

Abstract
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a natural product class that has undergone significant expansion due to the rapid growth in genome sequencing data and recognition that they are made by biosynthetic pathways that share many characteristic features. Their mode of actions cover a wide range of biological processes and include binding to membranes, receptors, enzymes, lipids, RNA, and metals as well as use as cofactors and signaling molecules. This review covers the currently known modes of action (MOA) of RiPPs. In turn, the mechanisms by which these molecules interact with their natural targets provide a rich set of molecular paradigms that can be used for the design or evolution of new or improved activities given the relative ease of engineering RiPPs. In this review, coverage is limited to RiPPs originating from bacteria.
Graphical Abstract
1. Introduction
Natural products account for a remarkably rich reservoir of bioactive pharmaceutical leads, as nearly half of the drugs introduced over the past four decades were derived from molecules produced by bacteria, fungi, or plants.1–3 Historic approaches towards discovery of novel therapeutics typically focused on using target- or phenotype-based screening.4 While these strategies have been successful, each is bereft with significant limitations. Recent advances in analytical instrumentation, genome sequencing, and metabolomics have facilitated the discovery of new natural products with therapeutic potential at an exponential rate.5 However, the elucidation of biological targets and mode of action (MOA) of these newly discovered natural products have lagged. Decades-long advances in various methodologies have set the framework for detailed MOA studies and avails many future opportunities for such efforts.6–8
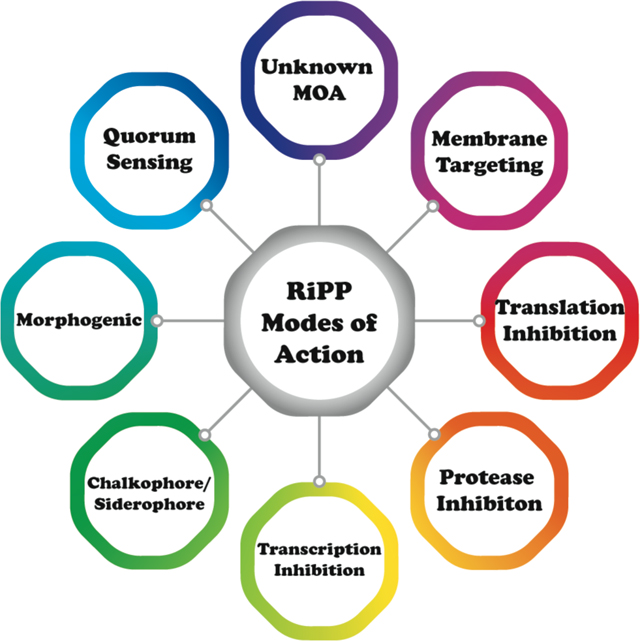
The ribosomally synthesized and post-translationally modified peptides (RiPPs) are a natural product class that has undergone significant expansion as a consequence of rapid growth in genome sequencing data.9,10 Biosynthetic gene clusters encoding RiPPs typically consist of one or more genes encoding a precursor peptide, which is modified by biosynthetic enzymes to yield the mature natural product or products. The co-localization of genes encoding the biosynthetic proteins with those encoding the substrate peptide facilitates prediction of the entire biosynthetic pathway. Growing interest in the potential of RiPP therapeutics has been spurred by the discovery of compounds with antibacterial,11 antifungal,12 antinociceptive,13,14 antiviral,15 and antitumor16–20 activities. The myriad activities of RiPPs serve to emphasize how post-translational modifications have allowed molecular evolution of structures that exploit a wide range of biological targets in different domains of life. These targets include the cell membrane, small molecule metabolites and biosynthetic intermediates, enzymes, receptors, RNA, and metal ions. In turn, the wide variety of natural targets and activities suggests that this class of compounds offers much promise for discovery and engineering of new activities. This review will provide a comprehensive discussion of the MOA of bacterial RiPPs for which such information is available, to complement earlier reviews that focused on specific compound classes or a subset of RiPPs.21–38 The review covers studies reported up to and including June 2022.
Despite a simplistic biosynthetic logic, RiPPs display an expansive diversity of molecular scaffolds. Based on the still limited information on RiPP MOA, the bioactivities associated with even closely related structural classes of RiPPs are rarely predictable. In some instances, elucidation of MOA and validation of biological targets for several compounds predate their characterization as members of a RiPP class.39,40 As both the substrate peptide and the enzymes that catalyze the post-translational modifications are direct gene products, RiPP biosynthetic gene clusters are excellent candidates for genome mining exercises, and several available robust bioinformatics platforms harness this capability.41–46 Likewise, the synteny of genes necessary for the production of the bioactive RiPP final product has enabled refactoring efforts for heterologous production. In several instances, an affinity tag was appended to the precursor peptide, allowing for facile purification of the modified precursor peptide prior to removal of the leader sequence (Figure 1) to yield high levels of purified compound as well as analogs.10 These advances in technologies that have enabled computational identification, high-level production in homologous/heterologous hosts, and relative ease of purification using affinity tags can empower studies on RiPPs for which MOA data have not yet been obtained.
Figure 1.

General biosynthetic scheme for RiPP maturation. The follower peptide is shown as dashes because it is not ubiquitous across known RiPP classes.
1.1. Preface - Organizational approach
In this review, we cover the MOA, identification, and validation of biological targets for all classes of currently known bacterial RiPPs for which such data are available. Reported values for minimal inhibitory concentration (MIC) have been converted to molar values to both maintain internal consistency across different studies and to allow for comparisons of potency against non-RiPP antibiotics. Generally, bioactivity and MOA cannot be predicted based on the RiPP class, which is defined by biosynthetic pathway and not activity. For example, many lanthipeptides share common biosynthetic origins but demonstrate widely varying bioactivities. The diversity of targets likely reflects the ability to rapidly evolve new RiPP structures and associated bioactivities. We organized this review based on structural and biosynthetic similarities rather than by MOA because of the well-established framework within the field.10 To ensure that similar MOAs across RiPP classes are recognized, we provide cross references when different RiPP classes target related processes and collated all activities in abbreviated format in Table 1.
Table 1. Summary of MOAs.
The table is organized to correspond to the order in which the RiPP classes are presented in this review.
| Bioactivity / Target or mechanism | Compound name | RiPP class |
|---|---|---|
| Antibacterial / lipid II; pore formation Autoinduction of biosynthesis | nisin | lanthipeptide |
| Antibacterial / lipid II; pore formation Autoinduction of biosynthesis | subtilin | lanthipeptide |
| Antibacterial / lipid II | microbisporicin (NAI-107) | lanthipeptide |
| Antibacterial / lipid II | mutacin 1140 | lanthipeptide |
| Antibacterial / lipid II; pore formation | epidermin | lanthipeptide |
| Antibacterial / lipid II; pore formation | geobacillin I | lanthipeptide |
| Antibacterial / lipid II; pore formation | gallidermin | lanthipeptide |
| Antibacterial / pore formation | Pep5 | lanthipeptide |
| Antibacterial / pore formation | epilancin 15X | lanthipeptide |
| Antifungal | pinensin A and B | lanthipeptide |
| Morphogenic peptide / facilitates aerial hyphae formation | SapT | lanthipeptide |
| Morphogenic peptide / facilitates aerial hyphae formation | SapB | lanthipeptide |
| Antibacterial / lipid II Autoinduction of biosynthesis | mersacidin | lanthipeptide |
| Antibacterial / lipid II | lacticin 481 | lanthipeptide |
| Antibacterial / lipid II | nukacin ISK-1 | lanthipeptide |
| Antibacterial / lipid II; pore formation Autoinduction of biosynthesis | bovicin HJ50 | lanthipeptide |
| Antibacterial / lipid II; pore formation | plantaricin C | lanthipeptide |
| Antibacterial / lipid II; pore formation | lacticin 3147 | lanthipeptide |
| Antibacterial / lipid II; pore formation | staphylococcin C55 | lanthipeptide |
| Antibacterial / lipid II; pore formation | haloduracin | lanthipeptide |
| Antibacterial Hemolytic Autoinduction of biosynthesis | cytolysin | lanthipeptide |
| Antibacterial / Phosphatidylethanolamine (PE) binding and membrane disruption Antiviral / PE binding | cinnamycin | lanthipeptide |
| Antibacterial / PE binding and membrane permeabilization. Antiviral / PE binding | duramycin | lanthipeptide |
| ACE inhibition | ancovenin | lanthipeptide |
| Antiviral / PE binding | divamide A and B | lanthipeptide |
| Antiviral / PE and gp120 binding | labyrinthopeptin A1 | lanthipeptide |
| Antiviral / PE binding Antiallodynic | labyrinthopeptin A2 | lanthipeptide |
| Antibacterial / cell wall biosynthesis Antiallodynic | NAI-112 | lanthipeptide |
| Antibacterial | avermipeptin B | lanthipeptide |
| Morphogenic peptide / facilitates aerial hyphae formation | AmfS | lanthipeptide |
| No identified activity | catenulipeptin | lanthipeptide |
| Anticancer Antibacterial | ammosamide C | lanthipeptide |
| Anticancer / myosin binding and QR2 inhibition | ammosamide A and B | lanthipeptide |
| Anticancer / myosin binding | ammosamide 272 | lanthipeptide |
| Anticancer / kinase inhibition | lymphostin | lanthipeptide |
| No identified activity | 3-thiaglutamate | lanthipeptide |
| Antibacterial / cell wall biosynthesis | cacaoidin | lanthipeptide/lanthidin |
| Antibacterial | lexapeptide | lanthipeptide/lanthidin |
| Antibacterial | microvionin | lipolanthine |
| Antibacterial | goadvionin | lipolanthine |
| Cytolytic | streptolysin S (SLS) | LAP |
| Hemolytic | clostridiolysin S (CLS) | LAP |
| Bactericidal (mildly hemolytic) | listeriolysin S (LLS) | LAP |
| Hemolytic | stapholysin S (StsA) | LAP |
| Antibacterial | plantazolicin | LAP |
| Antibacterial / DNA gyrase | microcin B17 (MccB17) | LAP |
| Antibacterial / block of 50S ribosomal subunit exit tunnel | klebsazolicin | LAP |
| Antibacterial / block of 50S ribosomal subunit exit tunnel | phazolicin | LAP |
| Antibacterial Induction of sporulation and secondary metabolite production | goadsporin | LAP |
| Antibacterial / protein synthesis (translocation) Antimalarial Anticancer / cytotoxic Reactivates latent HIV reservoirs | thiostrepton | pyritide |
| Antibacterial / protein synthesis (translocation) Antimalarial Anticancer / cytotoxic | micrococcin P1 | pyritide |
| Antibacterial / protein synthesis (EF-Tu) | GE2270A | pyritide |
| Antibacterial / protein synthesis (EF-Tu) | GE37468 | pyritide |
| Antibacterial / protein synthesis (EF-Tu) | amythiamicin A | pyritide |
| Antibacterial / protein synthesis (EF-Tu) | thiomuracin A | pyritide |
| Bacterial MDR activation | promothiocins A & B | pyritide |
| Antibacterial Bacterial MDR activation / TipA | nosiheptide | pyritide |
| MDR activation / TipA Anticancer/cytotoxic | berninamycins A & B | pyritide |
| Bacterial MDR activation / TipA | thioxamycin | pyritide |
| Bacterial MDR activation / TipA | thiotipin | pyritide |
| Anticancer / cytotoxic | siomycin A | pyritide |
| Anticancer / cytotoxic | thiocillin I | pyritide |
| Anticancer / cytotoxic | YM-266183 | pyritide |
| Bacteriophage RNA polymerase and human renin inhibitor | cyclothiazomycin | pyritide |
| Antibacterial | nocathiacins | pyritide |
| Antineoplastic / apoptosis inducer | thioviridamide | thioamitide |
| Antineoplastic / F1 subunit of ATP synthase; induces apoptosis | prethioviridamide | thioamitide |
| Antineoplastic / apoptosis inducer | thioalbamide | thioamitide |
| Antibacterial / protein synthesis; block of tRNA entry into ribosomal A-site. | bottromycin A2 | bottromycin |
| Antineoplastic / protein and RNA synthesis | ulithiacyclamide | cyanobactin |
| Antineoplastic / predicted to affect cytokinesis | cycloxazoline | cyanobactin |
| Antineoplastic | trunkamide A | cyanobactin |
| Reverses MDR in breast carcinoma model | dendroamide A | cyanobactin |
| Proposed chalkophore or siderophore | patellamide A | cyanobactin |
| Proposed chalkophore or siderophore MDR reversal in leukemic lymphoblasts | patellamide B | cyanobactin |
| Proposed chalkophore or siderophore MDR reversal in leukemic lymphoblasts | patellamide C | cyanobactin |
| MDR reversal in leukemic lymphoblasts | patellamide D | cyanobactin |
| Proposed chalkophore or siderophore | patellamide E | cyanobactin |
| Plasmin inhibitor | agardhipeptin A | cyanobactin |
| Antibacterial | sphaerocyclamide | cyanobactin |
| Antibacterial | kawaguchipeptin B | cyanobactin |
| Cytotoxic | microcyclamide | cyanobactin |
| Cytotoxic | lissoclinamide | cyanobactin |
| Cytotoxic | ascidiacyclamide | cyanobactin |
| Antibacterial / L-histidinol phosphate aminotransferase | pantocin A | pantocin |
| Antibacterial / Asp-tRNA synthetase | microcin C7 (McC) | microcin |
| Antibacterial / Asp-tRNA synthetase | McCYPs | microcin |
| Antibacterial / mannose PTS and pore formation | MccE492/MccE492m | microcin |
| Antibacterial / ATP synthase (proposed) | MccH47/MccH47m | microcin |
| Unknown | MccM/MccMm | microcin |
| Chalkophore | methanobactin | methanobactin |
| Signaling | AIPs (I, II, III, IV). | autoinducing peptide |
| Signaling | GBAP | autoinducing peptide |
| Competence activation | ComX168 | ComX |
| Competence activation | ComXRO-E-2 | ComX |
| Atrial natriuretic factor receptor antagonist | anantin | lasso peptide |
| Endothelin type B receptor antagonist | RES-701–1, 2, 3, and 4 | lasso peptide |
| Human glucagon receptor inhibitor | BI-32169 | lasso peptide |
| Antibacterial / prolyl endopeptidases | propeptin and propeptin-2 | lasso peptide |
| Antibacterial / ClpC1 protease | lassomycin | lasso peptide |
| Viral fusion inhibition Antibacterial / cell wall biosynthesis / Quorum sensing inhibition | siamycin I | lasso peptide |
| Viral fusion inhibition | siamyin II | lasso peptide |
| Quorum sensing inhibition | sviceucin | lasso peptide |
| Antibacterial / cell wall | streptomonomicin | lasso peptide |
| Antibacterial / RNA polymerase (RNAP); dissipates membrane potential Superoxide production inducer Antimitochondrial activity | microcin J25 (MccJ25) | lasso peptide |
| Unknown | microcin Y | lasso peptide |
| Antibacterial / RNAP | capistruin | lasso peptide |
| Antibacterial / RNAP | klebsidin | lasso peptide |
| Antibacterial / RNAP | citrocin | lasso peptide |
| Antibacterial / RNAP | ubonodin | lasso peptide |
| Antibacterial | lariatin A and B | lasso peptide |
| Antifungal / SakA kinase (proposed) | humidimycin | lasso peptide |
| Unknown | tryptorubin A | atroptide |
| Human tyrosinase and serine protease inhibition | microviridin A | graspetide |
| Serine protease inhibition | microviridin B-N | graspetide |
| Serine protease inhibition | plesiocin | graspetide |
| Serine protease inhibition Metalloprotease inhibition | marinostatin | graspetide |
| Trypsin-like protease inhibition Daphnid protease inhibition | microviridin J | graspetide |
| Subtilisin inhibition | microviridin K and L | graspetide |
| Antibacterial / pore formation | AS-48 | circular bacteriocin |
| Antibacterial / MalEFG implicated | garvicin ML | circular bacteriocin |
| Antibacterial / pore formation | carnocyclin A | circular bacteriocin |
| Antibacterial | circularin A | circular bacteriocin |
| Antibacterial | uberolysin A | circular bacteriocin |
| Antibacterial | lactocyclicin Q | circular bacteriocin |
| Antibacterial | amylocyclicin | circular bacteriocin |
| Antibacterial | enterocin NKR-5–3B | circular bacteriocin |
| Antibacterial | pumilarin | circular bacteriocin |
| Antibacterial | gassericin A | circular bacteriocin |
| Antibacterial | acidocin B | circular bacteriocin |
| Antibacterial | butyrivibriocin AR10 | circular bacteriocin |
| Antibacterial | plantaricyclin A | circular bacteriocin |
| Antibacterial / cell wall implicated | pheganomycins (including deoxypheganomocyin D) | amidinotide |
| Inhibitor of interaction of CsrA with its inhibitory RNA | crocagin A and B | crocagin |
| Antibacterial / glucose PTS implicated | sublancin 168 | glycocin |
| Bacteriostatic / GlcNAc PTS implicated | glycocin F | glycocin |
| Bacteriostatic | ASM1 | glycocin |
| Bacteriostatic | enterocin 96 and F4–9 | glycocin |
| Antibacterial | pallidocin | glycocin |
| Bacterial blight in rice crop / mimic of PSY1 plant hormone | RaxX | sulfatide |
| Cytotoxic / pore formation and lysosome neutralization | polytheonamide B | polytheonamide |
| Antibacterial / proteobacterial BamA; impedes protein insertion and assembly in the outer membrane | darobactin | darobactin |
| Antibacterial / membrane permeabilization Spermicidal Antiviral Biofilm formation inhibition | subtilosin A | sactipeptide |
| Antibacterial Biofilm formation inhibition | hyicin | sactipeptide |
| Antibacterial / pore formation | thuricin CD | sactipeptide |
| Antibacterial / membrane permeabilization | huazacin (thuricin Z) | Sactipeptide |
| Antibacterial Morphogenesis | thurincin H | sactipeptide |
| Antibacterial (lytic) | sporulation killing factor (SKF) | sactipeptide |
| Antibacterial | ruminococcin C | sactipeptide |
| Antibacterial (bactericidal) | streptosactin | sactipeptide |
| Antibacterial | EpeX* | epipeptide |
| Redox cofactor | mycofactocin | mycofactocin |
| Redox cofactor | pyrroloquinoline pyrrole | pyrroloquinoline pyrrole |
During RiPP biosynthesis, a portion of the precursor peptide is post-translationally modified in what is referred to as the “core” region. The biosynthetic enzymes necessary for modification are directed to the correct substrate by specific motifs known as recognition sequences present in a region known as the “leader peptide” if located N-terminal to the core peptide,47 or “follower peptide” if located C-terminal to the core region.9,10 The vast majority of characterized bacterial RiPPs utilize a leader-core paradigm, as opposed to a core-follower or a tripartite leader-core-follower paradigm.48 In all RiPPs, the recognition and modification sites are physically separated, thus allowing the biosynthetic pathways to maximize substrate selectivity via recognition of conserved leader/follower sequences, while tolerating variation in the core region. In this review, we use the standardized nomenclature for the precursor peptide9 where residues in the core peptide are indicated with a positive number starting from the junction with the leader sequence, while residues in the leader peptide are indicated with negative numbers counting back from this junction (Figure 1). Hence, the residue numbering of several RiPPs has been adapted to be consistent with this numbering scheme (which may differ from the numbering used in the primary literature). We do not discuss in further detail the biosynthetic pathways for the different classes of RiPPs. A number of excellent, comprehensive reviews detail the biosynthetic mechanisms for various RiPPs.9,10,29,33
2. Lanthipeptides and compounds made via related pathways
Lanthipeptides are characterized by the thioether crosslinked bis amino acids lanthionine (Lan) and methyllanthionine (MeLan; Figure 2). At present, these macrocyclic peptides can be generated by different sets of enzymes that are the basis for their classification scheme (class I-V). Lanthipeptides with antimicrobial activities have historically been termed lantibiotics.49 A salient feature of lantibiotics with a known MOA is that the targets are metabolites rather than macromolecules like proteins or RNA as discussed in the subsequent sections. In most currently characterized lanthipeptides, the (Me)Lan have DL stereochemistry (Figure 2), but compounds with LL- and D-allo-L-stereochemistry have been increasingly reported. In this review, the shorthand notations shown in Figure 2 will be used for lanthipeptides.
Figure 2.

Structures of thioether crosslinks and dehydrated amino acids commonly found in lanthipeptides. Shorthand notations are presented below the corresponding structure. Structures containing non-canonical LL-(methyl)lanthionine and D-allo-L-methyllanthionine stereochemistry are indicated with either a single or double asterisk in the shorthand notation, respectively.
2.1. Class I lanthipeptides
Nisin, a class I lanthipeptide produced by Lactococcus lactis, is the most extensively characterized family member and exhibits antimicrobial activity against many non-Proteobacteria often with submicromolar minimal inhibitory concentrations (MICs).49,50 Many nisin variants have been reported over the years, differing in a few amino acids but retaining the overall ring pattern. Unless specified otherwise, when discussing nisin in this review we refer to nisin Z (1). For more than 50 years, nisin has been used in the food industry to combat food-borne pathogens.51 The molecule exerts its bioactivity through binding of the cell wall precursor lipid II (2, Figure 3A) and the formation of relatively stable pores in the cell membrane that are composed of nisin and lipid II.52,53 Nisin binding is facilitated by the amide backbone of the A and B rings interacting with the pyrophosphate moiety of lipid II, as characterized by NMR spectroscopy using a soluble lipid II analog with a farnesyl chain instead of the full C55 undecaprenol tail (Figure 3C).54 The binding of lipid II is believed to act as an anchor for the formation of pores in the membrane involving a complex comprised of eight nisin and four lipid II molecules (Figure 3F).54,55 The 2:1 nisin:lipid II stoichiometry in the pore indicates that the 1:1 NMR structure (Figure 3B and C) only reveals part of the molecular interactions and that a second nisin molecule must bind to lipid II, nisin, or both. This aspect of pore formation is still poorly understood.
Figure 3.

(A) Chemical structure of nisin Z (1) and lipid II (2). The A and B rings of nisin interact with the pyrophosphate moiety of lipid II (green) and the C-E rings of nisin (orange) are involved in pore formation. A hinge region (purple) is critical for pore formation. (B) NMR characterization of nisin (space filling) binding to a lipid II analog (sticks). (C) Zoom-in view showing the interactions between the pyrophosphate of lipid II (green and red) with the amide backbone of the nisin A ring (carbon atoms in grey). (D-E) Proposed model for lipid II mediated pore formation wherein nisin first interacts with lipid II through the above-described interaction, followed by pore formation through a complex of eight nisin molecules and four lipid II molecules. The molecular details of the arrangement of nisin and lipid II in the pore remain unknown.
Lipid II and its variants have been used to investigate the molecular determinants of selectivity and affinity. Nisin binds to lipid I (lacking the GlcNAc, Figure 3A) and lipid II with affinities in the 10–50 nM range as determined by isothermal titration calorimetry (ITC), radiolabeling, and dye leakage experiments from vesicles.56–58 Hence, the GlcNAc moiety is not important for binding. Similarly, a lipid II analog in which the pentapeptide was removed still bound tightly to nisin.56 Thus, the pyrophosphate group appears the main recognition site on both lipid I and lipid II. However, pores are not formed with undecyl pyrophosphate and hence the MurNAc group is important for this activity.56,57 Several studies focusing on the length of the prenyl chain and the anchoring of this chain in the membrane have demonstrated that the interaction of nisin with lipid II is strongly dependent on the membrane environment.56,59 These experiments typically report on both the binding of nisin to lipid II and pore complex formation in the membrane, and deconvolution into specific molecular interactions is therefore difficult. The pores have been shown to be relatively stable (lifetime of seconds as opposed to milliseconds in the absence of lipid II) by electrophysiology, and once the complex is established in vesicles in model studies, nisin does not exchange when exposed to new vesicles containing lipid II and the complex appears stable for hours.56,60
The biosynthetic machinery of nisin is tolerant to changes in the precursor peptide and this feature has been leveraged to generate many variants. In-depth structure-activity relationships (SAR) for nisin have been extensively reviewed and will not be covered in detail here.10,51,61–68 These studies have identified a critical hinge region between the C and D-rings (Figure 3A) that is thought to allow the D and E rings to insert into the membrane to form the pore.55,69–74 Furthermore the N-terminus cannot be extended but the D and E rings can be removed without abolishing antibacterial activity.51,75–77 The latter truncates do, however, loose pore-forming activity. Several variants have been identified that are more potent than nisin against a subset of Gram-positive and/or Gram-negative bacteria.72,77–83
Many other class I lanthipeptides such as microbisporicin (NAI-107, 3), mutacin 1140 (4), epidermin (5), epilancin 15X (6) and geobacillin I (7) produced by Microbispora corallina, Streptococcus mutans, Staphylococcus epidermidis (5 and 6), and Geobacillus thermodinitrificans, respectively, contain variants of the lipid II-binding domain of nisin (i.e., the A and B rings; Figure 4).52,84–87 These compounds have a wide variety of C-terminal ring patterns and sequences and while all bind lipid II, several (e.g., microbisporicin and mutacin 1140) do not form pores.79,84,88–90 Studies on epidermin and the closely related gallidermin, which have shorter C-terminal tails compared to nisin, showed that pore formation depends on membrane thickness.91 Collectively, these studies showed that pore formation was not required for antibacterial activity and that lipid II binding was sufficient. In addition to inhibition of the transglycosylation step of cell wall biosynthesis (see also class II two-component lanthipeptides; section 2.2.1), nisin disrupts the localization of lipid II, taking it away from the well-controlled localization of cell wall biosynthetic enzyme complexes.92
Figure 4.

Shorthand notations for the structures of microbisporicin (3), mutacin 1140 (4), epidermin (5), epilancin 15X (6), geobacillin I (7), and subtilin (8). The nisin-like lipid II binding domains are indicated in blue, nisin-like pore forming domains are indicated in orange, and the hinge region, present in (8), is indicated in purple. The stereochemistry of the dihydroxyproline in (3) is not known. Throughout this review, in the shorthand notations the N-terminus is indicated by H- and the C-terminus by –OH.
Class I lanthipeptides often also display quorum-sensing activities. The biosynthetic gene clusters typically encode two-component signaling systems made up of a receptor histidine kinase and a response regulator.93 The best-studied examples are the NisRK autoinduction system that regulates nisin production and the corresponding SpaRK system involved in subtilin (8) autoinduction in Bacillus subtilis.51,94,95 Although the molecular details of lanthipeptide recognition are not known, NisK and SpaK are highly specific for their cognate ligand.96–98 Several SAR studies have demonstrated that residues important for signaling are different from those essential for antibacterial activity.99 For instance, the B-ring of nisin is critical for lipid II binding and antibacterial activity but is dispensable for autoinduction.77 Extensive mutagenesis studies along with investigation of crosstalk between the nisin and subtilin systems and construction of hybrid molecules identified the N-terminal Trp and Phe20 as critical residues in subtilin for SpaK recognition.97 Furthermore, whereas the B-ring and the native hinge region between C- and D-rings were shown to be required for subtilin signaling, the D- and E-rings were expendable.96
Many class I lanthipeptides do not contain the A/B ring lipid II-binding motif. Examples such as the structurally related Pep5 and epilancin 15X (6, Figure 4), produced by S. epidermis, induce pore formation, but do not appear to bind to lipid II.52 Given the low minimum inhibitory concentration (MIC) values of these compounds (nM against many Staphylococcus strains), they may also have a specific molecular target that remains undiscovered.52 Epilancin 15X is of particular interest because it lacks the lipid II-binding A and B rings of nisin but has a similar C-E ring pattern (Figure 4).100,101 Total synthesis and structural variants of epilancin 15X yielded insight into the importance of the C-terminal ring and the N-terminal region for activity,102 but without a known molecular target, these SAR studies are not fully interpretable.
Nisin and subtilin also inhibit the outgrowth of bacterial spores at sub-nanomolar concentrations, which are lower than their antibacterial activities.103–105 Initially Dha5 was reported to be important for this activity, but later studies refuted this finding.77 Mechanistic investigations suggest that inhibition of spore outgrowth is also mediated by lipid II binding. Nisin variants in the hinge region (N20P/M21P and M21P/K22P) capable of binding lipid II but deficient in pore formation retained antimicrobial activity against vegetative Bacillus anthracis cells but did not inhibit spore outgrowth. This observation suggests that pore formation is critical for the latter activity.106 Furthermore, nisin did not prevent spore germination and required germination to inhibit spore outgrowth.107
Several class I lanthipeptides exhibit weak or no antibacterial activity, but instead act as antifungal or morphogenetic peptides. Pinensin A and B (10 and 11) are the first characterized lanthipeptides produced by the Bacteroidetes Chitinophaga pinensis (Figure 5).109 The pinensins exhibit weak antibacterial activity but display antifungal activity with MICs in the micromolar range against yeast and filamentous fungi.109 Although the pinensin MOA remains unknown, it is hypothesized to have a novel biological target (for RiPPs) due to the specificity for fungi.109
Figure 5.

Chemical structures of SapT (9) and pinensin A (10) and B (11). The stereochemistry of the MeLan rings in SapT (9) was determined to be D-allo-L-methyllanthionine, the first lanthipeptide with such stereochemistry.108
SapT (9, Figure 5) produced by Streptomyces tendae, is another example of a lanthipeptide lacking robust antibacterial activity.110 The compound has morphogenetic activity that helps facilitate the formation of aerial hyphae in its producing organism and is required for spore formation.110 SapT is amphipathic, was the first lanthipeptide identified to contain D-allo-L-methyllanthionine,108 and acts as a biosurfactant. This activity is shared by a number of class III lanthipeptides including SapB (section 2.3), and SapT is able to restore hyphae formation and sporulation in Streptomyces coelicolor bld mutants that are unable to produce SapB.110,111
2.2. Class II lanthipeptides
Similar to the nisin group of class I lanthipeptides, a structurally unrelated subset of class II lanthipeptides target the cell wall biosynthetic intermediate lipid II. Class II lanthipeptide binding to lipid II can be predicted by the presence of a sequence motif that was first identified in the C-ring of mersacidin (12) produced by Bacillus sp HIL Y-85,54728 (Figure 6).112 The highly conserved Glu in this ring (Asp in some structurally related compounds) is critical for antibacterial activity, but unlike the NMR structure of nisin bound to lipid II, currently high-resolution structural information is not available for the binding mode of mersacidin and related compounds to lipid II.113 Using NMR spectroscopy, mersacidin was shown to undergo a conformational rearrangement upon lipid II binding, with the carboxyl group of Glu17 thought to interact with lipid II through a Ca2+ mediated bridge.114 This hypothesis is supported by the loss of activity upon mutagenesis of Glu17 to alanine as well as by the requirement of Ca2+ for activity.114,115 Unlike nisin, mersacidin requires the GlcNAc of lipid II for binding and does not bind to lipid I, and mersacidin lacks the ability to form pores.112,116
Figure 6.

Structures of the class II lanthipeptides mersacidin (12), lacticin 481 (13), bovicin HJ50 (14), nukacin ISK-1 (15), plantaricin C (16), and an alternative representation of plantaricin C (17). The 6-amino acid ring containing an Asp/Glu (orange) that is proposed to be the lipid II binding motif is shown in blue. The report on the structure of plantaricin C 16 deduced by NMR spectroscopy acknowledges that the distances between the β-carbons of the lanthionines in the proposed A and D-rings are ~8 Å, whereas these corresponding distances for the B and C rings are ~4 Å.127 We suggest an alternative possible ring pattern (17) based on the structures of mersacidin, lacticin 481, and nukacin ISK-1.128,129
Analogs of the C-ring of mersacidin are also found in other class II lantibiotics such as the lacticin 481 (13) group of compounds that include bovicin HJ50 (14) and nukacin ISK-1 (15) (Figure 6), and the α-peptides of many two-component lantibiotics (section 2.2.1).115,117,118 This conserved 6-amino acid containing ring with a Glu residue is also present in plantaricin C (16) in its originally reported structure. An alternative ring pattern would also contain a lipid II-binding ring (17, Figure 6). Although the overall ring patterns are varied, these compounds have all been shown to bind to lipid II using various biophysical techniques.119–124 As discussed for the nisin group of lantibiotics, lipid II binding by class II lantibiotics is often sufficient for antimicrobial activity with a subset of compounds also inducing pore formation and others not affecting the membrane potential.118,121,125,126
Variants of nukacin ISK-1, lacticin 481, and bovicin HJ50 have been investigated for bioactivity (Figure 6).121,130–136 As first shown for mersacidin, the highly conserved Glu/Asp in the ring C lipid II binding motif (A-ring for lacticin/nukacin/bovicin) is essential for bioactivity in all investigated compounds.118,119,136 In addition, Lys residues in these compounds are important for bioactivity, possibly by increasing the affinity for the negatively charged lipids in the bacterial membrane.118,130,137,138 Disruption of any of the rings in bovicin HJ50, including the macrocycle generated by disulfide formation, resulted in complete loss of bioactivity.136 The importance of these rings was also reported for lacticin 481,139 and synthetic studies showed that the stereochemistry of the three (Me)Lan crosslinks is also critical for bioactivity. Diastereomers in which each of the three lacticin 481 rings was individually changed from the DL to the LL-stereochemistry were inactive.102 Use of a fluorescently labeled lacticin 481 analog showed localization in rod-shaped bacteria that is consistent with the reported localization of lipid II.140
The MOA of nukacin ISK-1 (15) has been investigated by using the two-component antibiotic-sensing system LiaRS from B. subtilis. LiaRS is activated by cell wall-active antibiotics that interfere with the lipid II cycle, including nisin, nukacin ISK-1, and lacticin 481 (see also section 15.4 on epipeptides).137,141 Nukacin ISK-1 variants were tested for interference with the lipid II cycle using this reporter system, demonstrating the importance of the A-ring, the conserved Asp in this ring, and the positively charged amino acids at the N-terminus.137 NMR studies on nukacin ISK-1 showed the compound exists in two, interconverting conformations that differ in the relative orientation of the A and C rings (Figure 7).123,142 Only one of the two conformers binds to lipid II and a model was proposed in which amino acid residues in ring A are involved in lipid II binding via hydrogen bonding and that residues in ring C then engage in hydrophobic interactions, possibly with the prenyl chain of lipid II. Disruption of the C-ring abolished lipid II binding, and individual replacement of the three Phe in the C-ring with Ala also eliminated lipid II binding as suggested by the LiaRS reporter assay.137 It was hypothesized that similar movement of ring C could also explain the dynamic behavior reported for lacticin 481 and bovicin HJ50.118,128 Independently, incorporation of non-canonical amino acids in lacticin 481 resulted in variants with improved bioactivity and inhibition of the transglycosylation reaction by the penicillin-binding protein (PBP) 1b. These studies showed that the increased antibacterial activity was caused by improved binding to lipid II, the substrate for PBP1b.121,132 The positions at which improvements were observed were again the aromatic amino acids in the C-ring that were also identified in nukacin ISK-1s as important for lipid II binding, and the non-canonical amino acids that were incorporated at these sites had aromatic side chains, like the Phe residues in nukacin ISK-1.
Figure 7.

Two different conformations of nukacin ISK-1 that interconvert on the timescale of seconds. Both structures were solved by NMR spectroscopy (PDB IDs: 5Z5Q and 5Z5R).123
Like the nisin group of class I lanthipeptides, select class II lanthipeptides also have autoinduction activity as shown for mersacidin, bovicin HJ50, and other members of the lacticin 481 group, but not nukacin ISK-1.143–147 At present, the molecular details of mersacidin induction of its own biosynthesis are not known.143 Saturation mutagenesis on bovicin HJ50 (14) identified several charged and hydrophobic amino acids in ring B as well as two Gly residues at positions 4 and 23 as critical for recognition by its autoinducing receptor kinase BovK.136 Surprisingly, the A-ring, which is critical for bioactivity (see above), is dispensable for autoinduction, but the linear N-terminal sequence is required. Experiments with biotinylated bovicin HJ50 showed that the lanthipeptide interacts with the membrane domain of BovK and not the cytosolic domain. Additional mutagenesis studies suggested that a conserved hydrophobic region in the sixth transmembrane segment of BovK may be responsible for binding the lanthipeptide.136
2.2.1. Two-component class II lanthipeptides
A subset of class II lanthipeptides are two-component systems wherein two peptides work synergistically to elicit bioactivity. For most examples, these peptide pairs have been termed the α and β-peptides. The best-studied examples in terms of MOA are the enterococcal cytolysin (hereafter cytolysin), lacticin 3147 (18 and 19, Figure 8), and haloduracin (20 and 21). Although cytolysin is the longest-known two-component lanthipeptide,148 we will first discuss lacticin 3147 and haloduracin because their MOAs have similarities with the class II lanthipeptides discussed thus far.
Figure 8.

Structures of select two-component lanthipeptides. Lacticin 3147 composed of Ltnα (18, also called LtnA1) and Ltnβ (19, also called LtnA2), haloduracin composed of Halα (20) and Halβ (21), and staphylococcin C55 consisting of SacAα (22) and SacAβ (23). The proposed lipid II binding motifs are colored blue with the Glu that is critical for antibacterial activity in orange.
Lacticin 3147 (Figure 8) produced by L. lactis has antimicrobial activity against Gram-positive bacteria including other L. lactis strains, Listeria monocytogenes, and B. subtilis.149–151 Like many other lanthipeptides, lacticin 3147 disrupts the cell membrane potential.150 The α-peptide (Ltnα, 18) possesses the lipid II binding motif discussed above for mersacidin and has been shown to interact with lipid II.120,152 Although the α-peptide binds lipid II, it is not able to inhibit cell wall biosynthesis on its own. Upon the addition of the β-peptide (Ltnβ, 19), pores with 0.6 nm diameter are formed.120 Stoichiometry studies suggest that two-component lanthipeptides act in a 1:1 stoichiometry wherein the α peptide binds first followed by recruitment of the β-peptide. The structure of the pore forming complex has yet to be characterized.120,153 A model has been proposed in which Ltnα binds to the pyrophosphate moiety of lipid II via hydrogen bonds to the backbone amide groups in the lipid II-binding motif.152 An analog of Ltnβ in which the MeLan were replaced by Lan using chemical synthesis retained the ability to synergistically kill bacteria in the presence of wild type Ltnα, albeit with 100-fold reduced potency, but a synthetic analog in which the thioether bridges were replaced by ether linkages in Ltnβ lost the synergistic activity with wild type Ltnα.154,155 Extensive SAR (structure activity relationship) data has also been reported using variants generated by mutagenesis including Ala scanning and saturation mutagenesis of Ltnα and Ltnβ. Unfortunately, most of these studies did not distinguish between mutations that impacted biosynthesis and those that impacted antimicrobial activity.156,157 Nevertheless, mutations that disrupted the ring structures resulted in loss of activity with the exception of the A-ring of Ltnα.158 However, the A-ring does endow the peptide with enhanced resistance to thermal and proteolytic degradation.159 Furthermore, Glu24 in the lipid II-binding motif of Ltnα is critical for bioactivity.157 As for the single component mersacidin-like molecules discussed above, a molecular explanation for the importance of the Glu is currently not available.
Both peptides of lacticin 3147 contain D-Ala residues that are generated by post-translational modification of L-Ser. The importance of the three D-Ala in lacticin 3147 was confirmed by systematic substitution of these residues by L-Ala, which diminished both the production and the bioactivity. For Ltnα, the reduced production prevented activity determination. In Ltnβ, a single D to L conversion resulted in a 4-fold loss in activity, and a double mutation resulted in a 16-fold loss in activity.160 Additionally, combination of the individual peptides of lacticin 3147 with those from another two-component lanthipeptide, staphylococcin C55 (22 and 23), was examined. The lacticin 3147 and staphylococcin C55 peptides contain α-peptides with 86% identity and β-peptides with 55% identity (Figure 8). Peptides from both compounds displayed synergism within native pairs as well as cross synergism when a hybrid pair was spotted with similar, low nanomolar activity as the original pairs.161 Given the differences in sequence between Ltnβ (19) and SacAβ (23) (Figure 8), these data suggest that the synergistic activity has some flexibility with respect to the β-peptide.
Another well-studied, two-component lanthipeptide is haloduracin, the first RiPP discovered by genome mining produced by Bacillus halodurans C-125.162,163 Haloduracin has sequence homology with the lacticin 3147 peptides including the mersacidin lipid II-binding motif in Halα (20). The haloduracin α and β-peptides have optimal bioactivity when used in a 1:1 ratio and like the proposed lacticin 3147 model, the α-peptide binds first to a target with the β-peptide required for inducing pore formation.120,164 In vitro inhibition studies of the transglycosylation reaction catalyzed by PBP2b provided direct evidence for haloduracin α binding to lipid II and demonstrated that the binding ratio for pore formation is 1:2:2 lipid II:Halα:Halβ.122,164 Moreover, microscopy studies with fluorescently labeled Halα or Halβ resulted in localization of the α peptide primarily at sites of cell division and in punctate patterns along the long axis of bacilli.140 This localization is similar to that reported for lipid II. Conversely, Halβ was localized nonspecifically to the cell surface in the absence of Halα but formed the same specific patterns discussed above when co-administered with its partner. Using two-color labeling, colocalization of both components of the two-component lantibiotic was observed. These in vivo data support a model in which the α component first recognizes lipid II, followed by the recruitment of the β-component, which aids in pore formation.
Extensive SAR experiments have been reported for both the Halα and Halβ peptides.165 The C ring and Glu22 of Halα are essential for activity, whereas the A ring of Halα and the C and D rings of Halβ were determined to be important for activity but not essential. The B ring of Halα is not important despite a high level of conservation. Finally, unlike the findings with bovicin HJ50 described above, the disulfide of Halα was not important for activity but was important for stability.165
Cytolysin, produced by Enterococcus faecalis, was the first reported example of a two-component lanthipeptide.166 Cytolysin is comprised of a large and small subunit denoted as CylLL” (24) and CylLS” (25), respectively (Figure 9), which display synergistic bioactivity against both bacterial and eukaryotic cells.167 In addition, the CylLS” peptide serves as a quorum-sensing molecule that activates the cytolysin operon, leading to increased production of the cytolysin peptides.168 Cytolysin has been known to be a virulence factor since the 1930s and the presence of the biosynthetic locus contributes greatly to the lethality of E. faecalis infections.148 Recently, the toxin was directly correlated to the deleterious outcome of alcoholic liver disease.169 While the details of the mechanism of cell lysis remain unknown, SAR studies have provided insight into the features important for cytotoxicity. The rings of both components of cytolysin are required for activity.170 As noted previously, the canonical stereochemistry of (methyl)lanthionines is DL, which refers to D-stereochemistry at the α-carbon of the former Ser/Thr residues and L-stereochemistry at the former Cys residues (Figure 2). The cytolysin peptides were the first lanthipeptides reported to deviate from this canonical stereochemistry as the A ring of CylLS” and the A and B-rings of CylLL” were shown to contain LL-(Me)Lan.171 The stereochemistry of the A ring of the CylLS” peptide is important for bioactivity, as the epimer with DL-stereochemistry in the A-ring decreased its antimicrobial activity 20-fold when combined with CylLL”.172 However, its hemolytic activity was unaltered providing support for a model in which the two activities have different SAR and may involve different targets.173 A recent Ala-scanning study in which all residues in both peptides were individually substituted also suggests that the two activities of cytolysin have different SAR.170
Figure 9.

(A) Shorthand structures of cytolysin composed of CylLL” (24) and CylLS” (25). Sites with non-canonical LL-stereochemistry are annotated with asterisks (see Figure 2). (B) Two conformations of CylLL” in the bent and extended positions elucidated by solution NMR 28 spectroscopy (PDB ID: 6VGT; BMRB 30710). (C) NMR solution structure of CylLS” (PDB ID: 6VGT; BMRB 30702).
The large subunit CylLL” binds to erythrocytes with seven-fold higher affinity than the small subunit CylLS”; this may either suggest it recognizes a different molecular target or reflects a higher affinity for the hydrophobic membrane environment.170,174 Recently the NMR structures of CylLL” and CylLS” were determined in methanol showing that the large subunit forms two helices that are probably held in place by the lanthionine crosslinks.175 The two helices are separated by a flexible, Gly-rich sequence that results in a series of structures that differ in the relative orientation of the two helices (Figure 9B), with the colinear arrangement sufficient in length to form a membrane-spanning pore. This hinge arrangement is reminiscent of the hinge region between the C and D rings of nisin that is critical for pore formation (Figure 3A).
2.2.2. Phosphatidylethanolamine-binding class II lanthipeptides
A distinct group of lanthipeptides bind phosphatidylethanolamine (PE) in a 1:1 ratio.176,177 These compounds contain a rare lysinoalanine crosslink and usually also a hydroxylated Asp (Figure 10). One example is cinnamycin (26, previously Ro 09–0198), a class II lanthipeptide produced by various Streptomyces sp. Cinnamycin induces transbilayer phospholipid movement in a PE-concentration dependent manner, leading to membrane permeabilization and cytotoxicity.177,178 The mechanism by which cinnamycin accesses PE that is located in the inner leaflet of the membrane remains incompletely resolved. The interaction between PE and cinnamycin was characterized by NMR spectroscopy demonstrating that the hydrophobic pocket created by residues Phe7 through Cys14 as well as the erythro-3-hydroxy-L-aspartic acid at position 15 are important for the high selectivity of ligand recognition (Figure 11)179,180 and the high observed affinity (Kd = 20 nM by ITC).181 Experiments with other lipids showed that an acylated glycerol backbone and a primary amine group are necessary.176,182 The length of the acyl chains are not critical for affinity but at least one acyl chain is needed as no binding was observed with glycerophosphorylethanolamine by ITC.181 Recent modeling experiments suggest that more hydrogen bonds may be present than assigned in the NMR structure.183 Binding to PE also results in antiviral activity of cinnamycin and the structurally related duramycin (28) against herpes simplex virus type 1 (HSV-1) by inhibiting viral proliferation.184
Figure 10.

Chemical structure of cinnamycin (26), and its shorthand rendition (27). Structurally related molecules are shown with lysinoalanine crosslink shown in orange, (methyl)lanthionine rings shown in blue and hydroxyaspartate residues indicated in purple: duramycin (28), ancovenin (29), divamide A (30), and divamide B (31). Asp-OH, (3R)-hydroxy-aspartate.
Figure 11.

NMR structure of cinnamycin (26) bound to C12-lysophosphatidylethanolamine (labeled as PE). The carbons of hydroxyaspartate 15 (HyAsp15) are indicated in yellow and the carbon atoms of PE in pink (PDB ID: 2DDE). For clarity, only one carbon is shown of the C12 acyl chain of PE.179
Other lanthipeptides with similar ring patterns as cinnamycin have been reported to possess a wide array of bioactivities. These include the duramycins (e.g. 28), ancovenin (29), and the divamides (30, 31) (Figure 10). Duramycin was discovered as an inhibitor of phospholipase A2 with an IC50 around 1 μM. This activity is likely an indirect effect caused by the binding of the substrate PE rather than direct interaction with the protein itself.185–188 PE binding is also likely responsible for efflux of chloride from airway epithelium cells, which has led to investigation of duramycin as a potential treatment for cystic fibrosis.189–193 Similar to cinnamycin, the divamides and duramycin display antiviral activity. Duramycin efficiently inhibits cellular entry of West Nile, Dengue, and Ebola viruses in a mechanism mediated by phosphatidylserine (PS) receptors like T-cell immunoglobulin mucin domain protein 1 (TIM1). These various antiviral activities are likely directly related to PE binding because the virions of flaviviruses and filoviruses utilize PE for cell entry by TIM1.194 Disrupting PE association with PS receptors may serve as a promising broad-spectrum antiviral strategy. Divamides exhibit anti-human immunodeficiency virus (HIV) activity, thought to be mediated through lipid binding.195
The specific binding of PE by duramycin/cinnamycin and their analogs with high affinity has been used extensively to detect PE on cultured cells and in animal models.196,197 Fluorescently labeled analogs have been shown to be able to detect apoptotic cells,198 and analogs labeled with (99m)Tc have been used to detect various types of cell death,199–201 tumors,202,203 ionizing irradiation-induced tissue injuries,204,205 atherosclerotic plaques,206 and myocardial ischemic/reperfusion injury207 in various animal models.
Ancovenin is an inhibitor of the angiotensin 1-converting enzyme (ACE). Such inhibitors are commonly used in the treatment of high blood pressure, and the compound inhibits rat lung ACE with an IC50 of 870 nM.208 Ancovenin was not growth suppressive against B. subtilis ATCC 6633 or Staphylococcus aureus IFO 12732.209 The mechanism of this divergent activity within the cinnamycin family of peptides has not been investigated.
2.3. Class III lanthipeptides
In contrast to class I and II lanthipeptides, most known class III lanthipeptides exhibit weak or no antibacterial activity. Labyrinthopeptins are class III lanthipeptides that contain a labionin moiety first identified in 2010 (Figures 2 and 12).210 Labyrinthopeptin (Laby) A1 (32) and A2 (33) demonstrate antiviral activity against a variety of viruses, including Herpes simplex, Zika, and hepatitis C.210–212 The broad spectrum antiviral activity is thought to be in part exerted through the binding of PE leading to viral membrane disruption.211 N-terminally hexynoyl-derivatized LabyA1 and A2 fluorescently labeled using click chemistry was used to demonstrate PE binding. These modified peptides also displayed weak affinity for phosphatidylcholine (PC) and sphingomyelin, two molecules containing an ethanolamine-derived head group.211 Evaluation of hexynoyl-LabyA1 activity in the presence of PE-containing vesicles led to an 8-fold decrease in antiviral activity whereas vesicles containing PC had no effect on potency. Hexynoyl-LabyA1 and A2 peptides induced leakage in a PE-dependent and concentration dependent manner for vesicles of varying lipid compositions, monitored by release of fluorescent molecules within the vesicles, supporting the hypothesis that PE is a molecular target.211
Figure 12.

Structures of labyrinthopeptin A1 (32) and labyrinthopeptin A2 (33). For the structure of labionin, see Figure 2.
Hexynoyl-LabyA1 and A2 act weakly synergistically in a 1:1 ratio, featuring up to 2-fold higher PE-binding affinities than the individual peptides.211 While shown to increase activity when together, the molecular interaction between the Laby peptides has yet to be structurally characterized and it is unclear if these compounds interact directly or via PE in a sandwich-like manner. While it has been proposed that the virolytic activity of the Laby peptides could be exerted through PE binding, other PE-binding peptides such as duramycin, cinnamycin, and the divamides (section 2.2.2) are believed to exhibit antiviral activity by preventing PE-mediated infection.
Labyrinthopeptin A1 demonstrated anti-HIV and -HSV activity.213,214 In HIV transmission models, LabyA1 inhibited cell-free, cell-to-cell, and dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)-mediated viral infection. Time-of-drug addition studies and the lack of inhibition of viral binding to CD4+ T cells suggest that LabyA1 targets viral entry,213 like duramycin.194 Surface plasmon resonance experiments identified an interaction between LabyA1 and the gp120 protein of the HIV envelope, but the peptide did not interact with the HIV cellular receptors CXCR4 and CCR5. LabyA1 exhibits synergistic effects when given alongside a number of antiviral therapeutics such as tenovir or acyclovir and also maintains activity against acyclovir-resistant strains.213 The interaction between LabyA1 and gp120 has not yet been characterized; however, LabyA1 is active against HIV strains resistant to carbohydrate-binding agents, suggesting that it is not targeting the N-linked glycans of gp120.213 Time-of-drug addition studies indicate that it is also an entry inhibitor for HSV,213,214 most likely again mediated by PE binding.
In addition to antiviral activities, LabyA2 is effective in the treatment of neuropathic pain in animal models, making it the first lanthipeptide reported to have antiallodynic activity.210 NAI-112 (34), a glycosylated class III lanthipeptide produced in Actinoplanes sp. (Figure 13), also showed efficacy in the treatment of neuropathic pain.13 In mouse model studies, both compounds displayed a decrease in allodynia while NAI-112 also reduced hyperalgesia.13,210 While neither LabyA2 nor NAI-112 have a known MOA for these activities, it has been proposed that NAI-112 may interact with the vanilloid pathway,13 and that the molecule blocks pain sensation by decreasing the levels of lysophosphatidic acid (LPA) and increasing the levels of phosphatidic acid.215 LPA activates the vanilloid receptor 1 (also known as capsaicin receptor and TRPV1) triggering pain. NAI-112 also exhibits modest antibiotic activity with micromolar MIC values against various staphylococci and streptococci.13,216 NAI-112 was initially identified as a cell wall inhibitor,216 with resistance mutants suggesting that the peptidoglycan intermediate lipid II (Figure 3) may be a target.215
Figure 13.

The structure of NAI-112 (34). The sugar stereochemistry has not yet been determined.
Avermipeptin B from Streptomyces actuosus was identified by genome mining and is one of the few class III lanthipeptides exhibiting potent antibiotic activity.217 Avermipeptin B displays activity against S. aureus, E. faecalis and B. subtilis with MICs in the nanomolar range.217 To date, the mechanism by which this compound exerts its antibacterial activity has not been examined.
A subset of class III lanthipeptides exhibit morphogenic activity similar to the class I lanthipeptide SapT (see section 2.1). SapB (35) produced by S. coelicolor (Figure 14) from the bld locus facilitates aerial hyphae formation in the producing organism through reduction of surface tension.218–221 SapB-deficient mutants cannot form aerial hyphae and are unable to form spores.220 Restoration of aerial growth can be achieved with the addition of purified SapB; however, bld mutants fail to sporulate even with added SapB, indicating the timing of SapB production for spore development is critical.221,222 Molecular modeling of SapB suggests that the structure may be amphiphilic, supporting proposed biosurfactant functionality. Several attempts at structural characterization using NMR spectroscopy have been unsuccessful due to aggregation and insolubility of the compound.218
Figure 14.

Class III lanthipeptides. (A) Structures of SapB (35), AmfS (36), and catenulipeptin (37). (B) Sequence alignment of the core peptides of catenulipeptin and SapB illustrating the similar constellation of Cys and Ser residues, but different outcome of the cyclization process, resulting in labionins for the former and lanthionines for the latter.
Several other peptides with sequence homology to SapB have been identified including AmfS (36), produced in Streptomyces griseus (Figure 14).223,224 AmfS is also important for aerial hyphae formation and sporulation.223,225 Mutagenesis studies of AmfS revealed that all conserved post-translationally modified residues selected for replacement (Ser3, Cys10, Ser13, Ser16, and Cys20) were essential for activity.225 Ser6, although conserved and converted to Dha, was not targeted by mutagenesis in this study. Additional variants involving select non-conserved residues within the rings (L7A, L8V, V9S, and L19V) and the removal of the tail region (residues 21–22) still facilitated aerial hyphae formation, indicating that these residues do not play an important role in this process.225 However, SapB with V9L and L19T substitutions lost the ability to restore aerial mycelium formation in neighboring bld mutant colonies.
Catenulipeptin (37) has sequence homology with SapB but contains labionins instead of the lanthionines in SapB (Figure 14). Unlike SapB, catenulipeptin did not induce acceleration of aerial hyphae formation or restore formation in bld mutants in S. coelicolor, suggesting it may not function like SapB.224 Alternatively, catenulipeptin may be inactive in S. coelicolor as it is not its producing organism. Some biosurfactants are specific to the producing organism and are not cross compatible, with some even acting antagonistically to suppress aerial growth in other organisms,226 suggesting that a molecular target may be involved. It remains to be investigated if catenulipeptin shows morphogenetic activity in its producing organism, Catenulispora acidiphilia.
2.4. Pearlins
Pearlins are a recently defined RiPP class characterized by the post-translational nonribosomal addition of amino acids to the C-terminus of a precursor peptide in an aminoacyl-tRNA dependent process.227 These amino acids are then further enzymatically modified and eventually cleaved from the peptide to produce a variety of small molecule natural products including pyrroloquinoline alkaloids (38–41) and 3-thiaglutamate (42, Figure 15). Although pearlins do not contain (Me)Lan residues, they are discussed in the lanthipeptide section because their class-defining biosynthetic enzymes are related to class I lanthipeptide synthetases.
Figure 15.

Chemical structures of ammosamides A (38), B (39) and C (40), lymphostin (41), and 3-thiaglutamate (42).
The first pyrroloquinoline alkaloid discorhabdin C was isolated from the sea sponge Lantrunculia apicalis in the early 1980s.228,229 Since then, pyrroloquinoline alkaloids have been isolated from a wide variety of organisms including bacteria (ammosamides), marine sponges (damirone B), and terrestrial fungi (sanguinone A). The bacterial biosynthesis remained enigmatic until recent investigations determined that many of these natural products belong to the pearlin RiPP class.230–232 The two currently known bacterial pyrroloquinoline alkaloid families are the ammosamides (38-40) and lymphostin (41) (Figure 15).229
While a variety of ammosamide derivatives have been isolated (from Streptomyces CNR-698) they are believed to be all derived from ammosamide C (40), which is the naturally produced structure. All other ammosamides have been reported to be artifacts of isolation methods during which ammosamide C was exposed to oxidative conditions and various nucleophiles.233 While these compounds are derived from a single compound, the ammosamides exhibit diverse bioactivities.233 Ammosamides A and B (38 and 39) display cytotoxicity to a number of human cancer cell lines including colon carcinoma.234,235 Ammosamide B conjugated to a fluorescent probe underwent efficient cellular uptake and its molecular interaction with one or more targets was suggested to be either through strong noncovalent interactions or a covalent mechanism owing to the inability to remove the labeled compound through washing procedures. The labeled compound was then used for affinity co-purification from cell lysates, and protein MS/MS analysis identified the target as a member of the myosin family, a motor protein involved in muscle contraction and cytoskeletal structure.234 This finding was later confirmed through in vitro myosin II labeling.234 Further experiments indicated that ammosamides A and B interact with several other myosin family members.234,236 A crystal structure of ammosamide 272 (43) co-crystalized with the myosin 2 heavy chain (PDB 4AE3) from Dictyostelium discoideum revealed the molecular mechanism of binding as noncovalent in an allosteric region of the motor domain (Figure 16).
Figure 16.

(A) Chemical structure of ammosamide 272 (43). (B) Crystal structure showing the interaction between ammosamide 272 (carbons labeled in green) and the myosin 2 heavy chain (PDB ID: 4AE3). Side chains of myosin residues interacting with ammosamide 272 are shown in yellow.
Ammosamides A and B also inhibit quinone reductase 2 (QR2) with sub-micromolar IC50 values.237 QR2 is a flavin-dependent enzyme involved in cellular protection from quinone species. A co-crystal structure of ammosamide B and QR2 was obtained (PDB ID: 3UXE) (Figure 17), demonstrating that the compound engages in a stacking interaction with the bound flavin and forms a hydrogen bond network to Asn161 and Thr71 of QR2.238 Derivatives of ammosamide B have been synthesized and are potent inhibitors of human QR2 with IC50s as low as 4.1 nM.238
Figure 17.

Crystal structure of ammosamide B (carbon atoms in cyan) bound to QR2 (purple and green; PDB 3UXE). Asn161 is located on monomer A (purple), while Thr71 is located on monomer B (green). The FAD carbons are shown in orange.
In addition to the reported cytotoxicity of ammosamide C against mammalian cells, it also exhibits antimicrobial activity against Bacillus oceanisediminis. No antibacterial activity was detected for ammosamide A or B against this strain.233 Neither the biological target nor the MOA has been determined.
Lymphostin (41, Figure 15) produced by Streptomyces sp. KY11783, is another pyrroloquinoline alkaloid identified by activity-based screening for inhibitors of lymphocyte kinase (Lck), a tyrosine kinase produced in lymphoid cells involved in T-cell activation.239 Lymphostin inhibited Lck with an IC50 of 50 nM with the IC50 value against a murine myeloid leukemia cell line of 0.16 μM.239 In a mixed lymphocyte reaction measuring T-cell activation, lymphostin inhibited Lck with an IC50 of 9 nM. The compound also inhibited phosphatidylinositol 3-kinase with an IC50 of 1 nM.240 During efforts to characterize the lymphostin biosynthetic gene cluster, analogs were generated and investigated for inhibition of another prominent kinase, mammalian target of rapamycin (mTOR). Lymphostin and derivatives showed 0.8–1.8 nM inhibitory activity against mTOR as well as 14–700 nM cytotoxicity against the human prostate and breast cancer cell lines, LNCap and MDA-468, respectively.241
3-Thiaglutamate (42) is the only other currently reported pearlin (Figure 15).231 The biosynthetic gene cluster was discovered in the plant pathogen Pseudomonas syringae as well as other pathogenic pseudomonads. The compound is unstable, and it has not been determined to be the active product. Glutamate was recently shown to be a messenger that is involved in systemic defense responses in plants that involves the glutamate receptor,242 and hence 3-thiaglutamate or a derivative thereof could be an antimetabolite to interfere with plant defenses.231,243
2.5. Lanthidins (Class V lanthipeptides)
Lanthidins contain post-translational modifications commonly present in linaridins and lanthipeptides giving rise to their name. The first reported member, cacaoidin (44) isolated from Streptomyces cacaoi, contains an N-terminal N,N-dimethyl lanthionine, O-glycosylation of a Tyr, and several D-amino acids (Figure 18). While linaridins commonly contain N-terminal bis-N-methylation, they are defined by the presence of dehydrobutyrine residues, which are not present in cacaoidin.244 This observation combined with the presence of lanthionine and isolation of additional compounds that share these structural features led to the renaming of lanthidins as class V lanthipeptides.10,245–247
Figure 18.

Class V lanthipeptides cacaoidin (44) and lexapeptide (45). The D-Ala that was demonstrated to be important for antibacterial activity of lexapeptide is highlighted in red.
Cacaoidin was isolated via bioactivity-guided screening and showed activity against Firmicutes including methicillin-resistant S. aureus (MRSA) and Clostridium difficile. Investigation of the MOA of cacaoidin utilizing a lacZ reporter assay suggested the compound targets cell wall biosynthesis due to the induction of the LiaRS bioreporter (sections 2.2 and 15.4). This hypothesis was supported by experiments showing that the response was mitigated by supplementation with exogenous lipid II. The authors used these studies to suggest a lipid II binding mechanism, similar to other known lanthipeptides (see sections 2.1 and 2.2).244 Structural studies have yet to be performed on the proposed binding of cacaoidin to lipid II.
Another class V lanthipeptide, lexapeptide (45, Figure 18), was isolated from Streptomyces rochei, and exhibits activity against MRSA and methicillin-resistant S. epidermidis (MRSE).246 Mutational studies identified the D-Ala in the compound as important for activity with the L-isomer having reduced activity against a subset of tested bacterial strains. No further MOA studies have been published to date.
2.6. Lipolanthines
Lipolanthines are lipopeptides in which the peptide portion is ribosomally synthesized. This class is characterized by the presence of labionin/avionin(s) (Figure 2) and an N-terminal lipid moiety. Microvionin (46), the first lipolanthine isolated from Microbacterium arborescens (Figure 19), has activity against MRSA and Streptococcus pneumoniae.248 Goadvionin is another lipidated class III lanthipeptide and shows similar antibacterial activity.249 The precise MOA of lipolanthines requires further study.
Figure 19.

Structure of microvionin (46).
3. RiPPs generated by YcaO superfamily enzymes
YcaO enzymes activate the amide backbone of peptides/proteins to catalyze diverse reactions through the intermediacy of a phosphorylated hemiorthoamide.250 These enzymes induce an intra- or inter-molecular nucleophilic attack at an amide carbonyl to generate an oxyanion, which is then O-phosphorylated in an ATP-dependent manner.251 The nature of the final product depends on the identity of the nucleophile. Intramolecular attack by the side chains of Ser/Thr/Cys results in azoline formation and is involved in the biosynthesis of the compounds discussed in sections 3.1, 3.2 and 4. Intramolecular attack by the N-terminal amine results in lactamidine formation as observed in the bottromycins discussed in section 3.3. Finally, intermolecular attack, by an external sulfur donor, results in thioamide formation, exemplified by the thioamitides (section 3.4).
3.1. Linear azoline/azole-containing peptides (LAPs)
Linear azoline/azole-containing peptides (LAPs) are a class of non-macrocyclic RiPPs characterized by the presence of thiazol(in)e and/or (methyl)oxazol(in)e heterocycles.250 The cognate YcaO enzymes catalyze the cyclodehydration of Cys, Ser, and Thr residues to the corresponding azoline heterocycle, some of which undergo oxidation to the corresponding azole by an FMN-dependent dehydrogenase (Figure 20). As detailed in section 1.1, the peptide numbering scheme used for all RiPPs in this section will denote the first residue in the core peptide as residue “1”.
Figure 20.

Generic scheme for the biosynthesis of azol(in)e on a peptide substrate by a trifunctional heterocyclase. In LAP biosynthesis, the C-D complex is composed of an E1-like protein (yellow) and an ATP-dependent YcaO protein (green), which together perform cyclodehydration of Cys, Ser, and/or Thr residues. The B protein (orange) is an FMN-dependent dehydrogenase that generates the azole. Abbreviations for (methyl)azol(in)es heterocycles used in this review are shown.
The toxic virulence factor of Streptococcus pyogenes, streptolysin S (SLS), is a seminal LAP. The ability of certain streptococci to hemolyze erythrocytes was first observed in the 1890s, and four decades later, SLS was identified as one of two toxins from Group A Streptococcus (GAS) that could lyse mammalian erythrocytes.252 The other toxin, streptolysin O (SLO), is a larger protein that forms pores in cholesterol-containing membranes. The cytolytic activity of SLS is broad and includes erythrocytes, leukocytes, thrombocytes, as well as membrane-defined organelles such as the mitochondria and lysosomes. SLS is bactericidal against streptococci, staphylococci, and bacilli, but does not affect bacteria that possess an outer membrane (e.g. Pseudomonas, Vibrio, Rhodospirillum).253–257 This broad but nuanced cytolytic spectrum suggests SLS activity depends on membrane composition and/or access to the membrane.256,257
Though the detailed MOA of SLS is not known, it has been proposed to form pores in target membranes. Early studies demonstrated that treatment of artificial liposomes with SLS made these membranes permeable to cations.258 SLS was later shown to induce the formation of large pores in sheep erythrocyte membranes,259 suggesting that cytolysis could be caused by shredding of the membrane. Beta-hemolysis was shown to be temperature dependent, as erythrocytes treated with SLS at 17 °C did not lyse, while treatment at warmer temperatures resulted in lysis.260 Additionally, complexes of SLS pre-mixed with erythrocytes were insensitive to protease degradation at elevated temperatures. However, at lower temperatures, the SLS-erythrocyte complex was sensitive to proteolytic degradation. These results suggest a MOA in which SLS localizes to the membrane surface, but only inserts into the membrane and forms pores at permissive temperatures.
Three mechanisms have been proposed to account for SLS-mediated virulence: (1) soft tissue damage, (2) host phagocyte damage, and (3) GAS translocation across the epithelial barrier. A transposon insertion variant GAS was unable to induce hemolysis in a mouse model of infection.261 Further analysis mapped the insertion to the sagA promoter sequence, suggesting that expression of the SagA precursor peptide was essential for virulence. Subsequently, the role of SLS in tissue damage was shown using clinical isolates with group G Streptococcus (GGS) infections, whose SagA precursor peptide highly resembles that of GAS (Figure 21). In a mouse model of infection, injection with the GGS isolate resulted in necrotic ulcers, while injection of strains with a sagA deletion did not show this phenotype.262
Figure 21.

