

Abstract
Rationale
To date, it remains unclear whether recent changes in the management of patients with systemic sclerosis–associated pulmonary hypertension (SSc-PH) have improved survival.
Objectives
To describe a cohort of patients with SSc-PH and compare their characteristics and survival between the last two decades.
Methods
Patients with SSc-PH prospectively enrolled in the Johns Hopkins Pulmonary Hypertension Center Registry were grouped into two cohorts based on the date of diagnostic right heart catheterization: cohort A included patients whose disease was diagnosed between 1999 and 2010, and cohort B included those whose disease was diagnosed between 2010 and 2021. Patients’ characteristics were compared between the two cohorts.
Measurements and Main Results
Of 504 patients with SSc-PH distributed almost equally between the two cohorts, 308 (61%) had World Symposium on Pulmonary Hypertension group 1, 43 (9%) had group 2, and 151 (30%) had group 3 disease. Patients with group 1 disease in cohort B had significantly better clinical and hemodynamic characteristics at diagnosis, were more likely to receive upfront combination pulmonary arterial hypertension therapy, and had a nearly 4-year increase in median transplant-free survival in univariable analysis than those in cohort A (P < 0.01). Improved transplant-free survival was still observed after adjusting for patients’ baseline characteristics. In contrast, for group 2 or 3 patients with SSc-PH, there were no differences in baseline clinical, hemodynamic, or survival characteristics between the two cohorts.
Conclusions
This is the largest single-center study that compares clinical characteristics of patients with SSc-PH between the last two decades. Transplant-free survival has improved significantly for those with group 1 disease over the last decade, possibly secondary to earlier detection and better therapeutic management. Conversely, those with group 2 or 3 disease continue to have dismal prognosis.
Keywords: pulmonary hypertension, systemic sclerosis, pulmonary arterial hypertension, transplant-free survival
At a Glance Commentary
Scientific Knowledge on the Subject
Pulmonary hypertension, a common and serious complication of systemic sclerosis (SSc), may present in various forms and can be classified in any of the five groups of the World Symposium on Pulmonary Hypertension (WSPH) classification. Patients afflicted with SSc-associated pulmonary arterial hypertension (SSc-PAH, WSPH group 1) have notoriously refractory response and poor survival on modern PAH targeted therapy compared with other WSPH group 1 patients.
What This Study Adds to the Field
This is the largest single-center study comparing two cohorts of patients with SSc-PAH distributed over the last two consecutive decades. A significant improvement in survival was noted over the last compared to the previous decade. Survival remains dismal, however, in patients with WSPH groups 2 and 3 SSc.
Pulmonary hypertension (PH) is a frequent and serious complication of systemic sclerosis (SSc) (1). Because of the multiorgan involvement in SSc, PH can develop through different mechanisms (1, 2). Severe remodeling of small pulmonary arteries can result in World Symposium on Pulmonary Hypertension (WSPH) group 1 PH or pulmonary arterial hypertension (PAH). Myocardial fibrosis can precipitate systolic or diastolic dysfunction, leading to WSPH group 2 disease. Parenchymal lung involvement, most commonly manifesting as interstitial lung disease (ILD), causes hypoxia-induced pulmonary vasoconstriction that can chronically lead to group 3 PH. Increased risk of venous thromboembolism, most commonly in the presence of antiphospholipid antibodies, renders patients with SSc more susceptible to chronic thromboembolic disease and group 4 PH (1, 2).
Once diagnosed, SSc-PH carries a dismal prognosis, with some differences between WSPH phenotypes. Although SSc-PAH generally has a better prognosis compared with group 2 and 3 SSc-PH (3, 4), mortality remains remarkably higher than that reported for idiopathic PAH (5) and even other connective tissue disease–associated PAH (CTD-PAH) (6). In addition, survival did not improve for patients with SSc-PAH in the period immediately after the advent of oral PAH therapy in 2002, and median survival continued to be 3–4 years (7, 8).
However, since 2010, several studies and randomized controlled trials aiming at reducing morbidity and mortality for patients with PH, particularly PAH, were conducted. Over the last decade, robust screening algorithms were developed (9–13), powerful risk stratification approaches were outlined (5, 13–19), new therapeutic options were discovered (20–25), and superior therapeutic strategies were devised (26–28). Importantly, most of these studies included patients with SSc-PH and/or demonstrated interventional benefit for these patients in subgroup analyses. It remains unclear whether these changes have improved survival for patients with SSc-PH, with some recent studies reporting continued poor outcomes (5, 29, 30) and others reporting improved outcomes (31, 32). We hypothesized that survival may have improved for patients with SSc-PH in our registry, and we conducted this study with the following aims: 1) to describe a cohort of patients with SSc-PH and compare their clinical and hemodynamic characteristics between the last two decades; 2) to describe survival patterns and assess for any changes in survival over the last two decades; and 3) to identify factors associated with survival and explore possible reasons for changes in survival patterns, if present.
Methods
Study Population
This study was a retrospective analysis of patients with SSc-PH prospectively enrolled in the Johns Hopkins Pulmonary Hypertension Center Registry (institutional review board no. NA_00027124). Informed consent for enrollment into the registry was obtained from all patients. Diagnosis of SSc was confirmed by rheumatologists according to contemporary American College of Rheumatology/European League Against Rheumatism criteria (33, 34). PH was defined as resting mean pulmonary arterial pressure (mPAP) ⩾ 25 mm Hg on right heart catheterization (RHC), and patients were classified into WSPH groups by PH specialists according to contemporary PH guidelines (2, 13, 35). For the sake of PH classification, ILD was defined as total lung capacity <70% predicted combined with moderate-severe (grade 3–4) fibrosis on high-resolution computed tomography (8, 36). Patients were stratified into two cohorts based on the date of diagnostic RHC: cohort A included patients whose disease was diagnosed between October 1999 and September 2010, and cohort B included those whose disease was diagnosed between October 2010 and September 2021. Patients <18 years old at diagnosis, with missing diagnostic RHC, with mPAP < 25 mm Hg, with unknown vital status, or with disease diagnosed before October 1999 were excluded.
Outcomes of Interest
The primary outcome was transplant-free survival, defined from the date of PH diagnosis by RHC until death, lung transplantation, or the censoring date (October 1, 2010 for cohort A, October 1, 2021 for cohort B), whichever came earliest. Death was ascertained by reviewing medical records, death notifications, or social security death index or by phone contact.
Variables of Interest
Baseline clinical and hemodynamic variables routinely evaluated at the time of PH diagnosis (13) and/or with reported implications on the prognosis of patients with SSc-PH (13, 37–39) were assessed and compared between cohorts A and B. These variables included: World Health Organization functional class (WHO FC), 6-minute-walk distance (6MWD), serum N-terminal prohormone brain natriuretic peptide, echocardiographic parameters, pulmonary function testing (PFT) parameters, and RHC parameters (right atrial pressure, mPAP, cardiac index [CI], stroke volume index [SVI], pulmonary vascular resistance [PVR], pulmonary arterial compliance [PAC], and mixed venous oxygen saturation). In addition, PAH therapy received during the follow-up period was assessed. Upfront dual therapy was defined as that received within the first 4 months of diagnosis.
