

Abstract
Purpose:
This study aimed to identify baseline clinical features associated with the outcomes of patients enrolled in the COMBI-MB phase II study of dabrafenib and trametinib treatment in patients with V600 BRAF-mutant metastatic melanoma with melanoma brain metastases (MBM). Exploratory biomarker analysis was also conducted as part of the synergistic COMBI-BRV trial (BRV116521), to identify molecular and immunological changes associated with dabrafenib in MBMs and extracranial (ECM) metastases.
Patients and Methods:
Post-hoc analysis was performed for baseline features of patients (n=125) enrolled in COMBI-MB. Analyses were performed to identify baseline clinical features associated with ICRR, PFS, and overall survival (OS).
Exploratory biomarker analysis was performed on biospecimen collected in the COMBI-BRV trial in which patients with BRAF-mutant, resectable MBM were treated with dabrafenib for 10–14 days prior to craniotomy. Accessible ECM were resected or biopsied at the time of craniotomy. Biospecimens underwent molecular and immunological profiling for comparative analyses.
Results:
In COMBI-MB baseline treatment with corticosteroids was independently associated with lower ICRR (39% versus 63%, OR 0.323, CI 0.105–0.996, P= 0.049) and shorter PFS (HR 1.93, CI 1.06–3.51, P=0.031). Additional significant associations identified in the multivariate analysis were improved PFS in patients with a BRAFV600E genotype (HR 0.565, CI 0.321–0.996, P=0.048) and improved OS in patients with ECOG 0 (HR 0.44, CI 0.25–0.78, P=0.005).
Conclusion:
Corticosteroid treatment was associated with reduced ICRR and PFS in COMBI-MB, similar to results with immunotherapy for MBMs. Baseline corticosteroid treatment is a key factor to consider in MBM patient management and clinical trial design/interpretation.
Trial registration
ClinicalTrials.gov Identifier: NCT01266967 and NCT01978236
Keywords: COMBI-MB, brain metastases, dabrafenib, trametinib, biomarker
Introduction
Melanoma brain metastases (MBM) are present in up to 20% of patients at the time of diagnosis of stage IV disease and in up to 60% of patients by the time of melanoma-specific death(1–3). Historically, the median overall survival (OS) for patients with MBM was 4–5 months before the development of contemporary immune and targeted therapies(2). Markedly improved outcomes have been reported in recent years, particularly with combined anti-PD-1 and anti-CTLA-4 immune checkpoint blockade (ICB) therapies in patients with asymptomatic MBM(4,5). However, worse outcomes have been reported with both single-agent and combination ICB therapy regimens in patients who require corticosteroid treatment to control their symptoms from MBM(4–7). Thus, improving outcomes in patients with MBM remains a major unmet need.
Approximately 50% of cutaneous melanomas harbor a hotspot mutation affecting the V600 codon in the BRAF serine-threonine kinase(8,9). The presence of a BRAFV600 mutation results in marked hyperactivation of the MAPK signaling pathway(10). This molecular event was therapeutically exploited with the development of mutant-specific BRAF inhibitors, including vemurafenib (FDA-approval 2011), dabrafenib (FDA-approval, 2013), and encorafenib. Subsequent clinical trials demonstrated that combined treatment with BRAF and MEK inhibitors results in superior outcomes and tolerability, and ultimately led to the regulatory approval of dabrafenib and trametinib (2014), vemurafenib and cobimetinib (2015), and encorafenib and binimetinib (2018) for patients with BRAFV600 mutant, metastatic melanoma (11–15). Notably, none of the registration trials of the aforementioned agents allowed for participation of patients with untreated or progressing MBM.
The safety and activity of single-agent dabrafenib was confirmed in early phase trials, finding the treatment was active and could cross the blood brain barrier (16). With phase 2 trials confirming the activity of single-agent dabrafenib or vemurafenib treatment in patients with treated and untreated melanoma brain metastases (17,18). The COMBI-MB was the first prospective clinical trial reported to evaluate the safety and efficacy of an approved combination BRAF and MEK inhibitor targeted therapy regimen (dabrafenib and trametinib) in patients with MBM(15). The trial included a total of 125 patients with a BRAFV600 mutation and untreated or progressing MBM, who were enrolled into 4 cohorts based on BRAFV600 mutation status (BRAFV600E versus BRAFV600D/K/R), neurological symptoms, and prior local (brain) therapy. In Cohort A (BRAFV600E mutation, asymptomatic brain metastases, no prior local therapy), the largest (n=76) cohort in the study, treatment with dabrafenib and trametinib achieved an intracranial response rate (ICRR, objective intracranial complete or partial response as a proportion of all evaluable patients) of 58% and an intracranial disease control rate (IDCR, objective intracranial complete, partial or stable disease as a proportion of all evaluable patients) of 78%, and no new or unexpected toxicities were observed(15). While these results were promising, the median intracranial duration of response was only 6.5 months. In contrast, pooled analysis of the COMBI-D and COMBI-V studies of dabrafenib plus trametinib in metastatic melanoma patients (n= 563), which excluded patients with active MBMs, identified a median progression-free survival (PFS) of 11.1 months,(11,12,19). Notably the pooled analysis of COMBI-D/-V identified several factors associated with longer progression-free (PFS) and overall survival, including older age, female gender, BRAFV600E genotype, ECOG performance status (PS) of 0, normal serum lactate dehydrogenase (LDH) level and less than 3 organ sites of metastases(19).
It is currently unknown what clinical or biological factors are associated with clinical outcomes with dabrafenib and trametinib in patients with MBM. The identification of such factors may help to guide the use of dabrafenib and trametinib in these patients, inform the design and interpretation of future clinical trials, and perhaps provide insights into the observed difference in response duration between intracranial and extracranial metastases. Thus, we report here clinical features associated with ICRR, PFS and OS in patients with BRAF-mutant and MBM who were treated with dabrafenib and trametinib in COMBI-MB. This study also presents the exploratory biomarker analysis of COMBI-BRV trial (NCT01978236) of dabrafenib treatment in patient-matched MBM and extracranial metastases (ECM) as part of the previously un-reported phase II clinical trial of pre-operative treatment with dabrafenib in patients undergoing surgical resection of BRAF-mutant MBM. Synergistic exploratory analyses of the COMBI-BRV cohort were conducted to identify molecular and immunological differences in the response of intracranial and extracranial melanoma metastases in patients treated with dabrafenib, to determine if site specific responses contribute to these inferior outcomes seen in patients with MBM
Materials and Methods
Study populations
Post hoc analysis was performed on the COMBI-MB open-label, multicohort, phase 2 trial which evaluated the activity and safety of dabrafenib plus trametinib in patients with MBM (Figure 1A)(15). Briefly, cohort A included patients with BRAFV600E mutant, asymptomatic MBM, without previous local brain-directed therapy, and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; cohort B included patients with BRAFV600E mutant, asymptomatic MBM, with previous local therapy, and an ECOG performance status of 0 or 1; cohort C included patients with BRAFV600D/K/R mutant, asymptomatic MBM, with or without previous local therapy, and an ECOG performance status of 0 or 1; and cohort D included patients with BRAFV600D/E/K/R mutant, symptomatic MBM, with or without previous local therapy, and an ECOG PS of 0, 1, or 2(15). Analysis was conducted across all cohorts.
Figure 1. Consort diagram of the COMBI-MB and COMBI-BRV Trial.
