

Abstract
Small cell lung cancer (SCLC) is a recalcitrant malignancy that urgently needs new therapies. Four master transcription factors (ASCL1, NEUROD1, POU2F3, and YAP1) have been identified in SCLC, and each defines the transcriptome landscape of one molecular subtype. However, these master transcription factors have not been found directly druggable. We hypothesized that blocking their transcriptional coactivator(s) could provide an alternative approach to target these master transcription factors. Here, we identify that BET proteins physically interact with NEUROD1 and function as transcriptional coactivators. Using CRISPR knockout and ChIP-seq, we demonstrate that NEUROD1 plays a critical role in defining the landscapes of BET proteins in the SCLC genome. Blocking BET proteins by inhibitors led to broad suppression of the NEUROD1-target genes, especially those associated with superenhancers, resulting in the inhibition of SCLC growth in vitro and in vivo. LSAMP, a membrane protein in the IgLON family, was identified as one of the NEUROD1-target genes mediating BET inhibitor sensitivity in SCLC. Altogether, our study reveals that BET proteins are essential in regulating NEUROD1 transactivation and are promising targets in SCLC-N subtype tumors.
Keywords: Epigenetic, Superenhancer, Biomarker, LSAMP
Introduction
Small cell lung cancer (SCLC) is a recalcitrant malignancy with few treatment options (1). Despite recent advances in therapeutics, durable responses are typically infrequent in extensive-stage SCLC, and the prognosis remains dismal (2,3). Growing evidence has suggested that SCLC is a heterogeneous group of cancers. A recently proposed classification divided it into four molecular subtypes (SCLC-A, SCLC-N, SCLC-P, and SCLC-Y) on the basis of the expression of several master transcription factors (TFs), namely ASCL1, NEUROD1, POU2F3, and YAP1 (4,5). While the existence of the SCLC-Y subtype tumors is still debatable, several studies have confirmed the presence of the tumors in three other molecular subtypes (6,7). Emerging evidence suggests that various molecular subtypes of SCLC have different therapeutic susceptibilities, likely because of their distinct transcriptome profiles dictated by the master TFs (8,9). There has been substantial interest in targeting specific SCLC subtypes (8,10), but these master TFs have not been found directly druggable.
NEUROD1 is a neurogenic basic helix-loop-helix TF and plays a critical role in neuronal differentiation (11). It is vital for tumorigenesis by promoting migration and invasion of SCLC through NTRK2 (previously known as TrkB) and NCAM (12). Although both regulate neuroendocrine genes in SCLC, NEUROD1 and ASCL1 share only 5-7% of the occupied genomic regions (13). It is still unclear how NEUROD1 activates its downstream gene transcription and what genes, and to what degree, are dependent on it in SCLC. Such information is critical for developing new strategies to target this master TF.
The bromodomain and extraterminal domain (BET) family of proteins, including BRD2, BRD3, BRD4, and BRDT, are involved in multiple aspects of transcriptional regulation (14). Each of these proteins has two bromodomains (BDs) at the N-terminus essential for binding to active chromatin (14,15). BET inhibitors (BETis) selectively bind to these BDs and block BET proteins from accessing active chromatin, causing suppression of gene transcription (16,17). Although BET proteins are broadly associated with gene promoters and enhancers, BETi only inhibits a subset of genes, particularly those with unusually high levels of BRD4 occupancy at their enhancer sites known as superenhancers (SEs) (18,19).
We hypothesized that targeting the transcriptional cofactors could effectively block the transcriptional activity of the master TFs in SCLC. Here we demonstrated that BET proteins function as the transcriptional coactivators of NEUROD1 and are promising therapeutic targets in the SCLC-N subtype tumors.
Materials and Methods
Cell Culture.
All SCLC lines used in this study were purchased from commercial vendors (Supplementary Table S1). Cells were cultured in recommended media and maintained in a humidified incubator at 37 °C with 5% CO2. Cells were used for experiments within 15 passages after revival from cryopreservation. LX33 SCLC line was established by culturing tumor cells isolated from an LX33 SCLC PDX in HITES media (see Supplementary Table S1 for media composition). All cell lines tested negative for mycoplasma, and the short tandem repeat analysis was performed to authenticate commercial cell lines (last tested in November 2021).
CRISPR Knockout.
CRISPR was performed using a standard two-step method (20). Briefly, a Cas9 expression vector, lentiCas9-Blast (Addgene, #52962, RRID: Addgene_52962; a gift from Feng Zhang (21)), was transfected into H446 and DMS-273 cells by lipofection. The cells were selected with 1 (DMS-273) and 2.5 (H446) μg/ml blasticidin (Gibco). Subsequently, the Cas9-expressing cells were infected with lentiviral particles containing gRNA-carrying vectors targeting NEUROD1 (Sigma): HS5000024297 (abbreviated as #297), GAGTCCTCCTCTGCGTTCATGG; HS5000024298 (#298), GAGGAGGAGGACGAAGATGAGG. Transduction was performed at a multiplicity of infection (MOI) of 2 in the presence of 5 μg/ml polybrene (Sigma). Three days after transduction, the cells were selected with 0.5 (DMS-273) and 2.5 (H446) μg/ml puromycin (Gibco) for 2-3 months to enable clone expansion. Afterward, the KO of NEUROD1 was verified by western blot and Sanger sequencing and then maintained in the complete media without puromycin before any subsequent experiments.
RNA Sequencing (RNA-seq).
Total RNA libraries (COR-L279) and mRNA libraries (H446 and LX22) were constructed using a TruSeq Stranded Total RNA Kit (Illumina, RS-122-2201) and an mRNA Library Prep kit V2 (Illumina, RS-122-2001), respectively. Paired-end sequencing of the constructed libraries was performed on a HiSeq 4000 system (Illumina), and the raw fastq data were analyzed using the CCBR/Pipeliner (22). A false-discovery rate of 0.05 was set as a cutoff to define significant alterations in gene expression.
Gene Set Enrichment Analysis (GSEA).
GSEA was performed using GSEA (v.4.0.2; RRID:SCR_003199) under the setting of gene_set permutation at 1,000 times, with the DESeq2 normalized counts as data input. The NEUROD1 gene signature comprises 384 NEUROD1-target genes that showed at least 4-fold alterations in gene expression upon NEUROD1 KO in H446 cells (Supplementary Table S2).
ChIP-sequencing (ChIP-seq).
ChIP-seq was performed as described previously (23). For each assay, 4 – 10 μg antibodies were used: α-NEUROD1, Cell Signaling Technology, #4373, RRID:AB_10549071; α-BRD2, Cell Signaling Technology, #5848, RRID:AB_10835146; α-BRD3, Bethyl, A302-368A, RRID:AB_1907251; α-BRD4, Bethyl, A700-004, RRID:AB_2631885; α-H3K27Ac, Abcam, ab4729, RRID:AB_2118291. The ChIP-seq analysis was detailed in supplemental methods.
siRNA Transfection and Screen.
LSAMP siRNAs (#S8299 and #S8301; ThermoFisher Scientific) were transfected into cells using Lipofectamine RNAiMax (Thermo Scientific) at a final concentration of 20nM. The siRNA library that contained 90 siRNA targeting 45 genes of interest was custom synthesized by Qiagen.
Plasmid Transfection and Establishment of Stable Transformant.
Plasmids were transfected into cells using Lipofectamine 3000 reagent (ThermoFisher Scientific) following a standard protocol. LSAMP stable transformants were established by transfecting COR-L279 cells with a tagged ORF clone of this gene (Origene; # RC207618), followed by selection with G418 (1mg/ml; Roche) in semi-solid media (ClonaCell™-TCS medium, Stem Cell Technology). The surviving cell clones were expanded and maintained in complete media with 0.4 mg/ml G418.
Animal Studies.