Statistical Analysis
All statistical analyses were conducted using Stata version 17.0. For comparative analyses, we used the independent t test for normally distributed continuous variables, the Wilcoxon rank-sum test for skewed continuous variables, and the chi-square or Fisher exact tests for categorical variables. Univariable and multivariable Cox proportional hazards (CPH) models were used to assess associations between patients’ characteristics and transplant-free survival. Time-to-event analysis was performed using the Kaplan-Meier product-limit estimator, and the log-rank test was used to compare survival between the two cohorts. Transplant-free survival was assessed at 1, 3, and 5 years. The proportional hazards assumption was examined using Schoenfeld residuals. All comparisons were two-sided, and a P value < 0.05 was used to represent statistical significance.
Results
Study Population and Characteristics
A total of 504 patients met the eligibility criteria of the study (Figure 1) and were distributed almost equally between cohort A (n = 246) and cohort B (n = 258). Nearly 60% of patients had WSPH group 1 disease, 30% had group 3 disease, and 9% had group 2 disease (see Figure E1 in the online supplement). Only two patients had group 4 disease. Group 1 PH was slightly more common in cohort A (67% vs. 55%), and group 3 PH was more common in cohort B (34% vs. 26%). Further analyses were stratified by WSPH groups excluding group 4 (n = 2). In addition, considering the relatively limited number of patients with group 2 PH (n = 43), their findings are included in the online supplement.
Figure 1.

Flow diagram of the cohort. mPAP = mean pulmonary arterial pressure; PH = pulmonary hypertension; RHC = right heart catheterization; SSc = systemic sclerosis.
Patients’ demographic characteristics and comorbidities are shown in Table 1 (Table E1 for group 2 patients). Patients with group 1 PH were predominantly white (75%) and female (83%), with limited SSc subtype (86%) and an average age of 61 ± 13 years at diagnostic RHC. Patients with group 3 PH were predominantly female (75%), with an average age of 58 ± 11 years and a significant proportion of African Americans (38%) and diffuse SSc subtypes (38%). Systemic hypertension, hyperlipidemia, and thyroid disease were common comorbidities in both PH groups.
Table 1.
Demographic Characteristics and Comorbidities for World Symposium on Pulmonary Hypertension Group 1 and 3 Patients
| All Patients | Cohort A | Cohort B | ||
|---|---|---|---|---|
| WSPH Group 1 | (n = 308) 100% | (n = 165) 53.6% | (n = 143) 46.4% | P Value |
| Age at PH diagnosis, yr | 63 (53–69) | 61 (53–68) | 65 (54–72) | 0.029 |
| Female | 257 (83.4) | 145 (87.9) | 112 (78.3) | 0.024 |
| Race | ||||
| White | 231 (75.0) | 129 (78.2) | 102 (71.3) | 0.381 |
| African American | 66 (21.4) | 31 (18.8) | 35 (25.5) | |
| Other | 11 (3.8) | 5 (3.0) | 6 (4.2) | |
| Comorbidities | ||||
| Limited SSc* | 266 (86.4) | 145 (87.9) | 121 (84.6) | 0.405 |
| Hypertension | 123 (39.9) | 51 (30.9) | 72 (50.4) | 0.001 |
| Hyperlipidemia | 89 (28.9) | 36 (21.8) | 53 (37.1) | 0.003 |
| Diabetes mellitus | 26 (8.4) | 15 (9.1) | 11 (7.7) | 0.660 |
| CAD | 44 (14.3) | 22 (13.3) | 22 (15.4) | 0.608 |
| Atrial fibrillation | 43 (14.0) | 25 (15.2) | 18 (12.6) | 0.517 |
| MCTD (+ SLE, PM, DM) | 24 (7.8) | 11 (6.7) | 13 (9.1) | 0.429 |
| Thyroid disease | 111 (36.0) | 66 (40.0) | 45 (31.5) | 0.120 |
| Deaths | 143 (46.4) | 87 (52.7) | 56 (39.2) | |
| Lung transplant | 2 (0.6) | 0 (0.0) | 2 (1.4) |
| WSPH Group 3 | (n = 151) 100% | (n = 64) 42.4% | (n = 87) 57.6% | |
|---|---|---|---|---|
| Age at PH diagnosis, yr | 60 (50–66) | 59 (50–66) | 61 (51–65) | 0.946 |
| Female | 113 (74.8) | 49 (76.6) | 64 (73.6) | 0.675 |
| Race | ||||
| White | 82 (54.3) | 37 (57.8) | 45 (51.7) | 0.785 |
| African American | 58 (38.4) | 23 (35.9) | 35 (40.2) | |
| Other | 11 (7.3) | 4 (6.3) | 7 (8.1) | |
| Comorbidities | ||||
| Limited SSc* | 93 (61.6) | 40 (62.5) | 53 (60.9) | 0.844 |
| Hypertension | 54 (35.8) | 18 (28.1) | 36 (41.4) | 0.093 |
| Hyperlipidemia | 33 (21.9) | 12 (18.8) | 21 (24.1) | 0.429 |
| Diabetes mellitus | 13 (8.6) | 3 (4.7) | 10 (11.5) | 0.239 |
| CAD | 9 (6.0) | 4 (6.3) | 5 (5.8) | 1.000 |
| Atrial fibrillation | 16 (10.6) | 7 (10.9) | 9 (10.3) | 1.000 |
| MCTD (+ SLE, PM, DM) | 27 (17.9) | 9 (14.1) | 18 (20.7) | 0.294 |
| Thyroid disease | 34 (22.5) | 17 (26.6) | 17 (19.5) | 0.307 |
| Deaths | 88 (58.3) | 29 (45.3) | 59 (67.8) | |
| Lung transplant | 7 (4.6) | 2 (3.1) | 5 (5.7) |
Definition of abbreviations: CAD = coronary artery disease; DM = dermatomyositis; MCTD = mixed connective tissue disease; PH = pulmonary hypertension; PM = polymyositis; SLE = systemic lupus erythematosus; SSc = systemic sclerosis; WSPH = World Symposium on Pulmonary Hypertension.
For the Age variable (continuous, skewed variable), data are presented as median (lower quartile Q1 - upper quartile Q3).
For all other variables (categorical variables), data are presented as percentage.
Patients with systemic sclerosis sine scleroderma (WSPH group 1 n = 5; WSPH group 3 n = 5) were included with limited subtype.
Table 2 outlines PAH therapy received in each cohort. Among patients with group 1 disease, the majority (>90%) received PAH therapy during the follow-up period in both cohorts. WSPH group 1 patients in cohort B were more likely to be started on upfront dual combination therapy compared with those in cohort A (38% vs. 10%; P < 0.01). The majority (90%) of those started on upfront dual therapy in cohort B received an oral combination of a phosphodiesterase type 5 inhibitor or soluble guanylate cyclase stimulator and an endothelin receptor antagonist. Twenty-eight patients with group 1 PH (16 in cohort A and 12 in cohort B) did not receive any PAH therapy over the follow-up period for various socioeconomic and insurance-related issues. Likewise, most patients with group 3 PH (78%) received PAH therapy; however, there were no differences in the type of therapy received between the two cohorts.
Table 2.
