

Abstract
A catalytic and highly enantioselective dearomative alkynylation of chromanones has been discovered that enables the construction of biologically relevant tertiary ether stereogenic centers. This methodology is robust, accommodating a variety of alkynes and chromanones. More than 40 substrates tested gave rise to >90% ee. Computational studies have indicated that the optimal indanyl ligand identified for most cases likely affords a network of supportive, non-covalent interactions that drive the enantioselective nature of the reaction.
Keywords: tertiary ether stereocenter, benzopyrylium triflate, bis(oxaolines), alkynes, copper
Graphical Abstract

Chromanones can frequently be found in the heart of medicinal agents and bioactive compounds.1 Naturally occurring molecules containing a chromanone core often possess enticing biological properties. For instance, the blennolides are monomeric tetrahydroxanthones with antibacterial activity.2 Rugolotrosin A3 is also an antibacterial agent while phomoxanthone A4, dicerandrol C5, and versixanthone F6 possess anticancer activity. Gonytolide A is a dimeric chromanone with immune response promoter activity (Figure 1).7
Figure 1.

Naturally occurring bioactive compounds with chromanone cores containing challenging tertiary ether stereocenter in 2-position.
Chromanone-containing targets of interest are often host to stereogenic centers that can be a struggle to synthesize with high enantiocontrol.8 The chemical synthesis of highly enantioenriched chromanones containing a tertiary ether stereogenic center is a particularly daunting prospect (highlighted in purple, Figure 1).9 We encountered the synthetic challenge of constructing highly substituted 2-alkyl chromanones as part of a program dedicated toward the development of efficient methodologies for the enantioselective functionalization of oxygen heterocycles. Herein, we detail the development of a catalytic, highly enantioselective synthesis of chromanones containing 2-tertiary ether stereocenters by the alkynylation of benzopyrylium ions under the influence of copper (bis)oxazoline catalysis.
In 2014, when our research program began flowing in the direction of oxygen heterocycle functionalization, we were inspired by the power of dearomatizing heterocyclic ions generated in situ for the development of new catalytic enantioselective chemical technologies. The idea for the enantioselective functionalization of chromanones was rooted in the discoveries of Akiba and coworkers.10 Simply put, Akiba’s team demonstrated how to functionalize racemic chromanones by reacting a variety of carbon nucleophiles with benzopyrylium triflates (3, Scheme 1). Inspired by Akiba’s work, the Mattson group sought to control the facial selectivity of reactions with benzopyrylium triflates. Our first approach, published in 2016, was to take advantage of the cationic nature of the benzopyrylium triflates to control the stereochemical outcome of 4 from 1 through anion-binding catalysis.11 Specifically, our BINOL-based family of silanediols emerged as uniquely effective catalysts for the addition of silylketene acetals to benzopyrylium triflates that had been generated in situ via the reaction of various chromanones and silyl triflates.12 The desired adducts were isolated in up to 56% enantiomeric excess. Notably, this work inspired additional investigations into anion-binding catalysis as applied to benzopyrylium ion functionalization.13 While the modest enantioselectivity in the synthesis of 4 was a promising start we eventually learnt that there may be inherent challenges associated with silanediol-catalyzed functionalization of benzopyrylium triflates. Unfortunately, there was no enantiomeric excess observed in our hands with anion-binding catalysis when the benzopyrylium ion contained any substituent in the 2-position that was not hydrogen.
Scheme 1.

Mattson group’s enantioselective approaches to benzopyrylium ion functionalization.
The limitations of our silanediol catalyst system shifted us into problem-solving mode with our sights lasered in on achieving a highly enantioselective synthesis of chromanones containing the 2-tertiary ether stereocenter. To boost our odds of successfully executing the mission, we initiated two pursuits: (1) we began exploring new silanediol catalyst systems and scaffolds that may offer improved reactivity and (2) we launched an effort to identify transition metal catalysts systems with suitable reactivity. While, to-date, three separate publications have emerged in the last four years highlighting some of our key findings from these pursuits,12b,14 several heretofore untold insights are described below.
Our early attempts in the design of new catalyst platforms included the development of a silanediol and Cu(OTf)2 hybrid catalyst system.12b In 2017 we were delighted to find that the combination of a chiral silanediol and Cu(OTf)2 was uniquely effective and enabled the activation of alkylidene malonates for reaction with indoles giving rise to desirable adducts with excellent levels of enantiocontrol (Scheme 2). Curious as to how the hydrogen bond donor and Lewis acid could join forces, we probed the mechanism through experimentation. Based upon the results of judiciously chosen control experiments and associated UV-Vis data we reasoned that the silanediol may be hydrogen bonding with a triflate from the Cu(OTf)2 thereby generating a copper Lewis acid with enhanced activity.
Scheme 2.

