

Abstract
Purpose:
Cetuximab is a standard-of-care treatment for head and neck squamous cell carcinoma (HNSCC). Well-defined correlative markers of therapeutic responses are still lacking. Characterizing dynamic changes of T cell receptor (TCR) repertoire in peripheral blood and tumor tissue may facilitate developing markers for cetuximab response in HNSCCs.
Experimental Design:
We analyzed high-throughput TCRβ sequencing data generated with ImmunoSEQ platform using peripheral blood and tumor infiltrating lymphocytes (TIL) from HNSCC patients before and after cetuximab treatment (pre-/post-PBMC vs. pre-/post-TIL). Multiple analytic approaches were employed to normalize sequencing data.
Results:
Normalized TCR richness was significantly lower in post-TIL than pre-TIL, suggesting that cetuximab reduced TCR diversity and promoted TCR expansion in TIL samples, regardless of response status. The magnitude of clonal expansion (defined as expansion rate) in top-20 TCR clonotypes was significantly higher in responder PBMC with or without normalization, and in responder TIL upon normalization, than non-responder ones. Notably, the expanded top-20 or top-50 TCR clonotypes overlapped between PBMC and TIL samples, which occurred significantly more frequently in responders than non-responders.
Conclusions:
Cetuximab-treated HNSCC patients harbor dynamic changes of TCR repertoires correlative to therapeutic responses. The expansion rate of top TCR clonotypes in peripheral blood may serve as a minimally invasive, readily accessible, and feasible marker for predicting cetuximab responses in HNSCCs and beyond, and the expansion rate of top TCR clonotypes in TILs and their overlapping probability between PBMC and TIL may serve as additional predictive markers. Our study also highlights the importance of data normalization for TCR repertoire analysis.
Keywords: Cancer immunotherapy, head and neck cancers, T cell receptor (TCR) clonotype, cetuximab, clonal expansion of TCR, TCR repertoire analysis
Introduction
About 90% of head and neck cancers are head and neck squamous cell carcinoma (HNSCC) (1,2). HNSCCs have a high morbidity and mortality rate, with only 50–60% patients surviving 5 years (3). The risk factors for HNSCCs include carcinogens, such as alcohol and tobacco use, or oncogenic human papillomavirus (HPV) (2,4); thus, HNSCCs can be categorized into two main subtypes as HPV− or HPV+ HNSCCs, according to distinct etiology. HNSCCs often overexpress epidermal growth factor receptor (EGFR) (5), and agents have been approved by FDA to target EGFR in HNSCCs, including cetuximab, a monoclonal antibody (mAb) against EGFR. Response rate to cetuximab in HNSCCs remains modest (<15% in recurrent/metastatic (R/M) HNSCCs) (5) and markers correlated to clinical response remain poorly characterized.
T cells play an essential role in mediating anti-tumor immunity by recognizing tumor-specific or tumor-associated antigens via clonotypic T cell receptors (TCRs). TCRs are generated through a somatic DNA recombination process, termed V(D)J recombination (6,7), occurring in a random and stochastic manner. Most conventional T cells express TCRs consisting of an alpha (α) chain and a beta (β) chain, encoded by TRA and TRB, respectively, and linked by disulfide bonds. T cells can be grouped into different “clonotypes” based on their unique TCRα and/or TCRβ chains, constituting specific V(D)J gene segments and complementarity-determining region 3 (CDR3). The latter is of particular interest for T cell function because antigen specificity of each TCR is largely determined by CDR3 that encompasses the highly divergent junction of V(D)J recombination and encodes for the antigen binding pocket of TCR. Therefore, clonal diversity representing how many unique clones per sample and expansion features of the TCR repertoire can be assessed by analyzing TCRα and/or TCRβ CDR3 sequences of a given patient sample using high-throughput sequencing (8,9). Moreover, CDR3 analysis allows for assessing dynamic changes for a given TCR clone between pre- and post-treatment in a patient-specific manner and for identifying difference or similarity between peripheral and tumor-infiltrating T cells in their TCR repertoires. Hence, characterizing dynamic changes in the TCR repertoire of the peripheral blood mononuclear cells (PBMC) and tumor tissue may facilitate developing minimally invasive markers for immunotherapy efficacy, including clinical responses to cetuximab treatment.