Pulmonary Arterial Hypertension Therapy Received by World Symposium on Pulmonary Hypertension Group 1 and 3 Patients
| All Patients | Cohort A | Cohort B | ||
|---|---|---|---|---|
| WSPH Group 1 | (n = 308) 100% | (n = 165) 53.6% | (n = 143) 46.4% | P Value |
| Received PAH therapy | 280 (90.9) | 149 (90.3) | 131 (91.6) | 0.876 |
| Therapy type* | <0.001 | |||
| Single | 99 (35.4) | 60 (40.2) | 39 (29.8) | |
| PDE5I/sGCS† | 63 (63.6) | 31 (51.7) | 32 (82.1) | |
| ERA† | 28 (28.3) | 22 (36.7) | 6 (15.4) | |
| PCA† | 8 (8.1) | 7 (11.6) | 1 (2.5) | |
| Upfront dual | 65 (23.2) | 15 (10.1) | 50 (38.2) | |
| PDE5I + ERA (vs. PDE5I + PCA or ERA + PCA)‡ | 53 (81.5) | 8 (53.3) | 45 (90.0) | |
| Sequential dual | 60 (21.4) | 42 (28.2) | 18 (13.7) | |
| PDE5I + ERA (vs. PDE5I + PCA or ERA + PCA)§ | 45 (75.0) | 30 (71.4) | 15 (53.3) | |
| Triple (PDE5I + ERA + PCA) | 56 (20.0) | 32 (21.5) | 24 (18.3) |
| WSPH Group 3 | (n = 151) 100% | (n = 64) 42.4% | (n = 87) 57.6% | |
|---|---|---|---|---|
| Received PAH therapy | 117 (77.5) | 52 (81.3) | 65 (74.7) | 0.144 |
| Therapy type* | 0.693 | |||
| Single | 81 (69.2) | 35 (67.3) | 46 (70.8) | |
| PDE5I/sGCS† | 68 (84.0) | 27 (77.2) | 41 (89.1) | |
| ERA† | 11 (13.6) | 6 (17.1) | 5 (10.9) | |
| PCA† | 2 (2.4) | 2 (5.7) | 0 (0.0) | |
| Upfront dual | 10 (8.6) | 5 (9.6) | 5 (7.7) | |
| PDE5I + ERA (vs. PDE5I + PCA or ERA + PCA)‡ | 7 (70.0) | 4 (80.0) | 3 (60.0) | |
| Sequential dual | 16 (13.7) | 6 (11.5) | 10 (15.4) | |
| PDE5I + ERA (vs. PDE5I + PCA or ERA + PCA)§ | 13 (81.3) | 6 (100.0) | 7 (70.0) | |
| Triple (PDE5I + ERA + PCA) | 10 (8.6) | 6 (11.5) | 4 (6.2) |
Definition of abbreviations: ERA = endothelin receptor antagonist; PAH = pulmonary arterial hypertension; PCA = prostacyclin analogue; PDE5I = phosphodiesterase type 5 inhibitor; sGCS = soluble guanylate cyclase stimulator; WSPH = World Symposium on Pulmonary Hypertension.
For the Age variable (continuous, skewed variable), data are presented as median (lower quartile Q1 - upper quartile Q3).
For all other variables (categorical variables), data are presented as percentage.
Among those who received PAH therapy.
Among those who received monotherapy.
Among those who received upfront dual therapy.
Among those who received sequential dual therapy.
Clinical, functional, echocardiographic, and hemodynamic characteristics at the time of diagnosis were compared between the two cohorts (Table 3; Table E1 for group 2 PH). Among WSPH group 1 patients, those in cohort A were more likely to have WHO FC 3 or 4 limitations (P = 0.04) and higher serum N-terminal prohormone brain natriuretic peptide concentrations (P < 0.01) and tended to have lower 6MWD (P = 0.18) than those in cohort B. Furthermore, WSPH group 1 patients in cohort A had worse echocardiographic parameters, including right atrial dilation (P < 0.01), right ventricular dilation (P = 0.03), higher estimated pulmonary artery systolic pressure (P < 0.01), moderate or severe tricuspid regurgitation (P < 0.01), and presence of pericardial effusion (P = 0.04). In addition, all measured hemodynamic parameters, except CI (P = 0.08), were worse for WSPH group 1 patients in cohort A, including right atrial pressure (P < 0.01), mPAP (P < 0.01), SVI (P < 0.01), PVR (P = 0.03), PAC (P < 0.01), and mixed venous oxygen saturation (P < 0.01). In contrast, these characteristics were similar between the two cohorts for WSPH group 2 and 3 patients. There were no significant differences in PFT parameters between the two cohorts for both group 1 and group 3 disease patients (Table E2).
Table 3.
Clinical, Functional, Echocardiographic, and Hemodynamic Characteristics at the Time of Diagnosis for World Symposium on Pulmonary Hypertension Group 1 and 3 Patients
| All Patients | Cohort A | Cohort B | ||
|---|---|---|---|---|
| WSPH Group 1 | (n = 308) 100% | (n = 165) 53.6% | (n = 143) 46.4% | P Value |
| Clinical and functional | ||||
| WHO FC III/IV | 176 (62.2) (n = 283) | 100 (68.0) (n = 147) | 76 (55.9) (n = 136) | 0.035 |
| 6MWD, m | 306 ± 124 (n = 247) | 296 ± 122 (n = 124) | 317 ± 125 (n = 123) | 0.179 |
| NT-proBNP, ng/L | 894 (281–3,225) (n = 242) | 1584 (393–5,099) (n = 117) | 547 (204–1,965) (n = 125) | <0.001 |
| Echocardiographic | ||||
| RA dilation | 139 (64.4) (n = 216) | 81 (73.6) (n = 110) | 58 (54.7) (n = 106) | 0.004 |
| RV dilation | 139 (61.0) (n = 228) | 82 (71.3) (n = 115) | 57 (50.4) (n = 113) | 0.001 |
| Estimated PASP, mm Hg | 63 (48–80) (n = 198) | 66 (53–84) (n = 101) | 55 (43–73) (n = 97) | 0.002 |
| LVEF, % | 60 ± 8 (n = 200) | 59 ± 9 (n = 96) | 61 ± 6 (n = 104) | 0.101 |
| Moderate/severe TR | 70 (34.0) (n = 206) | 44 (44.0) (n = 100) | 26 (24.5) (n = 106) | 0.003 |
| Pericardial effusion | 72 (34.8) (n = 207) | 42 (41.6) (n = 101) | 30 (28.3) (n = 106) | 0.045 |
| Hemodynamic | ||||
| RAP, mm Hg | 8 ± 5 (n = 282) | 9 ± 6 (n = 149) | 7 ± 5 (n = 133) | 0.002 |
| mPAP, mm Hg | 40 ± 12 (n = 308) | 42 ± 13 (n = 165) | 38 ± 12 (n = 143) | 0.005 |
| CI, L/min/m2 | 2.46 ± 0.79 (n = 272) | 2.37 ± 0.75 (n = 138) | 2.54 ± 0.82 (n = 134) | 0.078 |
| SVI, mL/m2 | 31.9 ± 10.9 (n = 223) | 29.9 ± 10.2 (n = 111) | 33.9 ± 11.3 (n = 112) | 0.006 |
| PVR, WU | 6.8 (4.1–10.4) (n = 282) | 7.1 (4.7–11.6) (n = 144) | 6.0 (3.7–9.9) (n = 138) | 0.03 |
| PAC, ml/mm Hg | 1.4 (0.9–2.3) (n = 196) | 1.2 (0.8–1.7) (n = 108) | 1.7 (1.1–2.6) (n = 88) | <0.001 |
| SvO2, % | 65 ± 10 (n = 240) | 63 ± 9 (n = 123) | 67 ± 10 (n = 117) | 0.003 |
| WSPH Group 3 | (n = 151) 100% | (n = 64) 42.4% | (n = 87) 57.6% | |
|---|---|---|---|---|
| Clinical and functional | ||||
| WHO FC III/IV | 96 (73.9) (n = 130) | 36 (69.2) (n = 52) | 60 (76.9) (n = 78) | 0.328 |
| 6MWD, m | 278 ± 118 (n = 130) | 288 ± 122 (n = 51) | 272 ± 115 (n = 79) | 0.43 |
| NT-proBNP, ng/L | 510 (133–1894) (n = 133) | 348 (156–2484) (n = 53) | 547 (116–1731) (n = 80) | 0.605 |
| Echocardiographic | ||||
| RA dilation | 58 (51.3) (n = 113) | 24 (55.8) (n = 43) | 34 (48.6) (n = 70) | 0.455 |