Early attempt to use Cu(OTf)2 and silanediol to influence enantioselective alkynylation of 1a.
Excited by the discovery of an enhanced Lewis acid catalyst system with a built-in hydrogen bond donor, we applied the silanediol and Cu(OTf)2 hybrid toward the addition of phenyl acetylene to chromone (Scheme 2). A variety of reaction conditions were tested but never one hint of desired alkynylation was observed. On the other hand, immediately upon subjection of chromone 1a to TBSOTf, Cu(OTf)2, and (bis)oxazoline ligand 8, we observed the desired product 5a in 44% yield and with an encouraging 14% enantiomeric excess.
Encouraged by the discovery of an enantioselective alkynylation of chromone that was working in our hands, we zoned in on optimizing the reaction conditions. We found that we could tune the ligand, base, and reaction temperature to achieve excellent levels of enantiocontrol (up to 97% ee). The scope of the alkynylation reaction proved broad with respect to both the chromone and alkyne, giving rise to useful, nearly enantiopure chromanones in high yield.14a
Rejuvenated with the discovery of a robust, enantioselective alkynylation of 2-H chromones, we put our heart into tackling the synthesis of more sterically encumbered chromanones. Unfortunately, the benzyl ligand 8 that was identified as optimal for the 2-H chromanones afforded underwhelming selectivity in the case of 2-CO2Me chromanones (1, X = CO2Me Scheme 3). A re-evaluation of the ligand backbone in the alkynylation of chromanone 1 generated some promising trends. Specifically, while the benzyl ligand gave rise to 6 with 34% ee, the t-butyl ligand 12 afforded 6a in −67% enantiomeric excess (Scheme 3). With our collaborators in the Hadad group at the Ohio State University, computational modeling of potential transition states suggested that the t-butyl groups may control the orientation of the substrates in the enantiodetermining step through steric hindrance (Scheme 4). Specifically, transition state A has fewer interactions between the t-butyl substituents on the ligand and the benzopyrylium ion than transition state B. It was hypothesized that the enantioswitch occurring between use of ligand 8 and ligand 12 may be due to the benzyl substituent contributing additional supportive non-covalent interactions (e.g., π stacking) to the transition state whereas the t-butyl substituent of 12 controlled the transition state through primarily steric hindrance.
Scheme 3.

Promising enantiomeric excess formed with tertiary ether stereocenter.
Scheme 4.

Plausible transition states of enantiodetermining steps for 6a with ligand 12 as determined at the B3LYP/SDD/6–31+G(d) level of theory.
With the insight into the influence of ligand structure on controlling the stereochemical outcome, alongside of the promising 67% enantiomeric excess, we initiated a (bis)oxazoline ligand screen to identify a set of reaction conditions and ligand scaffolds that would enable the creation of the desired tertiary ether stereocenter with greater than 90% enantiomeric excess. Several data points from a selection of the ligands that were tested are described in Scheme 5.
Scheme 5.

Effect of ligand structure on enantomeric excess in alkynylation reaction.
While the t-butyl ligand 12 gave rise to a promising 87% yield and −38% enantiomeric excess of 6a at 0 °C, we reached a maximum enantiomeric excess of −67% ee upon cooling the reaction mixture to −78 °C. We were unable to identify reaction conditions to reach enantiomeric excesses in the 90% range with ligand 12. Moreover, we could not identify strategies to further increase the steric bulk easily of the t-butyl substituents in order to better control the facial selectivity of the enantiodetermining step. Having reached the end of the road with respect to ligand 12, we reasoned that perhaps instead of relying on steric hindrance to control the transition state of the alkynylation, we could control the non-covalent interactions offered by the aromatic ligands on (bis)oxazoline backbones. This choice was motivated by the ability to tailor the aromatic nature of the (bis)oxazoline scaffold through judicious choice of aromatic substituents. Building from the previously observed formation of 6a in 29% yield and 34% ee with ligand 8, we prepared 1-naphthyl and 2-naphthyl (bis)oxazolines 13 and 14. While these ligands offered a substantial improvement in yield, we were disappointed to find that the enantiomeric excess remained low. Finally a breakthrough came upon testing the indanyl-derived (bis)oxazoline 9. At 0°C we achieved a 42% enantiomeric excess and 26% yield after 48 h. Lowering the reaction temperature and extending the reaction time gave rise to a scenario where excellent levels of enantiocontrol (90% ee) were reached, along with reasonable yields (68%).
Ecstatic with the optimization of the stereocontrol of the alkynylation of benzopyrylium triflates, calculations were performed to better understand the “magic” associated with the indanyl (bis)oxazoline ligand (Scheme 6). The results of our computational investigations suggest that added non-covalent interactions in the R transition state resulted in a lower energy (Scheme 6, A). Non-covalent interaction analysis supported these findings (Scheme 6, B).
Scheme 6.