Amino acid sequences of the precursor peptides encoding SLS-like cytolysins. Shown are the predicted leader and core regions for SLS, clostridiolysin S (CLS), listeriolyin S (LLS), stapholysin S (StsA), and the group G Streptococcus (GGS) SLS-like cytolysin. Residues in the SLS sequence that when replaced with Ala abolished cytolytic activity are shown in blue. The exact structure of any of these RiPPs has yet to be determined.
Beyond hemolysis and tissue damage, S. pyogenes producing both SLS and SLO can induce macrophage cell-death through depolarization of the mitochondrial membrane and accumulation of reactive oxygen species.263 However, the individual contributions of each compound to these effects have not been characterized. Lastly, SLS has been implicated in more complex bioactivities observed during infection including paracellular invasion across the epithelial barrier. In vitro studies utilizing a Caco-2 cell permeability assay showed that SLS-producing GAS disrupted tight junctions and epithelial translocation, while SLS-deficient GAS did not demonstrate this translocation activity.264 These studies further demonstrated that intercellular junctions were not disrupted as a consequence of SLS-induced cell lysis or inflammatory cytokine production. The intercellular junctions were in fact degraded by the SLS-mediated recruitment of the endogenous cysteine protease calpain to the plasma membrane. The molecular mechanism for calpain recruitment to the plasma membrane by SLS has not yet been characterized. Given its role in promoting paracellular invasion, there may be some future promise in exploiting SLS scaffolds to develop oral therapeutics that would otherwise be poorly absorbed.
The mechanism of hemolysis has also been partially elucidated.265 In hypotonic phosphate buffered solution, introduction of SLS induced cellular swelling, indicating significant influx of water preceding lysis. Use of 6-methoxy-N-ethylquinolinium iodide as a fluorescent indicator also demonstrated that SLS triggered an influx of chloride ions. Subsequent experiments demonstrated that SLS-induced hemolysis occurred in a membrane cation transporter-dependent fashion. Anion exchanger 1 (AE1), also called band 3, is the most abundant protein in the erythrocyte membrane and mediates the exchange of chloride and bicarbonate anions. Treatment of erythrocytes with the AE1 inhibitors 4,4’-diisothiocyanatostilbene-2,2’-disulphonate (DIDS) and 4-acetamido-4’-isothiocyanato-2,2’-stilbenedisulphonic acid significantly reduced the hemolytic activity of SLS. Pretreatment of erythrocytes with antibodies specific to the exposed C-terminal epitope of AE1 also protected the cells from hemolysis. Synthetic peptides corresponding to this C-terminal fragment were also shown to block SLS activity in a dose-dependent fashion, supporting the hypothesis that SLS directly interacts with this region of AE1 to induce hemolysis. Furthermore, in mouse models of SLS-producing GAS bacteria, administration of DIDS significantly reduced the size of necrotic lesions. These findings suggest that SLS likely interacts with homologous ion channels to promote invasion of other cell types.265
The SLS core peptide contains 15 residues that can potentially undergo heterocyclization into (methyl)azol(in)es, and mutational studies have been used to dissect the SAR of the SLS core (Figure 21). The site-directed variants C1A, C4A and K30A of SLS did not demonstrate cytolytic activity (positions have been renumbered using standardized RiPP nomenclature; see Introduction).266 Follow-up studies identified additional residues that contributed to lytic activity.267 Ala substitutions at Cys8 or Ser16 abolished in vitro or in vivo bioactivity. Notably, substitutions with Pro (which like azol(in)es also possesses a five-membered ring) at these two positions rescued lytic activity in vitro. Activity was retained in an SLS variant in which all Cys residues were replaced with Pro, highlighting the importance of five-membered rings in imparting cytolytic activity. Truncation analysis of the SagA precursor demonstrated that the last 17 residues of the SLS core peptide were dispensable for hemolytic activity, but removal of the last 20 residues abolished activity.268 Thus, the minimal bioactive fragment of SLS consists of core residues Cys1– Ser11.
SLS-like virulence factors have also been identified in other bacteria (Figure 21) and each of these compounds show similarity in core sequences, exhibit similar bioactivities, and are likely to possess the same MOA. Clostridiolysin S (CLS), isolated from Clostridium botulinum showed in vitro hemolytic activity.269,270 S. aureus RF112 is predicted to produce a LAP termed stapholysin S (StsA).269 Though the compound has yet to be isolated from the producing organism, it has been produced using a chimeric substrate approach and was found to be cytolytic.267 More recently, SLS-like virulence factors were identified in certain Borrelia burgdorferi sensu lato strains, which are responsible for Lyme disease.268 The precursor peptides identified in Borrelia (Spirochaetes) are shorter than their Firmicute homologs by lacking ~15 residues of the most C-terminal portion of the core peptide. The N-terminal portion of the core peptide, consisting of contiguous residues capable of azol(in)e formation are retained.
A subset of L. monocytogenes lineage I strains produce a hemolytic compound termed listeriolysin S (LLS).271 LLS is expressed solely in the intestines of orally infected mice, and was shown to affect the growth of gut-associated bacteria and alter the composition of the gut microbiome.272 Cellular fractionation experiments demonstrated that LLS was contained with the cell envelope of producing bacteria.273 Further evidence of cell-envelope association was provided using immunostaining of producing cells expressing LLS tagged with a human influenza hemagglutinin-derived epitope. Treatment of L. lactis with LLS-containing membrane fractions did not induce lysis, suggesting further metabolic activity is necessary for bactericidal activity. Co-culture experiments were performed wherein LLS-producers and L. lactis were separated by semipermeable membranes. Membranes with a pore size of 0.4 μm prevented L. lactis killing, while a pore size of 8 μm allowed passage of LLS and induced lysis of L. lactis cells. Fluorescent reporter strains further demonstrated that L. lactis cells in contact with LLS-producing cells had drastically increased doubling times, suggesting a general decrease in metabolic activity. Imaging studies with SYTOX blue and DiBAC4 dyes demonstrated that LLS-treated cells lost membrane potential in a time dependent manner. Mass spectrometry-based proteomics on LLS-treated cells further demonstrated up-regulation of proteins involved in ATP synthesis, indicating that these cells increase ATP production to counteract membrane depolarization and ATP efflux.
The Nα,Nα-dimethylated LAP plantazolicin (47, Figure 22) also contains heterocyclic residues and similarly targets membranes. First isolated from Bacillus velezensis (reclassified from Bacillus amyloliquefaciens), plantazolicin was initially described as an antibiotic that inhibited the growth of B. subtilis, Bacillus cereus, and Bacillus megaterium at an extremely high dosage (1 mg in a spot-on-lawn assay).274,275 The spectrum of activity was later reevaluated using precise microbroth dilution assays, which demonstrated plantazolicin is specific towards B. anthracis (MIC = 0.88–14.1 μM), while possessing no or weak activity against B. subtilis or B. cereus (> 57 μM).276,277 Bioactivity was dependent on the presence of the Nα,Nα-dimethylation and the most N-terminal azole heterocycle. Acid hydrolysis of the lone MeOxH (residue 13) of plantazolicin led to a ~8-fold increase in MIC against B. anthracis Sterne, while an otherwise fully modified peptide lacking the N-terminal methyl groups negated all bioactivity.276 In liquid culture, the producing organism produces a plantazolicin side-product containing two azolines, which lacks antibiotic activity. The position of the azoline residues was not determined. The minimal fragment with a comparable MIC to plantazolicin is an N-terminal fragment (48) with Nα,Nα-dimethylation and five heterocycles (Figure 22). Surprisingly, this derivative demonstrated a wider growth inhibition spectrum that included additional strains of Bacilli and MRSA.278
Figure 22.

(A) Chemical structure of plantazolicin (47). The plantazolicin precursor peptide is shown above, and the core peptide is shown in bold. The numbering scheme for (47) is reflected in the labels above the core peptide. The sole methyloxazoline (blue) can be selectively acid hydrolyzed, which results in an 8-fold reduction in MIC against B. anthracis. The Nα-dimethylations (orange) are essential for activity. (B) Minimal synthetic plantazolicin fragment (48) (the C-terminal methyl ester was utilized to facilitate chemical synthesis and to better serve as a mimic of the amino acid that follows).
Observation of B. anthracis cells by differential interference contrast microscopy demonstrated that plantazolicin-treated cells underwent lysis, while the few remaining cells displayed compromised cell walls.276 Assays utilizing radiolabeled precursors revealed that plantazolicin treatment induced general perturbation of protein, fatty acid, and nucleic acid biosynthesis, but did not specifically inhibit cell wall biosynthesis.277 RNA-seq analysis of plantazolicin-treated cells showed significant upregulation of two genes, homologous to B. subtilis genes liaI and liaH, which are involved in the cell-envelope stress response (section 2.2 and 15.4). Two homologs of the B. subtilis membrane fluidity sensors DesK and DesR were also upregulated.277,279 The identity of the upregulated genes implicate the cell membrane as the target of plantazolicin, and this hypothesis was supported by observation of a marked loss in membrane potential in B. anthracis treated with plantazolicin, even at concentrations 100-fold below the MIC.277 The localization of fluorescently modified plantazolicin was determined to be the cellular envelope by sub-diffraction limit microscopy.277
Spontaneous mutations in the cardiolipin synthase gene conferred resistance to plantazolicin in B. anthracis Sterne.277 Cardiolipin is a cone-shaped phospholipid that is found in bacterial membranes and the mitochondrial inner membrane.280 The role of cardiolipin in the plantazolicin MOA was further supported by findings that pre-treatment of B. anthracis with cardiolipin increased the MIC by up to 16-fold and that plantazolicin co-localized with cardiolipin in the cell membrane.277 However, exogenously supplied cardiolipin did not render B. subtilis or B. cereus susceptible to plantazolicin. Atomic level details of plantazolicin-mediated membrane perturbation remain unknown, but these data strongly implicate cardiolipin in the plantazolicin MOA.
Other LAPs are known to inhibit bacterial growth through membrane-independent MOAs. For instance, microcin B17 (49, MccB17, Figure 23) inhibits DNA synthesis and induces the bacterial SOS response in E. coli.281 More specifically, treatment of target cells with MccB17 elicits rapid termination of DNA synthesis, and results in significant DNA degradation within 30 minutes of treatment. A resistance-conferring mutation was identified in the E. coli gyrB gene (corresponding to GyrB W751R), implicating GyrB as the molecular target of MccB17.40 E. coli DNA gyrase is a tetramer composed of two copies of both GyrA and GyrB. GyrA is responsible for DNA binding and handling, while GyrB contains the site of ATP hydrolysis and binds both GyrA and DNA.
Figure 23.

Chemical structure of microcin B17 (MccB17) (49). The MccB17 precursor peptide is shown above with the core peptide shown in bold. The numbering scheme for (49) is reflected in the labels above the core peptide.
Subsequently, in vitro and in vivo studies revealed that MccB17 induces DNA cleavage in a DNA gyrase-dependent fashion.40 Like other known gyrase inhibitors (e.g., fluoroquinolones), MccB17 stabilizes a transient gyrase-DNA-cleavage complex that blocks DNA polymerase movement.282 However, MccB17 has a unique MOA compared to the fluoroquinolones, given that MccB17 does not influence the ATPase activity or DNA supercoiling/relaxation activities of DNA gyrase in vitro. Moreover, MccB17 requires multiple turnovers before DNA cleavage occurs, and unlike the fluoroquinolones, MccB17 slows DNA supercoiling and relaxation, but does not completely halt both processes.282,283 Lastly, inhibition of gyrase cleavage and re-ligation reactions by MccB17 was found to be ATP dependent. MccB17 cleaves only 8% of closed-circular DNA in the absence of ATP, while DNA cleavage levels with ATP are comparable to those observed for ciprofloxacin (~30% vs. 40%).283 Kinetic simulations of the cleavage-ligation equilibrium suggest that MccB17 increases the rate of cleavage of the second strand of DNA, while ciprofloxacin increases the rate of cleavage on both strands.
The bioactivity of MccB17 is contingent on successful import into the cell. Spontaneous resistance mutations occurred in genes encoding the outer membrane protein ompF and the inner membrane peptide transporter sbmA.284 These data suggest that OmpF and SbmA are involved in the import of MccB17. SbmA has been implicated in the translocation of other antibacterial peptides including bleomycin, microcin J25 (a lasso peptide, see section 8.1), the mammalian peptide bactenecin-7 (Bac7), peptide nucleic acids (PNAs), and synthetic peptide phosphoramidate morpholino oligomers.285–290 Strains deficient in SbmA are non-susceptible to MccB17.284
SAR studies of MccB17 demonstrated the importance of the two 4,2 thiazole-oxazole bis-heterocycles (Figure 23). The first bis-heterocycle (the A site) may serve as a biosynthetic checkpoint, as the C15S variant resulted in lower titers of MccB17.291 The second bis-heterocycle (the B site) was crucial for bioactivity.291 DNA gyrase supercoiling kinetic experiments conducted in vitro confirmed the importance of these bis-heterocycles.292 Replacement of the B site heterocycles (with glycine) decreased the affinity of the MccB17-DNA gyrase interaction.293 Small heterocyclic derivatives of MccB17 mimicking the A and B sites, did not induce DNA cleavage by GyrB.292 To further isolate the region responsible for bioactivity, MccB17 N27K was digested to produce two fragments consisting of Val1–Ser26 and Gly28–Ile43. Each fragment retained about ~20 to 50% of gyrase inhibition as compared to full length MccB17. The C-terminus of MccB17 was shown to be important for DNA cleavage, as removal of the C-terminal Ile43 reduced in vitro activity, while deletion of Ser41–Ile43 abrogated all activity. Conversely, removal of eight N-terminal core residues yielded an MccB17 fragment that retained wild-type DNA cleavage activity.292
A longer fragment of MccB17 encompassing residues Gly20–Ile43 displayed greater inhibitory activity and more significantly stabilized the gyrase-DNA cleavage complex in vitro (as determined by measuring the amount of single-stranded DNA).292 However, this C-terminal fragment did not inhibit E. coli growth, implicating the N-terminus of MccB17 as important for membrane translocation. Independent studies elaborated on other residues required for cellular import. Removal of two unmodified C-terminal residues increased the MIC from 0.06 μM to 150 μM.294 The most critical residue was Ile43 as MccB17 variants ΔIle43, I43T, I43K, and I43E resulted in MIC increases of 10–500-fold. In contrast, the I43L variant retained wild-type bioactivity. In vivo studies of SbmA-overexpressing E. coli demonstrated that replacement of I43 with a stop codon (ΔI43), I43K, and I43E reduced transport across the membrane. In vitro gyrase inhibition assays also showed that ΔI43, I43T, and I43K possessed reduced inhibitory activity (MIC = 0.6–6 μM), while the I43E variant was inactive (MIC = 30 μM). Additional studies, such as those utilizing DNA gyrase point variants, are needed to elaborate the molecular details of the MccB17-DNA gyrase interaction.
A class of structurally related LAPs has demonstrated activity against the bacterial ribosome. Klebsazolicin (50, Figure 24) was discovered through genome mining from the opportunistic human pathogen Klebsiella pneumonia subsp. ozaenae. Heterologously produced klebsazolicin exhibited modest growth suppressive activity against various Proteobacteria including E. coli, K. pneumoniae, and Yersinia pseudotuberculosis with MICs in the 16–65 μM range.295 Klebsazolicin sensitivity was dependent on OmpF and SbmA, suggesting a role in cellular import, an uptake mechanism similar to that of MccB17 described previously. An E. coli fluorescent reporter assay indicated that klebsazolicin targeted the ribosome in vivo and inhibited protein translation in vitro with an IC50 of 340 nM. Mutational analysis identified the N-terminal 14-residue fragment as the minimal bioactive core of klebsazolicin (Figure 24). Genetic sequencing of spontaneous resistance mutants identified two mutations in the 23S rRNA gene, U2609G and U2609A, which reside at an exposed site at the peptide exit tunnel.295
Figure 24.

Chemical structure of klebsazolicin (50). Ser1 to Ala14 comprises the minimal bioactive core of klebsazolicin (blue). The klebsazolicin precursor peptide is shown above with the core peptide shown in bold. The numbering scheme for (50) is reflected in the labels above the core peptide.
Further evidence for the MOA of klebsazolicin was provided by the co-crystal structure of the molecule bound to the Thermus thermophilus 70S ribosome (Figure 25).295 Klebsazolicin occludes the peptide exit tunnel and binds in a similar manner to the streptogramin antibiotics. Translation terminates as the nascent di- or tri- peptide cannot be accommodated. Klebsazolicin generates extensive van der Waals contacts with U2609, which also is a site of spontaneous mutation after selection of klebsazolicin-resistant strains. Direct contacts were also observed for A2058, A2059, U2506, and U2585, which were previously demonstrated to be protected from chemical modification by dimethyl sulfate, 1-cyclohexyl-3-(2-morpholinoethyl)-carbodiimide, and methyl-p-toluenesulfonate in the presence of klebsazolicin. Furthermore, Thz7, Thz10 and the N-terminal amidine ring form key stacking interactions with nucleobases in the 23S rRNA (C2586, A2062, and C2610, respectively). The C-terminal portion of the peptide (Ser15–Gly23) occupies the exit tunnel.
Figure 25.

(A) Positioning of klebsazolicin (Kleb, dark purple) bound to the complex of the ribosome with tRNA (PDB ID: 5W4K). tRNA is bound in the A-site by A-tRNA (A: aminoacyl); P-site tRNA by P-tRNA (P: peptidyl); and E-site by E-tRNA (E: exit). (B) Overview of klebsazolicin blocking the exit tunnel. (C) Interactions of the 23S rRNA (carbon atoms in yellow) with klebsazolicin (carbon atoms in purple). Stacking interactions are shown as blue regions between regions of interest.
Rhizobium sp. Pop5, a symbiont of the common bean plant Phaseolus vulgaris, produces a related LAP phazolicin (51, Figure 26). Phazolicin inhibits the growth of members of the genera Rhizobium, Sinorhizobium, and Azorhizobium sp. (MIC = 1–4 μM).296 However, no activity was observed against other plant-associated bacteria from Proteobacteria, Firmicutes, and Actinobacteria (e.g. Erwina amylovora, Pseudomonas, Bacillus, and Arthobacter sp.). Growth inhibition of E. coli was only observed at very high concentrations of 5–10 mM. Phazolicin insensitivity was overcome using an E. coli strain with a deletion in tolC, which encodes a major outer membrane multidrug efflux porin, suggesting that phazolicin is subject to TolC-dependent efflux. As with klebsazolicin, in vivo fluorescent reporter systems were used to show that the ribosome is the target, which was then confirmed using in vitro translation assays. The cryoEM structure of phazolicin bound to the E. coli ribosome complex demonstrated that phazolicin binds within the ribosomal exit tunnel in a “curled” conformation (Figure 27). Numerous intramolecular interactions stabilize the exit tunnel placement and π-stacking interactions occur between Thz3, Oxz15, and Oxz18 of phazolicin and A751, C2611 and U2609 of the ribosome. Although both phazolicin and klebsazolicin occlude the ribosome exit tunnel, the set of peptide/ribosome interactions is different.
Figure 26.

Chemical structure of phazolicin (51). Residues in blue are involved in binding to nucleotides in the exit tunnel. The phazolicin precursor peptide is shown above with the core peptide shown in bold. The numbering scheme for (51) is reflected in the labels above the core peptide.
Figure 27.

Close-up of interactions between E. coli 23S rRNA (carbon atoms in pink) and phazolicin (carbon atoms in blue) (PDB ID: 6U48). Interactions and residues are shown. Stacking interactions are shown as yellow regions between residues of interest. The view in panel B has been rotated by 90°.
To understand the narrow spectrum of activity, the structure of E. coli ribosome-bound phazolicin was superimposed onto the structure of the T. thermophilus 70S ribosome. Phazolicin binds between uL4 and uL22 loops of the E. coli 50S ribosome (Figure 28). However, in the T. thermophilus ribosome, this site is occupied by uL4-His69 and uL22-Arg90, which would not permit phazolicin to bind.296 The E. coli ribosome possesses a smaller residue in the uL4 loop (Gly64), which permits binding although it has a positively charged and large residue in the uL22 loop (Lys90). In complementation studies, the uL4-G68H variant rendered Sinorhizobium melliloti resistant to phazolicin, while complementation of S. melliloti with uL22-K90R did not confer resistance. These results were rationalized by the observation that position 90 of the uL22 has more space in the phazolicin-bound structure, hence substituted side chains are not as conformationally restricted as in substitution at His69 of the uL4 loop.
Figure 28.

Position of phazolicin (blue carbon residues) in relation to E. coli 50S ribosome uL4 (light green), uL22 (gold) loops (PDB ID: 6U48), and E. coli 23S rRNA (carbon atoms in pink) Key residues on both proteinaceous loops are highlighted.
In addition to serving as antibiotics, other known LAPs function as signaling molecules. Goadsporin (52, Figure 29) was originally isolated in a screening effort to identify compounds that induced morphological changes and secondary metabolite production in Streptomyces lividans.297 S. lividans cultures treated with fermentation broths of the goadsporin producer Streptomyces sp. TP-A0584 induced pigmentation, aerial hyphae formation, and sporulation. Goadsporin possessed antibiotic activity against S. lividans, S. coelicolor and S. scabies with MICs of 4.0, 2.0 and 0.12 μM, respectively, but lacked antibiotic activity against all non-streptomycetes tested. Goadsporin also lacked cytotoxic, apoptotic, and anti-protein kinase C activities. In a disk diffusion assay using 42 randomly selected streptomycetes, goadsporin was growth suppressive in 32 of the strains and induced pigmentation in 20 strains. Goadsporin also induced the production of an anti-B. subtilis antibiotic in strain TP-A0593. Structural studies demonstrated that goadsporin is a 19 residue, N-terminally acetylated LAP containing four oxazoles, two thiazoles, and two alkenes (Figure 29).298,299
Figure 29.

Chemical structure of goadsporin (52). Dehydroamino acids are shown in blue; azoles that can be substituted without loss of activity are shown in orange; Gly10 is shown in green. The goadsporin precursor peptide is shown above with the core peptide shown in bold. The numbering scheme for (52) is reflected in the labels above the core peptide.
Introduction of the self-immunity gene godI into S. lividans endows the strain with goadsporin immunity.300 Sequence analysis demonstrated that GodI is ~50% identical to the E. coli Ffh signal recognition particle protein.300–302 Ffh binds the signal peptide of membrane proteins to facilitate membrane integration.303 These findings suggest that Ffh inhibition by goadsporin is responsible for the morphological and metabolic changes observed in susceptible bacteria.301,302 Genomic analyses of previously reported goadsporin-susceptible strains for the absence of GodI/Ffh has yet to be carried out. Such studies may inform on how changes in the primary sequence of Ffh across susceptible and non-susceptible strains affects the response to goadsporin.
SAR studies of goadsporin utilizing gene deletions have partially elucidated important functional groups for bioactivity. Deletion of godF and godG resulted in production of a goadsporin variant that lacked both alkenes (derived from Ser4 and Ser14), which lacked antibiotic activity.304 The G10A variant did not produce a zone of inhibition against S. lividans, but retained activity against S. scabies.300 Individual switching of methyloxazoles and oxazoles (T5S and S15T variants) did not diminish bioactivity, suggesting the importance of the heterocycle itself rather than methylation status of the ring. Interestingly, addition of a single Lys residue at the C-terminus of goadsporin eliminated activity. More detailed studies are needed to shed light on the MOA of this LAP.
3.2. Pyritides (including thiopeptides)
Pyritides are highly modified peptides defined by the presence of a six-membered, nitrogenous heterocycle that can exist in one of several oxidation states (most often pyridine). The pyritide class includes thiopeptides, which contain thiazoles and dehydroamino acids in addition to the central nitrogenous heterocycle, although many other secondary modifications have also been documented.305,306 Thiopeptides can be classified into one of five series (a through e), which are distinguished based on the oxidation state of the 6-membered, nitrogenous heterocycle (Figure 30).29 Many thiopeptides show impressive activity against Firmicutes by inhibiting ribosomal protein synthesis, though other bioactivities have been reported. Broadly speaking, the MOA of thiopeptides can be divided into one of four main categories: 1) binding of ribosomal RNA (rRNA) L11-binding domain (L11BD), 2) binding to Elongation Factor Thermo unstable (EF-Tu), 3) TipAL transcription factor binding, and 4) FOXM1 transcription factor inhibition. We will devote most of our discussion to these four categories; other bioactivities will only be briefly mentioned.
Figure 30.

(A) Classification of thiopeptides based on the oxidation state of the central heterocyclic ring. (B) A generalized scheme of prokaryotic translation. Thiopeptides known to inhibit any steps in the process are noted. E, exit site; P, peptidyl site; A, aminoacyl site; EF-G, elongation factor G.
3.2.1. Ribosome inhibitors
Thiostrepton (53, Figure 31), the prototypical and best-known thiopeptide, contains a 26-member primary macrocycle (series b) and was originally isolated in the 1950s from Streptomyces azureus ATCC 14921.307 Thiostrepton demonstrated potent activity against Firmicutes, such as S. aureus (MIC = 0.06 μM), B. subtilis (MIC = 0.06 μM), and hemolytic streptococci (MIC = 6 nM). Growth of Proteobacteria (such as E. coli, Salmonella, and Shigella) and fungi were not affected by treatment with thiostrepton at concentrations up to 12 μM.308 Early studies established that thiostrepton inhibits bacterial protein synthesis.309,310
Figure 31.

Structures of thiostrepton (53) and micrococcin P1 (54) and P2 (55). Residues that project into the cavity between uL11 and the 23S rRNA are shown in blue. The C-terminal dehydroalanine tail of thiostrepton that is required for activity is highlighted in red. Conjugating chemical species to the penultimate Dha16 (magenta), as opposed to Dha17 results in an analog devoid of activity.
Thiostrepton inhibits protein synthesis by binding to the 50S ribosomal subunit and preventing both mRNA-tRNA translocation by the GTPase Elongation Factor G (EF-G), and EF-Tu-catalyzed amino-acylated tRNA delivery (Figure 31).310–314 The exact site of thiostrepton binding on the 50S subunit was identified based on the self-resistance mechanism of the producer S. azureus ATCC 14921, through identification of spontaneous resistance mutations, and through biophysical studies.309,312,315–317 Self-immunity in S. azureus is conferred by an RNA methyltransferase that modifies A1067 of the 23S ribosomal RNA component of the 50S subunit.318 This thiostrepton resistance mechanism was also observed in several other streptomycetes, as well as two producers of the structurally related thiopeptides siomycin and sporangiomycin.319 Resistance was also observed in variants of B. subtilis and B. megaterium that either possessed point mutations in uL11 or lacked this protein altogether.320,321 uL11, along with (uL7/uL12)4 and uL10, form the GTPase-associated center in the 50S ribosomal subunit that serves as a binding site for GTPases on the ribosome.322 uL11 was also shown to be necessary for thiostrepton inhibition of the E. coli translational machinery in vitro.320,323–326 These data demonstrate that thiostrepton interacts with both the protein and RNA subunits of the ribosome.
The findings that uL11 bound the 23S rRNA along the same sequence that is targeted for methylation or mutation in resistant organisms provided a unifying rationale for the observed resistance determinants.327–329 Subsequently, thiostrepton was demonstrated to directly bind at this rRNA sequence, suggesting that binding disrupted the native uL11–23S rRNA interaction.330 Thiostrepton binding was proposed to occur through allosteric cooperativity, as thiostrepton inhibited EF-G-dependent and -independent translocation and had no effect on the P site binding of tRNAs. An allosteric model was further supported by the observation that micrococcin P1 (54, Figure 31), a related 26-membered macrocyclic thiopeptide (series d), bound the same region of the 23S rRNA but stimulated EF-G GTPase activity, which is the opposite of the response observed for thiostrepton.330,331 Structure-function studies of the 23S rRNA subunit demonstrated significant conformational changes upon uL11 binding, which stabilized select conformations and allowed for the cooperative binding of thiostrepton.331–334
Structural studies of the uL11–23S rRNA interface revealed a cleft that can accommodate a thiopeptide (Figure 32).335–338 These studies showed thiostrepton binding prevents any conformational transitions of uL11.321,339–342 Furthermore, thiostrepton was shown to induce tightening of the uL11–23S rRNA junction. Additional interactions between the uL11 N-terminal domain (NTD) and thiostrepton contribute to further stabilization of a conformation that does not allow the interactions with elongation and release factors necessary for translation.339,343–347 Disruption of these conformational changes has been shown to inhibit complexation of EF-G with amino-acylated tRNA during elongation.348 Studies on thiostrepton-resistant T. thermophilus uL11 variants demonstrated that amino acid substitutions increased the flexibility of uL11, potentially overcoming the conformational stabilizing effects of thiostrepton.349 These findings were further supported by NMR studies of Thermotoga maritima uL11 variants, demonstrating a compact, stable uL11-thiostrepton-23S rRNA structure. Substitution at conserved Pro sites increased uL11 flexibility and permitted translation to proceed in the presence of thiostrepton.350
Figure 32.

(A) Isolated view of interactions of thiostrepton with uL11 (green) and helices 43 and 44 of 23S rRNA (pink) from the structure of thiostrepton bound to the 50S ribosomal subunit from D. radiodurans (PDB ID: 3CF5). Residues in the primary macrocycle of thiostrepton that bind between uL11 and the 23S rRNA are shown in cyan. CTD, C-terminal domain; NTD, N-terminal domain. (B) Close-up of the interactions between thiostrepton, uL11 and the 23S rRNA. Key residues on uL11 and the 23S rRNA are shown in yellow. Thz6 is stacked between Pro21 and Pro25 of uL11. The quinaldic acid moiety (QA, carbon atoms in olive) engages in stacking interactions with A1078 of 23S rRNA and Thz1 is stacked against A1106. These positions correspond to A1067 and A1095 respectively in E. coli 23S rRNA.
Partial elucidation of the disparate activities of thiopeptides on the GTPase-associated center in the 50S subunit showed that thiostrepton increases the dissociation rate of GTP, thereby slowing GTP hydrolysis, while micrococcin P1 increases the dissociation of GDP+Pi from EF-G, subsequently increasing the rate of GTP hydrolysis.343 These observations are reconciled by structural studies on micrococcin P1 bound to the 50S ribosome of Deinococcus radiodurans.351 Micrococcin P1 does not share the thiostrepton mode of binding and instead stabilizes interactions between the uL11 and uL7 ribosomal proteins.351 It was hypothesized that in this conformation, uL7 is strategically poised to interact with EF-G, thus promoting GTP hydrolysis. As only GTP bound EF-G can associate with the ribosome, translocation, and protein synthesis ceases. These effects were rationalized by the presence or absence of the quinaldic acid (QA) heterocycle that directly interacts with the D. radiodurans 23S rRNA at A1078, which corresponds to A1067 in E. coli 23S rRNA (Figure 32).351,352 In addition to targeting elongation steps, these thiopeptides also possess inhibitory activity toward translation initiation by destabilizing the interactions between the ribosome, tRNA, and initiation factor 2.353–358
The discovery that extrachromosomal organellar DNA in Plasmodium sp. plastids encode rRNAs inspired efforts to target Plasmodium falciparum organellar protein synthesis using thiostrepton.359,360 The plasmodial 23S rRNA contains adenine at the position equivalent to E. coli A1067. Consequently, thiostrepton binds to plastid rRNA to terminate organellar, but not nuclear, protein translation and arrests the trophozoite to schizont transition of the parasite life cycle.361–363 Using the Malstat viability assay, thiostrepton displayed modest antimalarial activity (IC50 = 8.9 μM).364 Thiostrepton induced conformational changes in plastid rRNA fragments analogous to those that occur in bacterial rRNA, despite only 60% sequence identity between P. falciparum and E. coli rRNA.365 A single nucleotide substitution at the position equivalent to A1067 in the plastid rRNA prevented thiostrepton binding.365 Similar anti-plasmodial activity was observed for micrococcin P1, and is presumed to occur through a thiostrepton-like mechanism.366 Growth suppression towards other plastid organelle-containing parasites, specifically Babesia sp., has been reported for thiostrepton.367 Further exploration of the anti-parasitic activity of thiostrepton identified the P. falciparum 20S proteasome as a target.364 Treatment of trophozoites with the thiopeptide resulted in accumulation of ubiquitinated proteins, similar to the phenotype observed when treated with known proteasome inhibitor MG132, suggesting that thiostrepton impaired proteasome activity. Thiostrepton was found to inhibit both the chymotrypsin and caspase-like activities of human 20S proteasomes (IC50 = 5.2 μM and 3.8 μM, respectively).368 The dual-targeting ability of thiostrepton reduces the risk of developing resistance, and thus makes it an attractive anti-malarial drug.
3.2.2. Inhibitors of Elongation Factor Tu (EF-Tu)
The discovery of the d series thiopeptide GE2270A (56, 29-membered ring; Figure 33) in Planobispora rosea ATCC 53733 further expanded the notion of thiopeptides as inhibitors of the translational apparatus.369,370 GE2270A inhibits the growth of many Firmicutes, including Staphylococcus, Streptococcus, Enterococcus, and Clostridium (MIC = 23–190 nM), but shows no activity against Proteobacteria.369 Early studies determined that GE2270A inhibits protein synthesis369 by specifically targeting the GTP-bound form of EF-Tu (Kd = 1 nM) and preventing the aminoacyl-tRNAaa (aa-tRNAaa) interaction.371,372 Other thiopeptides known to bind EF-Tu include GE37468,373 thiomuracin A (57, Figure 33),374, and amythiamicin A (58, Figure 33),375 each of which contains a 29-membered macrocycle.
Figure 33.

Chemical structure of GE2270A (56), thiomuracin A (57), and amythiamycin (58).
The MOA of GE2270A was revealed by structural elucidation of E. coli (Ec) EF-Tu-GDP and T. thermophilis (Tt) EF-Tu-GNP (GNP is 5’-guanylyl imidodiphosphate, a non-hydrolyzable GTP analog) bound to GE2270A.376,377 The GE2270A-Ec EF-Tu-GDP structure showed that GE2270A binds primarily to domain 2 of EF-Tu, and is locked into a pocket by a salt bridge formed by Arg223 (Arg234 in Tt EF-Tu) and Glu259 (Glu271 in Tt EF-Tu) from Ec EF-Tu (Figure 34). The interactions between GE2270A and EF-Tu are dominated by van der Waals contacts rather than hydrogen-bonding interactions. Comparison of the hydrogen-bonding residues in GE2270A with the peptide sequences of GE37468 and thiomuracin A suggests that the following interactions may be found across other 29-membered ring thiopeptides and EF-Tu (E. coli numbering used here): Phe261EF-Tu and Tyr7; Asp216EF-Tu and Thr228EF-Tu with the b-hydroxyl group on Phe8 of thiomuracin.378 As these residues are not conserved in amythiamicin A (58) (Figure 33), the precepts of its interactions with EF-Tu have yet to be determined. Studies on the native resistance mechanism of the GE2270A producer P. rosea ATCC 53733 showed that this organism contains an alternate EF-Tu (EF-Tu1) which was resistant to GE2270A and other classes of EF-Tu-targeting antibiotics.379 EF-Tu1 has six amino acid substitutions in the GE2270A binding pocket as compared to Ec EF-Tu, and these substitutions limit the number of van der Waals contacts with GE2270A (Figure 34).
Figure 34.

(A) Crystal structure of Ec EF-Tu (PDB ID: 1D8T) bound to GE2270A (orange carbon atoms) and GDP. Ec EF-Tu residues involved in hydrogen bonding are shown in yellow. Salt bridge that locks GE2270A into the binding pocket is shown in red dashed lines. The six amino acids that differ between P. rosea EF-Tu1 and Ec EF-Tu are shown in pink. (B) Overlay of the position of GE2270A (spheres) bound to Tt EF-Tu (PDB ID 2C77) superimposed onto the structure of T. aquaticus EF-Tu bound to GTP (PDB ID: 1EFT). GE2270A intrudes into the region between domains 1 and 2 of “on” state EF-Tu. (C) Overlay of the position of GE2270A (spheres) as bound to Tt EF-Tu superimposed onto the structure of T. aquaticus EF-Tu GNP (light green) bound to yeast Phe-tRNA (yellow, PDB ID: ITTT). GE2270A clashes with the 3’ end of the tRNA molecule (red arrow). Tt EF-Tu in panels (B) and (C) are otherwise hidden from view.
EF-Tu adopts a compact structure when bound to GTP (“on state”) that is competent for binding to aa-tRNAaa. Upon GTP hydrolysis, the nucleotide-binding domain of EF-Tu (domain 1) undergoes a conformational change (“off state”) resulting in aa-tRNAaa dissociation (Figure 35).380,381 Superimposition of the structure of Tt EF-Tu-GDP bound to GE2270A with the structure of Thermus aquaticus-EF-Tu-GTP reveals that bound GE2270A would sterically clash with domain 1, suggesting that GE2270A prevents the conformational switching of EF-Tu that allows binding of charged tRNA (Figure 35).371 These observations are supported by the superimposition of the GE2270A-Tt EF-Tu-GDP structure with that of T. aquaticus EF-Tu bound to Phe-tRNA,382 which showed that GE2270A makes contacts with residues that would otherwise bind the aminoacylated 3’ end of tRNA (Figure 34).376,377
Figure 35.

EF-Tu in “on” (A) and “off” (B) states. EF-Tu adopts a compact state when bound to GTP or non-hydrolysable analogs such as GNP (PDB ID: 1EFT). EF-Tu adopts a relaxed state when bound to GDP (PDB ID: 1EFC).
3.2.3. Induction of the TipA promoter
In addition to stalling protein synthesis, thiopeptides are also known to affect prokaryotic pan-thiopeptide resistance pathways. This activity was first observed when thiostrepton (at concentrations as low as 0.3 μM) was shown to induce protein expression in S. lividans.383 TipAL and TipAS were among the most highly upregulated proteins during this response. TipAL belongs to the MerR family of stress response regulators. The N-terminus of TipAL is a winged-helix-turn-helix DNA-binding domain, while the C-terminal domain binds thiostrepton.384 An alternative start codon within the tipA gene encodes for TipAS, a discrete thiostrepton-binding domain. The TipAL-thiostrepton complex induces expression from the TipA promoter (ptipA). Binding of this complex, in turn, increases expression of TipAL and TipAS, with the latter expressed at a ~20-fold excess over TipAL. It is hypothesized that increased levels of TipAS sequester thiostrepton, serving as a resistance mechanism against thiostrepton and related antibiotics.384 Though TipA homologs are present across streptomycetes and other species (e.g. B. subtilis, Caulobacter crescentus, and Klebsiella oxytoca), the occurrence of non-thiopeptide producers that utilize this resistance mechanism has not yet been evaluated. Further studies along these avenues are warranted and may reveal that this resistance mechanism is employed against other classes of antibiotics.
Many other thiopeptides including thioxamycin (60), thiotipin (61), promothiocins A (62), nosiheptide (63) (Figures 36–38) are known to induce ptipA,383,385–387 which prompted efforts to discern the nature of interactions between thiopeptides and TipAL/AS. Detailed studies on TipAS demonstrated the formation of a lanthionine linkage between Cys214 in TipAS and a dehydroalanine residue in the C-terminal tail of thiostrepton.388,389 Thiopeptides including promothiocins A & B, nosiheptide, and berninamycins A & B (68, 69, Figure 40) were also able to form covalent linkages in vitro with TipAS,389 suggesting that this interaction is independent of macrocycle size. Tail-deleted variants of thiostrepton and promothiocin A were unable to form TipAS-antibiotic complexes, underscoring the necessity of the tail to form this covalent linkage.
Figure 36.

(A) Chemical structure of cyclothiazomycin (59), a thiopeptide that does not elicit the TipA response (i.e. lacks dehydroamino acids in the tail region), and (B) structures of those that do elicit the TipA response, thioxamycin (60), and thiotipin (61). The stereochemistry at Ala8 of thioxamycin is not defined.
Figure 38.

(A) Close up of TipAS bound to promothiocin A (carbon atoms shown in green, PDB ID: 2MBZ). (B) Close up of TipAS bound to nosiheptide (carbon atoms shown in teal, PDB ID: 2MC0). Key interacting residues on TipAs are shown in yellow. π-π stacking interactions are shown as opaque yellow regions. “Pyr” denotes the pyridine ring in panels (A) and (B). Each thiopeptide and TipAS interact mainly through hydrophobic packing. Limited hydrogen bonding contacts are made between the protein and the ligand. (C) Chemical structure of nosiheptide (63). (D) Generalized motif in various thiopeptides that is used for recognition by TipAS.
Figure 40.

Chemical structures of thiopeptides that do not act as proteasome inhibitors. Thiocillin I (67), berninamycins A (68) and B (69), and YM-266183 (70).
Studies on the requirements for TipAL-dependent ptipA induction showed that induction levels increased upon treatment with compounds with greater numbers of dehydroamino acids in the tail region.389 Thiopeptides that lacked a dehydroamino acid containing tail, such as GE2270A (56, Figure 33), amythiamicin A (58, Figure 33), and cyclothiazomycin (59, Figure 36) were unable to activate expression from ptipA. The importance of a C-terminal carboxamide is also underscored by the observation that higher dosages of thiopeptides with a C-terminal carboxylate (e.g., thioxamycin and thiotipin, Figure 36) were required for ptipA induction, suggesting that a charge at the C-terminal residue may disfavor interactions between the thiopeptide and TipAL. As tail-deleted variants of GE2270A and promothiocin A could still activate expression from ptipA at low levels, a secondary structural motif or a non-covalent mechanism may play a role.389
Thiopeptide recognition by TipAS/AL was structurally interrogated through NMR spectroscopy of TipAS-thiopeptide complexes.390,391 The structure of apo-TipAS revealed an antiparallel helical arrangement, which forms a V-shaped cleft with Cys214 located in a solvent-exposed region (Figure 37). The N-terminal region of apo-TipAS, encompassing helices α6 through α8, was disordered. Chemical shift mapping of promothiocin A-treated TipAS revealed stabilization of the N-terminal region, and suggested a hydrophobic cavity in which the thiopeptide was bound.390 These observations were validated by the co-crystal structures of TipAS bound to promothiocin A and nosiheptide where half of the “A ring” of both compounds are accommodated by helices a10–13, while the disordered N-terminus of TipAS is stabilized by binding the other half of the “A ring” (Figure 37).391 Extensive hydrophobic contacts are observed, with few if any specific interactions between the protein and thiopeptide ligand (Figure 38). Comparison of the structures reveals a common motif required for TipAS binding that includes the pyridine ring, Thz2, Oxz5, and Oxz9 (promothiocin A numbering; Figure 38). This motif explains the broad substrate tolerance of TipAS/AL, as it is found in micrococcin P1 (54), thioxamycin (60), thiotipin (61), nosiheptide (63), siomycin A (64), and many others.
Figure 37.

Promothiocin binding to TipAS. (A) Chemical structure of promothiocin A (62). (B) Structure of apo TipAS (PDB ID: 1N79). (C) Structure of TipAS bound to promothiocin A (green, PDB ID: 2MBZ). Helices α6-α8 are disordered in the apo structure and become ordered upon thiopeptide binding.
3.2.4. Anticancer Properties of Thiopeptides
Selected series a-e thiopeptides display low micromolar IC50 values against gastric and hepatocellular carcinomas.392 Investigations into anticancer MOAs have largely focused on the quinaldic acid containing compounds thiostrepton (53) and siomycin A (64) (Figures 31 and 39). Each of these thiopeptides targets the oncogenic transcription factor Forkhead box M1 (FOXM1), which activates the expression of proteins involved in cell cycle progression, angiogenesis, and metastasis.393,394 FOXM1 has received considerable interest as a chemotherapeutic target as it is overexpressed in breast tumors,395 hepatocellular carcinomas,396 non-small cell lung carcinomas,397 and other cancers.
Figure 39.

Structures of siomycin A (64), thiostrepton A hydrolyzed or methanolyzed at the ester in the B ring (65), and bisdehydroalanine fragment with anticancer activity (66).
Two independent studies revealed the anticancer potential of thiopeptides, with thiostrepton shown to inhibit FOXM1 mRNA and protein expression in a breast cancer cell line398 and siomycin A reducing FOXM1 mRNA levels in C3-Luc cells (embryonic mouse cells transformed with the human papillomavirus 16 genome, with luminescence reporter).399 Both thiostrepton and siomycin A exhibit cytotoxic activity in cancer cell lines (IC50 ~0.5–5.0 μM), but not in non-transformed MRC-5 cells (human lung fibroblasts). Multiple studies have correlated a decrease in FOXM1 expression to an increase in apoptosis, thus a pro-apoptotic action of thiopeptides has become the prevailing theory for their anticancer activity.18,398–401
The transcriptional activity of FOXM1 can be inhibited by direct inactivation or by preventing interaction with promoters. FOXM1 is activated via Thr596 phosphorylation402 and immunoblotting studies show that siomycin A-treated C3-Luc cells display lower levels of phosphorylated FOXM1. These data support a direct inactivation model wherein FOXM1 cannot be activated to promote transcription of its targets.399 Other biophysical studies support a promoter blockade model as FOXM1 binding to promoters decreases with increasing concentrations of thiostrepton.398 ITC experiments verified that thiostrepton bound to both oncogenic splice variants of FOXM1, but preferred FOXM1c.403 Thiostrepton inhibits the binding of FOXM1c to its DNA target with a KI of 11 μM (as compared to a KI of 31 μM for FOXM1b and its DNA target). In the same study, chromatin immunoprecipitation experiments demonstrated that thiostrepton-treatment of MCF-7 breast cancer cells reduced FOXM1 binding to genes implicated in cellular proliferation, i.e. Myc, Cdc25B, and cyclin B1.
Though specific interactions between thiostrepton and FOXM1 have yet to be determined, regions of the thiopeptide that are critical for antitumor activity have been identified. Truncation of the Dha terminus (Dha16 and Dha17) resulted in an analog that is unable to inhibit cell growth.403 A variant with Dha16 as the terminal residue displayed reduced activity as compared to the native compound (GI50 = 22 μM vs. 3.7 μM), whereas addition of a thiol-linked biotin tag selectively to Dha16 and not Dha17, resulted in a 10-fold reduction in activity as compared to thiostrepton. This observation suggests that the presence of at least one Dha is required for bioactivity. The importance of these residues should also hold for siomycin A, as the core peptides only differ at positions 1 and 2 (Ile1 vs. Ala1, and Val2 vs. Ser2). Lastly, SAR studies of the antitumor properties of thiostrepton suggest that the Dha-containing fragment 66 (Figure 39) is critical for bioactivity. This fragment exhibits an LC50 of 0.6 – 1.2 μM against various cell lines.404 However, the molecular target of this fragment has not been determined.
The cytotoxic activity of thiostrepton and siomycin A has also been attributed to proteasome inhibition18,405 and inhibition of mitochondrial translation.406 Treatment with siomycin A or thiostrepton resulted in stabilization of several marker proteins including p21, Mcl-1, p53, and hdm2, indicative of a non-functional proteasome.18,405 Other thiopeptides including micrococcin P1/P2 (Figure 31), thiocillin I (67), berninamycins (68, 69), YM-266183 (70), and thiostrepton that was hydrolyzed at the ester in the B-ring (65) (Figures 39–40) were unable to stabilize levels of these marker proteins nor were they able to inhibit FOXM1 (up to 10 μM).405 However, higher dosages of berninamycin (20 μM) reduced FOXM1 transcript and protein levels in prostate-derived epithelial cells.407 Apart from a strong dose-dependent effect, these results suggest that thiopeptides that only contain a primary macrocycle do not exert these bioactivities. This hypothesis is also supported by data on the methanolysis product of thiostrepton that converted the B ring to the methyl ester 65 (Figure 39).405 Taken together, these observations highlight that the presence of both primary and secondary macrocycles in addition to Dha16 and Dha17 are important for FOXM1-mediated bioactivity.
3.2.5. Additional bioactivities
In addition to the bioactivities discussed in sections 3.2.1-3.2.4, other therapeutic targets of thiopeptides have been explored. Cyclothiazomycin is an inhibitor of bacteriophage RNA polymerase408 and human renin (IC50 = 1.7 μM).409 Thiostrepton reactivates latent HIV-I cells in CD4+ T cells, which provides a means to target latent HIV infection.410,411 Thiostrepton also has been reported to rescue misfolded a-sarcoglycan (a major cause of limb-girdle muscular dystrophy type 2D),412 inhibit psoriasis-like inflammation induced by TLR7–9,413 and repress the sonic hedgehog signaling pathway in a triple-negative breast cancer model.414 Lastly, the antibacterial effects of nosiheptide415 (formerly multhiomycin) and the nocathiacins extends to clinically-relevant drug resistant strains including vancomycin-resistant S. aureus (VRSA), vancomycin-resistant Enterococci (VRE), and MRSA.416,417
3.3. Bottromycins
The bottromycins (71-74) are a group of macrocyclic RiPPs that possess growth-suppressive activity against several multidrug-resistant bacteria.33 The chemical structure of the bottromycins has undergone several revisions since its original isolation but its identity has been unequivocally established by total synthesis (Figure 41).418 Bottromycins contain a class-defining macrolactamidine and a suite of other modifications, including a decarboxylated C-terminal thiazole as well as several C- and O-methylations on the side chains (Figure 41). Studies on the MOA of the bottromycins were carried out primarily by studying bottromycin A2 (71), which was among the first two isolated members of this class and the subject of the total synthetic effort.418,419 The antibacterial activity of bottromycin has been demonstrated for Mycoplasma, MRSA, VRE, and the rice plant pathogen Xanthomonas oryzae pv. oryzae with MIC values in the low μM range.418,420,421
Figure 41.

Chemical structure of bottromycin A2 (71), and its congeners B2 (72), C2 (73), and D (74). The methyl group on Cβ of Phe6 (blue background) is critical for activity. The methyl ester (orange) is important because the corresponding carboxylic acid is inactive; however, replacement of the methyl ester with amides, ethyl/propyl esters, and propyl/isopropyl thioesters resulted in compounds with improved activity.
The bottromycins were discovered from soil-dwelling Streptomyces bottropensis and identified by the antibacterial activity present in spent media.419,422 Subsequently, isolation of bottromycin from Streptomyces No. 3668-L2 identified major and minor congeners (A2 and B2 respectively, Figure 41) that demonstrated similar antibacterial activities towards Firmicutes.423 Despite promising in vitro activity, bottromycins are not efficacious under physiological conditions owing to the hydrolytic instability of the methyl ester.
Though bottromycins are known protein synthesis inhibitors, their target was difficult to resolve. Isotopic labeling studies showed that bottromycin A2 inhibits protein synthesis in vitro and in vivo.420 Inhibition was dependent on base composition as significant translational stalling was observed for poly-C but not poly-U or poly-A. These early studies also showed that bottromycin did not affect tRNA charging, and based on these data, the mechanism for inhibition was proposed to occur through direct interaction of bottromycin with ribosome-bound mRNA in a nucleotide specific manner.
A series of subsequent studies pointed towards inhibition of translocation as a mechanism of action. In vitro translation experiments using poly-U mRNA demonstrated that bottromycin A2 inhibited the formation of triphenylalanine more than it did the formation of diphenylalanine, suggestive that translocation may be affected.424 To further study the MOA of bottromycin A2, interference of the puromycin reaction was employed. Puromycin is an aminonucleoside antibiotic that resembles aminoacylated Tyr but contains a non-hydrolyzable amide bond between the amino acid and the ribose sugar.425,426 Puromycin can enter the ribosomal A-site and be incorporated into a growing peptide; however, a second molecule of puromycin cannot be attached to the growing peptidyl-puromycin chain as the first molecule contains an inert bond. This results in the dissociation of the ribosome subunits, and chain termination.
Bottromycin A2 partially inhibited the puromycin reaction in the presence of EF-G and GTP, which are both needed for peptidyl-tRNA translocation (see Figure 30 for an overview of ribosome catalysis). In these studies, translocation of A-site loaded 3H-labeled initiator N-formylmethionine-tRNA (fMet-tRNA) and polylysyl-tRNA in the presence of EF-G, GTP, and puromycin was examined. More radioactivity was retained when bottromycin was added in the presence of GTP and EF-G than in the puromycin reaction alone, suggesting that bottromycin prevents translocation, and subsequent chain termination by puromycin.427 Similar experiments utilizing 3H-tRNA and 14C-polylysine revealed that bottromycin does not affect tRNA release from the P site, but is able to reduce the level of puromycin-dependent chain termination in the presence of GTP and EF-G. Based on these studies, bottromycin A2 was proposed to interfere with translocation and movement of mRNA on the ribosome,428 which was subsequently supported by mapping of the bottromycin binding site to the 50S ribosomal subunit.429
Subsequent studies cast doubt on inhibition of translocation as the mode of action of bottromycin A2. Experiments using E. coli ribosomes revealed that bottromycin A2 inhibited N-acetyl-14C-Phe-puromycin formation and also inhibited polyphenylalanine synthesis in the presence and absence of EF-G and GTP to similar degrees.424,430 Thus, it was concluded that bottromycin A2 inhibits peptidyltransferase activity. Additional studies complicate understanding of the mode of action, as bottromycin did not inhibit the puromycin reaction with native E. coli polysomes,431 and also failed to inhibit puromycin-dependent peptide release in protoplasts from B. megaterium.432
These conflicting results regarding the MOA of bottromycin A2 were resolved in a series of three papers. Bottromycin A2 was shown to inhibit the puromycin reaction, but increasing concentrations of puromycin reduced the levels of bottromycin-dependent inhibition suggesting that puromycin and bottromycin A2 occupy the same site on the ribosome.433 Next, bottromycin A2 was demonstrated to release peptidyl- and aminoacylated-tRNA from the ribosome A site,434,435 but not induce radiolabeled (oligo)-Phe-tRNA from the P site. These data suggest a model where bottromycin inhibits translation by releasing bound aminoacyl tRNAs from the A site. Translation studies utilizing E. coli polysomes reaffirmed that bottromycin A2 releases A site bound aminoacyl-tRNAs. This MOA also resolved conflicting studies regarding the inhibitory effect against polysomes with nascent polypeptides.431 Large peptidyl-tRNA binds more tightly to the A site than aminoacyl-tRNA, thus impeding bottromycin-dependent release.435 Thus, the conclusion of ~20 years of research established that bottromycin acts by binding the ribosomal A site to prevent recognition of charged tRNAs. An avenue for future research is to gain structural insights into this interaction for bottromycin A2 or its congeners.
The total syntheses of bottromycin A2 and its analogs have revealed which structural features are important for antibiotic activity. Bottromycin A2 and B2 differ by a methyl group at Pro2, with the latter compound four times less potent as an inhibitor against methicillin resistant S. aureus and vancomycin resistant Enterococci strains (MIC = 1.2 μM vs. 4.9 μM).436 Analogs of bottromycin A2 and B2 lacking the methyl group on Cβ of Phe6 (Figure 41) demonstrated no antibiotic activity (MIC > 32.5 μM). Likewise, removal of the Thz-β-Ala-OMe moiety from bottromycin A2 was detrimental to activity. However, a derivative in which the Thz-β-Ala-OMe was replaced with benzylamine showed effectiveness similar to bottromycin against MRSA and VRE strains (MIC = 1.3–2.7 μM). An analog that only possesses the β-Ala-OMe group (no Thz) had slightly poorer activity compared to the native compound or the benzylamine analog, suggesting that the terminal Thz can be altered. The methyl ester is also critical for bioactivity as the carboxylic acid of bottromycin A2 displayed MICs > 98 μM against MRSA and VRE.437 The hydrolytic lability of the Asp7 methyl ester to yield the inactive acid product led to synthetic efforts to generate a more stable compound in vivo. Substitution of the methyl-ester with various amides yielded bottromycin A2 analogs with improved in vivo efficacy against a mouse model of S. aureus infection compared to the parent compound; however, in vitro efficacy suffered.438 Replacement of the methyl ester with a propyl/isopropyl thioester or with an ethyl/propyl ester resulted in bottromycin A2 analogs with comparable, if not better, activity against S. aureus strains, MRSA, and VRE.437
3.4. Thioamitides
Thioamitides are RiPPs defined by the presence of a backbone thioamide(s) in place of amide(s), although other secondary modifications are typically also present. The first thioamitides to be discovered were the thioviridamides (75), and related compounds have been reported more recently (Figure 42).439 Generally, these compounds are cytotoxic against various cancer cell lines, but do not demonstrate significant antibacterial activity.20,439–442 The structural diversity of the thioamitides is reflected in the MOAs, which can impact primary and secondary metabolism.
Figure 42.

Chemical structures of thioviridamide (75), pre-thioviridamide (76), thioalbamide (77), and thiostreptamide s4 (78).
Thioviridamide (Tvr) from Streptomyces olivoviridis was first identified in a screen for antitumor compounds.439 Although thioviridamide is an acetone adduct of the true natural product prethioviridamide (pTvr, 76) (Figure 42), both compounds have comparable cytotoxic activities.439,441 Both Tvr and pTvr selectively induce apoptosis in cells transformed with the adenovirus type 12 (Ad12) or the adenoviral E1A apoptosis-suppressing oncogene with IC50 values in the low nM range. pTvr also inhibits proliferation of human cervical carcinoma cells (IC50 = 0.36 μM),443 but similar studies have not been extended to Tvr.
The cytotoxicity of pTvr was attributed to activation of the integrated stress response (ISR).443 pTvr induced significant cellular morphological changes, dissipation of mitochondrial membrane potential, a time-dependent reduction in O2 consumption as well as global translation inhibition. Screening of shRNA libraries revealed that pTvr induced the GCN2-ATF4 metabolic stress response pathway, which is central to the ISR and apoptotic induction.443,444 Photoaffinity labeling studies identified the target of pTvr as the ATP5B subunit of the mitochondrial F1 ATP synthase.443 Inhibition of F1Fo-ATPase activity was responsible for the upregulation of the GCN2-ATF4 pathway. The selectivity for E1A-expressing cells was due to increased upregulation of ATF4 upon treatment with pTvr as compared to cells that did not express E1A.
Thioalbamide (Tab, 77) and thiostrepamide s4 (78, Figure 42) are related to pTvr. Tab is similar to pTvr in that it also induces apoptosis in human cancer cell lines.20,445 Mechanistic studies on Tab demonstrated that treated cells were arrested at the G1 stage, and confirmed that both intrinsic and extrinsic apoptotic pathways were activated. Tab-induced apoptosis was attributed to increased production of reactive oxygen species (ROS). Perplexingly, superoxide dismutase isoform 2 was overexpressed in cells treated with Tab, suggesting that the mitochondria are responding to increased levels of ROS, but are failing to neutralize excess ROS.445 Additionally, exposure to Tab in vitro led to a >60% reduction of glycolysis and oxidative respiration in breast cancer cell lines. These findings support a general mechanism of metabolic disruption.
Mechanistic studies on both Tab and pTvr suggest that they perturb oxidative respiration, though a comprehensive understanding of the mechanism is lacking. Similarly, the molecular-level details regarding the pTvr:F1 ATP synthase β-subunit interaction is unknown. Derivatization studies identified pTvr analogs with improved cytotoxicity,446 and further studies on understanding of interactions between pTvr (and analogs) with ATP synthase are warranted.
4. Cyanobactins
Cyanobactins are RiPPs that are produced by cyanobacteria in both marine and freshwater environments.447–450 The majority of isolated cyanobactins are macrocyclic and contain (methyl)(thi/ox)azoline(s) installed by YcaO enzymes (section 3). However, linear cyanobactins451 and macrocyclic cyanobactins lacking azoles452 have also been characterized. Cyanobactins are often decorated with isoprenoid groups.48 Members of this class possess diverse bioactivities including antimalarial,453 anti-HIV,454 antifungal,16,455 and antitumor16,17,19,456,457 activity, though few MOA studies have been performed. The bioactivity of cyanobactins has been reviewed,19,458 and thus we only cover here bioactivities with known or preliminary MOAs.
Antineoplastic activity has been documented for several cyanobactins.16,17,19,456,457 Of these, ulithiacyclamide (79, Figure 43) is among the most potent, possessing an in vitro IC50 of 0.05 – 0.47 μM against L-1210 murine leukemia cells.459,460 Mechanistic work revealed that ulithiacyclamide inhibits protein and RNA synthesis, but detailed study of the inhibition mechanism has yet to be performed. Preliminary MOA studies have also been performed on cycloxazoline (80, Figure 43), which is cytotoxic against simian virus 40 transformed fetal lung (MRC5-CV1) and human bladder carcinoma (T24) cells at 0.66 μM.461 Cycloxazoline increased the proportion of cells arrested at the G2/M phase as well as the population of polyploid cells in a dose-dependent manner.462 This observation suggests the target of cycloxazoline is involved in cytokinesis. Trunkamide A (86) is a low micromolar growth inhibitor of cancer cell lines (IC50 = 0.8 – 1.5 μM) and has been pursued in clinical trials as an anticancer agent.463 Owing to the structural similarity to telomestatin (83),464 future studies could evaluate macrocyclic cyanobactins as telomerase inhibitors.
Figure 43.

Chemical structures of the cyanobactins ulithiacyclamide (79), cycloxazoline (80), dendroamide A (81), agardhipeptin A (82), telomestatin (83), (R,R,R) QZ59 (84), (S,S,S) QZ59 (85), trunkamide (86), haliclonamide A (87), haliclonamide B (88), sphaerocyclamide (89), kawaguchipeptin A (90), and kawaguchipeptin B (91).
Certain cyanobactins possess the ability to reverse multidrug resistance (MDR) phenotypes.465–467 MDR can arise from overexpression of ABC efflux pumps, e.g. P-glycoprotein (Pgp), which prevent the intracellular accumulation of chemotherapeutic compounds. Pgp accommodates a myriad of substrates due to its ability to repack its transmembrane helices upon substrate binding.468,469 Dendroamide A (81, Figure 43) chemosensitizes Pgp-overexpressing breast carcinomas (MCF-7/ADR) cells to daunomycin and actinomycin D.466 Cotreatment of MCF-7/ADR cells with either aforementioned chemotherapeutic and dendroamide A increased the percentage of cells killed versus treatment with chemotherapeutic alone. Direct interaction of dendroamide A with Pgp was confirmed using photoaffinity-labeling studies. Dendroamide A was able to outcompete the MDR modulator 3[H]-azidopine for binding to Pgp, suggesting the two compounds occupy a similar binding site.466,470 Pgp was proposed to contain at least three binding sites.471,472 The H-site binds Hoechst 3342, quercetin, and colchicine,471,473,474 the R-site binds anthracyclins such as daunomycin and doxorubicin,471,475 whereas the P-site binds prazosin and progesterone.472 Co-crystal structures and modeling studies of murine Pgp with stereoisomers of an analog of dendroamide A containing 1,3-selenazoles termed QZ59 (Figure 43) revealed that (S,S,S)-QZ59 (85) occupies both the H- and R-sites, while (R,R,R)-QZ59 (84) occupies a distinct site from the H- and R-site.476–478 Patellamides B and C (93, 94, Figure 44) were reported to chemosensitize human leukemic lymphoblasts to vinblastine, while patellamide D (95, Figure 44) chemosensitizes the same cell line to colchicine.465,467 Whether the patellamides interact with Pgp or with another efflux pump has yet to be determined.
Figure 44.

Chemical structure of the patellamide A (92), and patellamides B-E (93–96).
The resemblance of cyanobactins to porphyrins and cyclic crown ethers have led to the hypothesis that cyanobactins may coordinate metals and function as siderophores or chalkophores.479,480 Examples of metal-coordinating cyanobactins include haliclonamide A (87) (Fe3+-binding),481 and patellamides A, B, C, and E (Figure 44, Cu2+-binding).482,483 Furthermore, the cyanobacterial symbiotic Prochloron sp. has been shown to uptake Cu2+-complexed patellamide,484 suggesting that these organisms produce and reabsorb these molecules. However, the physiological relevance of metal binding by cyanobactins has not been investigated in detail.
Although cyanobactins have been associated with various bioactivities, the exact MOAs remain unknown. For example, agardhipeptin A (82, Figure 43) produced by Plankothrix sp. displays modest plasmin inhibitory activity.485 Similarly, sphaerocyclamide (89), a prenylated cyanobactin produced by Sphaerospemopsis sp. LEGE 00249 (Figure 43), showed only antimicrobial activity against the fouling bacterium Halomonas aquamarina CECT 5000.486 The kawaguchipeptins (90, 91, Figure 43) inhibit the growth of S. aureus (MIC ~0.7 μM) through a yet to be determined mechanism.487 Given the promising bioactivities observed, further characterization of the MOAs of these compounds is needed. Lastly, additional studies are necessary on cytotoxic cyanobactins such as microcyclamide,488 the lissoclinamides456,489 and ascidiacyclamide,490 to identity the cellular targets of these compounds.
5. Post-translationally modified microcins
Microcins are a class of ribosomally produced antimicrobial peptides that are produced predominantly by Enterobacteriaceae and demonstrate activity against a narrow range of related bacteria. The microcins are divided into two classes; class I microcins are post-translationally modified (i.e., RiPPs) and generally <5 kDa in size.37,491 Examples of a subset of class I microcins are discussed in other sections of this review, such as MccB17 (section 3.1 on LAPs) and MccJ25 (section 8.1 on lasso peptides). This section will review the known the MOA of all other class I microcins. Class II microcins are larger, ranging from 5–10 kDa in size.37,491 Class II microcins are further demarcated into subclasses IIa, which do not contain post-translational modifications, and IIb, which are post-translationally modified through conjugation with a siderophore.37,491 These class IIb siderophore-peptides will be also discussed in this section. Several prior reviews focus on the unmodified subclass IIa microcins that are not RiPPs.28,492,493
5.1. Microcin C7
Microcin C7 (McC) is a RiPP produced by E. coli and other Proteobacteria, which is active against strains of E. coli, Klebsiella, Salmonella, and Shigella.494 The MccA precursor peptide is encoded by one of the shortest known genes (21 nucleotides) and this length allows for facile iterative peptide synthesis via ribosome recycling without mRNA dissociation.495 Unprocessed McC contains a formylated N-terminus and an adenosine monophosphate (AMP) linked to a C-terminal Asp through a P-N bond (Figure 45, 97). Additional modification includes the attachment of an aminopropyl moiety on the phosphate group of AMP (Figure 45, 97).496–498 The final bioactive compound, termed here processed McC is formed after removal of the peptide by intracellular proteases in the susceptible cell (Figure 45, 98).
Figure 45.