We previously showed that cetuximab can significantly enhance antigen presentation and priming of adaptive immune responses in tumor-infiltrating lymphocytes (TIL) (10). To further delineate the actual changes in adaptive immunity in relation to cetuximab response, we examined the effect of cetuximab on the TCR repertoires of peripheral and tumor-infiltrating T cells from 14 HNSCC patients before and after treatment, taking advantage of a prospective neoadjuvant “window of opportunity” clinical trial of neoadjuvant cetuximab therapy (NCT01218048) (11). Some uncertainty and controversy exist about whether TCR dynamic changes correlate with clinical response to immunotherapy such as anti-EGFR. Further studies are needed to evaluate different analytic methods to overcome technical variations in the number of total productive TCR sequences obtained in different samples.
In the current study, we applied a new analytic method to TCR sequencing data and tested different normalization approaches that account for the variations in the number of total productive TCR sequences obtained in different samples. Using this method, we did not detect differences between responders and non-responders in TCR richness, defined by the number of unique productive TCR sequences. However, we did identify a few parameters that correlated with cetuximab response, including the expansion rate of top TCR clonotypes in PBMC or TIL and the overlapping probability of expanded top TCR clonotypes between PBMC and TIL. We propose that these parameters may serve as useful markers for predicting clinical response to cetuximab. Our study also highlights the importance of data normalization for TCR repertoire analysis, based on total number of productive TCR sequences amplified in a given sample.
Materials and Methods
Patient sample collection
Detailed information for HNSCC patient and clinical trial was previously described (11). Briefly, treatment naïve stage III/IV HNSCC patients who were candidates for surgical resection were recruited to the prospective phase II “window of opportunity” clinical trial (NCT01218048). The clinical trial was started in 2011 and completed in 2017. The prior study was approved by the Institutional Review Board (IRB) at the University of Pittsburgh and conducted in accordance with Good Clinical Practice guidelines and the ethical principles outlined in the Declaration of Helsinki. Written informed consent was obtained from all subjects before enrollment (11). Anti-EGFR treatment composed of 3–4 weekly doses of neoadjuvant cetuximab, followed by surgical resection within 36–48 hours of the last cetuximab dose received. Radiological response was evaluated according to RECIST criteria. The basic clinical characteristic of patients is included in Supplemental File 1. Patient sample collection was performed in the prior study (11). Briefly, tumor tissue and peripheral blood samples were collected at initial screening and again at the time of surgery under the IRB-approved tissue banking protocol (HCC 99–069). PBMCs were isolated from peripheral blood samples as described previously (11) and immediately used for experiments or stored at −80°C for further analysis. TILs were isolated as described previously and used for TCRβ sequencing with ImmunoSEQ platform by Adaptive Biotechnologies. Clinical samples were collected upon approval of IRB at the University of Pittsburgh (UPCI 99–069) in a prior study. The current study only involves analysis of existing dataset.
Analysis of TCR sequencing data and statistical analysis
Each patient has 4 samples sequenced including pre- and post-PBMC and pre- and post TIL (Tissue) before and after four weeks of cetuximab treatment. In total, 56 samples were sequenced for 14 HNSCC patients. ImmunoSEQ Analyzer was used to retrieve, process, and track the TCRβ sequencing data. The data retrieved from immunoSEQ analyzer contained two columns that are relevant to TCR repertoire analysis, “Total Productive Templates” representing the number of total productive TCR DNA sequences obtained per sample and “Productive Rearrangements” representing the number of unique productive TCR DNA sequences in each sample. We combined the productive rearrangements that encode the same CDR3 amino acid (a.a.) and contain the same V, D, J gene usage into the same TCR clonotypes using R. TCR richness was defined as the number of unique productive TCR a.a. sequences in each sample. Normalized TCR richness was calculated by dividing the number of unique productive TCR a.a. sequences with the number of total productive TCR DNA sequences. Differences between two independent groups (e.g., pre-PBMC vs. post-PBMC) were compared by the Wilcoxon Signed-Rank tests for the number of total productive TCR sequences, TCR richness or normalized TCR richness.