| RV dilation | 69 (58.5) (n = 118) | 26 (55.3) (n = 47) | 43 (60.6) (n = 71) | 0.571 |
| Estimated PASP, mm Hg | 63 (45–74) (n = 105) | 64 (45–80) (n = 46) | 63 (45–71) (n = 59) | 0.336 |
| LVEF, % | 59 ± 10 (n = 111) | 57 ± 10 (n = 42) | 60 ± 10 (n = 69) | 0.096 |
| Moderate/severe TR | 34 (30.9) (n = 110) | 14 (32.6) (n = 43) | 20 (29.9) (n = 67) | 0.764 |
| Pericardial effusion | 28 (26.2) (n = 107) | 13 (31.7) (n = 41) | 15 (22.7) (n = 66) | 0.304 |
| Hemodynamic | ||||
| RAP, mm Hg | 6 ± 4 (n = 141) | 7 ± 6 (n = 57) | 6 ± 5 (n = 84) | 0.256 |
| mPAP, mm Hg | 36 ± 9 (n = 151) | 36 ± 10 (n = 64) | 35 ± 8 (n = 87) | 0.364 |
| CI, L/min/m2 | 2.57 ± 0.65 (n = 141) | 2.63 ± 0.72 (n = 59) | 2.52 ± 0.60 (n = 82) | 0.322 |
| SVI, mL/m2 | 31.4 ± 8.0 (n = 120) | 32.1 ± 9.6 (n = 50) | 30.9 ± 6.6 (n = 70) | 0.431 |
| PVR, WU | 5.3 (3.5–7.4) (n = 142) | 5.1 (3.3–7.0) (n = 58) | 5.8 (3.7–7.6) (n = 84) | 0.377 |
| PAC, ml/mm Hg | 1.6 (1.2–2.2) (n = 115) | 1.7 (1.2–2.3) (n = 50) | 1.6 (1.2–2.0) (n = 65) | 0.417 |
| SvO2, % | 66 ± 9 (n = 128) | 67 ± 9 (n = 52) | 66 ± 8 (n = 76) | 0.256 |
Definition of abbreviations: 6MWD = 6-minute-walking distance; CI = cardiac index; LVEF = left ventricular ejection fraction; mPAP = mean pulmonary arterial pressure; NT-proBNP = N-terminus prohormone brain natriuretic peptide; PAC = pulmonary arterial compliance; PASP = pulmonary arterial systolic pressure; PVR = pulmonary vascular resistance; RA = right atrial; RAP = right atrial pressure; RV = right ventricle; SVI = stroke volume index; SvO2 = mixed venous oxygen saturation; TR = tricuspid regurgitation; WHO FC = World Health Organization functional class; WSPH = World Symposium on Pulmonary Hypertension.
Data are presented as median (interquartile range, 25th percentile–75th percentile), Wilcoxon rank-sum test.
For categorical variable (e.g., presence of absence of RA dilation, RV dilation etc), data are presented as percentage.
For continuous, normally distributed variables (e.g., LVEF and RAP), data are presented as mean ± SD.
For continuous, skewed variables (e.g., NT-proBNP and PVR), data are presented as median (lower quartile Q1 - upper quartile Q3).
Comparison of Transplant-Free Survival between the Two Cohorts at Univariable Analysis Level
During the follow-up period, a total of 250 patients with SSc-PH (50%) died, and 10 patients (2%) underwent lung transplant. Right ventricular failure, respiratory failure, sudden cardiac death or arrhythmia, and sepsis were the most common reported causes of death (50%, 24%, 12%, and 5%, respectively). For patients with group 1 disease, median follow-up was 2.9 years (2.4 years for cohort A, 3.2 years for cohort B). A total of 87 (53%) patients with group 1 disease died in cohort A. In cohort B, 56 (39%) patients with group 1 disease died and 2 (1%) underwent lung transplant. For patients with group 3 disease, median follow-up was 2 years (1.3 yr for cohort A, 2.2 yr for cohort B). A total of 29 (45%) group 3 patients died and 2 (3%) underwent lung transplant in cohort A. In cohort B, 59 (68%) group 3 patients died and 5 (6%) underwent lung transplant.
There was no significant difference in transplant-free survival between the two cohorts considering all WSPH groups combined (P = 0.10; Figure E2). However, different patterns were observed upon stratifying patients by WSPH group. Among patients with group 1 disease, cohort B had significantly better transplant-free survival than cohort A (estimated 1-, 3-, 5-, and 8-year survival rates: 91%, 74%, 60%, and 51% vs. 85%, 62%, 37%, and 27%, respectively; median survival, 8.8 yr vs. 4.0 yr; P < 0.01; Figure 2A). In addition, median death or transplant age was significantly higher for patients with group 1 PH in cohort B than in cohort A (74 yr vs. 65 yr; P < 0.01). In contrast, there was no difference in transplant-free survival between the two cohorts for patients with group 3 PH (estimated 1-, 3-, and 5-year survival rates: 83%, 44%, and 31%, respectively, for cohort B vs. 74%, 51%, and 40%, respectively, for cohort A; P = 0.79; Figure 2B) or for those with group 2 PH (estimated 3-year survival: 60% in cohort B vs. 68% in cohort A; P = 0.35; Figure E3).
Figure 2.

Comparison of transplant-free survival between cohort A and cohort B for patients with (A) World Symposium on Pulmonary Hypertension (WSPH) group 1 disease and (B) WSPH group 3 disease.
Risk-Adjusted Transplant-Free Survival of WSPH Group 1
The primary outcome models satisfied the proportional hazards assumption as assessed by Schoenfeld residuals. Several clinical, hemodynamic, and therapy-related characteristics were associated with transplant-free survival for patients with both group 1 and 3 disease in univariate analysis (Table E3). Notably, upfront, but not sequential, dual therapy with a phosphodiesterase type 5 inhibitor or soluble guanylate cyclase stimulator and an endothelin receptor antagonist was associated with lower hazard of death or transplant for patients with group 1 PH compared with monotherapy (hazard ratio, 0.52; 95% confidence interval, 0.30–0.91; P = 0.02; Figure E4).
Among clinical and hemodynamic parameters associated with transplant-free survival at univariable level, several were different between the two cohorts for patients with group 1 PH. To account for these differences, multivariable CPH models were used to adjust for clinically relevant variables (Table 4). Further details on the choice of the included covariates were added to the online supplement. Adjusting for age, sex, WHO FC, mPAP, and SVI, WSPH group 1 patients in cohort B had significantly better transplant-free survival than those in cohort A (hazard ratio, 0.6; 95% confidence interval, 0.41–0.88; P < 0.01). In addition, this association was still observed after replacing SVI with other associated hemodynamic variables, such as PVR, PAC, or CI, and WHO FC with 6MWD.