The animal experiments were approved and performed according to the regulations set by the National Cancer Institute-Bethesda Animal Care and Use Committee. SCLC-N subtype PDX LX22 and LX33 models were generously provided by John T. Poirier and Charles M. Rudin from MSKCC (24, 25). Freshly isolated PDX tumors were dissociated into single-cell suspension using a Human Tumor Dissociation Kit (Miltenyi Biotec) and a gentleMACS™ Octo Dissociator (Miltenyi Biotec). After lyzing red blood cells with ACK lysis buffer (Quality Biological), the remaining cells were washed with ample PBS three times and resuspended in PBS at 5x107 viable cells per ml. One hundred μl of cell suspension (~ 5x106 viable cells) was injected subcutaneously into the right flanks of NOD-SCID mice (6-week-old; both males and females; Charles River Laboratories, RRID:IMSR_JAX:005557). Once the tumor volume reached between 50 and 100 mm3 (average, 14-21 days), mice were randomized and drug treatments were given at specified doses and frequencies. Animals were euthanized if 1) tumor volume was ≥1500 mm3; 2) tumor became ulcerated; 3) 75 days had elapsed after a tumor became palpable.
For the transcriptome analysis of LX22 tumors, a magnetic-activated cell sorting method (26) was employed to separate tumor cells from mouse stromal cells using a Mouse Cell Depletion Kit (Miltenyi Biotec). Subsequently, RNA was extracted from the isolated tumor cells using an RNeasy Mini Kit (Qiagen).
For tumorigenicity assay, H446 NEUROD1-KO cells (gRNA-297, cl.12 and cl.15) and control cells (cl.10) were mixed with equal volumes of Matrigel (Corning). 1x106 cells in 100 μl were inoculated subcutaneously into each flank of athymic mice. Tumors were resected at the specified time intervals.
Statistical Analysis.
Statistical analyses and graphing were performed using GraphPad Prism (RRID: SCR_005012). All tests were performed with a two-sided significance level of 0.05.
Data availability.
RNA-seq and ChIP-seq datasets generated in this study are deposited at NCBI GEO (GSE210114).
Results
NEUROD1 is critical for the growth and tumorigenicity of SCLC-N cell lines.
To determine whether NEUROD1 is necessary for the growth of SCLC-N tumors, we used CRISPR to knock out this gene in H446 cells (an SCLC-N cell line) with two distinct guide RNAs (gRNAs). Western blots confirmed a complete loss of NEUROD1 protein expression in the KO clones (Figs. 1A and S1A). We validated NEUROD1 KO by identifying indels in the targeted NEUROD1 genomic sequences (Supplementary Fig. S1B–C) and finding a drastically decreased expression of two known NEUROD1-target genes - NEUROD2 and NHLH1 (Figs. 1B and S1D).
Figure 1. NEUROD1 is critical for the growth and tumorigenicity of the SCLC-N cell lines.
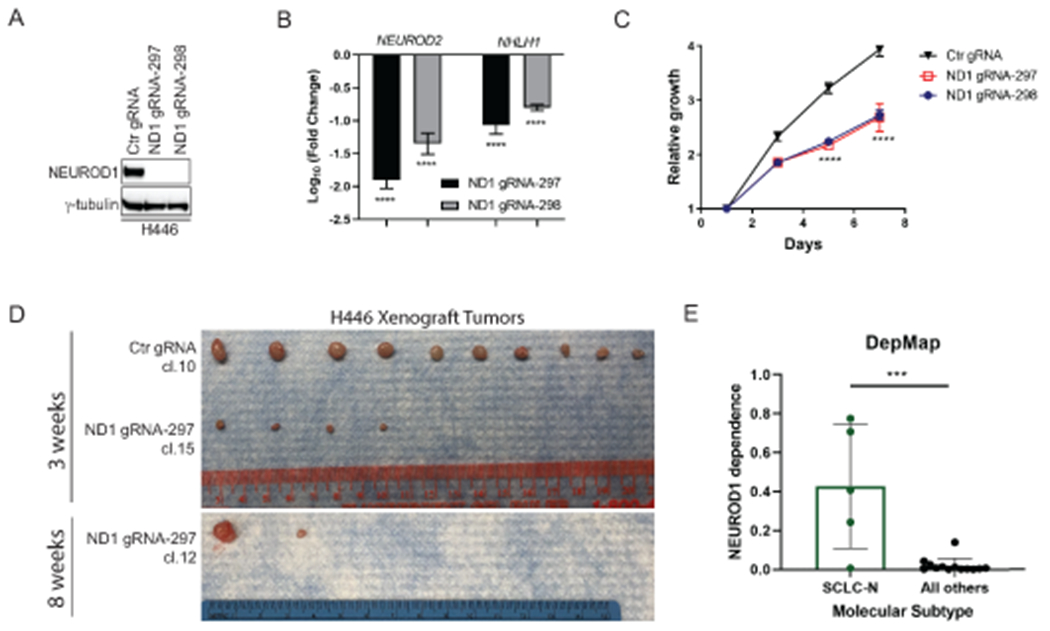
A) Western blot shows a complete loss of NEUROD1 protein expression in two H446 NEUROD1-KO clones, with one representative clone from each gRNA (#297 cl. 15 and #298 cl. S3). B) Expression of two NEUROD1-target genes, NEUROD2 and NHLH1, in two H446 NEUROD1-KO clones. The gene expression was measured by qRT-PCR and plotted relative to control cells. C) The H446 NEUROD1-KO cells (#297 cl. 15 and #298 cl. S3) proliferated substantially slower than control cells. Cell numbers were indirectly measured using the CellTiter Glo 2.0 assay every other day and compared to day one. Error bar represents SD of 4 replicates. D) NEUROD1 KO impaired the tumorigenicity of H446 cells. Top, the xenografts formed three weeks after injection (1x106 H446 control or NEUROD1-KO cells, #297 cl. 15; n=10 injections per group). Bottom, xenografts formed eight weeks after injection (1x106 H446 NEUROD1-KO cells, #297 cl. 12; n=10 injections per group). E) NEUROD1 gene dependency in the SCLC-N versus other molecular subtypes of SCLC lines in the CRISPR dataset (Version 2020, Q2) of the Cancer Dependency Map (DepMap) project (RRID:SCR_017655). The significance of two-group comparisons was determined using the ANOVA test with Dunnett’s multiple test correction (B), the Student’s t-test with multiple test correction (C), or the Student’s t-test (E). ***, P < 0.001; ****, P < 0.0001. Ctr, Control; gRNA, guide RNA; ND1, NEUROD1.
We next assessed the impact of NEUROD1 KO on the growth and tumorigenicity of H446 cells. As shown in Figure 1C, KO of NEUROD1 significantly decreased the proliferation of H446 cells. We profiled the transcriptome of three KO clones and three control clones and compared the expression of cell-cycle-specific genes (27). Supplementary Figure S1E shows almost identical expression patterns of these cell-cycle-specific genes between the KO and control cells, suggesting that the decreased proliferation was not due to cell cycle arrest in the KO cells. Next, we tested the tumorigenicity of the KO cells by injecting them subcutaneously into athymic mice. Compared to control cells, fewer and smaller xenografts formed in the KO group (Fig. 1D, top), and similar results were found when the KO cells were given five additional weeks to grow in vivo (Fig. 1D, bottom). Consistent with these findings, the CRISPR screens in the Dependency Map (DepMap) project confirmed that NEUROD1 is a high-dependency gene in the SCLC-N lines (Fig. 1E). Collectively, these results demonstrate that NEUROD1 is critical for the growth and tumorigenicity of the SCLC-N cell lines.
BET proteins interact with NEUROD1.