Expansion rate was calculated by dividing the frequency of a given TCR sequence between post- and pre-treatment samples (expansion rate for PBMC= % in post-PBMC / % in pre-PBMC; expansion rate for TIL= % in post-TIL / % in pre-TIL). If expansion rate is >1 for a given TCR clonotype, it will be counted as an expanded clone. Singleton clonotype was defined as the TCR sequences that only occurred once in a given PBMC or TIL sample. The sum of a singleton clonotype frequency was calculated by adding up the clonal frequency of all singleton clonotypes in a given sample. Normalized expansion rate was calculated in two different ways: (1) using clonal frequency of a singleton clonotype, or ; (2) using the sum (Σ) of total singleton clonotype frequency, or (n is the total number of singleton clonotypes). Differences between two independent groups (R vs. NR) were compared by the Wilcoxon rank sum test for expansion rate or normalized expansion rate. Data analyses were carried out using the R version 4.2.0. All tests are two-sided and a p-value of 0.05 or less is considered statistically significant.
Data availability
Adaptive ImmunoSEQ sequencing data are available via clients.adaptivebiotech.com/pub/ge-2022-ccr. The immuneACCESS DOI is https://doi.org/10.21417/HG2022CCR. Additional analysis information is available upon request to Dr. Jing H. Wang at jhw51@pitt.edu
Results
Variations in the number of total productive TCR sequences influence TCR richness analysis
We found that the number of total productive TCR sequences, which could serve as a surrogate for how many T cells were sequenced per sample, varied significantly in different samples; in particular, it appeared to be predominantly higher in post-PBMC samples (Figure 1A), regardless of responder vs. non-responder status (Figure 1B, C). Consistently, we showed that the number of total productive TCR sequences was significantly higher in post-PBMC than pre-PBMC samples (p=0.0006) (Figure 1D). These data suggest that there were many more T cells sequenced or many more productive TCR sequences obtained in post-PBMC group, raising a question about whether variations in the number of total productive TCR sequences may influence TCR richness analysis.
Figure 1. Influence of sample size variation on TCR richness analysis.
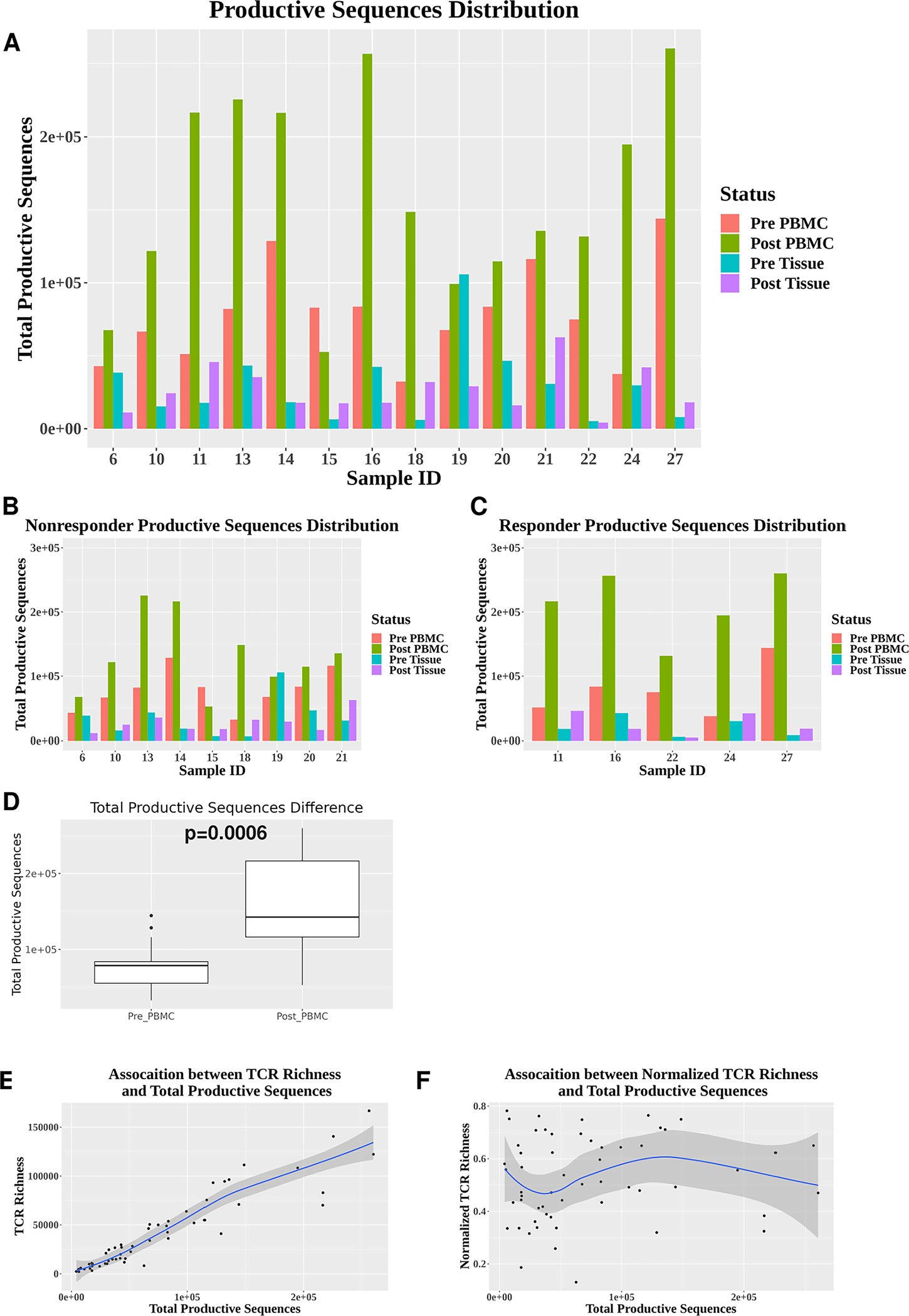