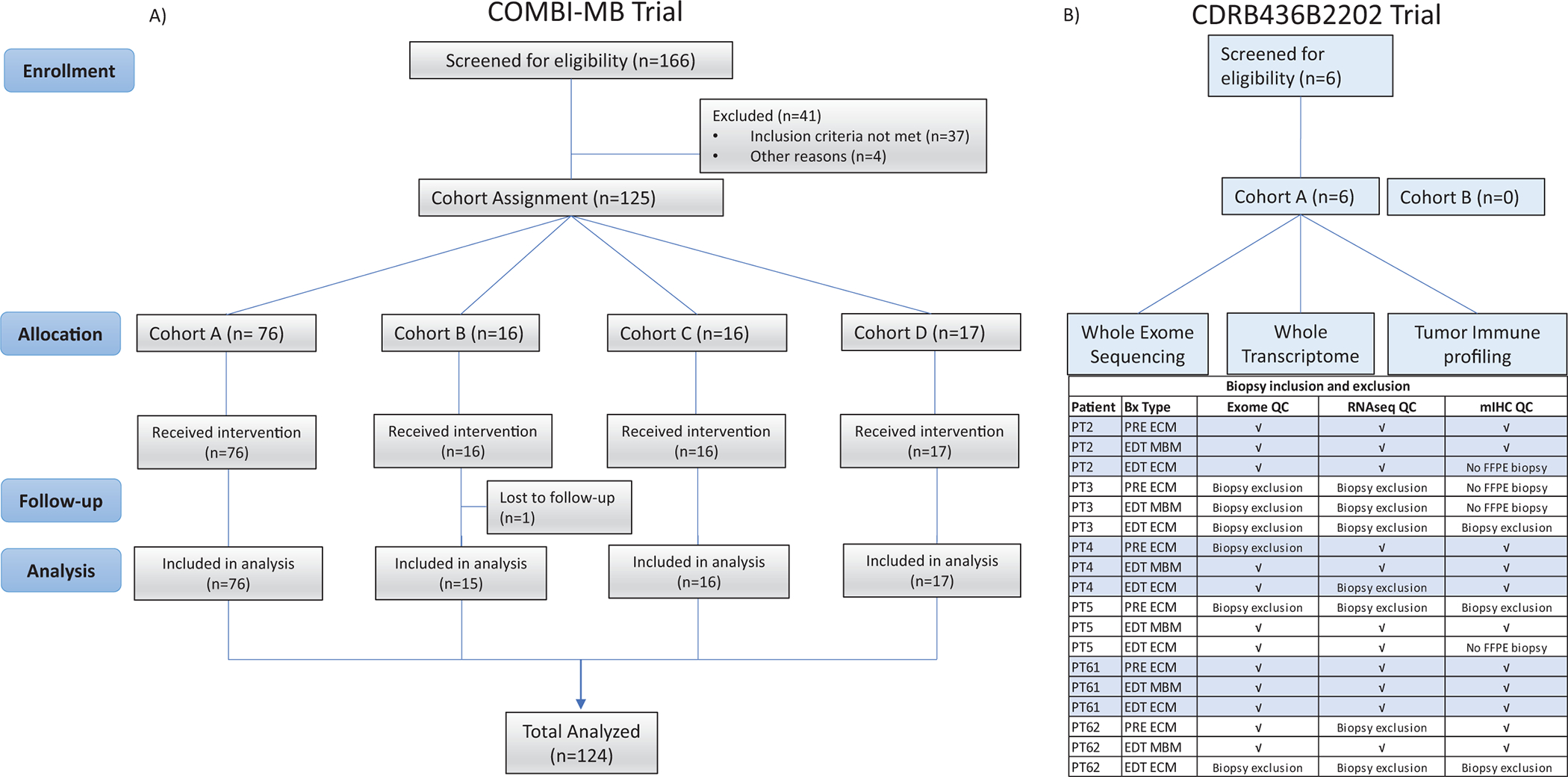
A) COMBI-MB trial profile; Cohort A=BRAFV600E-mutant, asymptomatic melanoma brain metastases, without previous local brain-directed therapy, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Cohort B=BRAFV600E-mutant, asymptomatic melanoma brain metastases, with previous local therapy, ECOG performance status of 0 or 1. Cohort C=BRAFV600D/K/R-mutant, asymptomatic melanoma brain metastases, with or without previous local therapy, ECOG performance status of 0 or 1. Cohort D=BRAFV600D/E/K/R-mutant, symptomatic melanoma brain metastases, with or without previous local therapy ECOG performance status of 0, 1, or 2, B) COMBI-BRV trial profile. Of the 18 biopsies, whole exome sequencing (WES) analyses were performed on 12 samples after quality control analyses, with samples excluded for lack of tumor content (n=5) or failure in DNA extraction in small biopsies (n=1). While transcriptomic analyses were performed on 11 samples after quality control analyses (3 samples excluded for lack of viable tumor; 4 samples excluded due to insufficient tumor content based on BRAF mutation frequency) (Supplementary Table 4).”Multiplex immunohistochemistry was performed on 11 biopsies, with samples excluded when no FFPE biopsies was collected (n=4) or lack of tumor cells (>100 melanoma cells, n=3). PT = patient ID, PRE = baseline biopsy, ECM = extracranial metastasis, MBM = melanoma brain metastasis, √ = included in analysis.
In addition, analyses were conducted on biospecimens collected in an international, open-label study of stage IV BRAFV600E/K metastatic melanoma with untreated, resectable MBM (1–4 cm) and ECM (NCT01978236, Figure 1B). The first cohort (Cohort A) of 15 patients were planned to receive single agent dabrafenib orally 150 mg bid for 7 to 14 days prior to surgery; the second cohort (Cohort B) of 15 patients was planned to receive the combination of dabrafenib 150 mg bid and trametinib 2 mg once daily for 7 to 14 days prior to surgery, followed by resection of MBM and safely accessible ECM. Patients with active disease after surgery received dabrafenib and trametinib until disease progression. Study sites included the University of Texas M D Anderson Cancer Center, Melanoma Institute Australia, and University of Pittsburgh Cancer Institute.
The study protocols were approved by the institutional review board or human research ethics committee at each participating institution. Furthermore, the study was conducted in accordance with both the Declaration of Helsinki and the International Conference of Harmonization Good Clinical Practice. Written informed consent was obtained for all patients.
Exome sequencing
DNA and RNA were isolated from FFPE tissue sections using the AllPrep DNA/RNA FFPE Kit (Qiagen) according to the manufacturer’s instructions and quality controls conducted as previously described(20). Library preparation was performed using the Nextera Flex Enrichment (Illumina) using IDT XGen Exome Research panel probes and sequenced on a NovaSeq 6000 S1 2×100bp. Sequenced reads were quality-checked using FastQC v0.11.8 (RRID:SCR_014583). DNA reads were aligned against the hg38 version of the human genome using bwa-mem v0.7.17 BWA (RRID:SCR_010910). Aligned reads were further processed for marking of duplicates with Picard v2.18.23 (RRID:SCR_006525) and base recalibration with GATK v4.1.5.0 (GATK, RRID:SCR_001876). For all samples with available exome sequencing, point mutations and small indels were called using HaplotypeCaller as provided in GATK( RRID:SCR_001876). The initial variant calling was used to confirm that sequenced tumors belonged to the same patient by measuring the jaccard index of putative heterozygote germline SNPs between samples. SNVs and small InDels with a minimum of 10 reads of coverage and supported by at least two reads were kept for further processing. Variants were annotated using ANNOVAR (RRID:SCR_012821) version dated April 16th, 2018 and those with an Exome Aggregation Consortium version 0.3 (RRID:SCR_004068). ExAC_ALL values below 1% were kept. Mutational hotspots were identified using the Cancer Hotspots database(21).
RNA sequencing
Libraries were prepared using the TruSeq RNA Exome kit and sequenced on a NovaSeq S2 2×100bp. Reads were aligned following the SCANB pipeline(22) and counts were generated using the prepDE.py script provided by the Stringtie team(23). Gene counts were normalized using edgeR (TMM) (RRID:SCR_012802). Gene set scores were generated using single-sample gene set enrichment analysis (SingScore)(24), based on the Hallmark gene sets from the Molecular Signatures Database v7.4 and the Pratilas MEK dependent signaling gene set(25).