Table 4.
Associations Between Patients’ Characteristics and Transplant-Free Survival for World Symposium on Pulmonary Hypertension Group 1 Patients: Multivariable Analysis
| HR (95% CI) | P Value | |
|---|---|---|
| Cohort B (vs. cohort A) | 0.60 (0.41–0.88) | 0.009 |
| Age at PH diagnosis (per 5-yr increase) | 1.12 (1.04–1.22) | 0.005 |
| Male | 1.71 (1.10–2.66) | 0.017 |
| WHO FC III/IV (vs. I/II) | 1.53 (1.02–2.30) | 0.04 |
| mPAP (per 5 mm Hg increase) | 1.04 (0.95–1.13) | 0.389 |
| SVI (per 5 ml/m2 decrease) | 1.28 (1.15–1.43) | <0.001 |
Definition of abbreviations: CI = confidence interval; HR = hazard ratio; mPAP = mean pulmonary arterial pressure; PH = pulmonary hypertension; SVI = stroke volume index; WHO FC = World Health Organization functional class.
To compare survival patterns across the two decades by risk group, patients with group 1 PH were stratified into three risk categories. The approach devised by the COMPERA (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension) investigators was used to stratify patients (5). A rounded average was computed for every patient based on six baseline clinical and hemodynamic parameters (cutoffs shown in Table E4) and was used to assign risk: one for low risk, two for intermediate risk, and three for high risk. Patients with fewer than two parameters were excluded from the analysis.
Most patients (87%) had at least four parameters (Figure E5). Nearly half of the patients had an intermediate-risk profile at diagnosis in both cohorts. Patients in cohort A were more likely to present with a high-risk profile than those in cohort B, who were more likely to present with a low-risk profile (chi-square P < 0.01; Figure E6). Transplant-free survival was compared between the two cohorts for each risk stratum; survival was better in cohort B for patients with low (P = 0.03; Figure 3A) and intermediate risk (P = 0.01; Figure 3B) at diagnosis but not for those with high risk (P = 0.95; Figure 3C).
Figure 3.
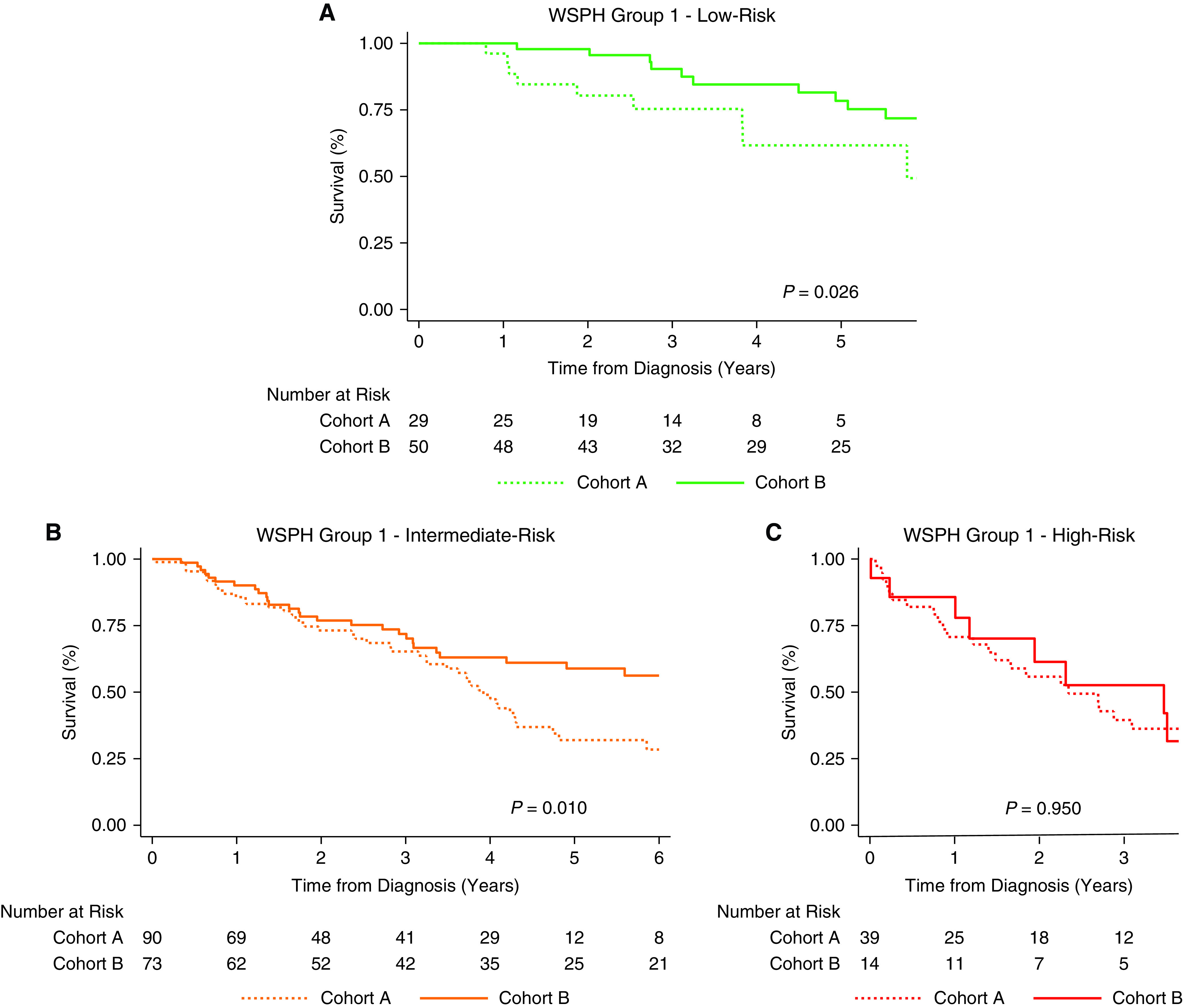
Comparison of transplant-free survival between cohort A and cohort B for WSPH group 1 patients with baseline (A) low-risk profile, (B) intermediate-risk profile, and (C) high-risk profile. WSPH = World Symposium on Pulmonary Hypertension.
Discussion
To our knowledge, this is the largest U.S.-based, single-center study that compares clinical, hemodynamic, and survival characteristics of patients with SSc-PH between the last two decades. The main findings are: 1) transplant-free survival has improved significantly for patients with SSc-PAH over the last decade, with an estimated 5-year rate of 60% (vs. 37% in the previous decade); 2) patients with SSc-PAH diagnosed in the last decade had significantly better baseline clinical and hemodynamic characteristics and were more likely to receive upfront combination PAH therapy than those diagnosed in the previous decade; 3) improved transplant-free survival was still observed after adjusting for baseline patient risk using multivariable CPH models and a validated multiparametric risk stratification tool; survival has improved for low- and intermediate-risk patients, but not for high-risk patients; 4) for group 2 and 3 patients with SSc-PH, there was no improvement in baseline clinical and hemodynamic characteristics in the last decade, and mortality remains high, with a 3-year survival rate of 60% and 44%, respectively; and 5) in addition to the type of PAH therapy received, several clinical and hemodynamic characteristics were associated with transplant-free survival for patients in group 1 and group 3 with SSc-PH.