A) Plausible transition states of enantiodetermining steps for 6a with ligand 9 as determined at the B3LYP/SDD/6–31+G(d) level of theory. B) NCI plots of plausible transition states (blue=attraction; green=weak interaction; red=steric effect)
With the ability to construct the tertiary ether stereocenter of 2-CO2Me chromanone 6a with excellent enantiocontrol, our efforts next focused on exploring the tolerance of the reaction to a variety of alkynes and chromones. Several well-tolerated substrates are listed in Scheme 7. The reaction scope with respect to the type of ester found at the 2-position is broad. The methyl ester gave rise to 6a in 90% ee while the ethyl ester gave rise to 6b in 89% ee. The isopropyl ester afforded 6c in 91% ee whereas the t-butyl ester may be too bulky generating 6d in 71% ee. The benzyl ester produced the desired product in 83% ee. The ester with the highest enantiomeric excess was the substrate with the trichloroethyl substituent, giving rise to 6e in 92% ee.
Scheme 7.

Chromone Substrate Scope.
Pleased with the substrate tolerance of the ester in the 2-position, we moved on to probe the effect of substituents in the 5-, 6-, 7-, and 8-position on the chromanone backbone (Scheme 8). We were delighted to find that a methoxy group in the 5-position gave rise to 6g with 93% ee. This particular substrate maps nicely onto several naturally occurring molecules, a few of which are depicted in Figure 1. A methoxy group was also well tolerated in the 7-position, affording 6h in 99% ee. The 7-methyl and 7-bromo chromanones generated products 6i and 6j in 82% ee and 85% ee, respectively. Product 6k, containing a bromo substituent in the 8-position was created in 95% ee. The tolerance of the reaction with respect to substituents in the 7-position proved to be remarkably broad. 7-Fluoro (6l), 7-trifluoromethyl (6m) and 7-allyl (6n) gave rise to the corresponding products in 93–97% enantiomeric excesses. Excellent levels of enantiocontrol (99% ee) were achieved when 7-phenyl (6o), 7-cyclopropylalkynyl (6p), and 7-vinyl (6q) chromanones were used as starting reagents.
Scheme 8.

Chromanone substrate scope.
During the course of our studies we did learn of two locations on the chromanone backbone that created problems with yield and/or enantiocontrol (bottom of Scheme 8). Specifically, we found that halogens in the 6-position typically gave rise to alkynylated products with low enantiomeric excess. Likewise, when chromanones containing select substituents in the 8-position, such as phenyl and methoxy, were tested in the reaction both low yields and low enantiomeric excesses were observed.
With the boundaries of the alkynylation reaction with respect to substitution patterns on the chromanone core well identified, our attention turned toward exploring the scope of the acetylene reaction partner (Scheme 9). We were delighted to find that a variety of phenyl acetylenes gave rise to desirable alkynylated products in excellent levels of enantiocontrol regardless of the substitution pattern. For example, para-substituted phenyl acetylenes of all electronic varieties participated well in the process, including methyl, methoxy, chloro, bromo, trifluoromethyl and phenyl, giving rise to the desired products 6s-6x in 89–91% ee. 3-, or 2-methyl phenyl acetylenes generated products 6y and 6z in 90–92% ee. 1-Naphthyl and 2-naphthyl acetylenes were also well tolerated substrates, affording 6aa and 6ab in 92% ee and 88% ee respectively. Alkynes containing aromatic heterocycles, such as thiophenes and furans, were easily accommodated as substrates. For instance, 6ac, 6ad, and 6ae were isolated in 93% ee, 92% ee, and 91% ee, respectively.
Scheme 9.

Alkyne substrate scope.
Aliphatic alkynes were more challenging substrates in the reaction. While cyclopropryl alkyne gave rise to 6af in 89% ee, cyclohexylalkyne afforded 6ag with a maximum enantiomeric excess of 81%. The reaction of t-butyl alkyne with 1 afforded 6ah in 91% ee when the t-butyl ligand 12 was employed. Reasonable enantiomeric excesses (86%) were observed in the synthesis of 6ai. On the other hand, despite significant attempts at optimization, low enantiomeric excess was observed in the syntheses of 6ah and 6ai, no matter what ligand was applied. There were also several alkynes that gave rise to no reaction under the optimized alkynylation conditions (see bottom of Scheme 9).
Finally, the scope of the reaction with respect to the carbonyl substituent in the 2-position was put to the test (Scheme 10). We were delighted to find that thioesters proceeded well in the process, giving rise to 15 in 52% yield and 92% ee. Selected amides were tolerated in the alkynylation event. For example, 16 was prepared in 41% yield and 75% ee. Chromanones containing ketones in the 2-position are also viable reaction partners. Specifically, 17 was synthesized in 37% yield and 71% ee.
Scheme 10.