Chemical structures of unprocessed McC (97) and processed McC (98). Essential residues required for recognition by YejA are highlighted in blue.
Early MOA studies suggested that unprocessed McC was an inhibitor of protein translation.499 However, this hypothesis was disproven, as neither 10 μM of unprocessed McC nor the MetArgThrGlyAsnAlaAsp heptapeptide inhibited in vitro translation.500 Treatment of unprocessed McC with crude cell extracts yields processed McC, a translation inhibitor that blocks the aminoacylation of aspartyl tRNA.500 Structural elucidation revealed that processed McC is an inert analog of the aspartyl-adenylate intermediate generated by aspartyl tRNA synthetase (AspRS). Inhibition of AspRS by processed McC disrupts tRNAAsp charging, and eventually leads to cell death via the inhibition of protein synthesis. Processed McC is a nanomolar competitive inhibitor of prokaryotic AspRS.496,500 Additionally, processed McC inhibited in vitro translation in a wheat germ extract system, suggesting that the compound is effective against plant AspRS; however, import of McC into plant cells was not tested.500
Active McC is only matured by cell extracts, suggesting that unprocessed McC must be imported into the cytosol of target organisms.500 This observation was confirmed as the E. coli YejABEF transport system was shown to actively transport unprocessed McC by recognition of the peptide portion of the conjugate (Figure 46).501 The N-terminal formyl methionine improves uptake by YejABEF, and conjugates shorter than the fMetArgThrGlyAsnAlaAsp heptapeptide are not imported.502 Further studies identified that the R2H, R2Y, A6F, and A6M variants of McC produced smaller zones of growth inhibition against a susceptible E. coli strain, but retained in vitro inhibitory activity, suggesting that Arg2 and Ala6 are required for import.503 Once inside the cytoplasm, unprocessed McC is matured into its bioactive form, wherein endogenous peptide deformylase removes the N-terminal formyl group from Met1,504 which is then removed by methionine aminopeptidase.505 The remaining polypeptide is digested up to the C-terminal Asp by the endogenous oligopeptidases PepA, PepB, or PepN.505 The processed mature aspartyl-adenylate analog can then bind to AspRS. The presence of the aminopropyl group improves inhibition of AspRS in an E. coli B test strain by 10-fold as compared to McC lacking the post-translational modification.496 This finding suggests that more favorable interactions are made between the aminopropylated compound and AspRS than the unmodified compound. Lastly, studies utilizing sulfamoyl analogs of McC demonstrate that replacement of Asp7 with other amino acids resulted in analogs that targeted other aaRSs.506,507 Substitutions to hydrophobic amino acids resulted in less E. coli growth inhibition, suggestive of poor YejABEF uptake or slower metabolism inside the target cell.507
Figure 46.

Schematic of McC processing. Unprocessed McC (97) is synthesized in the cytoplasm of the producer. The adenylation reaction converts the C-terminal Asn to an Asp amide. Met1-Ala6 are shown as blue circles, while the decorated Asp amide is shown as an orange circle with a star. After synthesis and export (left portion), unprocessed McC is imported into a susceptible cell via outer membrane transporters OmpC and OmpF, and the inner membrane transporter YejABEF (right portion). Unprocessed McC (97) is deformylated in the cytoplasm of the target cell, allowing for downstream proteolysis. Cellular proteases PepA, PepB, and PepN then remove the hexapeptide (blue circles) revealing the bioactive compound. Processed McC (98) inhibits the aminoacylation reaction carried out by AspRS.
Peptide-cytidinylates are a related class of protein synthesis inhibitors (Figure 47, 99). The first peptide-cytidinylate isolated was from B. velezensis (formerly, B. amyloliquefaciens) and was shown to possess a carboxymethyl modification on the cytidine.508 The unprocessed peptide-cytidinylate (pro-McCBam) was not bioactive against B. subtilis 168 or several strains of E. coli. Reported reasons for the lack of bioactivity were poor recognition by transporters, poor intracellular transport and processing, or lack of an appropriate cellular target. Subsequently, a second peptide-cytidinylate was isolated from Y. pseudotuberculosis IP32953 (termed McCYPs), which contains both an aminopropyl and a carboxymethyl moiety (Figure 47, 99).509 Like processed McCBam, processed McCYPs showed no bioactivity against an E. coli test strain. Intriguingly, bioactivity was demonstrated after proteolytic processing by TldD/E proteases in the producer strain, which reduces the 42-residue precursor peptide to 11 residues attached to the antibiotic warhead (MIC = 3 μM). The 11-residue conjugate (Figure 47, 100) can be imported by E. coli YejABEF, and the compound undergoes further processing by non-specific proteases in the target cell to generate McCYPs which inhibits AspRS. Hence, it is probable that the lack of bioactivity observed for pro-McCBam could be attributable to either a failure of import of the conjugate or a lack of proteolytic processing in the host strain.
Figure 47.

Peptide-cytidinylate moiety found on McCYps/Bam showing the cytidine and carboxymethyl moieties in blue (99). pro-McCYps (100) produced after TldD/E cleavage, the 11 amino acids that remain after cleavage are shown in orange. Neither the site of carboxymethylation nor the stereochemistry at phosphorus of these compounds have been established.
4.2. Siderophore-peptide conjugates
Siderophore-conjugated microcins, such as microcin E492 (MccE492) produced by Klebsiella pneumoniae RYC492, consist of linear peptides to which a linearized catecholate siderophore is attached via a bridging b-D-glucose.510–512 A trimer of N-(2,3-dihydroxybenzoyl)-L-serine (DHBS) anchored at the C-terminus of these peptide-sugar conjugate mimics the siderophores enterobactin and salmochelin, both of which are used in ferric iron acquisition.513 Discussion of the MOA of these peptides is complicated by the fact that early studies utilized the unmodified form of these peptides (i.e. the peptide lacking the DHBS and bridging glucose).514–516 Thus, we will use the “m” notation to denote the fully modified peptide (e.g., “MccE492m” refers to the form that is glycosylated and siderophore-conjugated), while “MccE492” is not modified or partially modified. Examples of siderophore-peptides include MccE492m (101), MccH47m, MccMm (Figure 48), and the uncharacterized MccI47m. These conjugates are hybrids of NRPS and RiPP pathways, similar to the amidinotides (section 11), as the siderophores are likely derived from an NRPS.
Figure 48.

Peptide sequences of characterized siderophore peptides. Residues colored in blue are predicted to be membrane spanning. The glycosylated trimer of N-(2,3 dihydroxybenzoyl)-L-serine (DHBS) is attached to the C-terminal serine carboxylate on MccE492m (101), and the bridging glucose is shown in orange. MccH47m and MccMm are thought to contain the same post-translational modification as MccE492m.
Of these peptides, MccE492 and MccE492m are the most extensively studied. Prior to 2004, the addition of a glycosylated siderophore was not a known modification in MccE492. These earlier studies utilized a recombinant over-producing E. coli strain that produced a compound with a mass consistent with that of Mcc492 (lacking the glycosidic linkage and the siderophore). Despite the lack of modification, Mcc492 was growth-suppressive against various E. coli (MIC = 0.02–0.14 μM), Salmonella ser. Enteritidis (MIC = 0.60 μM) and Salmonella ser. Typhimurium (MIC = 1.2 μM) strains.516 However, Mcc492 did not inhibit the growth of Klebsiella, Staphylococcus, or Vibrio at 10 μM. Though MccE492 was known to depolarize the E. coli membrane,514,516 clues related to means of cellular uptake were provided by evidence that enterobactin antagonized the activity of MccE492.517 This suggested that uptake of MccE492 occurred through the catecholate siderophore receptors Fiu, Cir, and FepA. These receptors bind to the siderophore enterobactin and its hydrolysis products (i.e. DHBS).
Optimization of the growth medium of the same recombinant overproducing E. coli strain revealed that MccE492 could be isolated with a 831 Da adduct at the C-terminus, which was confirmed to be a glycosylated trimer of DHBS (Figure 48).510,511 This product (namely MccE492m) was detected in the culture medium of the natural producer, K. pneumoniae RYC492.510 Heterologous production studies and enzymatic studies demonstrate that unmodified or partially modified forms of MccE492 (containing a glycosylated monomer/dimer of DHBS) are the result of incomplete processing.511,512,518 The antibacterial activity of fully modified MccE492m was 4–8 times greater than that of unmodified peptide against the same panel of Proteobacteria.510
Additional studies confirmed the role of the FepA, Cir and Fiu as the outer membrane receptors that import the peptide conjugate in a “Trojan horse” manner into target cells (Figure 49). The FepA, Cir, and Fiu receptors serve redundant function as MccE492m import is not compromised if any one or two of these receptors are deleted. The fepA−, cir−, or fiu− single deletion mutants, and the double mutants cir−/fiu− and cir−/fepA− remained susceptible to MccE492m. However, the fepA−/fiu− double mutant and cir−/fiu−/fepA− triple mutant were 8- fold or 200-fold more resistant to MccE492m, respectively.510 Unexpectedly, MccE492 could also be imported by the FepA, Fiu, and Cir outer membrane receptors, albeit the MICs are two to four times higher for MccE492 as compared to MccE492m for various receptor deletion strains.516,519 These findings underscore that MccE492 and MccE492m are recognized by the same receptors, although activity is notably enhanced in the presence of the DHBS trimer. However, another report found that secreted MccE492 lacking the C-terminal Ser and by consequence the siderophore, was unable to produce zones of inhibition on a lawn of susceptible E. coli.520
Figure 49.

Cartoon schematic of the MOAs for MccE492m and MccH47m. The FepA, Fiu, and Cir outer membrane receptors are responsible for recognizing the siderophore-peptide conjugate. The action of TonB is required to translocate the siderophore-peptide into the periplasm. MccE492(m) targets ManYZ in the occluded state to impede mannose import and utilizes the ManYZ as a scaffold for subsequent membrane insertion. Oligomerization of embedded MccE492 leads to pore formation, leakage of solutes (labeled M+) out of the cell, and eventual cell death. MccH47m targets ATP synthase and affects proton translocation.
Screening of E. coli K12 resistant mutants to MccE492 suggested that the cytoplasmic membrane components TonB, ExbB, and SemA could be involved in the mode of action.521 TonB/ExbB/SemA are inner membrane proteins that transduce the proton motive force of the cytoplasmic membrane to drive active transport by high-affinity outer membrane transporters, such as FepA, Cir, and Fiu.522 A TonB-deficient strain of E. coli was resistant to MccE492516 and MccE492m510 (MICs >10 μM, as compared to 0.02 μM for the wild-type strain). Furthermore, depolarization of the cytoplasmic membrane was not detected in the TonB-deficient strain treated with MccE492.516 The TonB-deficient strain of E. coli was 500 times more resistant to MccE492 than ExbB-deficient E. coli.516 These results support a model in which MccE492 or MccE492m binds to the catechol receptors, and require energy transduction by TonB/ExbB/SemA for translocation across the outer membrane.
Transposon mutagenesis revealed that the mannose permease complex (man-PTS) is required for the bactericidal activity of MccE492/MccE492m. Mutations in the manY and manZ genes, which encode for the inner membrane components of the man-PTS, confer resistance to both MccE492 and MccE492m.520 Furthermore, MccE492 co-purifies with ManZ, suggesting that the two may form a physiologically relevant complex.520,523 Thus, it was hypothesized that ManYZ may serve as a docking protein for MccE492/MccE492m for insertion into the membrane (Figure 49).520
The mechanism of action of MccE492 or MccE492m has been resolved with the cryoEM structure of MccE492 bound to ManYZ.524 The structure of apo-ManYZ revealed an outward facing transporter conformation,525 which upon binding to substrate will shift to an inward facing orientation. This sequence of steps is referred to as the “elevator mechanism”.526 The structure of MccE492 bound to ManYZ reveals the transporter in an occluded state wherein residues Leu61 – Thr77 of MccE492 are embedded in a pocket that prevent the core domain from rotating, thus impeding the ability of the transporter to engage in mannose import. The structure also reveals that the C-terminal Gly63 – Ser71 of MccE492 forms a hairpin structure that anchors ManYZ, while Asp4 – His51 forms an amphipathic α-helical domain that can insert into the inner membrane,524 cause pore formation, and disrupt the proton motive force, leading to cell death.
Additional bioactivities have been reported for MccE492, including cytotoxic activity against some human cell lines.527 MccE492 induces apoptosis in HeLa cells by disrupting mitochondrial function. It was proposed that once inside the cell, MccE492 inserts into the mitochondrial membrane leading to depolarization, release of cytochrome c, and activation of the intrinsic apoptosis pathway. Additional studies are needed to identify the cell surface receptor for MccE492. Lastly, MccE492 was reported to form amyloids-like fibrils which demonstrate reduced bactericidal activity.528 The post-translationally modified MccE492m demonstrates decreased amyloid formation.529 As soluble MccE492m is toxic to cells, these amyloids have been proposed to serve a functional role by serving as a reservoir of active microcins, which can be released in response to small environmental or intracellular changes.528,530,529
Studies on the MOA of MccH47 (Figure 48) are similarly complicated by difficulties in obtaining pure siderophore-modified peptide (MccH47m). A mixture of MccH47 and MccH47m inhibited growth of E. coli, Shigella, and Salmonella at concentrations < 2 μM, but was inactive against K. pneumoniae, K. oxytoca, Acinetobacter baumannii, Pseudomonas aeruginosa, and S. aureus up to ~100 μM.531 Like MccE492m, MccH47m relies on FepA, Fiu, Cir and additionally, the IroN receptors for cellular import.532,533 Further work on MccH427m demonstrated that TonB- and ATP synthase-deficient strains of E. coli were resistant, suggesting that ATP synthase may be the biological target.534 ATP synthase consists of a membrane-bound component (F0) and a cytoplasmic component (F1). Mutations that conferred resistance to MccH47m were localized to the a, b, or c subunits of F0, which corresponds to the proton channel of ATP synthase. These results suggested that MccH47m affects proton translocation and it was proposed that MccH47m dissipates the proton motive force by dysregulating proton transport (Figure 59).532
Figure 59.

Sequences and connectivity of lasso peptides referred to in this review. Green lines represent the isopeptide bond, while orange lines represent disulfide linkages.
Lastly, MccM (Figure 48) was shown to be most potent against strains of E. coli, while possessing some activity against Salmonella.535 The bactericidal effect of this peptide is also dependent on the FepA, Fiu, and Cir outer membrane receptors, and TonB.518 The molecular target of this RiPP is unknown.
5.3. Pantocin
Pantocin A (102) can be classified as a class I microcin as it is a post-translationally modified peptide produced by Pantoea agglomerans and shows biological activity only against related Enterobacteriaceae. Pantocin A, which contains a fused bicyclic core structure formed from two glutamic acid residues to yield an active tripeptide product, was the first identified member of this class and has served as a family prototype (Figure 50).10 The best studied target of pantocin A is the fire blight pathogen Erwinia amylovora, but the compound is also known to target other plant pathogenic Erwinia species (such as E. rhapontici and Dickeya dadantii).26,536 Cellular uptake is proposed to occur via unidentified tripeptide transporters as evidenced by suppression of activity in the presence of Ala-Gly-Gly tripeptides during cross feeding experiments.26,537 This hypothesis is supported by the requirement of a nitrogen-poor environment for bioactivity, wherein the activity of these transporters is essential. Pantocin A activity can be reversed by L-histidine and L-histidinol, and the compound has been shown to inhibit L-histidinol phosphate aminotransferase, a histidine biosynthetic enzyme. This interaction has not been structurally characterized. Other examples of such histidine-reversible antibiotics include herbicolin O538 and MccEh252,539 which likely share the same bioactive warhead as pantocin A, and are all active against plant pathogens. Moreover, the identity of the tripeptide transporters involved in import of these molecules has not yet been identified.
Figure 50.

(A) Structure of pantocin A (102). (B) Reaction catalyzed by L-histidinol phosphate aminotransferase.
6. Methanobactin
Methanobactins (mtbs) are involved in Cu(I) chelation and import by methanotrophic bacteria. These chalkophores were originally identified because of accumulation of an unidentified copper binding ligand in media containing methanotrophs with copper uptake mutations.540–543 In cultures of wild type cells, methanobactin was shown to accumulate extracellularly and copper binding initiates cellular uptake of the mtb-Cu+ complex. A crystal structure of the mtb-Cu+ complex (PDB ID: 2XJH) as well as NMR spectroscopy indicated that mtb interacted with the copper ion through two oxazolone-thioamide groups (103, Figure 51).544,545 Additional characterization demonstrated that mtb binds Cu(I). However, copper loading has been shown to occur with the addition of Cu(I) or Cu(II), and it has been hypothesized that reduction of Cu(II) may occur after loading onto mtb.546 Kinetic and spectral data were used to support this hypothesis and to develop a proposed model for mtb binding and reduction of Cu(II) to Cu(I) (Figure 51C). Methanobactins have a high degree of variability in the precursor sequences,547 but the mature structures maintain a similar ligand constellation, which is thought to be crucial for copper binding.548
Figure 51.

Structure of and proposed model for methanobactin Cu(II) binding. (A) Chemical structure of methanobactin from Methylosinus trichosporium bound to Cu+ (103). (B) Crystal structure of methanobactin binding copper (PDB ID: 2XJH). (C) Proposed model for the process of copper binding to methanobactin in the structure shown in panel A.546
Because of its toxicity, copper transport is tightly regulated. Methanotrophic bacteria have high copper requirements because one of the methane-converting enzymes is copper dependent. It has been proposed that methanobactin provides an acquisition mechanism, particularly from insoluble copper sources.549 Copper uptake and transport experiments concluded that while free copper ions undergo passive uptake through porins, the mtb-Cu complex and apo-mtb undergo active transport. Apo-mtb can act as an inhibitor of mtb-Cu uptake indicating that the same transport system is likely used for both forms.550 This transport system was later identified to be a TonB-dependent transporter, MbnT, ensuring import of mtb, and a periplasmic binding protein, MbnE, transporting periplasmic mbt-Cu.550–552
In addition to its involvement in copper acquisition, it has been proposed that mtb also plays a larger role for copper use within the cell. Two enzymes in methanotrophs are used in the process of methane oxidation, particulate methane monooxygenase (pMMO, a copper-dependent enzyme encoded at the pmo locus) and soluble methane monooxygenase (sMMO, an iron-dependent enzyme encoded at the mmo locus).553,554 The mtb-Cu complex has been shown to upregulate pmo gene expression. Initial purification attempts of pMMO resulted in the copurification of mtb leading to the conclusion that mtb could be acting as a copper-chelating cofactor.543 A series of structural and spectral characterizations of pMMO demonstrated that the enzyme did not co-crystallize with mtb but instead had three mononuclear copper centers suggesting that mtb does not act as a cofactor but may play a role in Cu delivery to the enzyme.555 EPR studies were performed that indicate mtb-Cu increases electron flow to the copper centers of pMMO although the mechanism remains to be fully elucidated.556
In addition to copper transport and roles in enzyme activity, mtb and another protein, the transcription factor MmoD, have been proposed to be involved in regulation of the pMMO and sMMO operons. A “copper switch” that upregulates pMMO and down regulates sMMO in the presence of copper557,558 was hypothesize to directly involve mtb. However, this hypothesis has been questioned because bacteria lacking the mtb biosynthetic genes still exhibit a copper switch.24
7. Signaling peptides
Quorum sensing (QS) is a phenomenon in which small molecules are utilized to modulate gene expression in response to changes in cell density. In Firmicutes, peptide hormones are used to facilitate the QS response, and examples include unmodified peptides and post-translationally modified peptides. Unmodified QS peptides are ribosomally synthesized as propeptides that are cleaved to elaborate their bioactive form.559,560 As these peptides do not undergo further modifications, they will not be covered in this review.
7.1. Autoinducing Peptides (AIPs) and gelatinase biosynthesis-activating pheromone (GBAP)
Peptides that are post-translationally modified to become signaling hormones belong to one of two classes: staphylococci utilize the macrocyclic thiolactone-containing AIPs (section 7.1.1), while enterococci utilize the macrocyclic lactone-containing GBAP (section 7.1.2). The QS response associated with these two peptides results in the up-regulation and expression of virulence factors, leading to disease states and the ability of the pathogen to evade host immune responses. An understanding of how these compounds interact with their cognate receptors could lead to the development of antagonists that subvert pathogenicity.561,562
7.1.1. AIPs from S. aureus
S. aureus is a human pathogen that causes a wide range of clinical pathologies including endocarditis and osteomyelitis.563 Among the many S. aureus virulence regulatory systems, a major one is encoded by the accessory gene regulatory locus (agrBDCA) that elaborates the peptide signal in a QS system (Figure 52).559 In this locus, agrD encodes the precursor peptide, and AgrB is an integral membrane endopeptidase that macrocyclizes the core peptide to form a thiolactone and cleaves off the C-terminal region of the precursor peptide. The peptide is presumably transferred to the extracellular face of the membrane by AgrB. The peptide is released from the membrane after the leader peptide is removed by the signal peptidase SpsB.564 Extracellular accumulation of the AIP leads to signal transduction through the Agr two-component regulatory system (TCS). Binding of the AIP to the AgrC histidine receptor kinase results in autophosphorylation, and subsequent phosphorylation on the transcriptional regulator AgrA (Figure 52). Phosphorylated AgrA can either bind to the P2 promoter, which up-regulates the agrBDCA operon, or to the P3 promoter,565 which up-regulates transcription of virulence factors including hemolysins,566 enterotoxins,567 and proteases.568 A complete list of genes and their protein functions regulated by the agr locus has been reviewed.567,569
Figure 52.

Schematic representation of the (A) agr and (B) fsr signaling pathways. The precursor peptide is synthesized with a leader region (orange). AgrB or FsrB is thought to install the thiolactone or lactone linkage (green) on the core peptide. The C-terminal portion of the AIP precursor peptide is removed and cyclized by AgrB. The AIP is secreted into the extracellular environment by an unknown mechanism. SpsB facilitates removal of the leader peptide. The GBAP leader peptide is thought to be removed by FsrB. Secreted proteases GelE (gelatinase, brown) and SprE (serine protease, blue) are factors associated with pathogenicity.
Allelic variations in the agrBDCA locus have given rise to at least four agr specificity groups (denoted by the roman numerals I, II, III, or IV; Figure 53) amongst different S. aureus strains.570–572 Each specificity group forms a cognate AIP signal:AgrC receptor pair. The autoinducing cascade is elicited only when a cognate signal:AgrC pair is formed. AIPs from other specificity groups generally inhibit agr activity, as do AIPs from other Staphylococcus sp.570,573 For certain toxin-mediated diseases, there is a strong correlation between the agr specificity group and the disease phenotype.574
Figure 53.

Chemical structures of the AIPs: AIP-I (104), AIP-II (105), AIP-III (106), and AIP-IV (107). Critical hydrophobic residues, which are part of the “triangular knob” motif, are shown in blue (see Figure 54). Substitution of the residues shown in orange greatly perturbs the three-dimensional structure of the AIPs.
SAR studies have focused on delineating the roles of the thiolactone and other residues of the AIPs with respect to agonist/antagonist effects on transcriptional activity. Comparison of synthetic, linear free acid and a linear thioester analog with cyclized AIP-II demonstrated that neither elicited or inhibited the agr response,575 highlighting that the macrocycle is functionally indispensable. The necessity of the thiolactone linkage has been probed using lactone or lactam containing analogs of AIP-II (105), each of which failed to activate AgrC-II. Similar behavior was also observed for the AIP-I lactam analog, wherein half-maximal activation was only achieved at 24 μM (~1000-fold higher than the EC50 of AIP-I).576 However, the thiolactone linkage is not critical for cross group inhibition, as both lactone and lactam variants of AIP-II were low nanomolar inhibitors of group I agr systems.575
Two key features govern the ability of an AIP to bind its receptor: the presence of a hydrophobic patch within the macrocycle and the identity of the residue following the ring-forming cysteine. Alanine scanning of residues within the thiolactone revealed that hydrophobic residues are critical for receptor activation and cross-inhibition. The F6A and I7A variants of AIP-I resulted in an increase in the EC50 of ~500-fold relative to wild-type AIP-I.576 AIP-II-L8A and -F9A were unable to activate AgrC-II or inhibit AgrC-I.575 AIP-III-F5A, -L6A, and -L7A also failed to illicit cross-group inhibition against AgrC-I, -II, and -IV (IC50 > 200 nM).577 The hydrophobic residues in AIPs can tolerate substitution to other bulky non-polar amino acids without loss of activity, as the activity profiles for the AIP-I-M8I or AIP-IV-Y5F did not differ from wild-type.578 These observations are reconcilable from the NMR structures of AIP-I (104), AIP-III (106), and AIP-IV (107), which identified a triangular knob formed by the hydrophobic patch. This knob may serve as a point of steric engagement with the receptor (Figure 54).579 AIP-II is unstructured in solution579 and contains only two hydrophobic residues, as opposed to the three possessed by the other AIPs. Presumably, AIP-II may engage its cognate receptor in a different manner than AIP-I, III, or IV.
Figure 54.

NMR structures of native AIPs. (A) AIP-I (carbon atoms in purple), (B) AIP-II (carbon atoms in light green), (C) AIP-III (carbon atoms in cyan), and (D) AIP-IV (carbon atoms in orange). Hydrophobic residues inside the macrocycle are labeled. Orientations of the triangular knobs are shown in dark blue.
SAR studies have also demonstrated that the residue following the ring-forming Cys is critical for activity. AIPs I, III, and IV possess Asp at this position. AIP-I-D5A and AIP-III-D4A inhibited AgrC across all four specificity groups.577,578 However, as AIP-I-D5N could still activate AgrC-I, (EC50 = 90 nM vs. 28 nM for native AIP-I), suggesting that receptor activation does not require ionic interactions. The NMR structure of AIP-III-D4A revealed that the Ala substitution collapses the triangular knob giving a more globular conformation (Figure 55).579 As a result, AIP-III-D4A presumably cannot make the necessary activation contacts. On the other hand, the NMR structures of AIP-I-D5A and -D5N retain the hydrophobic knob, but reveal a different orientation for the exocyclic tail than the wild-type peptide.580 These data suggest that in the context of AIP-I, and the highly similar AIP-IV, the exocyclic Asp is required for positioning the tail in a activation-inducing conformation. Structures of AIPs bound to their cognate receptors would directly answer what AIP conformations are necessary for receptor activation.
Figure 55.

NMR structures of (A) AIP-III, (B) AIP-III D4A, depicting the mutated residue in gold, (C) AIP-I, and (D) AIP-I D5A, depicting the mutated residue in gold. The triangular knob is lost in the AIP-III D4A variant and yields a more globular fold (orange line). The triangular knob is maintained in the AIP-I D5A variant, but the position of the exocyclic tail is different (green vs. blue line are pointing in opposite directions). These changes are likely to impact receptor binding.
The size of the macrocycle is also critical for activity of the AIPs. Contraction (via deletions) or additions (via introduction of L-b-homo amino acids) in the size of the macrocycle proved to be detrimental to agonist activity, without affecting antagonist activity.581 Taken together with previous observations, these data suggest that receptor agonism is intolerant to changes in AIP structure, while receptor antagonism is generally forgiving. The exocyclic tail also contributes to receptor activation. For instance, tail-less AIP-I (tAIP-I) is a poor activator of its cognate receptor (EC50 = 5.8 μM vs. 28 nM for AIP-I), while tail-less analogs of AIP-II (tAIP-II) and AIP-IV (tAIP-IV) were unable to activate their cognate receptors.578 Removal of the tail region results in weak inhibition against the cognate AgrC receptor, e.g. tAIP-I (IC50 = 4.5 μM), tAIP-II (IC50 = 230 nM), tAIP-III (IC50 > 200 nM), and tAIP-IV (IC50 = 4.1 μM).577,578 As the inhibitory concentrations of the tail-less variants against non-cognate receptors do not significantly differ, it can be surmised that the exocyclic tail is critical for agonist activity.
Little is known about what residues of AgrC mediate AIP binding. AgrC is a transmembrane protein that possesses receptor and signaling domains. To evaluate AIP recognition, chimeric AgrC receptor domains containing the N-terminal half from one class and the C-terminal half from another class were constructed. These studies revealed that the C-terminal region (residues 90–205) of the AgrC receptor is responsible for cognate AIP recognition.582 Further investigations of orthologous receptors AgrC-I and AgrC-IV, which only differ by 27 amino acids, revealed that swapping AgrC-I residues 104, 107, and 116 to their equivalents in AgrC-IV switched the activation profile upon presentation with AIP-I and -IV. These substitutions correspond to residues in the predicted second extracellular loop of AgrC receptors,583 which may directly engage the AIP. The first extracellular loop may also play a role in recognition, as swapping residues in this region of AgrC-IV for equivalent residues from AgrC-I, resulted in a 100-fold increase in EC50 when presented with AIP-IV.584 AgrC-II and AgrC-III are more sequence divergent than AgrC-I and IV, and further investigation is required to identify if their cognate ligands bind to the same extracellular loops. It should be noted that recognition may not be restricted to a single locus on the protein, and that conformational changes upon peptide binding may also occur. Structural data of ArgC or homologous receptors bound to the cognate signal would help provide insights into the determinants of signal specificity.
7.1.2. Fsr system from Enterococcus faecalis
E. faecalis is an opportunistic pathogen that can cause urinary tract infections, endocarditis, and prosthetic joint infections, which can be life threatening if left untreated.585,586 Analogous to the agr system in S. aureus, E. faecalis possess the fsr system that contributes to virulence (Figure 52B). An integral membrane protease/transporter installs a lactone (rather than the thiolactone found in the agr system) onto the FsrD precursor peptide to form the mature peptide hormone (GBAP, 108). Binding of extracellular GBAP to the histidine kinase receptor FsrC activates a two component regulatory system in which the phosphorylated FsrA response regulator activates the expression of genes encoding for gelatinase (GelE) and the serine protease SprE (Figure 52B).587–589 The fsr QS system has been shown to govern the pathogenicity of E. faecalis by regulating the formation of biofilms.590,591 GelE-positive E. faecalis strains have been reported to induce disease phenotypes in mice and rabbit models.592,593 In contrast, GelE deletion strains have been associated with increased survival in nematode models.594 Moreover, the expression of GelE is positively correlated with biofilm formation.595,596 For more details we refer to a recent review on virulence factors and E. faecalis pathogenicity.597
Two SAR studies have been conducted on GBAP (Figure 56) in which the role of the lactone, the residues in the macrocycle, and the necessity of the tail for agonist activity were tested.598,599 Replacement of the lactone linkage with a lactam maintained similar levels of agonist activity but replacement by a thiolactone or a disulfide was not tolerated.598 The tail region was found to be dispensable for agonist activity, as its complete removal, single alanine substitutions (Q1A and N2A), or substitutions with the corresponding D-amino acid resulted in similar EC50 values as compared to GBAP. Interestingly, the D-Phe7 variant demonstrated only a four-fold increase in EC50 over GBAP, suggesting that the FsrC binding pocket can accommodate the epimer. Alanine substitutions of residues in the macrocycle severely reduced agonist activity, as the I6A, F7A, and G8A variants failed to activate the QS response at 10 μM, suggesting that these residues are the most critical for interactions with the receptor. The NMR structure of wild type GBAP reveals that Ile6-Phe7-Gly8 forms a triangular knob similar to that reported in the structure of AIP-III (Figure 56).579,598 Hence, it is likely that the GBAP:FsrC engagement is governed by similar interactions.
Figure 56.

(A) Chemical structure of GBAP (108). Critical hydrophobic residues are shown in blue. (B) NMR structure of GBAP highlighting the triangular knob formed by Ile6-Phe7-Gly8.
Although specific residues of FsrC that engage GBAP have yet to be identified, the FsrC receptor-binding domain is predicted to consist of six transmembrane helices and three extracellular loops (akin to AgrC). Synchrotron-radiation circular dichroism spectroscopy on FsrC and FsrC bound to GBAP was used to study the interactions between these receptor-signal pairs.600 While no changes to the secondary structure occurred upon GBAP binding, changes to the tertiary structure of FsrC were inferred from spectral shifts in the near UV region (corresponding to signals from Tyr and Trp). Notably, several Tyr and Trp residues reside in the first or second extracellular loops of FsrC, suggesting movement of these residues upon signal binding. Further studies are necessary to further demarcate the regions and residues in FsrC required for GBAP binding.
7.2. ComX
B. subtilis also utilizes peptide pheromones for intra-species signaling. B. subtilis possesses multiple master regulatory proteins including Spo0A, DegU, and ComA. These regulators coordinate the ability of the organisms to differentiate into different subpopulations, as well as to enter states of natural competence.601 Both responses are important for proper niche establishment and survival. One of the most studied quorum-sensing pathways in B. subtilis comprises the ComQXPA system, in which ComX encodes for a ribosomally synthesized and post-translationally modified peptide signal.
The first ComX isolated was from B. subtilis 168, and was termed ComX168 (Figure 57, 109).602 Analysis of the ComX168 open reading frame revealed that the gene encodes for a 55-residue peptide, while the secreted product is a prenylated 10-residue peptide. A farnesyltransferase, ComQ, attaches a C15 lipid (farnesene) to the precursor peptide. Alanine scanning of the last nine amino acids of the core peptide revealed that the unmodified W53A variant was nonfunctional as a signal.603 The NMR structure of a related geranylated pheromone ComXRO-E-2 from B. subtilis strain RO-E-2 (Figure 57, 110) revealed that the mature compound features a tricyclic tryptophan modification geranylated at the C3 position.604 SAR studies on ComXRO-E-2 revealed that the minimal peptide that retained activity was the geranylated tripeptide [Phe-Trp-Glu] ComX RO-E-2.605 The length of the isoprenyl chain (i.e. C10 vs C15) may dictate specificity groups within this class of molecules.606 Currently, there are no studies on the effect of isoprene substitution on bioactivity of ComX peptides.
Figure 57.

Chemical structures of ComX168 (109) and ComXRO-E-2 (110). The minimal structural features required for ComXRO-E- activity are highlighted in blue. The inset shows a schematic of ComX pheromone production and activity. ComX peptides activate phosphorylation of ComA mediated by the ComP receptor. Phosphorylated ComA activates expression of surfactin biosynthesis, which is necessary for cellular differentiation in response to environmental changes and production of secreted proteases that are used for cells to escape biofilms.
The interactions that dictate specificity between ComX and its transmembrane receptor are not known, but the biological effects of the signaling cascade have been determined. ComX is sensed by the ComP-ComA two-component regulatory system. Upon binding of ComX to its second extracellular loop, the histidine kinase ComP undergoes autophosphorylation.607,608 The phosphate group is then transferred to the response regulator, ComA, and phosphorylated ComA activates the transcription of genes found in the sfrA operon.609–611 One of the proteins encoded in this operon is ComS,612 which prevents the degradation of transcription factor ComA613 permitting the expression of genes related to competence, surfactin biosynthesis, and biofilm formation (Figure 57, inset).614,615 Surfactin is secreted as a QS signal for neighboring cells to upregulate genes required for cellular differentiation, but is not directly involved in regulating competence.616 Phosphorylated ComA also activates the transcription of the DegQ transcriptional regulator that enhances the rate of DegU phosphorylation. High concentrations of phosphorylated DegU leads to the production of secreted proteases, which may allow individual cells to escape the biofilm.617,618 Lastly, as with other classes of autoinducing peptide receptors (section 7.1), polymorphisms in the comQ, comX, and N-terminal portion of comP reveal that B. subtilis strains can be classified into one of four pherotypes,619 wherein members of the same group can interact effectively with each other, but not between groups.
8. Peptide atropisomers
Atropisomerism is a type of conformational chirality that results from hindered rotation about a single bond, when the energetic barrier for rotation is high enough to allow for the isolation of individual conformers. Non-canonical atropisomers have identical connectivities and configurations at all stereogenic centers but cannot be converted by rotation along a single bond. For example, threaded lasso peptides may be considered as non-canonical atropisomers of the unthreaded counterpart.620
8.1. Lasso peptides
The lasso peptides are a class of RiPP with a class-defining macrolactam formed between the N-terminus and an internal Asp or Glu residue (an isopeptide bond). This macrolactam ring encircles the peptide forming a lariat knot topology. The C-terminal tail is locked within the ring and held in place either sterically by the presence of a large residue (often referred to as “plugs”) or by one or more disulfide bonds. This threaded, rotaxane-like conformation is known colloquially as the “lasso fold”, as opposed to an unthreaded, “branched-cyclic” conformation (Figure 58). Formally, lasso peptides may be considered as members of the class I microcins (see Section 5 on microcins), and microcin J25 is one of the first and among the best studied lasso peptides. Lasso peptides are divided into four subclasses based on the presence and connectivity of disulfide bonds (Figure 58). The sequence diversity of the lasso peptides and their attendant MOAs are broad, including antibacterial activity, receptor antagonism, and antiviral activity; these features make the lasso peptides a promising class of RiPPs for further discovery and development.621,622
Figure 58.

(A) Schematic structure and classification of lasso peptides. Important regions of a generic lasso peptide are shown. (B) Class I lasso peptides have two disulfide bonds (orange) that covalently join ring-to-loop and ring-to-tail. A large majority of lasso peptides belong to class II, which lack disulfide linkages; instead, the loop is sterically thought to be locked into place by the side chains of “plug” residues (orange circles) in the tail that are positioned below the ring and sometimes also above the ring. Class III lasso peptides are joined by ring-to-loop disulfides while class IV display a tail-to-tail disulfide linkage. The numbering of the residues in the schematic structure of class I shows that the lasso peptide is right-handed, which is the natural isomer for all currently known lasso peptides.
Prior to the genomics revolution, virtually all natural products were discovered via bioassay-guided approaches. Lasso peptides discovered during this time (pre-2000) are primarily active against cell-surface receptors. The first, anantin (Figure 59), is an antagonist of the atrial natriuretic factor receptor, with a reported Kd of 600 nM and an in vitro IC50 of 1.0 μM.623 Currently, little is known about the molecular level details of the peptide-receptor interaction. Subsequent discovery and evaluation of RES-701–1 (Figures 59, 60) demonstrated selective antagonism of the endothelin type B receptor (IC50 = 10 nM) over endothelin type A receptors.624 Related lasso peptide congeners RES-701–2, −3, and −4 were also selective antagonists of the endothelin type B receptor.625 This activity was dependent on a threaded conformation as chemically synthesized, branched-cyclic RES-701 lacked the selectivity/activity of naturally isolated threaded RES-701–1.626,627
Figure 60.

(A) Structure of endothelin type B receptor bound to the human hormone endothelin-1 peptide (PDB ID: 5GLH). The transmembrane (TM) domain is shown in green, while the intracellular domain is shown in orange. (B) Close up of endothelin-1 bound to the endothelin type B receptor. Portions of the linear tail of endothelin-1 (Ile19-Trp21) project into the cavity of the receptor. RES-701–1 and its congeners may bind in a similar way. Peptide sequences of the hormone endothelin 1 and the lasso peptide RES-701–1 are shown (the isopeptide linkage is shown in green and disulfide links in orange).
SAR studies demonstrated that the C-terminal Trp of RES-701–1 is directly involved in the selectivity for the endothelin type B receptor. Methyl esterification of the C-terminus yielded a dual selective analog that demonstrated affinity for both endothelin type B and type A receptors. Deletion of the C-terminal carboxylate to yield tryptamine resulted in a 5-fold loss in IC50 against the endothelin B receptor (IC50 = 10 nM vs. 49 nM).628 Further studies demonstrated that substitution of the C-terminal Trp to Phe, Tyr, or removal of Trp had only a modest effect on the binding activity against endothelin B.629 Substitution with Gly or Ala substantially weakened the in vitro IC50 against endothelin B by 12- and 50-fold, respectively. Substitution at the C-terminus with β−2-naphthyl-alanine modestly enhanced the IC50 from 10 nM to 6.3 nM. These SAR studies suggest that larger/aromatic sidechains at the C-terminus are involved in receptor binding, and replacement with smaller residues destabilized the interaction of RES-701–1 with the endothelin B receptor. The bioactivity of RES-701–1 was also demonstrated in isolated tissue samples.630,631 There are no structures of a complex between RES-701–1 and the endothelin B receptor, which may provide a framework for further optimization of this compound.
The lasso peptide BI-32169 (Figure 59) inhibits glucagon-dependent cAMP production by the human glucagon receptor with an IC50 of 440 nM.632,633 The activity of BI-32169 was enhanced by methyl esterification of the C-terminus. The first, and to date only, successful total chemical synthesis of a lasso peptide (BI-32169) in a threaded conformation was achieved using a cryptand-imidazolium support.634 The threaded conformation of synthetic BI-32169 was confirmed using a number of analytical and biophysical methods, and using the natural product as a standard. Moreover, synthetic BI-32169 afforded opportunities for SAR studies. Specifically, anti-glucagon receptor antagonism was enhanced from 520 nM to 160 nM when a BI-32169 variant consisting of all D-amino acids was used against BHK-21 cells expressing the human glucagon receptor. To date, no detailed SAR studies have shed light on the glucagon receptor:BI-32169 interaction.
Several characterized lasso peptides exhibit protease inhibitory activity. Propeptin (Figure 59) displays low micromolar activity against human, bovine, and flavobacterial prolyl endopeptidases (IC50 ~0.35 μM), and antibacterial activity against Mycobacterium phlei.635,636 Propeptin-2, which lacks the two most C-terminal residues of propeptin (Ser-Pro), displays two-fold decreased activity against flavobacterial prolyl endopeptidase and loses antibacterial activity against M. phlei.637
The lasso peptide lassomycin (Figure 59) is growth-suppressive against multiple clinical and drug-resistant strains of M. tuberculosis (MICs = 0.83–1.65 μM) and Mycobacterium sp.638 Spontaneous lassomycin-resistant bacteria possessed mutations to the molecular chaperone casinolytic protease C (ClpC1), which unfolds misfolded or aggregated proteins in an ATP-dependent manner and translocates these proteins to ClpP1/P2 proteases for protein recycling.638–640 In the presence of lassomycin, purified mycobacterial ClpC1 exhibited increased ATPase activity but a decrease in the ability to degrade the studied substrate casein. These data suggested that lassomycin inhibited translocation of a bound protein substrate to the proteolytic active site. Structural modeling studies indicated lassomycin likely interacts with the N-terminal half of ClpC1 (Figure 61). Resistance mutations against lassomycin were localized to the N-terminal region of ClpC1. The NMR structure of lassomycin was initially and incorrectly assigned as a branched-cyclic conformer (PDB ID: 2MAI), leading to questions on whether the threaded lasso conformation was necessary for bioactivity. Chemically synthesized lassomycin was isobaric to the naturally occurring compound, but exhibited markedly different NMR chemical shifts and lacked any anti-M. tuberculosis bioactivity up to 55 μM.641 These results proved that the synthetic, branched-cyclic form of natural lassomycin was incorrect and the threaded conformation is now presumed to be essential for bioactivity.
Figure 61.

Crystal structure of ClpC1 (PDB ID: 3WDB). The N-terminal portion of the protein (circled and labeled) is proposed to interact with lassomycin.
Siamycin I and II (Figure 59)642 were originally identified in a syncytia inhibition (SI) screen for inhibitors of viral fusion; both compounds showed inhibitory activity against HIV and HSV.643 In the SI assay, a cell line expressing the HIV envelope proteins gp160 (or the cleavage products gp120 and gp41) is co-cultured with HeLa cells expressing the CD4 receptor.644 Cell fusion generates polynuclear cells (syncytia), the formation of which is prevented by viral fusion inhibitors. Anti-HIV activity (ID50) as measured in a cell viability assay for siamycin I and II were determined to be 3.2 and 4.1 μM, respectively; while anti-HSV-1 ID50 for siamycin I was 22 μM and siamycin II was 12 μM.643 Furthermore, siamycin I possesses antiviral activity against laboratory strains of HIV-I (ED50 = 0.05–4.5 μM) as well as clinical isolates that are resistant to HIV protease and reverse transcriptase inhibitors (ED50 = 0.89–5.7 μM).645
Investigations of siamycin I-resistant HIV strains revealed multiple point mutations in both gp160 and gp41. Utilizing the SI assay, it was demonstrated that preincubation with siamycin I, followed by removal of the compound restored syncytia formation, suggestive of non-covalent inhibition. A third anti-HIV lasso peptide, siamycin III (originally RP 71955) was discovered using a fluorometric assay to screen for inhibitors of HIV-1 protease.646,647 Solution structure determination of siamycin III revealed a structure similar to that of siamycin I/II.647 Detailed mechanistic analyses on the anti-HIV activities of these peptides have not yet been reported.
In addition to inhibition of viral fusion, siamycin I showed antibacterial activity. S. aureus ATCC 29213-resistant mutants against siamycin I exhibited cross-resistance to both vancomycin and nisin (see section 2), which are both antibiotics that target the cell wall.648 Treatment of B. subtilis with siamycin I induced the liaI (lipid II-interacting antibiotic) cell wall stress response, which is generally indicative of a cell wall target (sections 2.2 and 15.4).649 Follow-up studies revealed that fluorescently labeled siamycin I localizes to the division septum of S. aureus, and in doing so, delocalizes penicillin-binding protein 2 (PBP2) from its usual site of action.648 In vitro studies determined that siamycin I inhibits the transglycosylation reaction catalyzed by PBP2, as detected by reduced undecaprenyl pyrophosphate release from lipid II.
Siamycin I at sub-lethal concentrations attenuates the fsr quorum-sensing system of E. faecalis. This quorum-sensing system is autoinduced by the peptide GBAP (section 7.1.2) and leads to the expression of two virulence factors: the gelatinase GelE and a serine protease SprE. Siamycin I inhibits GelE expression by decreasing GBAP production, which was attributed to disruption of the fsr two-component signal transduction proteins FsrC-FsrA (Figure 52B).650 Subsequent work showed siamycin I inhibited the autophosphorylation activity of FsrC,651 and spectroscopic studies supported a direct interaction between siamycin I and the FsrC kinase at a site distinct from the binding site of the native GBAP ligand.652 Siamycin I also possessed in vitro inhibitory activity against several ATP-dependent enzymes, including membrane sensor kinases, protein kinases, and ATPases of both prokaryotes and eukaryotes.651 Despite these in vitro activities, siamycin I lacks anti-Proteobacterial activity in vivo, suggesting that this lasso peptide cannot cross the bacterial outer membrane.643,651 The related class I lasso peptide sviceucin also demonstrated anti-fsr activity.653 Sviceucin has an MIC of 10 μM against E. faecalis, but treatment with sublethal dosages, below 1 μM, reduced GelE production by 50–70%.653 It has yet to be determined if sviceucin also targets FsrC.
Streptomonomicin (Figure 59), produced by Streptomonospora alba, is a cell wall-targeting lasso peptide that showed low μM MICs against multiple Bacillus sp., but little to no activity against fungi or Proteobacteria.654 Sequencing of spontaneously resistant B. anthracis colonies revealed five mutations to the walR gene and a single mutation in a regulatory region directly upstream. WalR is a response regulator that functions along with the histidine kinase WalK to form a two-component signal transduction system that controls the expression of proteins involved in cell wall metabolism and division.655,656 Of the isolated WalR variants, three contained amino acid substitutions in the DNA-binding domain, whereas the other two had substitutions in the phosphate group receiver domain.654 These B. anthracis mutants demonstrated a chaining phenotype, indicative of a septal cleavage defect, where daughter cells do not separate after division.654 The WalRK system regulates the expression of LytE, an autolysin involved in septal cleavage in Bacillus sp,654,657 and decreased expression of LytE would be consistent with the elongated phenotype. Downregulation of lytE was demonstrated for streptomonomicin-resistant mutants, leading to the hypothesis that streptomonomicin targets cell wall biosynthesis through the WalR protein.654 Despite these studies of resistant mutants, the details of the MOA of streptomonomicin remains unknown. Further study is needed to confirm or refute WalR as the target of streptomonomicin.
Several lasso peptides target proteobacterial RNA polymerase (RNAP). The best-studied of these is the class II lasso peptide microcin J25 (MccJ25), which demonstrates in vivo and in vitro activities against clinically relevant strains of E. coli, Salmonella sp., and Shigella sp.658–660 Studies of spontaneously resistant E. coli mutants identified a variant possessing a T391I substitution in RpoC, the β’ subunit of E. coli RNAP.661 RNAP as the target for MccJ25 was supported by complementation studies and in vitro and in vivo transcription assays. MccJ25 did not demonstrate in vitro activity against non-proteobacterial RNAP, including homologs from T. aquaticus, B. subtilis, and yeast.662 The site of MccJ25 binding was suggested to be the E. coli RNAP secondary channel, which was supported by identification of resistant mutations that mapped within and near this channel (Figure 62). Biophysical characterization demonstrated that MccJ25 binds to and occludes the secondary channel, preventing entry of nucleotides to the polymerase active site and locking RNAP into an inactive conformation.663,664 Saturation mutagenesis of the secondary channel demonstrated that substitutions at multiple sites within this channel endowed resistance to MccJ25.664 The co-crystal structure of MccJ25 bound to E. coli RNAP revealed that MccJ25 is bound primarily through hydrophobic contacts in the region between the bridge helix, activation loop, and F loop.665 MccJ25-Tyr9 is essential for RNAP inhibition as substitutions to any other amino acid abolished activity.666 The C-terminal tail is necessary for RNAP interaction and inhibition as confirmed by structural studies.665 Amidation of the C-terminal Gly of MccJ25 inhibited RNAP interaction in vitro but did not abolish peptide uptake.667
Figure 62.

(A) Surface model of E. coli RNAP β’ subunit bound to MccJ25 (PDB ID: 6N60). (B) Close up of bound MccJ25 (carbon atoms in green). MccJ25 interacts with the E. coli RNAP β’ subunit primarily through hydrophobic interactions. The activation loop (purple), F loop (red), and bridge helix (tan) and active site Mg2+ are also shown. (C) Location of resistance-conferring variants are shown as spheres in a wire model of E. coli RNAP β’ subunit. Spheres corresponding to residues in the activation loop (purple), F loop (red), or bridge helix (tan) are colored as in panel B. Sites of other variants are shown in blue.
As MccJ25 shows in vivo activity against Proteobacteria, the compound must cross both the inner and outer membranes to bind to the intracellular RNAP target. Transposon mutagenesis endowing MccJ25 resistance identified insertions in the fhuA gene, which encodes a multifunctional outer membrane protein largely involved in siderophore transport.668 A model of MccJ25 internalization was established with the identification of additional resistant E. coli strains that possessed mutations in tonB, exbB/D, and sbmA genes.286 These genes encode components of the TonB membrane transport pathway. Binding of TonB, ExbB, ExbD, and SbmA to an unidentified proton channel induces a conformational change in FhuA, causing it to open and presumably import MccJ25.
S. ser. Typhimurium contains an FhuA homolog but is otherwise resistant to MccJ25. However, S. ser. Typhimurium can be sensitized to MccJ25 when provided with E. coli fhuA. This result provides a framework for understanding how sequence differences in FhuA may account for MccJ25 import.669,670 Subsequently, mutational studies on E. coli FhuA suggested that MccJ25 bound the exterior loops of the protein, as the removal of these loops abolished MccJ25 bioactivity.671 MccJ25 binds to E. coli FhuA with a Kd of 1.2 μM and stoichiometry of 2:1 MccJ25 to FhuA.672 The co-crystal structure of E. coli FhuA-bound MccJ25 provided definitive evidence of a direct interaction of MccJ25 with the exterior cavity of FhuA, which is similar to the binding mode of the native ferrichrome ligand (Figure 63).673 However, ferrichrome interacts with FhuA through extensive polar contacts while MccJ25 makes limited contacts with FhuA via the MccJ25-His5 imidazole and the MccJ25-Ala3 backbone carbonyl. MccJ25-His5 engages in a critical hydrogen bonding contact with the backbone amide of FhuA-Phe115.
Figure 63.

Co-crystal structures of FhuA-MccJ25 and FhuA-ferrichrome. (A) The co-crystal structure of MccJ25 (green spheres) bound to FhuA (light blue ribbons) demonstrates that the lasso peptide is bound to the exterior cavity of FhuA (PDB ID: 4CU4). Ferrichrome (brown spheres) bound to FhuA (PDB ID: 1BY5) occupies a similar region. (B) Close-up of the ferrichrome binding pocket. FhuA residues engaging in hydrogen-bonding interactions are shown in yellow (black numbering), while carbon atoms of ferrichrome are shown in brown. (C) Close-up of the MccJ25 binding pocket. Residues engaging in hydrogen-bonding interactions are shown in yellow (black numbering for FhuA, green borders for MccJ25; carbon atoms of MccJ25 shown in green).
SAR studies of MccJ25 established the role of the threaded conformation for bioactivity. Thermolysin cleavage of the β-hairpin loop region of MccJ25 (between Phe10 and Val11) generated an interlocked dual-chain species (t-MccJ25), which increased the MICs, as measured by growth inhibition in liquid culture, to 80–150 nM against S. enteritidis (MccJ25, <2 nM) and 300–600 nM against a TolC-deficient strain of E. coli (MccJ25, 2–5 nM).674,675 In vitro transcription studies demonstrated that both MccJ25 and t-MccJ25 inhibited E. coli RNAP and that losses in bioactivity resulted from impaired FhuA-dependent uptake.676,677 Modification of MccJ25-His5 by carbethoxylation also reduced activity, likely by interfering with peptide uptake.678 Additional studies on the role of MccJ25-His5 suggested that this residue was important for internalization by the SbmA transporter.679 Bypass of FhuA by osmotic shock did not restore antibacterial activity to the MccJ25-H5A or -H5R variants, supporting the importance of the inner membrane transporter SbmA for MccJ25 to gain cytosolic access. Recently, the cryo-EM structure of SbmA bound to a monoclonal antibody has been determined to 3.2 Å resolution, albeit in the absence of bound ligand such as MccJ25, klebsazolicin or MccB17 (see section 3.1 on LAPs).680 The structure shows a previously unknown fold in an outward-open conformation exposing a large cavity. This transport cavity is of sufficient size (~23 Å) to accommodate MccJ25 (or a LAP), and targeted mutation of residues lining the transport cavity decreased translocation of MccJ25. Currently, no resistance-conferring mutations for sbmA have been reported, which would shed additional light on the interaction of MccJ25 and the SbmA inner membrane transporter.
Microcin Y(MccY) is a recently discovered lasso peptide produced by S. enterica serovar Birkenhead which possesses 52% sequence identity (13 of 25 residues) with MccJ25 (Figure 59).681 Despite this high relatedness, the antibacterial spectrum of MccY differs from MccJ25. MccY inhibits the growth of Salmonella infantis while MccJ25 is inactive (MIC = 0.04 μM vs. >200 μM).681 Unlike MccJ25, MccY does not inhibit the growth of E. coli (tested upwards of 200 μM).681 E. coli transformed with FhuA from S. enterica serovar Typhimurium became susceptible to MccY (MIC = 12.5 μM), suggesting that MccY and MccJ25 share an outer membrane uptake mechanism, though the specific intracellular target of MccY is unknown. Furthermore, while MccJ25 is translocated across the inner membrane by SbmA, it is currently unknown if MccY also utilizes this protein, as spontaneously resistance mutants have not been studied. Additional studies are necessary to determine if receptor (FhuA/SbmA) or putative target (RNAP) amino acid sequences are the determinate factor for MccY/MccJ bioactivity spectra.681
Secondary MOAs for MccJ25 have been proposed, including dissipation of the cytoplasmic membrane potential, superoxide production, and antimitochondrial activity. In S. enterica serovar Newport, MccJ25 was shown to inhibit respiration, as measured by oxygen consumption in the presence of 10 mM D-glucose.682 Assays using the tetrazolium dye MTT demonstrated a marked decrease in the activities of reduced nicotinamide adenine dinucleotide (NADH) and succinate dehydrogenases in isolated membrane fractions of MccJ25-treated S. ser. Newport, though these observations were not observed in assays against E. coli. Effects on oxygen consumption were observed in S. ser. Typhimurium, S. ser. Derby, and S. ser. Enteritidis strains transformed with the E. coli FhuA protein.670,677 Studies using bacteriomimetic vesicles showed that MccJ25 inserted into and increased the fluidity and permeability of these membranes in a FhuA-independent manner.683,684 2,4-Dinitrophenol-dependent dissipation of the proton motive force, which FhuA uses for active transport of MccJ25, did not drastically impair the antibacterial activity of this lasso peptide, suggesting that internalization by FhuA is not the only means of import.685
Reactive oxygen species (ROS) production has been observed in E. coli overexpressing FhuA treated with MccJ25.686 Utilizing a C-terminally amidated analog of MccJ25, which does not interact with RNAP (discussed above), E. coli FhuA-overexpression strains were sensitized to MccJ25 through the production of ROS.687 E. coli strains expressing FhuA with a deletion of either cytochrome bd-I or bo3 remained resistant to MccJ25, implicating these two enzymes as potential targets of MccJ25. MccJ25 was shown to weakly inhibit purified cytochrome bd-I, slowing the rate of electron transfer by the enzyme, and resulting in a net increase of superoxide production.688 Follow up studies confirmed the role of MccJ25-Tyr9 in superoxide production, and electron paramagnetic resonance studies demonstrated the formation of a tyrosyl radical and its participation in the redox reaction cycle.689,690
Antimitochondrial activities of MccJ25 have been observed in rat heart, wherein MccJ25 inserted into the membrane, dissipated the proton motive force, reduced intracellular ATP, and inhibited complex III of the cytochrome c reductase respiratory chain.691 Through ROS production, MccJ25 was shown to activate the mitochondrial permeability transition pore, leading to mitochondrial swelling and cytochrome c release.692 Furthermore, MccJ25 was shown to induce carbonylation of mitochondrial proteins and oxidation of mitochondrial lipids, both of which would induce the release of cytochrome c.693 MccJ25-dependent effects on the mitochondrial permeability transition pore and membrane were only observed in the presence of 20 μM MccJ25, suggesting this may not be a physiologically relevant MOA.692,693 Despite this, interference with oxidative respiratory cycles demonstrates the mechanistic plasticity of lasso peptides, which could be improved through engineering efforts.
Though MccJ25 has been extensively investigated in terms of its bioactivity, other lasso peptides including capistruin,694 acinetodin,695 klebsidin,695 citrocin,696 and ubonodin (Figure 59) are also known to inhibit E. coli RNAP.697 Capistruin, a class II lasso peptide, displays growth-suppressive activity against certain Proteobacteria (e.g., Burkholderia sp. and Pseudomonas sp.).698 Capistruin inhibited transcription activity of E. coli in vitro and MccJ25-resistant RNAP was also resistant to capistruin.694 Structural work on capistruin bound to RNAP demonstrated MccJ25-like binding within the secondary channel (Figure 64).665 However, capistruin is only weakly active (MIC = 25 μM) against E. coli, suggesting that capistruin is not able to cross the E. coli outer membrane as effectively as MccJ25.698 In support of this hypothesis, structural studies of E. coli FhuA demonstrated that capistruin could not be modeled into the exterior cavity without steric clashes.673
Figure 64.

Capistruin binding to RNAP. Capistruin (blue carbon atoms) occupies a similar binding site as MccJ25 (green carbon atoms, see Figure 62) but is not positioned as close to the RNAP active site. The closest residue to the catalytically requisite Mg2+ ion is Arg15 (12.8 Å away), as compared to His5 in MccJ25 (5.9 Å).
Capistruin is active against Burkholderia suggesting that Burkholderia FhuA can bind the lasso peptide.673 This proposal is speculative as currently no data suggest that capistruin is imported by a TonB-dependent siderophore receptor. Like capistruin, acinetodin and klebsidin are active against E. coli RNAP, and MccJ25-resistant variants exhibited cross-resistance to both lasso peptides.695 Additionally, while these compounds were inactive against E. coli, klebsidin was active against K. pneumonia. E. coli could be sensitized to klebsidin through expression of the K. pneumonia FhuA protein, which possesses 60% amino acid sequence identity to E. coli FhuA. These findings support a mechanism where these lasso peptides are imported by a FhuA/TonB-dependent receptor and rationalize the relatively narrow ranges of bioactivity.
The lasso peptides citrocin produced by Citrobacter pasteurii type strain CIP 55.13 and ubonodin produced by Burkholderia ubonesis MSMB2207 (Figure 59) display RNAP inhibitory activity in vitro, although the physiological relevance of this activity has not yet been established.696 At 1 μM, citrocin reduced transcription levels of E. coli RNAP to 15%. E. coli mutants resistant to citrocin contained alterations in sbmA but not in fhuA, indicating that citrocin is imported into susceptible cells through a different transporter than MccJ25, which engages FhuA. Notably, citrocin has more potent E. coli RNAP inhibitory activity despite possessing a higher MIC against E. coli BW25113, which expresses TolC (citrocin, 16–125 μM; MccJ25, 1 μM as determined in the same study by spot-on-lawn assays). This finding is likely due to differences in ability for citrocin to cross the outer membrane of Proteobacteria. The activity of ubonodin was initially tested against E. coli RNAP, revealing only 60% transcription inhibition at a 100 μM dose.696,697 The high concentration required for inhibitory activity suggests that E. coli RNAP may not be a physiologically relevant target of ubonodin. Moreover, ubonodin is unable to produce zones of inhibition on E. coli lawns but inhibits growth of several Burkholderia sp., particularly Burkholderia cepacia (MIC = 4 μM).697 Information on the viability of Burkholderia RNAP as the target of ubonodin is not currently available.
Lariatin A and B (Figure 59) were isolated from Rhodococcus sp. K01-B0171, and were initially observed to be growth-suppressive against M. smegmatis and M. tuberculosis.699 SAR studies of lariatin A have been performed, identifying residues necessary for bioactivity.700,701 Tyr6 could be replaced with other aromatic residues (Trp, Phe) without impacting the size of the zone of growth inhibition. Alanine substitutions at Gly11 and Asn14 resulted in the complete loss of activity. Lastly, Lys17 could be replaced with Arg, but not Ala, without any loss of activity, suggesting that a positively charged residue is critical for activity.701 The molecular target(s) of lariatin A/B have not been reported.
Humidimycin (Figure 59) is an antifungal lasso peptide that was identified by high-throughput screening for potentiators of the antifungal drug caspofungin against pathogenic Aspergillus fumigatus and Candida albicans.702 Humidimycin, when introduced with caspofungin, prevented phosphorylation/activation of SakA, the primary controller kinase of the high osmolarity glycerol pathway. Deletion mutants of SakA exhibited higher sensitivity to humidimycin, suggesting that this pathway is the target of the lasso peptide, though no details have been reported. These findings are of interest given the previously observed kinase inhibitory activity observed in the closely related lasso peptide siamycin I.651
8.2. Atroptides
Tryptorubin A (111), the only reported atroptide, was isolated from the fungus-derived bacterium Streptomyces sp. CLI2509 (Figure 65).703 Structural studies demonstrated that tryptorubin A contained a Trp-Trp crosslink and an unusual Tyr-Trp linkage. These two features give rise to a highly strained macrocycle. Initial bioinformatics predictions suggested that a hybrid type III PKS/NRPS encoded the hexapeptide, but subsequent analysis suggested that tryptorubin A is the product of a cytochrome P450 modification of a genetically encoded precursor peptide. P450 enzymes are primarily associated with hydroxylation reactions, but they are known to install C-C and C-N bonds,704,705 but experimental evidence of the biosynthetic mechanism has yet to be established. Structural elucidation of tryptorubin A revealed that it could exist as one of two non-canonical atropisomers that cannot be interconverted by rotation along a single bond;620 chemical synthesis was used to access both atropisomers. There are no reports associated with the bioactivity of tryptorubin A.
Figure 65.

Chemical structure of tryptorubin A (111). The macrocycle formed by Trp2, Tyr3, and Trp5 is shown in blue.
9. Graspetides
The graspetides are a class of RiPPs defined by the presence of one or more lactone/lactam rings formed between the ω-carboxy groups of Glu/Asp and the hydroxy groups of Ser/Thr (macrolactone) or the ε-amino group of Lys (macrolactam) (Figure 66).706 The formation of the ester or amide linkages are catalyzed by enzymes of the ATP grasp ligase superfamily, hence the classification of these products as graspetides.10 Initially, graspetides were largely identified in various genera of freshwater Cyanobacteria, including the bloom-forming Microcystis and Planktothrix. More recent genome mining efforts have identified graspetides in phyla outside of Cyanobacteria, including numerous terrestrial species.707–709 In general, the graspetides are known serine protease inhibitors (Table 2).
Figure 66.

Alignment of the microviridin core sequences. Residues that are involved in macrolactone (orange and purple background) and macrolactam (green background) formation are shown. The identity of residue at position 5 (blue background) largely dictates target protease specificity. A similar figure is reported in ref (34) but has errors at positions 12 and 13 in the last two entries.
Table 2. IC50 values of characterized graspetides.
IC50 (μM) values of characterized graspetides, unless otherwise indicated. N.D. indicates not determined.
| Graspetide | P1 Residue | Trypsin | Elastase | Chymotrypsin | Ref. |
|---|---|---|---|---|---|
| Microviridin A | F | >58 | >58 | >58 | 710 |
| Microviridin B | L | 34 | 0.03 | 1.5 | 711 |
| Microviridin C | L | 18 | 0.05 | 2.8 | 711 |
| Microviridin D | M | >55 | 0.4 | 0.7 | 712 |
| Microviridin E | Y | >60 | 0.4 | 0.7 | 712 |
| Microviridin F | Y | >59 | 3.5 | >59 | 712 |
| Microviridin G | L | >55 | 0.01 | 0.77 | 713 |
| Microviridin H | L | >55 | 0.017 | 1.57 | 713 |
| Microviridin I | L | N.D. | 0.19 | N.D. | 714 |
| Microviridin J | R | 0.02–0.09 | >6 | 1.7 | 715 |
| Microviridin K | M | >100 | N.D. | N.D. | 716 |
| Microviridin L | F | 58 | >58 | 42 | 717 |
| Microviridin M | L | >34 | 2.9 | >34 | 718 |
| Microviridin N3 | L | >30 | 2.5 | >30 | 718 |
| Microviridin SD1634 | R | 8.2 | N.D. | 15.7 | 719 |
| Microviridin 1777 | L | >10 | 0.16 | 0.10 | 720 |
| Plesiocina | L | >100 | 0.016 | 0.0075 | 721 |
| Marinostatinb | M | >50 | N.D. | N.D. | 722 |
Reported as Ki; Produced by heterologous expression in E. coli.
Reported as Ki; Performed using chemically synthesized compound. For IC50 values against a wider panel of proteases, see a recent review.34
The majority of characterized graspetides belong to the microviridin family. Group I microviridins (e.g., microviridins A, B, G, I, J, K, L) contain two macrolactones (Thr-Asp and Ser-Glu) and one macrolactam (Lys-Asp/Glu). Groups II and III microviridins (e.g., microviridins C, D, E, H and microviridin SD1652, respectively) retain the macrolactam linkage displayed by group I but only possess one macrolactone (group II: Thr-Asp; group III: Ser-Glu). Group IV microviridins also retain the macrolactam linkage of groups I-III but do not possess any other crosslinks (e.g. microviridin F, 122, Figure 69).34 The tricyclic compound microviridin A (112, Figure 67) was the first characterized graspetide, and was identified during a screen for human tyrosinase inhibitors (IC50 = 330 μM).710 Mammalian tyrosinase plays a key role in the development of melanin.723 Excessive melanin production (hyperpigmentation) has been linked to Parkinson’s disease and some skin cancers, and tyrosinase has been validated as a therapeutic target for treating hyperpigmentation.724 The discovery of microviridin A as an inhibitor of tyrosinase suggested the possibility of utilizing the tricyclic caged scaffold for therapeutic purposes. However, to date, there are no additional details on the MOA and no structural data of tyrosinase bound to microviridin A has been reported.
Figure 69.