Several factors may have contributed to better transplant-free survival of patients with SSc-PAH over the last decade in our study. First, PAH was detected at an earlier stage, as is evident by better clinical and hemodynamic parameters and a higher proportion of low-risk patients in cohort B. In the last decade, more robust PAH early-detection algorithms were devised (9–13, 40) and have influenced practice at our center. Nevertheless, improved survival should be interpreted cautiously in the context of earlier disease detection (9). Lead-time bias, wherein survival appears to be better based on earlier diagnosis, is unlikely to explain alone the observed difference in survival for the following reasons: 1) better survival was still observed in cohort B after adjusting for patients’ risk (i.e., upon comparing patients with similar baseline characteristics between the two cohorts); and 2) patients with SSc-PAH in cohort B were slightly older (average age 62 yr vs. 60 yr), and their median death or transplant age was significantly higher (74 yr vs. 65 yr) than cohort A, both of which are inconsistent with lead-time bias. Thus, our results suggest an actual change in disease course from earlier initiation of PAH therapy, resulting in better survival (41). A second factor that could have partially contributed to improved transplant-free survival is better clinical management of patients with SSc-PAH. Over the last decade, better risk stratification approaches were developed and validated in SSc-PAH (5, 13–19), and more treatment options became available, such as riociguat (20, 25), oral (42) and inhaled (43) treprostinil, macitentan (22), and selexipag (21, 23). Moreover, superior therapeutic strategies emerged, including early combination therapy (26, 27). This is partially reflected by the higher proportion of patients with SSc-PAH receiving upfront oral combination PAH therapy in cohort B. Furthermore, better public measures and more effective SSc management guidelines could have contributed to improved survival for patients with SSc-PAH. However, the absence of such improvement in transplant-free survival for the rest of patients with SSc-PH (groups 2 and 3) and the magnitude of the observed difference for patients with group 1 disease (nearly 4-year increase in median survival and 9-year increase in median death age) suggest that the contribution of those measures is likely minor.
An important finding in this study is that transplant-free survival improved for low- and intermediate-risk patients with SSc-PAH but not for high-risk patients, who continued to have a poor prognosis. Although this emphasizes the importance of developing or applying better therapeutic strategies for high-risk patients, it also highlights the need for cost-effective and efficient tools for screening patients with asymptomatic or mildly symptomatic SSc, with the aim of detecting PAH before any irreversible right ventricular change (9, 41, 44, 45). This is particularly relevant in SSc, considering the relatively high prevalence of PAH in this population (9).
Poor survival of patients with SSc-PAH has been consistently reported in previous cohort studies and registries, including the COMPERA registry (N = 216) (5), the FPHN (French Pulmonary Hypertension Network) registry (N = 513) (37), REVEAL (Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Disease Management) (N = 166) (6), SPAHR (Swedish PAH Registry) (N = 161) (29), the UK Pulmonary Hypertension Service (N = 259) (46), the Australian Scleroderma Cohort Study (N = 132) (30), as well as other SSc-PAH cohorts (47–49). Reported 1-, 3-, and 5-year survival rates in these cohorts were 78–88%, 47–62%, and 32–38%, respectively. The PHAROS (Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma) registry (N = 160), however, reported survival rates that were comparable to those of cohort B in our study (1-, 3-, 5-, and 8-year survival rates of 95%, 75%, 63%, and 49%, respectively) (32). Patients enrolled in the PHAROS registry, however, had a milder disease at diagnosis evident by a lower proportion of patients with WHO FC 3 or 4 limitations (41% vs. 56% in cohort B in our study). Although most of these studies described dismal outcomes, enrolled patients with SSc-PAH were diagnosed and managed in the last two decades (2001–2017), so it is unclear from these studies whether survival has changed over the last decade.
A recent study compared the survival of patients with SSc-PAH diagnosed in 2006–2011 vs. 2012–2017 (31). The study included 306 patients from the FPHN registry and the French National Systemic Sclerosis Cohort. Survival was not different between the two periods; however, a stratified analysis showed that patients aged ⩽70 years diagnosed in 2012–2017 (n = 70) had better survival than those diagnosed in 2006–2011 (n = 82). Our study, however, suggests that transplant-free survival improvement was observed in the overall cohort independently of age and reported a slightly higher overall 3-year survival (74% vs. 67%). Differences could possibly be attributed to earlier detection of PAH in our cohort, as well as longer follow-up (median of 3.2 yr vs. 1.3 yr), especially considering that the benefit of upfront dual oral therapy was only established in 2015 (26) but was started earlier in our patients with SSc-PAH (27). Two other studies assessed mortality trends among patients with PAH before and after 2015. The first study included 119 patients with CTD-PAH and reported no survival benefit for patients whose disease was diagnosed after 2015, despite increased use of combination therapy (50). Patients’ clinical and RHC-derived hemodynamic characteristics were similar between those diagnosed before and after 2015, with nearly 70% having WHO FC 3 or 4 limitations at diagnosis (vs. 56% in our cohort). This suggests that patients were still presenting at a relatively advanced disease stage, unlike those in our cohort whose disease was detected earlier and who received PAH therapy early on along the disease course. This again would emphasize the importance of early PAH detection and initiation of therapy. The second study also reported more frequent use of upfront combination therapy and a slight, nonsignificant trend toward better 3-year survival for patients with PAH diagnosed after 2015 (51). This was particularly true for patients with CTD-PAH (n = 536) whose 3-year survival increased from 57% to 67%. Although our study compared transplant-free survival across two different eras (before and after 2010 vs. 2015), our findings support the existence of such a trend in survival. A recent meta-analysis of 19 PH registries that included patients with CTD-PAH reported better survival for patients whose disease was diagnosed between 2010 and 2019 than those diagnosed between 2000 and 2009 (52). However, this comparison had two major limitations: 1) inclusion of registries (10 out of 19) that had significant overlap between the two periods; and 2) a higher proportion of patients with systemic lupus erythematosus–associated PAH in the 2010–2019 period (19% vs. 5% of reported CTD subtypes), who have a better prognosis than patients with SSc-PAH (6).
In contrast to patients with SSc-PAH, WSPH group 3 patients continue to have poor outcomes, with a 3-year transplant-free survival rate of 44%, similar to previously reported rates (4). Unlike group 1 PH, group 3 PH in cohort B was not detected earlier, possibly because these patients initially present with relatively less severe PH (13). In addition, therapeutic options remain limited for patients with SSc with parenchymal lung disease, particularly for those with SSc-ILD, who constitute the majority of group 3 patients in our cohort. Moreover, it was not until 2021 that the benefit of inhaled treprostinil was demonstrated in patients with ILD (24). In the current study, being on PAH therapy did not provide any survival benefit for group 3 PH patients in univariate analysis.
This is the experience of a single PH referral center, which may limit the generalizability of the findings. Although patients were referred to our program from the Johns Hopkins Scleroderma Center over the past two decades, referral bias was an unlikely concern, as the referral process for PH, based on a long-adopted algorithm, remains unchanged. Furthermore, because of the longitudinal nature of the study that spanned over two decades, definitions and classifications of PH and SSc may have slightly changed over time. Nevertheless, the major change in the definition of PH as mPAP > 20 mm Hg introduced in 2018 was not used in this study, to minimize the discrepancy between the two cohorts (2). In addition, the classification of patients into WSPH groups at our center has been consistent over the last two decades, particularly the distinction between SSc-PAH and SSc-ILD-PH using routine PFT screening and high-resolution computed tomography scans of the chest (8, 36). Although we highlighted the main factors that could have contributed to improved survival in patients with SSc-PAH, we may have missed other factors, such as closer follow-up and better overall healthcare quality and access in the last decade. An additional limitation is that our findings were not adjusted to patients’ comorbidities and administered background therapies, such as immunosuppressive therapy; nevertheless, most of these comorbidities were more common in cohort B. Finally, for patients who were lost to follow-up, PAH therapy was assumed to be the same as the one received on last patient contact; this may have created some inconsistency between the two cohorts if there were differential period-dependent adjustments to therapy for those patients.