Tolerance of thioester, amide, and ketone substituents in the reaction.
A proposed reaction pathway for this process is depicted in Scheme 11. This (bis)oxazoline and the CuI interact to give rise to complex I. The formation of the chiral copper acetylide II is proposed to occur upon exposure of I to Hunig’s base and phenyl acetylene. The addition of the copper acetylide II to the benzopyrylium ion 20, generated in situ from 1 and t-butyldimethylsilyl triflate (TBSOTf), affords 21 and regenerates the chiral copper complex I. It is proposed that the absolute stereochemistry of the tertiary ether is set during this step. Finally, the loss of TBSOTf gives rise to the isolated product 6.
Scheme 11.
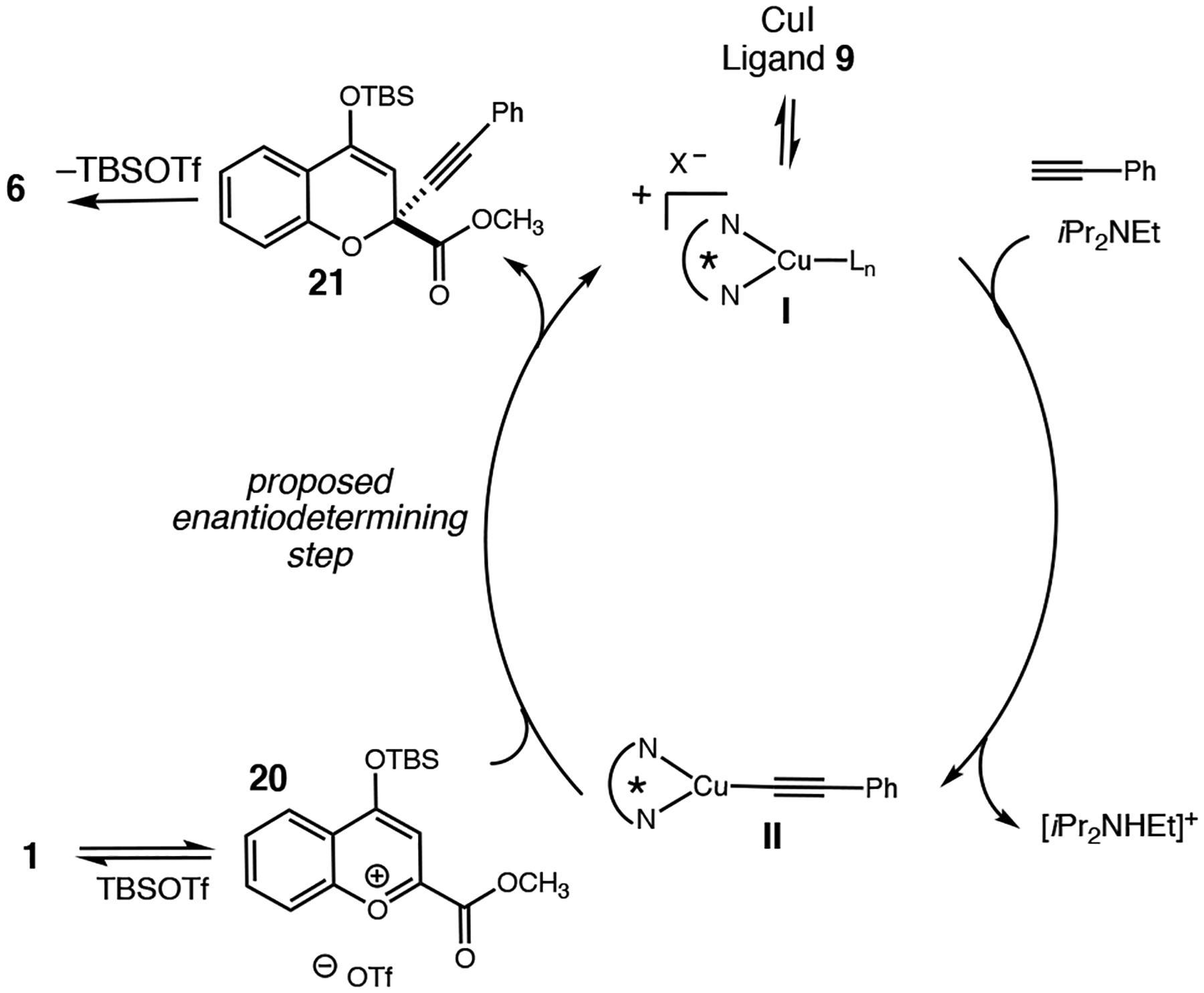
Plausible reaction pathway
To conclude, a robust and highly enantioselective method for the synthesis of biologically relevant tertiary ether stereocenters has been identified. The process proved to be extremely tolerant of a variety of reaction partners, including long lists of chromanones and alkynes. Also included within this manuscript are the substrates found to be sub-optimal and non-compatible with the new transformation. Our computational studies suggest that the indanyl ligand, the most optimal ligand for the majority of the substrates included herein, is likely driving the selectivity of the reaction through a network of supportive non-covalent interactions that result in preferential formation of the R stereoisomer with ligand 9. Given the large number of naturally occurring chromanones and tetrahydroxanthones with a tertiary ether in the 2-position, this method may find utility in natural product and bioactive target synthesis. Ongoing efforts in the Mattson group are dedicated toward uncovering the potential of the methodology in complex molecule construction.
Anhydrous toluene, dichloromethane, diethyl ether and THF were dried using a pure process technologies solvent system. Anhydrous DCE, chlorobenzene, m-xylene, and o-xylene were used as received. Substituted chromone-2-carboxylate esters were prepared according to literature. 14 CuI was used as received and stored in a desiccator under ambient lab conditions. TBSOTf was vacuum distilled and stored under dry nitrogen. DIPEA was used as received. Alkynes were used as received or prepared according to literature.14 All bis(oxazoline) ligands were used as received from Sigma Aldrich or TCl or prepared according to literature.14 All other reagents were used directly as received from the manufacturer unless otherwise noted. Preparative silica gel chromatography was performed using SiliaFlash F60 silica gel (40 – 63 μm). Analytical thin layer chromatography was performed using Analtech 250 μm silica gel HLF plates and visualized under UV 254nm or 