Chemical structures of microviridin F (122), marinostatin (123), and plesiocin (124). The hairpin motifs in plesiocin are outlined in green.
Figure 67.

Structures of group I microviridins A (112), B (113), G (114), J (115), K (116), and L (117). Group I microviridins contain two macrolactones (shown in blue) and one macrolactam (shown in orange) which are installed by ATP grasp ligases. Residues involved in the microviridin L mutagenesis study are labeled.
Microviridins B (113, Figure 67) and C (118, Figure 68) were identified through screening efforts to identify inhibitors of the serine protease elastase.711 Elastase inhibitors are of therapeutic interest for the treatment of pulmonary emphysema and cystic fibrosis.725 Similar screening efforts identified microviridins D-H (Figure 67–69), which show inhibitory activity towards other serine proteases.712,713 The initial studies of microviridin G/H lead to the speculation that the identity of the amino acid at position 5 of the core peptide might dictate protease specificity. Lastly, the bicyclic marinostatin (121, Figure 69) was identified during a screen for subtilisin inhibitors with inhibitory activity against several serine, cysteine, and metalloproteases.726,727
Figure 68.

Structures of the group II microviridins C (118), D (119), E (120), and H (121). Group II microviridins contain two macrolactones (shown in blue) and one macrolactam (shown in orange) which are installed by ATP grasp ligases.
The polycyclic structures of the graspetides suggest that they may bind to the active sites of target enzymes as conformationally restrained structural mimics of substrates. Support for this hypothesis emerged from studies of Microcystis UWOCC MRC, which induced a lethal, molting defect when fed to the cyanobacterial predator Daphnia sp., a species of planktonic crustacean.728 The cyanobacterial metabolite that induced toxicity in daphnids was identified as microviridin J, an inhibitor of trypsin-like proteases (115, Figure 67).715 Subsequently, purified microviridin J was shown to induce the same molting defect in Daphnia as cells of Microcystis UWOCC MRC, demonstrating daphnid protease inhibition as a likely mechanism of toxicity.729
Atomic details of the MOA were established through structural elucidation of microviridin J bound to bovine trypsin.730 In the structure, Thr4, Arg5, and Lys6 in microviridin J adopted a binding conformation similar to that of bona fide trypsin substrates, and this conformation is rigidified by the macrolactone (Figure 70). It was proposed that the rigid, compact structure of the microviridin prevented cleavage of the target amide bond despite the binding of Arg5 of microviridin J in the S1 specificity pocket of the enzyme.
Figure 70.

Co-crystal structure of bovine trypsin (teal) bound to microviridin J (PDB ID:4KTU). Residues Thr4, Arg5, and Leu6 of microviridin J (purple carbon atoms) mimic the substrate of trypsin. The Ser195 nucleophile is proximal to the scissile bond (black arrow), but peptide cleavage is prevented by the rigid microviridin J structure.
Biochemical studies of the related microviridin L (117, Figure 67) supported the hypothesis that the identity of the residues at position 5 of the graspetide core peptide contributed to protease specificity. Naturally occurring microviridin L is a selective inhibitor of subtilisin (IC50 = 5.8 μM) and contains Phe5 at the P1 site.731 Random mutational analysis identified a microviridin L G2A variant that was produced at high levels with few unprocessed side-products, and this backbone was used for further SAR studies. Microviridin L variant G2A/F5R demonstrated a marked inhibition of trypsin (IC50 = 2.5 μM). Likewise, variant G2A/F5I inhibited elastase (IC50 = 5.2 μM).730 Activity of additional microviridin L variants is summarized in Table 3. The specificity of these variants could be further tuned to accommodate substrate preference for a particular protease target. For example, plasmin is a serine protease with a preference for basic residues at the P1 site but biases Lys over Arg at this position.732 The variant G2A/F5K demonstrated inhibitory activity against plasmin (IC50 = 2.0 μM), while the G2A/F5R variant was a far less effective plasmin inhibitor (IC50 = 47 μM).
Table 3. Protease inhibition profile of microviridin L variants.
The microviridin core sequence was altered at position 5 (the P1 site) and also contained a G2A substitution that facilitated high-level production of the tricyclic product.
| Inhibitory activity (IC50, μM) | ||||||
|---|---|---|---|---|---|---|
| Microviridin L variant | Elastase | Chymotrypsin | Trypsin | Subtilisin | Thrombin | Plasmin |
| G2A/F5L | 0.65 | 14 | >65 | 0.78 | >65 | >65 |
| G2A/F5I | 5.2 | >33 | >65 | >33 | >65 | >65 |
| G2A/F5V | 0.16 | 66 | >66 | >0.4 | >66 | >66 |
| G2A/F5K | >65 | >65 | 2.5 | 0.32 | >65 | 2 |
| G2A/F5R | 64 | >64 | 1.8 | 0.64 | >64 | 47 |
Microviridin K (116, Figure 67) contains Met5 at the specificity-determining P1 site and is an inhibitor of the serine protease subtilisin (IC50 = 5.2 μM).716 Notably, the M5R variant strongly inhibited trypsin (IC50 = 0.41 μM), consistent with the preference of this protease for a basic residue at the P1 site. The cage-like tricyclic structure was shown to be important but not critical for inhibitory activity as a bicyclic variant of microviridin K showed a modest decrease in inhibitory activity against subtilisin (IC50 = 8.3 μM), and the bicyclic M5R microviridin K variant showed a near five-fold loss of potency against trypsin (IC50 = 1.8 μM). These studies highlight the importance of both the specificity determining residue, as well as the tricyclic ring structure for protease inhibition.
Structural studies of the subtilisin inhibitor marinostatin (123, Figure 69) by solution NMR spectroscopy demonstrated a tricyclic structure like the microviridins. The structure also revealed a Thr3-Met4-Arg5-Tyr6 motif that is proposed to structurally mimic the P2-P2’ residues of a protease substrate (Figure 71) and bind to the active site of subtilisin as a non-cleavable inhibitor.733 Replacement of the native ester linkages with disulfide bonds generated an analog that mimicked the structure of a P2-P2’ substrate binding motif. The backbone amides of residues Thr3-Met4-Arg5 exhibited similar rates of hydrogen-deuterium exchange for marinostatin and the disulfide linked variant.733 Though the disulfide variant exhibits similar activity against subtilisin as the native compound (3.4 pM vs. 1.5 pM), it possesses differences in chemical shifts (|δ| > 0.2 ppm) from native marinostatin, suggestive of differences around the Met4-Arg5-Tyr6 reactive center.733 These findings suggest that conformational restraints arising from intramolecular linkages involving specific residues play a significant role in stabilizing a non-cleavable conformation of marinostatin and other graspetides.
Figure 71.

Four peptidic protease inhibitors. (A) Marinostatin, (B) microviridin J, (C) SSI, and (D) OMTKY3. All four inhibitors adopt similar structural conformations of residues that mimic the P2-P2’ sites in substrates.
The sequence of marinostatin that is believed to mimic the P2-P2’ residues of a protease substrate is similar to that found in members of the Streptomyces subtilisin inhibitor protein (SSI) family and to the Kazal protein domains which function as non-classical serine protease inhibitors (Figure 71).734 Based on structural alignment, the average dihedral angles of the residues in the sequence Thr3-Met4-Arg5-Tyr6 of marinostatin were close to those observed for the structure of SSI bound to subtilisin BPN,735 and to the Kazal protein turkey ovomucoid third domain (OMTKY3) bound to chymotrypsin.736 In the co-crystal structures with each of these proteinaceous inhibitors, the scissile peptide bond (P1-P1’) is intact but the carbonyl carbon of the P1 residue is distorted towards a tetrahedral geometry.737 The region encompassing the P2-P2’ residues in both SSI and OMTKY3 is flanked by a C-terminal β-sheet and an N-terminal disulfide bond, which stabilizes the protease substrate region (Figure 71).735,736 It has been suggested that the compounds may undergo proteolysis but are not released from the active site.738 Similarly, the macrolactone in marinostatin and the microviridins provide conformational restraints accounting for their activity as protease inhibitors (Figure 71). The importance of the crosslinks is reflected in experiments demonstrating that tricyclic microviridins generally show better inhibition than variants containing fewer rings.707,716,730 Additional experimental and computational studies are needed to provide a conclusive mechanistic rationale as to how these graspetides act as protease inhibitors.
Plesiocin (124, Figure 69) is the first characterized bicyclic graspetide that contains four hairpin loops, each of which are formed by residues participating in macrolactone linkages (Figure 69).721 Plesiocin inhibits the serine proteases elastase and chymotrypsin. This preference is rationalized as both proteases prefer hydrophobic residues at the P1 position, which corresponds to Leu in the Leu-Ala-Ile-Gly motif found in each hairpin (Figure 66). The number of hairpin repeats has been demonstrated to directly affect inhibitory activity. In vitro produced plesiocin containing four hairpin motifs possessed Ki values of 16 nM against elastase and 7.5 nM against chymotrypsin. In contrast, inhibition assays against an analog with only the first of four hairpin motifs displayed weaker inhibitory activity (Ki = 380 nM against elastase and 63 nM against chymotrypsin). The P1 Leu within each hairpin motif was critical for protease inhibition. Substitution of all four Leu residues to Ala increased the Ki against chymotrypsin to 3.3 μM.739 Variants that retained Leu in a single hairpin, whilst the others were replaced with Ala, also demonstrated lowered chymotrypsin inhibitory activity (Ki = 7.2–230 nM). This result demonstrates that each individual hairpin can act independently as a protease inhibitor. It remains unknown if plesiocin is cleaved into smaller fragments.
In vitro produced plesiocin demonstrated near-complete inhibition of chymotrypsin at a plesiocin:chymotrypsin ratio of ~0.16, where a ratio of ~1.0 would indicate a 1:1 stoichiometry.739 A 1:1 inhibitor-to-chymotrypsin ratio was demonstrated for a single bicyclic hairpin motif, consistent with super-stoichiometric inhibition by the four-hairpin plesiocin.739 Protease inhibition plasticity similar to that of the microviridins was also shown for plesiocin, which shows better inhibition against elastase/chymotrypsin over trypsin due to the hydrophobicity of the bicyclic motif. The introduction of a basic residue to the scissile site of the hairpin can bias inhibition towards trypsin over chymotrypsin.721,739
10. Circular bacteriocins
Circular bacteriocins contain a circular peptide backbone created by a macrolactam linkage between the N- and C-terminus.25 These compounds range from 58–70 amino acids and can be divided into two subclasses based on their physicochemical properties.21 The majority of circular bacteriocins fall under subclass I, which possess a high isoelectric point (pI > 9). Subclass II circular bacteriocins tend to feature lower isoelectric points (pI < 7).25 Examples of structurally characterized circular bacteriocins include enterocin AS-48 (hereafter AS-48), carnocyclin A, and acidocin B.740–742 Bacterial growth inhibition studies have been carried out with other circular bacteriocins such as enterocin NKR-5–3B, lactocyclicin Q, and leucocyclicin Q.743–745
The growth-suppression spectrum of known circular bacteriocins is primarily directed against Firmicutes (Table 4). Notable genera inhibited include Lactobacillus, Lactococcus, and Enterococcus. The range of activity also encompasses food spoilage genera within Firmicutes, such as Clostridium, Staphylococcus, Bacillus, and Listeria.746 These compounds have no or weak bioactivity against Proteobacteria, including Klebsiella, Acinetobacter, Pseudomonas, or Enterobacter. High concentrations of AS-48 (>6.6 μM), lactocyclicin Q (~17–34 μM), or leucocyclicin Q (34–38 μM) were required to observe growth inhibition of E. coli.743,744,747
Table 4. Bacterial growth-suppression activity of circular bacteriocins.
+ indicates that a zone of inhibition was observed (activity), − indicates that no activity was observed, w (“weak”) indicates that a zone of inhibition was observed only at higher concentrations (91 μM); N.D., not determined.
| Bacteriocin | Class | Lactobacillus | Lactococcus | Enterococcus | Staphylococcus | Bacillus | Listeria | Ref |
|---|---|---|---|---|---|---|---|---|
| AS-48 | I | N.D. | N.D. | + | + | + | N.D. | 747 |
| Garvicin ML | I | + | + | + | N.D. | N.D. | + | 759 |
| Carnocyclin A | I | + | + | + | + | N.D. | + | 761 |
| Circularin A | I | + | N.D. | + | N.D. | N.D. | N.D. | 762 |
| Uberolysin A | I | N.D. | + | + | + | N.D. | N.D. | 763 |
| Lactocyclicin Q | I | + | + | + | w | + | + | 743 |
| Amylocyclicin | I | N.D. | N.D. | N.D. | N.D. | + | 764 | |
| Enterocin NKR-5–3B | I | + | + | + | N.D. | + | + | 745 |
| Pumilarin | I | N.D. | + | − | + | N.D. | 765 | |
| Gassericin A | II | N.D. | N.D. | N.D. | + | + | + | 766 |
| Acidocin B | II | + | N.D. | N.D. | − | − | − | 767 |
| Butyrivibriocin AR10 | II | + | N.D. | N.D. | N.D. | + | N.D. | 768 |
| Plantaricyclin A | II | + | + | − | − | − | − | 769 |
Early work on AS-48 demonstrated that the toxin forms pores of ~7 Å in diameter in artificial lipid bilayers, thus allowing cations, anions, and small molecules to pass through in a voltage-independent manner.747 Carnocyclin A demonstrated the same ability to form pores in lipid bilayers, but these channels were only permeable to small anions in a voltage-dependent fashion.741,748 Differences in ion selectivity amongst these peptides suggest that the circular bacteriocins may not share a universal MOA.
AS-48 was the first circular bacteriocin to be structurally characterized,749 revealing the same fold as NK-lysin,750,751 a non-RiPP cationic antibacterial and tumor lysing peptide.751,752 Both peptides adopt a helical bundle conformation, and thus were predicted to exhibit the same MOA. As NK-lysin and AS-48 both lack membrane-spanning helices, pore formation was hypothesized to result from a phenomenon known as molecular electroporation. When a peptide with sufficient charge density binds to a membrane, the peptide local electric field can induce destabilization of the membrane, resulting in pore formation. This mechanism does not necessarily imply that the peptide inserts into the membrane.
Further characterization of AS-48 provided further insight into its MOA. Crystallographic studies of AS-48 bound to decyl-β-D-maltoside highlighted the amphipathic nature of the molecule.18 These structures reveal that AS-48 exists as one of two possible dimers, one associated through its hydrophobic face, or one associated by its hydrophilic face (Figure 72). The aliphatic tail of the detergent is bound to the hydrophobic core of AS-48, suggesting that AS-48 would similarly associate with membrane lipids. AS-48 has an amino acid distribution and surface properties often found in membrane inserted proteins.740 The highly cationic α4 helix (AS-8449–69) alone does not possess bactericidal activity, indicating that its charge density alone is not sufficient to induce pore formation.754 However, the bactericidal effect of AS-48 against Listeria monocytogenes CECT 4032 decreased when cultures were co-treated with AS-8449–69 indicating that the cationic helix competes for binding at the membrane. These findings suggest the antibacterial action of AS-48 requires all its aliphatic helices to be present. Finally, coarse-grained simulations reveal that AS-48 fully inserts into the membrane. In 9 out of 15 simulations, a toroidal pore was observed (Figure 73).755 In this model, the membrane adopts a highly curved arrangement (due to the association of helices a4–5 with hydrophilic phospholipid head groups) that leads to membrane destabilization, and eventual cell death.755,756
Figure 72.

(A) Crystal structure of dimeric AS-48 obtained in Tris-HCl pH 7.5 (left, PDB ID: 1O83). Hydrophobic helices are shown in orange, while hydrophilic helices are shown in blue. Crystal structure of dimeric AS-48 in the presence of the detergent decyl-β-D-maltoside (right, PDB ID: 1084). (B) Hydrophobic surface models of AS-48 in aqueous solution (left) and in the presence of detergent (right). Hydrophobic residues are shown in orange; hydrophilic residues are shown in blue. (C) Structure of carnocyclin A (PDB ID: 2KJF). Hydrophobic helices are shown in silver, while hydrophilic helices are shown in green. (D) NMR solution structure of acidocin B obtained in deuterated sodium dodecyl sulfate and D2O (PDB ID: 2MWR). Amphipathic helices are shown in salmon, while the hydrophobic helix is shown in cyan.
Figure 73.

Cartoon representation of the toroidal pore arrangement of AS-48 (shown as blue cylinders) in the lipid bilayer. Three copies of AS-48 are hypothetically required for pore formation.755
The NMR structure of the anion-selective peptide carnocyclin A shows an electropositive helix, which may direct the peptide to the membrane (Figure 72).13 However, currently available data do not explain why carnocyclin A is anion selective and why it exerts voltage-dependent activity. The NMR structure of acidocin B,742 a class II circular bacteriocin, has also been elucidated (Figure 72). Unlike AS-48 and carnocyclin A, acidocin B possesses one hydrophobic helix and three weakly amphipathic helices. The largely hydrophobic helix of acidocin B may facilitate membrane interaction. MOA studies have yet to be reported on the anionic class II bacteriocins. One testable hypothesis is that the MOA may involve lipid extraction and membrane solubilization, similar to the negatively charged saposin-like proteins.757,758
Though pore formation and membrane disruption is thought to be a primary MOA for the circular bacteriocins, garvicin ML (GarML)759 exerts its bactericidal activity by targeting the maltose ABC transporter.760 Resistant mutants to GarML possessed a large chromosomal level deletion that included the genes for the maltose ABC transporter (malEFG).760 A direct correlation between increased expression levels of the maltose ABC transporter and the increased sensitivity of cells to GarML was shown.760 The transporter is proposed to become constitutively open upon GarML binding, leading to the efflux of intracellular solutes, followed by cell death.760 It is worth noting that other RiPPs also target sugar transport systems, including MccE492/MccE492m (section 4.2) the glycocins (section 13).
11. Amidinotides
Amidinotides are characterized by the presence of a phenyl-2-guanidinoacetic acid-containing molecule linked to a ribosomally synthesized peptide. The pheganomycins (PGMs, 125–129) were first isolated in the late 1980s from Streptomyces cirratus (Figure 74).770 The amidinotides are hybrid NRPS/PKS-RiPP products owing to the presence of the nonproteinogenic (S)-2-amino-2-(3,5-dihydroxy-phenyl)acetic acid moiety.771,772 An ATP-grasp ligase links a precursor peptide with this non-proteinogenic amino acid to yield the final product. Although the related compounds resorcinomycins A and B773 also possess the phenyl-2-guanidinoacetic acid moiety, they are not true RiPPs, as they contain a single Gly residue instead of a ribosomally synthesized peptide. Known amidinotides display a narrow spectrum of activity, primarily inhibiting the growth of Mycobacterium sp.
Figure 74.

Chemical structures of the pheganomycins (125–128) and deoxypheganomycin D (129). A substituent at position 4 of the aromatic ring appears important for bioactivity of these compounds (blue). The terminal aspartate (orange) of deoxypheganomycin D may contribute to its bioactivity. The proteinogenic portion of both compounds (boxed) is labeled with the original residues above the structures prior to post-translational modification.
Early mechanistic studies performed on a chemically derivatized pheganomycin with increased stability termed deoxypheganomycin D (129, Figure 74) suggested that the molecule acts on the mycobacterial cell wall. Mycobacterium smegmatis ATCC 607 treated with 0.27 μM of deoxypheganomycin D retained only 13% of 14C-glycerol label in its purified cell wall extracts as compared to an untreated sample.770 Deoxypheganomocyin D was proposed to affect cell wall synthesis and lead to increased permeability. Notably, treatment of mycobacteria with the compound increased both influx and efflux of radiolabeled amino acids and nucleotides, consistent with increased permeability. Deoxypheganomycin D does not inhibit protein or DNA/RNA synthesis. No additional MOA studies have been reported on any amidinotide.
SAR studies revealed that the peptide portion of the PGMs alone did not possess any inhibitory effects.774 Additionally, substitution of the 4-hydroxymethyl group with a hydrogen in PGM abolished bioactivity. However, deoxypheganomycin D lacking the 4-hydroxy group but containing a C-terminal Asp inhibited the growth of M. smegmatis.770 Additional MOA studies on amidinotides are warranted.
12. Crocagins
Crocagins A and B (130, 131, Figure 75) from the soil-dwelling Myxococcales Chondromyces crocatus Cm c5 were first identified in a screen for compounds that disrupt the interaction between CsrA and its small noncoding inhibitory RNA (see below).775 They possess a highly substituted tetrahydropyrimidone moiety that is derived from the tripeptide Ile-Tyr-Trp (by the formation of two C-N bonds) and a carbamate moiety.776 The compounds are structurally alike but crocagin B (131) contains a 2,3-dehydro-Tyr residue (Figure 75).
Figure 75.

Chemical structure of crocagin A (130) and crocagin B (131). Crocagin B is dehydrogenated between Cα and Cβ of Tyr (C=C bond shown in blue).
Pathogens can alter their physiology and virulence properties in response to changing environmental conditions. Regulation of gene expression is predominantly controlled at transcription initiation.777,778 However, certain bacteria can regulate gene expression at the post-transcriptional level by controlling translation initiation efficiency, transcript elongation efficiency, and/or rates of RNA decay.779 Bacterial post-transcriptional regulation is mediated by small RNAs and RNA-binding proteins (RBPs) that engage the nascent RNA transcript. Upon binding to transcripts, these effectors can induce changes to the secondary structure of RNA. Examples include facilitating or preventing access to the Shine-Dalgarno sequence,780,781 Rho-utilization sites,782 and preventing mRNA degradation.783 CsrA/RsmA (carbon storage regulator A and repressor of secondary metabolites A, respectively) are RBPs that can either activate or inhibit translation from a transcript.780,784–786 CsrA/RsmA systems have been extensively reviewed.787 Crocagin A inhibits the interaction of CsrA from Y. pseudotuberculosis YPIII with its inhibitory RNA ligand at a modest IC50 of 35.8 μM, and was shown to directly bind to CsrA to elicit these effects (Kd = 12.9 μM). Crocagin A was also screened for antibacterial and cytotoxic effects, but no such activities were observed.776 Bioactivity for crocagin B has not been reported.
13. Glycocins
Currently characterized glycosylated bacteriocins (glycocins) contain at least one mono-or disaccharide moiety installed on Cys or Ser of peptides containing two disulfide bonds.9,23,788 Whereas all known glycocins are glycoactive (i.e. require the sugar for bioactivity), bioinformatic studies suggest currently uncharacterized members will not always contain disulfides and thus the glycosylation serves as the class-defining post-translational modification.789 The glycocin class has been divided into four subclasses based on sequence homology and named for the original glycocin identified from the respective subclass: sublancin (132), glycocin F (133), enterocin 96, and enterocin F4–9 (134) (Figure 76).23 The exact structure of enterocin 96 has not been established. Several glycocins possess growth-suppressive activity against Firmicutes and in rare cases Proteobacteria. The mechanism(s) through which glycocins impart this bioactivity remains unknown, but several studies have made progress towards understanding the MOA.23 Despite having similar three-dimensional structures, characterized glycocins have either bacteriostatic or bactericidal activities and display considerable SAR differences. Taken together, these observations suggest the potential for multiple MOAs within the glycocin class.790
Figure 76.

Glycocin subclasses and NMR structures. (A) Chemical structures of sublancin (132), glycocin F (133), and enterocin F4–9 (134). (B) NMR three-dimensional structures of sublancin (PDB ID: 2MIJ) and (C) glycocin F (PDB ID: 2KUY).
Sublancin 168 (132, Figure 76) is the archetypical glycocin with growth-suppressive activity against multi-drug resistant human pathogens including MRSA. The compound is produced by B. subtilis 168, and while initially reported to be a lantibiotic,791 a structural revision corrected the earlier report.792 The first MOA insights came from studies showing that mutations in two different regions of the B. subtilis genome confer resistance to sublancin. The first region encodes a large mechanosensitive channel, MscL, and the second is an operon (ptsGHI) encoding the phosphoenolpyruvate:glucose phosphotransferase system (PTS) involved in sugar uptake.793,794 To gain further insight, variants of sublancin were prepared via heterologous expression and utilized for SAR studies.795,796 These investigations revealed that in addition to the glycosylation post-translational modification, Asn31 and Arg33 are important for activity. A positively charged residue at position 33 is likely important for activity, as replacement of Arg33 with Lys resulted in similar activity as native sublancin. Although Arg33 is important for sublancin activity, charged residues are not conserved at this position across all glycocins suggesting niche-specialization.790 Indeed, several glycocins have a narrow spectrum of antibacterial activity, and are only active against organisms within the same genus as the producing organism.790
In addition to the amino acid sequence, the sugar moiety is essential for antibacterial activity.795,796 The glucose on sublancin has been substituted with several other sugars, including galactose, N-acetylglucosamine, mannose, and xylose with minimal influence on the MICs.796,797 Notably, whereas mannose and N-acetylglucosamine are imported by the PTS, galactose and xylose are not. When testing bioactivity under minimal media conditions, addition of free glucose suppressed the bioactivity of sublancin. Other tested sugars had no effect on the activity of wt sublancin nor analogs carrying different sugars. Therefore, it appears that glucose competes with sublancin and its analogs for the glucose PTS, and that changing the sugar on sublancin does not direct the compound to different sugar import systems. Consistent with this conclusion, resistance mutations in ptsGHI imparted resistance against sublancin analogs carrying different sugars.796 The glucose PTS phosphorylates the C6 hydroxy group of glucose during import raising the question whether the glucose on sublancin must be phosphorylated for activity. However, the high antibacterial activity of a sublancin analog carrying xylose argues against phosphorylation at the C6 hydroxy group because xylose does not possess such a group (Figure 77).797
Figure 77.

(A) Schematic representation of the glucose PTS used to import glucose into B. subtilis. A relay mechanism transfers a phosphate from phosphoenolpyruvate (PEP) via several protein carriers to the hydroxyl at C6 of the incoming glucose. A separate phosphorylation mechanism is involved in catabolite repression. (B) Replacement of the glucose on sublancin with xylose (which lacks C6) did not compromise antimicrobial activity.797 R = sublancin aglycon.
To gain further insight into the sublancin MOA, macromolecular synthesis assays were performed. Radiolabeled precursors were utilized to measure incorporation into the cell wall, DNA, RNA, and proteins.796 Sublancin affected DNA, RNA, and protein biosynthesis, suggestive of a process upstream of macromolecule biosynthesis that is interrupted by sublancin. This observation is consistent with the hypothesis that the PTS is involved in the bioactivity of sublancin through a yet unknown MOA. However, several findings argue against a simple blockage of glucose import. First, sublancin is bactericidal, not bacteriostatic. Second, sublancin is bioactive even when bacteria are grown on a non-PTS carbon source. Finally, mutations that inactivate the glucose PTS provide resistance, but overexpression of ptsG, ptsH, or ptsI renders cells hypersensitive.797 Collectively, these observations suggest sublancin acts through a mechanism that requires an active glucose PTS, and that possibly induces a lethal gain of function in the glucose PTS. For other natural products that target the sugar PTS (e.g. garvicin ML, section 10 and MccE492, section 4.2), such gain of function has been shown to involve pore formation or constitutive channel opening, but such a mechanism was not observed for sublancin.797
Another study focused on the effect of sublancin on the mouse host immune system.798 Sublancin induced macrophage migration, upregulates production of select anti-inflammatory chemokines, downregulated pro-inflammatory cytokines, and modulated the gut microbiota populations compared to control samples.798 All of these effects could be mechanisms by which sublancin may be effective in treatment of bacterial infections in a host.
Glycocin F (133, Figure 76) is produced by Lactobacillus plantarum and exhibits bacteriostatic activity. A model for glycocin F-receptor interactions was proposed after SAR studies with analogs prepared by solid-phase synthesis (Figure 78).799 These investigations identified the disulfide bond nearest the interhelical loop, the size of the interhelical loop, and the O-linked N-acetyl-D-glucosamine (GlcNAc) as essential for activity.799,800 The S-linked GlcNAc and C-terminal tail contribute to the activity, but were not essential. Similar to sublancin, even when the identity of the C-terminal sugar was changed, bacteriostasis was abolished with the addition of free GlcNAc, but not other sugars, suggesting that changing the identity of the C-terminal sugar did not alter the PTS system being targeted.788,799 Similar SAR studies were also performed for ASM1, a glycocin F-type family member with ~91% sequence identity to glycocin F, yielding the same conclusions on the importance of these structural features for bioactivity.801 The results led to a model in which the C-terminal tail is utilized to localize glycocin F to the correct cell surface receptor. This initial interaction is proposed to facilitate binding of the O-linked GlcNAc and the interhelical loop to a currently unknown primary target to initiate bacteriostasis.799
Figure 78.

Proposed model for glycocin F-receptor interactions. The O-linked GlcNAc in the loop between the helices is black while the C-terminal S-linked GlcNAc is white. (A) Both sugars bind the GlcNAc-specific PTS membrane spanning EIIC domain (purple) followed by (B) the loop GlcNAc binding its primary target (blue) that has yet to be identified.799 Disulfides are shown in orange.
Enterocin 96 and enterocin F4–9 (134, Figure 76) produced by E. faecalis also exert bacteriostatic activity. SAR studies on enterocin 96 indicated that unlike other glycocins, the disulfides were dispensable. Furthermore, the di-glucosylated form was more active than the mono-glucosylated form, and replacement with galactose at the terminal position eliminated activity.802 For enterocin F4–9, the sugar moieties, proposed to be GlcNAc, and disulfides were essential for activity.803 These studies suggest that the details of the MOAs of different glycocins may differ in subtle but important ways.
A glycocin produced by a thermophilic bacterium, Aeribacillus pallidus (also referred to as Geobacillus sp. 8), was independently identified by two research groups and termed pallidocin and geocillicin, herein referred to as pallidocin (135, Figure 79).790,804 Similar to all other glycocins, pallidocin was glycoactive, and bioactivity could be reversed by the addition of glucose. However, no reduction in bioactivity upon glucose addition was observed for bacillicin BAG2O and bacillicin CER074 even though these glycocins are glucosylated.790 This finding further suggests that glycocins display unique MOAs.
Figure 79.

Schematic structure of pallidocin (135).
14. Sulfatides
Xanthomonas oryzae pv. Oryzae (Xoo) is a plant pathogen that causes bacterial blight disease in rice.805,806 Early efforts to understand the pathogenicity of Xoo were focused on identifying strains of rice that were resistant to bacterial blight.807,808 One such strain, IRBB21 possesses a gene encoding a histidine kinase receptor, XA21, which confers pathogen immunity to transgenic rice.808,809 Subsequently, the genes required for Xoo virulence have been identified, and were termed raxXSTAB (required for activation of XA21).810,811 RaxX, the product of the rax gene cluster, is a RiPP featuring a sulfated tyrosine (136, Figure 80). RaxX activates the XA21-mediated immune response, which suppresses infection in rice.812
Figure 80.

Chemical structure of RaxX21 (136). Post-translational modiications are colored in blue. The C-terminus of the peptide contains residues that are important for recognition by XA21 (orange). Amino acid sequences of Arabidopsis PSY1, RaxX21 and Rax13 are shown (top), highlighting the position of the tyrosine residue in each (green). Additional chemical modifications are found on Arabidopsis PSY1 (Pro-4-hydroxylation and O-glycosylation).
Knowledge of the XA21-mediated immune response is incomplete but is briefly summarized below. XA21 is a cell-surface receptor that provides the first line of defense in plants against bacterial pathogens.813 XA21 consists of an extracellular ligand binding domain, a transmembrane domain, a juxtamembrane domain, and an intracellular Ser/Thr kinase domain.808,814,815 RaxX activates the XA21-mediated immune response by inducing autophosphorylation of XA21 and/or phosphorylation of downstream proteins, many of which are unidentified.816,817 Although the order of the signaling cascade is not known, several XA21-binding (XB) proteins and regulators have been identified.809,818–822 For example, both XB3 and XB25 have been shown to be required for XA21-mediated immunity.818–822 The former protein is hypothesized to induce the MAPK cascade, which leads to transcriptional activation of defense-related genes.823 Several studies have investigated the details of the XA21-mediated immune response and interactome.823–825
SAR studies of RaxX have identified residues required for XA21 activation. The RaxX precursor peptide contains 60 amino acids and undergoes sulfation on Tyr41. A putative leader peptide cleavage site has been identified, but has not been validated in vitro.826 The unmodified precursor peptide is not bioactive. Though the sulfated 60-mer precursor peptide activates XA21 at an EC50 of 100 nM, a shorter construct termed RaxX21 (precursor peptide residues 35–55) is a more potent activator (EC50 = 20 nM). An even shorter construct, encompassing precursor residues 32–49, activates XA21 at an EC50 of > 10 μM, suggesting that key binding residues may reside near the C-terminus of the peptide. The binding dissociation constant (Kd) of RaxX21 to the extracellular domain of XA21 is 16 nM, whereas non-sulfated RaxX21 binds with a Kd of 205 nM.826 The specific interactions between the receptor and the peptide have yet to be investigated.
RaxX possesses additional bioactivity. RaxX21 resembles a sulfotyrosine-containing plant peptide hormone, PSY1, which induces growth in Arabidopsis (Figure 80).827,828 Treatment of an Arabidopsis mutant that lacks the ability to produce sulfated and bioactive PSY1 with low nanomolar concentrations of either RaxX21 or PSY1 increased root growth by at most two-fold. The minimal peptide required to promote root growth encompasses precursor peptide residues 40–52 (termed RaxX13). Unlike RaxX21, PSY1 does not induce an immune response in XA21 expressing rice, indicating that RaxX21 is recognized by both hormone and XA21 receptors, whereas PSY1 only targets the former.828 The growth promoting property of RaxX is rationalized by the fact that Xoo is a biotrophic pathogen that requires a living host to obtain nutrients for its survival. Thus, by mimicking of the growth stimulating activity of PSY1, RaxX reprograms the host environment to benefit the pathogen. It is likely that RaxX evolved specifically to mimic PSY1.828 Consequently, rice may have evolved the XA21 receptor, which specifically invokes the immune response in the presence of RaxX, but not in the presence of PSY1 and related peptides.828
15. RiPPs with class-defining features installed by radical SAM enzymes
The radical SAM (rSAM) enzymes form a large and diverse superfamily with more than a million predicted members.829,830 These enzymes employ a [4Fe-4S] center to reductively cleave S-adenosylmethionine, generating L-Met as a byproduct and a 5’-deoxyadenosyl (5’-dA) radical. In most known examples, the 5’-dA radical abstracts a hydrogen atom from the substrate, which then undergoes further chemistry to yield many different outcomes for individual members of the superfamily.831
15.1. Polytheonamides
Originally discovered in the early 1990s, the polytheonamides were not immediately recognized as RiPPs owing to their extensive modifications, which include N-acylation, β-hydroxylation, N-methylation, C-methylation, and a large number of epimerizations.832–834 Polytheonamides are potent cytotoxins against cultured murine cells (IC50 = ~13 pM). Biochemical and analytical chemical studies have largely focused on polytheonamide B (137, Figure 80), which is structurally homologous to the other polytheonamides, and will be the focus for the remainder of this discussion.
Polytheonamide B was proposed to act as a cationic membrane channel.835 Detailed solution NMR spectroscopy revealed that polytheonamide B adopted a b-helical structure 45 Å in length with a central pore diameter of ~4 Å.836 Upon insertion into membranes, polytheonamide B affords selective permeation of monovalent cations. The β-helical structure is stabilized by hydrogen bonding interactions between five side chain N-methylated D-Asn and one D-Asn, which all reside on the same face of the helix (Figure 81). The N-terminal half of polytheonamide B is largely nonpolar while the C-terminus is relatively polar leading to the hypothesis that insertion of polytheonamide B into the membrane is facilitated by its N-terminal portion. As the structure was acquired in a solvent (1:1 methanol/chloroform) that mimics the dielectric constant in a cell membrane, the reported conformation is believed to reflect the native conformation of polytheonamide B in its active form.
Figure 81.

Structure of polytheonamide B (137, pTB). The N-terminal 5,5-dimethyl-2-oxohexanoate is indicated in pink. L-amino acids are shown in blue, while D-amino acids are shown in orange. SAR studies utilized derivatives of pTB (137a-i) that were modified at the N-terminal 5, 5-dimethyl-2-oxohexanoate group (R1), residue 44 (R2), β-substituents of residues 2, 22, 29, and 37 (X, and Y), and residue 47 (Z).
In synthetic planar membranes, polytheonamide B treatment led to permeabilization of K+ and Cs+ ions in a periodic manner. Opening and closing events were monitored by voltammetry on the side of the membrane that contained polytheonamide B.837 The probability of an opening event was lower at −200 mV than at +200 mV, indicating a voltage-dependent gating mechanism. These findings suggest an asymmetric polytheonamide B structure orients in the membrane, as ion permeability was unidirectional i.e., permeabilization of K+ and Cs+ ions only occurred in one direction across the membrane. Transmembrane current amplitude was directly proportional to polytheonamide B concentration, indicating that this compound forms a unimolecular ion channel instead of a channel composed of multiple polytheonamide B subunits. The current recordings demonstrated ion selectivity for monovalent cations and impermeability to divalent cations, and that polytheonamide B is not released from the membrane once it is incorporated.837
The selective orientation of polytheonamide B is attributed to its asymmetric polarity, as the N-terminus is more hydrophobic than the C-terminus (Figure 82). Addition of polytheonamide B to eukaryotic membrane mimetic liposomes (10:1 phosphatidylcholine:cholesterol) encapsulating carboxyfluorescein as a pH indicator (internal pH = 6.5; bulk solution pH = 5.5) dissipated the pH gradient leading to liposomal fluorescence.838 However, polytheonamide B did not disrupt the membrane sufficiently to allow transmembrane passage of carboxyfluorescein, which with a molecular radius of ~5 Å in solution, is larger than the polytheonamide B pore diameter of ~4 Å.
Figure 82.

(A) Solution NMR structure of polytheonamide B (PDB ID: 2RQO). The hydrogen-bonding network (dashed lines) occurring between D-Asn residues is presumed to stabilize a pore-forming conformation. (B) Polytheonamide B exhibits a relatively polar C-terminus (left) and a more nonpolar N-terminus (right). Hydrophobic residues are colored from orange (hydrophobic) to blue (hydrophilic).
Synthetic studies supported this mechanism and localized the membrane disruption activity to the C-terminus and membrane insertion/orientation to the hydrophobic N-terminus of the peptide.838–841 Smaller fragments exhibited lower H+/Na+ exchange across liposomal membranes than full length polytheonamide B with exchange increasing as fragments were added from the C-towards the N-terminus (Figure 83).838 In planar membrane channel current assays, the B+C+D fragment demonstrated a similar open/close gating mechanism to native polytheonamide B, though with a lower voltage dependency.
Figure 83.

Bioactivity of polytheonamide B fragments. The C-terminal fragment D exhibited the highest membrane perturbation measured by carboxyfluorescein (CF) release from egg yolk phosphatidylcholine and cholesterol (10:1) liposomes but lacked ion transport ability measured by H+/Na+ exchange. Conversely, N-terminal fragments lacked membrane perturbation activity. Additions of N-terminal fragments to fragment D yielded a compound that lost membrane disruption activity but gained ion transport activity. Fragment IC50 values were determined in vitro against murine p388 leukemia cells. Ion exchange and CF release values were estimated from the maximal percentage of each variable released over the time course of the experiment. Percentage exchange monitors loss of the starting material from inside the liposome, i.e. 5% exchange means 5% left the liposome interior. Figure adapted from reference.838
These data support a mechanism where the C-terminus functions as a membrane disruption domain, while the N-terminus forms the ion channel. These N-terminal fragments add sufficient length to span the entire lipid bilayer.838 Conversion of the N-terminal acylamide group (5,5-dimethyl-2-oxohexanamide) to more polar groups like amino, acetylamide, or trimethyl ammonium increased IC50 values by 240-, 480-, or 2,500-fold respectively (137a, 137b, 137c, Figure 81 inset).839 Conversely, introducing a hydrophobic octanamide (137d) functionality only decreased toxicity by 5-fold while a palmitamide (137e) decreased IC50 by a factor of three. Current measurements in planar bilayers demonstrated ion channel activity for all variants except the trimethyl ammonium analog, which was proposed to engage in a channel-blocking mechanism caused by the quaternary cation.
Derivatization studies have also been performed to elucidate SAR of polytheonamide B. These studies were performed using a synthetically accessible analog of polytheonamide B, which retained all intramolecular hydrogen bonding interactions and chiral centers, but the β-substituents of residues 2, 22, 29, and 37 were removed (Figure 81), and residues 44 and 47 were exchanged for propargyl glycine, and D-Thr respectively. A dansyl group was installed at position 44 via Huisgen cycloaddition to serve as a fluoregenic marker. These six modifications resulted in an analog that possessed an IC50 of 12 nM against p388 mouse leukemia cells, approximately 100-fold less potent than polytheonamide B (Figure 81, analog 137f).842 Despite the bulky fluorophore substituent, the dansylated analog could insert into liposomal membranes, which permitted its use in further SAR studies. Improper insertion in the membrane, e.g. perpendicular to the membrane, would have decreased bioactivity of 137f. In vitro analyses of liposomal H+/Na+ exchange also demonstrated this mimic can efficiently permeabilize phospholipid bilayers to small cations with a gating mechanism like that of polytheonamide B.
Additional SAR studies were performed using dansylated polytheonamide B (Figure 80, 137f). Truncation studies demonstrated that removal of 12 N-terminal residues (Δ1–12) resulted in an IC50 of 3.7 nM against murine p388 mouse leukemia cells, compared to 12 nM for the full length dansylated polytheonamide B.843 The Δ1–11 and Δ1–13 constructs possessed IC50 values of >420 and 81 nM, respectively. Ion transport assays performed using liposomes demonstrated that Δ1–11 is unable to permeabilize membranes to H+/Na+. Utilizing pH-indicator dyes, Δ1–11 was also unable to induce a pH shift in murine P388 leukemia cells, further supporting an inability to induce ion transport across membranes. This finding was consistent with truncation studies on native polytheonamide B, where N-terminal truncations reduced membrane disruption and cation transport ability (Figure 83).838 Despite an inability to efficiently transport cations, the Δ1–11 dansylated polytheonamide B analog suggests a novel MOA.
Modification of the N-terminus of full length dansylated polytheonamide B further increased potency. Introduction of a palmitamide on the N-terminus improved the IC50 from 12 to 0.84 nM (Figure 81, 137g).841 This change was attributed to the increased logP of this analog, which was determined to be 4.9, compared to 4.5 for polytheonamide B. Introduction of a more polar N-terminal functional group like tetramethylammonium (Figure 81, 137h) or a free amine (Figure 81, 137i) increased the IC50 to 83 and 25 nM, respectively, likely due to the decreased logP of these compounds (2.6 for the trimethylammonium, and 3.2 for the free amine derivatives). These studies highlight that the relative hydrophobicity of the N-terminus is also critical to the bioactivity of polytheonamide B.
The published NMR structure has served as a starting point to perform molecular dynamics simulations. The network of hydrogen bonds between Asn and methyl-Asn side chains served to stabilize the channel structure, allowing polytheonamide B to be “trapped” in the membrane and to retain channel activities over long residence times, contributing to its potency.836,837,844 Cation transport through the channel is proposed to occur through coordination by backbone carbonyls and is additionally facilitated by the movement of water molecules through the channel.845,846 Water molecules residing within the polytheonamide B channel at resting state function as a water wire, which transport H+ during membrane depolarization.847 This ion transport mechanism was demonstrated to be reversible depending on the voltage potential, as proton transport is at equilibrium at ~0 mV, confirming the ability of polytheonamide B to depolarize cell membranes regardless of potential direction.
In addition to membrane depolarization, cell localization studies demonstrated the ability of polytheonamide B to neutralize lysosomes through a micropinocytosis pathway.848,849 Chemically-synthesized polytheonamide B was functionalized with an N-terminal BODIPY group, facilitating studies in live cells. The dye bis-(1,3-dibutylbarbituric acid) trimethine oxonol [DiBAC4(3)] was used to study changes in membrane potential. Treatment of DiBAC4(3)-exposed cells with 5 nM polytheonamide B led to complete membrane depolarization in MCF-7 cells within 1 h.848 Imaging studies with BODIPY-functionalized polytheonamide B demonstrated that this compound did not localize to the plasma membrane but instead to smaller organelles.848
Colocalization studies with LysoTracker (which accumulates in acidic lysosomes) demonstrated BODIPY-polytheonamide B was located within lysosomal membranes.848 The lysosomal pH-indicator fluorescein-tetramethylrhodamine-tagged dextran (FRD) demonstrated a pH increase from 4 to 7 in lysosomes after treatment with polytheonamide B, presumably as a consequence of proton transport. Internalization of polytheonamide B was shown to be reduced by 5-(N-ethyl-N-isopropyl)-amiloride, which is an inhibitor of micropinocytosis, suggesting polytheonamide B is actively internalized by a native cellular pathway. Flow cytometry studies with polytheonamide B demonstrated induction of the apoptotic pathway due to membrane depolarization and dissipation of the lysosomal pH gradient. This secondary effect was observed at concentrations near the measured IC50 values, supporting the hypothesis that this secondary MOA is potentially physiologically relevant.
15.2. Darobactin
Darobactin (138) is a first-in-class, heptapeptide RiPP that features a C-O linkage between the C7 indole of Trp1 and the β-carbon of Trp3, and a C-C bond between the C6 indole of Trp3 and the β-carbon of Lys5 (Figure 84). The C-O and C-C bonds are remarkably installed by the same rSAM enzyme.850 Darobactin is selectively active against several clinically relevant Proteobacteria such as P. aeruginosa and K. pneumoniae (MICs = 2–16.5 μM). Conversely, the compound was found to possess little activity against Firmicutes. The first clues regarding the darobactin MOA were uncovered when resistant mutants from E. coli MG1655 were selected and found to map to the bamA gene. Given that Firmicutes lack BamA, darobactin is a rare example of a Proteobacteria-selective antibiotic.
Figure 84.

(A) Structure of darobactin (138). Strands β1 (blue) and β16 (orange) surround the lateral pore of BamA. (B) The open state of the lateral pore shown in ribbons. (C) The open state of the lateral pore highlights the hydrogen bonding contacts between β1 and β16. (D) The closed state of the lateral pore. (E) Location of resistance-conferring mutations in BamA, noting proximity to the lateral pore. (F) Close-up of β-strand mimicry interactions between darobactin (pink carbon atoms) and β1 of BamA (blue carbon atoms).
BamA is an essential β-barrel outer membrane protein (OMP) that functions as a chaperone that assists other OMPs to properly fold and insert into the OM of Proteobacteria. A critical feature of BamA is the lateral pore, which is an opening between the N- and C-terminal β-strands.851 Structural analyses have captured the lateral pore in an “open” and “closed” state (Figure 84).852–855 The open state allows for proper folding of the nascent OMP polypeptide, and subsequent insertion into the OM.856 Sequencing of darobactin-resistant mutants revealed three strains that possessed two to three substitutions located in or near the lateral pore.850 Isothermal titration calorimetry was used to demonstrate darobactin binding to BamA (Kd = 1.2 μM), and NMR analysis revealed that BamA was stabilized in the closed state upon binding darobactin.850,857,858
The structure of the BamA-darobactin complex reveals that the antibiotic mimics a β-strand that binds to the open conformation of BamA along β1 (Asn422-Phe428, Figure 84).859 Consequently, binding of darobactin occludes the lateral pore such that BamA is unable to fold or insert proteins into the OM. The unmodified core peptide of the darobactin precursor peptide does not possess any inhibitory effects, presumably as it cannot serve as a β-strand mimic.850 Alanine scanning analyses and binding experiments revealed that the most critical residue of BamA in strand β1 for darobactin binding is Asn427, which makes direct side chain contacts with Asn2 in darobactin.859 Single alanine substitutions of Asn422 through Phe426 did not significantly affect binding to darobactin.
Molecular dynamic simulations suggest how three distinct BamA substitutions (G429V, E435K, and Q445P) confer resistance to darobactin.859 The BamA-G429V replacement appears to disrupt the interactions between darobactin Asn2 and β16 of BamA. The BamA E435K variant introduces unfavorable allosteric effects with nearby negatively charged residues on BamA, leading to the destabilization of β1-β2 of BamA, and a 32-fold increase in the MIC over wild-type. Lastly, the Q445P substitution induces a conformational change in β2, which in turn leads to the destabilization of the β1-darobactin interaction.
Heterologous expression has been independently pursued by two groups to produce darobactin and derivatives thereof.860,861 Pseudomonas heterorhabitis VMG was predicted to produce darobactin B, which differs only by two substitutions (S4T and S6R). The MICs for darobactin B against various Proteobacteria were approximately the same as darobactin A: including P. aeruginosa (5.7–15.2 μM), K. pneumoniae (1.9–3.8 μM), A. baumanii (7.6–121 μM), and E. coli (0.95–3.8 μM). Analogs with a K5R or a N2S substitution were 8–16 fold less potent than darobactin A.860 Another study produced a new-to-nature F7W analog with 2–4 fold greater inhibitory activity against clinically relevant Proteobacteria.861 These studies demonstrated that the darobactin scaffold is amendable to engineering.
15.3. Sactipeptides
Sactipeptides are RiPPs with a class-defining, intramolecular sulfur-to-alpha carbon thioether (sactionine) crosslink installed by a rSAM enzyme. Most structurally characterized sactipeptides display a hairpin conformation arising from the cysteine donor residues being more N-terminal than the sactionine acceptor residues (Figure 85). Subtilosin A (139, Figure 85) and sporulation killing factor (SKF, 144, Figure 87) are further head-to-tail cyclized. All characterized sactipeptides possess antibacterial activities. The majority display growth-suppressive activity against closely related bacteria, typically other Firmicutes, such as the human pathogens C. difficile, L. monocytogenes, and Staphylococcus sp., or the actinobacterium Gardnerella vaginalis.306,862–872 MOA studies on sactipeptides strongly support the plasma membrane as the principle target.863,868,873–875
Figure 85.

(A) Chemical structure of subtilosin A (139). (B) Hydrophobicity surface map of subtilosin A (PDB ID: 1PXQ). Hydrophobic regions are shown in orange, while hydrophilic regions are shown in blue. The hydrophilic patch composed of Asn1, Lys2 and Trp34 is highlighted by the red curved line. The right panel shows the same surface rotated 180° about the vertical axis.
Figure 87.

Chemical structures of sporulation killing factor (144), RumC (145), and streptosactin (146). NMR solution structure of RumC is shown in the inset (PDB ID: 6T33). Stereocenters marked with * do not have an assigned absolute stereochemistry.
Subtilosin A (Figure 85) was first isolated from B. subtilis 168, and was found to inhibit the growth of various Bacilli strains and other Gram-positive bacteria such as L. monocytogenes ATCC 19115 and E. faecium (formerly S. faecium IFO 3181).864,876 Though subtilosin A is reported to produce zones of inhibition against Gram-negative bacteria including K. pneumoniae and Porphyromonas gingivalis, the susceptibility of Gram-negative strains is dependent on the absence of a polysaccharide capsule.876 Though subtilosin A is not hemolytic, a T6I variant demonstrated hemolytic activity and proved to be more potent against a number of bacteria tested (Table 5).877
Table 5.
MICs/MLCs of subtilosin A and variant T6I.
| Indicator | Subtilosin A MIC/MLC (μM) | Subtilosin A T6I MIC/MLC (μM) |
|---|---|---|
| Bacillus subtilis | 250 | 100 |
| Bacillus anthracis | 16 | 2.56 |
| Bacillus cereus | 40 | 6.4 |
| Bacillus thuringiensis | 40 | 6.4 |
| Enterococcus faecalis | 100 | 16 |
| Staphylococcus carnosus | 100 | 16 |
| Listeria monocytogenes | 40 | 6.4 |
| Streptococcus pyogenes | 100 | 6.4 |
| Rabbit blood agar* | >250 | 16 |
Reported as a minimum lytic concentration (MLC). Hemolysis was assessed using rabbit blood agar, as opposed to purified erythrocytes. Table adapted from (877).
Subtilosin A permeabilizes membranes to small molecules and ions. Treatment of G. vaginalis with subtilosin A at 0.6 μM induced ATP release, causing extracellular ATP content to increase to 40% of the total (intracellular + extracellular) ATP concentration.868 Utilizing the fluorophore 3,3’-dipropylthia-dicarbocyanine and the K+/H+ ionophore exchanger nigericin, subtilosin A was found to not affect the transmembrane potential (ΔΨ). Though subtilosin A did not deplete ΔΨ, it was shown to completely deplete the transmembrane pH gradient (ΔpH).868 The fact that subtilosin A only dissipated the ΔpH component of the proton motive force suggests the formation of transient pores in the bacterial membrane. Additional studies with L. monocytogenes demonstrated minimal effect on the pH gradient and ATP efflux, further suggesting that subtilosin A activity depends on the composition of the cell membrane.869
Fluorescence spectroscopy studies confirm that subtilosin A targets the membrane.873,878 The intrinsic fluorescence of Trp34 in subtilosin A shifted to shorter wavelengths in the presence of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol (POPG) small unilamellar vesicles (SUVs), indicating the Trp residue is buried inside the phospholipid bilayer in each case.873 In a separate experiment, carboxyfluorescein-containing SUVs were exposed to subtilosin A and dye leakage was observed in a concentration-dependent manner. No dye leakage was observed at concentrations lower than 16.4 μM, which is the lowest concentration at which subtilosin A exists as a monomer. It was hypothesized that subtilosin A has detergent-like behavior, in which a critical concentration is required for aggregation and membrane permeabilization. Subtilosin A exhibits selectivity for membrane composition as no dye leakage was observed for SUVs with compositions mimicking mammalian membranes.
NMR studies of subtilosin A in lipid bilayers further refine the model of membrane association. NMR studies of 15N-labeled subtilosin A in aligned POPC lipid bilayers demonstrated that the compound exists in two conformations, one perpendicular to the membrane surface and a smaller population aligned nearly parallel to the membrane surface.873 Utilizing 31P NMR spectroscopy to study lipid conformation, upfield shifts of ~2–4 ppm of POPC, POPG, and 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC) bilayers in the presence of subtilosin A indicate an altered lipid head group orientation. This conformational change was supported in studies of quadrupolar splitting of the α- and β-methylenes of choline in DMPC membranes, indicating a subtilosin A-dependent conformational change as well as a modest increase in lipid acyl chain conformational exchange. The NMR solution structure of subtilosin A reveals a hydrophilic patch consisting of Asn1, Lys2 and Trp34 (Figure 85),879 which is posited to associate with phospholipid head groups.873 Therefore it was proposed that subtilosin A does not fully insert and span the plasma membranes, but rather binds at the bilayer surface creating local permeabilization defects. However, as the concentrations necessary to induce membrane permeability were higher than the MIC of subtilosin A (Table 5), it was hypothesized that bactericidal activity may depend on interaction with a surface receptor.873,877 Further study will be required to determine if there is a cell-surface receptor for subtilosin A.
Subtilosin A also demonstrates spermicidal activity and inhibits late-stage replication of HSV-1.880–882 Subtilosin A has in vitro activity against human spermatozoa with an IC50 of ~19 μM, and treatment of ectocervical cell cultures with 40 μM subtilosin A reduced cell viability by a modest 5–10% over 24 h.881 Subtilosin A inhibited the forward progression of spermatozoa and induced tail coiling in a dose dependent manner, though no detailed mechanism for this observation has been proposed. Concerning HSV-1, subtilosin inhibited viral replication in vitro by ~92% at concentrations as low as 1.0 μM.882 Pretreatment of viral particles with 17.3 μM subtilosin A reduced the viral titer by 99%. Inactivation of HSV-1 was shown to occur not in the early stages of viral replication but instead the compound likely inhibited viral assembly or release.
Additional MOA studies of subtilosin A indicated inhibition of quorum sensing-dependent biofilm formation.883 At sub-lethal concentrations, subtilosin A inhibited biofilm formation of G. vaginalis (229 nM, >90% reduction), L. monocytogenes (4.4 μM, 80% reduction) and E. coli (4.4 μM, 60% reduction). Subtilosin A was also implicated as a potential inhibitor of quorum sensing in Chromobacterium violaceum. C. violaceum produces the pigment violacein only when neighboring cells secrete N-acyl homoserine lactone (AHL) quorum sensing molecules.884 Subtilosin A at concentrations of 2–37 μM reduced C. violaceum violacein production by >70%, suggesting that subtilosin A inhibits AHL production or secretion.883 Similarly, autoinducer-2 (AI-2) is an AHL found in a wide variety of bacteria.885 Production of AI-2 in G. vaginalis was inhibited by subtilosin A at 882 nM and 1.2 μM with no influence on bacterial growth. However, inhibition of AI-2 production was not observed for L. monocytogenes at 82 nM–1.3 μM subtilosin A, but cell viability declined to ~50% when treated with the highest concentration (1.3 μM).883 To summarize, in addition to membrane disruption, the anti-biofilm activity of subtilosin A suggests a second molecular target, providing opportunities for further MOA studies. A recently described sactipeptide hyicin was isolated from Staphylococcus hyicus 4244 and possesses 68% and 85% sequence identity and similarity to subtilosin A, respectively. This peptide demonstrated bactericidal activity against S. aureus and Staphylococcus saprophyticus, and was able to inhibit biofilm formation and disrupt preformed biofilms.870 Currently, the structure of hyicin is unknown, and more detailed MOA studies have not been performed, but given the sequence similarity to subtilosin A, it is reasonable to assume these two sactipeptides share an MOA.
MOA studies have also been undertaken for thuricin CD, thuricin Z/huazacin, and thurincin H (140, Figure 86). B. thuringiensis DPC 6431 produces thuricin CD, a two-component antibiotic composed of the separate sactipeptides thuricin α (141) and β (142), both of which are necessary for full activity.866,886 Thuricin CD irreversibly depolarizes the membrane and induces pore-formation in Bacillus firmus at 125 nM.875 Membrane polarization was studied using the dye 3,3’-diethyloxacarbocyanine iodide, which loses fluorescence and serves as a probe for bacterial membrane potential. Flow cytometry experiments revealed that both thuricin α and thuricin β (individually at 3 μM) induced depolarization and reduced cell size in isolation. At higher concentrations (9 μM), thuricin CD rapidly elicited depolarization to a greater degree than its individual components. Light microscopy showed B. firmus contained low-density regions of the plasma membrane after thuricin CD treatment. The individual components of thuricin CD seem to act synergistically to disrupt the bacterial membrane, though the potential role of a native cell surface receptor has not been ruled out. Furthermore, the membrane-bound thuricin CD structure is unknown, and higher-order structures beyond heterodimerization may be involved in membrane disruption.
Figure 86.

The structures of the sactipeptides thuricin H (140), thuricin CD, composed of two components thuricin α and thuricin β (141, 142), and huazicin/thuricin Z (143). Thuricin Z/huazacin stereocenters marked with * do not have assigned stereochemistry.
Similarly, huazacin306 (143, Figure 86; synonymous with thuricin Z, produced by B. thuringiensis serovar Huazhongensis) interacts directly with the bacterial cell membrane and induces ion permeability of artificial bacterial membranes.863 Confocal fluorescence microscopy demonstrated B. cereus uptake of propidium iodide upon huazacin treatment, indicating membrane permeabilization. Transmission electron microscopy of treated B. cereus showed an intact cell envelope with partial lysis of the inner bilayer. Scanning electron micrographs additionally demonstrated a collapsed bacterial cell surface and membrane perforations. Huazacin possesses a relatively narrow spectrum of activity, with MICs of 2–8 μM against B. cereus sp. and no notable activity towards other bacteria. The narrow spectrum activity is likely a consequence of species-specific differences in cell envelope composition. Bacteriomimetic large unilamellar vesicles (LUVs) (9:1 1,2-dipalmitoyl-sn-glycero-3-phosphatidyl glycerol to 1,2-dipalmitoyl-sn-glycero-3phosphatidylethanolamine) were permeabilized to K+/H+ upon huazacin treatment. Additionally, fungi-like LUVs (4:1 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine to ergosterol) did not demonstrate permeabilization in a huazacin-dependent manner.
Thurincin H (140, Figure 86), produced by B. thuringiensis SF36, also possesses potent activity against Bacillus and Listeria sp.887 with reported MICs against B. cereus strains 0.5–1 nM.888 Preliminary studies of thurincin H against B. cereus demonstrated a reduction in cell viability of ~3-log CFU/mL at 16 × MIC, but no decrease in optical density even at 256 × MIC, suggesting that thurincin H lacks lytic activity. B. cereus treated with 256 × MIC of thurincin H demonstrated a collapsed rod-shaped morphology. Moreover, surviving B. cereus did not display membrane punctures, as was the case when cells were treated with nisin (section 2.1). In assays utilizing propidium iodide, treatment with thurincin H did not permeabilize the membrane.874
Several other bioactive sactipeptides have been reported, though with no associated MOA. The gene cluster encoding sporulation killing factor (SKF, 144, Figure 87) was originally identified in B. subtilis as a target of the Spo0A transcriptional regulator.889,890 The Spo0A protein is activated by phosphorylation as the terminal regulator of the Stage 0 sporulation phospho-relay pathway, which is responsible for initiating sporulation in B. subtilis. During times of nutrient limitation, Spo0A activates transcription of SKF, which induces lysis of other B. subtilis cells, presumably to release nutrients for surviving cells. SKF was subsequently determined to be a secreted compound containing sactionine linkages.862,891 The molecular target of SKF remains unknown.
Two recently discovered sactipeptides further expand their structural diversity.892–895 Ruminococcin C (RumC, 145, Figure 87) was identified as an anti-clostridial antibiotic in a gnotobiotic mouse model. When colonized with Ruminococcus gnavus, sterile mice generated feces that inhibited the growth of Clostridium perfringens.892,896 RumC was purified from the fecal material, and shown to have antibacterial activity against C. perfringens, B. subtilis, and L. monocytogenes.892 The solution NMR structure of RumC demonstrated a bi-hairpin structure (Figure 87), which is unique among sactipeptides, as these RiPPs generally contain only a single hairpin motif.894 Bacterial cytological profiling demonstrated that RumC does not exhibit pore-forming activity, but did induce phenotypes similar to metronidazole, which is known to inhibit nucleic acid synthesis.897 RumC has also demonstrated antibiofilm activity and can even disrupt biofilms at high (>2 × MIC) concentrations.898 Furthermore, RumC exhibits antifungal activity against Heterobasidion annosum, a property that is unique among known sactipeptides.898
The recently characterized streptosactin (146, Figure 87) from Streptococcus sp. exhibited narrow spectrum bactericidal activity against Streptococcus thermophilus LMD-9 and LMG 18311 at 1 μM.895 Streptosactin also displayed bactericidal activity against the producing strain S. thermophilus JIM 8232 at 1 μM. In all cases of bactericidal activity, streptosactin was dosed in the pre-logarithmic growth phase against strains possessing identical streptosactin biosynthetic gene clusters relative to the S. thermophilus producing strain leading to the hypothesis that this compound acts as a fratricidal agent. Previous studies also demonstrated that expression of the streptosactin gene cluster correlates with the development of competence in S. thermophilus.899 Currently, the fratricidal mechanism of streptosactin is proposed as a strategy for competent S. thermophilus cells to incorporate genomic DNA from non-competent neighboring cells.
15.4. Epipeptides
Epipeptides are RiPPs with D-amino acids installed by a rSAM enzyme. B. subtilis produces the epipeptide EpeX* (147) (formerly YddF*, where * indicates the epimerized peptide; Figure 88) that has potent bactericidal effects against the producing strain.900,901 Such intra-species competition is believed to aid the survival of a sub-population at the expense of other members of the same population. Coordinated cell death induces lysis of susceptible cells, which release nutrients and transferrable genetic traits that can be utilized by the toxin-producing cells.902–904 Although the precise function of EpeX* is not currently known, we focus on the bioactivity of EpeX*, and its possible role in cannibalism and fratricide - two distinct outcomes of coordinated cell death.
Figure 88.

Structure of EpeX* (147). Val4 and Ile12 are epimerized to the D-configuration (blue).
The LiaFRS system is a three-component regulatory system that senses cell envelope stress in B. subtilis (Figure 89). These proteins are encoded by the liaIH-liaGFSR locus,141 wherein LiaS is a membrane-bound histidine kinase, LiaR is a DNA-binding response regulator, and LiaF a negative regulator of LiaRS-mediated signal transduction.905 Upon cell wall perturbations caused by antibiotics like bacitracin, nisin (section 2.1), nukacin ISK-1 (section 2.2), cacaoidin (section 2.5), siamycin I (section 8.1), and vancomycin, the LiaFRS cell-envelope stress response is triggered.141,900,906 LiaR binds to its primary target, the liaI promoter, and induces expression of the liaIH genes,141,907 which encode for a transmembrane protein (LiaI) and a homolog of phage-shock protein A (LiaH). These two proteins are hypothesized to mediate cell membrane stress,907,908 although the molecular-level details are unknown. The EpeX* encoding operon epeXEPAB (formerly yddFGHIJ) was identified in a search to uncover additional regulators of LiaFRS signaling. Cells deficient in the EpeAB ABC transporter showed increased expression from the PliaI promoter.900 This observation indicates that the cell envelope stress response was triggered by the inability to export a self-produced toxic compound. Thus, the EpeAB transporter likely exports EpeX*.900
Figure 89.

