, L. and Krüger, R. (2020) Missing heritability in Parkinson’s disease: the emerging role of non-coding genetic variation. J. Neural Transm. Suppl., 127, 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Visscher, P.M., Brown, M.A., McCarthy, M.I. and Yang, J. (2012) Five years of GWAS discovery. Am. J. Hum. Genet., 90, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pasaniuc, B., Zaitlen, N., Shi, H., Bhatia, G., Gusev, A., Pickrell, J., Hirschhorn, J., Strachan, D.P., Patterson, N. and Price, A.L. (2014) Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics, 30, 2906–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang, J., Ferreira, T., Morris, A.P., Medland, S.E., Madden, P.A., Heath, A.C., Martin, N.G., Montgomery, G.W., Weedon, M.N., Loos, R.J.et al. (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet., 44, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nicolae, D.L., Gamazon, E., Zhang, W., Duan, S., Dolan, M.E. and Cox, N.J. (2010) Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet., 6, e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maurano, M.T., Humbert, R., Rynes, E., Thurman, R.E., Haugen, E., Wang, H., Reynolds, A.P., Sandstrom, R., Qu, H., Brody, J.et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., Hasz, R., Walters, G., Garcia, F., Young, N.et al. (2013) The genotype-tissue expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li, Y.I., Wong, G., Humphrey, J. and Raj, T. (2019) Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat. Commun., 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kia, D.A., Zhang, D., Guelfi, S., Manzoni, C., Hubbard, L., Reynolds, R.H., Botía, J., Ryten, M., Ferrari, R., Lewis, P.A.et al. (2021) Identification of candidate Parkinson disease genes by integrating genome-wide association study, expression, and epigenetic data sets. JAMA Neurol., 78, 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yao, S., Zhang, X., Zou, S.C., Zhu, Y., Li, B., Kuang, W.P., Guo, Y., Li, X.S., Li, L. and Wang, X.Y. (2021) A transcriptome-wide association study identifies susceptibility genes for Parkinson’s disease. NPJ Parkinsons, 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noyce, A.J., Bandres-Ciga, S., Kim, J., Heilbron, K., Kia, D., Hemani, G., Xue, A., Lawlor, D.A., Smith, G.D., Duran, R.et al. (2019) The Parkinson's disease Mendelian randomization research portal. Mov. Disord., 34, 1864–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chouhan, A.K., Guo, C., Hsieh, Y.C., Ye, H., Senturk, M., Zuo, Z., Li, Y., Chatterjee, S., Botas, J., Jackson, G.R.et al. (2016) Uncoupling neuronal death and dysfunction in Drosophila models of neurodegenerative disease. Acta Neuropathol. Commun., 4, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rousseaux, M.W., Vázquez-Vélez, G.E., Al-Ramahi, I., Jeong, H.H., Bajić, A., Revelli, J.P., Ye, H., Phan, E.T., Deger, J.M., Perez, A.M.et al. (2018) A druggable genome screen identifies modifiers of α-synuclein levels via a tiered cross-species validation approach. J. Neurosci. Res., 38, 9286–9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gusev, A., Ko, A., Shi, H., Bhatia, G., Chung, W., Penninx, B.W., Jansen, R., De Geus, E.J., Boomsma, D.I., Wright, F.A.et al. (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet., 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raj, T., Li, Y.I., Wong, G., Humphrey, J., Wang, M., Ramdhani, S., Wang, Y.C., Ng, B., Gupta, I., Haroutunian, V.et al. (2018) Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet., 50, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gockley, J., Montgomery, K.S., Poehlman, W.L., Wiley, J.C., Liu, Y., Gerasimov, E., Greenwood, A.K., Sieberts, S.K., Wingo, A.P., Wingo, T.S.et al. (2021) Multi-tissue neocortical transcriptome-wide association study implicates 8 genes across 6 genomic loci in Alzheimer’s disease. Genome Med., 13, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wingo, A.P., Liu, Y., Gerasimov, E.S., Gockley, J., Logsdon, B.A., Duong, D.M., Dammer, E.B., Robins, C., Beach, T.G., Reiman, E.M.et al. (2021) Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat. Genet., 53, 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hammond, R.K., Pahl, M.C., Su, C., Cousminer, D.L., Leonard, M.E., Lu, S., Doege, C.A., Wagley, Y., Hodge, K.M., Lasconi, C.et al. (2021) Biological constraints on GWAS SNPs at suggestive significance thresholds reveal additional BMI loci. elife, 10, e62206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giambartolomei, C., Vukcevic, D., Schadt, E.E., Franke, L., Hingorani, A.D., Wallace, C. and Plagnol, V. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mancuso, N., Freund, M.K., Johnson, R., Shi, H., Kichaev, G., Gusev, A. and Pasaniuc, B. (2019) Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet., 51, 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Finucane, H.K., Reshef, Y.A., Anttila, V., Slowikowski, K., Gusev, A., Byrnes, A., Gazal, S., Loh, P.R., Lareau, C., Shoresh, N.et al. (2018) Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet., 50, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Craig, D.W., Hutchins, E., Violich, I., Alsop, E., Gibbs, J.R., Levy, S., Robison, M., Prasad, N., Foroud, T., Crawford, K.L.et al. (2021) RNA sequencing of whole blood reveals early alterations in immune cells and gene expression in Parkinson’s disease. Nat. Aging, 1, 734–747. [DOI] [PubMed] [Google Scholar]