365nm. All 1H NMR spectra were acquired using a Bruker BioSpin 500 MHz Avance III Digital NMR spectrometer and calibrated using the solvent signal (CDCl3 7.26 ppm). J Coupling constants are reported in Hz. Multiplicities are reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; hept, heptet; m, multiplet; b, broad; dd, doublet of doublets; ddd, doublet of doublet of doublets; td, triplet of doublets; ddt, doublet of doublet of triplets; dtd, doublet of triplet of doublets. All 13C NMR spectra were acquired using a Bruker BioSpin 126MHz Avance III Digital NMR spectrometer and calibrated using the solvent signal (CDCl3 77.16 ppm). Infrared spectra were acquired using a Bruker Vertex 70 with an ATR accessory. High resolution mass spectra were acquired using an Agilent 6520 Q-TOF mass spectrometer. Chiral HPLC analysis was performed using an Agilent 1260 equip with a diode array detector using Chiralcel OD-H or AD-H columns. Optical Rotations were acquired on a JASCO P-2000 Digital Polarimeter with a sodium lamp (λ = 589 nm).
Procedure
General procedure for synthesis of 2-ethynyl chromanones (6a – 6z, 6aa – 6ak, 15 – 17)
To an 8 mL screw top vial was added chromone-2-carboxylate (0.2 mmol, 1.0 eq), CuI (0.02 mmol, 10 mol%), (S)-Indanyl-BOX (0.024 mmol, 12 mol%), chlorobenzene (2 mL), i-Pr2NEt (0.3 mmol, 1.5 eq), and phenyl acetylene (0.26 mmol, 1.3 eq) in that order at room temperature. This mixture was allowed to stir for 30 minutes. The vial was purged with dry N2 and then cooled to −78 °C. TBSOTf (0.26 mmol, 1.3 eq) was added at −78 °C, then the reaction was transferred to the lab chiller at −35 °C and allowed to react for 96h. The reaction was quenched by the addition of 6N HCl (2 mL) and stirred for 2 hours. The reaction mixture was extracted with EtOAc (3 × 2 mL), washed with saturated NaHCO3 solution, dried over anhydrous NaSO4, and the solvent removed under vacuum to obtain the crude product. The crude product was purified by column chromatography on silica gel.
S-Ethyl (R)-4-oxo-2-(phenylethynyl)chromane-2-carbothioate (15):
Prepared according to general procedure, using PhCl, (S)-Indanyl-BOX, chromone (46.8 mg, 0.20 mmol) and phenyl acetylene (29 μl, 0.26 mmol). was isolated as a yellow oil (35.1 mg, 0.104 mmol, 52% yield). Rf = 0.37 (9:1, Hexanes:EtOAc). IR (neat, ATR) ѵ: 3029, 2930, 2266, 1738, 1688, 1510, 1341, 1121, 754. Chiral HPLC: 97.3:2.6 e.r., 95% ee, Chiralcel OD-H column (5% iPrOH/Hexanes, 1 mL/min, 254 nm); tR (minor) = 7.0 min, tR (major) = 7.8 min. HRMS (ESI+) m/z calculated for C20H16O3S [M+H]+ 337.0893, found 337.0883.
1H NMR (500 MHz, CDCl3) δ 7.91 (dd, J = 7.8, 1.7 Hz, 1H), 7.56 (ddd, J = 8.3, 7.2, 1.8 Hz, 1H), 7.42 − 7.36 (m, 2H), 7.36 − 7.31 (m, 1H), 7.31 − 7.25 (m, 2H), 7.15 (dd, J = 8.3, 1.0 Hz, 1H), 7.10 (ddd, J = 8.1, 7.2, 1.1 Hz, 1H), 3.37 (d, J = 16.6 Hz, 1H), 3.25 (d, J = 16.6 Hz, 1H), 2.96 (qd, J = 7.5, 4.5 Hz, 2H), 1.29 (t, J = 7.4 Hz, 3H).