(A) Schematic of the lia cell stress response signaling pathway. (B) Schematic of the lia signaling pathway in response to EpeX*. EpeP is a transmembrane protease that processes and exports EpeX*.909 EpeAB is an ABC transporter involved in providing self-immunity against the antibacterial compound.
Subsequent characterization of EpeX* has revealed it triggers the LiaRS response in stationary B. subtilis cells when exogenously supplied.910 LiaIH mediates resistance against EpeX*, as a ΔliaIH strain was 3-fold more susceptible to EpeX* than wild type cells. EpeX* inhibits growth of B. subtilis (MIC < 0.94 μM), whereas synthetic analogs containing either all L-amino acids, or only one of the two modifications (D-Val4 or D-Ile12), did not inhibit growth of B. subtilis.901 Investigation into the MOA of EpeX* revealed depolarization of the cell membrane, leading to membrane permeation as monitored by fluorescence dyes.910 Furthermore, EpeX* rigidifies the membrane, which may account for its delayed polarization kinetics.
One proposal for EpeX* is that it functions as a cannibalism toxin, a strategy in which a toxin-producing sub-population benefits from nutrients released by the lysis of susceptible cells.891,902,903 Two characterized cannibalism toxins in B. subtilis are sporulation delaying protein (SDP) and sporulation killing factor (SKF, section 15.3).891,911–913 Expression of both SDP and SKF are controlled by Spo0A, the master transcriptional regulator of sporulation.889,890 Expression of EpeX* is correlated with the expression of SDP, suggesting that the two peptides could act synergistically.901 In support, it was noted that the SDP and EpeX* producing operons are controlled by the same transcriptional repressor.914 There is no evidence that the epe (formerly yyd) operon is regulated by Spo0A.910
An alternative hypothesis is that EpeX* functions as a fratricidal toxin. Fratricide is a process that initiates along with natural competence, wherein non-competent sub-populations are exposed to antimicrobials for which they lack immunity. Any DNA resultant from cell lysis can be naturally incorporated into the competent population.915,916 This process is suggested to enhance genetic diversity via exchange of extracellular DNA.917 Deletion of ComA (section 7.2), the transcriptional regulator responsible for inducing natural competence in B. subtilis, indirectly leads to decreased levels of the epeXEPAB mRNA levels.611,918 Further characterization of EpeX* is needed to validate whether EpeX* indeed functions as a fratricidal toxin.
15.5. Mycofactocin
Mycofactocin was bioinformatically predicted when co-occurrence was observed between genes encoding a specific set of redox-active enzymes and the mycofactocin biosynthetic gene cluster in mycobacteria.919 These findings led to the hypothesis that mycofactocin could be a new redox cofactor for these enzymes.920 In vitro studies indicated that the putative oxidoreductases had non-exchangeable NAD cofactors and had low, single turnover activity, but that activity could be increased through the introduction of redox mediators.919,921 Furthermore, disruption of the mycofactocin biosynthetic genes inhibited the growth of the producing strains on minimal media as first shown for M. tuberculosis919,922 and M. smegmatis.923,924 Additionally, mycofactocin-related genes were shown to be strongly upregulated in the presence of ethanol.925 Collectively, these studies were consistent with a possible redox cofactor role of the product of the mycofactocin biosynthetic gene cluster.
Because efforts to isolate mycofactocin from producing organisms were unsuccessful, the first direct evidence of mycofactocin acting as a cofactor was not obtained until 2019.924 Cell lysates from wild-type M. smegmatis were utilized to illustrate that mycofactocin can reduce dichlorophenolidonphenol (DCPIP), while mycofactocin-deficient lysates could not. A null variant was also generated for methanol dehydrogenase (Δmno), one of the enzymes that was thought to require mycofactocin as a cofactor, and the Δmno lysate was also unable to reduce DCPIP. However, DCPIP reduction was observed when lysates of the two mutants were mixed, suggesting mycofactocin was supplied by the Δmno strain and the Mno protein was provided by the mycofactocin deficient strain.924
Since these discoveries, the structure of pre-mycofactocin (148) has been elucidated by reconstitution of its biosynthetic enzymes (Figure 90).925,926 The final structure of mycofactocin has yet to be confirmed and is believed to be further modified, potentially via glycosylation.927 Notably, pre-mycofactocin and glycosylated pre-mycofactocin were accepted as substrates for enzymatic catalysis by carveol dehydrogenase.925,926
Figure 90.