Multiplex immunohistochemistry
Multiplex immunohistochemistry was performed on the lesion matching FFPE melanoma biopsies used for RNA and DNA sequencing. Tissue sections were cut and stained for multiple markers using and following the instructions outlined in the Opal 7 plex IHC kit (Akoya Biosciences: NEL811001KT, RRID:AB_2890927) for three separate panels as previously described(26,27). Firstly, an oncogenic signaling panel included phospho-Akt (Cell Signaling Technology Cat# 3787, RRID:AB_331170) phospho-Erk1/2 (Cell Signaling Technology Cat # 4370, RRID:AB_2315112), phospho-p90RSK (Cell Signaling Technology Cat# 11989, RRID:AB_2687613), phospho-S6 Ribosomal Protein (Cell Signaling Technology Cat# 4858, RRID:AB_916156), and SOX10 (Biocare Cat# ACI3099C, RRID:AB_2861289). Secondly, a T-cell specific panel included antibodies specific to PD-1 (Cell Marque Cat# 315M-95, RRID:AB_1160824), FoxP3 (Abcam Cat# ab20034, RRID:AB_445284), CD8 (Cell Marque Cat# 108R-15, RRID:AB_2892088), CD3 (Cell Marque Cat# CM103R95, RRID:AB_1158162), SOX10 (Biocare Cat# ACI3099C, RRID:AB_2861289). Finally, B cell and macrophage panel included CD20 (Dako Cat# M075501–2, RRID:AB_2282030), CD68 (Cell Marque Cat# CM168M95, RRID:AB_1158188), PD-L1 (Cell Signaling Technology Cat# 13684, RRID:AB_2687655) and SOX10 (Biocare Cat# ACI3099C, RRID:AB_2861289). Multispectral images were acquired using a Vectra 3 multispectral microscope (Akoya Biosciences). Individual markers were spectrally unmixed and expression in each cell was quantified using the Inform software (Akoya Biosciences). The quantitative data was exported and analyzed in Spotfire (Tibco). Samples with less than 100 melanoma (SOX10 positive) cells were excluded from further analysis.
Statistical analysis
Exploratory biomarker data was generated blinded to all clinical data. The associations between categorical variables and treatment type were tested using either the two-tailed Fisher exact test or the χ2 test as appropriate. Clinical outcomes analyzed were intracranial response (ICRR) determined as per RECIST V.1.1. Duration of intracranial, extracranial, and overall response, defined as the time from first documented complete or partial response until the time of disease progression; PFS, defined as the interval between the first dose of study treatment and the earliest date of disease progression or death from any cause; OS, defined as the time from first dose until death due to any cause. Summaries for response, PFS and OS were generated using Kaplan-Meier estimates along with two-sided 95% CIs and log-rank tests. Univariate and multivariate analysis was conducted on clinical features to test for associations with outcomes using Cox models with Brookmeyer and Crowley method to calculate confidence intervals. Associations with ICR were identified using logistic regression modelling. Exploratory biomarker data was assessed with a linear mixed-effect model with a random intercept to account for intra-patient correlation. To control the Type I error rate given the small sample size(28), the models were fitted with the restricted maximum likelihood (REML) method using the lme4 package in R v4.1.0(29) and p-values derived using the Satterthwaite approximation as provided in the sjPlot R package(30). No correction for multiple testing was pursued due to the exploratory nature of this analysis and small sample size. A p-value less than 0.05 was considered statistically significant
Data Availability
The data generated in this study is available in the European Genome-Phenome Archive (EGAC00001002614) database.
Results
COMBI-MB Population
The baseline characteristics of all 125 patients enrolled in COMBI-MB were pooled together (Table 1 and Figure 1A) to identify patient and disease factors associated with clinical outcomes in patients with melanoma brain metastases treated with dabrafenib and trametinib. The representativeness of the study participants is summarized in supplementary table 1. The clinical outcomes for the patients have been reported previously(15). Standard clinical features assessed for associations with clinical outcomes included baseline age, gender, ECOG performance status, BRAF mutation type (V600E or V600D/K/R), prior systemic anti-cancer treatment, and serum LDH. The number of brain metastases, size of brain metastases (largest intracranial lesion; sum of long diameters of target lesions), prior CNS-directed treatment (any; radiation; surgery), presence of uncontrolled symptoms from brain metastases, and treatment with corticosteroids at baseline were also assessed. At baseline, 33 patients were recorded as receiving steroids - 16 on dexamethasone (median dose 4 mg/day), 12 on prednisone (median dose 40 mg/day) and for 5 patient details for the specific corticosteroid treatment were not available. All patients were treated with the standard dose of dabrafenib (150 mg twice daily) and trametinib (2 mg daily).
Table 1.
Baseline characteristics of COMBI-MB trial.
| Cohort A (n=76) | Cohort B (n=16) | Cohort C (n=16) | Cohort D (n=17) | Total (n=125) | ||
|---|---|---|---|---|---|---|
|
|
||||||
| Age | Median (range) | 52·0 (23–84) | 54·5 (36–74) | 63·0 (44–84) | 46·0 (23–68) | |
| <65 | 60 (79%) | 12 (75%) | 9 (56%) | 16 (94%) | 97 (78%) | |
| ≥65 | 16 (21%) | 4 (25%) | 7 (44%) | 1 (6%) | 28 (22%) | |
|
| ||||||
| Sex | Male | 40 (53%) | 10 (63%) | 11 (69%) | 11 (65%) | 72 (58%) |
| Female | 36 (47%) | 6 (38%) | 5 (31%) | 6 (35%) | 53 (42%) | |
|
| ||||||
| ECOG | 0 | 50 (66%) | 11 (69%) | 12 (75%) | 9 (53%) | 82 (66%) |
| 1 | 25 (33%) | 5 (31%) | 4 (25%) | 6 (35%) | 40 (32%) | |
| 2 | 1 (1%)* | 0 | 0 | 2 (12%) | 3 (2%) | |
|
| ||||||
| BRAF genotype | V600E | 73 (96%) | 16 (100%) | 0 | 15 (88%) | 104 (83%) |
| V600K | 3 (4%)† | 0 | 14 (88%) | 1 (6%) | 18 (14%) | |
| V600R | 0 | 0 | 2 (13%) | 1 (6%) | 3 (2.4%) | |
| V600D | 0 | 0 | 0 | 0 | 0 | |
|
| ||||||
| Target brain metastases | 1 | 41 (54%) | 7 (44%) | 7 (44%) | 7 (41%) | 62 (49.6%) |
| 2 | 20 (26%) | 7 (44%) | 6 (38%) | 7 (41%) | 40 (32%) | |
| 3 | 7 (9%) | 2 (13%) | 2 (13%) | 1 (6%) | 12 (9.6%) | |
| 4 | 4 (5%) | 0 | 0 | 1 (6%) | 5 (4%) | |
| 5 | 4 (5%) | 0 | 1 (6%) | 1 (6%) | 6 (4.8%) | |
|
| ||||||
| SLD of target intracranial lesions (mm) | 20 (6–117) | 14 (5–40) | 20 (5–61) | 33 (10–84) | ||
|
| ||||||
| Extracranial metastases | No | 8 (11%) | 4 (25%) | 0 | 5 (29%) | 17 (14%) |
| Yes | 68 (89%) | 12 (75%) | 16 (100%) | 12 (71%) | 108 (86%) | |
|
| ||||||
| Lactate dehydrogenase concentration | Normal (≤ULN) | 48 (63%) | 13 (81%) | 10 (63%) | 12 (71%) | 83 (66%) |
| Elevated (>ULN) | 28 (37%) | 3 (19%) | 6 (38%) | 5 (29%) | 42 (34%) | |
|
| ||||||
| Receiving steroid therapy | Yes | 13 (17%) | 3 (19%) | 3 (19%) | 14 (82%) | 33 (26%) |
| No | 63 (83%) | 13 (81%) | 13 (81%) | 3 (18%) | 92 (74%) | |
|
| ||||||
| Previous systemic anticancer treatment | No | 59 (78%) | 11 (69%) | 13 (81%) | 10 (59%) | 93 (74%) |
| Yes | 17 (22%) | 5 (31%) | 3 (19%) | 7 (41%) | 32 (26%) | |
Data are median (range) or n (%). Cohort A= BRAF V600E -mutant, asymptomatic melanoma brain metastases, without previous local brain-directed therapy, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Cohort B= BRAF V600E -mutant, asymptomatic melanoma brain metastases, with previous local therapy, ECOG performance status of 0 or 1. Cohort C= BRAF V600D/K/R -mutant, asymptomatic melanoma brain metastases, with or without previous local therapy, ECOG performance status of 0 or 1. Cohort D= BRAF V600D/E/K/R -mutant, symptomatic melanoma brain metastases, with or without previous local therapy ECOG performance status of 0, 1, or 2. ECOG=Eastern Cooperative Oncology Group. SLD=sum of lesion diameters. ULN=upper limit of normal.