In conclusion, our study demonstrates improved transplant-free survival of patients with SSc-PAH over the last decade that is unlikely explained by lead-time bias alone. Better survival could be attributed to earlier disease detection, allowing for earlier intervention along the disease course, as well as better therapeutic options and approaches, notably upfront combination PAH therapy. This emphasizes the importance of robust screening and optimization of PAH therapy in the management of patients with SSc-PAH. In contrast, the prognosis of group 2 and 3 SSc-PH remains dismal and warrants further studies to explore better treatment and management strategies.
Footnotes
Supported by The National Institutes of Health/National Heart, Lung, and Blood Institute awards R01 HL114910 and U01HL125175-01 (P.M.H.). NIH/NHLBI K23HL153781 (S.H.) and NIH/NHLBI K23HL153781 (C.E.S.).
Author Contributions: Each author contributed substantially to patient enrollment. They all reviewed and edited the manuscript. All authors approved the submission of the manuscript for publication and take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202204-0731OC on September 29, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Launay D, Sobanski V, Hachulla E, Humbert M. Pulmonary hypertension in systemic sclerosis: different phenotypes. Eur Respir Rev . 2017;26:170056. doi: 10.1183/16000617.0056-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J . 2019;53:1801913. doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bourji KI, Kelemen BW, Mathai SC, Damico RL, Kolb TM, Mercurio V, et al. Poor survival in patients with scleroderma and pulmonary hypertension due to heart failure with preserved ejection fraction. Pulm Circ . 2017;7:409–420. doi: 10.1177/2045893217700438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lefèvre G, Dauchet L, Hachulla E, Montani D, Sobanski V, Lambert M, et al. Survival and prognostic factors in systemic sclerosis-associated pulmonary hypertension: a systematic review and meta-analysis. Arthritis Rheum . 2013;65:2412–2423. doi: 10.1002/art.38029. [DOI] [PubMed] [Google Scholar]
- 5. Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer J, Benjamin N, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J . 2017;50:1700740. doi: 10.1183/13993003.00740-2017. [DOI] [PubMed] [Google Scholar]
- 6. Chung L, Farber HW, Benza R, Miller DP, Parsons L, Hassoun PM, et al. Unique predictors of mortality in patients with pulmonary arterial hypertension associated with systemic sclerosis in the REVEAL registry. Chest . 2014;146:1494–1504. doi: 10.1378/chest.13-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rubenfire M, Huffman MD, Krishnan S, Seibold JR, Schiopu E, McLaughlin VV. Survival in systemic sclerosis with pulmonary arterial hypertension has not improved in the modern era. Chest . 2013;144:1282–1290. doi: 10.1378/chest.12-0653. [DOI] [PubMed] [Google Scholar]
- 8. Campo A, Mathai SC, Le Pavec J, Zaiman AL, Hummers LK, Boyce D, et al. Hemodynamic predictors of survival in scleroderma-related pulmonary arterial hypertension. Am J Respir Crit Care Med . 2010;182:252–260. doi: 10.1164/rccm.200912-1820OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weatherald J, Montani D, Jevnikar M, Jaïs X, Savale L, Humbert M. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev . 2019;28:190023. doi: 10.1183/16000617.0023-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coghlan JG, Denton CP, Grünig E, Bonderman D, Distler O, Khanna D, et al. DETECT study group Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis . 2014;73:1340–1349. doi: 10.1136/annrheumdis-2013-203301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thakkar V, Stevens WM, Prior D, Moore OA, Byron J, Liew D, et al. N-terminal pro-brain natriuretic peptide in a novel screening algorithm for pulmonary arterial hypertension in systemic sclerosis: a case-control study. Arthritis Res Ther . 2012;14:R143. doi: 10.1186/ar3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thakkar V, Stevens W, Prior D, Youssef P, Liew D, Gabbay E, et al. The inclusion of N-terminal pro-brain natriuretic peptide in a sensitive screening strategy for systemic sclerosis-related pulmonary arterial hypertension: a cohort study. Arthritis Res Ther . 2013;15:R193. doi: 10.1186/ar4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J . 2015;46:903–975. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 14. Benza RL, Gomberg-Maitland M, Elliott CG, Farber HW, Foreman AJ, Frost AE, et al. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL risk score calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest . 2019;156:323–337. doi: 10.1016/j.chest.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 15. Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL) Circulation . 2010;122:164–172. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 16. Kylhammar D, Kjellström B, Hjalmarsson C, Jansson K, Nisell M, Söderberg S, et al. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur Heart J . 2018;39:4175–4181. doi: 10.1093/eurheartj/ehx257. [DOI] [PubMed] [Google Scholar]
- 17. Weatherald J, Boucly A, Sitbon O. Risk stratification in pulmonary arterial hypertension. Curr Opin Pulm Med . 2018;24:407–415. doi: 10.1097/MCP.0000000000000510. [DOI] [PubMed] [Google Scholar]
- 18. Boucly A, Weatherald J, Savale L, Jaïs X, Cottin V, Prevot G, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J . 2017;50:1700889. doi: 10.1183/13993003.00889-2017. [DOI] [PubMed] [Google Scholar]
- 19. Mercurio V, Diab N, Peloquin G, Housten-Harris T, Damico R, Kolb TM, et al. Risk assessment in scleroderma patients with newly diagnosed pulmonary arterial hypertension: application of the ESC/ERS risk prediction model. Eur Respir J . 2018;52:1800497. doi: 10.1183/13993003.00497-2018. [DOI] [PubMed] [Google Scholar]
- 20. Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC, et al. PATENT-1 Study Group Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med . 2013;369:330–340. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- 21. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, et al. GRIPHON Investigators Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med . 2015;373:2522–2533. doi: 10.1056/NEJMoa1503184. [DOI] [PubMed] [Google Scholar]
- 22. Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, et al. SERAPHIN Investigators Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med . 2013;369:809–818. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 23. Gaine S, Chin K, Coghlan G, Channick R, Di Scala L, Galiè N, et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Respir J . 2017;50:1602493. doi: 10.1183/13993003.02493-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waxman A, Restrepo-Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, et al. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med . 2021;384:325–334. doi: 10.1056/NEJMoa2008470. [DOI] [PubMed] [Google Scholar]
- 25. Humbert M, Coghlan JG, Ghofrani HA, Grimminger F, He JG, Riemekasten G, et al. Riociguat for the treatment of pulmonary arterial hypertension associated with connective tissue disease: results from PATENT-1 and PATENT-2. Ann Rheum Dis . 2017;76:422–426. doi: 10.1136/annrheumdis-2015-209087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, et al. AMBITION Investigators Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med . 2015;373:834–844. doi: 10.1056/NEJMoa1413687. [DOI] [PubMed] [Google Scholar]