We hypothesized that targeting transcriptional coactivator(s) can block the transactivation of NEUROD1. To identify its transcriptional partner(s), we first performed ChIP-sequencing (ChIP-seq) to map NEUROD1 genome occupancy in the H446 control and KO cells. As expected, the NEUROD1/NEUROG2 binding motif was enriched in the genomic sequences corresponding to NEUROD1 peaks (Supplementary Fig. S2A). Most of the NEUROD1 peaks locate in intronic (45.8%) and intergenic regions (45.9%), and the remaining was present at transcription start sites (TSS; 1.12%), promoter-TSS sites (3.88%), and exons + 3’ or 5’ untranslated regions (UTRs; 3.28%) (Fig. 2A). Compared to the NEUROD1 ChIP-seq reported in two other SCLC-N lines (13), H446 shared a substantial number of NEUROD1-occupied genomic regions with H82 and H524, despite each having a high percentage of private binding sites (Supplementary Fig. S2B). A metagene analysis showed that NEUROD1 binds to the genomic regions within seven kilobases upstream of TSS and downstream of transcription end sites, suggesting that it binds to both promoters and distal regulatory elements (e.g., enhancers) (Fig. 2B).
Figure 2. NEUROD1 interacts with BET proteins and defines their genomic landscapes in SCLC.
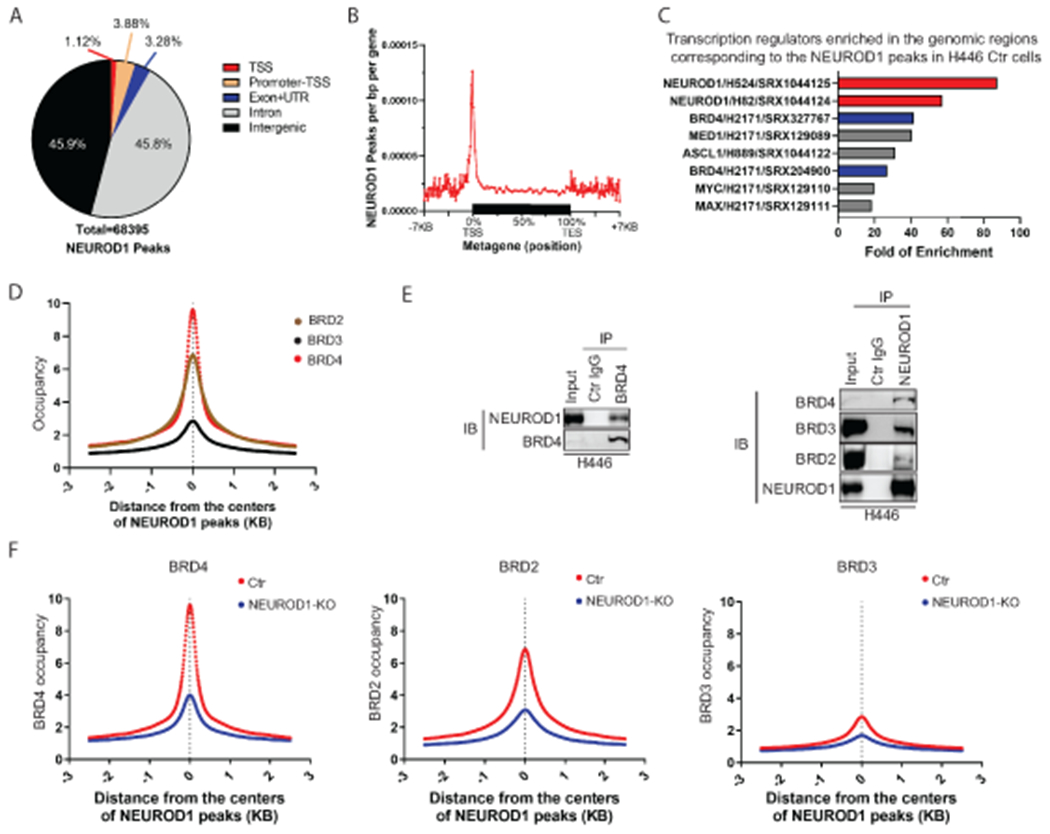
A) A breakdown of the genomic locations of the NEUROD1 peaks identified by ChIP-seq in H446 control cells (control gRNA). B) A metagene profile of NEUROD1 occupancy in H446 control cells. C) The bar graph shows the top eight TF candidates identified by an Enrichment Analysis (ChIP-Atlas, RRID:SCR_015511) that occupy the genomic regions in other lung cells corresponding to the NEUROD1 peaks in H446 control cells. NEUROD1 and BRD4 were highlighted. D) BRD2, BRD3, and BRD4 occupancy near the centers of NEUROD1-occupied regions in H446 control cells (2.5 kilobases [KB] bilateral). E) Co-IP assays using H446 nuclear extracts. Left, detection of NEUROD1 after BRD4 IP; right, detection of BRD2, BRD3, and BRD4 after NEUROD1 IP. F) Effect of NEUROD1 KO on BRD4, BRD2, and BRD3 occupancy near the centers of the NEUROD1-occupied regions in H446 control cells. Ctr, control; KO, knockout; IB, immunoblotting; IP, immunoprecipitation; TSS, transcription start site; TES, transcription end site; UTR, untranslated region.
To identify transcription regulators in the NEUROD1-occupied genomic regions, we performed enrichment analysis using the lung-related ChIP-seq datasets archived at ChIP-atlas.org (28). Among the eight identified candidates, the top two are NEUROD1 in H524 and H82, demonstrating the validity of this approach (Fig. 2C). BRD4 and MED1, two markers of active enhancers, showed high degrees of enrichment. ASCL1, which shares 5-7% of the genomic binding regions with NEUROD1 (13), is also among the identified candidates. We chose BRD4 for further study because there are inhibitors targeting this gene and its association with NEUROD1 has not been previously characterized.
To confirm the colocalization of BRD4 and NEUROD1 in the H446 genome, we performed ChIP-seq to map the occupancies of BRD4 and its two analogs - BRD2 and BRD3. Figure 2D shows that all three BET family proteins colocalized with NEUROD1, with their occupancies peaking at the centers of the NEUROD1-occupied genomic regions. To determine whether NEUROD1 physically interacts with BET proteins, we performed co-immunoprecipitation (co-IP) assays using the nuclear extracts from H446 and H524 cells. Figure 2E and Supplemental Figure S3 show that endogenous NEUROD1 was immunoprecipitated with the BET proteins and vice versa in both cell lines, indicating physical interaction between these proteins. To assess whether BET proteins interact with other master TFs in SCLC, we performed a co-IP assay using the nuclear extract from H1963 cells, an SCLC-A line. Supplementary Figure S4 shows that BRD4 was immunoprecipitated with ASCL1 and vice versa in H1963 cells, demonstrating that BET proteins also interact with ASCL1. The associations between BET proteins and two other master TFs – POU2F3 and YAP1, remain to be determined due to a lack of suitable antibodies for IP.
We next assessed the impact of NEUROD1 KO on the genome occupancies of BET proteins. As shown in Figure 2F, the occupancy of BET proteins decreased substantially in the NEUROD1-occupied genomic regions upon NEUROD1 KO, suggesting that NEUROD1 directs the binding of BET proteins in the SCLC genome. To determine whether these changes are restricted to the NEUROD1-occupied genomic regions, we grouped all BRD4 peaks in H446 control cells on the basis of the presence or absence of NEUROD1 co-occupancy (Supplementary Fig. S5A, density plot 1 vs. 4). NEUROD1 KO abrogated BRD4 occupancy in the genomic regions co-occupied by BRD4 and NEUROD1 (Supplementary Fig. S5A, density plot 3 vs. 2) but did not affect BRD4 occupancy in the genome singly occupied by BRD4 (Supplementary Fig. S5A, density plot 6 vs. 5). Similarly, in supplementary Figure S5B, the aggregate signal plots show that BRD4 occupancy was only affected by NEUROD1 KO in the genomic regions co-occupied by BRD4 and NEUROD1, but not in the locations singly occupied by either of these two proteins. Together these results demonstrate that NEUROD1 defines the landscapes of BET proteins in SCLC-N cell lines.
Targeting BET proteins leads to the suppression of NEUROD1-target genes.