13C NMR (126 MHz, CDCl3) δ 195.92, 188.70, 158.55, 136.56, 132.27, 129.63, 128.44, 126.98, 122.75, 121.05, 120.96, 118.69, 89.29, 83.60, 82.13, 45.84, 23.99, 14.26.
(R)-2-(Phenylethynyl)-2-(pyrrolidine-1-carbonyl)chroman-4-one (16):
Prepared according to general procedure, using PhCl, (S)-Indanyl-BOX, chromone (48.6 mg, 0.20 mmol) and phenyl acetylene (29 μl, 0.26 mmol). was isolated as a yellow oil (28.5 mg, 0.082 mmol, 41% yield). Rf = 0.40 (2:1, Hexanes:EtOAc). IR (neat, ATR) ѵ: 3031, 2966, 2194, 1720, 1687, 1512, 1355, 714. Chiral HPLC: 86.7:13.2 e.r., 74% ee, Chiralcel AD-column (5% iPrOH/Hexanes, 1 mL/min, 254 nm); tR (minor) = 36.5 min, tR (major) = 31.7 min. HRMS (ESI+) m/z calculated for C22H19NO3 [M+H]+ 346.1438, found 346.1424.
1H NMR (500 MHz, Chloroform-d) δ 7.95 (dd, J = 7.9, 1.7 Hz, 1H), 7.54 − 7.47 (m, 1H), 7.40 − 7.32 (m, 3H), 7.30 (ddt, J = 8.2, 6.4, 1.3 Hz, 2H), 7.09 (td, J = 7.6, 1.0 Hz, 1H), 7.02 (dd, J = 8.4, 1.1 Hz, 1H), 4.16 (dt, J = 12.0, 6.3 Hz, 1H), 3.95 (dt, J = 11.2, 7.1 Hz, 1H), 3.64 − 3.51 (m, 2H), 3.46 (ddd, J = 12.7, 8.1, 5.5 Hz, 1H), 3.23 (d, J = 16.8 Hz, 1H), 2.08 − 1.94 (m, 2H), 1.94 − 1.77 (m, 2H).
13C NMR (126 MHz, CDCl3) δ 190.08, 163.81, 158.02, 136.04, 132.06, 129.56, 128.54, 127.26, 122.72, 121.52, 121.14, 117.71, 89.23, 83.69, 78.16, 47.72, 47.61, 46.64, 26.81, 23.64.
(R)-2-(Phenylethynyl)-2-(3-phenylpropioloyl)chroman-4-one (17):
Prepared according to general procedure, using PhCl, (S)-Indanyl-BOX, chromone (54.8 mg, 0.20 mmol) and 1-phenylpenta-1,4-diyn-3-one (28.5 μL, 0.26 mmol). 7w was isolated as a colorless oil (23.4 mg, 0.132 mmol, 37% yield). Rf = 0.46 (4:1, Hexanes:EtOAc). IR (neat, ATR) ѵ: 3051, 2919, 2341, 1758, 1617. Chiral HPLC: 85.5:14.1 e.r., 71% ee, Chiralcel AD-H column (5% iPrOH/Hexanes, 1 mL/min, 254 nm); tR (minor) = 12.6 min, tR (major) = 14.5 min. HRMS (ESI+) m/z calculated for C26H16O3 [M+H]+ 377.1172, found 377.1164.
1H NMR (500 MHz, Chloroform-d) δ 7.95 (dd, J = 8.0, 1.7 Hz, 3H), 7.53 − 7.46 (d, 1H), 7.41 − 7.30 (m, 10H), 7.29 (ddt, J = 8.5, 6.4, 1.2 Hz, 1H), 7.1 (td, J = 7.0, 1.0 Hz, 1H), 6.98 (dd, J = 8.3, 1.0 Hz, 1H) 3.46 (dd, J = 12.9, 8.4, 5.6 Hz, 1H), 3.23 d, J = 17 Hz, 1H)
13C NMR (126 MHz, CDCl3) δ 188.60, 178.87, 158.71, 136.51, 133.54, 132.19, 131.56, 129.58, 128.78, 128.36, 126.95, 122.61, 120.98, 120.84, 119.34, 118.56, 97.42, 90.16, 85.18, 82.94, 81.39, 44.72.
Supplementary Material
Funding Information
We gratefully acknowledge the National Institutes of Health for funding these studies through 1R35GM124804-01. C.M.H acknowledges generous computational resources as provided by the Ohio Supercomputer Center.
Biographies