Structure of pre-mycofactocin (148).
Pyrroloquinoline pyrrole is another RiPP cofactor that has been extensively studied. We refer the reader to previous reviews for its function.928–932
16. Concluding remarks and outlook
Studies of the mechanisms of action of almost all major classes of RiPPs predate their identification as post-translationally modified peptides. Such studies have been foundational in enabling research advances in RiPP identification, characterization, and biosynthetic enzymology. No doubt spurred by the discovery of classes of RiPPs with coveted bioactivities, the identification and dissection of targets for these compounds continue to stimulate further advances including engineering and structure-activity relationship studies. The synteny of genes encoding all necessary precursors and biosynthetic proteins for RiPP production allows for a comparatively straightforward pipeline for prediction, production, and engineering.
While analytical, biochemical, and biosynthetic studies of RiPPs have largely been fueled by the genomic revolution and advances in instrumentation, efforts on identification of MOAs have not advanced as quickly. The challenge in this area is the fact that, despite the simplistic biosynthetic logic, the diversity of molecular scaffolds that can be elaborated by RiPP pathways is expansive. Thus, the bioactivities associated with even closely related structural classes of RiPPs cannot be easily predicted. Conversely, the slow rates at which MOAs are identified provide numerous opportunities for further investigations not only of newly discovered RiPPs but also for known compounds for which such studies are either limited in scope or have yet to be carried out. This review compiles what is currently known about the MOA of RiPP natural products and highlights the many gaps in our current understanding and thus the opportunities for further studies. Considering the staggering pace at which new RiPP scaffolds continue to be discovered, a large number of compounds are available for targeted MOA studies. Given that many known classes of RiPPs can be genetically configured for affinity purification from a heterologous source, it is entirely practical for the community to establish a common source collection to enable such future efforts.
Another area with much potential for future exploration is the various microbiomes, such as in the human gut and oral cavity or the plant rhizosphere. RiPPs are encoded in the genomes of a remarkably wide spectrum of different phyla and applications of either the producing organisms or the compounds as growth promotors or probiotics are exciting prospects. However, such applications will require much more investigation of aspects of RiPP research that have received much less attention. These include assessment of toxicity and immunogenicity, pharmacokinetics, and gastrointestinal stability.38,933–937
One caveat with agnostic approaches towards MOA studies of RiPPs is that a number of these natural products may not have easily assayable activities or have pleiotropic activities. This aside, advancements in the technological state of instrumentation and computing have set the framework necessary for further in-depth studies. In-depth studies of each RiPP class could also result in more quantitative analysis of MOA and allow for useful comparisons across different classes of RiPPs that share the same target. Such comparisons are currently lacking and opportunities to fill this knowledge gap are ample. Given what is already known about the MOAs that are recapitulated here, additional studies will likely continue to provide unexpected insights for translating these compounds into modalities that are useful for society.
ACKNOWLEDGEMENTS
We thank the National Institutes of Health (R37 GM058822 to W.A.V, R01 AI144967 to D.A.M and W.A.V, and GM131347 and GM079038 to S.K.N.) and the Howard Hughes Medical Institute (W.A.V) for funding our programs on RiPPs.
Funding Sources
This work was supported by the National Institutes for Health (GM123998 to D.A.M; R37 GM058822 to W.A.V., R01 AI144967 to D.A.M. and W.A.V., and GM131347 and GM079038 to S.K.N.), and the Howard Hughes Medical Institute (W.A.V.).
D.A.M. is a cofounder of Lassogen, Inc.
Biographies
Emily K. Desormeaux
Emily was born near Houston, Texas in 1994. She received her B.S. in Chemistry from Texas A&M University in 2017. The following year, she began her graduate career at the University of Illinois Urbana-Champaign working in the lab of Prof. Wilfred van der Donk. Her doctoral work is focused on the mechanistic enzymology of lanthipeptide synthetases and the identification of lanthipeptide gene clusters.
Adam J. DiCaprio
Adam was born in 1990 in Poughkeepsie, New York and received a B.S. in Biology from Ursinus College in 2012. After 3 years as an NMR research associate at Process NMR Associates, he joined the group of Prof. Doug Mitchell and obtained his Ph.D. in Chemistry from the University of Illinois at Urbana-Champaign in 2021. His thesis work centered on applications of NMR spectroscopy to elucidate protein-protein interfaces of biosynthetic importance as well as natural product structure determination. He recently joined Merck & Co. as a Senior Scientist within the NMR Structure Elucidation Group.
Chayanid Ongpipattanakul
Chayanid (Ing) was born in Bangkok, Thailand in 1994. She earned a B.S. in Biochemistry from the University of California, Los-Angeles in 2016. In 2021, she received her Ph.D. in Biochemistry from the University of Illinois at Urbana-Champaign having conducted research in Prof. Satish Nair’s laboratory. Her thesis work was focused on the structural and biochemical characterization of natural product biosynthetic enzymes. She is currently a postdoctoral researcher in Prof. Charles Craik’s group at the University of California, San Francisco.
Wilfred A. van der Donk
Wilfred was born in Culemborg, the Netherlands. He obtained his B.S. and M.S. at Leiden University under the direction of Jan Reedijk and Willem Driessen. In 1989 he moved to Rice University where he completed his Ph.D. in organic chemistry in 1994 with Kevin Burgess. After postdoctoral studies with JoAnne Stubbe at MIT, he started his independent career in 1997 at the University of Illinois at Urbana-Champaign, where he currently holds the Richard E. Heckert chair in the Department of Chemistry. Since 2008, he is an Investigator of the Howard Hughes Medical Institute. He is affiliated with the Departments of Biochemistry and Bioengineering and is a faculty member of the Carle R. Woese Institute for Genomic Biology. Research in his laboratory uses chemistry, enzymology and molecular biology to better understand enzyme catalysis and to use that knowledge for synthetic biology.
Douglas A. Mitchell
Doug was born near Pittsburgh, Pennsylvania in 1980. He received a B.S. in Chemistry from Carnegie Mellon University in 2002. After a short internship in medicinal chemistry at Merck Research Laboratories, he obtained his Ph.D. from the University of California, Berkeley in 2006 while working with Prof. Michael Marletta. For postdoctoral studies, he worked with Prof. Jack Dixon at the University of California, San Diego. Prof. Mitchell joined the Department of Chemistry at the University of Illinois at Urbana-Champaign in 2009 and currently holds the John and Margaret Witt Professorship. Mitchell is affiliated with the Department of Microbiology and is a faculty member of the Carle R. Woese Institute for Genomic Biology. The Mitchell lab employs a wide variety of approaches to identify and characterize novel natural products. The lab also has strong interests in bioinformatics, biosynthetic enzymology, and new reaction discovery.
Satish K. Nair
Satish was born in Bihar but comes from Kerala, India. His family emigrated to the U.S. during the bicentennial (1976), and he was educated in the New York City public school system. He received his B.S. in Chemistry (with Honors) from Brown University in 1989 and carried out his doctoral work with David Christianson at the University of Pennsylvania where he received his Ph.D. in Chemistry in 1994. He was a Leukemia Society of America post-doctoral fellow in the laboratory of Stephen K. Burley at Rockefeller University. Satish joined the Department of Biochemistry at the University of Illinois at Urbana-Champaign in 2001. He currently serves at the Head of Biochemistry and holds the Gregorio Weber Chair. He is also the Director for the Center of Biophysics and Co-Director of the Macromolecular CryoEM and MicroED facility. The Nair lab is interested in structure-function studies of enzymes involved in the biosynthesis of primary and secondary metabolites.
References
- (1).Newman DJ; Cragg GM Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod 2020, 83 (3), 770–803. [DOI] [PubMed] [Google Scholar]
- (2).Atanasov AG; Zotchev SB; Dirsch VM; International Natural Product Sciences Taskforce; Supuran, C. T. Natural products in drug discovery: advances and opportunities. Nat Rev. Drug Discov 2021, 20 (3), 200–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Miethke M; Pieroni M; Weber T; Brönstrup M; Hammann P; Halby L; Arimondo PB; Glaser P; Aigle B; Bode HB; Moreira R; Li Y; Luzhetskyy A; Medema MH; Pernodet J-L; Stadler M; Tormo JR; Genilloud O; Truman AW; Weissman KJ; Takano E; Sabatini S; Stegmann E; Brötz-Oesterhelt H; Wohlleben W; Seemann M; Empting M; Hirsch AKH; Loretz B; Lehr C-M; Titz A; Herrmann J; Jaeger T; Alt S; Hesterkamp T; Winterhalter M; Schiefer A; Pfarr K; Hoerauf A; Graz H; Graz M; Lindvall M; Ramurthy S; Karlén A; van Dongen M; Petkovic H; Keller A; Peyrane F; Donadio S; Fraisse L; Piddock LJV; Gilbert IH; Moser HE; Müller R. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem 2021, 5 (10), 726–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Drewry DH; Macarron R. Enhancements of screening collections to address areas of unmet medical need: an industry perspective. Curr. Opin. Chem. Biol 2010, 14 (3), 289–298. [DOI] [PubMed] [Google Scholar]
- (5).Harvey AL; Edrada-Ebel R; Quinn RJ The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov 2015, 14 (2), 111–129. [DOI] [PubMed] [Google Scholar]
- (6).Salvador-Reyes LA; Luesch H. Biological targets and mechanisms of action of natural products from marine cyanobacteria. Nat. Prod. Rep 2015, 32 (3), 478–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Maksimov AY; Balandina SY; Topanov PA; Mashevskaya IV; Chaudhary S. Organic antifungal drugs and targets of their action. Curr. Top. Med. Chem 2021, 21 (8), 705–736. [DOI] [PubMed] [Google Scholar]
- (8).Porras G; Chassagne F; Lyles JT; Marquez L; Dettweiler M; Salam AM; Samarakoon T; Shabih S; Farrokhi DR; Quave CL Ethnobotany and the role of plant natural products in antibiotic drug discovery. Chem. Rev 2021, 121 (6), 3495–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian K-D; Fischbach MA; Garavelli JS; Göransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Müller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJT; Rebuffat S; Ross RP; Sahl H-G; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Süssmuth RD; Tagg JR; Tang G-L; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep 2013, 30 (1), 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Montalbán-López M; Scott TA; Ramesh S; Rahman IR; van Heel AJ; Viel JH; Bandarian V; Dittmann E; Genilloud O; Goto Y; Grande Burgos MJ; Hill C; Kim S; Koehnke J; Latham JA; Link AJ; Martínez B; Nair SK; Nicolet Y; Rebuffat S; Sahl H-G; Sareen D; Schmidt EW; Schmitt L; Severinov K; Süssmuth RD; Truman AW; Wang H; Weng J-K; van Wezel GP; Zhang Q; Zhong J; Piel J; Mitchell DA; Kuipers OP; van der Donk WA New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep 2021, 38 (1), 130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tracanna V; de Jong A; Medema MH; Kuipers OP Mining prokaryotes for antimicrobial compounds: from diversity to function. FEMS Microbiol. Rev 2017, 41 (3), 417–429. [DOI] [PubMed] [Google Scholar]
- (12).Buda De Cesare G; Cristy SA; Garsin DA; Lorenz MC Antimicrobial peptides: a new frontier in antifungal therapy. mBio 2020, 11 (6), e02123–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Iorio M; Sasso O; Maffioli SI; Bertorelli R; Monciardini P; Sosio M; Bonezzi F; Summa M; Brunati C; Bordoni R; Corti G; Tarozzo G; Piomelli D; Reggiani A; Donadio S. A glycosylated, labionin-containing lanthipeptide with marked antinociceptive activity. ACS Chem. Biol 2014, 9 (2), 398–404. [DOI] [PubMed] [Google Scholar]
- (14).Green BR; Olivera BM Venom peptides from cone snails: pharmacological probes for voltage-gated sodium channels. Curr. Top. Membr 2016, 78, 65–86. [DOI] [PubMed] [Google Scholar]
- (15).Fu Y; Jaarsma AH; Kuipers OP Antiviral activities and applications of ribosomally synthesized and post-translationally modified peptides (RiPPs). Cell. Mol. Life Sci 2021, 78 (8), 3921–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kobayashi J; Tsuda M; Nakamura T; Mikami Y; Shigemori H. Hymenamides A and B, new proline-rich cyclic heptapeptides from the Okinawan marine sponge Hymeniacidon sp. Tetrahedron 1993, 49 (12), 2391–2402. [Google Scholar]
- (17).Rudi A; Aknin M; Gaydou EM; Kashman Y. Four new cytotoxic cyclic hexa- and heptapeptides from the marine ascidian Didemnum molle. Tetrahedron 1998, 54 (43), 13203–13210. [Google Scholar]
- (18).Bhat UG; Halasi M; Gartel AL Thiazole antibiotics target FoxM1 and induce apoptosis in human cancer cells. PLoS One 2009, 4 (5), e5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sivonen K; Leikoski N; Fewer DP; Jokela J. Cyanobactins—ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biotechnol 2010, 86 (5), 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Frattaruolo L; Lacret R; Cappello AR; Truman AW A genomics-based approach identifies a thioviridamide-like compound with selective anticancer activity. ACS Chem. Biol 2017, 12 (11), 2815–2822. [DOI] [PubMed] [Google Scholar]
- (21).Gabrielsen C; Brede DA; Nes IF; Diep DB Circular bacteriocins: biosynthesis and mode of action. Appl. Environ. Microbiol 2014, 80 (22), 6854–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Oppedijk SF; Martin NI; Breukink E. Hit ‘em where it hurts: The growing and structurally diverse family of peptides that target lipid-II. Biochim. Biophys. Acta 2016, 1858 (5), 947–957. [DOI] [PubMed] [Google Scholar]
- (23).Norris GE; Patchett ML The glycocins: in a class of their own. Curr. Opin. Struct. Biol 2016, 40, 112–119. [DOI] [PubMed] [Google Scholar]
- (24).Kenney GE; Rosenzweig AC Chalkophores. Annu. Rev. Biochem 2018, 87, 645–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Perez RH; Zendo T; Sonomoto K. Circular and leaderless bacteriocins: biosynthesis, mode of action, applications, and prospects. Front. Microbiol 2018, 9 (2085). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smits THM; Duffy B; Blom J; Ishimaru CA; Stockwell VO Pantocin A, a peptide-derived antibiotic involved in biological control by plant-associated Pantoea species. Arch. Microbiol 2019, 201 (6), 713–722. [DOI] [PubMed] [Google Scholar]
- (27).Tan S; Moore G; Nodwell J. Put a bow on It: knotted antibiotics take center stage. Antibiotics 2019, 8 (3), E117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Telhig S; Ben Said L; Zirah S; Fliss I; Rebuffat S. Bacteriocins to thwart bacterial resistance in Gram negative bacteria. Front. Microbiol 2020, 11, 586433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Vinogradov AA; Suga H. Introduction to thiopeptides: biological activity, biosynthesis, and strategies for functional reprogramming. Cell Chem. Biol 2020, 27 (8), 1032–1051. [DOI] [PubMed] [Google Scholar]
- (30).Barbour A; Wescombe P; Smith L. Evolution of lantibiotic salivaricins: new weapons to fight infectious diseases. Trends. Microbiol 2020, 28 (7), 578–593. [DOI] [PubMed] [Google Scholar]
- (31).Cao L; Do T; Link AJ Mechanisms of action of ribosomally synthesized and posttranslationally modified peptides (RiPPs). J. Ind. Microbiol. Biotechnol 2021, 48 (3–4), kuab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chan DCK; Burrows LL Thiopeptides: antibiotics with unique chemical structures and diverse biological activities. J. Antibiot 2021, 74 (3), 161–175. [DOI] [PubMed] [Google Scholar]
- (33).Franz L; Kazmaier U; Truman AW; Koehnke J. Bottromycins - biosynthesis, synthesis and activity. Nat. Prod. Rep 2021, 38 (9), 1659–1683. [DOI] [PubMed] [Google Scholar]
- (34).do Amaral SC; Monteiro PR; Neto J. da SP; Serra GM; Gonçalves EC; Xavier LP; Santos AV Current knowledge on microviridin from cyanobacteria. Marine Drugs 2021, 19 (1), 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Travin DY; Severinov K; Dubiley S. Natural trojan horse inhibitors of aminoacyl-tRNA synthetases. RSC. Chem. Biol 2021, 2 (2), 468–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bailly C. The bacterial thiopeptide thiostrepton. An update of its mode of action, pharmacological properties and applications. Eur. J. Pharmacol 2022, 914, 174661. [DOI] [PubMed] [Google Scholar]
- (37).Parker JK; Davies BW Microcins reveal natural mechanisms of bacterial manipulation to inform therapeutic development. Microbiology 2022, 168 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rebuffat S. Ribosomally synthesized peptides, foreground players in microbial interactions: recent developments and unanswered questions. Nat. Prod. Rep 2022, 39 (2), 273–310. [DOI] [PubMed] [Google Scholar]
- (39).Bernheimer AW Cytolytic toxins of bacterial origin. The nature and properties of cytolytic proteins are discussed with emphasis on staphylococcal alpha-toxin. Science 1968, 159 (3817), 847–851. [DOI] [PubMed] [Google Scholar]
- (40).Vizán JL; Hernández-Chico C; del Castillo I; Moreno F. The peptide antibiotic microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. EMBO J. 1991, 10 (2), 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Skinnider MA; Johnston CW; Edgar RE; Dejong CA; Merwin NJ; Rees PN; Magarvey NA Genomic charting of ribosomally synthesized natural product chemical space facilitates targeted mining. Proc. Natl. Acad. Sci. U.S.A 2016, 113 (42), E6343–E6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tietz JI; Schwalen CJ; Patel PS; Maxson T; Blair PM; Tai H-C; Zakai UI; Mitchell DA A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol 2017, 13 (5), 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).de Los Santos ELC NeuRiPP: Neural network identification of RiPP precursor peptides. Sci. Rep 2019, 9 (1), 13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Merwin NJ; Mousa WK; Dejong CA; Skinnider MA; Cannon MJ; Li H; Dial K; Gunabalasingam M; Johnston C; Magarvey NA DeepRiPP integrates multiomics data to automate discovery of novel ribosomally synthesized natural products. Proc. Natl. Acad. Sci. U.S.A 2020, 117 (1), 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Blin K; Shaw S; Kloosterman AM; Charlop-Powers Z; van Wezel GP; Medema MH; Weber T. AntiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49 (W1), W29–W35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Moffat AD; Santos-Aberturas J; Chandra G; Truman AW A user guide for the identification of new RiPP biosynthetic gene clusters using a RiPPER-based workflow. Methods Mol. Biol 2021, 2296, 227–247. [DOI] [PubMed] [Google Scholar]
- (47).Oman TJ; van der Donk WA Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol 2010, 6 (1), 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Gu W; Dong S-H; Sarkar S; Nair SK; Schmidt EW The biochemistry and structural biology of cyanobactin pathways: enabling combinatorial biosynthesis. Meth. Enzymol 2018, 604, 113–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Schnell N; Entian K-D; Schneider U; Götz F; Zähner H; Kellner R; Jung G. Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature 1988, 333 (6170), 276–278. [DOI] [PubMed] [Google Scholar]
- (50).Rogers LA The inhibiting effect of Streptococcus lactis on Lactobacillus balgaricus. J. Bacteriol 1928, 16 (5), 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lubelski J; Rink R; Khusainov R; Moll GN; Kuipers OP Biosynthesis, immunity, regulation, mode of action and engineering of the model lantibiotic nisin. Cell. Mol. Life Sci 2008, 65 (3), 455–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Brötz H; Josten M; Wiedemann I; Schneider U; Götz F; Bierbaum G; Sahl H-G Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol. Microbiol 1998, 30 (2), 317–327. [DOI] [PubMed] [Google Scholar]
- (53).Breukink E; Wiedemann I; Kraaij C. van; Kuipers OP; Sahl H-G; Kruijff B. de. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 1999, 286 (5448), 2361–2364. [DOI] [PubMed] [Google Scholar]
- (54).Hsu S-TD; Breukink E; Tischenko E; Lutters MAG; de Kruijff B; Kaptein R; Bonvin AMJJ; van Nuland NAJ The nisin–lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat. Struct. Mol. Biol 2004, 11 (10), 963–967. [DOI] [PubMed] [Google Scholar]
- (55).Hasper HE; de Kruijff B; Breukink E. Assembly and stability of nisin−lipid II pores. Biochemistry 2004, 43 (36), 11567–11575. [DOI] [PubMed] [Google Scholar]
- (56).t Hart P; Oppedijk SF; Breukink E; Martin NI New insights into nisin’s antibacterial mechanism revealed by binding studies with synthetic lipid II analogues. Biochemistry 2016, 55 (1), 232–237. [DOI] [PubMed] [Google Scholar]
- (57).Bonev BB; Breukink E; Swiezewska E; Kruijff BD; Watts A. Targeting extracellular pyrophosphates underpins the high selectivity of nisin. FASEB J. 2004, 18 (15), 1862–1869. [DOI] [PubMed] [Google Scholar]
- (58).Wiedemann I; Breukink E; van Kraaij C; Kuipers OP; Bierbaum G; de Kruijff B; Sahl H-G Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem 2001, 276 (3), 1772–1779. [DOI] [PubMed] [Google Scholar]
- (59).Breukink E; van Heusden HE; Vollmerhaus PJ; Swiezewska E; Brunner L; Walker S; Heck AJR; de Kruijff B. Lipid II Is an intrinsic component of the pore induced by nisin in bacterial membrane. J. Biol. Chem 2003, 278 (22), 19898–19903. [DOI] [PubMed] [Google Scholar]
- (60).Wiedemann I; Benz R; Sahl H-G Lipid II-mediated pore formation by the peptide antibiotic nisin: a black lipid membrane study. J. Bacteriol 2004, 186 (10), 3259–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Cotter PD; Hill C; Ross RP Bacterial lantibiotics: strategies to improve therapeutic potential. Curr. Protein Pept. Sci 2005, 6 (1), 61–75. [DOI] [PubMed] [Google Scholar]
- (62).Chatterjee C; Paul M; Xie L; van der Donk WA Biosynthesis and mode of action of lantibiotics. Chem. Rev 2005, 105, 633–684. [DOI] [PubMed] [Google Scholar]
- (63).Bierbaum G; Sahl H-G Lantibiotics: mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol 2009, 10 (1), 2–18. [DOI] [PubMed] [Google Scholar]
- (64).Asaduzzaman SM; Sonomoto K. Lantibiotics: diverse activities and unique modes of action. J. Biosci. Bioeng 2009, 107 (5), 475–487. [DOI] [PubMed] [Google Scholar]
- (65).Knerr PJ; van der Donk WA Discovery, biosynthesis, and engineering of lantipeptides. Annu. Rev. Biochem 2012, 81 (1), 479–505. [DOI] [PubMed] [Google Scholar]
- (66).Field D; Cotter PD; Ross RP; Hill C. Bioengineering of the model lantibiotic nisin. Bioengineered 2015, 6 (4), 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Breukink E; de Kruijff B. Lipid II as a target for antibiotics. Nat. Rev. Drug Disc 2006, 5 (4), 321–323. [DOI] [PubMed] [Google Scholar]
- (68).Martin NI; Breukink E. The expanding role of lipid II as a target for lantibiotics. Future Microbiol. 2007, 2 (5), 513–525. [DOI] [PubMed] [Google Scholar]
- (69).Wiedemann I; Breukink E; van Kraaij C; Kuipers OP; Bierbaum G; de Kruijff B; Sahl HG Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem 2001, 276 (3), 1772–1779. [DOI] [PubMed] [Google Scholar]
- (70).Healy B; Field D; O’Connor PM; Hill C; Cotter PD; Ross RP Intensive mutagenesis of the nisin hinge leads to the rational design of enhanced derivatives. PLoS One 2013, 8 (11), e79563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Field D; Connor PMO; Cotter PD; Hill C; Ross RP The generation of nisin variants with enhanced activity against specific Gram-positive pathogens. Mol. Microbiol 2008, 69 (1), 218–230. [DOI] [PubMed] [Google Scholar]
- (72).Yuan J; Zhang Z-Z; Chen X-Z; Yang W; Huan L-D Site-directed mutagenesis of the hinge region of nisin Z and properties of nisin Z mutants. Appl. Microbiol. Biotechnol 2004, 64 (6), 806–815. [DOI] [PubMed] [Google Scholar]
- (73).Zhou L; van Heel AJ; Kuipers OP The length of a lantibiotic hinge region has profound influence on antimicrobial activity and host specificity. Front. Microbiol 2015, 6 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Zaschke-Kriesche J; Reiners J; Lagedroste M; Smits SHJ Influence of nisin hinge-region variants on lantibiotic immunity and resistance proteins. Bioorg. Med. Chem 2019, 27 (17), 3947–3953. [DOI] [PubMed] [Google Scholar]
- (75).Demel RA; Peelen T; Siezen RJ; Kruijff BD; Kuipers OP Nisin Z, mutant nisin Z and lacticin 481 interactions with anionic lipids correlate with antimicrobial activity. Eur. J. Biochem 1996, 235 (1–2), 267–274. [DOI] [PubMed] [Google Scholar]
- (76).Meer JR van der; Polman J; Beerthuyzen MM; Siezen RJ; Kuipers OP; Vos WMD, Characterization of the Lactococcus lactis nisin A operon genes nisP, encoding a subtilisin-like serine protease involved in precursor processing, and nisR, encoding a regulatory protein involved in nisin biosynthesis. J. Bacteriol 1993, 175 (9), 2578–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Rink R; Wierenga J; Kuipers A; Kluskens LD; Driessen AJM; Kuipers OP; Moll GN Dissection and modulation of the four distinct activities of nisin by mutagenesis of rings A and B and by C-terminal truncation. Appl. Environ. Microbiol 2007, 73 (18), 5809–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Field D; Begley M; O’Connor PM; Daly KM; Hugenholtz F; Cotter PD; Hill C; Ross RP Bioengineered nisin A derivatives with enhanced activity against both Gram positive and Gram negative pathogens. PLoS One 2012, 7 (10), e46884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Smith MK; Draper LA; Hazelhoff P-J; Cotter PD; Ross RP; Hill C. A bioengineered nisin derivative, M21A, in combination with food grade additives eradicates biofilms of Listeria monocytogenes. Front. Microbiol 2016, 7 (1939). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Li Q; Montalban-Lopez M; Kuipers OP Increasing the antimicrobial activity of nisin-based lantibiotics against Gram-negative pathogens. Appl. Environ. Microbiol 2018, 84 (12), e00052–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Molloy EM; Field D; Connor PMO; Cotter PD; Hill C; Ross RP Saturation mutagenesis of lysine 12 leads to the identification of derivatives of nisin A with enhanced antimicrobial activity. PLoS One 2013, 8 (3), e58530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Cebrián R; Macia-Valero A; Jati AP; Kuipers OP Design and expression of specific hybrid lantibiotics active against pathogenic Clostridium spp. Front. Microbiol 2019, 10, 2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Zaschke-Kriesche J; Behrmann LV; Reiners J; Lagedroste M; Gröner Y; Kalscheuer R; Smits SHJ Bypassing lantibiotic resistance by an effective nisin derivative. Bioorg. Med. Chem 2019, 27 (15), 3454–3462. [DOI] [PubMed] [Google Scholar]
- (84).Castiglione F; Lazzarini A; Carrano L; Corti E; Ciciliato I; Gastaldo L; Candiani P; Losi D; Marinelli F; Selva E; Parenti F. Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chem. Biol 2008, 15 (1), 22–31. [DOI] [PubMed] [Google Scholar]
- (85).Pokhrel R; Bhattarai N; Baral P; Gerstman BS; Park JH; Handfield M; Chapagain PP Molecular mechanisms of pore formation and membrane disruption by the antimicrobial lantibiotic peptide Mutacin 1140. Phys. Chem. Chem. Phys 2019, 21 (23), 12530–12539. [DOI] [PubMed] [Google Scholar]
- (86).Garg N; Tang W; Goto Y; Nair SK; Donk WA van der. Lantibiotics from Geobacillus thermodenitrificans. Proc. Natl. Acad. Sci. U.S.A 2012, 109 (14), 5241–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Garg N; Oman TJ; Andrew Wang T-S; De Gonzalo CVG; Walker S; van der Donk WA Mode of action and structure–activity relationship studies of geobacillin I. J. Antibiot 2014, 67 (1), 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Smith L; Hasper H; Breukink E; Novak J; Cerkasov J; Hillman JD; Wilson-Stanford S; Orugunty RS Elucidation of the antimicrobial mechanism of mutacin 1140. Biochemistry 2008, 47 (10), 3308–3314. [DOI] [PubMed] [Google Scholar]
- (89).Brunati C; Thomsen TT; Gaspari E; Maffioli S; Sosio M; Jabes D; Løbner-Olesen A; Donadio S. Expanding the potential of NAI-107 for treating serious ESKAPE pathogens: synergistic combinations against Gram-negatives and bactericidal activity against nondividing cells. J. Antimicrob. Chemother 2018, 73 (2), 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Münch D; Müller A; Schneider T; Kohl B; Wenzel M; Bandow JE; Maffioli S; Sosio M; Donadio S; Wimmer R; Sahl H-G The lantibiotic NAI-107 binds to bactoprenol-bound cell wall precursors and impairs membrane functions. J. Biol. Chem 2014, 289 (17), 12063–12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Bonelli RR; Schneider T; Sahl H-G; Wiedemann I. Insights into in vivo activities of lantibiotics from gallidermin and epidermin mode-of-action studies. Antimicrob. Agents Chemother 2006, 50 (4), 1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Hasper HE; Kramer NE; Smith JL; Hillman JD; Zachariah C; Kuipers OP; Kruijff B. de; Breukink E. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 2006, 313 (5793), 1636–1637. [DOI] [PubMed] [Google Scholar]
- (93).Stock AM; Robinson VL; Goudreau PN Two-component signal transduction. Annu. Rev. Biochem 2000, 69 (1), 183–215. [DOI] [PubMed] [Google Scholar]
- (94).Kuipers OP; Beerthuyzen MM; de Ruyter PGGA; Luesink EJ; de Vos WM Autoregulation of nisin biosynthesis in Lactococcus lactis by signal transduction. J. Biol. Chem 1995, 270 (45), 27299–27304. [DOI] [PubMed] [Google Scholar]
- (95).Kleerebezem M. Quorum sensing control of lantibiotic production; nisin and subtilin autoregulate their own biosynthesis. Peptides 2004, 25 (9), 1405–1414. [DOI] [PubMed] [Google Scholar]
- (96).Geiger C; Spieß T; Korn SM; Kötter P; Entian K-D Specificity of subtilin-mediated activation of histidine kinase SpaK. Appl. Environ. Microbiol 2017, 83 (18), e00781–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Spieß T; Korn SM; Kötter P; Entian K-D Autoinduction specificities of the lantibiotics subtilin and nisin. Appl. Environ. Microbiol 2015, 81 (22), 7914–7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Kraaij CV; Breukink E; Rollema HS; Siezen RJ; Demel RA; Kruijff BD; Kuipers OP Influence of charge differences in the C-Terminal part of nisin on antimicrobial activity and signaling capacity. Eur. J. Biochem 1997, 247 (1), 114–120. [DOI] [PubMed] [Google Scholar]
- (99).O’ Connor M; Field D; Grainger A; O’ Connor PM; Draper L; Ross RP; Hill C. Nisin M: a bioengineered nisin A variant that retains full induction capacity but has significantly reduced antimicrobial activity. Appl. Environ. Microbiol 2020, 86 (15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Ekkelenkamp MB; Hanssen M; Danny Hsu S-T; de Jong A; Milatovic D; Verhoef J; van Nuland NAJ Isolation and structural characterization of epilancin 15X, a novel lantibiotic from a clinical strain of Staphylococcus epidermidis. FEBS Lett. 2005, 579 (9), 1917–1922. [DOI] [PubMed] [Google Scholar]
- (101).Wang X; Gu Q; Breukink E. Non-lipid II targeting lantibiotics. Biochim. Biophys. Acta Biomembr 2020, 1862 (8), 183244. [DOI] [PubMed] [Google Scholar]
- (102).Knerr PJ; van der Donk WA Chemical synthesis and biological activity of analogues of the lantibiotic epilancin 15X. J. Am. Chem. Soc 2012, 134 (18), 7648–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Morris SL; Walsh RC; Hansen JN Identification and characterization of some bacterial membrane sulfhydryl groups which are targets of bacteriostatic and antibiotic action. J. Biol. Chem 1984, 259 (21), 13590–13594. [PubMed] [Google Scholar]
- (104).Chan WC; Dodd HM; Horn N; Maclean K; Lian LY; Bycroft BW; Gasson MJ; Roberts GC Structure-activity relationships in the peptide antibiotic nisin: role of dehydroalanine 5. Appl. Environ. Microbiol 1996, 62 (8), 2966–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Liu W; Hansen JN Enhancement of the chemical and antimicrobial properties of subtilin by site-directed mutagenesis. J. Biol. Chem 1992, 267 (35), 25078–25085. [PubMed] [Google Scholar]
- (106).Gut IM; Blanke SR; van der Donk WA Mechanism of inhibition of Bacillus anthracis spore outgrowth by the lantibiotic nisin. ACS Chem. Biol 2011, 6 (7), 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Gut IM; Prouty AM; Ballard JD; Donk WA van der; Blanke, S. R. Inhibition of Bacillus anthracis spore outgrowth by nisin. Antimicrob. Agents Chemother 2008, 52 (12), 4281–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Sarksian R; Hegemann JD; Simon MA; Acedo JZ; van der Donk WA Unexpected methyllanthionine stereochemistry in the morphogenetic lanthipeptide SapT. J. Am. Chem. Soc 2022, 144 (14), 6373–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Mohr KI; Volz C; Jansen R; Wray V; Hoffmann J; Bernecker S; Wink J; Gerth K; Stadler M; Müller R. Pinensins: the first antifungal lantibiotics. Angew. Chem. Int. Ed. Engl 2015, 54 (38), 11254–11258. [DOI] [PubMed] [Google Scholar]
- (110).Kodani S; Lodato MA; Durrant MC; Picart F; Willey JM SapT, a lanthionine-containing peptide involved in aerial hyphae formation in the streptomycetes. Mol. Microbiol 2005, 58 (5), 1368–1380. [DOI] [PubMed] [Google Scholar]
- (111).Kodani S; Hudson ME; Durrant MC; Buttner MJ; Nodwell JR; Willey JM The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor. Proc. Natl. Acad. Sci. U.S.A 2004, 101 (31), 11448–11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Brötz H; Bierbaum G; Markus A; Molitor E; Sahl HG Mode of action of the lantibiotic mersacidin: inhibition of peptidoglycan biosynthesis via a novel mechanism? Antimicrob. Agents Chemother 1995, 39 (3), 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Szekat C; Jack RW; Skutlarek D; Färber H; Bierbaum G. Construction of an expression system for site-directed mutagenesis of the lantibiotic mersacidin. Appl. Environ. Microbiol 2003, 69 (7), 3777–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Hsu S-TD; Breukink E; Bierbaum G; Sahl H-G; Kruijff B. de; Kaptein R; Nuland NAJ van; Bonvin AMJJ NMR study of mersacidin and lipid II interaction in dodecylphosphocholine micelles: conformational changes are key to antimicrobial activity. J. Biol. Chem 2003, 278 (15), 13110–13117. [DOI] [PubMed] [Google Scholar]
- (115).Böttiger T; Schneider T; Martínez B; Sahl H-G; Wiedemann I. Influence of Ca2+ ions on the activity of lantibiotics containing a mersacidin-like lipid II binding motif. Appl. Environ. Microbiol 2009, 75 (13), 4427–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Brötz H; Bierbaum G; Leopold K; Reynolds PE; Sahl H-G The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother 1998, 42 (1), 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Dufour A; Hindré T; Haras D; Le Pennec J-P The biology of lantibiotics from the lacticin 481 group is coming of age. FEMS Microbiol. Rev 2007, 31 (2), 134–167. [DOI] [PubMed] [Google Scholar]
- (118).Zhang J; Feng Y; Teng K; Lin Y; Gao Y; Wang J; Zhong J. Type AII lantibiotic bovicin HJ50 with a rare disulfide bond: structure, structure-activity relationships and mode of action. Biochem. J 2014, 461 (3), 497–508. [DOI] [PubMed] [Google Scholar]
- (119).Islam MR; Nishie M; Nagao J; Zendo T; Keller S; Nakayama J; Kohda D; Sahl H-G; Sonomoto K. Ring A of nukacin ISK-1: a lipid II-binding motif for type-A(II) lantibiotic. J. Am. Chem. Soc 2012, 134 (8), 3687–3690. [DOI] [PubMed] [Google Scholar]
- (120).Wiedemann I; Böttiger T; Bonelli RR; Wiese A; Hagge SO; Gutsmann T; Seydel U; Deegan L; Hill C; Ross P; Sahl H-G The mode of action of the lantibiotic lacticin 3147 – a complex mechanism involving specific interaction of two peptides and the cell wall precursor lipid II. Mol. Microbiol 2006, 61 (2), 285–296. [DOI] [PubMed] [Google Scholar]
- (121).Knerr PJ; Oman TJ; Garcia De Gonzalo CV; Lupoli TJ; Walker S; van der Donk WA Non-proteinogenic amino acids in lacticin 481 analogues result in more potent inhibition of peptidoglycan transglycosylation. ACS Chem. Biol 2012, 7 (11), 1791–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Oman TJ; Lupoli TJ; Wang T-SA; Kahne D; Walker S; van der Donk WA Haloduracin α binds the peptidoglycan precursor lipid II with 2:1 stoichiometry. J. Am. Chem. Soc 2011, 133 (44), 17544–17547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Fujinami D; Mahin A-A; Elsayed KM; Islam MR; Nagao J; Roy U; Momin S; Zendo T; Kohda D; Sonomoto K. The lantibiotic nukacin ISK-1 exists in an equilibrium between active and inactive lipid-II binding states. Commun. Biol 2018, 1 (150), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Wiedemann I; Böttiger T; Bonelli RR; Schneider T; Sahl H-G; Martínez B. Lipid II-based antimicrobial activity of the lantibiotic plantaricin C. Appl. Environ. Microbiol 2006, 72 (4), 2809–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Roy U; Islam MR; Nagao J; Iida H; Mahin A-A; Li M; Zendo T; Nakayama J; Sonomoto K. Bactericidal activity of nukacin ISK-1: an alternative mode of action. Biosci. Biotechnol. Biochem 2014, 78 (7), 1270–1273. [DOI] [PubMed] [Google Scholar]
- (126).Asaduzzaman SM; Nagao J; Iida H; Zendo T; Nakayama J; Sonomoto K. Nukacin ISK-1, a bacteriostatic lantibiotic. Antimicrob. Agents Chemother 2009, 53 (8), 3595–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Turner DL; Brennan L; Meyer HE; Lohaus C; Siethoff C; Costa HS; Gonzalez B; Santos H; Suárez JE Solution structure of plantaricin C, a novel lantibiotic. Eur. J. Biochem 1999, 264 (3), 833–839. [DOI] [PubMed] [Google Scholar]
- (128).van den Hooven HW; Lagerwerf FM; Heerma W; Haverkamp J; Piard JC; Hilbers CW; Siezen RJ; Kuipers OP; Rollema HS The structure of the lantibiotic lacticin 481 produced by Lactococcus lactis: location of the thioether bridges. FEBS Lett. 1996, 391 (3), 317–322. [DOI] [PubMed] [Google Scholar]
- (129).Sashihara T; Kimura H; Higuchi T; Adachi A; Matsusaki H; Sonomoto K; Ishizaki A. A novel lantibiotic, nukacin ISK-1, of Staphylococcus warneri ISK-1: cloning of the structural gene and identification of the structure. Biosci. Biotechnol. Biochem 2000, 64 (11), 2420–2428. [DOI] [PubMed] [Google Scholar]
- (130).Asaduzzaman SM; Nagao J; Aso Y; Nakayama J; Sonomoto K. Lysine-oriented charges trigger the membrane binding and activity of nukacin ISK-1. Appl. Environ. Microbiol 2006, 72 (9), 6012–6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Islam MR; Shioya K; Nagao J; Nishie M; Jikuya H; Zendo T; Nakayama J; Sonomoto K. Evaluation of essential and variable residues of nukacin ISK-1 by NNK scanning. Mol. Microbiol 2009, 72 (6), 1438–1447. [DOI] [PubMed] [Google Scholar]
- (132).Levengood MR; Knerr PJ; Oman TJ; van der Donk WA In vitro mutasynthesis of lantibiotic analogues containing nonproteinogenic amino ccids. J. Am. Chem. Soc 2009, 131 (34), 12024–12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Oman TJ; Knerr PJ; Bindman NA; Velásquez JE; van der Donk WA An engineered lantibiotic synthetase that does not require a leader peptide on its substrate. J. Am. Chem. Soc 2012, 134 (16), 6952–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Kakkar N; Perez JG; Liu WR; Jewett MC; van der Donk WA Incorporation of nonproteinogenic amino acids in class I and II lantibiotics. ACS Chem. Biol 2018, 13 (4), 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Si T; Tian Q; Min Y; Zhang L; Sweedler JV; van der Donk WA; Zhao H. Rapid screening of lanthipeptide analogs via in-colony removal of leader peptides in Escherichia coli. J. Am. Chem. Soc 2018, 140 (38), 11884–11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Teng K; Zhang J; Zhang X; Ge X; Gao Y; Wang J; Lin Y; Zhong J. Identification of ligand specificity determinants in lantibiotic bovicin HJ50 and the receptor BovK, a multitransmembrane histidine kinase. J. Biol. Chem 2014, 289 (14), 9823–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Elsayed KM; Islam MR; Abdullah-Al-Mahin; Nagao J; Zendo T; Sonomoto K LiaRS reporter assay: A simple tool to identify lipid II binding moieties in lantibiotic nukacin ISK-1. J. Biosci. Bioeng 2017, 123 (3), 398–401. [DOI] [PubMed] [Google Scholar]
- (138).Bindman NA; Bobeica SC; Liu WR; van der Donk WA Facile removal of leader peptides from lanthipeptides by incorporation of a hydroxy acid. J. Am. Chem. Soc 2015, 137 (22), 6975–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Chatterjee C; Patton GC; Cooper L; Paul M; van der Donk WA Engineering dehydro amino acids and thioethers into peptides using lacticin 481 synthetase. Chem. Biol 2006, 13 (10), 1109–1117. [DOI] [PubMed] [Google Scholar]
- (140).Bindman NA; van der Donk WA A general method for fluorescent labeling of the N-termini of lanthipeptides and its application to visualize their cellular localization. J. Am. Chem. Soc 2013, 135 (28), 10362–10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Mascher T; Zimmer SL; Smith T-A; Helmann JD Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob. Agents Chemother 2004, 48 (8), 2888–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Fujinami D; Motomura H; Oshima H; Mahin A-A; Elsayed KM; Zendo T; Sugita Y; Sonomoto K; Kohda D. Mosaic cooperativity in slow polypeptide topologicalisomerization revealed by residue-specific NMR thermodynamic analysis. J. Phys. Chem. Lett 2020, 11 (5), 1934–1939. [DOI] [PubMed] [Google Scholar]
- (143).Schmitz S; Hoffmann A; Szekat C; Rudd B; Bierbaum G. The lantibiotic mersacidin is an autoinducing peptide. Appl. Environ. Microbiol 2006, 72 (11), 7270–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Ni J; Teng K; Liu G; Qiao C; Huan L; Zhong J. Autoregulation of lantibiotic bovicin HJ50 biosynthesis by the BovK-BovR two-component signal transduction system in Streptococcus bovis HJ50. Appl. Environ. Microbiol 2011, 77 (2), 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (145).Wescombe PA; Upton M; Dierksen KP; Ragland NL; Sivabalan S; Wirawan RE; Inglis MA; Moore CJ; Walker GV; Chilcott CN; Jenkinson HF; Tagg JR Production of the lantibiotic salivaricin A and its variants by oral streptococci and use of a specific induction assay to detect their presence in human saliva. Appl. Environ. Microbiol 2006, 72 (2), 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Barbour A; Philip K; Muniandy S. Enhanced production, purification, characterization and mechanism of action of salivaricin 9 lantibiotic produced by Streptococcus salivarius NU10. PLoS One 2013, 8 (10), e77751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Aso Y; Sashihara T; Nagao J-I; Kanemasa Y; Koga H; Hashimoto T; Higuchi T; Adachi A; Nomiyama H; Ishizaki A; Nakayama J; Sonomoto K. Characterization of a gene cluster of Staphylococcus warneri ISK-1 encoding the biosynthesis of and immunity to the lantibiotic, nukacin ISK-1. Biosci. Biotechnol. Biochem 2004, 68 (8), 1663–1671. [DOI] [PubMed] [Google Scholar]
- (148).Van Tyne D; Martin MJ; Gilmore MS Structure, function, and biology of the Enterococcus faecalis cytolysin. Toxins 2013, 5 (5), 895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Ryan MP; Jack RW; Josten M; Sahl HG; Jung G; Ross RP; Hill C. Extensive post-translational modification, including serine to D-alanine conversion, in the two-component lantibiotic, lacticin 3147. J. Biol. Chem 1999, 274 (53), 37544–37550. [DOI] [PubMed] [Google Scholar]
- (150).McAuliffe O; Ryan MP; Ross RP; Hill C; Breeuwer P; Abee T. Lacticin 3147, a broad-spectrum bacteriocin which selectively dissipates the membrane potential. Appl. Environ. Microbiol 1998, 64 (2), 439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Suda S; Cotter PD; Hill C; Ross RP Lacticin 3147--biosynthesis, molecular analysis, immunity, bioengineering and applications. Curr. Protein. Pept. Sci 2012, 13 (3), 193–204. [DOI] [PubMed] [Google Scholar]
- (152).Bakhtiary A; Cochrane SA; Mercier P; McKay RT; Miskolzie M; Sit CS; Vederas JC Insights into the mechanism of action of the two-peptide lantibiotic lacticin 3147. J. Am. Chem. Soc 2017, 139 (49), 17803–17810. [DOI] [PubMed] [Google Scholar]
- (153).Morgan SM; O’Connor PM; Cotter PD; Ross RP; Hill C. Sequential actions of the two component peptides of the lantibiotic lacticin 3147 explain its antimicrobial activity at nanomolar concentrations. Antimicrob. Agents Chemother 2005, 49 (7), 2606–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Pattabiraman VR; McKinnie SMK; Vederas JC Solid-supported synthesis and biological evaluation of the lantibiotic peptide bis(desmethyl) lacticin 3147 A2. Angew. Chem. Int. Ed. Engl 2008, 47 (49), 9472–9475. [DOI] [PubMed] [Google Scholar]
- (155).Liu H; Pattabiraman VR; Vederas JC Synthesis and biological activity of oxa-lacticin A2, a lantibiotic analogue with sulfur replaced by oxygen. Org. Lett 2009, 11 (24), 5574–5577. [DOI] [PubMed] [Google Scholar]
- (156).Field D; Collins B; Cotter PD; Hill C; Ross RP A system for the random mutagenesis of the two-peptide lantibiotic lacticin 3147: analysis of mutants producing reduced antibacterial activities. J. Mol. Microbiol. Biotechnol 2007, 13 (4), 226–234. [DOI] [PubMed] [Google Scholar]
- (157).Deegan LH; Suda S; Lawton EM; Draper LA; Hugenholtz F; Peschel A; Hill C; Cotter PD; Ross RP Manipulation of charged residues within the two-peptide lantibiotic lacticin 3147. Microb. Biotechnol 2010, 3 (2), 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Cotter PD; Deegan LH; Lawton EM; Draper LA; O’Connor PM; Hill C; Ross RP Complete alanine scanning of the two-component lantibiotic lacticin 3147: generating a blueprint for rational drug design. Mol. Microbiol 2006, 62 (3), 735–747. [DOI] [PubMed] [Google Scholar]
- (159).Suda S; Westerbeek A; O’Connor PM; Ross RP; Hill C; Cotter PD Effect of bioengineering lacticin 3147 lanthionine bridges on specific activity and resistance to heat and proteases. Chem. Biol 2010, 17 (10), 1151–1160. [DOI] [PubMed] [Google Scholar]
- (160).Cotter PD; O’Connor PM; Draper LA; Lawton EM; Deegan LH; Hill C; Ross RP Posttranslational conversion of L-serines to D-alanines is vital for optimal production and activity of the lantibiotic lacticin 3147. Proc. Natl. Acad. Sci. U.S.A 2005, 102 (51), 18584–18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).O’Connor EB; Cotter PD; O’Connor P; O’Sullivan O; Tagg JR; Ross RP; Hill C. Relatedness between the two-component lantibiotics lacticin 3147 and staphylococcin C55 based on structure, genetics and biological activity. BMC Microbiol. 2007, 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).McClerren AL; Cooper LE; Quan C; Thomas PM; Kelleher NL; van der Donk WA Discovery and in vitro biosynthesis of haloduracin, a two-component lantibiotic. Proc. Natl. Acad. Sci. U.S.A 2006, 103 (46), 17243–17248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Lawton EM; Cotter PD; Hill C; Ross RP Identification of a novel two-peptide lantibiotic, Haloduracin, produced by the alkaliphile Bacillus halodurans C-125. FEMS Microbiol. Lett 2007, 267 (1), 64–71. [DOI] [PubMed] [Google Scholar]
- (164).Oman TJ; van der Donk WA Insights into the mode of action of the two-peptide lantibiotic haloduracin. ACS Chem. Biol 2009, 4 (10), 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Cooper LE; McClerren AL; Chary A; van der Donk WA Structure-activity relationship studies of the two-component lantibiotic haloduracin. Chem. Biol 2008, 15 (10), 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Booth MC; Bogie CP; Sahl HG; Siezen RJ; Hatter KL; Gilmore MS Structural analysis and proteolytic activation of Enterococcus faecalis cytolysin, a novel lantibiotic. Mol. Microbiol 1996, 21 (6), 1175–1184. [DOI] [PubMed] [Google Scholar]
- (167).Coburn PS; Gilmore MS The Enterococcus faecalis cytolysin: a novel toxin active against eukaryotic and prokaryotic cells. Cell. Microbiol 2003, 5 (10), 661–669. [DOI] [PubMed] [Google Scholar]
- (168).Haas W; Shepard BD; Gilmore MS Two-component regulator of Enterococcus faecalis cytolysin responds to quorum-sensing autoinduction. Nature 2002, 415 (6867), 84–87. [DOI] [PubMed] [Google Scholar]
- (169).Duan Y; Llorente C; Lang S; Brandl K; Chu H; Jiang L; White RC; Clarke TH; Nguyen K; Torralba M; Shao Y; Liu J; Hernandez-Morales A; Lessor L; Rahman IR; Miyamoto Y; Ly M; Gao B; Sun W; Kiesel R; Hutmacher F; Lee S; Ventura-Cots M; BosquesPadilla F; Verna EC; Abraldes JG; Brown RS; Vargas V; Altamirano J; Caballería J; Shawcross DL; Ho SB; Louvet A; Lucey MR; Mathurin P; Garcia-Tsao G; Bataller R; Tu XM; Eckmann L; van der Donk WA; Young R; Lawley TD; Stärkel P; Pride D; Fouts DE; Schnabl B. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575 (7783), 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Rahman IR; Sanchez A; Tang W; van der Donk WA Structure-activity relationships of the enterococcal cytolysin. ACS Infect. Dis 2021, 7 (8), 2445–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Tang W; van der Donk WA The sequence of the enterococcal cytolysin imparts unusual lanthionine stereochemistry. Nat. Chem. Biol 2013, 9 (3), 157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Mukherjee S; Huo L; Thibodeaux GN; van der Donk WA Synthesis and bioactivity of diastereomers of the virulence lanthipeptide cytolysin. Org. Lett 2016, 18 (23), 6188–6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Cox CR; Coburn PS; Gilmore MS Enterococcal cytolysin: a novel two component peptide system that serves as a bacterial defense against eukaryotic and prokaryotic cells. Curr. Protein Pept. Sci 2005, 6 (1), 77–84. [DOI] [PubMed] [Google Scholar]
- (174).Coburn PS; Pillar CM; Jett BD; Haas W; Gilmore MS Enterococcus faecalis senses target cells and in response expresses cytolysin. Science 2004, 306 (5705), 2270–2272. [DOI] [PubMed] [Google Scholar]
- (175).Bobeica SC; Zhu L; Acedo JZ; Tang W; Donk WA van der. Structural determinants of macrocyclization in substrate-controlled lanthipeptide biosynthetic pathways. Chem. Sci 2020, 11 (47), 12854–12870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Choung SY; Kobayashi T; Takemoto K; Ishitsuka H; Inoue K. Interaction of a cyclic peptide, Ro09–0198, with phosphatidylethanolamine in liposomal membranes. Biochim. Biophys. Acta 1988, 940 (2), 180–187. [DOI] [PubMed] [Google Scholar]
- (177).Wakamatsu K; Choung SY; Kobayashi T; Inoue K; Higashijima T; Miyazawa T. Complex formation of peptide antibiotic Ro09–0198 with lysophosphatidylethanolamine: 1H NMR analyses in dimethyl sulfoxide solution. Biochemistry 1990, 29 (1), 113–118. [DOI] [PubMed] [Google Scholar]
- (178).Makino A; Baba T; Fujimoto K; Iwamoto K; Yano Y; Terada N; Ohno S; Sato SB; Ohta A; Umeda M; Matsuzaki K; Kobayashi T. Cinnamycin (Ro 09–0198) promotes cell binding and toxicity by inducing transbilayer lipid movement. J. Biol. Chem 2003, 278 (5), 3204–3209. [DOI] [PubMed] [Google Scholar]
- (179).Hosoda K; Ohya M; Kohno T; Maeda T; Endo S; Wakamatsu K. Structure determination of an immunopotentiator peptide, cinnamycin, complexed with lysophosphatidylethanolamine by 1H-NMR1. J. Biochem 1996, 119 (2), 226–230. [DOI] [PubMed] [Google Scholar]
- (180).Ökesli A; Cooper LE; Fogle EJ; van der Donk WA Nine post-translational modifications during the biosynthesis of cinnamycin. J. Am. Chem. Soc 2011, 133 (34), 13753–13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Machaidze G; Ziegler A; Seelig J. Specific binding of Ro 09–0198 (cinnamycin) to phosphatidylethanolamine: a thermodynamic analysis. Biochemistry 2002, 41 (6), 1965–1971. [DOI] [PubMed] [Google Scholar]
- (182).Choung SY; Kobayashi T; Inoue J; Takemoto K; Ishitsuka H; Inoue K. Hemolytic activity of a cyclic peptide Ro09–0198 isolated from Streptoverticillium. Biochim. Biophys. Acta 1988, 940 (2), 171–179. [DOI] [PubMed] [Google Scholar]
- (183).Vestergaard M; Berglund NA; Hsu P-C; Song C; Koldsø H; Schiøtt B; Sansom MSP Structure and dynamics of cinnamycin–lipid complexes: mechanisms of selectivity for phosphatidylethanolamine lipids. ACS Omega 2019, 4 (20), 18889–18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Naruse N; Tenmyo O; Tomita K; Konishi M; Miyaki T; Kawaguchi H; Fukase K; Wakamiya T; Shiba T. Lanthiopeptin, a new peptide antibiotic. Production, isolation and properties of lanthiopeptin. J. Antibiot 1989, 42 (6), 837–845. [DOI] [PubMed] [Google Scholar]
- (185).Märki F; Hänni E; Fredenhagen A; van Oostrum J. Mode of action of the lanthionine-containing peptide antibiotics duramycin, duramycin B and C, and cinnamycin as indirect inhibitors of phospholipase A2. Biochem. Pharmacol 1991, 42 (10), 2027–2035. [DOI] [PubMed] [Google Scholar]
- (186).Oliynyk I; Varelogianni G; Roomans GM; Johannesson M. Effect of duramycin on chloride transport and intracellular calcium concentration in cystic fibrosis and non-cystic fibrosis epithelia. APMIS 2010, 118 (12), 982–990. [DOI] [PubMed] [Google Scholar]
- (187).Zebedin E; Koenig X; Radenkovic M; Pankevych H; Todt H; Freissmuth M; Hilber K. Effects of duramycin on cardiac voltage-gated ion channels. Naunyn-Schmied. Arch. Pharmacol 2008, 377 (1), 87–100. [DOI] [PubMed] [Google Scholar]
- (188).Sheth TR; Henderson RM; Hladky SB; Cuthbert AW Ion channel formation by duramycin. Biochimica et Biophysica Acta (BBA) - Biomembranes 1992, 1107 (1), 179–185. [DOI] [PubMed] [Google Scholar]
- (189).Cloutier MM; Guernsey L; Mattes P; Koeppen B. Duramycin enhances chloride secretion in airway epithelium. Am. J. Physiol 1990, 259 (3 Pt 1), C450–454. [DOI] [PubMed] [Google Scholar]
- (190).Cloutier MM; Guernsey L; Sha’afi RI Duramycin increases intracellular calcium in airway epithelium. Membr. Biochem 1993, 10 (2), 107–118. [DOI] [PubMed] [Google Scholar]
- (191).Grasemann H; Stehling F; Brunar H; Widmann R; Laliberte TW; Molina L; Döring G; Ratjen F. Inhalation of Moli1901 in patients with cystic fibrosis. Chest 2007, 131 (5), 1461–1466. [DOI] [PubMed] [Google Scholar]
- (192).Oliynyk I; Varelogianni G; Roomans GM; Johannesson M. Effect of duramycin on chloride transport and intracellular calcium concentration in cystic fibrosis and non-cystic fibrosis epithelia. APMIS 2010, 118 (12), 982–990. [DOI] [PubMed] [Google Scholar]
- (193).Steiner I; Errhalt P; Kubesch K; Hubner M; Holy M; Bauer M; Müller M; Hinterberger S; Widmann R; Mascher D; Freissmuth M; Kneussl M. Pulmonary pharmacokinetics and safety of nebulized duramycin in healthy male volunteers. Naunyn Schmiedebergs Arch Pharmacol 2008, 378 (3), 323–333. [DOI] [PubMed] [Google Scholar]
- (194).Richard AS; Zhang A; Park S-J; Farzan M; Zong M; Choe H. Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proc. Natl. Acad. Sci. U.S.A 2015, 112 (47), 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Smith TE; Pond CD; Pierce E; Harmer ZP; Kwan J; Zachariah MM; Harper MK; Wyche TP; Matainaho TK; Bugni TS; Barrows LR; Ireland CM; Schmidt EW Accessing chemical diversity from the uncultivated symbionts of small marine animals. Nat. Chem. Biol 2018, 14 (2), 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Zhao M. Lantibiotics as probes for phosphatidylethanolamine. Amino Acids 2011, 41 (5), 1071–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Zhao M; Li Z; Bugenhagen S. 99mTc-labeled duramycin as a novel phosphatidylethanolamine-binding molecular probe. J. Nucl. Med 2008, 49 (8), 1345–1352. [DOI] [PubMed] [Google Scholar]
- (198).Emoto K; Toyama-Sorimachi N; Karasuyama H; Inoue K; Umeda M. Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp. Cell Res 1997, 232 (2), 430–434. [DOI] [PubMed] [Google Scholar]
- (199).Audi SH; Jacobs ER; Zhao M; Roerig DL; Haworth ST; Clough AV In vivo detection of hyperoxia-induced pulmonary endothelial cell death using (99m)Tc-duramycin. Nucl. Med. Biol 2015, 42 (1), 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Delvaeye T; Wyffels L; Deleye S; Lemeire K; Goncalves A; Decrock E; Staelens S; Leybaert L; Vandenabeele P; Krysko D. Noninvasive whole-body imaging of phosphatidylethanolamine as a cell death marker using (99m)Tc-duramycin during TNF-induced SIRS. J. Nucl. Med 2018, 59 (7), 1140–1145. [DOI] [PubMed] [Google Scholar]
- (201).Luo R; Niu L; Qiu F; Fang W; Fu T; Zhao M; Zhang YJ; Hua ZC; Li XF; Wang F. Monitoring apoptosis of breast cancer xenograft after paclitaxel treatment with 99mTc-Labeled duramycin SPECT/CT. Mol. Imaging 2016, 15, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Elvas F; Vangestel C; Rapic S; Verhaeghe J; Gray B; Pak K; Stroobants S; Staelens S; Wyffels L. Characterization of [(99m)Tc]duramycin as a SPECT imaging agent for early assessment of tumor apoptosis. Mol. Imaging Biol 2015, 17 (6), 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Elvas F; Boddaert J; Vangestel C; Pak K; Gray B; Kumar-Singh S; Staelens S; Stroobants S; Wyffels L. (99m)Tc-duramycin SPECT imaging of early tumor response to targeted therapy: a comparison with (18)F-FDG PET. J. Nucl. Med 2017, 58 (4), 665–670. [DOI] [PubMed] [Google Scholar]
- (204).Johnson SE; Li Z; Liu Y; Moulder JE; Zhao M. Whole-body imaging of high-dose ionizing irradiation-induced tissue injuries using 99mTc-duramycin. J. Nucl. Med 2013, 54 (8), 1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Medhora M; Haworth S; Liu Y; Narayanan J; Gao F; Zhao M; Audi S; Jacobs ER; Fish BL; Clough AV Biomarkers for radiation pneumonitis using noninvasive molecular imaging. J. Nucl. Med 2016, 57 (8), 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Liu Z; Larsen BT; Lerman LO; Gray BD; Barber C; Hedayat AF; Zhao M; Furenlid LR; Pak KY; Woolfenden JM Detection of atherosclerotic plaques in ApoE-deficient mice using (99m)Tc-duramycin. Nucl. Med. Biol 2016, 43 (8), 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Wang L; Wang F; Fang W; Johnson SE; Audi S; Zimmer M; Holly TA; Lee DC; Zhu B; Zhu H; Zhao M. The feasibility of imaging myocardial ischemic/reperfusion injury using (99m)Tc-labeled duramycin in a porcine model. Nucl. Med. Biol 2015, 42 (2), 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Wakamiya T; Ueki Y; Shiba T; Kido Y; Motoki Y. Structural determination of ancovenin, a peptide inhibitor of angiotensin I converting enzyme. BCSJ 1990, 63 (4), 1032–1038. [Google Scholar]
- (209).Kido Y; Hamakado T; Yoshida T; Anno M; Motoki Y; Wakamiya T; Shiba T. Isolation and characterization of ancovenin, a new inhibitor of angiotensin I converting enzyme, produced by actinomycetes. J. Antibiot 1983, 36 (10), 1295–1299. [DOI] [PubMed] [Google Scholar]
- (210).Meindl K; Schmiederer T; Schneider K; Reicke A; Butz D; Keller S; Gühring H; Vértesy L; Wink J; Hoffmann H; Brönstrup M; Sheldrick GM; Süssmuth RD Labyrinthopeptins: a new class of carbacyclic lantibiotics. Angew. Chem. Int. Ed 2010, 49 (6), 1151–1154. [DOI] [PubMed] [Google Scholar]
- (211).Prochnow H; Rox K; Birudukota NVS; Weichert L; Hotop S-K; Klahn P; Mohr K; Franz S; Banda DH; Blockus S; Schreiber J; Haid S; Oeyen M; Martinez JP; Süssmuth RD; Wink J; Meyerhans A; Goffinet C; Messerle M; Schulz TF; Kröger A; Schols D; Pietschmann T; Brönstrup M. Labyrinthopeptins exert broad-spectrum antiviral activity through lipid-binding-mediated virolysis. J. Virol 2020, 94 (2), e01471–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Blockus S; Sake SM; Wetzke M; Grethe C; Graalmann T; Pils M; Le Goffic R; Galloux M; Prochnow H; Rox K; Hüttel S; Rupcic Z; Wiegmann B; Dijkman R; Rameix-Welti M-A; Eléouët J-F; Duprex WP; Thiel V; Hansen G; Brönstrup M; Haid S; Pietschmann T. Labyrinthopeptins as virolytic inhibitors of respiratory syncytial virus cell entry. Antiviral Res. 2020, 177, 104774. [DOI] [PubMed] [Google Scholar]
- (213).Férir G; Petrova MI; Andrei G; Huskens D; Hoorelbeke B; Snoeck R; Vanderleyden J; Balzarini J; Bartoschek S; Brönstrup M; Süssmuth RD; Schols D. The lantibiotic peptide labyrinthopeptin A1 demonstrates broad anti-HIV and anti-HSV activity with potential for microbicidal applications. PLoS One 2013, 8 (5), e64010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Varella Coelho ML; de Souza Duarte AF; do Carmo de Freire Bastos M. Bacterial labionin-containing peptides and sactibiotics: unusual types of antimicrobial peptides with potential use in clinical settings (a review). Curr. Top. Med. Chem 2017, 17 (10), 1177–1198. [DOI] [PubMed] [Google Scholar]
- (215).Tocchetti A; Iorio M; Hamid Z; Armirotti A; Reggiani A; Donadio S. Understanding the mechanism of action of NAI-112, a lanthipeptide with potent antinociceptive activity. Molecules 2021, 26 (22), 6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Monciardini P; Iorio M; Maffioli S; Sosio M; Donadio S. Discovering new bioactive molecules from microbial sources. Microb. Biotechnol 2014, 7 (3), 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Liu W; Sun F; Hu Y. Genome mining-mediated discovery of a new avermipeptin analogue in Streptomyces actuosus ATCC 25421. ChemistryOpen 2018, 7 (7), 558–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Kodani S; Hudson ME; Durrant MC; Buttner MJ; Nodwell JR; Willey JM The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor. Proc. Natl. Acad. Sci. U.S.A 2004, 101 (31), 11448–11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Nguyen KT; Willey JM; Nguyen LD; Nguyen LT; Viollier PH; Thompson CJ A central regulator of morphological differentiation in the multicellular bacterium Streptomyces coelicolor. Mol. Microbiol 2002, 46 (5), 1223–1238. [DOI] [PubMed] [Google Scholar]
- (220).Willey J; Santamaria R; Guijarro J; Geistlich M; Losick R. Extracellular complementation of a developmental mutation implicates a small sporulation protein in aerial mycelium formation by S. coelicolor. Cell 1991, 65 (4), 641–650. [DOI] [PubMed] [Google Scholar]
- (221).Tillotson RD; Wösten HA; Richter M; Willey JM A surface active protein involved in aerial hyphae formation in the filamentous fungus Schizophillum commune restores the capacity of a bald mutant of the filamentous bacterium Streptomyces coelicolor to erect aerial structures. Mol. Microbiol 1998, 30 (3), 595–602. [DOI] [PubMed] [Google Scholar]
- (222).Gaskell AA; Giovinazzo JA; Fonte V; Willey JM Multi-tier regulation of the streptomycete morphogenetic peptide SapB. Mol. Microbiol 2012, 84 (3), 501–515. [DOI] [PubMed] [Google Scholar]
- (223).Ueda K; Oinuma K-I; Ikeda G; Hosono K; Ohnishi Y; Horinouchi S; Beppu T. AmfS, an extracellular peptidic morphogen in Streptomyces griseus. J. Bacteriol 2002, 184 (5), 1488–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Wang H; van der Donk WA Biosynthesis of the class III lantipeptide catenulipeptin. ACS Chem. Biol 2012, 7 (9), 1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Takano H; Matsui Y; Nomura J; Fujimoto M; Katsumata N; Koyama T; Mizuno I; Amano S; Shiratori-Takano H; Komatsu M; Ikeda H; Ueda K. High production of a class III lantipeptide AmfS in Streptomyces griseus. Biosci. Biotechnol. Biochem 2017, 81 (1), 153–164. [DOI] [PubMed] [Google Scholar]
- (226).Straight PD; Willey JM; Kolter R. Interactions between Streptomyces coelicolor and Bacillus subtilis: role of surfactants in raising aerial structures. J. Bacteriol 2006, 188 (13), 4918–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Zhang Z; van der Donk WA Nonribosomal peptide extension by a peptide amino-acyl tRNA ligase. J. Am. Chem. Soc 2019, 141 (50), 19625–19633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Perry NB; Blunt JW; McCombs JD; Munro MHG Discorhabdin C, a highly cytotoxic pigment from a sponge of the genus Latrunculia. J. Org. Chem 1986, 51 (26), 5476–5478. [Google Scholar]
- (229).Colosimo DA; MacMillan JB Ammosamides unveil novel biosynthetic machinery. Cell Chem. Biol 2016, 23 (12), 1444–1446. [DOI] [PubMed] [Google Scholar]
- (230).Jordan PA; Moore BS Biosynthetic pathway connects cryptic ribosomally synthesized posttranslationally modified peptide genes with pyrroloquinoline alkaloids. Cell Chem. Biol 2016, 23 (12), 1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Ting CP; Funk MA; Halaby SL; Zhang Z; Gonen T; Donk WA van der. Use of a scaffold peptide in the biosynthesis of amino acid–derived natural products. Science 2019, 365 (6450), 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Daniels PN; Lee H; Splain RA; Ting CP; Zhu L; Zhao X; Moore BS; van der Donk WA A biosynthetic pathway to aromatic amines that uses glycyl-tRNA as nitrogen donor. Nat. Chem 2022, 14 (1), 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Reimer D; Hughes CC Thiol-based probe for electrophilic natural products reveals that most of the ammosamides are artifacts. J. Nat. Prod 2017, 80 (1), 126–133. [DOI] [PubMed] [Google Scholar]
- (234).Hughes CC; MacMillan JB; Gaudêncio SP; Fenical W; La Clair JJ Ammosamides A and B target myosin. Angew. Chem. Int. Ed 2009, 48 (4), 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Hughes CC; MacMillan JB; Gaudêncio SP; Jensen PR; Fenical W. The ammosamides: structures of cell cycle modulators from a marine-derived Streptomyces species. Angew. Chem. Int. Ed 2009, 48 (4), 725–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Beuzer P; La Clair JJ; Cang H. Color-coded super-resolution small-molecule imaging. ChemBioChem 2016, 17 (11), 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Pan E; Oswald NW; Legako AG; Life JM; Posner BA; MacMillan JB Precursor-directed generation of amidine containing ammosamide analogs: ammosamides E-P. Chem. Sci 2013, 4 (1), 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Reddy PVN; Jensen KC; Mesecar AD; Fanwick PE; Cushman M. Design, synthesis, and biological evaluation of potent quinoline and prrroloquinoline ammosamide analogues as inhibitors of quinone reductase 2. J. Med. Chem 2012, 55 (1), 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Nagata H; Ochiai K; Aotani Y; Ando K; Yoshida M; Takahashi I; Tamaoki T. Lymphostin (LK6-A), a novel immunosuppressant from Streptomyces sp. KYI 1783: taxonomy of the producing organism, fermentation, isolation and biological activities. J. Antibiot 1997, 50 (7), 537–542. [DOI] [PubMed] [Google Scholar]
- (240).Nagata H; Yano H; Sasaki K; Sato S; Nakanishi S; Takahashi I; Tamaoki T. Inhibition of lymphocyte kinase Lck and phosphatidylinositol 3-kinase by a novel immunosuppressant, lymphostin. Biosci. Biotechnol. Biochem 2002, 66 (3), 501–507. [DOI] [PubMed] [Google Scholar]
- (241).Miyanaga A; Janso JE; McDonald L; He M; Liu H; Barbieri L; Eustáquio AS; Fielding EN; Carter GT; Jensen PR; Feng X; Leighton M; Koehn FE; Moore BS Discovery and assembly-line biosynthesis of the lymphostin pyrroloquinoline alkaloid family of mTOR inhibitors in Salinispora bacteria. J. Am. Chem. Soc 2011, 133 (34), 13311–13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Toyota M; Spencer D; Sawai-Toyota S; Jiaqi W; Zhang T; Koo AJ; Howe GA; Gilroy S. Glutamate triggers long-distance, calcium-based plant defense signaling. Science 2018, 361 (6407), 1112–1115. [DOI] [PubMed] [Google Scholar]
- (243).Arrebola E; Cazorla FM; Perez-García A; de Vicente A. Chemical and metabolic aspects of antimetabolite toxins produced by Pseudomonas syringae pathovars. Toxins 2011, 3 (9), 1089–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Ortiz-López FJ; Carretero-Molina D; Sánchez-Hidalgo M; Martín J; González I; Román-Hurtado F; Cruz M. de la García-Fernández S; Reyes F; Deisinger JP; Müller A; Schneider T; Genilloud O. Cacaoidin, first member of the new lanthidin RiPP family. Angew. Chem. Int. Ed. Engl 2020, 59 (31), 12654–12658. [DOI] [PubMed] [Google Scholar]
- (245).Kloosterman AM; Cimermancic P; Elsayed SS; Du C; Hadjithomas M; Donia MS; Fischbach MA; van Wezel GP; Medema MH Expansion of RiPP biosynthetic space through integration of pan-genomics and machine learning uncovers a novel class of lanthipeptides. PLoS Biol. 2020, 18 (12), e3001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Xu M; Zhang F; Cheng Z; Bashiri G; Wang J; Hong J; Wang Y; Xu L; Chen X; Huang S-X; Lin S; Deng Z; Tao M. Functional genome mining reveals a class V lanthipeptide containing a D-amino acid introduced by an F420 H2 -dependent reductase. Angew. Chem. Int. Ed. Engl 2020, 59 (41), 18029–18035. [DOI] [PubMed] [Google Scholar]
- (247).Román-Hurtado F; Sánchez-Hidalgo M; Martín J; Ortiz-López FJ; Genilloud O. Biosynthesis and heterologous expression of cacaoidin, the first member of the lanthidin family of RiPPs. Antibiotics 2021, 10 (4), 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Wiebach V; Mainz A; Siegert M-AJ; Jungmann NA; Lesquame G; Tirat S; Dreux-Zigha A; Aszodi J; Le Beller D; Süssmuth RD The anti-staphylococcal lipolanthines are ribosomally synthesized lipopeptides. Nat Chem Biol 2018, 14 (7), 652–654. [DOI] [PubMed] [Google Scholar]
- (249).Kozakai R; Ono T; Hoshino S; Takahashi H; Katsuyama Y; Sugai Y; Ozaki T; Teramoto K; Teramoto K; Tanaka K; Abe I; Asamizu S; Onaka H. Acyltransferase that catalyses the condensation of polyketide and peptide moieties of goadvionin hybrid lipopeptides. Nat. Chem 2020, 12 (9), 869–877. [DOI] [PubMed] [Google Scholar]
- (250).Burkhart BJ; Schwalen CJ; Mann G; Naismith JH; Mitchell DA YcaO-dependent posttranslational amide activation: biosynthesis, structure, and function. Chem. Rev 2017, 117 (8), 5389–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Dunbar KL; Melby JO; Mitchell DA YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nat Chem Biol 2012, 8 (6), 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Todd EW The differentiation of two distinct serological varieties of streptolysin, streptolysin O and streptolysin S. J. Pathol. Bacteriol 1938, 47 (3), 423–445. [Google Scholar]
- (253).Bernheimer AW; Schwartz LL Lysosomal disruption by bacterial toxins. J. Bacteriol 1964, 87 (5), 1100–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Keiser H; Weissmann G; Bernheimer AW Studies on lysosomes: IV. Solubilization of enzymes during mitochondrial swelling and disruption of lysosomes by streptolysin S and other hemolytic agents. J. Cell. Biol 1964, 22 (1), 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Bernheimer AW; Schwartz LL Effects of staphylococcal and other bacterial toxins on platelets in vitro. J. Pathol. Bacteriol 1965, 89 (1), 209–223. [DOI] [PubMed] [Google Scholar]
- (256).Bernheimer AW Disruption of wall-less bacteria by streptococcal and staphylococcal toxins. J. Bacteriol 1966, 91 (5), 1677–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Hryniewicz W; Pryjma J. Effect of streptolysin S on human and mouse T and B lymphocytes. Infect. Immun 1977, 16 (3), 730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Bangham AD; Standish MM; Weissmann G. The action of steroids and streptolysin S on the permeability of phospholipid structures to cations. J. Mol. Biol 1965, 13 (1), 253–259. [DOI] [PubMed] [Google Scholar]
- (259).Buckingham L; Duncan JL Approximate dimensions of membrane lesions produced by streptolysin S and streptolysin O. Biochim. Biophys. Acta Biomembr 1983, 729 (1), 115–122. [DOI] [PubMed] [Google Scholar]
- (260).Carr A; Sledjeski DD; Podbielski A; Boyle MDP; Kreikemeyer B. Similarities between complement-mediated and streptolysin S-mediated hemolysis. J. Biol. Chem 2001, 276 (45), 41790–41796. [DOI] [PubMed] [Google Scholar]
- (261).Betschel SD; Borgia SM; Barg NL; Low DE; Azavedo JCSD Reduced virulence of group A streptococcal Tn916 mutants that do not produce streptolysin S. Infect. Immun 1998, 66 (4), 1671–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Humar D; Datta V; Bast DJ; Beall B; De Azavedo JC; Nizet V. Streptolysin S and necrotising infections produced by group G streptococcus. The Lancet 2002, 359 (9301), 124–129. [DOI] [PubMed] [Google Scholar]
- (263).Goldmann O; Sastalla I; Wos-Oxley M; Rohde M; Medina E. Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell. Microbiol 2009, 11 (1), 138–155. [DOI] [PubMed] [Google Scholar]
- (264).Sumitomo T; Nakata M; Higashino M; Jin Y; Terao Y; Fujinaga Y; Kawabata S. Streptolysin S contributes to group A streptococcal translocation across an epithelial barrier. J. Biol. Chem 2011, 286 (4), 2750–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Higashi DL; Biais N; Donahue DL; Mayfield JA; Tessier CR; Rodriguez K; Ashfeld BL; Luchetti J; Ploplis VA; Castellino FJ; Lee SW Activation of band 3 mediates group A Streptococcus streptolysin S-based beta-haemolysis. Nat. Microbiol 2016, 1 (2), 15004. [DOI] [PubMed] [Google Scholar]
- (266).Datta V; Myskowski SM; Kwinn LA; Chiem DN; Varki N; Kansal RG; Kotb M; Nizet V. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol. Microbiol 2005, 56 (3), 681–695. [DOI] [PubMed] [Google Scholar]
- (267).Mitchell DA; Lee SW; Pence MA; Markley AL; Limm JD; Nizet V; Dixon JE Structural and functional dissection of the heterocyclic peptide cytotoxin streptolysin S. J. Biol. Chem 2009, 284 (19), 13004–13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Molloy EM; Casjens SR; Cox CL; Maxson T; Ethridge NA; Margos G; Fingerle V; Mitchell DA Identification of the minimal cytolytic unit for streptolysin S and an expansion of the toxin family. BMC Microbiol. 2015, 15, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Lee SW; Mitchell DA; Markley AL; Hensler ME; Gonzalez D; Wohlrab A; Dorrestein PC; Nizet V; Dixon JE Discovery of a widely distributed toxin biosynthetic gene cluster. Proc. Natl. Acad. Sci. U.S.A 2008, 105 (15), 5879–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Gonzalez DJ; Lee SW; Hensler ME; Markley AL; Dahesh S; Mitchell DA; Bandeira N; Nizet V; Dixon JE; Dorrestein PC Clostridiolysin S, a post-translationally modified biotoxin from Clostridium botulinum. J. Biol. Chem 2010, 285 (36), 28220–28228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Cotter PD; Draper LA; Lawton EM; Daly KM; Groeger DS; Casey PG; Ross RP; Hill C. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 2008, 4 (9), e1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Quereda JJ; Dussurget O; Nahori M-A; Ghozlane A; Volant S; Dillies M-A; Regnault B; Kennedy S; Mondot S; Villoing B; Cossart P; Pizarro-Cerda J. Bacteriocin from epidemic Listeria strains alters the host intestinal microbiota to favor infection. Proc. Natl. Acad. Sci. U.S.A 2016, 113 (20), 5706–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Meza-Torres J; Lelek M; Quereda JJ; Sachse M; Manina G; Ershov D; Tinevez J-Y; Radoshevich L; Maudet C; Chaze T; Giai Gianetto Q; Matondo M; Lecuit M; Martin-Verstraete I; Zimmer C; Bierne H; Dussurget O; Cossart P; Pizarro-Cerdá J. Listeriolysin S: A bacteriocin from Listeria monocytogenes that induces membrane permeabilization in a contact-dependent manner. Proc. Natl. Acad. Sci. U.S.A 2021, 118 (40), e2108155118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Scholz R; Molohon KJ; Nachtigall J; Vater J; Markley AL; Süssmuth RD; Mitchell DA; Borriss R. Plantazolicin, a novel microcin B17/streptolysin S-like natural product from Bacillus amyloliquefaciens FZB42. J. Bacteriol 2011, 193 (1), 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Kalyon B; Helaly SE; Scholz R; Nachtigall J; Vater J; Borriss R; Süssmuth RD Plantazolicin A and B: structure elucidation of ribosomally synthesized thiazole/oxazole peptides from Bacillus amyloliquefaciens FZB42. Org. Lett 2011, 13 (12), 2996–2999. [DOI] [PubMed] [Google Scholar]
- (276).Molohon KJ; Melby JO; Lee J; Evans BS; Dunbar KL; Bumpus SB; Kelleher NL; Mitchell DA Structure determination and interception of biosynthetic intermediates for the plantazolicin class of highly discriminating antibiotics. ACS Chem. Biol 2011, 6 (12), 1307–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Molohon KJ; Blair PM; Park S; Doroghazi JR; Maxson T; Hershfield JR; Flatt KM; Schroeder NE; Ha T; Mitchell DA Plantazolicin Is an ultranarrow-spectrum antibiotic that targets the Bacillus anthracis membrane. ACS Infect. Dis 2016, 2 (3), 207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Hao Y; Blair PM; Sharma A; Mitchell DA; Nair SK Insights into methyltransferase specificity and bioactivity of derivatives of the antibiotic plantazolicin. ACS Chem. Biol 2015, 10 (5), 1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Albanesi D; Mansilla MC; de Mendoza D. The membrane fluidity sensor DesK of Bacillus subtilis controls the signal decay of Its cognate response regulator. J. Bacteriol 2004, 186 (9), 2655–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Sohlenkamp C; Geiger O. Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiol. Rev 2016, 40 (1), 133–159. [DOI] [PubMed] [Google Scholar]
- (281).Herrero M; Moreno F. Microcin B17 blocks DNA replication and induces the SOS system in Escherichia coli. J. Gen. Microbiol 1986, 132 (2), 393–402. [DOI] [PubMed] [Google Scholar]
- (282).Heddle JG; Blance SJ; Zamble DB; Hollfelder F; Miller DA; Wentzell LM; Walsh CT; Maxwell A. The antibiotic microcin B17 is a DNA gyrase poison: characterisation of the mode of inhibition. J. Mol. Biol 2001, 307 (5), 1223–1234. [DOI] [PubMed] [Google Scholar]
- (283).Pierrat OA; Maxwell A. The action of the bacterial toxin microcin B17. Insight into the cleavage-religation reaction of DNA gyrase. J. Biol. Chem 2003, 278 (37), 35016–35023. [DOI] [PubMed] [Google Scholar]
- (284).Lavina M; Pugsley AP; Moreno F. Identification, mapping, cloning and characterization of a gene (sbmA) required for microcin B17 action on Escherichia coli K12. J. Gen. Microbiol 1986, 132, 1685–1693. [DOI] [PubMed] [Google Scholar]
- (285).Yorgey P; Lee J; Kördel J; Vivas E; Warner P; Jebaratnam D; Kolter R. Posttranslational modifications in microcin B17 define an additional class of DNA gyrase inhibitor. Proc. Natl. Acad. Sci. U.S.A 1994, 91 (10), 4519–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Salomón RA; Farías RN The peptide antibiotic microcin 25 is imported through the TonB pathway and the SbmA protein. J. Bacteriol 1995, 177 (11), 3323–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Mattiuzzo M; Bandiera A; Gennaro R; Benincasa M; Pacor S; Antcheva N; Scocchi M. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol 2007, 66 (1), 151–163. [DOI] [PubMed] [Google Scholar]
- (288).Pränting M; Negrea A; Rhen M; Andersson DI Mechanism and fitness costs of PR-39 resistance in Salmonella enterica Serovar Typhimurium LT2. Antimicrob. Agents Chemother 2008, 52 (8), 2734–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Puckett SE; Reese KA; Mitev GM; Mullen V; Johnson RC; Pomraning KR; Mellbye BL; Tilley LD; Iversen PL; Freitag M; Geller BL Bacterial resistance to antisense peptide phosphorodiamidate morpholino oligomers. Antimicrob. Agents Chemother 2012, 56 (12), 6147–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Ghosal A; Vitali A; Stach JEM; Nielsen PE Role of SbmA in the uptake of peptide nucleic acid (PNA)-peptide conjugates in E. coli. ACS Chem. Biol 2013, 8 (2), 360–367. [DOI] [PubMed] [Google Scholar]