We posited that BET proteins function as the transcriptional coactivators of NEUROD1 and that inhibiting the former proteins would suppress NEUROD1-target genes. To assess the transcriptional activity of NEUROD1, we first identified its target genes in H446 (N=1,216) through an integrated analysis of the gene expression data and the ChIP-seq data using the Binding and Expression Target Analysis (BETA) algorithm (29) (Fig. 3A). This analysis also found that NEUROD1 functions as a transcriptional activator (Fig. 3B). Consistent with a previous report that NEUROD1 plays a crucial role in neuron differentiation (11), Ingenuity pathway analysis identified multiple neuronal-related pathways enriched in the NEUROD1-target genes (Supplementary Fig. S6).
Figure 3. BETi suppresses NEUROD1-target genes, particularly those associated with superenhancers.
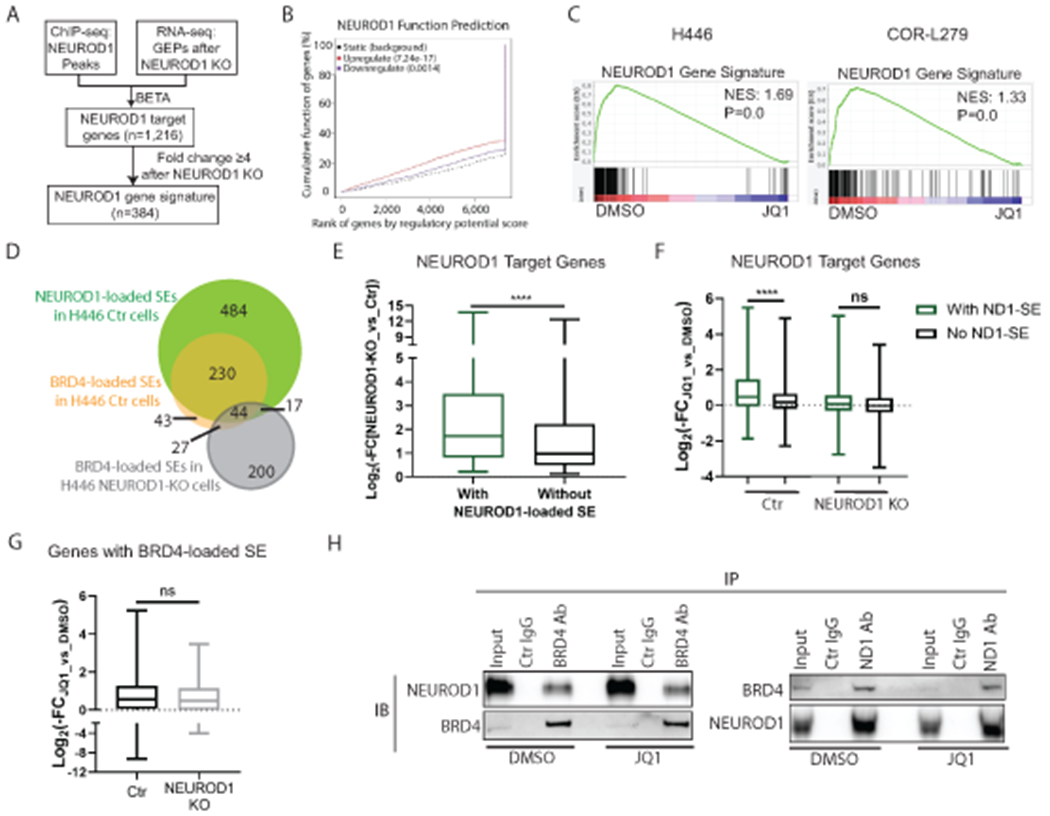
A) A flow chart illustrating the procedures to identify NEUROD1-target genes and to constitute a NEUROD1 gene signature. B) The BETA algorithm predicts that NEUROD1 primarily functions as a transcriptional activator (red curve). C) GSEA plots showing depletion of NEUROD1 gene signature after 24-hr JQ1 treatment in H446 (left; 1 μM) and COR-L279 cells (right; 0.5 μM). D) Venn diagrams show the overlaps between the NEUROD1- and BRD4-loaded SEs in H446 control and NEUROD1-KO cells. E) Comparing the effects of NEUROD1 KO on the expression of the NEUROD1-target genes with NEUROD1-loaded SEs and those without in H446 control cells. F) Differential effects of JQ1 (1μM, 24 hrs) on the expression of the NEUROD1-target genes (with or without the NEUROD1-loaded SEs) in H446 control cells (left two bars) versus the NEUROD1-KO cells (right two bars). G) JQ1 (1μM, 24 hrs) suppressed the genes with BRD4-loaded SEs in H446 control cells (n=307) and those in NEUROD1-KO cells (n=258). H) co-IP of BRD4 or NEUROD1 using the nuclear extract from H446 cells treated with JQ1 (1μM, 6 hrs). Left, detection of NEUROD1 after IP of BRD4. Right, detection of BRD4 after IP of NEUROD1. Whiskers represent minimum to maximum, and boxes show the first, median, and third quartile (E-G). The significance of two-group comparisons was determined using the Student’s t-test (E-G). ****, P<0.0001; ns: not significant. BETA, the Binding and Expression Target Analysis; FC, fold change; GEP, gene expression profile; IB, immunoblotting; IP, immunoprecipitation; NES, normalized enrichment score; SE; superenhancer.
We next selected a subset of NEUROD1-target genes with at least 4-fold alterations in gene expression upon NEUROD1 KO to constitute a NEUROD1 gene signature (n=384; Fig. 3A and Supplemental Table S2). Using the transcriptome data of the SCLC cell lines in the CCLE dataset, we found a significant enrichment of this signature in the SCLC-N lines versus all others (Supplementary Fig. S7). JQ1 significantly depleted this NEUROD1 gene signature in H446 and COR-L279 (an SCLC-N line) (Fig. 3C) without decreasing NEUROD1 expression (Supplementary Fig. S8), supporting our hypothesis that the transcriptional activity of NEUROD1 could be blocked by targeting its coactivators - BET proteins.
BETi preferentially suppresses the NEUROD1-target genes associated with superenhancers.
To determine what subset of NEUROD1-target genes is more susceptible to BETi, we assessed the effect of JQ1 on the genes with SEs. We compared the BRD4- or NEUROD1-loaded SEs in H446 control cells to the BRD4-loaded SEs in the KO cells (Supplementary Fig. S9A–C, and Fig. 3D). Consistent with our conclusion that NEUROD1 defines the landscape of BET proteins in SCLC, we found a substantial overlap between the BRD4- and NEUROD1-loaded SEs in H446 control cells (Fig. 3D), as well as a strong correlation between the NEUROD1 and BRD4 signals in these SE regions (Supplementary Fig. S9D). In contrast, the KO and control cells only shared a few SEs (Fig. 3D).
Among the NEUROD1-target genes, KO of NEUROD1 caused a more substantial suppression of the genes with NEUROD1-loaded SEs than those without (Fig. 3E), suggesting that these SEs are critical in the regulation of gene expression. Similarly, JQ1 suppressed the genes with the NEUROD1-loaded SEs to a greater degree than those without in H446 control cells (Fig. 3F, left two). In contrast, neither of these two lists of genes was affected by JQ1 in the NEUROD1-KO cells (Fig. 3F, right two), suggesting these genes became less dependent on the BET proteins after NEUROD1 KO. To rule out the possibility that BET proteins become unresponsive to BETi in the KO cells due to an unknown cause (e.g., posttranslational modifications), we identified the genes associated with BRD4-loaded SEs in the H446 control cells (n=307) and those in KO cells (n=258). Despite these two lists of genes being vastly different, both were suppressed by JQ1 to the same degree (Fig. 3G), indicating that BET proteins remain sensitive to JQ1 after NEUROD1 KO.
BETi is known to block BET proteins from accessing active chromatin by binding to their BD domains. To evaluate whether BETi disrupts the interaction between BET proteins and NEUROD1, we performed a co-IP assay after treating H446 cells with JQ1. As shown in Figure 3H, the physical interaction between NEUROD1 and BRD4 remained intact despite JQ1 treatment, suggesting that the interaction between these two proteins depends on neither the BD domains of BRD4 nor a chromatin scaffold. Collectively, these results demonstrate that BETi preferentially suppresses the NEUROD1-target genes associated with SEs.