Yong Guan obtained his B.S. and M.S. from Wuhan University in 2004 and 2006. He completed doctoral research on construction of a vaulted biaryl ligand library for the aziridination reaction with Prof. William Wulff and earned a Ph.D. in 2012 from Michigan State University. He performed his postdoctoral research with Prof. Christopher Douglas at University of Minnesota on one-pot Sonogashira coupling and regioselective tetradehydro-Diels–Alder reaction to synthesis rubicenes (2013–2014), and with Prof. Steven Townsend at Vanderbilt University on metal-free synthesis of unsymmetrical organoselenides and selenoglycosides (2015–2016). In 2017, he joined Mattson group at Worcester Polytechnic Institute as a postdoctoral fellow. His current projects are on asymmetric copper catalysis and drug discovery.

Tadas Buivydas received his B.S. and M.S. from Worcester State University in 2017 and 2018 where his research focused on elucidating the reaction mechanism of nitroxyl (HNO) and GAPDH via electronic structure/molecular mechanics calculations. In 2018, he joined the Mattson group at Worcester Polytechnic Institute and his projects are currently on asymmetric copper catalysis and methodology development in the context of drug discovery.

Remy F. Lalisse received his B.S. degree from the State University of New York (SUNY) Geneseo in 2017. In 2017, he joined Hadad group at The Ohio State University as a Ph.D student and is projected to graduate in 2022. His current projects are on dynamics and mechanistic Studies of organophosphorus inhibited and aged forms of cholinesterases. In collaborative efforts, he has studied reaction mechanism calculations for small molecule catalysis in method development of novel copper and cobalt catalysts for synthetic applications.

Rameez Ali received his BSc. Honors in Chemistry in 2009 from Mahidol University International College, Thailand. He then moved to United States and completed his doctoral research in 2015 under Prof. Thomas W. Bell at University of Nevada Reno. His doctoral research is primarily focused on developing and modulating activities for small molecule entry inhibitors for Human Immunodeficiency Virus (HIV-1). In 2017, he joined Mattson group at Worcester Polytechnic Institute as a postdoctoral fellow. His current projects are on synthesis of small molecules for drug discovery.

Christopher M. Hadad received his B.S. degree from the University of Delaware in 1987 and then completed his Ph.D. as a Hertz Foundation fellow under the guidance of Professor Kenneth B. Wiberg at Yale University. After a NSF post-doctoral fellowship at the University of Colorado (Boulder) with Professor Charles H. DePuy, he joined the faculty of The Ohio State University as an assistant professor of Chemistry in 1994. In 2006, he was promoted to full professor, was the vice chair for undergraduate studies in the Department of Chemistry from 2006–2011, served the department as the interim chair in 2007, and served the College as Associate Dean (2011–2014) and then Dean (2014–2018) of the Natural and Mathematical Sciences division of the College of Arts and Sciences. Our research team explores diverse areas in chemistry, but almost always with a mechanistic focus, using both experimental and computational approaches.