Patient had ECOG performance status 1 at time of screening and enrolment.
Patients were enrolled based on BRAF V600E status but were found to be BRAF V600K on central confirmation.
Factors associated with clinical outcomes in COMBI-MB
Univariate and multivariate analysis identified a significant association of ICRR with baseline corticosteroid treatment. The associations between corticosteroid treatment and LDH levels and outcome are summarized in Figure 2. Treatment with dabrafenib and trametinib achieved an ICRR of 39% in patients treated with corticosteroids (n=33) and 63% in patients not on corticosteroids (n=92) (Figure 2A and Figure 2B respectively). This difference in ICRR was significant on both univariate (OR 0.381, CI 0.168–0.862, P= 0.02) and multivariate analyses with all clinical variables (OR 0.323, CI 0.105–0.996, P= 0.0491). No other feature was significantly associated with ICRR (Table 2). Features tested included age at treatment, gender, ECOG PS, BRAF mutation status (V600E vs other), number of brain metastases, intracranial lesion size, presence of extracranial metastases, LDH, prior treatment or uncontrolled symptoms from the intracranial lesions. Intracranial response duration (ICRD) was shorter for patients with uncontrolled symptoms from brain metastasis (n=24, median 4.4 months) compared to those without symptoms on univariate analysis (n=101, median 5.6 months, HR 2.365, CI 1.184–4.726, P= 0.0148), but no clinical features were significantly associated with ICRD on multivariate analysis (Supplementary Table 2).
Figure 2. Outcome analysis of the COMBI-MB trial.

Waterfall plot of best intracranial response in A) MBM patient on steroids at baseline, and B) MBM patients not on steroids at baseline. Kaplan-meier curves demonstrate C) progression-free survival stratified by baseline steroid treatment status, D) overall survival stratified by baseline steroid treatment status, E) progression-free survival stratified by baseline lactate dehydrogenase, and F) overall survival stratified by baseline lactate dehydrogenase.
Table 2.
Univariate and multivariate analyses of baseline factors associated with overall intracranial response rate
| Univariate | Multivariate | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Category | Patients (N) | N responding (%) | OR (95% CI) | p value | OR (95% CI) | p value | |
|
| |||||||
| Age | <54 | 59 | 34 (58) | 1.066 (0.524–2.166) | 0.8599 | 1.143 (0.512–2.554) | 0.7443 |
| >=54 | 66 | 37 (56) | |||||
|
| |||||||
| Gender | Female | 53 | 28 (53) | 0.755 (0.369–1.546) | 0.4425 | 0.735 (0.334–1.618) | 0.4447 |
| Male | 72 | 43 (60) | |||||
|
| |||||||
| ECOG | 0 | 82 | 50 (61) | 1.637 (0.777–3.447) | 0.1946 | 1.034 (0.399–2.681) | 0.9456 |
| >=1 | 43 | 21 (49) | |||||
|
| |||||||
| BRAF mutation status | V600E | 104 | 61 (59) | 1.56 (0.609–3.999) | 0.3541 | 1.686 (0.574–4.957) | 0.3421 |
| Other | 21 | 10 (48) | |||||
|
| |||||||
| Brain metastases | 1 | 62 | 37 (60) | 1.138 (0.432–2.997) | 0.568 | 1.287 (0.355–4.668) | 0.5891 |
| 2 | 40 | 21 (53) | 0.85 (0.303–2.386) | 0.5717 | 0.988 (0.286–3.410) | 0.7598 | |
| >=3 | 23 | 13 (57) | |||||
|
| |||||||
| SLD of target intracranial lesion | <median | 62 | 35 (56) | 0.972 (0.479–1.973) | 0.9378 | 0.632 (0.184–2.167) | 0.4655 |
| >=median | 63 | 36 (57) | |||||
|
| |||||||
| Largest intracranial lesion | <median | 60 | 35 (58) | 1.128 (0.555–2.291) | 0.7396 | 1.177 (0.392–3.530) | 0.7717 |
| >=median | 65 | 36 (55) | |||||
|
| |||||||
| Extracranial metastases | Yes | 97 | 55 (57) | 0.982 (0.420–2.297) | 0.9669 | 1.212 (0.435–3.379) | 0.7135 |
| No | 28 | 16 (57) | |||||
|
| |||||||
| Elevated serum LDH | Yes | 42 | 21 (50) | 0.660 (0.312–1.394) | 0.2761 | 0.71 (0.302–1.676) | 0.436 |
| No | 83 | 50 (60) | |||||
|
| |||||||
| Steroid use at baseline | Yes | 33 | 13 (39) | 0.381 (0.168–0.862) | 0.0206 | 0.323 (0.105–0.996) | 0.0491 |
| No | 92 | 58 (63) | |||||
|
| |||||||
| Previously treatment anti-cancer | Yes | 32 | 21 (66) | 1.642 (0.712–3.786) | 0.2449 | 1.831 (0.710–4.719) | 0.2105 |
| No | 93 | 50 (54) | |||||
|
| |||||||
| Yes | 28 | 14 (50) | 0.702 (0.302–1.632) | 0.4108 | 1.186 (0.385–3.654) | 0.7669 | |
| Previous treatment to brain | No | 97 | 57 (59) | ||||
|
| |||||||
| XRT to brain | Yes | 34 | 15 (44) | 0.493 (0.222–1.096) | 0.0827 | 0.477 (0.156–1.464) | 0.196 |
| No | 91 | 56 (62) | |||||
|
| |||||||
| Previous Surgery to brain | Yes | 121 | 69 (57) | 1.327 (0.181–9.734) | 0.7809 | 1.487 (0.172–12.850) | 0.7182 |
| No | 4 | 2 (50) | |||||
|
| |||||||
| Presence of uncontrolled symptoms from brain | Yes | 24 | 13 (54) | 0.876 (0.358–2.143) | 0.7721 | 1.603 (0.479–5.367) | 0.4438 |
| No | 101 | 58 (57) | |||||
Consistent with the association with ICRR, patients on corticosteroids had a shorter PFS (median 4.3 versus 6.2 months) on both univariate (HR 1.788, CI 1.152–2.774, P= 0.0095) and multivariate (HR 1.931, CI 1.061–3.513, P= 0.0312) analyses (Table 3 and Figure 2C). The median PFS was significantly longer for patients with an ECOG PS of 0 (6.5 months) compared to those with ECOG ≥1 on univariate analysis only (3.8 months, HR 0.617, CI 0.411–0.927, P= 0.02) (Table 3). While the median PFS was not significantly different for patients with elevated or normal LDH (HR, 1.16, CI, 0.768–1.749, p=0.4821, Figure 2E). Multivariate analysis identified longer PFS in patients with a BRAFV600E (5.9 months) mutation vs BRAFV600D/K/R mutant disease (4.2 months, HR 0.565, CI 0.321–0.996, P= 0.0483).
Table 3.