The SCLC-N cell lines are more susceptible to BETi.
On the basis of the above findings, we predicted that the SCLC-N tumors are more sensitive to BETi. We measured NEUROD1 expression and the sensitivity to JQ1 and OTX-015 (a BETi) in 50 SCLC lines. Supplementary Figure S10A shows a strong correlation between the IC50 values of JQ1 and OTX-015, demonstrating that these measurements were reliable. Consistent with our prediction, NEUROD1 is only expressed in the cell lines sensitive to JQ1 using the cutoff of IC50 to JQ1 = 5μM (Fig. 4A). By choosing a different cutoff (IC50 to JQ1 = 1μM), we found NEUROD1 expression remains significantly elevated in the sensitive lines compared to the resistant lines (Supplementary Fig. S10B). By grouping the SCLC lines according to their molecular subtypes, we found that the SCLC-N lines were more susceptible to BETi than the SCLC-A lines (Fig. 4B). The differences between the SCLC-N lines and the SCLC-P or SCLC-Y lines had no statistical significance, likely because of too few cell lines in the latter two groups. Figure 4C shows the JQ1 IC50 values as a function of NEUROD1 expression in all SCLC lines. Using a public high-throughput drug screen dataset (30), we confirmed that the SCLC-N lines are more sensitive to JQ1 than the SCLC-A lines (Supplementary Fig. S10C).
Figure 4. The SCLC-N lines are more susceptible to BETi.
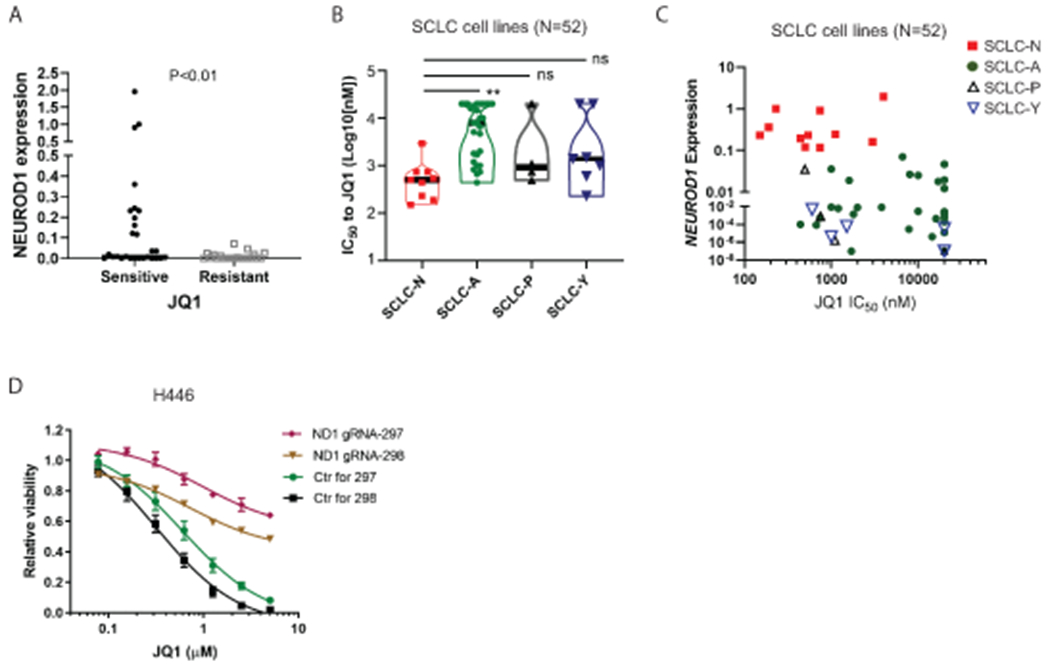
A) NEUROD1 expression was compared between the JQ1-sensitive and -resistant SCLC lines (N=52). An IC50 = 5 μM JQ1 was used as the cutoff to define sensitive and resistant SCLC lines. B) Differential sensitivity to JQ1 (IC50) among four molecular subtypes of SCLC lines (N=52). C) A plot showing NEUROD1 expression at the function of JQ1 IC50 in the 52 SCLC lines included in (A-B). D) Viability of the H446 NEUROD1-KO and control cells after 72-hr JQ1 treatment. The significance of the two-group comparisons was determined using the Student’s t-test (A) and the ANOVA test with Dunnett’s multiple test correction (B). **, P<0.01; ns: not significant.
To further assess the role of NEUROD1 in BETi sensitivity, we compared the response to JQ1 treatment between H446 KO and control cells. Figure 4D shows that NEUROD1 KO led to a more than 20-fold increase in the JQ1’s IC50 in H446 cells. To ensure that these findings are not cell-type specific, we knocked out NEUROD1 in DMS-273 (an SCLC-N line) and found a similar increase in the JQ1’s IC50 value in the KO cells (2.5 μM in the KO cells versus 0.15 μM in control cells; Supplementary Fig. S11A–B). These findings confirm that NEUROD1 is critical in determining BETi sensitivity in SCLC.
MYC expression has been found to predict BETi sensitivity in some but not all tumor types. To examine whether MYC mediates NEUROD1’s regulation of BETi sensitivity, we first assessed MYC expression after NEUROD1 KO. KO of NEUROD1 modestly suppressed MYC gene and protein expression in H446 but not in DMS-273 cells (Supplementary Figure S12A–B). Next, we utilized a reporter to assess the effect of BETi on MYC’s transactivation and found a similar dose- and time-dependent suppression by JQ1 in the H446 KO and control cells (Supplementary Figure S12C). These findings indicate that NEUROD1 regulates BETi sensitivity independent of MYC. Collectively, these results demonstrate that the SCLC-N lines are more susceptible to BETi due to the dependence of NEUROD1 on BET proteins for transcriptional activation.
LSAMP is one of the NEUROD1-target genes mediating BETi sensitivity.
To identify the NEUROD1-target gene(s) that mediates BETi sensitivity, we reasoned that both NEUROD1 and BET proteins should regulate such a gene(s). Therefore, we focused on the NEUROD1-target genes that showed > 2-fold change in expression after BETi treatment, among which we selected 45 genes that had substantial losses of BET proteins occupancy upon NEUROD1 KO for further analysis (Fig. 5A). Through siRNA screens, we found 13 and 14 candidate genes that consistently showed ≥ 20% growth inhibition upon knockdown by two distinct siRNAs in H446 and COR-L279 cells (Fig. 5B; Supplementary Table S3). Among the ten shared candidate genes between these two cell lines (highlighted in red in Fig. 5B), we chose LSAMP (limbic system-associated membrane protein) for further investigation because a higher expression of this gene was associated with a worse prognosis in SCLC patients (Fig. 5C). Next, we confirmed that both NEUROD1 and BET proteins regulate LSAMP gene transcription. Supplementary Figure S13A shows that NEUROD1 occupied the TSS site of LSAMP in H446, H524, and H82 cells. Consistent with these ChIP-seq results, NEUROD1 KO substantially decreased LSAMP gene and protein expression in H446 and DMS-273 cells (Supplementary Fig. S13B–D). Finally, JQ1 decreased LSAMP protein expression in a dose-dependent manner in H446 cells (Supplementary Fig. S13E).
Figure 5. LSAMP, a NEUROD1-target gene, partially mediates BETi sensitivity.