- 27. Hassoun PM, Zamanian RT, Damico R, Lechtzin N, Khair R, Kolb TM, et al. Ambrisentan and tadalafil up-front combination therapy in scleroderma-associated pulmonary arterial hypertension. Am J Respir Crit Care Med . 2015;192:1102–1110. doi: 10.1164/rccm.201507-1398OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fischer A, Denton CP, Matucci-Cerinic M, Gillies H, Blair C, Tislow J, et al. Ambrisentan response in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): a subgroup analysis of the ARIES-E clinical trial. Respir Med . 2016;117:254–263. doi: 10.1016/j.rmed.2016.06.018. [DOI] [PubMed] [Google Scholar]
- 29. Hjalmarsson C, Kjellström B, Jansson K, Nisell M, Kylhammar D, Kavianipour M, et al. Early risk prediction in idiopathic versus connective tissue disease-associated pulmonary arterial hypertension: call for a refined assessment. ERJ Open Res . 2021;7:854. doi: 10.1183/23120541.00854-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morrisroe K, Stevens W, Huq M, Prior D, Sahhar J, Ngian GS, et al. Australian Scleroderma Interest Group (ASIG) Survival and quality of life in incident systemic sclerosis-related pulmonary arterial hypertension. Arthritis Res Ther . 2017;19:122. doi: 10.1186/s13075-017-1341-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hachulla E, Launay D, Boucly A, Mouthon L, de Groote P, Cottin V, et al. Survival improved in patients aged ≤ 70 years with systemic sclerosis-associated pulmonary arterial hypertension during the period 2006 to 2017 in France. Chest . 2020;157:945–954. doi: 10.1016/j.chest.2019.10.045. [DOI] [PubMed] [Google Scholar]
- 32. Kolstad KD, Li S, Steen V, Chung L, PHAROS Investigators Long-term outcomes in systemic sclerosis-associated pulmonary arterial hypertension from the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma Registry (PHAROS) Chest . 2018;154:862–871. doi: 10.1016/j.chest.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Masi AT, Subcommittee For Scleroderma Criteria of the American Rheumatism Association Diagnostic and Therapeutic, Criteria Committee Preliminary criteria for the classification of systemic sclerosis (scleroderma): subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum . 1980;23:581–590. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 34. van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Ann Rheum Dis . 2013;72:1747–1755. doi: 10.1136/annrheumdis-2013-204424. [DOI] [PubMed] [Google Scholar]
- 35. Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol . 2013;62:D42–D50. doi: 10.1016/j.jacc.2013.10.032. [DOI] [PubMed] [Google Scholar]
- 36. Mathai SC, Bueso M, Hummers LK, Boyce D, Lechtzin N, Le Pavec J, et al. Disproportionate elevation of N-terminal pro-brain natriuretic peptide in scleroderma-related pulmonary hypertension. Eur Respir J . 2010;35:95–104. doi: 10.1183/09031936.00074309. [DOI] [PubMed] [Google Scholar]
- 37. Weatherald J, Boucly A, Launay D, Cottin V, Prévot G, Bourlier D, et al. Haemodynamics and serial risk assessment in systemic sclerosis associated pulmonary arterial hypertension. Eur Respir J . 2018;52:1800678. doi: 10.1183/13993003.00678-2018. [DOI] [PubMed] [Google Scholar]
- 38. Ghio S, Mercurio V, Fortuni F, Forfia PR, Gall H, Ghofrani A, et al. TAPSE in PAH investigators A comprehensive echocardiographic method for risk stratification in pulmonary arterial hypertension. Eur Respir J . 2020;56:2000513. doi: 10.1183/13993003.00513-2020. [DOI] [PubMed] [Google Scholar]
- 39. Winstone TA, Assayag D, Wilcox PG, Dunne JV, Hague CJ, Leipsic J, et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: a systematic review. Chest . 2014;146:422–436. doi: 10.1378/chest.13-2626. [DOI] [PubMed] [Google Scholar]
- 40. Glynn P, Hale S, Hussain T, Freed BH. Cardiovascular imaging for systemic sclerosis monitoring and management. Front Cardiovasc Med . 2022;9:846213. doi: 10.3389/fcvm.2022.846213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vachiéry JL, Yerly P, Huez S. How to detect disease progression in pulmonary arterial hypertension. Eur Respir Rev . 2012;21:40–47. doi: 10.1183/09059180.00009011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tapson VF, Torres F, Kermeen F, Keogh AM, Allen RP, Frantz RP, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest . 2012;142:1383–1390. doi: 10.1378/chest.11-2212. [DOI] [PubMed] [Google Scholar]
- 43. McLaughlin VV, Benza RL, Rubin LJ, Channick RN, Voswinckel R, Tapson VF, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol . 2010;55:1915–1922. doi: 10.1016/j.jacc.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 44. Vonk Noordegraaf A, Chin KM, Haddad F, Hassoun PM, Hemnes AR, Hopkins SR, et al. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J . 2019;53:1801900. doi: 10.1183/13993003.01900-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Humbert M, Yaici A, de Groote P, Montani D, Sitbon O, Launay D, et al. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: clinical characteristics at diagnosis and long-term survival. Arthritis Rheum . 2011;63:3522–3530. doi: 10.1002/art.30541. [DOI] [PubMed] [Google Scholar]
- 46. Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JS, Vrapi F, et al. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med . 2009;179:151–157. doi: 10.1164/rccm.200806-953OC. [DOI] [PubMed] [Google Scholar]
- 47. Keogh A, Strange G, McNeil K, Williams TJ, Gabbay E, Proudman S, et al. The Bosentan Patient Registry: long-term survival in pulmonary arterial hypertension. Intern Med J . 2011;41:227–234. doi: 10.1111/j.1445-5994.2009.02139.x. [DOI] [PubMed] [Google Scholar]
- 48. McLaughlin VV, Langer A, Tan M, Clements PJ, Oudiz RJ, Tapson VF, et al. Pulmonary Arterial Hypertension-Quality Enhancement Research Initiative (PAH-QuERI) Investigators Contemporary trends in the diagnosis and management of pulmonary arterial hypertension: an initiative to close the care gap. Chest . 2013;143:324–332. doi: 10.1378/chest.11-3060. [DOI] [PubMed] [Google Scholar]
- 49. Yaylalı YT, Başarıcı I, Kılıçkıran Avcı B, Meriç M, Sinan ÜY, Şenol H, et al. Risk assessment and survival of patients with pulmonary hypertension: multicenter experience in Turkey. Anatol J Cardiol . 2019;21:322–330. doi: 10.14744/AnatolJCardiol.2019.53498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zelt JGE, Sugarman J, Weatherald J, Partridge ACR, Liang JC, Swiston J, et al. Mortality trends in pulmonary arterial hypertension in Canada: a temporal analysis of survival per ESC/ERS guideline era. Eur Respir J . 2022;59:2101552. doi: 10.1183/13993003.01552-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hoeper MM, Pausch C, Grünig E, Staehler G, Huscher D, Pittrow D, et al. Temporal trends in pulmonary arterial hypertension: results from the COMPERA registry. Eur Respir J . 2022;59:2102024. doi: 10.1183/13993003.02024-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khanna D, Zhao C, Saggar R, Mathai SC, Chung L, Coghlan JG, et al. Long-term outcomes in patients with connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era: meta-analyses of randomized, controlled trials and observational registries. Arthritis Rheumatol. 2021;73:837–847. doi: 10.1002/art.41669. [DOI] [PMC free article] [PubMed] [Google Scholar]