Univariate and multivariate analyses of baseline factors associated with progression-free survival
| Univariate | Multivariate | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Category | (N) | events | Median Months | HR | 95% CI | pval | HR | 95% CI | pval | |
|
| ||||||||||
| Age | <54 | 59 | 47 | 5.6(5.3–7.3) | 1.096 | (0.740–1.623) | 0.6479 | 1.093 | (0.705–1.696) | 0.6908 |
| >=54 | 66 | 54 | 5.7(5.4–7.3) | |||||||
|
| ||||||||||
| Gender | Female | 53 | 43 | 5.6(4.2–7.4) | 1.056 | (0.711–1.568) | 0.7884 | 0.869 | (0.557–1.356) | 0.5368 |
| Male | 72 | 58 | 5.8(5.5–7.3) | |||||||
|
| ||||||||||
| ECOG | 0 | 82 | 63 | 6.5(5.6–7.5) | 0.617 | (0.411–0.927) | 0.02 | 0.692 | (0.401–1.197) | 0.1881 |
| >=1 | 43 | 38 | 3.8(3.5–5.9) | |||||||
|
| ||||||||||
| BRAF mutation status | V600E | 104 | 83 | 5.9(5.5–7.3) | 0.638 | (0.381–1.066) | 0.0859 | 0.565 | (0.321–0.996) | 0.0483 |
| Other | 21 | 18 | 4.2(3.5–9.1) | |||||||
|
| ||||||||||
| Brain metastases | 1 | 62 | 48 | 7.2(5.5–9.1) | 0.592 | (0.348–1.006) | 0.0528 | 0.673 | (0.322–1.403) | 0.2903 |
| 2 | 40 | 33 | 5.5(4.2–6.8) | 0.808 | (0.463–1.411) | 0.4537 | 1.014 | (0.503–2.045) | 0.9685 | |
| >=3 | 23 | 20 | 5.5(3.6–7.4) | |||||||
|
| ||||||||||
| SLD of target intracranial lesion | median | 62 | 49 | 5.9(5.3–7.3) | 0.996 | (0.674–1.472) | 0.9841 | 1.418 | (0.667–3.014) | 0.3637 |
| >=median | 63 | 52 | 5.6(4.7–7.3) | |||||||
|
| ||||||||||
| Largest intracranial lesion | median | 60 | 46 | 5.9(5.5–7.3) | 0.951 | (0.642–1.408) | 0.801 | 1.018 | (0.523–1.983) | 0.9572 |
| 65 | 55 | 5.6(4.3–7.3) | ||||||||
|
| ||||||||||
| Extracranial metastases | Yes | 97 | 82 | 5.6(4.7–6.7) | 1.45 | (0.879–2.391) | 0.1452 | 1.249 | (0.722–2.162) | 0.4261 |
| No | 28 | 19 | 7.3(5.6–14.6) | |||||||
|
| ||||||||||
| Elevated serum LDH | Yes | 42 | 35 | 5.6(3.7–7.5) | 1.159 | (0.768– 1.749) | 0.4821 | 0.811 | (0.514–1.280) | 0.368 |
| No | 83 | 66 | 5.7(5.5–7.2) | |||||||
|
| ||||||||||
| Steroid use at baseline | Yes | 33 | 29 | 4.3(3.5–6.4) | 1.788 | (1.152–2.774) | 0.0095 | 1.931 | (1.061–3.513) | 0.0312 |
| No | 92 | 72 | 6.2(5.6–7.3) | |||||||
|
| ||||||||||
| Previously treatment anti-cancer | Yes | 32 | 27 | 7.4(5.6–12.0) | 0.72 | (0.768– 1.749) | 0.1485 | 0.716 | (0.435–1.179) | 0.1895 |
| No | 93 | 74 | 5.5(4.7–6.2) | |||||||
|
| ||||||||||
| Previous treatment to brain | Yes | 28 | 21 | 7.2(5.5–13.4) | 0.778 | (0.481– 1.260) | 0.3077 | 0.796 | (0.441–1.435) | 0.4473 |
| No | 97 | 80 | 5.6(5.3–6.7) | |||||||
|
| ||||||||||
| XRT to brain | Yes | 34 | 26 | 5.3(4.3–12.2) | 0.781 | (0.497– 1.227) | 0.2838 | 0.766 | (0.422–1.390) | 0.3804 |
| No | 91 | 75 | 5.9(5.5–7.2) | |||||||
|
| ||||||||||
| Previous Surgery to brain | Yes | 121 | 99 | 5.6(5.4–7.2) | 1.795 | (0.441– 7.302) | 0.4141 | 1.817 | (0.420–7.859) | 0.4243 |
| No | 4 | 2 | 9.1(6.2–9.1) | |||||||
|
| ||||||||||
| Presence of uncontrolled symptoms from brain | Yes | 24 | 21 | 5.3(3.7–7.5) | 1.504 | (0.921– 2.456) | 0.103 | 1.171 | (0.611–2.241) | 0.6346 |
| No | 101 | 80 | 5.8(5.5–7.2) | |||||||
Several factors were associated with OS on univariate analysis (Supplementary Table 3). OS was shorter in patients treated with corticosteroids (median 9.3 versus 13.5 months, HR 1.642, CI 1.037– 2.598, P= 0.0343; Figure 2D) and those with elevated LDH (median 10.6 versus 12.7 months, HR 1.595, CI 1.045– 2.434, P= 0.0305; Figure 2F). OS was longer in patients with ECOG PS of 0 compared to >=1 (median 18.9 versus 8.2 months, HR 0.435, CI 0.285–0.664, P= 0.0001). Only ECOG PS remained significant on multivariate analysis (HR 0.441, CI 0.249–0.779, P= 0.0048).
Molecular and immune analysis of melanoma brain and extracranial metastases during dabrafenib treatment on the COMBI-BRV trial
COMBI-BRV was a clinical trial designed to evaluate and compare the molecular and immune effects of dabrafenib +/− trametinib in MBMs versus ECMs. The study enrolled patients with BRAFV600E/K mutant metastatic melanoma, with a planned craniotomy for 1 or more previously untreated MBM(s). Safely accessible ECM were biopsied prior to the start of treatment (“PRE”). Patients were then treated for 7 to 14 days with dabrafenib (Cohort A) or dabrafenib + trametinib (Cohort B) prior to craniotomy (last dose given the night before surgery). Samples collected on the day of craniotomy were designated as early during treatment (“EDT”). Six patients were enrolled in the study, all of whom entered cohort A and received dabrafenib monotherapy (Supp Table 3). Due to slow enrollment the trial was halted after 6 patients completed treatment, and as such no patients received dabrafenib plus trametinib prior to craniotomy (Figure 1B). All patients underwent planned craniotomy; safely accessible ECM were also biopsied or excised. Two patients on the COMBI-BRV trial (PT4 and PT62) received corticosteroids prior to craniotomy. Biospecimens were available for PRE ECM, EDT ECM and EDT MBM for 6 patients (PT3 did not have a PRE ECM biopsy). Response data, biospecimen availability and variables used for analysis are summarized in Supplementary Table 4 and Supplementary Table 5.
Oncogenic signaling was examined within the PRE and EDT melanoma biopsies via whole transcriptome sequencing (Figure 3A). Of the total 18 biopsies, transcriptomic analysis were performed on 11 samples after quality control analyses (3 samples excluded for lack of viable tumor; 4 samples excluded due to insufficient tumor content based on BRAF mutation frequency) (Supplementary Table 5). Subsequent single sample pathway analysis (Figure 3B, Supplementary Table 6) showed a trend of decreased MAPK (MEK dependent) signaling from PRE ECM to EDT ECM biopsy sites (Mean difference= −0.14, P=0.081, Supplementary Figure 1A and B) and a significant decrease from PRE ECM to EDT MBM (Mean difference= −0.11, P=0.03, Supplementary Figure 1A and B). MAPK signaling did not differ between EDT ECM and EDT MBM biopsy sites (Mean difference= 0.01, P=0.678). A significant increase in MTOR signaling pathway score from PRE ECM to EDT ECM biopsy sites (Mean difference= 0.04, P=0.024); and a significant decrease of cell cycle score from PRE ECM to EDT ECM (Mean difference= −0.08, P=0.015) and a trend versus EDT MBM biopsy sites (Mean difference= −0.07, P=0.061), were observed. Whilst oxidative phosphorylation and PI3K/MTOR signaling were higher in EDT MBM than EDT ECM, no significant differences were observed between any biopsy site/timepoint in this small cohort (Supplementary Table 6, Supplementary Figure 1A).
Figure 3. Analysis of biospecimen from the COMBI-BRV trial.

A) Unsupervised clustering of gene expression signatures, B) Changes in MEK dependent gene expression and phosphorylated S6 protein expression. C) Changes in HALLMARK interferon gamma gene expression and intratumoral cytotoxic T-cell (CD8+) densities. D) Waterfall plot depicting changes in intratumoral T-cell (CD3+) densities between treatment timepoints and colored via steroid treatment status.