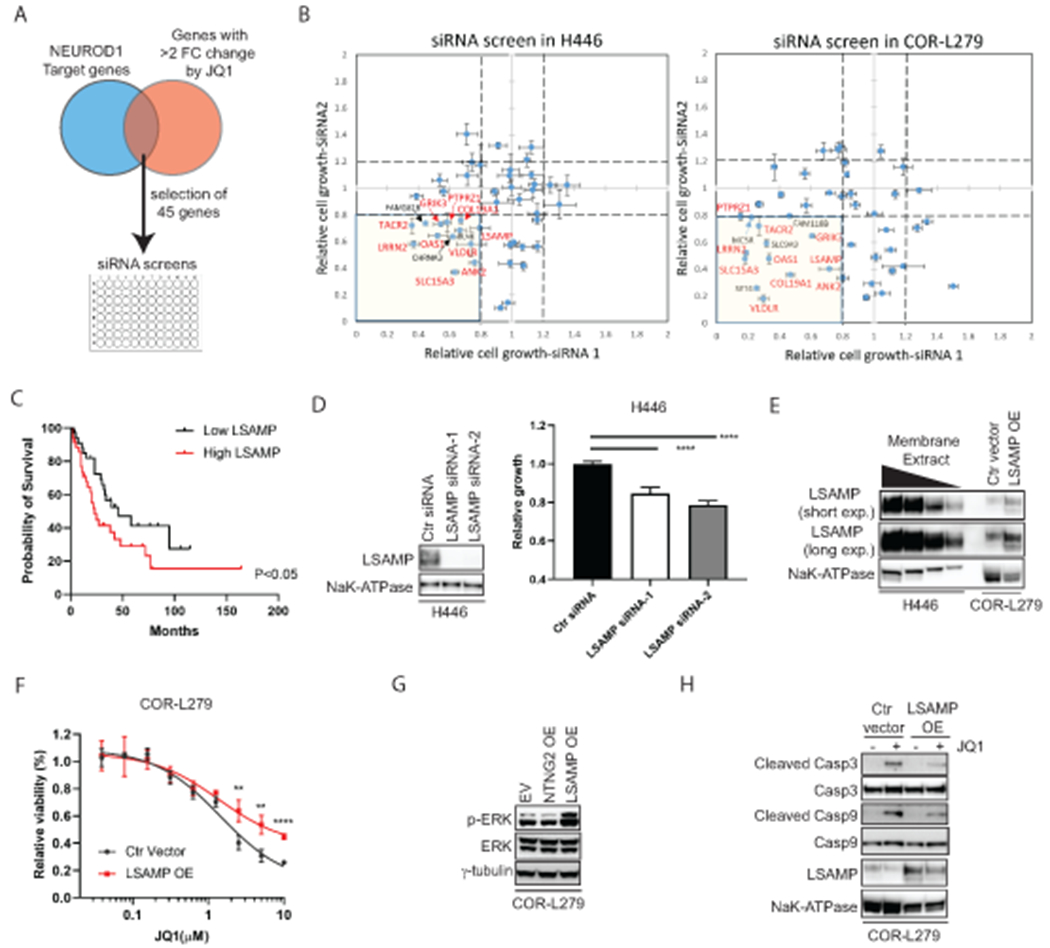
A) A diagram depicts the selection of 45 genes regulated by both NEUROD1 and BET proteins for siRNA screens. B) The results of siRNA screens (72 hrs) in H446 (left) and COR-L279 cells (right). The genes highlighted in red are the shared candidate genes between H446 and COR-L279 cells showing ≥ 20% growth inhibition upon knockdown with each of two distinct siRNAs. C) Kaplan-Meier curves show an inverse correlation between LSAMP expression and the overall survival in SCLC patients (N=69; ref. (39)). The patients were dichotomized on the basis of LSAMP transcript levels in the tumors (top 50% vs. bottom 50%). D) H446 proliferation was suppressed by LSAMP knockdown using two siRNAs distinct from the ones in the initial screens (B). Left, LSAMP expression in the membrane fractions of the knockdown cells. NaK-ATPase serves as a loading control. Right, the effect of LSAMP knockdown on H446 proliferation (measured by the CellTiter Glo 2.0 assay) at 72 hrs post-transfection. E) Stable expression of ectopic LSAMP in COR-L279 cells. A tapering amount of membrane protein from H446 (20, 10, 5, 2.5 μg) was loaded and compared with 20 μg membrane protein from the COR-L279 cells with or without ectopic LSAMP. F) Differential sensitivity to JQ1 (72 hrs) between the COR-L279 cells with ectopic LSAMP and those with control vector. G) Western blots show increased ERK phosphorylation in the COR-L279 cells following the overexpression of LSAMP, but not NTNG2. H) JQ1 (5μM, 24 hrs) induced less caspase 3 and 9 cleavage in the COR-L279 cells with ectopic LSAMP than in control. The significance of two-group comparisons was determined using the Log-rank test (C), the ANOVA test with Dunnett’s multiple test correction (D, right panel), and the Student’s t-test with multiple test correction (F). **, P<0.01; ****, P<0.0001. Ctr, control; FC, fold change; OE, overexpression.
To assess the function of LSAMP in SCLC, we knocked down this gene using two siRNAs distinct from the ones included in the initial screens (Fig. 5D left panel) and found a similar degree of growth inhibition in H446 cells (Fig. 5D right panel). Next, we stably overexpressed LSAMP in COR-L279, which has a much lower basal level of this gene than H446 (Fig. 5E). Figure 5F shows that the COR-L279 cells with ectopic LSAMP were less susceptible to high doses of BETi than control cells. Because a recent study found that LSAMP regulates the expression of Sprouty – a negative regulator of ERK signaling (31), we examined the effect of LSAMP overexpression on ERK phosphorylation. As shown in Figure 5G, overexpression of LSAMP, but not NTNG2 (another NEUROD1-target gene), increased ERK1/2 phosphorylation in COR-L279 cells. Because ERK signaling plays a vital role in survival, we examined the effects of LSAMP on apoptosis. As shown in Figure 5H, ectopic expression of LSAMP lessened the JQ1-induced cleavage of caspase 3 and 9, indicating that LSAMP mitigates the JQ1-induced apoptosis in SCLC. Together these results support that LSAMP is one of the NEUROD1-target genes mediating BETi sensitivity.
BETi inhibits the NEUROD1 gene network and suppresses SCLC growth in vivo.
To assess the therapeutic effects of BETi in vivo, we chose NHWD-870 (a potent BETi in early-phase clinical trials) over JQ1 because the latter has unfavorable pharmacokinetic properties in vivo (32). We first confirmed that NHWD-870 has a similar sensitivity profile as JQ1 among SCLC lines (Supplementary Fig. S14). Using LX22 - an SCLC-N PDX model, we found NHWD-870 significantly decreased tumor burden and extended median survival of the tumor-bearing mice from 27 to 42 days (Fig. 6A). To assess how NHWD-870 affected NEUROD1-target genes in LX22 tumors, we performed RNA-seq to profile the transcriptome changes in the tumor cells isolated from the xenografts following one week of NHWD-870 treatment. As shown in Figure 6B–C, NHWD-870 significantly depleted the NEUROD1 gene signature and decreased LSAMP transcript levels in the xenograft tumors without affecting NEUROD1 expression.
Figure 6. BETi suppresses NEUROD1 transactivation and inhibits SCLC growth in vivo.