- 25. Bennett, A.D., Schneider, J.A., Arvanitakis, Z. and Wilson, S.R. (2012) Overview and findings from the religious orders study. Curr. Alzheimer Res., 9, 628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett, D.A., Wilson, R.S., Arvanitakis, Z., Boyle, P.A., deToledo-Morrell, L. and Schneider, J.A. (2013) Selected findings from the religious orders study and rush memory and aging project. J. Alzheimers Dis., 33, S397–S403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoffman, G.E. and Schadt, E.E. (2016) variance Partition: interpreting drivers of variation in complex gene expression studies. BMC Bioinform., 17, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Al-Ramahi, I., Lu, B., Di Paola, S., Pang, K., deHaro, M., Peluso, I., Gallego-Flores, T., Malik, N.T., Erikson, K., Bleiberg, B.A.et al. (2018) High-throughput functional analysis distinguishes pathogenic, nonpathogenic, and compensatory transcriptional changes in neurodegeneration. Cell Syst., 7, 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Onur, T.S., Laitman, A., Zhao, H., Keyho, R., Kim, H., Wang, J., Mair, M., Wang, H., Li, L., Perez, A.et al. (2021) Downregulation of glial genes involved in synaptic function mitigates Huntington's disease pathogenesis. elife, 10, e64564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim, Y.W., Al-Ramahi, I., Koire, A., Wilson, S.J., Konecki, D.M., Mota, S., Soleimani, S., Botas, J. and Lichtarge, O. (2021) Harnessing the paradoxical phenotypes of APOE ɛ2 and APOE ɛ4 to identify genetic modifiers in Alzheimer's disease. Alzheimers Dement., 17, 831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wainberg, M., Sinnott-Armstrong, N., Mancuso, N., Barbeira, A.N., Knowles, D.A., Golan, D., Ermel, R., Ruusalepp, A., Quertermous, T., Hao, K.et al. (2019) Opportunities and challenges for transcriptome-wide association studies. Nat. Genet., 51, 592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Novarino, G., El-Fishawy, P., Kayserili, H., Meguid, N.A., Scott, E.M., Schroth, J., Silhavy, J.L., Kara, M., Khalil, R.O., Ben-Omran, T.et al. (2012) Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science, 338, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reynolds, R.H., Botía, J., Nalls, M.A., Hardy, J., Gagliano Taliun, S.A. and Ryten, M. (2019) Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. NPJ Parkinsons, 5, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szklarczyk, D., Gable, A.L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., Simonovic, M., Doncheva, N.T., Morris, J.H., Bork, P.et al. (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res., 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cherubini, M. and Wade-Martins, R. (2018) Convergent pathways in Parkinson’s disease. Cell Tissue Res., 373, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bandres-Ciga, S., Saez-Atienzar, S., Kim, J.J., Makarious, M.B., Faghri, F., Diez-Fairen, M., Iwaki, H., Leonard, H., Botia, J., Ryten, M.et al. (2020) Large-scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta Neuropathol., 140, 341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vidyadhara, D.J., Lee, J.E. and Chandra, S.S. (2019) Role of the endolysosomal system in Parkinson’s disease. J. Neurochem., 150, 487–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang, J., Lee, S.H., Goddard, M.E. and Visscher, P.M. (2011) GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet., 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schneider, J.A., Arvanitakis, Z., Bang, W. and Bennett, D.A. (2007) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology, 69, 2197–2204. [DOI] [PubMed] [Google Scholar]
- 40. Wilson, R.S., Yu, L., Schneider, J.A., Arnold, S.E., Buchman, A.S. and Bennett, D.A. (2011) Lewy bodies and olfactory dysfunction in old age. Chem. Senses, 36, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wan, Y.W., Al-Ouran, R., Mangleburg, C.G., Perumal, T.M., Lee, T.V., Allison, K., Swarup, V., Funk, C.C., Gaiteri, C., Allen, M.et al. (2020) Meta-analysis of the Alzheimer’s disease human brain transcriptome and functional dissection in mouse models. Cell Rep., 32, 107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu, Y., Flockhart, I., Vinayagam, A., Bergwitz, C., Berger, B., Perrimon, N. and Mohr, S.E. (2011) An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform., 12, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The reduced GWAS summary statistic of PD was shared by Nalls and colleagues (https://bit.ly/2ofzGrk) (2). Pre-computed predictive models for GTEx v7 multi-tissue expression are available to download from TWAS/FUSION datahub (http://gusevlab.org/projects/fusion/) (16). Single-tissue cis-QTL data are available from the GTEx portal (8). The PD blood transcriptomic dataset is available from Craig et al. (24). The ROS/MAP mapped RNA-seq data that support the findings of this study are available from the AMP-AD Knowledge Portal (https://www.synapse.org/!Synapse:syn2580853) upon authentication.