- (291).Roy RS; Kelleher NL; Milne JC; Walsh CT In vivo processing and antibiotic activity of microcin B17 analogs with varying ring content and altered bisheterocyclic sites. Chem. Biol 1999, 6 (5), 305–318. [DOI] [PubMed] [Google Scholar]
- (292).Collin F; Thompson RE; Jolliffe KA; Payne RJ; Maxwell A. Fragments of the bacterial toxin microcin B17 as gyrase poisons. PLoS One 2013, 8 (4), e61459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Zamble DB; Miller DA; Heddle JG; Maxwell A; Walsh CT; Hollfelder F. In vitro characterization of DNA gyrase inhibition by microcin B17 analogs with altered bisheterocyclic sites. Proc. Natl. Acad. Sci. U.S.A 2001, 98 (14), 7712–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Shkundina I; Serebryakova M; Severinov K. The C-terminal part of microcin B is crucial for DNA gyrase inhibition and antibiotic uptake by sensitive cells. J. Bacteriol 2014, 196 (9), 1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Metelev M; Osterman IA; Ghilarov D; Khabibullina NF; Yakimov A; Shabalin K; Utkina I; Travin DY; Komarova ES; Serebryakova M; Artamonova T; Khodorkovskii M; Konevega AL; Sergiev PV; Severinov K; Polikanov YS Klebsazolicin inhibits 70S ribosome by obstructing the peptide exit tunnel. Nat. Chem. Biol 2017, 13 (10), 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Travin DY; Watson ZL; Metelev M; Ward FR; Osterman IA; Khven IM; Khabibullina NF; Serebryakova M; Mergaert P; Polikanov YS; Cate JHD; Severinov K. Structure of ribosome-bound azole-modified peptide phazolicin rationalizes its species-specific mode of bacterial translation inhibition. Nat. Comm 2019, 10 (1), 4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Onaka H; Tabata H; Igarashi Y; Sato Y; Furumai T. Goadsporin, a chemical substance which promotes secondary metabolism and morphogenesis in Streptomycetes I. Purification and characterization. J. Antibiot 2001, 54 (12), 1036–1044. [DOI] [PubMed] [Google Scholar]
- (298).Igarashi Y; Kan Y; Fujii K; Fujita T; Harada K-I; Naoki H; Tabata H; Onaka H; Furumai T. Goadsporin, a chemical substance which promotes secondary metabolism and morphogenesis in Streptomycetes II. Structure determination. J. Antibiot 2001, 54 (12), 1045–1053. [DOI] [PubMed] [Google Scholar]
- (299).Fujii K; Yahashi Y; Nakano T; Imanishi S; Baldia SF; Harada K. Simultaneous detection and determination of the absolute configuration of thiazole-containing amino acids in a peptide. Tetrahedron 2002, 58 (34), 6873–6879. [Google Scholar]
- (300).Onaka H; Nakaho M; Hayashi K; Igarashi Y; Furumai T. 2005. Cloning and characterization of the goadsporin biosynthetic gene cluster from Streptomyces sp.TP-A0584. Microbiology 2005, 151 (12), 3923–3933. [DOI] [PubMed] [Google Scholar]
- (301).Onaka H. Biosynthesis of heterocyclic antibiotics in actinomycetes and an approach to synthesize the natural compounds. Actinomycetologica 2006, 20 (2), 62–71. [Google Scholar]
- (302).Onaka H. Biosynthesis of indolocarbazole and goadsporin, two different heterocyclic antibiotics produced by actinomycetes. Biosci. Biotechnol. Biochem 2009, 73 (10), 2149–2155. [DOI] [PubMed] [Google Scholar]
- (303).Park S-K; Jiang F; Dalbey RE; Phillips GJ Functional analysis of the signal recognition particle in Escherichia coli by characterization of a temperature-sensitive ffh mutant. J. Bacteriol 2002, 184 (10), 2642–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Ozaki T; Kurokawa Y; Hayashi S; Oku N; Asamizu S; Igarashi Y; Onaka H. Insights into the biosynthesis of dehydroalanines in goadsporin. ChemBioChem 2016, 17 (3), 218–223. [DOI] [PubMed] [Google Scholar]
- (305).Schwalen CJ; Hudson GA; Kille B; Mitchell DA Bioinformatic expansion and discovery of thiopeptide antibiotics. J. Am. Chem. Soc 2018, 140 (30), 9494–9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Hudson GA; Burkhart BJ; DiCaprio AJ; Schwalen CJ; Kille B; Pogorelov TV; Mitchell DA Bioinformatic mapping of radical S-adenosylmethionine-dependent ribosomally synthesized and post-translationally modified peptides identifies new Cα, Cβ, and Cγ-linked thioether-containing peptides. J. Am. Chem. Soc 2019, 141 (20), 8228–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Dutcher JD; Vandeputte J. Thiostrepton, a new antibiotic. II. Isolation and chemical characterization. Antibiot. Annu 1955, 3, 560–561. [PubMed] [Google Scholar]
- (308).Pestka S; Bodley JW The thiostrepton group of antibiotics. In Mechanism of Action of Antimicrobial and Antitumor Agents; Corcoran JW, Hahn FE, Snell JF, Arora KL, Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 1975; pp 551–573. [Google Scholar]
- (309).Weisblum B; Demohn V. Inhibition by thiostrepton of the formation of a ribosome-bound guanine nucleotide complex. FEBS Letters 1970, 11 (3), 149–152. [DOI] [PubMed] [Google Scholar]
- (310).Weisblum B; Demohn V. Thiostrepton, an inhibitor of 50S ribosome subunit function. J. Bacteriol 1970, 101 (3), 1073–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Pestka S. Thiostrepton: A ribosomal inhibitor of translocation. Biochem. Biophys. Res. Commun 1970, 40 (3), 667–674. [DOI] [PubMed] [Google Scholar]
- (312).Sopori ML; Lengyel P. Components of the 50S ribosomal subunit involved in GTP cleavage. Biochem. Biophys. Res. Commun 1972, 46 (1), 238–244. [DOI] [PubMed] [Google Scholar]
- (313).Cundliffe E. The mode of action of thiostrepton in vivo. Biochem. Biophys. Res. Commun 1971, 44 (4), 912–917. [DOI] [PubMed] [Google Scholar]
- (314).Modolell JM; Cabrer B; Parmeggiani A; Azquez DV Inhibition by siomycin and thiostrepton of both aminoacyl-tRNA and factor G binding to ribosomes. Proc. Natl. Acad. Sci. U.S.A 1971, 68 (8), 1796–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Bodley JW; Lin L; Highland JH Studies on translocation. VI. Thiostrepton prevents the formation of a ribosome-G factor-guanine nucleotide complex. Biochem. Biophys. Res. Commun 1970, 41, 1406–1411. [DOI] [PubMed] [Google Scholar]
- (316).Highland JH; Lin L; Bodley JW Translocation. VIII. Protection of ribosomes from thiostrepton inactivation by the binding of G factor and guanosine diphosphate. Biochemistry 1971, 10 (24), 4404–4409. [DOI] [PubMed] [Google Scholar]
- (317).Pestka S; Hintikka H. Studies on the formation of ribonucleic acid-ribosome complexes XVI. Effect of ribosomal translocation inhibitors on polyribosomes. J. Biol. Chem 1971, 246 (24), 7723–7730. [PubMed] [Google Scholar]
- (318).Cundliffe E; Thompson J. Ribose methylation and resistance to thiostrepton. Nature 1979, 278, 859–861. [DOI] [PubMed] [Google Scholar]
- (319).Thompson J; Cundliffe E. Resistance to thiostrepton, siomycin, and sporangiomycin in actinomycetes that produce them. J. Bacteriol 1980, 142 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Pestka S; Weiss D; Vince R; Wienen B; Stöffler G; Smith I. Thiostrepton-resistant mutants of Bacillus subtilis: localization of resistance to the 50S subunit. Molec. Gen. Genet 1976, 144 (3), 235–241. [DOI] [PubMed] [Google Scholar]
- (321).Baumann S; Schoof S; Harkal SD; Arndt H-D Mapping the binding site of thiopeptide antibiotics by proximity-induced covalent capture. J. Am. Chem. Soc 2008, 130 (17), 5664–5666. [DOI] [PubMed] [Google Scholar]
- (322).Wilson DN; Nierhaus KH Ribosomal proteins in the spotlight. Crit. Rev. Biochem. Mol. Biol 2005, 40 (5), 243–267. [DOI] [PubMed] [Google Scholar]
- (323).Highland JH; Howard GA; Ochsner E; Hasenbank R; Gordon J; Stöffler G. Identification of a ribosomal protein necessary for thiostrepton binding to Escherichia coli ribosomes. J. Biol. Chem 1975, 250 (3), 1141–1145. [PubMed] [Google Scholar]
- (324).Smith I; Weiss D; Pestka S. A micrococcin-resistant mutant of Bacillus subtilis: localization of resistance to the 50S subunit. Mol. Gen. Genet 1976, 144 (3), 231–233. [DOI] [PubMed] [Google Scholar]
- (325).Cundliffe E; Dixon P; Stark M; Stöffler G; Ehrlich R; Stöffler-Meilicke M; Cannon M. Ribosomes in thiostrepton-resistant mutants of Bacillus megaterium lacking a single 50S subunit protein. J. Mol. Biol 1979, 132 (2), 235–252. [DOI] [PubMed] [Google Scholar]
- (326).Wienen B; Ehrlich R; Stöffler-Meilicke M; Stöffler G; Smith I; Weiss D; Vince R; Pestka S. Ribosomal protein alterations in thiostrepton- and micrococcin-resistant mutants of Bacillus subtilis. J. Biol. Chem 1979, 254 (16), 8031–8041. [PubMed] [Google Scholar]
- (327).Thompson J; Schmidt F; Cundliffe E. Site of action of a ribosomal RNA methylase conferring resistance to thiostrepton. J. Biol. Chem 1982, 257 (14), 7915–7917. [PubMed] [Google Scholar]
- (328).Hummel H; Böck A. Thiostrepton resistance mutations in the gene for 23S ribosomal RNA of Halobacteria. Biochimie 1987, 69 (8), 857–861. [DOI] [PubMed] [Google Scholar]
- (329).Thompson J; Cundliffe E; Dahlberg AE Site-directed mutagenesis of Escherichia coli 23S ribosomal RNA at position 1067 within the GTP hydrolysis centre. J. Mol. Biol 1988, 203 (2), 457–465. [DOI] [PubMed] [Google Scholar]
- (330).Egebjerg J; Douthwaite S; Garrett RA Antibiotic interactions at the GTPase-associated centre within Escherichia coli 23S rRNA. EMBO J. 1989, 8 (2), 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Ryan PC; Lu M; Draper DE Recognition of the highly conserved GTPase center of 23S ribosomal RNA by ribosomal protein L11 and the antibiotic thiostrepton. J. Mol. Biol 1991, 221 (4), 1257–1268. [DOI] [PubMed] [Google Scholar]
- (332).Thompson J; Cundliffe E; Stark M. Binding of thiostrepton to a complex of 23S rRNA with ribosomal protein L11. Eur. J. Biochem 1979, 98 (1), 261–265. [DOI] [PubMed] [Google Scholar]
- (333).Thompson J; Cundliffe E. The binding of thiostrepton to 23S ribosomal RNA. Biochimie 1991, 73 (7), 1131–1135. [DOI] [PubMed] [Google Scholar]
- (334).Egebjerg J; Douthwaite SR; Liljas A; Garrett RA Characterization of the binding sites of protein L11 and the L10.(L12)4 pentameric complex in the GTPase domain of 23S ribosomal RNA from Escherichia coli. J. Mol. Biol 1990, 213 (2), 275–288. [DOI] [PubMed] [Google Scholar]
- (335).Rosendahl G; Douthwaite S. Ribosomal proteins L11 and L10.(L12)4 and the antibiotic thiostrepton interact with overlapping regions of the 23S rRNA backbone in the ribosomal GTPase centre. J. Mol. Biol 1993, 234 (4), 1013–1020. [DOI] [PubMed] [Google Scholar]
- (336).Rosendahl G; Douthwaite S. The antibiotics micrococcin and thiostrepton interact directly with 23S rRNA nucleotides 1067A and 1095A. Nucleic Acids Res. 1994, 22 (3), 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Xing Y; Draper DE Cooperative interactions of RNA and thiostrepton antibiotic with two domains of ribosomal protein L11. Biochemistry 1996, 35 (5), 1581–1588. [DOI] [PubMed] [Google Scholar]
- (338).Conn GL; Draper DE; Lattman EE; Gittis AG Crystal structure of a conserved ribosomal protein-RNA complex. Science 1999, 284 (5417), 1171–1174. [DOI] [PubMed] [Google Scholar]
- (339).Porse BT; Leviev I; Mankin AS; Garrett RA The antibiotic thiostrepton inhibits a functional transition within protein L11 at the ribosomal GTPase centre. J. Mol. Biol 1998, 276 (2), 391–404. [DOI] [PubMed] [Google Scholar]
- (340).Wimberly BT; Guymon R; McCutcheon JP; White SW; Ramakrishnan V. A detailed view of a ribosomal active site: the structure of the L11–RNA complex. Cell 1999, 97 (4), 491–502. [DOI] [PubMed] [Google Scholar]
- (341).Ilin S; Hoskins A; Ohlenschläger O; Jonker HRA; Schwalbe H; Wöhnert J. Domain reorientation and induced fit upon RNA Binding: solution structure and dynamics of ribosomal protein L11 from Thermotoga maritima. ChemBioChem 2005, 6 (9), 1611–1618. [DOI] [PubMed] [Google Scholar]
- (342).Schoof S; Baumann S; Ellinger B; Arndt H-D A fluorescent probe for the 70S-ribosomal GTPase-associated center. ChemBioChem 2009, 10 (2), 242–245. [DOI] [PubMed] [Google Scholar]
- (343).Porse BT; Cundliffe E; Garrett RA The antibiotic micrococcin acts on protein L11 at the ribosomal GTPase centre. J. Mol. Biol 1999, 287 (1), 33–45. [DOI] [PubMed] [Google Scholar]
- (344).Blyn LB; Risen LM; Griffey RH; Draper DE The RNA-binding domain of ribosomal protein L11 recognizes an rRNA tertiary structure stabilized by both thiostrepton and magnesium ion. Nucleic Acids Res. 2000, 28 (8), 1778–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Seo H-S; Kiel M; Pan D; Raj VS; Kaji A; Cooperman BS Kinetics and thermodynamics of RRF, EF-G, and thiostrepton interaction on the Escherichia coli ribosome. Biochemistry 2004, 43 (40), 12728–12740. [DOI] [PubMed] [Google Scholar]
- (346).Bausch SL; Poliakova E; Draper DE Interactions of the N-terminal domain of ribosomal protein L11 with thiostrepton and rRNA. J. Biol. Chem 2005, 280 (33), 29956–29963. [DOI] [PubMed] [Google Scholar]
- (347).Bowen WS; Dyke NV; Murgola EJ; Lodmell JS; Hill WE Interaction of thiostrepton and elongation factor-G with the ribosomal protein L11-binding domain. J. Biol. Chem 2005, 280 (4), 2934–2943. [DOI] [PubMed] [Google Scholar]
- (348).Gonzalez RL; Chu S; Puglisi JD Thiostrepton inhibition of tRNA delivery to the ribosome. RNA 2007, 13 (12), 2091–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Cameron DM; Thompson J; Gregory ST; March PE; Dahlberg AE Thiostrepton-resistant mutants of Thermus thermophilus. Nucleic Acids Res. 2004, 32 (10), 3220–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Jonker HRA; Ilin S; Grimm SK; Wöhnert J; Schwalbe H. L11 domain rearrangement upon binding to RNA and thiostrepton studied by NMR spectroscopy. Nucleic Acids Res. 2007, 35 (2), 441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Harms JM; Wilson DN; Schluenzen F; Connell SR; Stachelhaus T; Zaborowska Z; Spahn CMT; Fucini P. Translational regulation via L11: molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Mol. Cell 2008, 30 (1), 26–38. [DOI] [PubMed] [Google Scholar]
- (352).Lentzen G; Klinck R; Matassova N; Aboul-ela F; Murchie AIH Structural basis for contrasting activities of ribosome binding thiazole antibiotics. Chem. Biol 2003, 10 (8), 769–778. [DOI] [PubMed] [Google Scholar]
- (353).Naaktgeboren N; Roobol K; Gubbens J; Voorma HO The mode of action of thiostrepton in the initiation of protein synthesis. Eur. J. Biochem 1976, 70 (1), 39–47. [DOI] [PubMed] [Google Scholar]
- (354).Sarkar P; Stringer EA; Maitra U. Thiostrepton inhibition of initiation factor 1 activity in polypeptide chain initiation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A 1974, 71 (12), 4986–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Lockwood AH; Sarkar P; Maitra U; Brot N; Weissbach H. Effect of thiostrepton on polypeptide chain initiation in Escherichia coli. J. Biol. Chem 1974, 249 (18), 5831–5834. [PubMed] [Google Scholar]
- (356).Cameron DM; Thompson J; March PE; Dahlberg AE Initiation factor IF2, thiostrepton and micrococcin prevent the binding of elongation factor G to the Escherichia coli ribosome. J. Mol. Biol 2002, 319 (1), 27–35. [DOI] [PubMed] [Google Scholar]
- (357).Brandi L; Marzi S; Fabbretti A; Fleischer C; Hill WE; Gualerzi CO; Stephen Lodmell J. The translation initiation functions of IF2: targets for thiostrepton inhibition. J. Mol. Biol 2004, 335 (4), 881–894. [DOI] [PubMed] [Google Scholar]
- (358).Masuda T; Petrov AN; Iizuka R; Funatsu T; Puglisi JD; Uemura S. Initiation factor 2, tRNA, and 50S subunits cooperatively stabilize mRNAs on the ribosome during initiation. Proc. Natl. Acad. Sci. U.S.A 2012, 109 (13), 4881–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (359).McConkey GA; Rogers MJ; McCutchan TF Inhibition of Plasmodium falciparum protein synthesis. Targeting the plastid-like organelle with thiostrepton. J. Biol. Chem 1997, 272 (4), 2046–2049. [DOI] [PubMed] [Google Scholar]
- (360).Chaubey S; Kumar A; Singh D; Habib S. The apicoplast of Plasmodium falciparum is translationally active. Mol. Microbiol 2005, 56 (1), 81–89. [DOI] [PubMed] [Google Scholar]
- (361).Clough B; Strath M; Preiser P; Denny P; Wilson IR Thiostrepton binds to malarial plastid rRNA. FEBS Lett. 1997, 406 (1–2), 123–125. [DOI] [PubMed] [Google Scholar]
- (362).Goodman CD; Su V; McFadden GI The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol 2007, 152 (2), 181–191. [DOI] [PubMed] [Google Scholar]
- (363).Tarr SJ; Nisbet RER; Howe CJ Transcript-level responses of Plasmodium falciparum to thiostrepton. Mol. Biochem. Parasitol 2011, 179 (1), 37–41. [DOI] [PubMed] [Google Scholar]
- (364).Aminake MN; Schoof S; Sologub L; Leubner M; Kirschner M; Arndt H-D; Pradel G. Thiostrepton and derivatives exhibit antimalarial and gametocytocidal activity by dually targeting parasite proteasome and apicoplast. Antimicrob. Agents Chemother 2011, 55 (4), 1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Rogers MJ; Bukhman YV; McCutchan TF; Draper DE Interaction of thiostrepton with an RNA fragment derived from the plastid-encoded ribosomal RNA of the malaria parasite. RNA 1997, 3 (8), 815–820. [PMC free article] [PubMed] [Google Scholar]
- (366).Rogers MJ; Cundliffe E; McCutchan TF The antibiotic micrococcin Is a potent inhibitor of growth and protein synthesis in the malaria parasite. Antimicrob. Agents Chemother 1998, 42 (3), 715–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).AbouLaila M; Munkhjargal T; Sivakumar T; Ueno A; Nakano Y; Yokoyama M; Yoshinari T; Nagano D; Katayama K; El-Bahy N; Yokoyama N; Igarashi I. Apicoplast-targeting antibacterials inhibit the growth of Babesia parasites. Antimicrob. Agents Chemother 2012, 56 (6), 3196–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (368).Schoof S; Pradel G; Aminake MN; Ellinger B; Baumann S; Potowski M; Najajreh Y; Kirschner M; Arndt H-D Antiplasmodial thiostrepton derivatives: proteasome inhibitors with a dual mode of action. Angew. Chem. Int. Ed. Engl 2010, 49 (19), 3317–3321. [DOI] [PubMed] [Google Scholar]
- (369).Selva E; Beretta G; Montanini N; Saddler GS; Gastaldo L; Ferrari P; Lorenzetti R; Landini P; Ripamonti F; Goldstein BP; Berti M; Montanaro L; Denaro M. Antibiotic GE2270A: a novel inhibitor of bacterial protein synthesis. I. Isolation and characterization. J. Antibiot 1991, 44 (7), 693–701. [DOI] [PubMed] [Google Scholar]
- (370).Selva E; Ferrari P; Kurz M; Tavecchia P; Colombo L; Stella S; Restelli E; Goldstein BP; Ripamonti F; Denaro M. Components of the GE2270 complex produced by Planobispora rosea ATCC 53773. J. Antibiot 1995, 48 (9), 1039–1042. [DOI] [PubMed] [Google Scholar]
- (371).Anborgh PH; Parmeggiani A. New antibiotic that acts specifically on the GTP-bound form of elongation factor Tu. EMBO J. 1991, 10 (4), 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Anborgh PH; Parmeggiani A. Probing the reactivity of the GTP- and GDP-bound conformations of elongation factor Tu in complex with the antibiotic GE2270 A. J. Biol. Chem 1993, 268 (33), 24622–24628. [PubMed] [Google Scholar]
- (373).Stella S; Montanini N; Le Monnier F; Ferrari P; Colombo L; Marinelli F; Landini P; Ciciliato I; Goldstein BP; Selva E. Antibiotic GE37468 A: a new inhibitor of bacterial protein synthesis. I. Isolation and characterization. J. Antibiot 1995, 48 (8), 780–786. [DOI] [PubMed] [Google Scholar]
- (374).Morris RP; Leeds JA; Naegeli HU; Oberer L; Memmert K; Weber E; LaMarche MJ; Parker CN; Burrer N; Esterow S; Hein AE; Schmitt EK; Krastel P. Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J. Am. Chem. Soc 2009, 131 (16), 5946–5955. [DOI] [PubMed] [Google Scholar]
- (375).Shimanaka K; Kinoshita N; Iinuma H; Hamada M; Takeuchi T. Novel antibiotics, amythiamicins. I. Taxonomy, fermentation, isolation, physico-chemical properties, and antimicrobial activity. J. Antibiot 1994, 47 (6), 668–674. [DOI] [PubMed] [Google Scholar]
- (376).Heffron SE; Jurnak F. Structure of an EF-Tu complex with a thiazolyl peptide antibiotic determined at 2.35 Å resolution: atomic basis for GE2270A inhibition of EF-Tu,. Biochemistry 2000, 39 (1), 37–45. [DOI] [PubMed] [Google Scholar]
- (377).Parmeggiani A; Krab IM; Okamura S; Nielsen RC; Nyborg J; Nissen P. Structural basis of the action of pulvomycin and GE2270A on elongation factor Tu. Biochemistry 2006, 45 (22), 6846–6857. [DOI] [PubMed] [Google Scholar]
- (378).Young TS; Dorrestein PC; Walsh CT Codon randomization for rapid exploration of chemical space in thiopeptide antibiotic variants. Chem. Biol 2012, 19 (12), 1600–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Sosio M; Amati G; Cappellano C; Sarubbi E; Monti F; Donadio S. An elongation factor Tu (EF-Tu) resistant to the EF-Tu inhibitor GE2270 in the producing organism Planobispora rosea. Mol. Microbiol 1996, 22 (1), 43–51. [DOI] [PubMed] [Google Scholar]
- (380).Berchtold H; Reshetnikova L; Reiser COA; Schirmer NK; Sprinzl M; Hilgenfeld R. Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature 1993, 365 (6442), 126–132. [DOI] [PubMed] [Google Scholar]
- (381).Song H; Parsons MR; Rowsell S; Leonard G; Phillips SEV Crystal structure of intact elongation factor EF-Tu from Escherichia coli in GDP conformation at 2.05Å resolution. J. Mol. Biol 1999, 285 (3), 1245–1256. [DOI] [PubMed] [Google Scholar]
- (382).Nissen P; Kjeldgaard M; Thirup S. r.; Polekhina, G.; Reshetnikova, L.; Clark, B. F. C.; Nyborg, J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science 1995, 270 (5241), 1464–1472. [DOI] [PubMed] [Google Scholar]
- (383).Murakami T; Holt TG; Thompson CJ Thiostrepton-induced gene expression in Streptomyces lividans. J. Bacteriol 1989, 171 (3), 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (384).Holmes D. j.; Caso J. l.; Thompson C. j. Autogenous transcriptional activation of a thiostrepton-induced gene in Streptomyces lividans. EMBO J. 1993, 12 (8), 3183–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Yun BS; Hidaka T; Furihata K; Seto H. Microbial metabolites with tipA promoter inducing activity. II. Geninthiocin, a novel thiopeptide produced by Streptomyces sp. DD84. J. Antibiot 1994, 47 (9), 969–975. [DOI] [PubMed] [Google Scholar]
- (386).Yun B-S; Hidaka T; Furihata K; Seto H. Promothiocins A and B, novel thiopeptides with tipA promoter inducing activity produced by Streptomyces sp. SF2741. J. Antibiot 1994, 47 (4), 510–514. [DOI] [PubMed] [Google Scholar]
- (387).Yun B-S; Seto H. Promoinducin, a novel thiopeptide produced by Streptomyces sp. SF2741. Biosci. Biotechnol. Biochem 1995, 59 (5), 876–880. [DOI] [PubMed] [Google Scholar]
- (388).Chiu ML; Folcher M; Griffin P; Holt T; Klatt T; Thompson CJ Characterization of the covalent binding of thiostrepton to a thiostrepton-induced protein from Streptomyces lividans. Biochemistry 1996, 35 (7), 2332–2341. [DOI] [PubMed] [Google Scholar]
- (389).Chiu ML; Folcher M; Katoh T; Puglia AM; Vohradsky J; Yun B-S; Seto H; Thompson CJ Broad spectrum thiopeptide recognition specificity of the Streptomyces lividans TipAL protein and its role in regulating gene expression. J. Biol. Chem 1999, 274 (29), 20578–20586. [DOI] [PubMed] [Google Scholar]
- (390).Kahmann JD; Sass H-J; Allan MG; Seto H; Thompson CJ; Grzesiek S. Structural basis for antibiotic recognition by the TipA class of multidrug-resistance transcriptional regulators. EMBO J. 2003, 22 (8), 1824–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Habazettl J; Allan M; Jensen PR; Sass H-J; Thompson CJ; Grzesiek S. Structural basis and dynamics of multidrug recognition in a minimal bacterial multidrug resistance system. Proc. Natl. Acad. Sci. U.S.A 2014, 111 (51), E5498–E5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (392).Kim J; Lee W-H Cancer therapeutic agent comprising thiopeptide with multiple thiazole rings. WO 02/066046 A1.
- (393).Kalin TV; Wang I-C; Ackerson TJ; Major ML; Detrisac CJ; Kalinichenko VV; Lyubimov A; Costa RH Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006, 66 (3), 1712–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Myatt SS; Lam EW-F The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7 (11), 847–859. [DOI] [PubMed] [Google Scholar]
- (395).Bektas N; Haaf A. ten; Veeck J; Wild PJ; Lüscher-Firzlaff J; Hartmann A; Knüchel R; Dahl E. Tight correlation between expression of the forkhead transcription factor FOXM1 and HER2 in human breast cancer. BMC Cancer 2008, 8 (1), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Okabe H; Satoh S; Kato T; Kitahara O; Yanagawa R; Yamaoka Y; Tsunoda T; Furukawa Y; Nakamura Y. Genome-wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res. 2001, 61 (5), 2129–2137. [PubMed] [Google Scholar]
- (397).Garber ME; Troyanskaya OG; Schluens K; Petersen S; Thaesler Z; Pacyna-Gengelbach M; van de Rijn M; Rosen GD; Perou CM; Whyte RI; Altman RB; Brown PO; Botstein D; Petersen I. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl. Acad. Sci. U.S.A 2001, 98 (24), 13784–13789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (398).Kwok JM-M; Myatt SS; Marson CM; Coombes RC; Constantinidou D; Lam EW-F Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther 2008, 7 (7), 2022–2032. [DOI] [PubMed] [Google Scholar]
- (399).Radhakrishnan SK; Bhat UG; Hughes DE; Wang I-C; Costa RH; Gartel AL Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006, 66 (19), 9731–9735. [DOI] [PubMed] [Google Scholar]
- (400).Bhat UG; Zipfel PA; Tyler DS; Gartel AL Novel anticancer compounds induce apoptosis in melanoma cells. Cell Cycle 2008, 7 (12), 1851–1855. [DOI] [PubMed] [Google Scholar]
- (401).Halasi M; Zhao H; Dahari H; Bhat UG; Gonzalez EB; Lyubimo AV; Tonetti DA; Gartel AL Thiazole antibiotics against breast cancer. Cell Cycle 2010, 9 (6), 1214–1217. [DOI] [PubMed] [Google Scholar]
- (402).Major ML; Lepe R; Costa RH Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol. Cell. Biol 2004, 24 (7), 2649–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (403).Hegde NS; Sanders DA; Rodriguez R; Balasubramanian S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nature Chem. 2011, 3 (9), 725–731. [DOI] [PubMed] [Google Scholar]
- (404).Nicolaou KC; Zak M; Rahimipour S; Estrada AA; Lee SH; O’Brate A; Giannakakou P; Ghadiri MR Discovery of a biologically active thiostrepton fragment. J. Am. Chem. Soc 2005, 127 (43), 15042–15044. [DOI] [PubMed] [Google Scholar]
- (405).Pandit B; Bhat UG; Gartel AL Proteasome inhibitory activity of thiazole antibiotics. Cancer Biol. Ther 2011, 11 (1), 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Zhang L; Ging NC; Komoda T; Hanada T; Suzuki T; Watanabe K. Antibiotic susceptibility of mammalian mitochondrial translation. FEBS Lett. 2005, 579 (28), 6423–6427. [DOI] [PubMed] [Google Scholar]
- (407).Sayanjali B; Christensen GJM; Al-Zeer MA; Mollenkopf H-J; Meyer TF; Brüggemann H. Propionibacterium acnes inhibits FOXM1 and induces cell cycle alterations in human primary prostate cells. Intl. J. Med. Microbiol 2016, 306 (7), 517–528. [DOI] [PubMed] [Google Scholar]
- (408).Hashimoto M; Murakami T; Funahashi K; Tokunaga T; Nihei K; Okuno T; Kimura T; Naoki H; Himeno H. An RNA polymerase inhibitor, cyclothiazomycin B1, and its isomer. Bioorg. Med. Chem 2006, 14 (24), 8259–8270. [DOI] [PubMed] [Google Scholar]
- (409).Aoki M; Ohtsuka T; Yamada M; Ohba Y; Yoshizaki H; Yasuno H; Sano T; Watanabe J; Yokose K; Seto H. Cyclothiazomycin, a novel polythiazole-containing peptide with renininhibitory activity. J. Antibiot 1991, 44 (6), 582–588. [DOI] [PubMed] [Google Scholar]
- (410).Archin NM; Liberty AL; Kashuba AD; Choudhary SK; Kuruc JD; Crooks AM; Parker DC; Anderson EM; Kearney MF; Strain MC; Richman DD; Hudgens MG; Bosch RJ; Coffin JM; Eron JJ; Hazuda DJ; Margolis DM Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487 (7408), 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Peng W; Hong Z; Chen X; Gao H; Dai Z; Zhao J; Liu W; Li D; Deng K. Thiostrepton reactivates latent HIV-1 through the p-TEFb and NF-κB pathways mediated by heat shock response. Antimicrob. Agents Chemother 2020, 64 (5), e02328–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Hoch L; Henriques SF; Bruge C; Marsolier J; Benabides M; Bourg N; Tournois J; Mahé G; Morizur L; Jarrige M; Bigot A; Richard I; Nissan X. Identification of thiostrepton as a pharmacological approach to rescue misfolded alpha-sarcoglycan mutant proteins from degradation. Sci. Rep 2019, 9 (1), 6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Lai C-Y; Yeh D-W; Lu C-H; Liu Y-L; Huang L-R; Kao C-Y; Chen H-Y; Huang C-YF; Chang C-H; Luo Y; Xiang R; Chuang T-H Identification of thiostrepton as a novel inhibitor for psoriasis-like inflammation induced by TLR7–9. J. Immunol 2015, 195 (8), 3912–3921. [DOI] [PubMed] [Google Scholar]
- (414).Yang N; Zhou T-C; Lei X; Wang C; Yan M; Wang Z-F; Liu W; Wang J; Ming K-H; Wang B-C; Xu B-L; Liu Q. Inhibition of sonic hedgehog signaling pathway by thiazole antibiotic thiostrepton attenuates the CD44+/CD24-stem-like population and sphereforming capacity in triple-negative breast cancer. Cell. Physiol. Biochem 2016, 38 (3), 1157–1170. [DOI] [PubMed] [Google Scholar]
- (415).Tanaka T; Endo T; Shimazu A; Yoshida R; Suzuki Y; Otake N; Yonehara H. A new antibiotic, multhiomycin. J. Antibiot 1970, 23 (5), 231–237. [DOI] [PubMed] [Google Scholar]
- (416).Pucci MJ; Bronson JJ; Barrett JF; DenBleyker KL; Discotto LF; Fung-Tomc JC; Ueda Y. Antimicrobial evaluation of nocathiacins, a thiazole peptide class of antibiotics. Antimicrob. Agents Chemother 2004, 48 (10), 3697–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Haste NM; Thienphrapa W; Tran DN; Loesgen S; Sun P; Nam S-J; Jensen PR; Fenical W; Sakoulas G; Nizet V; Hensler ME Activity of the thiopeptide antibiotic nosiheptide against contemporary strains of methicillin-resistant Staphylococcus aureus. J. Antibiot 2012, 65 (12), 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).Shimamura H; Gouda H; Nagai K; Hirose T; Ichioka M; Furuya Y; Kobayashi Y; Hirono S; Sunazuka T; Ōmura S. Structure determination and total synthesis of bottromycin A2: a potent antibiotic against MRSA and VRE. Angew. Chem. Int. Ed. Engl 2009, 48 (5), 914–917. [DOI] [PubMed] [Google Scholar]
- (419).Waisvisz JM; van der Hoeven MG; van Peppen J; Zwennis WCM Bottromycin. I. A new sulfur-containing antibiotic. J. Am. Chem. Soc 1957, 79 (16), 4520–4521. [Google Scholar]
- (420).Tanaka N; Nishimura T; Nakamura S; Umezawa H. Biological studies on bottromycin A and its hydrazide. J. Antibiot 1966, 19 (4), 149–154. [PubMed] [Google Scholar]
- (421).Tanaka N; Nishimura T; Nakamura S; Umezawa H; Hayami T. Activity of bottromycin against Mycoplasma gallisepticum. J. Antibiot 1968, 21 (1), 75–76. [DOI] [PubMed] [Google Scholar]
- (422).Eisenman W; Minieri PP; Abbey A; Charlebois J; Moncrieff-Yeates M; Rigler NE A new sulfur-containing antibiotic, produced by a Streptomyces, active against bacteria and fungi. Antibiot. Chemother 1953, 3 (4), 385–392. [PubMed] [Google Scholar]
- (423).Nakamura S; Chikaike T; Karasawa K; Tanaka N; Yonehara H; Umezawa H. Isolation and characterization of bottromycins A and B. J. Antibiot 1965, 18, 47–52. [PubMed] [Google Scholar]
- (424).Pestka S. Studies on the formation of transfer ribonucleic acid-ribosome complexes: VIII. Survey of the effect of antibiotics on N-acetyl-phenylalanyl-puromycin formation: Possible mechanism of chloramphenicol action. Arch. Biochem. Biophys 1970, 136 (1), 80–88. [DOI] [PubMed] [Google Scholar]
- (425).Yarmolinsky MB; Haba GL Inhibition by puromycin of amino acid incorporation into protein. Proc. Natl. Acad. Sci. U.S.A 1959, 45 (12), 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Nathans D. Puromycin inhibition of protein synthesis: incorporation of puromycin into peptide chains. Proc. Natl. Acad. Sci. U.S.A 1964, 51 (4), 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Tanaka N; Lin Y-C; Okuyama A. Studies on translocation of F-met-tRNA and peptidyl-tRNA with antibiotics. Biochem. Biophys. Res. Commun 1971, 44 (2), 477–483. [DOI] [PubMed] [Google Scholar]
- (428).Lin Y-C; Kinoshita T; Tanaka N. Mechanism of protein synthesis inhibition by bottromycin A2: studies with puromycin. J. Antibiot 1968, 21 (8), 471–476. [DOI] [PubMed] [Google Scholar]
- (429).Kinoshita T; Tanaka N. On the site of action of bottromycin A2. J. Antibiot 1970, 23 (6), 311–312. [DOI] [PubMed] [Google Scholar]
- (430).Pestka S; Brot N. Studies on the formation of transfer ribonucleic acid-ribosome complexes XV. Effect of antibiotics on steps of bacterial protein synthesis: some new ribosomal inhibitors of translocation. J. Biol. Chem 1971, 246 (24), 7715–7722. [PubMed] [Google Scholar]
- (431).Pestka S. Studies on transfer ribonucleic acid-ribosome complexes XIX. Effect of antibiotics on peptidyl puromycin synthesis on polyribosomes from Escherichia coli. J. Biol. Chem 1972, 247 (14), 4669–4678. [PubMed] [Google Scholar]
- (432).Cundliffe E; McQuillen K. Bacterial protein synthesis: the effects of antibiotics. J. Mol. Biol 1967, 30 (1), 137–146. [DOI] [PubMed] [Google Scholar]
- (433).Otaka T; Kaji A. Mode of action of bottromycin A2: effect on peptide bond formation. FEBS Lett. 1981, 123, 173–176. [DOI] [PubMed] [Google Scholar]
- (434).Otaka T; Kaji A. Mode of action of bottromycin A2. Release of aminoacyl- or peptidyl-tRNA from ribosomes. J. Biol. Chem 1976, 251, 2299–2306. [PubMed] [Google Scholar]
- (435).Otaka T; Kaji A. Mode of action of bottromycin A2: effect of bottromycin A2 on polysomes. FEBS Lett. 1983, 153, 53–59. [DOI] [PubMed] [Google Scholar]
- (436).Yamada T; Yagita M; Kobayashi Y; Sennari G; Shimamura H; Matsui H; Horimatsu Y; Hanaki H; Hirose T; Omura S; Sunazuka T. Synthesis and evaluation of antibacterial activity of bottromycins. J. Org. Chem 2018, 83 (13), 7135–7149. [DOI] [PubMed] [Google Scholar]
- (437).Kobayashi Y; Ichioka M; Hirose T; Nagai K; Matsumoto A; Matsui H; Hanaki H; Masuma R; Takahashi Y; Ōmura S; Sunazuka T. Bottromycin derivatives: efficient chemical modifications of the ester moiety and evaluation of anti-MRSA and anti-VRE activities. Bioorg. Med. Chem. Lett 2010, 20 (20), 6116–6120. [DOI] [PubMed] [Google Scholar]
- (438).Miller W; Chaiet L; Rasmussen G; Christensen B; Hannah J; Miller AK; Wolfe F. Bottromycin. Separation of biologically active compounds and preparation and testing of amide derivatives. J. Med. Chem 1968, 11 (4), 746–749. [DOI] [PubMed] [Google Scholar]
- (439).Hayakawa Y; Sasaki K; Adachi H; Furihata K; Nagai K; Shin-ya K. Thioviridamide, a novel apoptosis inducer in transformed cells from Streptomyces olivoviridis. J. Antibiot 2006, 59 (1), 1–5. [DOI] [PubMed] [Google Scholar]
- (440).Kjaerulff L; Sikandar A; Zaburannyi N; Adam S; Herrmann J; Koehnke J; Müller R. Thioholgamides: thioamide-containing cytotoxic RiPP natural products. ACS Chem. Biol 2017, 12 (11), 2837–2841. [DOI] [PubMed] [Google Scholar]
- (441).Izawa M; Nagamine S; Aoki H; Hayakawa Y. Identification of essential biosynthetic genes and a true biosynthetic product for thioviridamide. J. Gen. Appl. Microbiol 2018, 64 (1), 50–53. [DOI] [PubMed] [Google Scholar]
- (442).Kawahara T; Izumikawa M; Kozone I; Hashimoto J; Kagaya N; Koiwai H; Komatsu M; Fujie M; Sato N; Ikeda H; Shin-ya K. Neothioviridamide, a polythioamide compound produced by heterologous expression of a Streptomyces sp. cryptic RiPP biosynthetic gene cluster. J. Nat. Prod 2018, 81 (2), 264–269. [DOI] [PubMed] [Google Scholar]
- (443).Takase S; Kurokawa R; Kondoh Y; Honda K; Suzuki T; Kawahara T; Ikeda H; Dohmae N; Osada H; Shin-ya K; Kushiro T; Yoshida M; Matsumoto K. Mechanism of action of prethioviridamide, an anticancer ribosomally synthesized and post-translationally modified peptide with a polythioamide structure. ACS Chem. Biol 2019, 14 (8), 1819–1828. [DOI] [PubMed] [Google Scholar]
- (444).Pakos-Zebrucka K; Koryga I; Mnich K; Ljujic M; Samali A; Gorman AM The integrated stress response. EMBO Rep. 2016, 17 (10), 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Frattaruolo L; Fiorillo M; Brindisi M; Curcio R; Dolce V; Lacret R; Truman AW; Sotgia F; Lisanti MP; Cappello AR Thioalbamide, a thioamidated peptide from Amycolatopsis alba, affects tumor growth and stemness by inducing metabolic dysfunction and oxidative stress. Cells 2019, 8 (11), 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Kudo K; Koiwai H; Kagaya N; Nishiyama M; Kuzuyama T; Shin-ya K; Ikeda H. Comprehensive derivatization of thioviridamides by heterologous expression. ACS Chem. Biol 2019, 14 (6), 1135–1140. [DOI] [PubMed] [Google Scholar]
- (447).Schmidt EW; Nelson JT; Rasko DA; Sudek S; Eisen JA; Haygood MG; Ravel J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl. Acad. Sci. U.S.A 2005, 102 (20), 7315–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Donia MS; Fricke WF; Ravel J; Schmidt EW Variation in tropical reef symbiont metagenomes defined by secondary metabolism. PLoS One 2011, 6 (3), e17897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Donia MS; Fricke WF; Partensky F; Cox J; Elshahawi SI; White JR; Phillippy AM; Schatz MC; Piel J; Haygood MG; Ravel J; Schmidt EW Complex microbiome underlying secondary and primary metabolism in the tunicate-Prochloron symbiosis. Proc. Natl. Acad. Sci. U.S.A 2011, 108 (51), E1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Schmidt EW; Donia MS; McIntosh JA; Fricke WF; Ravel J. Origin and variation of tunicate secondary metabolites. J. Nat. Prod 2012, 75 (2), 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Leikoski N; Liu L; Jokela J; Wahlsten M; Gugger M; Calteau A; Permi P; Kerfeld CA; Sivonen K; Fewer DP Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem. Biol 2013, 20 (8), 1033–1043. [DOI] [PubMed] [Google Scholar]
- (452).Murakami M; Itou Y; Ishida K; Shin HJ Prenylagaramides A and B, new cyclic peptides from two strains of Oscillatoria agardhii. J. Nat. Prod 1999, 62 (5), 752–755. [DOI] [PubMed] [Google Scholar]
- (453).Donia MS; Wang B; Dunbar DC; Desai PV; Patny A; Avery M; Hamann MT Mollamides B and C, cyclic hexapeptides from the Indonesian tunicate Didemnum molle. J. Nat. Prod 2008, 71 (6), 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Mitchell SS; Faulkner DJ; Rubins K; Bushman FD Dolastatin 3 and two novel cyclic peptides from a Palauan collection of Lyngbya majuscula. J. Nat. Prod 2000, 63 (2), 279–282. [DOI] [PubMed] [Google Scholar]
- (455).Tsuda M; Shigemori H; Mikami Y; Kobayashi J. Hymenamides C-E, new cyclic heptapeptides with two proline residues from the Okinawan marine sponge Hymeniacidon sp. Tetrahedron 1993, 49 (31), 6785–6796. [Google Scholar]
- (456).Hawkins CJ; Lavin MF; Marshall KA; Van den Brenk AL; Watters DJ Structure-activity relationships of the lissoclinamides: cytotoxic cyclic peptides from the ascidian Lissoclinum patella. J. Med. Chem 1990, 33 (6), 1634–1638. [DOI] [PubMed] [Google Scholar]
- (457).Carroll AR; Coll JC; Bourne DJ; MacLeod J,K; Zabrieske M,T; Ireland C,M; Bowden B,F Patellins 1–6 and trunkamide A: novel cyclic hexa-, hepta- and octa-peptides from colonial ascidians, Lissoclinum sp. Aust. J. Chem 1996, 49 (6), 659–667. [Google Scholar]
- (458).Gogineni V; Hamann MT Marine natural product peptides with therapeutic potential: chemistry, biosynthesis, and pharmacology. Biochim. Biophys. Acta Gen. Subj 2018, 1862 (1), 81–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Ireland CM; Durso AR; Newman RA; Hacker MP Antineoplastic cyclic peptides from the marine tunicate Lissoclinum patella. J. Org. Chem 1982, 47, 1807–1811. [Google Scholar]
- (460).Kohda K; Ohta Y; Yokoyama Y; Kawazoe Y; Kato T; Suzumura Y; Hamada Y; Shioiri T. Mechanistic aspects of the cytocidal action of ulithiacyclamide on mouse leukemia L1210 cells in vitro. Biochem. Pharmacol 1989, 38 (24), 4497–4500. [DOI] [PubMed] [Google Scholar]
- (461).Hambley TW; Hawkins CJ; Lavin MF; van den Brenk A; Watters DJ Cycloxazoline: A cytotoxic cyclic hexapeptide from the ascidian Lissoclinum bistratum. Tetrahedron 1992, 48 (2), 341–348. [Google Scholar]
- (462).Watters DJ; Beamish HJ; Marshall KA; Gardiner RA; Seymour GJ; Lavin MF Accumulation of HL-60 leukemia cells in G2/M and inhibition of cytokinesis caused by two marine compounds, bistratene A and cycloxazoline. Cancer Chemother. Pharmacol 1994, 33 (5), 399–409. [DOI] [PubMed] [Google Scholar]
- (463).Bowden BF; Garcia Gravalos D. Cyclic hepta-peptide derivative from colonial ascidians, Lissoclinum sp. 6025466.
- (464).Shin-ya K; Wierzba K; Matsuo K; Ohtani T; Yamada Y; Furihata K; Hayakawa Y; Seto H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc 2001, 123 (6), 1262–1263. [DOI] [PubMed] [Google Scholar]
- (465).Williams AB; Jacobs RS A marine natural product, patellamide D, reverses multidrug resistance in a human leukemic cell line. Cancer Lett. 1993, 71 (1–3), 97–102. [DOI] [PubMed] [Google Scholar]
- (466).Ogino J; Moore RE; Patterson GM; Smith CD Dendroamides, new cyclic hexapeptides from a blue-green alga. Multidrug-resistance reversing activity of dendroamide A. J. Nat. Prod 1996, 59 (6), 581–586. [DOI] [PubMed] [Google Scholar]
- (467).Fu X; Do T; Schmitz FJ; Andrusevich V; Engel MH New cyclic peptides from the ascidian Lissoclinum patella. J. Nat. Prod 1998, 61 (12), 1547–1551. [DOI] [PubMed] [Google Scholar]
- (468).Loo TW; Bartlett MC; Clarke DM Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrateinduced fit mechanism for drug binding. J. Biol. Chem 2003, 278 (16), 13603–13606. [DOI] [PubMed] [Google Scholar]
- (469).Safa AR Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr. Med. Chem. Anticancer Agents 2004, 4 (1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (470).Bruggemann EP; Currier SJ; Gottesman MM; Pastan I. Characterization of the azidopine and vinblastine binding site of P-glycoprotein. J. Biol. Chem 1992, 267 (29), 21020–21026. [PubMed] [Google Scholar]
- (471).Shapiro AB; Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem 1997, 250 (1), 130–137. [DOI] [PubMed] [Google Scholar]
- (472).Shapiro AB; Fox K; Lam P; Ling V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone: evidence for a third drug-binding site. Eur. J. Biochem 2001, 259 (3), 841–850. [DOI] [PubMed] [Google Scholar]
- (473).Qu Q; Sharom FJ Proximity of bound Hoechst 33342 to the ATPase catalytic sites places the drug binding site of P-glycoprotein within the cytoplasmic membrane leaflet. Biochemistry 2002, 41 (14), 4744–4752. [DOI] [PubMed] [Google Scholar]
- (474).Pajeva IK; Globisch C; Wiese M. Structure-function relationships of multidrug resistance P-glycoprotein. J. Med. Chem 2004, 47 (10), 2523–2533. [DOI] [PubMed] [Google Scholar]
- (475).Loo TW; Clarke DM Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem 2002, 277 (46), 44332–44338. [DOI] [PubMed] [Google Scholar]
- (476).Aller SG; Yu J; Ward A; Weng Y; Chittaboina S; Zhuo R; Harrell PM; Trinh YT; Zhang Q; Urbatsch IL; Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323 (5922), 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (477).Martinez L; Arnaud O; Henin E; Tao H; Chaptal V; Doshi R; Andrieu T; Dussurgey S; Tod M; Di Pietro A; Zhang Q; Chang G; Falson P. Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J. 2014, 281 (3), 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).Li J; Jaimes KF; Aller SG Refined structures of mouse P-glycoprotein: refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23 (1), 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (479).Wright SH; Raab A; Feldmann J; Krupp E; Jaspars M. Marine Metabolites and Metal Ion Chelation. In Handbook of Marine Natural Products; Fattorusso E, Gerwick WH, Taglialatela-Scafati O, Eds.; Springer Netherlands: Dordrecht, 2012; pp 861–892. [Google Scholar]
- (480).Bertram A; Pattenden G. Marine metabolites: metal binding and metal complexes of azole-based cyclic peptides of marine origin. Nat. Prod. Rep 2007, 24 (1), 18–30. [DOI] [PubMed] [Google Scholar]
- (481).Guan LL; Sera Y; Adachi K; Nishida F; Shizuri Y. Isolation and evaluation of nonsiderophore cyclic peptides from marine sponges. Biochem. Biophys. Res. Commun 2001, 283 (4), 976–981. [DOI] [PubMed] [Google Scholar]
- (482).Freeman DJ; Pattenden G; Drake AF; Siligardi G. Marine metabolites and metal ion chelation. circular dichroism studies of metal binding to Lissoclinum cyclopeptides. J. Chem. Soc., Perkin Trans. 2 1998, No. 1, 129–136.
- (483).Morris LA; Jaspars M. A Cu 2+ selective marine metabolite. In Special Publications; Chrystal EJT, Wrigley SK, Thomas R, Nicholson N, Hayes M, Eds.; Royal Society of Chemistry: Cambridge, 2007; pp 140–166.
- (484).Comba P; Eisenschmidt A; Gahan LR; Herten D-P; Nette G; Schenk G; Seefeld M. Is Cu(II) coordinated to patellamides inside prochloron cells? Chemistry 2017, 23 (50), 12264–12274. [DOI] [PubMed] [Google Scholar]
- (485).Shin HJ; Matsuda H; Murakami M; Yamaguchi K. Agardhipeptins A and B, two new cyclic hepta- and octapeptide, from the cyanobacterium Oscillatoria agardhii (NIES-204). Tetrahedron 1996, 52 (41), 13129–13136. [Google Scholar]
- (486).Martins J; Leikoski N; Wahlsten M; Azevedo J; Antunes J; Jokela J; Sivonen K; Vasconcelos V; Fewer DP; Leão PN Sphaerocyclamide, a prenylated cyanobactin from the cyanobacterium Sphaerospermopsis sp. LEGE 00249. Sci. Rep 2018, 8 (1), 14537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (487).Ishida K; Matsuda H; Murakami M; Yamaguchi K. Kawaguchipeptin B, an antibacterial cyclic undecapeptide from the cyanobacterium Microcystis aeruginosa. J. Nat. Prod 1997, 60 (7), 724–726. [DOI] [PubMed] [Google Scholar]
- (488).Ishida K; Nakagawa H; Murakami M. Microcyclamide, a cytotoxic cyclic hexapeptide from the cyanobacterium Microcystis aeruginosa. J. Nat. Prod 2000, 63 (9), 1315–1317. [DOI] [PubMed] [Google Scholar]
- (489).Degnan BM; Hawkins CJ; Lavin MF; McCaffrey EJ; Parry DL; van den Brenk AL; Watters DJ New cyclic peptides with cytotoxic activity from the ascidian Lissoclinum patella. J. Med. Chem 1989, 32 (6), 1349–1354. [DOI] [PubMed] [Google Scholar]
- (490).Hamamoto Y; Endo M; Nakagawa M; Nakanishi T; Mizukawa K. A new cyclic peptide, ascidiacyclamide, isolated from ascidian. J. Chem. Soc., Chem. Commun 1983, No. 6, 323.
- (491).Baquero F; Lanza VF; Baquero M-R; del Campo R; Bravo-Vázquez DA Microcins in Enterobacteriaceae: peptide antimicrobials in the eco-active intestinal chemosphere. Front. Microbiol 2019, 10, 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (492).Pons AM; Lanneluc I; Cottenceau G; Sable S. New developments in non-post translationally modified microcins. Biochimie 2002, 84 (5–6), 531–537. [DOI] [PubMed] [Google Scholar]
- (493).Duquesne S; Destoumieux-Garzón D; Peduzzi J; Rebuffat S. Microcins, gene-encoded antibacterial peptides from Enterobacteria. Nat. Prod. Rep 2007, 24 (4), 708–734. [DOI] [PubMed] [Google Scholar]
- (494).Garcia-Bustos JF; Pezzi N; Mendez E. Structure and mode of action of microcin 7, an antibacterial peptide produced by Escherichia coli. Antimicrob. Agents Chemother 1985, 27, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Zukher I; Pavlov M; Tsibulskaya D; Kulikovsky A; Zyubko T; Bikmetov D; Serebryakova M; Nair SK; Ehrenberg M; Dubiley S; Severinov K. Reiterative synthesis by the ribosome and recognition of the N-Terminal formyl group by biosynthetic machinery contribute to evolutionary conservation of the length of antibiotic microcin C peptide precursor. mBio 2019, 10 (2), e00768–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (496).Metlitskaya A; Kazakov T; Vondenhoff GH; Novikova M; Shashkov A; Zatsepin T; Semenova E; Zaitseva N; Ramensky V; Van Aerschot A; Severinov K. Maturation of the translation inhibitor microcin C. J. Bacteriol 2009, 191 (7), 2380–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (497).Severinov K; Nair SK Microcin C: biosynthesis and mechanisms of bacterial resistance. Future Microbiol. 2012, 7 (2), 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Kulikovsky A; Serebryakova M; Bantysh O; Metlitskaya A; Borukhov S; Severinov K; Dubiley S. The molecular mechanism of aminopropylation of peptide-nucleotide antibiotic microcin C. J. Am. Chem. Soc 2014, 136 (31), 11168–11175. [DOI] [PubMed] [Google Scholar]
- (499).Guijarro JI; González-Pastor JE; Baleux F; Millán JLS; Castilla MA; Rico M; Moreno F; Delepierre M. Chemical structure and translation inhibition studies of the antibiotic microcin C7. J. Biol. Chem 1995, 270 (40), 23520–23532. [DOI] [PubMed] [Google Scholar]
- (500).Metlitskaya A; Kazakov T; Kommer A; Pavlova O; Praetorius-Ibba M; Ibba M; Krasheninnikov I; Kolb V; Khmel I; Severinov K. Aspartyl-tRNA synthetase is the target of peptide nucleotide antibiotic Microcin C. J. Biol. Chem 2006, 281 (26), 18033–18042. [DOI] [PubMed] [Google Scholar]
- (501).Novikova M; Metlitskaya A; Datsenko K; Kazakov T; Kazakov A; Wanner B; Severinov K. The Escherichia coli Yej transporter is required for the uptake of translation inhibitor microcin C. J. Bacteriol 2007, 189 (22), 8361–8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (502).Vondenhoff GHM; Blanchaert B; Geboers S; Kazakov T; Datsenko KA; Wanner BL; Rozenski J; Severinov K; Van Aerschot A. Characterization of Peptide Chain Length and Constituency Requirements for YejABEF-Mediated Uptake of Microcin C Analogues. J Bacteriol 2011, 193 (14), 3618–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (503).Kazakov T; Metlitskaya A; Severinov K. Amino acid residues required for maturation, cell uptake, and processing of translation inhibitor microcin C. J. Bacteriol 2007, 189 (5), 2114–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Rajagopalan PT; Datta A; Pei D. Purification, characterization, and inhibition of peptide deformylase from Escherichia coli. Biochemistry 1997, 36 (45), 13910–13918. [DOI] [PubMed] [Google Scholar]
- (505).Kazakov T; Vondenhoff GH; Datsenko KA; Novikova M; Metlitskaya A; Wanner BL; Severinov K. Escherichia coli peptidase A, B, or N can process translation inhibitor microcin C. J. Bacteriol 2008, 190 (7), 2607–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (506).Van de Vijver P; Vondenhoff GHM; Kazakov TS; Semenova E; Kuznedelov K; Metlitskaya A; Van Aerschot A; Severinov K. Synthetic microcin C analogs targeting different aminoacyl-tRNA synthetases. J. Bacteriol 2009, 191 (20), 6273–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (507).Vondenhoff GHM; Dubiley S; Severinov K; Lescrinier E; Rozenski J; Van Aerschot A. Extended targeting potential and improved synthesis of microcin C analogs as antibacterials. Bioorg. Med. Chem 2011, 19 (18), 5462–5467. [DOI] [PubMed] [Google Scholar]
- (508).Serebryakova M; Tsibulskaya D; Mokina O; Kulikovsky A; Nautiyal M; Van Aerschot A; Severinov K; Dubiley S. A trojan-horse peptide-carboxymethyl-cytidine antibiotic from Bacillus amyloliquefaciens. J. Am. Chem. Soc 2016, 138 (48), 15690–15698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (509).Tsibulskaya D; Mokina O; Kulikovsky A; Piskunova J; Severinov K; Serebryakova M; Dubiley S. The product of Yersinia pseudotuberculosis mcc operon Is a peptide-cytidine antibiotic activated inside producing cells by the TldD/E protease. J. Am. Chem. Soc 2017, 139 (45), 16178–16187. [DOI] [PubMed] [Google Scholar]
- (510).Thomas X; Destoumieux-Garzón D; Peduzzi J; Afonso C; Blond A; Birlirakis N; Goulard C; Dubost L; Thai R; Tabet J-C; Rebuffat S. Siderophore peptide, a new type of post-translationally modified antibacterial peptide with potent activity. J. Biol. Chem 2004, 279 (27), 28233–28242. [DOI] [PubMed] [Google Scholar]
- (511).Nolan EM; Fischbach MA; Koglin A; Walsh CT Biosynthetic tailoring of microcin E492m: post-translational modification affords an antibacterial siderophore-peptide conjugate. J. Am. Chem. Soc 2007, 129 (46), 14336–14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Nolan EM; Walsh CT Investigations of the MceIJ-catalyzed posttranslational modification of the microcin E492 C-terminus: linkage of ribosomal and nonribosomal peptides to form “trojan horse” antibiotics. Biochemistry 2008, 47 (35), 9289–9299. [DOI] [PubMed] [Google Scholar]
- (513).Raymond KN; Dertz EA; Kim SS Enterobactin: an archetype for microbial iron transport. Proc. Natl. Acad. Sci. U.S.A 2003, 100 (7), 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).de Lorenzo V; Pugsley AP Microcin E492, a low-molecular-weight peptide antibiotic which causes depolarization of the Escherichia coli cytoplasmic membrane. Antimicrob. Agents Chemother 1985, 27 (4), 666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (515).Lagos R; Wilkens M; Vergara C; Cecchi X; Monasterio O. Microcin E492 forms ion channels in phospholipid bilayer membrane. FEBS Lett. 1993, 321 (2–3), 145–148. [DOI] [PubMed] [Google Scholar]
- (516).Destoumieux-Garzón D; Thomas X; Santamaria M; Goulard C; Barthélémy M; Boscher B; Bessin Y; Molle G; Pons A-M; Letellier L; Peduzzi J; Rebuffat S. Microcin E492 antibacterial activity: evidence for a TonB-dependent inner membrane permeabilization on Escherichia coli: TonB-dependent activity of MccE492. Mol. Microbiol 2003, 49 (4), 1031–1041. [DOI] [PubMed] [Google Scholar]
- (517).Orellana C; Lagos R. The activity of microcin E492 from Klebsiella pneumoniae is regulated by a microcin antagonist. FEMS Microbiol. Lett 1996, 136 (3), 297–303. [DOI] [PubMed] [Google Scholar]
- (518).Vassiliadis G; Peduzzi J; Zirah S; Thomas X; Rebuffat S; Destoumieux-Garzón D. Insight into siderophore-carrying peptide biosynthesis: enterobactin is a precursor for microcin E492 posttranslational modification. Antimicrob. Agents Chemother 2007, 51 (10), 3546–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Destoumieux-Garzón D; Peduzzi J; Thomas X; Djediat C; Rebuffat S. Parasitism of ironsiderophore receptors of Escherichia coli by the siderophore-peptide microcin E492m and its unmodified counterpart. Biometals 2006, 19 (2), 181–191. [DOI] [PubMed] [Google Scholar]
- (520).Bieler S; Silva F; Soto C; Belin D. Bactericidal activity of both secreted and nonsecreted microcin E492 requires the mannose permease. J. Bacteriol 2006, 188 (20), 7049–7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (521).Pugsley AP; Moreno F; de Lorenzo V. Microcin-E492-insensitive mutants of Escherichia coli K12. J. Gen. Microbiol 1986, 132 (12), 3253–3259. [DOI] [PubMed] [Google Scholar]
- (522).Noinaj N; Guillier M; Barnard TJ; Buchanan SK TonB-dependent transporters: regulation, structure, and function. Annu Rev Microbiol 2010, 64, 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Biéler S; Silva F; Belin D. The polypeptide core of Microcin E492 stably associates with the mannose permease and interferes with mannose metabolism. Res. Microbiol 2010, 161 (8), 706–710. [DOI] [PubMed] [Google Scholar]
- (524).Huang K; Zeng J; Liu X; Jiang T; Wang J. Structure of the mannose phosphotransferase system (man-PTS) complexed with microcin E492, a pore-forming bacteriocin. Cell Discov 2021, 7 (1), 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Liu X; Zeng J; Huang K; Wang J. Structure of the mannose transporter of the bacterial phosphotransferase system. Cell Res 2019, 29 (8), 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (526).Jeckelmann J-M; Erni B. The mannose phosphotransferase system (Man-PTS) - Mannose transporter and receptor for bacteriocins and bacteriophages. Biochimica et Biophysica Acta (BBA) - Biomembranes 2020, 1862 (11), 183412. [DOI] [PubMed] [Google Scholar]
- (527).Hetz C; Bono MR; Barros LF; Lagos R. Microcin E492, a channel-forming bacteriocin from Klebsiella pneumoniae, induces apoptosis in some human cell lines. Proc. Natl. Acad. Sci. U.S.A 2002, 99 (5), 2696–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Bieler S; Estrada L; Lagos R; Baeza M; Castilla J; Soto C. Amyloid formation modulates the biological activity of a bacterial protein. J. Biol. Chem 2005, 280 (29), 26880–26885. [DOI] [PubMed] [Google Scholar]
- (529).Marcoleta A; Marin M; Mercado G; Valpuesta JM; Monasterio O; Lagos R. Microcin E492 amyloid formation is retarded by posttranslational modification. J. Bacteriol 2013, 195 (17), 3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (530).Shahnawaz M; Soto C. Microcin amyloid fibrils are reservoir of toxic oligomeric species. J. Biol. Chem 2012, 287 (15), 11665–11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (531).Palmer JD; Mortzfeld BM; Piattelli E; Silby MW; McCormick BA; Bucci V. Microcin H47: a class IIb microcin with potent activity against multidrug resistant Enterobacteriaceae. ACS Infect. Dis 2020, 6 (4), 672–679. [DOI] [PubMed] [Google Scholar]
- (532).Rodríguez E; Laviña M. The proton channel is the minimal structure of ATP synthase necessary and sufficient for microcin H47 antibiotic action. Antimicrob. Agents Chemother 2003, 47 (1), 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (533).Patzer SI; Baquero MR; Bravo D; Moreno F; Hantke K. The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors FepA, Cir, Fiu and IroN. Microbiology 2003, 149 (Pt 9), 2557–2570. [DOI] [PubMed] [Google Scholar]
- (534).Trujillo M; Rodríguez E; Laviña M. ATP synthase is necessary for microcin H47 antibiotic action. Antimicrob. Agents Chemother 2001, 45 (11), 3128–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Vassiliadis G; Destoumieux-Garzón D; Lombard C; Rebuffat S; Peduzzi J. Isolation and characterization of two members of the siderophore-microcin family, microcins M and H47. Antimicrob. Agents Chemother 2010, 54 (1), 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (536).Wright SA; Zumoff CH; Schneider L; Beer SV Pantoea agglomerans strain EH318 produces two antibiotics that inhibit Erwinia amylovora in vitro. Appl. Environ. Microbiol 2001, 67 (1), 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (537).Jin M; Liu L; Wright SAI; Beer SV; Clardy J. Structural and functional analysis of pantocin A: an antibiotic from Pantoea agglomerans discovered by heterologous expression of cloned genes. Angew. Chem. Int. Ed 2003, 42 (25), 2898–2901. [DOI] [PubMed] [Google Scholar]
- (538).Ishimaru CA; Klos EJ; Brubaker RR Multiple antibiotic production by Erwinia herbicola. Phytopathology 78, 746–750. [Google Scholar]
- (539).Vanneste JL; Yu J; Cornish DA Presence of genes homologous to those necessary for synthesis of microcin MccEh252 in strains of Pantoea agglomerans. Acta. Hortic 2008, No. 793, 391–396.
- (540).Fitch MW; Graham DW; Arnold RG; Agarwal SK; Phelps P; Speitel GE; Georgiou G. Phenotypic characterization of copper-resistant mutants of Methylosinus trichosporium OB3b. Appl. Environ. Microbiol 1993, 59 (9), 2771–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Zahn JA; DiSpirito AA Membrane-associated methane monooxygenase from Methylococcus capsulatus (Bath). J. Bacteriol 1996, 178 (4), 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (542).Téllez CM; Gaus KP; Graham DW; Arnold RG; Guzman RZ Isolation of copper biochelates from Methylosinus trichosporium OB3b and soluble methane monooxygenase mutants. Appl. Environ. Microbiol 1998, 64 (3), 1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Choi D-W; Kunz RC; Boyd ES; Semrau JD; Antholine WE; Han J-I; Zahn JA; Boyd JM; de la Mora AM; DiSpirito AA The membrane-associated methane monooxygenase (pMMO) and pMMO-NADH:quinone oxidoreductase complex from Methylococcus capsulatus Bath. J. Bacteriol 2003, 185 (19), 5755–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Kim HJ; Graham DW; DiSpirito AA; Alterman MA; Galeva N; Larive CK; Asunskis D; Sherwood PMA Methanobactin, a copper-acquisition compound from nethaneoxidizing bacteria. Science 2004, 305 (5690), 1612–1615. [DOI] [PubMed] [Google Scholar]
- (545).Behling LA; Hartsel SC; Lewis DE; DiSpirito AA; Choi DW; Masterson LR; Veglia G; Gallagher WH NMR, mass spectrometry and chemical evidence reveal a different chemical structure for methanobactin that contains oxazolone rings. J. Am. Chem. Soc 2008, 130 (38), 12604–12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (546).Choi DW; Zea CJ; Do YS; Semrau JD; Antholine WE; Hargrove MS; Pohl NL; Boyd ES; Geesey GG; Hartsel SC; Shafe PH; McEllistrem MT; Kisting CJ; Campbell D; Rao V; de la Mora AM; DiSpirito AA Spectral, kinetic, and thermodynamic properties of Cu(I) and Cu(II) binding by methanobactin from Methylosinus trichosporium OB3b. Biochemistry 2006, 45 (5), 1442–1453. [DOI] [PubMed] [Google Scholar]
- (547).Krentz BD; Mulheron HJ; Semrau JD; DiSpirito AA; Bandow NL; Haft DH; Vuilleumier S; Murrell JC; McEllistrem MT; Hartsel SC; Gallagher WH A comparison of methanobactins from Methylosinus trichosporium OB3b and Methylocystis strain SB2 predicts methanobactins are synthesized from diverse peptide precursors modified to create a common core for binding and reducing copper ions. Biochemistry 2010, 49 (47), 10117–10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).El Ghazouani A; Basle A; Gray J; Graham DW; Firbank SJ; Dennison C. Variations in methanobactin structure influences copper utilization by methane-oxidizing bacteria. Proc. Natl. Acad. Sci. U.S.A 2012, 109 (22), 8400–8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (549).Balasubramanian R; Rosenzweig AC Copper methanobactin: a molecule whose time has come. Curr. Opin. Chem. Biol 2008, 12 (2), 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (550).Balasubramanian R; Kenney GE; Rosenzweig AC Dual pathways for copper uptake by methanotrophic bacteria. J. Biol. Chem 2011, 286 (43), 37313–37319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (551).Gu W; Farhan Ul Haque M; Baral BS; Turpin EA; Bandow NL; Kremmer E; Flatley A; Zischka H; DiSpirito AA; Semrau JD A TonB-dependent transporter is responsible for methanobactin uptake by Methylosinus trichosporium OB3b. Appl. Environ. Microbiol 2016, 82 (6), 1917–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (552).Dassama LMK; Kenney GE; Ro SY; Zielazinski EL; Rosenzweig AC Methanobactin transport machinery. Proc. Natl. Acad. Sci. U.S.A 2016, 113 (46), 13027–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (553).Banerjee R; Jones JC; Lipscomb JD Soluble methane monooxygenase. Annu. Rev. Biochem 2019, 88, 409–431. [DOI] [PubMed] [Google Scholar]
- (554).Koo CW; Rosenzweig AC Biochemistry of aerobic biological methane oxidation. Chem. Soc. Rev 2021, 50 (5), 3424–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (555).Lieberman RL; Rosenzweig AC Crystal structure of a membrane-bound metalloenzyme that catalyses the biological oxidation of methane. Nature 2005, 434 (7030), 177–182. [DOI] [PubMed] [Google Scholar]
- (556).Choi DW; Antholine WE; Do YS; Semrau JD; Kisting CJ; Kunz RC; Campbell D; Rao V; Hartsel SC; DiSpirito AA Effect of methanobactin on the activity and electron paramagnetic resonance spectra of the membrane-associated methane monooxygenase in Methylococcus capsulatus Bath. Microbiology 2005, 151 (10), 3417–3426. [DOI] [PubMed] [Google Scholar]
- (557).Semrau JD; Jagadevan S; DiSpirito AA; Khalifa A; Scanlan J; Bergman BH; Freemeier BC; Baral BS; Bandow NL; Vorobev A; Haft DH; Vuilleumier S; Murrell JC Methanobactin and MmoD work in concert to act as the ‘copper-switch’ in methanotrophs. Environ. Microbiol 2013, 15 (11), 3077–3086. [DOI] [PubMed] [Google Scholar]
- (558).Kenney GE; Sadek M; Rosenzweig AC Copper-responsive gene expression in the methanotroph Methylosinus trichosporium OB3b. Metallomics 2016, 8 (9), 931–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (559).Thoendel M; Kavanaugh JS; Flack CE; Horswill AR Peptide signaling in the staphylococci. Chem. Rev 2011, 111 (1), 117–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).Cook LC; Federle MJ Peptide pheromone signaling in Streptococcus and Enterococcus. FEMS Microbiol. Rev 2014, 38 (3), 473–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (561).Vasquez JK; Tal-Gan Y; Cornilescu G; Tyler KA; Blackwell HE Simplified AIP-II peptidomimetics are potent inhibitors of Staphylococcus aureus AgrC quorum sensing receptors. Chembiochem 2017, 18 (4), 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (562).Vasquez JK; Blackwell HE Simplified autoinducing peptide mimetics with singlenanomolar activity against the Staphylococcus aureus AgrC quorum sensing receptor. ACS Infect Dis 2019, 5 (4), 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Lowy FD Staphylococcus aureus infections. N. Engl. J. Med 1998, 339 (8), 520–532. [DOI] [PubMed] [Google Scholar]
- (564).Kavanaugh JS; Thoendel M; Horswill AR A role for type I signal peptidase in Staphylococcus aureus quorum sensing: a role for signal peptidase in S. aureus quorum sensing. Mol. Microbiol 2007, 65 (3), 780–798. [DOI] [PubMed] [Google Scholar]
- (565).Novick RP; Ross HF; Projan SJ; Kornblum J; Kreiswirth B; Moghazeh S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993, 12 (10), 3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (566).Recsei P; Kreiswirth B; O’Reilly M; Schlievert P; Gruss A; Novick RP Regulation of exoprotein gene expression in Staphylococcus aureus by agar. Mol. Gen. Genet 1986, 202 (1), 58–61. [DOI] [PubMed] [Google Scholar]
- (567).Dunman PM; Murphy E; Haney S; Palacios D; Tucker-Kellogg G; Wu S; Brown EL; Zagursky RJ; Shlaes D; Projan SJ Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol 2001, 183 (24), 7341–7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (568).Shaw L; Golonka E; Potempa J; Foster SJ The role and regulation of the extracellular proteases of Staphylococcus aureus. Microbiology 2004, 150, 217–228. [DOI] [PubMed] [Google Scholar]
- (569).Novick RP Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol 2003, 48 (6), 1429–1449. [DOI] [PubMed] [Google Scholar]
- (570).Ji G; Beavis R; Novick RP Bacterial interference caused by autoinducing peptide variants. Science 1997, 276 (5321), 2027–2030. [DOI] [PubMed] [Google Scholar]
- (571).Jarraud S; Lyon GJ; Figueiredo AMS; Gérard L; Vandenesch F; Etienne J; Muir TW; Novick RP Exfoliatin-producing strains define a fourth agr specificity group in Staphylococcus aureus. J. Bacteriol 2000, 182 (22), 6517–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (572).Dufour P; Jarraud S; Vandenesch F; Greenland T; Novick RP; Bes M; Etienne J; Lina G. High genetic variability of the agr locus in Staphylococcus species. J. Bacteriol 2002, 184 (4), 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (573).Otto M; Süßmuth R; Vuong C; Jung G; Götz F. Inhibition of virulence factor expression in Staphylococcus aureus by the Staphylococcus epidermidis agr pheromone and derivatives. FEBS Lett. 1999, 450 (3), 257–262. [DOI] [PubMed] [Google Scholar]
- (574).Jarraud S; Mougel C; Thioulouse J; Lina G; Meugnier H; Forey F; Nesme X; Etienne J; Vandenesch F. Relationships between Staphylococcus aureus genetic background, virulence factors, agr groups (alleles), and human disease. Infect. Immun 2002, 70 (2), 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (575).Mayville P; Ji G; Beavis R; Yang H; Goger M; Novick RP; Muir TW Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence. Proc. Natl. Acad. Sci. U.S.A 1999, 96 (4), 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (576).McDowell P; Affas Z; Reynolds C; Holden MT; Wood SJ; Saint S; Cockayne A; Hill PJ; Dodd CE; Bycroft BW; Chan WC; Williams P. Structure, activity and evolution of the group I thiolactone peptide quorum-sensing system of Staphylococcus aureus. Mol. Microbiol 2001, 41 (2), 503–512. [DOI] [PubMed] [Google Scholar]
- (577).Tal-Gan Y; Stacy DM; Foegen MK; Koenig DW; Blackwell HE Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J. Am. Chem. Soc 2013, 135 (21), 7869–7882. [DOI] [PubMed] [Google Scholar]
- (578).Lyon GJ; Wright JS; Muir TW; Novick RP Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus. Biochemistry 2002, 41 (31), 10095–10104. [DOI] [PubMed] [Google Scholar]
- (579).Tal-Gan Y; Ivancic M; Cornilescu G; Cornilescu CC; Blackwell HE Structural characterization of native autoinducing peptides and abiotic analogues reveals key features essential for activation and inhibition of an AgrC quorum sensing receptor in Staphylococcus aureus. J. Am. Chem. Soc 2013, 135 (49), 18436–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (580).Tal-Gan Y; Ivancic M; Cornilescu G; Blackwell HE Characterization of structural elements in native autoinducing peptides and non-native analogues that permit the differential modulation of AgrC-type quorum sensing receptors in Staphylococcus aureus. Org. Biomol. Chem 2016, 14 (1), 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (581).Johnson JG; Wang B; Debelouchina GT; Novick RP; Muir TW Increasing AIP macrocycle size reveals key features of agr activation in Staphylococcus aureus. Chembiochem 2015, 16 (7), 1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (582).Wright JS; Lyon GJ; George EA; Muir TW; Novick RP Hydrophobic interactions drive ligand-receptor recognition for activation and inhibition of staphylococcal quorum sensing. Proc. Natl. Acad. Sci. U.S.A 2004, 101 (46), 16168–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (583).Geisinger E; George EA; Chen J; Muir TW; Novick RP Identification of ligand specificity determinants in AgrC, the Staphylococcus aureus quorum-sensing receptor. J. Biol. Chem 2008, 283 (14), 8930–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (584).Jensen RO; Winzer K; Clarke SR; Chan WC; Williams P. Differential recognition of Staphylococcus aureus quorum-sensing signals depends on both extracellular loops 1 and 2 of the transmembrane sensor AgrC. J. Mol. Biol 2008, 381 (2), 300–309. [DOI] [PubMed] [Google Scholar]
- (585).Gristina AG; Costerton JW Bacterial adherence to biomaterials and tissue. The significance of its role in clinical sepsis. J. Bone Joint Surg. Am 1985, 67 (2), 264–273. [PubMed] [Google Scholar]
- (586).Garsin DA; Frank KL; Silanpää J; Ausubel FM; Hartke A; Shankar N; Murray BE Pathogenesis and Models of Enterococcal Infection. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore MS, Clewell DB, Ike Y, Shankar N, Eds.; Massachusetts Eye and Ear Infirmary: Boston, 2014. [PubMed] [Google Scholar]
- (587).Nakayama J; Cao Y; Horii T; Sakuda S; Akkermans AD; de Vos WM; Nagasawa H. Gelatinase biosynthesis-activating pheromone: a peptide lactone that mediates a quorum sensing in Enterococcus faecalis. Mol. Microbiol 2001, 41 (1), 145–154. [DOI] [PubMed] [Google Scholar]
- (588).Nakayama J; Chen S; Oyama N; Nishiguchi K; Azab EA; Tanaka E; Kariyama R; Sonomoto K. Revised model for Enterococcus faecalis fsr quorum-sensing system: the small open reading frame fsrD encodes the gelatinase biosynthesis-activating pheromone propeptide corresponding to Staphylococcal AgrD. J. Bacteriol 2006, 188 (23), 8321–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (589).Del Papa MF; Perego M. Enterococcus faecalis virulence regulator FsrA binding to target promoters. J. Bacteriol 2011, 193 (7), 1527–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (590).Qin X; Singh KV; Weinstock GM; Murray BE Effects of Enterococcus faecalis fsr genes on production of gelatinase and a serine protease and virulence. Infect. Immun 2000, 68 (5), 2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (591).Qin X; Singh KV; Weinstock GM; Murray BE Characterization of fsr, a regulator controlling expression of gelatinase and serine protease in Enterococcus faecalis OG1RF. J. Bacteriol 2001, 183 (11), 3372–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Thurlow LR; Thomas VC; Narayanan S; Olson S; Fleming SD; Hancock LE Gelatinase contributes to the pathogenesis of endocarditis caused by Enterococcus faecalis. Infect. Immun 2010, 78 (11), 4936–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (593).Steck N; Hoffmann M; Sava IG; Kim SC; Hahne H; Tonkonogy SL; Mair K; Krueger D; Pruteanu M; Shanahan F; Vogelmann R; Schemann M; Kuster B; Sartor RB; Haller D. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 2011, 141 (3), 959–971. [DOI] [PubMed] [Google Scholar]
- (594).Sifri CD; Mylonakis E; Singh KV; Qin X; Garsin DA; Murray BE; Ausubel FM; Calderwood SB Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and Mice. Infect. Immun 2002, 70 (10), 5647–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Hancock LE; Perego M. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J. Bacteriol 2004, 186 (17), 5629–5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (596).Soares RO; Fedi AC; Reiter KC; Caierão J; d’Azevedo PA Correlation between biofilm formation and gelE, esp, and agg genes in Enterococcus spp. clinical isolates. Virulence 2014, 5 (5), 634–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (597).Ali L; Goraya M; Arafat Y; Ajmal M; Chen J-L; Yu D. Molecular mechanism of quorum-sensing in Enterococcus faecalis: its role in virulence and therapeutic approaches. Int. J. Mol. Sci 2017, 18 (5), 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (598).Nishiguchi K; Nagata K; Tanokura M; Sonomoto K; Nakayama J. Structure-activity relationship of gelatinase biosynthesis-activating pheromone of Enterococcus faecalis. J. Bacteriol 2009, 191 (2), 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (599).McBrayer DN; Gantman BK; Cameron CD; Tal-Gan Y. An entirely solid phase peptide synthesis-based strategy for synthesis of gelatinase biosynthesis-activating pheromone (GBAP) analogue libraries: investigating the structure–activity relationships of the Enterococcus faecalis quorum sensing signal. Org. Lett 2017, 19 (12), 3295–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (600).Patching SG; Edara S; Ma P; Nakayama J; Hussain R; Siligardi G; Phillips-Jones MK Interactions of the intact FsrC membrane histidine kinase with its pheromone ligand GBAP revealed through synchrotron radiation circular dichroism. Biochim. Biophys. Acta Biomembr 2012, 1818 (7), 1595–1602. [DOI] [PubMed] [Google Scholar]
- (601).Lopez D; Vlamakis H; Kolter R. Generation of multiple cell types in Bacillus subtilis. FEMS Microbiol. Rev 2009, 33 (1), 152–163. [DOI] [PubMed] [Google Scholar]
- (602).Magnuson R; Solomon J; Grossman AD Biochemical and genetic characterization of a competence pheromone from B. subtilis. Cell 1994, 77 (2), 207–216. [DOI] [PubMed] [Google Scholar]
- (603).Bacon Schneider K; Palmer TM; Grossman AD Characterization of comQ and comX, two genes required for production of ComX pheromone in Bacillus subtilis. J. Bacteriol 2002, 184 (2), 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (604).Okada M; Sato I; Cho SJ; Iwata H; Nishio T; Dubnau D; Sakagami Y. Structure of the Bacillus subtilis quorum-sensing peptide pheromone ComX. Nat. Chem. Biol 2005, 1 (1), 23–24. [DOI] [PubMed] [Google Scholar]
- (605).Okada M; Yamaguchi H; Sato I; Cho SJ; Dubnau D; Sakagami Y. Structure-activity relationship studies on quorum sensing ComX(RO-E-2) pheromone. Bioorg. Med. Chem. Lett 2007, 17 (6), 1705–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (606).Okada M; Yamaguchi H; Sato I; Tsuji F; Dubnau D; Sakagami Y. Chemical structure of posttranslational modification with a farnesyl group on tryptophan. Biosci. Biotechnol. Biochem 2008, 72 (3), 914–918. [DOI] [PubMed] [Google Scholar]
- (607).Weinrauch Y; Penchev R; Dubnau E; Smith I; Dubnau D. A Bacillus subtilis regulatory gene product for genetic competence and sporulation resembles sensor protein members of the bacterial two-component signal-transduction systems. Genes Dev. 1990, 4 (5), 860–872. [DOI] [PubMed] [Google Scholar]
- (608).Piazza F; Tortosa P; Dubnau D. Mutational analysis and membrane topology of ComP, a quorum-sensing histidine kinase of Bacillus subtilis controlling competence development. J. Bacteriol 1999, 181 (15), 4540–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (609).Nakano MM; Xia LA; Zuber P. Transcription initiation region of the srfA operon, which is controlled by the ComP-ComA signal transduction system in Bacillus subtilis. J. Bacteriol 1991, 173 (17), 5487–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (610).Nakano MM; Magnuson R; Myers A; Curry J; Grossman AD; Zuber P. SrfA is an operon required for surfactin production, competence development, and efficient sporulation in Bacillus subtilis. J. Bacteriol 1991, 173 (5), 1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (611).Roggiani M; Dubnau D. ComA, a phosphorylated response regulator protein of Bacillus subtilis, binds to the promoter region of srfA. J. Bacteriol 1993, 175 (10), 3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (612).Hamoen LW; Eshuis H; Jongbloed J; Venema G; van Sinderen D. A small gene, designated comS, located within the coding region of the fourth amino acid-activation domain of srfA, is required for competence development in Bacillus subtilis. Mol. Microbiol 1995, 15 (1), 55–63. [DOI] [PubMed] [Google Scholar]
- (613).Turgay K; Hahn J; Burghoorn J; Dubnau D. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 1998, 17 (22), 6730–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (614).Berka RM; Hahn J; Albano M; Draskovic I; Persuh M; Cui X; Sloma A; Widner W; Dubnau D. Microarray analysis of the Bacillus subtilis K-state: genome-wide expression changes dependent on ComK. Mol. Microbiol 2002, 43 (5), 1331–1345. [DOI] [PubMed] [Google Scholar]
- (615).Ogura M; Yamaguchi H; Kobayashi K; Ogasawara N; Fujita Y; Tanaka T. Whole-genome analysis of genes regulated by the Bacillus subtilis competence transcription factor ComK. J. Bacteriol 2002, 184 (9), 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (616).Rahman FB; Sarkar B; Moni R; Rahman MS Molecular genetics of surfactin and its effects on different sub-populations of Bacillus subtilis. Biotechnol. Rep 2021, 32, e00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (617).Verhamme DT; Kiley TB; Stanley-Wall NR DegU co-ordinates multicellular behaviour exhibited by Bacillus subtilis. Mol. Microbiol 2007, 65 (2), 554–568. [DOI] [PubMed] [Google Scholar]
- (618).Špacapan M; Danevčič T; Štefanic P; Porter M; Stanley-Wall NR; Mandic-Mulec I. The ComX quorum sensing peptide of Bacillus subtilis affects biofilm formation negatively and sporulation positively. Microorganisms 2020, 8 (8), 1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (619).Tortosa P; Logsdon L; Kraigher B; Itoh Y; Mandic-Mulec I; Dubnau D. Specificity and genetic polymorphism of the Bacillus competence quorum-sensing system. J. Bacteriol 2001, 183 (2), 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (620).Reisberg SH; Gao Y; Walker AS; Helfrich EJN; Clardy J; Baran PS Total synthesis reveals atypical atropisomerism in a small-molecule natural product, tryptorubin A. Science 2020, 367 (6476), 458–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (621).Hegemann JD; Zimmermann M; Xie X; Marahiel MA Lasso peptides: an intriguing class of bacterial natural products. Acc. Chem. Res 2015, 48 (7), 1909–1919. [DOI] [PubMed] [Google Scholar]
- (622).Maksimov MO; Pan SJ; James Link A. Lasso peptides: structure, function, biosynthesis, and engineering. Nat. Prod. Rep 2012, 29 (9), 996–1006. [DOI] [PubMed] [Google Scholar]
- (623).Weber W; Fischli W; Hochuli E; Kupfer E; Weibel EK Anantin - a peptide antagonist of the atrial natriuretic factor (ANF). I. Producing organism, fermentation, isolation and biological activity. J. Antibiot 1991, 44 (2), 164–171. [DOI] [PubMed] [Google Scholar]
- (624).Morishita Y; Chiba S; Tsukuda E; Tanaka T; Ogawa T; Yamasaki M; Yoshida M; Kawamoto I; Matsuda Y. RES-701–1, a novel and selective endothelin type B receptor antagonist produced by Streptomyces sp. RE-701. J. Antibiot 1994, 47 (3), 269–275. [DOI] [PubMed] [Google Scholar]
- (625).Ogawa T; Ochiai K; Tanaka T; Tsukuda E; Chiba S; Yano K; Yamasaki M; Yoshida M; Matsuda Y. RES-701–2, −3 and −4, novel and selective endothelin type B receptor antagonists produced by Streptomyces sp. J. Antibiot 1995, 48 (11), 1213–1220. [DOI] [PubMed] [Google Scholar]