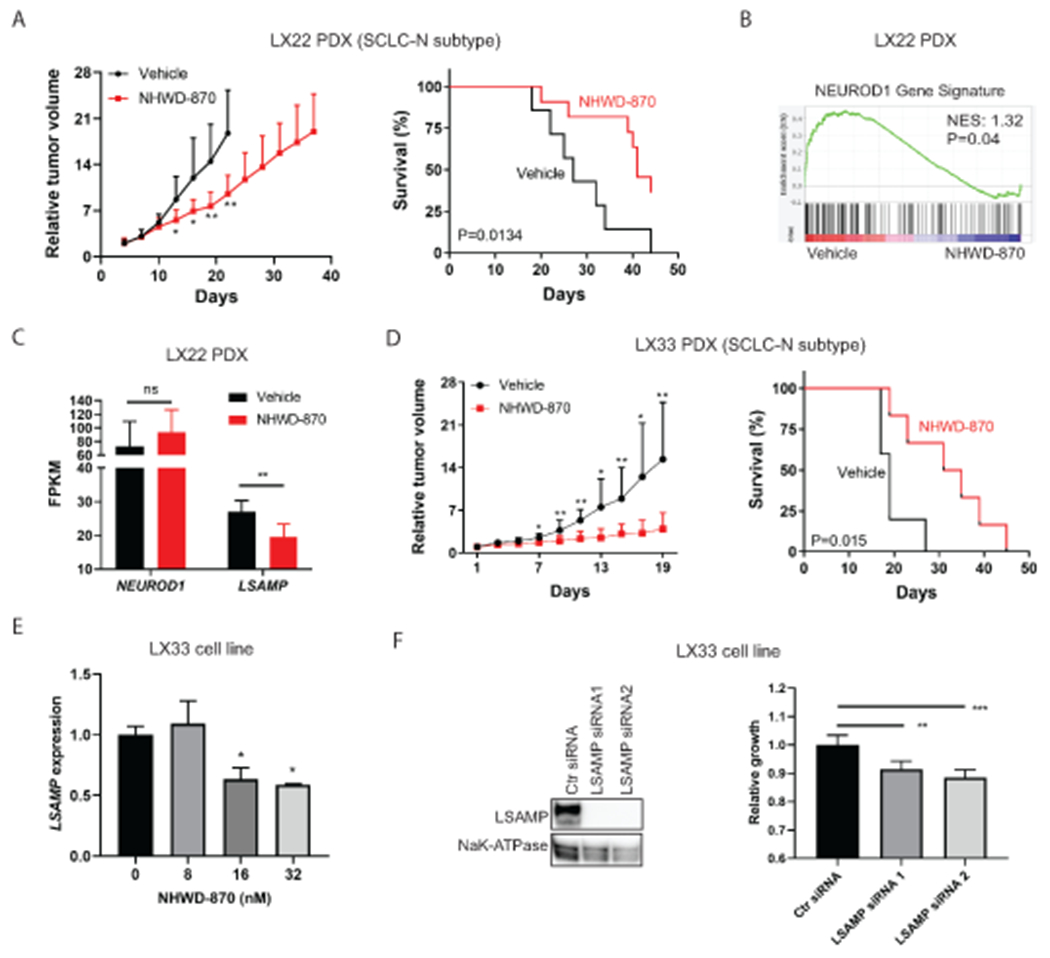
A) NHWD-870 (2mg/kg via gavage on a 5-day-on/2-day-off schedule) suppressed the tumor growth (left) and extended the survival (right) in the LX22 SCLC PDX model (an SCLC-N subtype). n=13 per group. B) A GSEA plot shows that NHWD-870 (2mg/kg via gavage daily for five days) significantly depleted the NEUROD1 gene signature in the LX22 PDX xenografts. n=6 per group. C) The effect of NHWD-870 on NEUROD1 and LSAMP transcript abundance (FPKM) in the LX22 model. D) NHWD-870 (3mg/kg via gavage daily) suppressed the growth (left) and extended the survival (right) in the LX33 SCLC PDX model (an SCLC-N subtype). n=6 per group. E) NHWD-870 (24 hrs) caused a dose-dependent decrease of LSAMP expression (qRT-PCR) in LX33 cells. F) LSAMP knockdown decreases LX33 proliferation. Left, knockdown of LSAMP by two siRNAs. Right, the effects of LSAMP knockdown on the proliferation of LX33 cells (72 hrs post-transfection). The significance of the two-group comparison was determined using the Student’s t-test with multiple test correction (A and D, left panels), the Student’s t-test (C), the Log-rank test (A and D, right panels), and the ANOVA test with Dunnett’s multiple test correction (E, and F right panel). *, P<0.05; **, P<0.01; ***, P<0.001; ns, not significant. Ctr, control; FPKM, Fragments Per Kilobase of transcript per Million mapped reads; NES, normalized enrichment score; PDX, patient-derived xenograft.
To demonstrate that NHWD-870 has antitumor activity in SCLC-N tumors in general, we used another SCLC-N PDX model - LX33. Like LX22, NHWD-870 significantly lessened the tumor burden and extended the median survival in the LX33 model (Fig. 6D). We attempted to establish tumor cell lines from both PDX models but could only succeed with LX33. In the LX33 SCLC line, NHWD-870 suppressed LSAMP expression as low as 16nM (Fig. 6E). LSAMP knockdown decreased LX33 cell proliferation, supporting that BETi inhibits SCLC growth by suppressing the NEUROD1 gene network in vivo (Fig. 6F).
Discussion
Because the master TFs define the transcriptome landscapes of SCLC, it is appealing to target these genes for therapeutic purposes. Here, we present evidence that BET proteins partner with NEUROD1 to activate the transcription of its downstream genes, resulting in the susceptibility of the SCLC-N tumors to BET inhibitors. Our results suggest that targeting transcriptional coactivators is a new approach to blocking the functions of the master TFs and can be potentially developed into a subtype-specific therapy.
Consistent with the role of NEUROD1 as a master TF in SCLC, our results demonstrated that it physically interacts with BET proteins and defines their genomic landscapes in H446 cells. The development of BETi resistance in the NEUROD1-KO cells is likely connected with the altered landscapes of BET proteins causing these factors to be associated with a distinct set of genes. Although BETi was equally effective in inhibiting BET proteins in the NEUROD1-KO cells, the resistance of these cells to BETi suggests that the new set of genes associated with the BRD4-loaded SEs are not critical for cell growth.
MYC is commonly amplified in SCLC-N cell lines (9,33), which raised the question that NEURDO1 expression may sensitize SCLC to BETi through MYC. Several lines of evidence suggest that NEUROD1 regulates BETi sensitivity independent of MYC, although MYC could be another factor contributing to BETi sensitivity in SCLC-N lines. In our study, H1694 - an SCLC-N line expressing N-MYC but not MYC, is also sensitive to BETi (Supplementary Table S4). Secondly, KO of NEUROD1 in H446 and DMS-273 substantially increased resistance to BETi without affecting MYC expression or transactivation.
In search for the genes mediating BETi sensitivity in SCLC-N tumors, we identified LSAMP as one of the potential candidates through the siRNA screens. LSAMP, a member of the IgLON family of immunoglobulin domain-containing cell adhesion molecules, is considered a tumor suppressor gene in ovarian cancer and osteosarcoma on the basis of its frequent deletion and a positive correlation between LSAMP expression and overall survival (34,35). However, in SCLC, a higher expression of this gene was correlated with a worse prognosis, suggesting that the functions of LSAMP could be tumor-type-specific. Consistent with a recent report that LSAMP and its homolog regulate Sprouty family genes in human glioblastoma cell lines and drosophila brain (31), we found overexpression of LSAMP increased ERK phosphorylation and lessened JQ1-induced apoptosis. Besides LSAMP, our siRNA screen results suggest that several other NEUROD1-target genes suppressed by BETi also play a role in tumor growth. Therefore, BETi inhibits SCLC growth likely by suppressing a panel of NEUROD1-target genes.
In contrast to a previous report that ASCL1 expression could predict BETi sensitivity in SCLC (36), the results from this study and a public high-throughput screen dataset (30) show that the SCLC-A lines are generally more resistant to BETi than the SCLC-N lines. Interestingly, BRD4 also interacts with ASCL1 in H1963 (a BETi-sensitive SCLC-A line with an IC50 to JQ1 = 0.83 μM; Supplemental Table S4), which may contribute to its sensitivity to BETi. However, whether BET proteins interact differently with ASCL1 in the BETi-resistant SCLC-A lines remains to be investigated. Recently, Wang et al. reported that, in SCLC-A lines, BETi activates IGF1 signaling by suppressing IGFBP5, an ASCL1-target gene and a negative regulator of IGF1 signaling (37). In contrast, such a feedback mechanism was not present in SCLC-N lines (37), which could contribute to the differential BETi sensitivity between SCLC-A and SCLC-N lines.