Lesion-matched formalin-fixed paraffin-embedded biopsies were available for all patients for multiplex immunohistochemistry and image analysis (Supplementary Table 5 and Supplementary Figure 1C). While the protein expression of pS6 and pp90RSK were reduced in melanoma cells from the PRE biopsies compared to both EDT biopsy sites (Supplementary Table 6), only the reduction in pS6 from pretreatment to EDT MBM reached significance (Mean difference= −0.39, P=0.043, Figures 3B, Supplementary Figure 1B). Other qualitative changes in expression, such as reduced proliferation (Ki-67) from PRE ECM to both EDT sites, were observed but did not reach statistical significance (Supplementary Table 5).
A trend of increased interferon gamma signaling activity was observed following treatment from PRE ECM to EDT ECM (Mean difference= 0.06, P=0.073) and to EDT MBM (Mean difference= 0.09, P=0.051) (Figure 3C, Supplementary Figure 1B, Supplementary Table 6). The total (CD3+) and cytotoxic (CD8+) T-cell densities measured by mIHC tended to increase from PRE ECM to both EDT sites, and macrophage densities (CD68+) tended to decrease, but no differences reached significance (Figure 3C, Supplementary Figure 1D). In addition, no significant differences were observed between EDT ECM and EDT MBM biospecimen. Interestingly, the two patients (PT4 and PT62) treated with corticosteroids in the pre-operative period experienced a reduction in immune cell densities (T-cells, cytotoxic T-cells and PD-L1 positivity) from their PRE to their EDT ECM biopsies (Figure 3D). PT4 had the highest densities of cytotoxic T-cells in the PRE biopsies, then experienced a reduction in T-cell and PD-L1 positive cell densities, down to the lowest levels of the cohort in both EDT ECM and EDT MBM biopsies (Supplementary Figure 1E and F, Supplementary Table 5).
Exome sequencing was possible for 5 patients’ tumors [1 sample failed to extract adequate DNA; 4 melanoma biopsies lacked enough tumor content to detect the BRAF mutation and were excluded from analysis (Supplementary Table 5). BRAFV600E mutations were detected in all remaining tumors except PT62, whose tumors carried a BRAFV600K mutation (Supplementary Table 7). Screening for known genetic mechanisms of MAPK inhibitor resistance(31,32) revealed a BRAF amplification in the PRE ECM biopsy of patient PT2, which was not present in the EDT biopsies, while the remaining mutations were concordant between all biopsies for this patient. PT4 contained MAP2K1P124S and CDKN2AQ50X mutations and loss of heterozygosity (LOH) of chromosome 10 in both EDT lesions, with an additional PTENR130X mutation detected exclusively in the MBM (Supplementary Table 7). In addition, the EDT MBM for PT62 (the only biopsy profiled for this patient) contained an allelic imbalance of the long arm of chromosome 10 and a deleterious mutation of PTEN, suggesting inactivation of this gene (Supplementary Table 7).
Discussion
There remains a critical need to improve our understanding of the determinants of benefit of BRAF plus MEK inhibitor targeted therapy for MBMs. This study reveals for the first time that baseline treatment with corticosteroids is associated with significantly reduced ICRR and shorter PFS in patients with BRAF-mutant melanoma brain metastases who were treated with dabrafenib and trametinib. Further, our analysis of biospecimens in the COMBI-BRV trial is the first to explore molecular and immune differences between intracranial and extracranial metastases in patients receiving a BRAF inhibitor (dabrafenib).
The need for corticosteroids has previously been shown to be associated with worse clinical outcomes in clinical trials of immune checkpoint inhibitors for MBM patients. In the phase II trial that evaluated ipilimumab 10 mg/kg, the ICRR was 18% in patients with asymptomatic melanoma brain metastases but only 5% in patients who required corticosteroids to control symptoms (7). In the ABC trial, the response rate with nivolumab in patients (n=25) with asymptomatic, previously untreated brain metastases was 20%(33,34). The ABC trial also evaluated nivolumab in a cohort (Cohort C) of MBM patients who either had previous CNS-directed therapy, neurological symptoms (n=10), or leptomeningeal disease (LMD; n=4). Only 1 patient in this cohort had an intracranial response, which was noted to be a patient with neurological symptoms (ICRR 10%). While the response rates even among asymptomatic patients in each of these trials were low, much more impressive results have been seen with combination immunotherapy with ipilimumab and nivolumab. The ICRR for ipilimumab and nivolumab in patients with asymptomatic brain metastases was 54% in CheckMate-204 (Cohort A; n=101) and 59% in ABC (Cohort A; n=27 drug-treatment naïve) studies(5,33,34). However, the ICRR in patients with symptomatic brain metastases, including in patients (n=12) requiring up to 4 mg/day of dexamethasone, in CheckMate-204 (Cohort B, n=18) was 22%(5).
In this study we show that the association of corticosteroids with poor clinical outcomes in melanoma patients with brain metastases also applies to targeted therapy. In the COMBI-MB trial, baseline treatment with corticosteroids was associated with a 2-month reduction in the median PFS that was independent of measures of tumor burden, including the number of MBM, ECM/MBM lesion size, or serum LDH(15). Although the specific mechanism behind corticosteroid-driven immunosuppression and impaired response to immunotherapies is elusive(35), it has been well established that host response also plays a critical role in augmenting the response to targeted therapies, albeit not specifically in patients with brain metastases(36,37). For example, treatment of metastatic melanoma with BRAF inhibitors has been shown to increase T-cell infiltration and upregulation of melanoma antigen expression (MART-1, TYRP1/2 and GP100) in the melanoma biopsies from patients early during treatment (10–14 days)(38). These studies have led to combination trials that aim to optimize the scheduling of combination molecular inhibitors with immunotherapies to take advantage of this immunogenic window (39).
Together the data suggests that patients with symptomatic MBMs who are treated with corticosteroids have poor outcomes, regardless of whether they receive immune or targeted. Whether use of corticosteroids is simply a surrogate for aggressive tumor biology or specifically antagonizes the effects of targeted therapy in MBMs remains to be determined. Notably, the available data strongly supports that symptomatic MBM patients who require corticosteroids need to be included in clinical trials, as there is an unmet need to identify strategies that will improve their poor outcomes. However, this patient cohort requires specific consideration in cohort design and trial analysis for both targeted and immune therapies. Interestingly, recently reported initial results for the TRICOTEL study of combined treatment with vemurafenib, cobimetinib and atezolizumab in patients with BRAF-mutant brain metastases showed comparable outcomes for patients with symptomatic (n=24, ICRR 46%) and asymptomatic (n=41, ICRR 39%) MBMs(40). These results, combined with the inferior outcomes reported here for targeted therapy alone, and previously for immunotherapy alone, support the rationale to continue to explore combinatorial approaches in the challenging setting.
The COMBI-BRV trial offered the unique opportunity to investigate oncogenic and immunological signaling at EDT in ECM and MBM. As the study was ultimately limited in recruitment, this analysis should be considered exploratory in nature due to the small sample size. Despite this limitation, the interrogation of the available samples following short-term treatment with dabrafenib demonstrated comparable inhibition of MEK dependent signaling, as well as stimulation of interferon gamma signaling, in both ECMs and MBM sites early on treatment. While prior studies in larger unmatched cohorts detected upregulation of PI3K/AKT/mTOR signaling (41) and increased oxidative phosphorylation in MBM compared to ECM(42), we observed trends but no significant differences from PRE to either EDT sites in the COMBI-BRV cohort. However, as noted above these analyses were limited by small numbers and the heterogeneity observed among these samples. In addition, the MBM samples within the COMBI-BRV cohort represent early on treatment (7–14 days) biopsies, with the expected decrease in viable tumor content and influx of immune cells potentially affecting the bulk gene expression signatures. Prior studies have also reported a decrease in interferon gamma gene signatures in MBMs versus same-patient ECMs(43). Whilst Fischer et al, observed an immunosuppressed TME, with lower T-cell densities and lower immunoscores, in MBM compared to ECM sites in untreated patients, the results here suggest that treatment with MAPKi may alter this suppressive TME in MBMs, at least at this early timepoint (42). Therefore, this data confirms the MEK dependent inhibition across both MBMs and ECMs, whilst highlighting increased immunogenicity of the MBM lesions early during BRAF inhibitor treatment.