Anita Mattson became a professor of chemistry at Worcester Polytechnic Institute in 2020 and she was promoted to department head in 2021. Prior to WPI, she received her BS from Northern Michigan University and a PhD from Northwestern University under the direction of Prof. Karl Scheidt. After postdoctoral training with Prof. Michael Crimmins at the University of North Carolina at Chapel Hill she began her independent career as an assistant professor of chemistry at The Ohio State University in 2009. She was promoted to associate professor of chemistry at OSU in 2015. Driven by a passion for improving human health through organic chemistry, she leads a research team in the areas of catalyst design, methodology development, bioactive target synthesis, and medicinal chemistry.
References
- (1).(a) Wezeman T; Brase S; Masters K Nat. Prod. Rep 2015, 32, 6. [DOI] [PubMed] [Google Scholar]; (b) Masters K; Brase S Chem. Rev 2012, 112, 3717. [DOI] [PubMed] [Google Scholar]
- (2).Zhang W; Kroh K; Zia-Ullah; Florke U; Pescitelli G; Di Bari L; Antus S; Kurtan T; Rheinheimer J; Draeger S; Schulz B Chem Eur. J 2008, 14, 4913. [DOI] [PubMed] [Google Scholar]
- (3).Stewart M; Capon RJ; White JM; Lacey E; Tennant S; Gill J; Shaddock M J. Nat. Prod 2004, 67, 728. [DOI] [PubMed] [Google Scholar]
- (4).(a) Rönsberg D; Debbab A; Mándi A; Vasylyeva V; Böhler P; Stork B; Engelke L; Hamcher A; Sawadogo R; Diederich M; Wray V; Lin W; Kassack MU; Janiak C; Scheu S; Wesselborg S; Kurtán T; Aly AH; Proksch PJ Org. Chem 2013, 78, 12409. [DOI] [PubMed] [Google Scholar]; (b) Böhler P; Stuhldreier F; Anand R; Kondadi A; Schlutermann D; Berleth N; Deitersen J; Wallot-Hieke N; Wu W; Frank M; Niemann H; Wesbuer E; Barbian A; Luyten T; Parys J; Weidtkamp-Peters S; Borchardt A; Reichert A; Pena-Blano A; Garcia-Saez A; Itskanov S; van der Bliek A; Proksch P; Wesselborg S; Stork B Cell Death & Disease 2018, 9, 286–303. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang C; Engelke L; Bickel D; Hamacher A; Frank M; Proksch P; Gohlke H; Kassack MU Bioorg. Med. Chem 2019, 27, 115044. [DOI] [PubMed] [Google Scholar]; (d) Yang R; Dong Q; Zu H; Gao X; Zhao Z; Qin J; Chen C; Luo D ACS Omega 2020, 5, 25927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kikuchi H; Isobe M; Sekiya M; Abe Y; Hoshikawa T; Ueda K; Kurata S; Katou Y; Oshima Y Org. Lett 2011, 13, 4624. [DOI] [PubMed] [Google Scholar]
- (6).Wu G; Yu G; Kurtan T; Mandi A; Peng J; Mo X; Liu M; Li H; Sun X; Li J; Zhu T; Gu Q; Li DJ Nat. Prod 2015, 78, 2691. [DOI] [PubMed] [Google Scholar]; Kolotilo NV; Sinitsa AA; Rassukana Yu. V.; Onys’ko PP Zh. Obshch. Khim 2006, 76, 1260; [Google Scholar]; Chem. Abstr 2006, 146, 316980. [Google Scholar]
- (7).Kikuchi H; Isobe M; Sekiya M; Abe Y; Hoshikawa T; Ueda K; Kurata S; Katou Y; Oshima Y Org. Lett 2011, 13, 4624. [DOI] [PubMed] [Google Scholar]
- (8).For a review, see:; Nibbs AE Scheidt KA Eur. J. Org. Chem 2012, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]; For select examples of enantioselective 2-alkylchromanone synthesis, see:; (a) Rao AV; Gaitonde AS; Prakash SP; Rao SP Tetrahedron Lett. 1994, 35, 6347. [Google Scholar]; (b) Kawasaki M; Kakuda H; Goto M; Kawabata S; Kometani T Tetrahedron: Asymmetry 2003, 14, 1529. [Google Scholar]; (c) Biddle MM; Lin M; Scheidt KA J. Am. Chem. Soc 2007, 129, 3830. [DOI] [PubMed] [Google Scholar]; (d) Boekl H; Mackert R; Muramann C; Schweickert N US66646136B1, 2013.; (e) Termath AO; Sebode H; Schlundt W; Stemmler RT; Netscher T; Bonrath W; Schmalz H-G Chem. Eur. J 2014, 20, 12051. [DOI] [PubMed] [Google Scholar]; (f) Brown MK; Degrado SJ; Hoveyda AH Angew. Chem. Int. Ed 2005, 44, 5306.; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2005, 117, 5440. [Google Scholar]; (g) Vila C; Hornillos V; Fananas-Mastral M; Feringa BL Chem. Commun 2013, 49, 5933. [DOI] [PubMed] [Google Scholar]; (h) Trost BM; Gnanamani E; Kalnmals CA; Hung C-I; Tracy JS J. Am. Chem. Soc 2019, 141, 1489. [DOI] [PubMed] [Google Scholar]
- (9).For select reports demonstrating the importance of constructing tertiary ether stereocenters from chromanones, see:; (a) Baek D; Ryu H; Ryu J; Lee J; Stoltz B; Hong S Chem. Sci 2020, 11, 4602. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gerten A; Stanley L Tetrahedron Lett. 2016, 57, 5460. [Google Scholar]
- (10).Iwasaki H; Kume T; Yamamoto T; Akiba K Tetrahedron Lett. 1987, 28, 6355. [Google Scholar]
- (11).Hardman-Baldwin A; Visco M; Wieting J; Stern C; Kondo S; Mattson A Org. Lett 2016, 18, 3766. [DOI] [PubMed] [Google Scholar]
- (12).(a) Attard J; Osawa K; Guan Y; Hatt J; Kondo S; Mattson A Synthesis 2019, 51, 2107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guan Y; Attard J; Visco M; Fisher T; Mattson A Chem. Eur. J 2018, 24, 7123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schafer AG; Wieting JM; Fisher TJ; Mattson AE Angew. Chem., Int. Ed 2013, 52, 11321. [DOI] [PubMed] [Google Scholar]; (d) Wieting JM; Fisher TJ; Schafer AG; Visco MD; Gallucci JC; Mattson AE Eur. J. Org. Chem 2015, 2015, 525. [Google Scholar]; (e) Schafer AG; Wieting JM; Mattson AE Org. Lett 2011, 13, 5228–5231. [DOI] [PubMed] [Google Scholar]
- (13).(a) Fischer T; Bamberger J; Gomez-Martinez M; Piekarski D; Mancheno O Angew. Chem. Int. Ed 2018, 58, 3217. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeRatt L; Pappoppula M; Aponick A Angew. Chem. Int. Ed 2019, 58, 8416. [DOI] [PubMed] [Google Scholar]
- (14).(a) Guan Y; Attard J; Mattson AE Chem. Eur. J 2020, 26,1742. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guan Y; Buivydas T; Lalisse R; Attard J; Ali R; Stern C; Hadad C; Mattson A ACS Catal. 2021, 11, 6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.