Substantial intratumoral heterogeneity is present in SCLC, which could be an obstacle for subtype-specific therapies. For instance, immunohistochemical analyses of the molecular subtype composition in SCLC tumors revealed that 20.4-37% of the tumors had two or more subtypes (6,7). Since our results showed that the non-SCLC-N subtypes are less sensitive to BETi, BETi single therapy may select for the subtypes other than SCLC-N and result in early recurrence. One potential solution to this obstacle is to use drug combinations that would be effective against two or more subtypes. For instance, in another study, we found that the mTOR inhibitors could potentiate the antitumor activities of BETi in the BETi-resistant SCLC lines (38).
In summary, our study demonstrates that SCLC-N subtype tumors are susceptible to BETi, likely due to their dependence on BET proteins in NEUROD1 transactivation. Our results suggest that blocking transcriptional coactivator(s) is an alternative approach to targeting the master TFs in SCLC and warrants further investigation.
Supplementary Material
Implications:
Our findings suggest that targeting transcriptional coactivators could be a novel approach to blocking the master transcription factors in SCLC for therapeutic purposes.
Acknowledgments
This study was supported by the Center for Cancer Research, the Intramural Research Program of the National Cancer Institute (NCI) (grant ZIA BC011787: Chen), and in part by an NCI FLEX award (grant ZIA BC011839: Chen and Schrump) and NCI U01 CA231844 (Oliver). The SCLC PDX models were made available through the support of NCI U24 CA213274 (Charles Rudin, MSKCC).
The authors thank Yulong Li, Yan-Jin Liu, Shih-Hsin Hsiao, Kaitlin C. McLoughlin, and Sichuan Xi for their assistance in this study; Nenghui Wang for providing NHWD-870; and John T. Poirier and Charles M. Rudin for sharing SCLC PDX models.
Footnotes
Competing Interest Statement: The authors declare no conflict of interest.
References
- 1.Scientific Framework for Small Cell Lung Cancer (SCLC). [cited 2014 June]. Available: https://deainfo.nci.nih.gov/advisory/ctac/workgroup/SCLC/SCLC%20Congressional%20Response.pdf
- 2.Ganti AKP, Loo BW, Bassetti M, Blakely C, Chiang A, D’Amico TA, et al. Small cell lung cancer, version 2.2022. NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021;19:1441–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howlader N, Forjaz G, Mooradian MJ, Meza R, Kong CY, Cronin KA, et al. The effect of advances in lung-cancer treatment on population mortality. N Engl J Med 2020;383:640–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019;19:289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poirier JT, George J, Owonikoko TK, Berns A, Brambilla E, Byers LA, et al. New approaches to SCLC therapy: from the laboratory to the clinic. J Thorac Oncol 2020;15:520–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baine MK, Hsieh MS, Lai WV, Egger JV, Jungbluth AA, Daneshbod Y, et al. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: a comprehensive immunohistochemical and histopathologic characterization. J Thorac Oncol 2020;15:1823–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qu S, Fetsch P, Thomas A, Pommier Y, Schrump DS, Miettinen MM, et al. Molecular subtypes of primary SCLC tumors and their associations with neuroendocrine and therapeutic markers. J Thorac Oncol 2022;17:141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021;39:346–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tlemsani C, Pongor L, Elloumi F, Girard L, Huffman KE, Roper N, et al. SCLC-CellMiner: A resource for small cell lung cancer cell line genomics and pharmacology based on genomic signatures. Cell Rep 2020;33:108296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frese KK, Simpson KL, Dive C. Small cell lung cancer enters the era of precision medicine. Cancer Cell 2021;39:297–9. [DOI] [PubMed] [Google Scholar]
- 11.Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, et al. Induction of human neuronal cells by defined transcription factors. Nature 2011;476:220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osborne JK, Larsen JE, Shields MD, Gonzales JX, Shames DS, Sato M, et al. NeuroD1 regulates survival and migration of neuroendocrine lung carcinomas via signaling molecules TrkB and NCAM. Proc Natl Acad Sci USA 2013;110:6524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, et al. ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep 2016;16:1259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donati B, Lorenzini E, Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol Cancer 2018;17:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 2014;54:728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010;468:1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cochran AG, Conery AR, Sims RJ. Bromodomains: a new target class for drug development. Nat Rev Drug Discov 2019;18:609–28. [DOI] [PubMed] [Google Scholar]
- 18.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013;24:777–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cong L, Zhang F. Genome engineering using CRISPR-Cas9 system. In: Pruett-Miller SM, editor. Chromosomal mutagenesis. New York: Springer; 2015. p 197–217. [DOI] [PubMed] [Google Scholar]
- 21.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.CCBR_Pipeliner - RNAseq gene expression pipeline documentation. [cited 2018 Mar 16]. Available: https://github.com/CCBR/Pipeliner/wiki/2.-RNA-Seq-Gene-Expression-Pipeline-Documentation.
- 23.Park YK, Ge K. Glucocorticoid receptor accelerates, but is dispensable for, adipogenesis. Mol Cell Biol 2017;37:e00260–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, et al. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell 2017;31:286–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hann CL, Daniel VC, Sugar EA, Dobromilskaya I, Murphy SC, Cope L, et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res 2008;68:2321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneeberger VE, Allaj V, Gardner EE, Poirier JT, Rudin CM. Quantitation of murine stroma and selective purification of the human tumor component of patient-derived xenografts for genomic analysis. PLoS ONE 2016;11:e0160587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizuno H, Nakanishi Y, Ishii N, Sarai A, Kitada K. A signature-based method for indexing cell cycle phase distribution from microarray profiles. BMC Genom 2009;10:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oki S, Ohta T, Shioi G, Hatanaka H, Ogasawara O, Okuda Y, et al. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang NS, Unkila MT, Reineks EZ, Distelhorst CW. Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J Biol Chem 2001;276:44117–28. [DOI] [PubMed] [Google Scholar]
- 30.Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, et al. Small cell lung cancer screen of oncology drugs, investigational agents, and gene and microRNA expression. J Natl Cancer Inst 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ariss MM, Terry AR, Islam ABMMK, Hay N, Frolov MV. Amalgam regulates the receptor tyrosine kinase pathway through Sprouty in glial cell development in the Drosophila larval brain. J Cell Sci 2020;133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin M, Guo Y, Hu R, Cai WL, Li Y, Pei S, et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat Commun 2020;11:1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 1983;306:194–6. [DOI] [PubMed] [Google Scholar]
- 34.Kresse SH, Ohnstad HO, Paulsen EB, Bjerkehagen B, Szuhai K, Serra M, et al. LSAMP, a novel candidate tumor suppressor gene in human osteosarcomas, identified by array comparative genomic hybridization. Genes Chromosomes Cancer 2009;48:679–93. [DOI] [PubMed] [Google Scholar]
- 35.Ntougkos E, Rush R, Scott D, Frankenberg T, Gabra H, Smyth JF, et al. The IgLON family in epithelial ovarian cancer: expression profiles and clinicopathologic correlates. Clin Cancer Res 2005;11:5764–8. [DOI] [PubMed] [Google Scholar]
- 36.Lenhart R, Kirov S, Desilva H, Cao J, Lei M, Johnston K, et al. Sensitivity of small cell lung cancer to BET inhibition is mediated by regulation of ASCL1 gene expression. Mol Cancer Ther 2015;14:2167–74. [DOI] [PubMed] [Google Scholar]
- 37.Wang XD, Hu R, Ding Q, Savage TK, Huffman KE, Williams N, et al. Subtype-specific secretomic characterization of pulmonary neuroendocrine tumor cells. Nat Commun 2019;10:3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumari A, Gesumaria L, Liu Y-J, Hughitt VK, Zhang X, Ceribelli M, et al. mTOR inhibition overcomes RSK3-mediated resistance to BET Inhibitors in small cell lung cancer. bioRxiv 2021.11.08.467833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and ChIP-seq datasets generated in this study are deposited at NCBI GEO (GSE210114).