Together these findings highlight the continued challenges to improving outcomes in patients with MBMs, particularly those with symptomatic disease. These results, combined with the inferior outcomes reported here for targeted therapy alone, and previously for immunotherapy alone, support the rationale for continued research and for further exploration of combinatorial approaches for patients who require corticosteroid treatment. These findings also raise the need for a restrained use of steroids to treat asymptomatic patients with peritumoral edema detected via medical imaging. Therefore, regardless of the treatment strategy, the use of corticosteroids to treat symptomatic MBM is associated with poor outcomes and highlights the urgent need for new, more effective strategies for these patients(44).
Supplementary Material
{kind=link}
Translational Relevance.
This study, along with emerging data from immunotherapy treated cohorts, identifies the clear need to develop more effective strategies for MBM patients who require steroids to control symptoms from these tumors. Such patients need to continue to be included in clinical trials, but likely should be considered and evaluated separately from MBM patients who do not require steroids. This consistent observation of worse outcomes also supports the strategy to minimize and/or avoid steroid treatment in patients with MBM when possible (i.e., patients with asymptomatic cerebral edema), regardless of systemic therapy to be given.
ACKNOWLEDGMENTS AND FINIANCIAL SUPPORT
This study was supported by a Team Science Grant from the Melanoma Research Alliance (#269996). GSK supported the 2 clinical trials and provided the data/samples we used in the study. RAS and GVL are supported by National Health and Medical Research Council of Australia (NHMRC) Practitioner Fellowships, and their research is supported by an NHMRC Program grant (APP2006415 and APP2018514). Support from CLEARbridge Foundation, The Cameron Family and colleagues at Melanoma Institute Australia and Royal Prince Alfred Hospital are also gratefully acknowledged. GVL is supported by the University of Sydney Medical Foundation. JSW is supported by an NHMRC investigator fellowship (APP1174325), Melanoma Research Alliance young investigator fellowship (#700455), Cancer Institute NSW (TPG2114) and the University of Sydney. MAD is supported by the National Cancer Institute of the National Institutes of Health under grant award number P50CA221703, Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the AIM at Melanoma Foundation, the American Cancer Society and the Melanoma Research Alliance, Cancer Fighters of Houston, the Anne and John Mendelsohn Chair for Cancer Research, and philanthropic contributions to the Melanoma Moon Shots Program of MD Anderson.
Footnotes
Conflict of interest statement
James S. Wilmott: None to disclose, Johnathan Engh : None to disclose, Hussein Tawbi: None to disclose, Nduka Amankulor: None to disclose, Brindha Shivalingam: None to disclose, Ismael A. Vergara: None to disclose, Hansol Lee: None to disclose, Peter A. Johansson: None to disclose, Peter M. Ferguson: None to disclose
Hiya Banerjee: Employment - Novartis Pharmaceuticals UK Ltd., Stock and Other Ownership Interests - Bristol-Myers Squibb (I)
Philippe Saiag: Consulting or Advisory Role - Bristol-Myers Squibb; MSD; Novartis; Pierre Fabre; Roche/Genentech; Sanofi, Research Funding - Pierre Farbre, Travel, Accommodations, Expenses - Bristol-Myers Squibb; MSD; Novartis; Pierre Farbre ; Roche
Caroline Robert: Stock and Other Ownership Interests - RiboNexus, Consulting or Advisory Role - AstraZeneca; Bristol-Myers Squibb; MSD; Novartis; Pfizer; Pierre Fabre; Roche; Sanofi, Research Funding - Novartis (Inst)
Jean-Jacques Grob: Consulting or Advisory Role - Amgen; BMS; Merck KGaA; MSD Oncology; Novartis; Philogen; Pierre fabre; Roche; Roche/Genentech; Sanofi; Sun Pharma, Speakers’ Bureau – Novartis, Travel, Accommodations, Expenses - BMS; MSD Oncology; Novartis; Pierre Fabre
Richard A Scolyer: Fees for professional services from MetaOptima Technology Inc., F. Hoffmann-La Roche Ltd, Evaxion, Provectus Biopharmaceuticals Australia, Qbiotics, Novartis, MSD Sharp & Dohme, NeraCare, AMGEN Inc., Bristol-Myers Squibb, Myriad Genetics, GlaxoSmithKline.
Lisa H Butterfield: Unrelated advisory activities: Calidi Scientific and Medical Advisory Board, April 6, 2017-present; Kalivir, Scientific Advisory Board, 2018–2021; Torque Therapeutics, Scientific Advisory Board, 2018–2020; Khloris, Scientific Advisory Board, 2019-present; Pyxis, Scientific Advisory Board, 2019-present; Cytomix, Scientific Advisory Board, 2019-present; DCprime, Scientific Advisory Board meeting, Nov. 2020; RAPT, Scientific Advisory Board, 2020-present; Takeda, Scientific Advisor, 2020-present; EnaraBio scientific advisor, Feb. 2021.
John M Kirkwood: Honoraria: Bristol Myers Squibb, Consulting or Advisory Role: Novartis, Iovance Biotherapeutics, Amgen, Checkmate Pharmaceuticals, Harbour BioMed, Istari Oncology, OncoSec, Scopus BioPharma, Pfizer, Oncocyte, Takeda, AXIO Research, Immunocore, Natera, DermTech, Ankyra Therapeutics, Becker Pharmaceutical Consulting, Fenix Group International, Intellisphere LLC, IQVIA, Merck, Replimune, SR One Capital Management, Research Funding: Amgen (Inst), Bristol Myers Squibb (Inst), Castle Biosciences (Inst), Checkmate Pharmaceuticals (Inst), Immunocore (Inst), Iovance Biotherapeutics (Inst), Novartis (Inst), Merck (Inst), Immvira (Inst), Iovance Biotherapeutics (Inst), Schering-Plough (Inst), Harbour BioMed (Inst), Takeda (Inst), Verastem (Inst)
Georgina V. Long: Honoraria - BMS; Pierre Fabre Consulting or Advisory Role - Agenus; Amgen; Array BioPharma; Boehringer Ingelheim; Bristol-Myers Squibb; Evaxion Biotech; Hexal; Highlight Therapeutics; Merck Sharpe & Dohme; Novartis; OncoSec; Pierre Fabre; Provectus; QBiotics; Regeneron; Skyline Diagnostics
Michael A. Davies: Consulting or Advisory Role - ABM; Apexigen; Array BioPharma; Bristol-Myers Squibb; Eisai; Genentech/Roche; NanoString Technologies; Novartis; Pfizer, Research Funding – ABM Therapautics (Inst); AstraZeneca (Inst); Genentech/Roche (Inst); GlaxoSmithKline (Inst); Lead Pharma (Inst); Merck (Inst); Myriad Genetics (Inst); Oncothyreon (Inst); Pfizer (Inst); Sanofi (Inst)
References
- 1.Patel JK, Didolkar MS, Pickren JW, Moore RH. Metastatic pattern of malignant melanoma: A study of 216 autopsy cases. The American Journal of Surgery 1978;135(6):807–10 doi 10.1016/0002-9610(78)90171-X. [DOI] [PubMed] [Google Scholar]
- 2.Davies MA, Liu P, McIntyre S, Kim KB, Papadopoulos N, Hwu WJ, et al. Prognostic factors for survival in melanoma patients with brain metastases. Cancer 2011;117(8):1687–96 doi 10.1002/cncr.25634. [DOI] [PubMed] [Google Scholar]
- 3.Pires da Silva I, Lo S, Quek C, Gonzalez M, Carlino MS, Long GV, et al. Site-specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti–PD-1 therapy. Cancer 2020;126(1):86–97 doi 10.1002/cncr.32522. [DOI] [PubMed] [Google Scholar]
- 4.Long GV, Atkinson V, Lo S, Sandhu S, Guminski AD, Brown MP, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. The Lancet Oncology 2018;19(5):672–81 doi 10.1016/s1470-2045(18)30139-6. [DOI] [PubMed] [Google Scholar]
- 5.Tawbi HA, Forsyth PA, Hodi FS, Lao CD, Moschos SJ, Hamid O, et al. Safety and efficacy of the combination of nivolumab plus ipilimumab in patients with melanoma and asymptomatic or symptomatic brain metastases (CheckMate 204). Neuro-Oncology 2021. doi 10.1093/neuonc/noab094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. New England Journal of Medicine 2018;379(8):722–30 doi 10.1056/NEJMoa1805453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. The Lancet Oncology 2012;13(5):459–65 doi 10.1016/S1470-2045(12)70090-6. [DOI] [PubMed] [Google Scholar]