- (626).He JX; Cody WL; Flynn MA; Welch KM; Reynolds EE; Doherty AM RES-701–1, synthesis and a reevaluation of its effects on the endothelin receptors. Bioorganic Med. Chem. Lett 1995, 5 (6), 621–626. [Google Scholar]
- (627).Katahira R; Shibata K; Yamasaki M; Matsuda Y; Yoshida M. RES-701–1, comparative study of the synthetic and the microbial-origin compounds. Bioorganic Med. Chem. Lett 1995, 5 (15), 1595–1600. [Google Scholar]
- (628).Shibata K; Yano K; Tanaka T; Matsuda Y; Yamasaki M. C-terminal modifications of an endothelin antagonist RES-701–1: production of ETA/ETB dual selective analogs. Lett. Pept. Sci 1997, 4 (3), 167–170. [Google Scholar]
- (629).Shibata K; Yano K; Tanaka T; Matsuda Y; Yamasaki M. Analogs of an endothelin antagonist RES-701–1: substitutions of C-terminal amino acid. Bioorg. Med. Chem. Lett 1996, 6 (7), 775–778. [Google Scholar]
- (630).Karaki H; Sudjarwo SA; Hori M; Tanaka T; Matsuda Y. Endothelin ETB receptor antagonist, RES-701–1: effects on isolated blood vessels and small intestine. Eur. J. Pharmacol 1994, 262 (3), 255–259. [DOI] [PubMed] [Google Scholar]
- (631).Ikemura T; Ohmori K; Tanaka T; Matsuda Y; Kitamura S. Effects of endothelin B antagonist RES−701−1 on endothelin-induced contractile responses in-vivo and in-vitro in guinea-pigs. J. Pharm. Pharmacol 1996, 48 (1), 100–105. [DOI] [PubMed] [Google Scholar]
- (632).Potterat O; Wagner K; Gemmecker G; Mack J; Puder C; Vettermann R; Streicher R. BI32169, a bicyclic 19-peptide with strong glucagon receptor antagonist activity from Streptomyces sp. J. Nat. Prod 2004, 67 (9), 1528–1531. [DOI] [PubMed] [Google Scholar]
- (633).Knappe TA; Linne U; Xie X; Marahiel MA The glucagon receptor antagonist BI-32169 constitutes a new class of lasso peptides. FEBS Lett. 2010, 584 (4), 785–789. [DOI] [PubMed] [Google Scholar]
- (634).Chen M; Wang S; Yu X. Cryptand-imidazolium supported total synthesis of the lasso peptide BI-32169 and its D-enantiomer. Chem. Comm 2019, 55 (23), 3323–3326. [DOI] [PubMed] [Google Scholar]
- (635).Kimura K-I; Kanou F; Takahashi H; Esumi Y; Uramoto M; Yoshihama M. Propeptin, a new inhibitor of prolyl endopeptidase produced by Microbispora. J. Antibiot 1997, 50 (5), 373–378. [DOI] [PubMed] [Google Scholar]
- (636).Kimura K; Kanou F; Yamashita Y; Yoshimoto T; Yoshihama M. Prolyl endopeptidase inhibitors derived from actinomycetes. Biosci. Biotechnol. Biochem 1997, 61 (10), 1754–1756. [DOI] [PubMed] [Google Scholar]
- (637).Kimura K; Yamazaki M; Sasaki N; Yamashita T; Negishi S; Nakamura T; Koshino H. Novel propeptin analog, propeptin-2, missing two amino acid residues from the propeptin C -terminus loses antibiotic potency. J. Antibiot 2007, 60 (8), 519–523. [DOI] [PubMed] [Google Scholar]
- (638).Gavrish E; Sit CS; Cao S; Kandror O; Spoering A; Peoples A; Ling L; Fetterman A; Hughes D; Bissell A; Torrey H; Akopian T; Mueller A; Epstein S; Goldberg A; Clardy J; Lewis K. Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-Dependent protease ClpC1P1P2. Chem. Biol 2014, 21 (4), 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (639).Leodolter J; Warweg J; Weber-Ban E. The Mycobacterium tuberculosis ClpP1P2 protease interacts asymmetrically with Its ATPase partners ClpX and ClpC1. PLoS One 2015, 10 (5), e0125345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (640).Malik IT; Brötz-Oesterhelt H. Conformational control of the bacterial Clp protease by natural product antibiotics. Nat. Prod. Rep 2017, 34 (7), 815–831. [DOI] [PubMed] [Google Scholar]
- (641).Lear S; Munshi T; Hudson S, A.; Hatton, C.; Clardy, J.; A. Mosely, J.; J. Bull, T.; S. Sit, C.; L. Cobb, S. Total chemical synthesis of lassomycin and lassomycin-amide. Org. Biomol. Chem 2016, 14 (19), 4534–4541. [DOI] [PubMed] [Google Scholar]
- (642).Nar H; Schmid A; Puder C; Potterat O. High-resolution crystal structure of a lasso peptide. ChemMedChem 2010, 5 (10), 1689–1692. [DOI] [PubMed] [Google Scholar]
- (643).Tsunakawa M; Hu S-L; Hoshino Y; Detlefson DJ; Hill SE; Furumai T; White RJ; Nishio M; Kawano K; Yamamoto S; Fukagawa Y; Oki T. Siamycins I and II, new anti-HIV peptides: I. Fermentation, isolation, biological activity and initial characterization. J. Antibiot 1995, 48 (5), 433–434. [DOI] [PubMed] [Google Scholar]
- (644).Oka M; Iimura S; Tenmyo O; Sawada Y; Sugawara M; Ohkusa N; Yamamoto H; Kawano K; Hu SL; Fukagawa Y. Terpestacin, a new syncytium formation inhibitor from Arthrinium sp. J. Antibiot 1993, 46 (3), 367–373. [DOI] [PubMed] [Google Scholar]
- (645).Lin PF; Samanta H; Bechtold CM; Deminie CA; Patick AK; Alam M; Riccardi K; Rose RE; White RJ; Colonno RJ Characterization of siamycin I, a human immunodeficiency virus fusion inhibitor. Antimicrob. Agents Chemother 1996, 40 (1), 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (646).Helynck G; Dubertret C; Mayaux J-F; Leboul J. Isolation of RP 71955, a new anti-HIV-1 peptide secondary metabolite. J. Antibiot 1993, 46 (11), 1756–1757. [DOI] [PubMed] [Google Scholar]
- (647).Frechet D; Guitton JD; Herman F; Faucher D; Helynck G; Monegier du Sorbier B; Ridoux JP; James-Surcouf E; Vuilhorgne M. Solution structure of RP 71955, a new 21 amino acid tricyclic peptide active against HIV-1 virus. Biochemistry 1994, 33 (1), 42–50. [DOI] [PubMed] [Google Scholar]
- (648).Tan S; Ludwig KC; Müller A; Schneider T; Nodwell JR The lasso peptide siamycin-I targets lipid II at the Gram-positive cell surface. ACS Chem. Biol 2019, 14 (5), 966–974. [DOI] [PubMed] [Google Scholar]
- (649).Daniel-Ivad M; Hameed N; Tan S; Dhanjal R; Socko D; Pak P; Gverzdys T; Elliot MA; Nodwell JR An engineered allele of afsQ1 facilitates the discovery and investigation of cryptic natural products. ACS Chem. Biol 2017, 12 (3), 628–634. [DOI] [PubMed] [Google Scholar]
- (650).Nakayama J; Tanaka E; Kariyama R; Nagata K; Nishiguchi K; Mitsuhata R; Uemura Y; Tanokura M; Kumon H; Sonomoto K. Siamycin attenuates fsr quorum sensing mediated by a gelatinase biosynthesis-activating pheromone in Enterococcus faecalis. J. Bacteriol 2007, 189 (4), 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (651).Ma P; Nishiguchi K; Yuille HM; Davis LM; Nakayama J; Phillips-Jones MK Anti-HIV siamycin I directly inhibits autophosphorylation activity of the bacterial FsrC quorum sensor and other ATP-dependent enzyme activities. FEBS Lett. 2011, 585 (17), 2660–2664. [DOI] [PubMed] [Google Scholar]
- (652).Phillips-Jones MK; Patching G, S.; Edara S; Nakayama J; Hussain R; Siligardi G. Interactions of the intact FsrC membrane histidine kinase with the tricyclic peptide inhibitor siamycin I revealed through synchrotron radiation circular dichroism. Phys. Chem. Chem. Phys 2013, 15 (2), 444–447. [DOI] [PubMed] [Google Scholar]
- (653).Li Y; Ducasse R; Zirah S; Blond A; Goulard C; Lescop E; Giraud C; Hartke A; Guittet E; Pernodet J-L; Rebuffat S. Characterization of sviceucin from Streptomyces provides insight into enzyme exchangeability and disulfide bond formation in lasso peptides. ACS Chem. Biol 2015, 10 (11), 2641–2649. [DOI] [PubMed] [Google Scholar]
- (654).Metelev M; Tietz JI; Melby JO; Blair PM; Zhu L; Livnat I; Severinov K; Mitchell DA Structure, bioactivity, and resistance mechanism of streptomonomicin, an unusual lasso peptide from an understudied halophilic actinomycete. Chem. Biol 2015, 22 (2), 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (655).Dhiman A; Bhatnagar S; Kulshreshtha P; Bhatnagar R. Functional characterization of WalRK: a two-component signal transduction system from Bacillus anthracis. FEBS Open Bio. 2014, 4, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (656).Dubrac S; Msadek T. Tearing down the wall: peptidoglycan metabolism and the WalK/WalR (YycG/YycF) essential two-component system. In Bacterial Signal Transduction: Networks and Drug Targets; Utsumi R, Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, 2008; pp 214–228. [DOI] [PubMed] [Google Scholar]
- (657).Hashimoto M; Ooiwa S; Sekiguchi J. Synthetic lethality of the lytE cwlO genotype in Bacillus subtilis is caused by lack of D,L-endopeptidase activity at the lateral cell wall. J. Bacteriol 2012, 194 (4), 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (658).Salomón RA; Farías RN Microcin 25, a novel antimicrobial peptide produced by Escherichia coli. J. Bacteriol 1992, 174 (22), 7428–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (659).Wilson K-A; Kalkum M; Ottesen J; Yuzenkova J; Chait BT; Landick R; Muir T; Severinov K; Darst SA Structure of microcin J25, a peptide inhibitor of bacterial RNA polymerase, is a lassoed tail. J. Am. Chem. Soc 2003, 125 (41), 12475–12483. [DOI] [PubMed] [Google Scholar]
- (660).Bayro MJ; Mukhopadhyay J; Swapna GVT; Huang JY; Ma L-C; Sineva E; Dawson PE; Montelione GT; Ebright RH Structure of antibacterial peptide microcin J25: a 21-residue lariat protoknot. J. Am. Chem. Soc 2003, 125 (41), 12382–12383. [DOI] [PubMed] [Google Scholar]
- (661).Delgado MA; Rintoul MR; Farías RN; Salomón RA Escherichia coli RNA polymerase Is the target of the cyclopeptide antibiotic microcin J25. J. Bacteriol 2001, 183 (15), 4543–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (662).Yuzenkova J; Delgado M; Nechaev S; Savalia D; Epshtein V; Artsimovitch I; Mooney RA; Landick R; Farias RN; Salomon R; Severinov K. Mutations of bacterial RNA polymerase leading to resistance to microcin J25. J. Biol. Chem 2002, 277 (52), 50867–50875. [DOI] [PubMed] [Google Scholar]
- (663).Adelman K; Yuzenkova J; La Porta A; Zenkin N; Lee J; Lis JT; Borukhov S; Wang MD; Severinov K. Molecular mechanism of transcription inhibition by peptide antibiotic microcin J25. Mol. Cell 2004, 14 (6), 753–762. [DOI] [PubMed] [Google Scholar]
- (664).Mukhopadhyay J; Sineva E; Knight J; Levy RM; Ebright RH Antibacterial peptide microcin J25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel. Mol. Cell 2004, 14 (6), 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (665).Braffman NR; Piscotta FJ; Hauver J; Campbell EA; Link AJ; Darst SA Structural mechanism of transcription inhibition by lasso peptides microcin J25 and capistruin. Proc. Natl. Acad. Sci. U.S.A 2019, 116 (4), 1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (666).Pavlova O; Mukhopadhyay J; Sineva E; Ebright RH; Severinov K. Systematic structure-activity analysis of microcin J25. J. Biol. Chem 2008, 283 (37), 25589–25595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (667).Vincent PA; Bellomio A; de Arcuri BF; Farías RN; Morero RD MccJ25 C-terminal is involved in RNA-polymerase inhibition but not in respiration inhibition. Biochem. Biophys. Res. Commun 2005, 331 (2), 549–551. [DOI] [PubMed] [Google Scholar]
- (668).Salomón RA; Farías RN The FhuA protein is involved in microcin 25 uptake. J. Bacteriol 1993, 175 (23), 7741–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (669).Killmann H; Braun M; Herrmann C; Braun V. FhuA barrel-cork hybrids are active transporters and receptors. J. Bacteriol 2001, 183 (11), 3476–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (670).Vincent PA; Delgado MA; Farías RN; Salomón RA Inhibition of Salmonella enterica serovars by microcin J25. FEMS Microbiol. Lett 2004, 236 (1), 103–107. [DOI] [PubMed] [Google Scholar]
- (671).Endriß F; Braun V. Loop deletions indicate regions important for FhuA transport and receptor functions in Escherichia coli. J. Bacteriol 2004, 186 (14), 4818–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (672).Destoumieux-Garzón D; Duquesne S; Peduzzi J; Goulard C; Desmadril M; Letellier L; Rebuffat S; Boulanger P. The iron–siderophore transporter FhuA is the receptor for the antimicrobial peptide microcin J25: role of the microcin Val11–Pro16 β-hairpin region in the recognition mechanism. Biochem J. 2005, 389 (3), 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (673).Mathavan I; Zirah S; Mehmood S; Choudhury HG; Goulard C; Li Y; Robinson CV; Rebuffat S; Beis K. Structural basis for hijacking siderophore receptors by antimicrobial lasso peptides. Nat. Chem. Biol 2014, 10 (5), 340–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (674).Rosengren KJ; Blond A; Afonso C; Tabet J-C; Rebuffat S; Craik DJ Structure of thermolysin cleaved microcin J25: extreme stability of a two-chain antimicrobial peptide devoid of covalent links. Biochemistry 2004, 43 (16), 4696–4702. [DOI] [PubMed] [Google Scholar]
- (675).Blond A; Cheminant M; Destoumieux-Garzón D; Ségalas-Milazzo I; Peduzzi J; Goulard C; Rebuffat S. Thermolysin-linearized microcin J25 retains the structured core of the native macrocyclic peptide and displays antimicrobial activity. Eur. J. Biochem 2002, 269 (24), 6212–6222. [DOI] [PubMed] [Google Scholar]
- (676).Semenova E; Yuzenkova Y; Peduzzi J; Rebuffat S; Severinov K. Structure-activity analysis of microcin J25: distinct parts of the threaded lasso molecule are responsible for interaction with bacterial RNA polymerase. J. Bacteriol 2005, 187 (11), 3859–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (677).Bellomio A; Vincent PA; de Arcuri BF; Salomón RA; Morero RD; Farías RN The microcin J25 β-hairpin region is important for antibiotic uptake but not for RNA polymerase and respiration inhibition. Biochem. Biophys. Res. Commun 2004, 325 (4), 1454–1458. [DOI] [PubMed] [Google Scholar]
- (678).Bellomio A; Rintoul MR; Morero RD Chemical modification of microcin J25 with diethylpyrocarbonate and carbodiimide: evidence for essential histidyl and carboxyl residues. Biochem. Biophys. Res. Commun 2003, 303 (2), 458–462. [DOI] [PubMed] [Google Scholar]
- (679).Cristóbal RE de Solbiati JO; Zenoff AM; Vincent PA; Salomón RA; Yuzenkova J; Severinov K; Farías RN Microcin J25 uptake: His5 of the MccJ25 lariat ring Is involved in interaction with the inner membrane MccJ25 transporter protein SbmA. J. Bacteriol 2006, 188 (9), 3324–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (680).Ghilarov D; Inaba-Inoue S; Stepien P; Qu F; Michalczyk E; Pakosz Z; Nomura N; Ogasawara S; Walker GC; Rebuffat S; Iwata S; Heddle JG; Beis K. Molecular mechanism of SbmA, a promiscuous transporter exploited by antimicrobial peptides. Sci. Adv 2021, 7 (37), eabj5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (681).Li Y; Han Y; Zeng Z; Li W; Feng S; Cao W. Discovery and bioactivity of the novel lasso peptide microcin Y. J. Agric. Food Chem 2021, 69 (31), 8758–8767. [DOI] [PubMed] [Google Scholar]
- (682).Rintoul MR; de Arcuri BF; Salomón RA; Farías, R. N.; Morero, R. D. The antibacterial action of microcin J25: evidence for disruption of cytoplasmic membrane energization in Salmonella newport. FEMS Microbiol. Lett 2001, 204 (2), 265–270. [DOI] [PubMed] [Google Scholar]
- (683).Rintoul MR; de Arcuri BF; Morero RD Effects of the antibiotic peptide microcin J25 on liposomes: role of acyl chain length and negatively charged phospholipid. Biochim. Biophys. Acta Biomembr 2000, 1509 (1), 65–72. [DOI] [PubMed] [Google Scholar]
- (684).Bellomio A; Oliveira RG; Maggio B; Morero RD Penetration and interactions of the antimicrobial peptide, microcin J25, into uncharged phospholipid monolayers. J. Colloid Interface Sci 2005, 285 (1), 118–124. [DOI] [PubMed] [Google Scholar]
- (685).Dupuy FG; Chirou MVN; de Arcuri BF; Minahk CJ; Morero RD Proton motive force dissipation precludes interaction of microcin J25 with RNA polymerase, but enhances reactive oxygen species overproduction. Biochim. Biophys. Acta Gen. Subj 2009, 1790 (10), 1307–1313. [DOI] [PubMed] [Google Scholar]
- (686).Bellomio A; Vincent PA; Arcuri BF de; Farías, R. N.; Morero, R. D. Microcin J25 has dual and independent mechanisms of action in Escherichia coli: RNA polymerase inhibition and increased superoxide production. J. Bacteriol 2007, 189 (11), 4180–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (687).Galván AE; Chalón MC; Schurig-Briccio LA; Salomón RA; Minahk CJ; Gennis RB; Bellomio A. Cytochromes bd-I and bo3 are essential for the bactericidal effect of microcin J25 on Escherichia coli cells. Biochim. Biophys. Acta Bioenerg 2018, 1859 (2), 110–118. [DOI] [PubMed] [Google Scholar]
- (688).Galván AE; Chalón MC; Ríos Colombo NS; Schurig-Briccio LA; Sosa-Padilla B; Gennis RB; Bellomio A. Microcin J25 inhibits ubiquinol oxidase activity of purified cytochrome bd-I from Escherichia coli. Biochimie 2019, 160, 141–147. [DOI] [PubMed] [Google Scholar]
- (689).Chalon MC; Bellomio A; Solbiati JO; Morero RD; Farias RN; Vincent PA Tyrosine 9 is the key amino acid in microcin J25 superoxide overproduction. FEMS Microbiol. Lett 2009, 300 (1), 90–96. [DOI] [PubMed] [Google Scholar]
- (690).Chalón MC; Wilke N; Pedersen J; Rufini S; Morero RD; Cortez L; Chehín RN; Farias RN; Vincent PA Redox-active tyrosine residue in the microcin J25 molecule. Biochem. Biophys. Res. Commun 2011, 406 (3), 366–370. [DOI] [PubMed] [Google Scholar]
- (691).Niklison Chirou MV; Minahk CJ; Morero RD Antimitochondrial activity displayed by the antimicrobial peptide microcin J25. Biochem. Biophys. Res. Commun 2004, 317 (3), 882–886. [DOI] [PubMed] [Google Scholar]
- (692).Chirou MN; Bellomio A; Dupuy F; Arcuri B; Minahk C; Morero R. Microcin J25 induces the opening of the mitochondrial transition pore and cytochrome c release through superoxide generation. FEBS J. 2008, 275 (16), 4088–4096. [DOI] [PubMed] [Google Scholar]
- (693).Niklison-Chirou MV; Dupuy F; Pena LB; Gallego SM; Barreiro-Arcos ML; Avila C; Torres-Bugeau C; Arcuri BE; Bellomio A; Minahk C; Morero RD Microcin J25 triggers cytochrome c release through irreversible damage of mitochondrial proteins and lipids. Int. J. Biochem. Cell Biol 2010, 42 (2), 273–281. [DOI] [PubMed] [Google Scholar]
- (694).Kuznedelov K; Semenova E; Knappe TA; Mukhamedyarov D; Srivastava A; Chatterjee S; Ebright RH; Marahiel MA; Severinov K. The antibacterial threaded-lasso peptide capistruin inhibits bacterial RNA polymerase. J. Mol. Biol 2011, 412 (5), 842–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (695).Metelev M; Arseniev A; Bushin LB; Kuznedelov K; Artamonova TO; Kondratenko R; Khodorkovskii M; Seyedsayamdost MR; Severinov K. Acinetodin and klebsidin, RNA polymerase targeting lasso peptides produced by human isolates of Acinetobacter gyllenbergii and Klebsiella pneumoniae. ACS Chem. Biol 2017, 12 (3), 814–824. [DOI] [PubMed] [Google Scholar]
- (696).Cheung-Lee WL; Parry ME; Jaramillo Cartagena A; Darst SA; Link AJ Discovery and structure of the antimicrobial lasso peptide citrocin. J. Biol. Chem 2019, 294 (17), 6822–6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (697).Cheung-Lee WL; Parry ME; Zong C; Cartagena AJ; Darst SA; Connell ND; Russo R; Link AJ Discovery of ubonodin, an antimicrobial lasso peptide active against members of the Burkholderia cepacia Complex. ChemBioChem 2020, 21 (9), 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (698).Knappe TA; Linne U; Zirah S; Rebuffat S; Xie X; Marahiel MA Isolation and structural characterization of capistruin, a lasso peptide predicted from the genome sequence of Burkholderia thailandensis E264. J. Am. Chem. Soc 2008, 130 (34), 11446–11454. [DOI] [PubMed] [Google Scholar]
- (699).Iwatsuki M; Uchida R; Takakusagi Y; Matsumoto A; Jiang C-L; Takahashi Y; Arai M; Kobayashi S; Matsumoto M; Inokoshi J; Tomoda H; Ōmura S. Lariatins, novel anti-mycobacterial peptides with a lasso structure, produced by Rhodococcus jostii K01-B0171. J. Antibiot 2007, 60 (6), 357–363. [DOI] [PubMed] [Google Scholar]
- (700).Iwatsuki M; Koizumi Y; Gouda H; Hirono S; Tomoda H; Ōmura S. Lys17 in the ‘lasso’ peptide lariatin A is responsible for anti-mycobacterial activity. Bioorg. Med. Chem. Lett 2009, 19 (10), 2888–2890. [DOI] [PubMed] [Google Scholar]
- (701).Inokoshi J; Koyama N; Miyake M; Shimizu Y; Tomoda H. Structure-activity analysis of Gram-positive bacterium-producing lasso peptides with anti-mycobacterial activity. Sci. Rep 2016, 6 (1), 30375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (702).Valiante V; Monteiro MC; Martín J; Altwasser R; Aouad NE; González I; Kniemeyer O; Mellado E; Palomo S; Pedro N. de; Pérez-Victoria I; Tormo JR; Vicente F; Reyes F; Genilloud O; Brakhage AA Hitting the caspofungin salvage pathway of human-pathogenic fungi with the novel lasso peptide humidimycin (MDN-0010). Antimicrob. Agents Chemother 2015, 59 (9), 5145–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (703).Wyche TP; Ruzzini AC; Schwab L; Currie CR; Clardy J. Tryptorubin A: A polycyclic peptide from a fungus-derived Streptomycete. J. Am. Chem. Soc 2017, 139 (37), 12899–12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (704).Zhang X; Li S. Expansion of chemical space for natural products by uncommon P450 reactions. Nat. Prod. Rep 2017, 34 (9), 1061–1089. [DOI] [PubMed] [Google Scholar]
- (705).Rudolf JD; Chang C-Y; Ma M; Shen B. Cytochromes P450 for natural product biosynthesis in Streptomyces: sequence, structure, and function. Nat. Prod. Rep 2017, 34 (9), 1141–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (706).Dittmann E; Gugger M; Sivonen K; Fewer DP Natural product biosynthetic diversity and comparative genomics of the cyanobacteria. Trends Microbiol. 2015, 23 (10), 642–652. [DOI] [PubMed] [Google Scholar]
- (707).Ahmed MN; Reyna-González E; Schmid B; Wiebach V; Süssmuth RD; Dittmann E; Fewer DP Phylogenomic analysis of the microviridin biosynthetic pathway coupled with targeted chemo-enzymatic synthesis yields potent protease inhibitors. ACS Chem. Biol 2017, 12 (6), 1538–1546. [DOI] [PubMed] [Google Scholar]
- (708).Lee H; Choi M; Park J-U; Roh H; Kim S. Genome mining reveals high topological diversity of ω-ester-containing peptides and divergent evolution of ATP-grasp macrocyclases. J. Am. Chem. Soc 2020, 142 (6), 3013–3023. [DOI] [PubMed] [Google Scholar]
- (709).Ramesh S; Guo X; DiCaprio AJ; De Lio AM; Harris LA; Kille BL; Pogorelov TV; Mitchell DA Bioinformatics-guided expansion and discovery of graspetides. ACS Chem. Biol 2021, 16 (12), 2787–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (710).Ishitsuka MO; Kusumi T; Kakisawa H; Kaya K; Watanabe MM Microviridin. A novel tricyclic depsipeptide from the toxic cyanobacterium Microcystis viridis. J. Am. Chem. Soc 1990, 112 (22), 8180–8182. [Google Scholar]
- (711).Okino T; Matsuda H; Murakami M; Yamaguchi K. New microviridins, elastase inhibitors from the blue-green alga Microcystis aeruginosa. Tetrahedron 1995, 51 (39), 10679–10686. [Google Scholar]
- (712).Shin HJ; Murakami M; Matsuda H; Yamaguchi K. Microviridins D-F, serine protease inhibitors from the cyanobacterium Oscillatoria agardhii (NIES-204). Tetrahedron 1996, 52 (24), 8159–8168. [Google Scholar]
- (713).Murakami M; Sun Q; Ishida K; Matsuda H; Okino T; Yamaguchi K. Microviridins, elastase inhibitors from the cyanobacterium Nostoc minutum (NIES-26). Phytochemistry 1997, 45 (6), 1197–1202. [Google Scholar]
- (714).Fujii K; Sivonen K; Naganawa E; Harada K. Non-toxic peptides from toxic cyanobacteria, Oscillatoria agardhii. Tetrahedron 2000, 56 (5), 725–733. [Google Scholar]
- (715).Rohrlack T; Christoffersen K; Hansen PE; Zhang W; Czarnecki O; Henning M; Fastner J; Erhard M; Neilan BA; Kaebernick M. Isolation, characterization, and quantitative analysis of microviridin J, a new microcystis metabolite toxic to daphnia. J. Chem. Ecol 2003, 29 (8), 1757–1770. [DOI] [PubMed] [Google Scholar]
- (716).Reyna-González E; Schmid B; Petras D; Süssmuth RD; Dittmann E. Leader peptide-free In vitro reconstitution of microviridin biosynthesis enables design of synthetic proteasetargeted libraries. Angew. Chem. Int. Ed. Engl 2016, 55 (32), 9398–9401. [DOI] [PubMed] [Google Scholar]
- (717).Ziemert N; Ishida K; Weiz A; Hertweck C; Dittmann E. Exploiting the natural diversity of microviridin gene clusters for discovery of novel tricyclic depsipeptides. Appl. Environ. Microbiol 2010, 76 (11), 3568–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (718).Gatte-Picchi D; Weiz A; Ishida K; Hertweck C; Dittmann E. Functional analysis of environmental DNA-derived microviridins provides new insights into the diversity of the tricyclic peptide family. Appl. Environ. Microbiol 2014, 80 (4), 1380–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (719).Reshef V; Carmeli S. New microviridins from a water bloom of the cyanobacterium Microcystis aeruginosa. Tetrahedron 2006, 62 (31), 7361–7369. [Google Scholar]
- (720).Sieber S; Grendelmeier SM; Harris LA; Mitchell DA; Gademann K. Microviridin 1777: a toxic chymotrypsin inhibitor discovered by a metabologenomic Aaproach. J. Nat. Prod 2020, 83 (2), 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (721).Lee H; Park Y; Kim S. Enzymatic cross-linking of side chains generates a modified peptide with four hairpin-like bicyclic repeats. Biochemistry 2017, 56 (37), 4927–4930. [DOI] [PubMed] [Google Scholar]
- (722).Taichi M; Yamazaki T; Kawahara K; Motooka D; Nakamura S; Harada S; Teshima T; Ohkubo T; Kobayashi Y; Nishiuchi Y. Structure–activity relationship of marinostatin, a serine protease inhibitor isolated from a marine organism. J. Pept. Sci 2010, 16 (7), 329–336. [DOI] [PubMed] [Google Scholar]
- (723).Lai X; Wichers HJ; Soler-Lopez M; Dijkstra BW Structure and function of human tyrosinase and tyrosinase-related proteins. Chemistry 2018, 24 (1), 47–55. [DOI] [PubMed] [Google Scholar]
- (724).Li J; Feng L; Liu L; Wang F; Ouyang L; Zhang L; Hu X; Wang G. Recent advances in the design and discovery of synthetic tyrosinase inhibitors. Eur. J. Med. Chem 2021, 224, 113744. [DOI] [PubMed] [Google Scholar]
- (725).Polverino E; Rosales-Mayor E; Dale GE; Dembowsky K; Torres A. The role of neutrophil elastase inhibitors in lung diseases. Chest 2017, 152 (2), 249–262. [DOI] [PubMed] [Google Scholar]
- (726).Imada C; Simidu U; Taga N. Isolation and characterization of marine bacteria producing alkaline protease inhibitor. Nippon Suisan Gakkaishi 1985, 51 (5), 799–803. [Google Scholar]
- (727).Imada C; Taga N; Maeda M. Cultivation conditions for subtilisin Inhibitor-producing bacterium and general properties of the inhibitor “marinostatin.” Nippon Suisan Gakkaishi 1985, 51 (5), 805–810. [Google Scholar]
- (728).Kaebernick M; Rohrlack T; Christoffersen K; Neilan BA A spontaneous mutant of microcystin biosynthesis: genetic characterization and effect on Daphnia. Environ. Microbiol 2001, 3 (11), 669–679. [DOI] [PubMed] [Google Scholar]
- (729).Rohrlack T; Christoffersen K; Kaebernick M; Neilan BA Cyanobacterial protease inhibitor microviridin J causes a lethal molting disruption in Daphnia pulicaria. Appl. Environ. Microbiol 2004, 70 (8), 5047–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (730).Weiz AR; Ishida K; Quitterer F; Meyer S; Kehr J-C; Müller KM; Groll M; Hertweck C; Dittmann E. Harnessing the evolvability of tricyclic microviridins to dissect protease–inhibitor interactions. Angew. Chem. Int. Ed. Engl 2014, 53 (14), 3735–3738. [DOI] [PubMed] [Google Scholar]
- (731).Schechter I; Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun 1967, 27 (2), 157–162. [DOI] [PubMed] [Google Scholar]
- (732).Harris JL; Backes BJ; Leonetti F; Mahrus S; Ellman JA; Craik CS Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. U.S.A 2000, 97 (14), 7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (733).Kanaori K; Kamei K; Taniguchi M; Koyama T; Yasui T; Takano R; Imada C; Tajima K; Hara S. Solution structure of marinostatin, a natural ester-linked protein protease inhibitor. Biochemistry 2005, 44 (7), 2462–2468. [DOI] [PubMed] [Google Scholar]
- (734).Rawlings ND; Tolle DP; Barrett AJ Evolutionary families of peptidase inhibitors. Biochem. J 2004, 378 (Pt 3), 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (735).Takeuchi Y; Satow Y; Nakamura KT; Mitsui Y. Refined crystal structure of the complex of subtilisin BPN’ and Streptomyces subtilisin inhibitor at 1.8 Å resolution. J. Mol. Biol 1991, 221 (1), 309–325. [PubMed] [Google Scholar]
- (736).Fujinaga M; Sielecki AR; Read RJ; Ardelt W; Laskowski M; James MNG Crystal and molecular structures of the complex of α-chymotrypsin with its inhibitor turkey ovomucoid third domain at 1.8 Å resolution. J. Mol. Biol 1987, 195 (2), 397–418. [DOI] [PubMed] [Google Scholar]
- (737).Laskowski M; Kato I. Protein inhibitors of proteinases. Annu Rev Biochem 1980, 49, 593–626. [DOI] [PubMed] [Google Scholar]
- (738).Sellers A; Murphy G; Meikle MC; Reynolds JJ Rabbit bone collagenase inhibitor blocks the activity of other neutral metalloproteinases. Biochem. Biophys. Res. Commun 1979, 87 (2), 581–587. [DOI] [PubMed] [Google Scholar]
- (739).Lee C; Lee H; Park J-U; Kim S. Introduction of bifunctionality into the multidomain architecture of the ω-ester-containing peptide plesiocin. Biochemistry 2020, 59 (3), 285–289. [DOI] [PubMed] [Google Scholar]
- (740).Sánchez-Barrena MJ; Martínez-Ripoll M; Gálvez A; Valdivia E; Maqueda M; Cruz V; Albert A. Structure of bacteriocin AS-48: from soluble state to membrane bound state. J. Mol. Biol 2003, 334 (3), 541–549. [DOI] [PubMed] [Google Scholar]
- (741).Martin-Visscher LA; Gong X; Duszyk M; Vederas JC The three-dimensional structure of carnocyclin A reveals that many circular bacteriocins share a common structural motif. J. Biol. Chem 2009, 284 (42), 28674–28681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (742).Acedo JZ; van Belkum MJ; Lohans CT; McKay RT; Miskolzie M; Vederas JC Solution structure of acidocin B, a circular bacteriocin produced by Lactobacillus acidophilus M46. Appl. Environ. Microbiol 2015, 81 (8), 2910–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (743).Sawa N; Zendo T; Kiyofuji J; Fujita K; Himeno K; Nakayama J; Sonomoto K. Identification and characterization of lactocyclicin Q, a novel cyclic bacteriocin produced by Lactococcus sp. strain QU 12. Appl. Environ. Microbiol 2009, 75 (6), 1552–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (744).Masuda Y; Ono H; Kitagawa H; Ito H; Mu F; Sawa N; Zendo T; Sonomoto K. Identification and characterization of leucocyclicin Q, a novel cyclic bacteriocin produced by Leuconostoc mesenteroides TK41401. Appl. Environ. Microbiol 2011, 77 (22), 8164–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (745).Himeno K; Rosengren KJ; Inoue T; Perez RH; Colgrave ML; Lee HS; Chan LY; Henriques ST; Fujita K; Ishibashi N; Zendo T; Wilaipun P; Nakayama J; Leelawatcharamas V; Jikuya H; Craik DJ; Sonomoto K. Identification, characterization, and three-dimensional structure of the novel circular bacteriocin, enterocin NKR-5–3B, from Enterococcus faecium. Biochemistry 2015, 54 (31), 4863–4876. [DOI] [PubMed] [Google Scholar]
- (746).Chen H; Hoover DG Bacteriocins and their food applications. Comp. Rev. Food Sci. Food Safety 2003, 2 (3), 82–100. [DOI] [PubMed] [Google Scholar]
- (747).Gálvez A; Maqueda M; Valdivia E; Quesada A; Montoya E. Characterization and partial purification of a broad spectrum antibiotic AS-48 produced by Streptococcus faecalis. Can. J. Microbiol 1986, 32 (10), 765–771. [DOI] [PubMed] [Google Scholar]
- (748).Gong X; Martin-Visscher LA; Nahirney D; Vederas JC; Duszyk M. The circular bacteriocin, carnocyclin A, forms anion-selective channels in lipid bilayers. Biochim. Biophys. Acta 2009, 1788 (9), 1797–1803. [DOI] [PubMed] [Google Scholar]
- (749).Gonzalez C; Langdon GM; Bruix M; Galvez A; Valdivia E; Maqueda M; Rico M. Bacteriocin AS-48, a microbial cyclic polypeptide structurally and functionally related to mammalian NK-lysin. Proc. Natl. Acad. Sci. U.S.A 2000, 97 (21), 11221–11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (750).Liepinsh E; Andersson M; Ruysschaert JM; Otting G. Saposin fold revealed by the NMR structure of NK-lysin. Nat. Struct. Biol 1997, 4 (10), 793–795. [DOI] [PubMed] [Google Scholar]
- (751).Andersson M; Gunne H; Agerberth B; Boman A; Bergman T; Olsson B; Dagerlind A; Wigzell H; Boman HG; Gudmundsson GH NK-lysin, structure and function of a novel effector molecule of porcine T and NK cells. Vet. Immunol. Immunopathol 1996, 54 (1–4), 123–126. [DOI] [PubMed] [Google Scholar]
- (752).Miteva M; Andersson M; Karshikoff A; Otting G. Molecular electroporation: a unifying concept for the description of membrane pore formation by antibacterial peptides, exemplified with NK-lysin. FEBS Lett. 1999, 462 (1–2), 155–158. [DOI] [PubMed] [Google Scholar]
- (753).Gutiérrez P; Li Y; Osborne MJ; Pomerantseva E; Liu Q; Gehring K. Solution structure of the carbon storage regulator protein CsrA from Escherichia coli. J. Bacteriol 2005, 187 (10), 3496–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (754).Jiménez MA; Barrachi-Saccilotto AC; Valdivia E; Maqueda M; Rico M. Design, NMR characterization and activity of a 21-residue peptide fragment of bacteriocin AS-48 containing its putative membrane interacting region. J. Pept. Sci 2005, 11 (1), 29–36. [DOI] [PubMed] [Google Scholar]
- (755).Cruz VL; Ramos J; Melo MN; Martinez-Salazar J. Bacteriocin AS-48 binding to model membranes and pore formation as revealed by coarse-grained simulations. Biochim. Biophys. Acta Biomembr 2013, 1828 (11), 2524–2531. [DOI] [PubMed] [Google Scholar]
- (756).Zhao H; Sood R; Jutila A; Bose S; Fimland G; Nissen-Meyer J; Kinnunen PKJ Interaction of the antimicrobial peptide pheromone plantaricin A with model membranes: implications for a novel mechanism of action. Biochim. Biophys. Acta Biomembr 2006, 1758 (9), 1461–1474. [DOI] [PubMed] [Google Scholar]
- (757).Ciaffoni F; Salvioli R; Tatti M; Arancia G; Crateri P; Vaccaro AM Saposin D solubilizes anionic phospholipid-containing membranes. J. Biol. Chem 2001, 276 (34), 31583–31589. [DOI] [PubMed] [Google Scholar]
- (758).Gebai A; Gorelik A; Nagar B. Crystal structure of saposin D in an open conformation. J. Struct. Biol 2018, 204 (2), 145–150. [DOI] [PubMed] [Google Scholar]
- (759).Borrero J; Brede DA; Skaugen M; Diep DB; Herranz C; Nes IF; Cintas LM; Hernández PE Characterization of garvicin ML, a novel circular bacteriocin produced by Lactococcus garvieae DCC43, isolated from mallard ducks (Anas platyrhynchos). Appl. Environ. Microbiol 2011, 77 (1), 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (760).Gabrielsen C; Brede DA; Hernández PE; Nes IF; Diep DB The maltose ABC transporter in Lactococcus lactis facilitates high-level sensitivity to the circular bacteriocin garvicin ML. Antimicrob. Agents Chemother 2012, 56 (6), 2908–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (761).Martin-Visscher LA; van Belkum MJ; Garneau-Tsodikova S; Whittal RM; Zheng J; McMullen LM; Vederas JC Isolation and characterization of carnocyclin A, a novel circular bacteriocin produced by Carnobacterium maltaromaticum UAL307. Appl. Environ. Microbiol 2008, 74 (15), 4756–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (762).Kemperman R; Kuipers A; Karsens H; Nauta A; Kuipers O; Kok J. Identification and characterization of two novel clostridial bacteriocins, circularin A and closticin 574. Appl. Environ. Microbiol 2003, 69 (3), 1589–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (763).Wirawan RE; Swanson KM; Kleffmann T; Jack RW; Tagg JR Uberolysin: a novel cyclic bacteriocin produced by Streptococcus uberis. Microbiology 2007, 153 (5), 1619–1630. [DOI] [PubMed] [Google Scholar]
- (764).Scholz R; Vater J; Budiharjo A; Wang Z; He Y; Dietel K; Schwecke T; Herfort S; Lasch P; Borriss R. Amylocyclicin, a novel circular bacteriocin produced by Bacillus amyloliquefaciens FZB42. J. Bacteriol 2014, 196 (10), 1842–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (765).van Heel AJ; Montalban-Lopez M; Oliveau Q; Kuipers OP Genome-guided identification of novel head-to-tail cyclized antimicrobial peptides, exemplified by the discovery of pumilarin. Microb. Genom 2017, 3 (10), e000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (766).Kawai Y; Saito T; Kitazawa H; Itoh T. Gassericin A; an uncommon cyclic bacteriocin produced by Lactobacillus gasseri LA39 linked at N - and C -terminal ends. Biosci. Biotechnol. Biochem 1998, 62 (12), 2438–2440. [DOI] [PubMed] [Google Scholar]
- (767).ten Brink B; Minekus M; van der Vossen JMBM; Leer RJ; Veld JHJH in’t. Antimicrobial activity of lactobacilli: preliminary characterization and optimization of production of acidocin B, a novel bacteriocin produced by Lactobacillus acidophilus M46. J. Appl. Microbiol 1994, 77 (2), 140–148. [DOI] [PubMed] [Google Scholar]
- (768).Kalmokoff ML; Teather RM Isolation and characterization of a bacteriocin (butyrivibriocin AR10) from the ruminal anaerobe Butyrivibrio fibrisolvens AR10: evidence in support of the widespread occurrence of bacteriocin-like activity among ruminal isolates of B. fibrisolvens. Appl. Environ. Microbiol 1997, 63 (2), 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (769).Borrero J; Kelly E; O’Connor PM; Kelleher P; Scully C; Cotter PD; Mahony J; van Sinderen D. Plantaricyclin A, a novel circular bacteriocin produced by Lactobacillus plantarum NI326: purification, characterization, and heterologous production. Appl. Environ. Microbiol 2018, 84 (1), e01801–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (770).Suzukake-Tsuchiya K; Hori M; Shimada N; Hamada M. Mode of action of deoxypheganomycin D on Mycobacterium smegmatis ATCC 607. J. Antibiot 1988, 41 (5), 675–683. [DOI] [PubMed] [Google Scholar]
- (771).Noike M; Matsui T; Ooya K; Sasaki I; Ohtaki S; Hamano Y; Maruyama C; Ishikawa J; Satoh Y; Ito H; Morita H; Dairi T. A peptide ligase and the ribosome cooperate to synthesize the peptide pheganomycin. Nat. Chem. Biol 2015, 11 (1), 71–76. [DOI] [PubMed] [Google Scholar]
- (772).Ooya K; Ogasawara Y; Noike M; Dairi T. Identification and analysis of the resorcinomycin biosynthetic gene cluster. Biosci. Biotechnol. Biochem 2015, 79 (11), 1833–1837. [DOI] [PubMed] [Google Scholar]
- (773).Masaki S; Konishi T; Tsuji N; Shoji J. New antibiotics, resorcinomycins A and B: antibacterial activity of resorcinomycin A against mycobacteria in vitro. J. Antibiot 1989, 42 (3), 463–466. [DOI] [PubMed] [Google Scholar]
- (774).Ogasawara Y; Ooya K; Fujimori M; Noike M; Dairi T. Structure and activity relationships of the anti-mycobacterium antibiotics resorcinomycin and pheganomycin. J. Antibiot 2016, 69 (2), 119–120. [DOI] [PubMed] [Google Scholar]
- (775).Maurer CK; Fruth M; Empting M; Avrutina O; Hoßmann J; Nadmid S; Gorges J; Herrmann J; Kazmaier U; Dersch P; Müller R; Hartmann RW Discovery of the first small-molecule CsrA-RNA interaction inhibitors using biophysical screening technologies. Future Med. Chem 2016, 8 (9), 931–947. [DOI] [PubMed] [Google Scholar]
- (776).Viehrig K; Surup F; Volz C; Herrmann J; Abou Fayad A; Adam S; Köhnke J; Trauner D; Müller R. Structure and biosynthesis of crocagins: polycyclic posttranslationally modified ribosomal peptides from Chondromyces crocatus. Angew. Chem. Int. Ed. Engl 2017, 56 (26), 7407–7410. [DOI] [PubMed] [Google Scholar]
- (777).McClure WR Mechanism and control of transcription initiation in prokaryotes. Annu. Rev. Biochem 1985, 54, 171–204. [DOI] [PubMed] [Google Scholar]
- (778).van Hijum SAFT; Medema MH; Kuipers OP Mechanisms and evolution of control logic in prokaryotic transcriptional regulation. Microbiol. Mol. Biol. Rev 2009, 73 (3), 481–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (779).Van Assche E; Van Puyvelde S; Vanderleyden J; Steenackers HP RNA-binding proteins involved in post-transcriptional regulation in bacteria. Front. Microbiol 2015, 6, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (780).Irie Y; Starkey M; Edwards AN; Wozniak DJ; Romeo T; Parsek MR Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol. Microbiol 2010, 78 (1), 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (781).Ren B; Shen H; Lu ZJ; Liu H; Xu Y. The phzA2-G2 transcript exhibits direct RsmA-mediated activation in Pseudomonas aeruginosa M18. PLoS One 2014, 9 (2), e89653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (782).Figueroa-Bossi N; Schwartz A; Guillemardet B; D’Heygère F; Bossi L; Boudvillain M. RNA remodeling by bacterial global regulator CsrA promotes Rho-dependent transcription termination. Genes Dev. 2014, 28 (11), 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (783).Yakhnin AV; Baker CS; Vakulskas CA; Yakhnin H; Berezin I; Romeo T; Babitzke P. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol. Microbiol 2013, 87 (4), 851–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (784).Romeo T; Gong M; Liu MY; Brun-Zinkernagel AM Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J. Bacteriol 1993, 175 (15), 4744–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (785).Cui Y; Chatterjee A; Liu Y; Dumenyo CK; Chatterjee AK Identification of a global repressor gene, rsmA, of Erwinia carotovora subsp. carotovora that controls extracellular enzymes, N-(3-oxohexanoyl)-L-homoserine lactone, and pathogenicity in soft-rotting Erwinia spp. J. Bacteriol 1995, 177 (17), 5108–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (786).Baker CS; Morozov I; Suzuki K; Romeo T; Babitzke P. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol. Microbiol 2002, 44 (6), 1599–1610. [DOI] [PubMed] [Google Scholar]
- (787).Vakulskas CA; Potts AH; Babitzke P; Ahmer BMM; Romeo T. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol. Mol. Biol. Rev 2015, 79 (2), 193–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (788).Stepper J; Shastri S; Loo TS; Preston JC; Novak P; Man P; Moore CH; Havlíček V; Patchett ML; Norris GE Cysteine S -glycosylation, a new post-translational modification found in glycopeptide bacteriocins. FEBS Lett. 2011, 585 (4), 645–650. [DOI] [PubMed] [Google Scholar]
- (789).Palaniappan K; Chen I-MA; Chu K; Ratner A; Seshadri R; Kyrpides NC; Ivanova NN; Mouncey NJ IMG-ABC v.5.0: an update to the IMG/atlas of biosynthetic gene clusters knowledgebase. Nucleic Acids Res. 2020, 48 (D1), D422–D430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (790).Ren H; Biswas S; Ho S; van der Donk WA; Zhao H. Rapid discovery of glycocins through pathway refactoring in Escherichia coli. ACS Chem. Biol 2018, 13 (10), 2966–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (791).Paik SH; Chakicherla A; Hansen JN Identification and characterization of the structural and transporter genes for, and the chemical and biological properties of, sublancin 168, a novel lantibiotic produced by Bacillus subtilis 168. J. Biol. Chem 1998, 273 (36), 23134–42. [DOI] [PubMed] [Google Scholar]
- (792).Oman TJ; Boettcher JM; Wang H; Okalibe XN; van der Donk WA Sublancin is not a lantibiotic but an S-linked glycopeptide. Nat. Chem. Biol 2011, 7 (2), 78–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (793).Kouwen TRHM; Trip EN; Denham EL; Sibbald MJJB; Dubois J-YF; van Dijl JM The large mechanosensitive channel MscL determines bacterial susceptibility to the bacteriocin sublancin 168. Antimicrob. Agents Chemother 2009, 53 (11), 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (794).Garcia De Gonzalo CV; Denham EL; Mars RAT; Stülke J; van der Donk WA; van Dijl JM The phosphoenolpyruvate:sugar phosphotransferase system Is involved in sensitivity to the glucosylated bacteriocin sublancin. Antimicrob. Agents Chemother 2015, 59 (11), 6844–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (795).Biswas S; Garcia De Gonzalo CV; Repka LM; van der Donk WA Structure–activity relationships of the S-linked glycocin sublancin. ACS Chem. Biol 2017, 12 (12), 2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (796).Wu C; Biswas S; Garcia De Gonzalo CV; van der Donk WA Investigations into the mechanism of action of sublancin. ACS Infect. Dis 2019, 5 (3), 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (797).Biswas S; Wu C; van der Donk WA The antimicrobial activity of the glycocin sublancin is dependent on an active phosphoenolpyruvate-sugar phosphotransferase system. ACS Infect. Dis 2021, 7 (8), 2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (798).Li J; Chen J; Yang G; Tao L. Sublancin protects against methicillin-resistant Staphylococcus aureus infection by the combined modulation of innate immune response and microbiota. Peptides 2021, 141, 170533. [DOI] [PubMed] [Google Scholar]
- (799).Bisset SW; Yang S-H; Amso Z; Harris PWR; Patchett ML; Brimble MA; Norris GE Using chemical synthesis to probe structure–activity relationships of the glycoactive bacteriocin glycocin F. ACS Chem. Biol 2018, 13 (5), 1270–1278. [DOI] [PubMed] [Google Scholar]
- (800).Amso Z; Bisset SW; Yang S-H; Harris PWR; Wright TH; Navo CD; Patchett ML; Norris GE; Brimble MA Total chemical synthesis of glycocin F and analogues: S -glycosylation confers improved antimicrobial activity. Chem. Sci 2018, 9 (6), 1686–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (801).Main P; Hata T; Loo TS; Man P; Novak P; Havlíček V; Norris GE; Patchett ML Bacteriocin ASM1 is an O/S -diglycosylated, plasmid-encoded homologue of glycocin F. FEBS Lett. 2020, 594 (7), 1196–1206. [DOI] [PubMed] [Google Scholar]
- (802).Nagar R; Rao A. An iterative glycosyltransferase EntS catalyzes transfer and extension of O- and S-linked monosaccharide in enterocin 96. Glycobiology 2017, 27 (8), 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (803).Maky MA; Ishibashi N; Zendo T; Perez RH; Doud JR; Karmi M; Sonomoto K. Enterocin F4–9, a novel O -linked glycosylated bacteriocin. Appl. Environ. Microbiol 2015, 81 (14), 4819–4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (804).Kaunietis A; Buivydas A; Čitavičius DJ; Kuipers OP Heterologous biosynthesis and characterization of a glycocin from a thermophilic bacterium. Nat. Commun 2019, 10 (1), 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (805).Mew TW; Alaverez AM; Leach JE; Swings J. Focus on Bacterial Blight of Rice. Plant Dis. 1993, 77 (1), 5–12. [Google Scholar]
- (806).Niño-Liu DO; Ronald PC; Bogdanove AJ Xanthomonas oryzae pathovars: model pathogens of a model crop. Mol. Plant Pathol 2006, 7 (5), 303–324. [DOI] [PubMed] [Google Scholar]
- (807).Khush G, S.; Tabien, R., E. A new resistance gene to bacterial blight derived from O. longistaminata. Rice Genet. Newsl 1990, 7, 121–122. [Google Scholar]
- (808).Song WY; Wang GL; Chen LL; Kim HS; Pi LY; Holsten T; Gardner J; Wang B; Zhai WX; Zhu LH; Fauquet C; Ronald P. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 1995, 270 (5243), 1804–1806. [DOI] [PubMed] [Google Scholar]
- (809).Wang GL; Song WY; Ruan DL; Sideris S; Ronald PC The cloned gene, Xa21, confers resistance to multiple Xanthomonas oryzae pv. oryzae isolates in transgenic plants. Mol. Plant Microbe Interact 1996, 9 (9), 850–855. [DOI] [PubMed] [Google Scholar]
- (810).da Silva FG; Shen Y; Dardick C; Burdman S; Yadav RC; de Leon AL; Ronald PC Bacterial genes involved in type I secretion and sulfation are required to elicit the rice Xa21 -mediated innate immune response. Mol. Plant Microbe Interact 2004, 17 (6), 593–601. [DOI] [PubMed] [Google Scholar]
- (811).Pruitt RN; Schwessinger B; Joe A; Thomas N; Liu F; Albert M; Robinson MR; Chan LJG; Luu DD; Chen H; Bahar O; Daudi A; De Vleesschauwer D; Caddell D; Zhang W; Zhao X; Li X; Heazlewood JL; Ruan D; Majumder D; Chern M; Kalbacher H; Midha S; Patil PB; Sonti RV; Petzold CJ; Liu CC; Brodbelt JS; Felix G; Ronald PC The rice immune receptor XA21 recognizes a tyrosine-sulfated protein from a Gram-negative bacterium. Sci. Adv 2015, 1 (6), e1500245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (812).Wei T; Chern M; Liu F; Ronald PC Suppression of bacterial infection in rice by treatment with a sulfated peptide: suppression of bacterial infection in rice. Mol. Plant Pathol 2016, 17 (9), 1493–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (813).Jones JDG; Dangl JL The plant immune system. Nature 2006, 444 (7117), 323–329. [DOI] [PubMed] [Google Scholar]
- (814).Shiu S-H; Bleecker AB Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol. 2003, 132 (2), 530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (815).Shiu S-H; Karlowski WM; Pan R; Tzeng Y-H; Mayer KFX; Li W-H Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell 2004, 16 (5), 1220–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (816).Ronald PC The molecular basis of disease resistance in rice. Plant Mol. Biol 1997, 35 (1–2), 179–186. [PubMed] [Google Scholar]
- (817).Xu W-H; Wang Y-S; Liu G-Z; Chen X; Tinjuangjun P; Pi L-Y; Song W-Y The autophosphorylated Ser686, Thr688, and Ser689 residues in the intracellular juxtamembrane domain of XA21 are implicated in stability control of rice receptor-like kinase. Plant J. 2006, 45 (5), 740–751. [DOI] [PubMed] [Google Scholar]
- (818).Peng Y; Bartley LE; Chen X; Dardick C; Chern M; Ruan R; Canlas PE; Ronald PC OsWRKY62 is a negative regulator of basal and XA21-mediated defense against Xanthomonas oryzae pv. oryzae in rice. Mol. Plant 2008, 1 (3), 446–458. [DOI] [PubMed] [Google Scholar]
- (819).Park C-J; Peng Y; Chen X; Dardick C; Ruan D; Bart R; Canlas PE; Ronald PC Rice XB15, a protein phosphatase 2C, negatively regulates cell death and XA21-mediated innate immunity. PLoS Biol. 2008, 6 (9), e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (820).Wang Y-S; Pi L-Y; Chen X; Chakrabarty PK; Jiang J; De Leon AL; Liu G-Z; Li L; Benny U; Oard J; Ronald PC; Song W-Y Rice XA21 binding protein 3 is a ubiquitin ligase required for full Xa21-mediated disease resistance. Plant Cell. 2006, 18 (12), 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (821).Jiang Y; Chen X; Ding X; Wang Y; Chen Q; Song W-Y The XA21 binding protein XB25 is required for maintaining XA21-mediated disease resistance. Plant J. 2013, 73 (5), 814–823. [DOI] [PubMed] [Google Scholar]
- (822).Lee I; Seo Y-S; Coltrane D; Hwang S; Oh T; Marcotte EM; Ronald PC Genetic dissection of the biotic stress response using a genome-scale gene network for rice. Proc. Natl. Acad. Sci. U.S.A 2011, 108 (45), 18548–18553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (823).Park C-J; Han S-W; Chen X; Ronald PC Elucidation of XA21-mediated innate immunity. Cell. Microbiol 2010, 12 (8), 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (824).Seo Y-S; Chern M; Bartley LE; Han M; Jung K-H; Lee I; Walia H; Richter T; Xu X; Cao P; Bai W; Ramanan R; Amonpant F; Arul L; Canlas PE; Ruan R; Park C-J; Chen X; Hwang S; Jeon J-S; Ronald PC Towards establishment of a rice stress response interactome. PLoS Genet. 2011, 7 (4), e1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (825).Sharma R; De Vleesschauwer D; Sharma MK; Ronald PC Recent advances in dissecting stress-regulatory crosstalk in rice. Mol. Plant 2013, 6 (2), 250–260. [DOI] [PubMed] [Google Scholar]
- (826).Luu DD; Joe A; Chen Y; Parys K; Bahar O; Pruitt R; Chan LJG; Petzold CJ; Long K; Adamchak C; Stewart V; Belkhadir Y; Ronald PC Biosynthesis and secretion of the microbial sulfated peptide RaxX and binding to the rice XA21 immune receptor. Proc. Natl. Acad. Sci. U.S.A 2019, 116 (17), 8525–8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (827).Amano Y; Tsubouchi H; Shinohara H; Ogawa M; Matsubayashi Y. Tyrosine-sulfated glycopeptide involved in cellular proliferation and expansion in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A 2007, 104 (46), 18333–18338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (828).Pruitt RN; Joe A; Zhang W; Feng W; Stewart V; Schwessinger B; Dinneny JR; Ronald PC A microbially derived tyrosine-sulfated peptide mimics a plant peptide hormone. New Phytol. 2017, 215 (2), 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (829).Holliday GL; Akiva E; Meng EC; Brown SD; Calhoun S; Pieper U; Sali A; Booker SJ; Babbitt PC Atlas of the radical SAM superfamily: divergent evolution of function using a “plug and play” domain. Methods Enzymol. 2018, 606, 1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (830).Oberg N; Precord TW; Mitchell DA; Gerlt JA RadicalSAM.org: a resource to interpret sequence-function space and discover new radical SAM enzyme chemistry. ACS Bio Med Chem Au 2022, 2 (1), 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (831).Mahanta N; Hudson GA; Mitchell DA Radical S-adenosylmethionine enzymes involved in RiPP biosynthesis. Biochemistry 2017, 56 (40), 5229–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (832).Hamada T; Sugawara T; Matsunaga S; Fusetani N. Polytheonamides, unprecedented highly cytotoxic polypeptides, from the marine sponge Theonella swinhoei: 1. Isolation and component amino acids. Tetrahedron Lett. 1994, 35 (5), 719–720. [Google Scholar]
- (833).Hamada T; Sugawara T; Matsunaga S; Fusetani N. Polytheonamides, unprecedented highly cytotoxic polypeptides from the marine sponge Theonella swinhoei 2. Structure elucidation. Tetrahedron Lett. 1994, 35 (4), 609–612. [Google Scholar]
- (834).Hamada T; Matsunaga S; Yano G; Fusetani N. Polytheonamides A and B, highly cytotoxic, linear polypeptides with unprecedented structural features, from the marine sponge, Theonella swinhoei. J. Am. Chem. Soc 2005, 127 (1), 110–118. [DOI] [PubMed] [Google Scholar]
- (835).Oiki S; Muramatsu I; Matsunaga S; Fusetani N. A channel-forming peptide toxin: polytheonamide from marine sponge Theonella swinhoei. Nihon Yakurigaku Zasshi 1997, 110 (Suppl 1), 195–198. [DOI] [PubMed] [Google Scholar]
- (836).Hamada T; Matsunaga S; Fujiwara M; Fujita K; Hirota H; Schmucki R; Güntert P; Fusetani N. Solution structure of polytheonamide B, a highly cytotoxic nonribosomal polypeptide from marine sponge. J. Am. Chem. Soc 2010, 132 (37), 12941–12945. [DOI] [PubMed] [Google Scholar]
- (837).Iwamoto M; Shimizu H; Muramatsu I; Oiki S. A cytotoxic peptide from a marine sponge exhibits ion channel activity through vectorial-insertion into the membrane. FEBS Lett. 2010, 584 (18), 3995–3999. [DOI] [PubMed] [Google Scholar]
- (838).Matsuoka S; Shinohara N; Takahashi T; Iida M; Inoue M. Functional analysis of synthetic substructures of polytheonamide B: a transmembrane channel-forming peptide. Angew. Chem. Int. Ed. Engl 2011, 123 (21), 4981–4985. [DOI] [PubMed] [Google Scholar]
- (839).Shinohara N; Itoh H; Matsuoka S; Inoue M. Selective modification of the N-terminal structure of polytheonamide B significantly changes its cytotoxicity and activity as an ion channel. ChemMedChem 2012, 7 (10), 1770–1773. [DOI] [PubMed] [Google Scholar]
- (840).Hayata A; Itoh H; Matsutaka S; Inoue M. Dual chemical modification of a polytheonamide mimic: rational design and synthesis of ion-channel-forming 48-mer peptides with potent cytotoxicity. Chemistry 2016, 22 (10), 3370–3377. [DOI] [PubMed] [Google Scholar]
- (841).Itoh H; Matsutaka S; Kuranaga T; Inoue M. Control of the cytotoxicity of dansylated polytheonamide mimic, an artificial peptide ion channel, by modification of the N-terminal structure. Tetrahedron Lett. 2014, 55 (3), 728–731. [Google Scholar]
- (842).Itoh H; Matsuoka S; Kreir M; Inoue M. Design, synthesis and functional analysis of dansylated polytheonamide mimic: an artificial peptide ion channel. J. Am. Chem. Soc 2012, 134 (34), 14011–14018. [DOI] [PubMed] [Google Scholar]
- (843).Itoh H; Inoue M. Structural permutation of potent cytotoxin, polytheonamide B: discovery of cytotoxic peptide with altered activity. ACS Med. Chem. Lett 2013, 4 (1), 52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (844).Renevey A; Riniker S. The importance of N-methylations for the stability of the β6·3-helical-helical conformation of polytheonamide B. Eur. Biophys. J 2017, 46 (4), 363–374. [DOI] [PubMed] [Google Scholar]
- (845).Iwamoto M; Matsunaga S; Oiki S. Paradoxical one-ion pore behavior of the long β-helical peptide of marine cytotoxic polytheonamide B. Sci. Rep 2014, 4 (1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (846).Kalathingal M; Sumikama T; Mori T; Oiki S; Saito S. Structure and dynamics of solvent molecules inside the polytheonamide B channel in different environments: a molecular dynamics study. Phys. Chem. Chem. Phys 2018, 20 (5), 3334–3348. [DOI] [PubMed] [Google Scholar]
- (847).Matsuki Y; Iwamoto M; Mita K; Shigemi K; Matsunaga S; Oiki S. Rectified proton grotthuss conduction across a long water-wire in the test nanotube of the polytheonamide B channel. J. Am. Chem. Soc 2016, 138 (12), 4168–4177. [DOI] [PubMed] [Google Scholar]
- (848).Hayata A; Itoh H; Inoue M. Solid-phase total synthesis and dual mechanism of action of the channel-forming 48-mer peptide polytheonamide B. J. Am. Chem. Soc 2018, 140 (33), 10602–10611. [DOI] [PubMed] [Google Scholar]
- (849).Xue Y-W; Hayata A; Itoh H; Inoue M. Biological effects of a simplified synthetic analogue of ion-channel-forming polytheonamide B on plasma membrane and lysosomes. Chemistry 2019, 25 (66), 15198–15204. [DOI] [PubMed] [Google Scholar]
- (850).Imai Y; Meyer KJ; Iinishi A; Favre-Godal Q; Green R; Manuse S; Caboni M; Mori M; Niles S; Ghiglieri M; Honrao C; Ma X; Guo JJ; Makriyannis A; Linares-Otoya L; Böhringer N; Wuisan ZG; Kaur H; Wu R; Mateus A; Typas A; Savitski MM; Espinoza JL; O’Rourke A; Nelson KE; Hiller S; Noinaj N; Schäberle TF; D’Onofrio A; Lewis K. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576 (7787), 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (851).Noinaj N; Kuszak AJ; Gumbart JC; Lukacik P; Chang H; Easley NC; Lithgow T; Buchanan SK Structural insight into the biogenesis of β-barrel membrane proteins. Nature 2013, 501 (7467), 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (852).Iadanza MG; Higgins AJ; Schiffrin B; Calabrese AN; Brockwell DJ; Ashcroft AE; Radford SE; Ranson NA Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM. Nat. Commun 2016, 7 (1), 12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (853).Han L; Zheng J; Wang Y; Yang X; Liu Y; Sun C; Cao B; Zhou H; Ni D; Lou J; Zhao Y; Huang Y. Structure of the BAM complex and its implications for biogenesis of outermembrane proteins. Nat. Struct. Mol. Biol 2016, 23 (3), 192–196. [DOI] [PubMed] [Google Scholar]
- (854).Ni D; Wang Y; Yang X; Zhou H; Hou X; Cao B; Lu Z; Zhao X; Yang K; Huang Y. Structural and functional analysis of the β-barrel domain of BamA from Escherichia coli. FASEB J. 2014, 28 (6), 2677–2685. [DOI] [PubMed] [Google Scholar]
- (855).Gu Y; Li H; Dong H; Zeng Y; Zhang Z; Paterson NG; Stansfeld PJ; Wang Z; Zhang Y; Wang W; Dong C. Structural basis of outer membrane protein insertion by the BAM complex. Nature 2016, 531 (7592), 64–69. [DOI] [PubMed] [Google Scholar]
- (856).Noinaj N; Kuszak AJ; Balusek C; Gumbart JC; Buchanan SK Lateral opening and exit pore formation are required for BamA function. Structure 2014, 22 (7), 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (857).Hartmann J-B; Zahn M; Burmann IM; Bibow S; Hiller S. Sequence-specific solution NMR assignments of the β-barrel insertase BamA to monitor its conformational ensemble at the atomic level. J. Am. Chem. Soc 2018, 140 (36), 11252–11260. [DOI] [PubMed] [Google Scholar]
- (858).Kaur H; Hartmann J-B; Jakob RP; Zahn M; Zimmermann I; Maier T; Seeger MA; Hiller S. Identification of conformation-selective nanobodies against the membrane protein insertase BamA by an integrated structural biology approach. J. Biomol. NMR 2019, 73 (6–7), 375–384. [DOI] [PubMed] [Google Scholar]
- (859).Kaur H; Jakob RP; Marzinek JK; Green R; Imai Y; Bolla JR; Agustoni E; Robinson CV; Bond PJ; Lewis K; Maier T; Hiller S. The antibiotic darobactin mimics a β-strand to inhibit outer membrane insertase. Nature 2021, 593 (7857), 125–129. [DOI] [PubMed] [Google Scholar]
- (860).Böhringer N; Green R; Liu Y; Mettal U; Marner M; Modaresi SM; Jakob RP; Wuisan ZG; Maier T; Iinishi A; Hiller S; Lewis K; Schäberle TF Mutasynthetic production and antimicrobial characterization of darobactin analogs. Microbiol. Spectr 2021, 9 (3), e01535–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (861).Groß S; Panter F; Pogorevc D; Seyfert CE; Deckarm S; Bader CD; Herrmann J; Müller R. Improved broad-spectrum antibiotics against Gram-negative pathogens via darobactin biosynthetic pathway engineering. Chem. Sci 2021, 12 (35), 11882–11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (862).Liu W-T; Yang Y-L; Xu Y; Lamsa A; Haste NM; Yang JY; Ng J; Gonzalez D; Ellermeier CD; Straight PD; Pevzner PA; Pogliano J; Nizet V; Pogliano K; Dorrestein PC Imaging mass spectrometry of intraspecies metabolic exchange revealed the cannibalistic factors of Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A 2010, 107 (37), 16286–16290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (863).Mo T; Ji X; Yuan W; Mandalapu D; Wang F; Zhong Y; Li F; Chen Q; Ding W; Deng Z; Yu S; Zhang Q. Thuricin Z: A narrow-spectrum sactibiotic that targets the cell membrane. Angew. Chem. Int. Ed. Engl 2019, 131 (52), 18969–18973. [DOI] [PubMed] [Google Scholar]
- (864).Babasaki K; Takao T; Shimonishi Y; Kurahashi K. Subtilosin A, a new antibiotic peptide produced by Bacillus subtilis 168: isolation, structural analysis, and biogenesis. J. Biochem 1985, 98 (3), 585–603. [DOI] [PubMed] [Google Scholar]
- (865).Zheng G; Slavik MF Isolation, partial purification and characterization of a bacteriocin produced by a newly isolated Bacillus subtilis strain. Lett. Appl. Microbiol 1999, 28 (5), 363–367. [DOI] [PubMed] [Google Scholar]
- (866).Rea MC; Sit CS; Clayton E; O’Connor PM; Whittal RM; Zheng J; Vederas JC; Ross RP; Hill C. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc. Natl. Acad. Sci. U.S.A 2010, 107 (20), 9352–9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (867).Rea MC; Dobson A; O’Sullivan O; Crispie F; Fouhy F; Cotter PD; Shanahan F; Kiely B; Hill C; Ross RP Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. U.S.A 2011, 108 (Supplement 1), 4639–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (868).Sutyak Noll K; Sinko PJ; Chikindas ML Elucidation of the molecular mechanisms of action of the natural antimicrobial peptide subtilosin against the bacterial vaginosisassociated pathogen Gardnerella vaginalis. Probiotics Antimicrob. Proteins 2011, 3 (1), 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (869).Kuijk S. van; Noll KS; Chikindas ML The species-specific mode of action of the antimicrobial peptide subtilosin against Listeria monocytogenes Scott A. Lett. Appl. Microbiol 2012, 54 (1), 52–58. [DOI] [PubMed] [Google Scholar]
- (870).Duarte AF de S; Ceotto-Vigoder H; Barrias ES; Souto-Padrón TCBS; Nes IF; do C. de F. Bastos M Hyicin 4244, the first sactibiotic described in staphylococci, exhibits an anti-staphylococcal biofilm activity. Int. J. Antimicrob. Agents 2018, 51 (3), 349–356. [DOI] [PubMed] [Google Scholar]
- (871).Sutyak KE; Wirawan RE; Aroutcheva AA; Chikindas ML Isolation of the Bacillus subtilis antimicrobial peptide subtilosin from the dairy product-derived Bacillus amyloliquefaciens. J. Appl. Microbiol 2008, 104 (4), 1067–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (872).Amrouche T; Sutyak Noll K; Wang Y; Huang Q; Chikindas ML Antibacterial activity of subtilosin alone and combined with curcumin, poly-lysine and zinc lactate against Listeria monocytogenes strains. Probiotics Antimicrob. Proteins 2010, 2 (4), 250–257. [DOI] [PubMed] [Google Scholar]
- (873).Thennarasu S; Lee D-K; Poon A; Kawulka KE; Vederas JC; Ramamoorthy A. Membrane permeabilization, orientation, and antimicrobial mechanism of subtilosin A. Chem. Phys. Lipids 2005, 137 (1), 38–51. [DOI] [PubMed] [Google Scholar]
- (874).Wang G; Feng G; Snyder AB; Manns DC; Churey JJ; Worobo RW Bactericidal thurincin H causes unique morphological changes in Bacillus cereus F4552 without affecting membrane permeability. FEMS. Microbiol. Lett 2014, 357 (1), 69–76. [DOI] [PubMed] [Google Scholar]
- (875).Mathur H; Fallico V; O’Connor PM; Rea MC; Cotter PD; Hill C; Ross RP Insights into the mode of action of the sactibiotic thuricin CD. Front. Microbiol 2017, 8, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (876).Shelburne CE; An FY; Dholpe V; Ramamoorthy A; Lopatin DE; Lantz MS The spectrum of antimicrobial activity of the bacteriocin subtilosin A. J. Antimicrob. Chemother 2006, 59 (2), 297–300. [DOI] [PubMed] [Google Scholar]
- (877).Huang T; Geng H; Miyyapuram VR; Sit CS; Vederas JC; Nakano MM Isolation of a variant of subtilosin A with hemolytic activity. J. Bacteriol 2009, 191 (18), 5690–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (878).Marx R; Stein T; Entian K-D; Glaser SJ Structure of the Bacillus subtilis peptide antibiotic subtilosin A determined by 1H-NMR and matrix assisted laser desorption/ionization time-of-flight mass spectrometry. J. Protein. Chem 2001, 20 (6), 501–506. [DOI] [PubMed] [Google Scholar]
- (879).Kawulka KE; Sprules T; Diaper CM; Whittal RM; McKay RT; Mercier P; Zuber P; Vederas JC Structure of subtilosin A, a cyclic antimicrobial peptide from Bacillus subtilis with unusual sulfur to α-carbon cross-links: formation and reduction of α-thio-α-amino acid derivatives,. Biochemistry 2004, 43 (12), 3385–3395. [DOI] [PubMed] [Google Scholar]
- (880).Silkin L; Hamza S; Kaufman S; Cobb SL; Vederas JC Spermicidal bacteriocins: lacticin 3147 and subtilosin A. Bioorganic Med. Chem. Lett 2008, 18 (10), 3103–3106. [DOI] [PubMed] [Google Scholar]
- (881).Sutyak KE; Anderson RA; Dover SE; Feathergill KA; Aroutcheva AA; Faro S; Chikindas ML Spermicidal activity of the safe natural antimicrobial peptide subtilosin. Infect. Dis. Obstet. Gynecol 2008, 2008, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (882).Torres NI; Noll KS; Xu S; Li J; Huang Q; Sinko PJ; Wachsman MB; Chikindas ML Safety, formulation and in vitro antiviral activity of the antimicrobial peptide subtilosin against herpes simplex virus type 1. Probiotics Antimicrob. Proteins 2013, 5 (1), 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (883).Algburi A; Zehm S; Netrebov V; Bren AB; Chistyakov V; Chikindas ML Subtilosin prevents biofilm formation by inhibiting bacterial quorum sensing. Probiotics Antimicrob. Proteins 2017, 9 (1), 81–90. [DOI] [PubMed] [Google Scholar]
- (884).McClean KH; Winson MK; Fish L; Taylor A; Chhabra SR; Camara M; Daykin M; Lamb JH; Swift S; Bycroft BW; Stewart GSAB; Williams P. Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology 143 (12), 3703–3711. [DOI] [PubMed] [Google Scholar]
- (885).Waters CM; Bassler BL Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol 2005, 21 (1), 319–346. [DOI] [PubMed] [Google Scholar]
- (886).Sit CS; McKay RT; Hill C; Ross RP; Vederas JC The 3D structure of thuricin CD, a two-component bacteriocin with cysteine sulfur to α-carbon cross-links. J. Am. Chem. Soc 2011, 133 (20), 7680–7683. [DOI] [PubMed] [Google Scholar]
- (887).Lee H; Churey JJ; Worobo RW Biosynthesis and transcriptional analysis of thurincin H, a tandem repeated bacteriocin genetic locus, produced by Bacillus thuringiensis SF361. FEMS. Microbiol. Lett 2009, 299 (2), 205–213. [DOI] [PubMed] [Google Scholar]
- (888).Wang G; Manns DC; Guron GK; Churey, John J; Worobo RW Large-scale purification, characterization, and spore outgrowth inhibitory effect of thurincin H, a bacteriocin produced by Bacillus thuringiensis SF361. Probiotics Antimicrob. Proteins 2014, 6 (2), 105–113. [DOI] [PubMed] [Google Scholar]
- (889).Burbulys D; Trach KA; Hoch JA Initiation of sporulation in B. subtilis is controlled by a multicomponent phosphorelay. Cell 1991, 64 (3), 545–552. [DOI] [PubMed] [Google Scholar]
- (890).Fawcett P; Eichenberger P; Losick R; Youngman P. The transcriptional profile of early to middle sporulation in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A 2000, 97 (14), 8063–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (891).González-Pastor JE; Hobbs EC; Losick R. Cannibalism by sporulating bacteria. Science 2003, 301 (5632), 510–513. [DOI] [PubMed] [Google Scholar]
- (892).Crost EH; Ajandouz EH; Villard C; Geraert PA; Puigserver A; Fons M. Ruminococcin C, a new anti-Clostridium perfringens bacteriocin produced in the gut by the commensal bacterium Ruminococcus gnavus E1. Biochimie 2011, 93 (9), 1487–1494. [DOI] [PubMed] [Google Scholar]
- (893).Balty C; Guillot A; Fradale L; Brewee C; Boulay M; Kubiak X; Benjdia A; Berteau O. Ruminococcin C, an anti-clostridial sactipeptide produced by a prominent member of the human microbiota Ruminococcus gnavus. J. Biol. Chem 2019, 294 (40), 14512–14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (894).Roblin C; Chiumento S; Bornet O; Nouailler M; Müller CS; Jeannot K; Basset C; Kieffer-Jaquinod S; Couté Y; Torelli S; Le Pape L; Schünemann V; Olleik H; De La Villeon B; Sockeel P; Di Pasquale E; Nicoletti C; Vidal N; Poljak L; Iranzo O; Giardina T; Fons M; Devillard E; Polard P; Maresca M; Perrier J; Atta M; Guerlesquin F; Lafond M; Duarte V. The Unusual Structure of Ruminococcin C1 Antimicrobial Peptide Confers Clinical Properties. Proc. Natl. Acad. Sci. U.S.A 2020, 117 (32), 19168–19177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (895).Bushin LB; Covington BC; Rued BE; Federle MJ; Seyedsayamdost MR Discovery and biosynthesis of streptosactin, a sactipeptide with an alternative topology encoded by commensal bacteria in the human microbiome. J. Am. Chem. Soc 2020, 142 (38), 16265–16275. [DOI] [PubMed] [Google Scholar]
- (896).Ramare F; Nicoli J; Dabard J; Corring T; Ladire M; Gueugneau AM; Raibaud P. Trypsin-dependent production of an antibacterial substance by a human Peptostreptococcus strain in gnotobiotic rats and in vitro. Appl. Environ. Microbiol 1993, 59 (9), 2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (897).Chiumento S; Roblin C; Kieffer-Jaquinod S; Tachon S; Leprètre C; Basset C; Aditiyarini D; Olleik H; Nicoletti C; Bornet O; Iranzo O; Maresca M; Hardré R; Fons M; Giardina T; Devillard E; Guerlesquin F; Couté Y; Atta M; Perrier J; Lafond M; Duarte V. Ruminococcin C, a promising antibiotic produced by a human gut symbiont. Sci. Adv 2019, 5 (9), eaaw9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (898).Roblin C; Chiumento S; Jacqueline C; Pinloche E; Nicoletti C; Olleik H; CourvoisierDezord E; Amouric A; Basset C; Dru L; Ollivier M; Bogey-Lambert A; Vidal N; Atta M; Maresca M; Devillard E; Duarte V; Perrier J; Lafond M. The Multifunctional Sactipeptide Ruminococcin C1 Displays Potent Antibacterial Activity In Vivo as Well as Other Beneficial Properties for Human Health. IJMS 2021, 22 (6), 3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (899).Fontaine L; Boutry C; de Frahan MH; Delplace B; Fremaux C; Horvath P; Boyaval P; Hols P. A novel pheromone quorum-sensing system controls the development of natural competence in Streptococcus thermophilus and Streptococcus salivarius. J. Bacteriol 2010, 192 (5), 1444–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (900).Butcher BG; Lin Y-P; Helmann JD The yydFGHIJ operon of Bacillus subtilis encodes a peptide that induces the LiaRS two-component system. J. Bacteriol 2007, 189 (23), 8616–8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (901).Benjdia A; Guillot A; Ruffié P; Leprince J; Berteau O. Post-translational modification of ribosomally synthesized peptides by a radical SAM epimerase in Bacillus subtilis. Nat. Chem 2017, 9 (7), 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (902).Claverys J-P; Håvarstein LS Cannibalism and fratricide: mechanisms and raisons d’être. Nat. Rev. Microbiol 2007, 5 (3), 219–229. [DOI] [PubMed] [Google Scholar]
- (903).González-Pastor JE Cannibalism: a social behavior in sporulating Bacillus subtilis. FEMS. Microbiol. Rev 2011, 35 (3), 415–424. [DOI] [PubMed] [Google Scholar]
- (904).Popp PF; Mascher T. Coordinated cell death in isogenic bacterial populations: sacrificing some for the benefit of many? J. Mol. Biol 2019, 431 (23), 4656–4669. [DOI] [PubMed] [Google Scholar]
- (905).Jordan S; Junker A; Helmann JD; Mascher T. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J. Bacteriol 2006, 188 (14), 5153–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (906).Cao M; Wang T; Ye R; Helmann JD Antibiotics that inhibit cell wall biosynthesis induce expression of the Bacillus subtilis σW and σM regulons: antibiotic induction of ECF σ factor regulons. Mol. Microbiol 2002, 45 (5), 1267–1276. [DOI] [PubMed] [Google Scholar]
- (907).Wolf D; Kalamorz F; Wecke T; Juszczak A; Mäder U; Homuth G; Jordan S; Kirstein J; Hoppert M; Voigt B; Hecker M; Mascher T. In-depth profiling of the LiaR response of Bacillus subtilis. J. Bacteriol 2010, 192 (18), 4680–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (908).Domínguez-Escobar J; Wolf D; Fritz G; Höfler C; Wedlich-Söldner R; Mascher T. Subcellular localization, interactions and dynamics of the phage-shock protein-like Lia response in Bacillus subtilis. Mol. Microbiol 2014, 92 (4), 716–732. [DOI] [PubMed] [Google Scholar]
- (909).Popp PF; Friebel L; Benjdia A; Guillot A; Berteau O; Mascher T. The epipeptide biosynthesis locus epeXEPAB Is widely distributed in Firmicutes and triggers intrinsic cell envelope stress. Microb. Physiol 2021, 31 (3), 306–318. [DOI] [PubMed] [Google Scholar]
- (910).Popp PF; Benjdia A; Strahl H; Berteau O; Mascher T. The epipeptide YydF intrinsically triggers the cell envelope stress response of Bacillus subtilis and causes severe membrane perturbations. Front. Microbiol 2020, 11, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (911).Zheng G; Yan LZ; Vederas JC; Zuber P. Genes of the sbo-alb locus of Bacillus subtilis are required for production of the antilisterial bacteriocin subtilosin. J. Bacteriol 1999, 181 (23), 7346–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (912).Zheng G; Hehn R; Zuber P. Mutational analysis of the sbo-alb locus of Bacillus subtilis: identification of genes required for subtilosin production and immunity. J. Bacteriol 2000, 182 (11), 3266–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (913).Ellermeier CD; Hobbs EC; Gonzalez-Pastor JE; Losick R. A three-protein signaling pathway governing immunity to a bacterial cannibalism toxin. Cell 2006, 124 (3), 549–559. [DOI] [PubMed] [Google Scholar]
- (914).Albano M; Smits WK; Ho LTY; Kraigher B; Mandic-Mulec I; Kuipers OP; Dubnau D. The Rok protein of Bacillus subtilis represses genes for cell surface and extracellular functions. J. Bacteriol 2005, 187 (6), 2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (915).Guiral S; Mitchell TJ; Martin B; Claverys J-P Competence-programmed predation of noncompetent cells in the human pathogen Streptococcus pneumoniae: Genetic requirements. Proc. Natl. Acad. Sci. U.S.A 2005, 102 (24), 8710–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (916).Gilmore MS; Haas W. The selective advantage of microbial fratricide. Proc. Natl. Acad. Sci. U.S.A 2005, 102 (24), 8401–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (917).Wei H; Håvarstein LS Fratricide Is essential for efficient gene transfer between Pneumococci in biofilms. Appl. Environ. Microbiol 2012, 78 (16), 5897–5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (918).Comella N; Grossman AD Conservation of genes and processes controlled by the quorum response in bacteria: characterization of genes controlled by the quorum-sensing transcription factor ComA in Bacillus subtilis: quorum sensing by ComX-ComP-ComA in B. subtilis. Mol. Microbiol 2005, 57 (4), 1159–1174. [DOI] [PubMed] [Google Scholar]
- (919).Ayikpoe R; Govindarajan V; Latham JA Occurrence, function, and biosynthesis of mycofactocin. Appl. Microbiol. Biotechnol 2019, 103 (7), 2903–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (920).Haft DH Bioinformatic evidence for a widely distributed, ribosomally produced electron carrier precursor, its maturation proteins, and its nicotinoprotein redox partners. BMC Genom. 2011, 12 (1), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (921).Haft DH; Pierce PG; Mayclin SJ; Sullivan A; Gardberg AS; Abendroth J; Begley DW; Phan IQ; Staker BL; Myler PJ; Marathias VM; Lorimer DD; Edwards TE Mycofactocin-associated mycobacterial dehydrogenases with non-exchangeable NAD cofactors. Sci. Rep 2017, 7 (1), 41074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (922).Griffin JE; Gawronski JD; DeJesus MA; Ioerger TR; Akerley BJ; Sassetti CM High-resolution phenotypic profiling defines genes essential for Mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7 (9), e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (923).Krishnamoorthy G; Kaiser P; Lozza L; Hahnke K; Mollenkopf H-J; Kaufmann SHE Mycofactocin Is associated with ethanol metabolism in Mycobacteria. mBio 2019, 10 (3), e00190–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (924).Dubey AA; Jain V. Mycofactocin is essential for the establishment of methylotrophy in Mycobacterium smegmatis. Biochem. Biophys. Res. Commun 2019, 516 (4), 1073–1077. [DOI] [PubMed] [Google Scholar]
- (925).Peña-Ortiz L; Graça AP; Guo H; Braga D; Köllner TG; Regestein L; Beemelmanns C; Lackner G. Structure elucidation of the redox cofactor mycofactocin reveals oligo-glycosylation by MftF. Chem. Sci 2020, 11 (20), 5182–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (926).Ayikpoe RS; Latham JA MftD catalyzes the formation of a biologically active redox center in the biosynthesis of the ribosomally synthesized and post-translationally modified redox cofactor mycofactocin. J. Am. Chem. Soc 2019, 141 (34), 13582–13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (927).Peña-Ortiz L; Graça AP; Guo H; Braga D; Köllner TG; Regestein L; Beemelmanns C; Lackner G. Structure elucidation of the redox cofactor mycofactocin reveals oligo-glycosylation by MftF. Chem. Sci 2020, 11 (20), 5182–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (928).Duine JA; Frank J; Jongejan JA Enzymology of quinoproteins. Adv. Enzymol. Relat. Areas. Mol. Biol 1987, 59, 169–212. [DOI] [PubMed] [Google Scholar]
- (929).Oubrie A; Dijkstra BW Structural requirements of pyrroloquinoline quinone dependent enzymatic reactions. Protein. Sci 2000, 9 (7), 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (930).Klinman JP; Bonnot F. Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem. Rev 2014, 114 (8), 4343–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (931).Takeda K; Umezawa K; Várnai A; Eijsink VG; Igarashi K; Yoshida M; Nakamura N. Fungal PQQ-dependent dehydrogenases and their potential in biocatalysis. Curr. Opin. Chem. Biol 2019, 49, 113–121. [DOI] [PubMed] [Google Scholar]
- (932).Sarmiento-Pavía PD; Sosa-Torres ME Bioinorganic insights of the PQQ-dependent alcohol dehydrogenases. J. Biol. Inorg. Chem 2021, 26 (2–3), 177–203. [DOI] [PubMed] [Google Scholar]
- (933).Ongey EL; Yassi H; Pflugmacher S; Neubauer P. Pharmacological and pharmacokinetic properties of lanthipeptides undergoing clinical studies. Biotechnol. Lett 2017, 39 (4), 473–482. [DOI] [PubMed] [Google Scholar]
- (934).Geng M; Ravichandran A; Escano J; Smith L. Efficacious analogs of the lantibiotic mutacin 1140 against a systemic methicillin-resistant Staphylococcus aureus Infection. Antimicrob. Agents Chemother 2018, 62 (12), e01626–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (935).Pulse ME; Weiss WJ; Kers JA; DeFusco AW; Park JH; Handfield M. Pharmacological, toxicological, and dose range assessment of OG716, a novel lantibiotic for the treatment of Clostridium difficile-associated infection. Antimicrob. Agents Chemother 2019, 63 (4), e01904–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (936).Sandiford SK Current developments in lantibiotic discovery for treating Clostridium difficile infection. Expert Opin. Drug Discov 2019, 14 (1), 71–79. [DOI] [PubMed] [Google Scholar]
- (937).Soltani S; Zirah S; Rebuffat S; Couture F; Boutin Y; Biron E; Subirade M; Fliss I. Gastrointestinal stability and cytotoxicity of bacteriocins From Gram-positive and Gram-negative bacteria: a comparative in vitro study. Front. Microbiol 2021, 12, 780355. [DOI] [PMC free article] [PubMed] [Google Scholar]