- 8.Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017;545(7653):175–80 doi 10.1038/nature22071. [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas N Genomic Classification of Cutaneous Melanoma. Cell 2015;161(7):1681–96 doi 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417(6892):949–54 doi 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 11.Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med 2019;381(7):626–36 doi 10.1056/NEJMoa1904059. [DOI] [PubMed] [Google Scholar]
- 12.Grob JJ, Amonkar MM, Karaszewska B, Schachter J, Dummer R, Mackiewicz A, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. The Lancet Oncology 2015;16(13):1389–98 doi 10.1016/s1470-2045(15)00087-x. [DOI] [PubMed] [Google Scholar]
- 13.Ascierto PA, McArthur GA, Dréno B, Atkinson V, Liszkay G, Di Giacomo AM, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. The Lancet Oncology 2016;17(9):1248–60 doi 10.1016/S1470-2045(16)30122-X. [DOI] [PubMed] [Google Scholar]
- 14.Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. The Lancet Oncology 2018;19(5):603–15 doi 10.1016/S1470-2045(18)30142-6. [DOI] [PubMed] [Google Scholar]
- 15.Davies MA, Saiag P, Robert C, Grob JJ, Flaherty KT, Arance A, et al. Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. The Lancet Oncology 2017;18(7):863–73 doi 10.1016/s1470-2045(17)30429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 2012;379(9829):1893–901 doi 10.1016/s0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. The Lancet Oncology 2012;13(11):1087–95 doi 10.1016/s1470-2045(12)70431-x. [DOI] [PubMed] [Google Scholar]
- 18.McArthur GA, Maio M, Arance A, Nathan P, Blank C, Avril MF, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicentre study. Ann Oncol 2017;28(3):634–41 doi 10.1093/annonc/mdw641. [DOI] [PubMed] [Google Scholar]
- 19.Long GV, Grob JJ, Nathan P, Ribas A, Robert C, Schadendorf D, et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: a pooled analysis of individual patient data from randomised trials. Lancet Oncol 2016;17(12):1743–54 doi 10.1016/s1470-2045(16)30578-2. [DOI] [PubMed] [Google Scholar]
- 20.De Paoli-Iseppi R, Johansson PA, Menzies AM, Dias KR, Pupo GM, Kakavand H, et al. Comparison of whole-exome sequencing of matched fresh and formalin fixed paraffin embedded melanoma tumours: implications for clinical decision making. Pathology 2016;48(3):261–6 doi 10.1016/j.pathol.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discovery 2018;8(2):174 doi 10.1158/2159-8290.CD-17-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brueffer C, Gladchuk S, Winter C, Vallon-Christersson J, Hegardt C, Häkkinen J, et al. The mutational landscape of the SCAN-B real-world primary breast cancer transcriptome. EMBO Mol Med 2020;12(10):e12118–e doi 10.15252/emmm.202012118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols 2016;11(9):1650–67 doi 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foroutan M, Bhuva DD, Lyu R, Horan K, Cursons J, Davis MJ. Single sample scoring of molecular phenotypes. BMC Bioinformatics 2018;19(1):404 doi 10.1186/s12859-018-2435-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, et al. V600EBRAF is associated with disabled feedback inhibition of RAF–MEK signaling and elevated transcriptional output of the pathway. Proceedings of the National Academy of Sciences 2009;106(11):4519–24 doi 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019;35(2):238–55.e6 doi 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017;545(7653):175–80 doi 10.1038/nature22071. [DOI] [PubMed] [Google Scholar]
- 28.Luke SG. Evaluating significance in linear mixed-effects models in R. Behav Res Methods 2017;49(4):1494–502 doi 10.3758/s13428-016-0809-y. [DOI] [PubMed] [Google Scholar]
- 29.Bates D, Mächler M, Bolker B, Walker S. Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software 2015;67(1):1 – 48 doi 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 30.Lüdecke D 2021. sjPlot: Data Visualization for Statistics in Social Science. <https://CRAN.R-project.org/package=sjPlot>.
- 31.Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact. Clinical Cancer Research 2014;20(7):1965 doi 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- 32.Carlino MS, Fung C, Shahheydari H, Todd JR, Boyd SC, Irvine M, et al. Preexisting MEK1P124 mutations diminish response to BRAF inhibitors in metastatic melanoma patients. Clin Cancer Res 2015;21(1):98–105 doi 10.1158/1078-0432.Ccr-14-0759. [DOI] [PubMed] [Google Scholar]
- 33.Long GV, Atkinson V, Lo S, Guminski AD, Sandhu SK, Brown MP, et al. Five-year overall survival from the anti-PD1 brain collaboration (ABC Study): Randomized phase 2 study of nivolumab (nivo) or nivo+ipilimumab (ipi) in patients (pts) with melanoma brain metastases (mets). Journal of Clinical Oncology 2021;39(15_suppl):9508– doi 10.1200/JCO.2021.39.15_suppl.9508. [DOI] [Google Scholar]
- 34.Long GV, Atkinson V, Lo S, Sandhu S, Guminski AD, Brown MP, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. The Lancet Oncology 2018;19(5):672–81 doi 10.1016/S1470-2045(18)30139-6. [DOI] [PubMed] [Google Scholar]
- 35.Giles AJ, Hutchinson M-KND, Sonnemann HM, Jung J, Fecci PE, Ratnam NM, et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. Journal for ImmunoTherapy of Cancer 2018;6(1):51 doi 10.1186/s40425-018-0371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. The Journal of clinical investigation 2013;123(3):1371–81 doi 10.1172/JCI66236. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012;18(5):1386–94 doi 10.1158/1078-0432.Ccr-11-2479. [DOI] [PubMed] [Google Scholar]
- 38.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19(5):1225–31 doi 10.1158/1078-0432.Ccr-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long GV, Carlino MS, Au-Yeung G, Spillane AJ, Shannon KF, Gyorki DE, et al. NeoTrio: Randomized trial of neoadjuvant (NAT) pembrolizumab (Pembro) alone, in sequence (SEQ) with, or concurrent (CON) with dabrafenib plus trametinib (D+T) in resectable BRAF-mutant stage III melanoma to determine optimal combination of therapy. Journal of Clinical Oncology 2022;40(16_suppl):9503– doi 10.1200/JCO.2022.40.16_suppl.9503. [DOI] [Google Scholar]
- 40.Dummer R, Queirolo P, Abajo Guijarro AM, Hu Y, Wang D, de Azevedo SJ, et al. Atezolizumab, vemurafenib, and cobimetinib in patients with melanoma with CNS metastases (TRICOTEL): a multicentre, open-label, single-arm, phase 2 study. The Lancet Oncology 2022. doi 10.1016/S1470-2045(22)00452-1. [DOI] [PubMed] [Google Scholar]
- 41.Chen G, Chakravarti N, Aardalen K, Lazar AJ, Tetzlaff MT, Wubbenhorst B, et al. Molecular Profiling of Patient-Matched Brain and Extracranial Melanoma Metastases Implicates the PI3K Pathway as a Therapeutic Target. Clinical Cancer Research 2014;20(21):5537 doi 10.1158/1078-0432.CCR-13-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer GM, Jalali A, Kircher DA, Lee W-C, McQuade JL, Haydu LE, et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discovery 2019;9(5):628 doi 10.1158/2159-8290.CD-18-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trembath DG, Davis ES, Rao S, Bradler E, Saada AF, Midkiff BR, et al. Brain Tumor Microenvironment and Angiogenesis in Melanoma Brain Metastases. Frontiers in Oncology 2021;10:2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banks PD, Lasocki A, Lau PKH, Sandhu S, McArthur G, Shackleton M. Bevacizumab as a steroid-sparing agent during immunotherapy for melanoma brain metastases: A case series. Health Science Reports 2019;2(3):e115 doi 10.1002/hsr2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study is available in the European Genome-Phenome Archive (EGAC00001002614) database.