(A) The number of total productive TCR sequences (y-axis) shown for all 56 samples collected from 14 HNSCC patients. Each patient (sample ID indicted on x-axis) has 4 samples sequenced including pre-PBMC, post-PBMC, pre-Tissue (TIL) and post-Tissue (TIL). (B-C) The number of total productive TCR sequences in responder samples (n=5) (B) and non-responder samples (n=9) (C). (D) Differences in the number of total productive TCR sequences between pre-PBMC (n=14) and post-PBMC (n=14). Statistical analysis was performed using Wilcoxon signed rank test (p=0.0006). (E) A strong positive association between the number of unique productive TCR sequences and the number of total productive TCR sequences (n=56) (R2=0.87). (F) Little association between normalized TCR richness (the number of unique productive TCR sequences) and the number of total productive TCR sequences (n=56) (R2=0.01).
Our current analysis revealed that TCR richness demonstrated a supporting strong positive association with the number of total productive TCR sequences (Figure 1E), meaning that if a given sample has more productive TCR sequences, it will have more unique productive TCR sequences. To reduce the influence of such sampling variation, we normalized the number of unique productive TCR sequences by dividing it by the number of total productive TCR sequences, defined as “normalized TCR richness”. After applying the normalization step, we did not detect a strong association between normalized TCR richness and the number of total productive TCR sequences (Figure 1F). These data suggest that we have minimized the effect of sampling variation on TCR richness analysis.
Normalized TCR richness is significantly different between pre- and post-TIL samples
After normalizing TCR richness with the number of total productive TCR sequences, we did not detect differences between pre-PBMC and post-PBMC samples in normalized TCR richness (Figure 2A). However, we found that the normalized TCR richness was significantly lower in post-TIL than pre-TIL samples (Figure 2B, p=0.05), indicating that clonal diversity was reduced in post-TIL, consistent with the notion that TCRs underwent clonal expansion at tumor site after cetuximab treatment. We did not observe significant differences in normalized TCR richness between responders and non-responders for pre-PBMC, post-PBMC, pre-TIL and post-TIL (Figure 2C, D). We also detected no significant differences in normalized TCR richness within responder or non-responder group by comparing pre-PBMC vs. post-PBMC or pre-TIL vs. post-TIL (data not shown). Lastly, we compared HPV+ vs. HPV− samples and did not identify significant differences for all paired comparison between HPV+ and HPV− or within HPV+ or HPV− samples by comparing pre- vs. post-PBMC or pre- vs. post-TIL (data not shown). In summary, we did not detect significant differences in normalized TCR richness in paired comparison among all samples except between pre-TIL and post-TIL.
Figure 2. Comparison of normalized TCR richness.
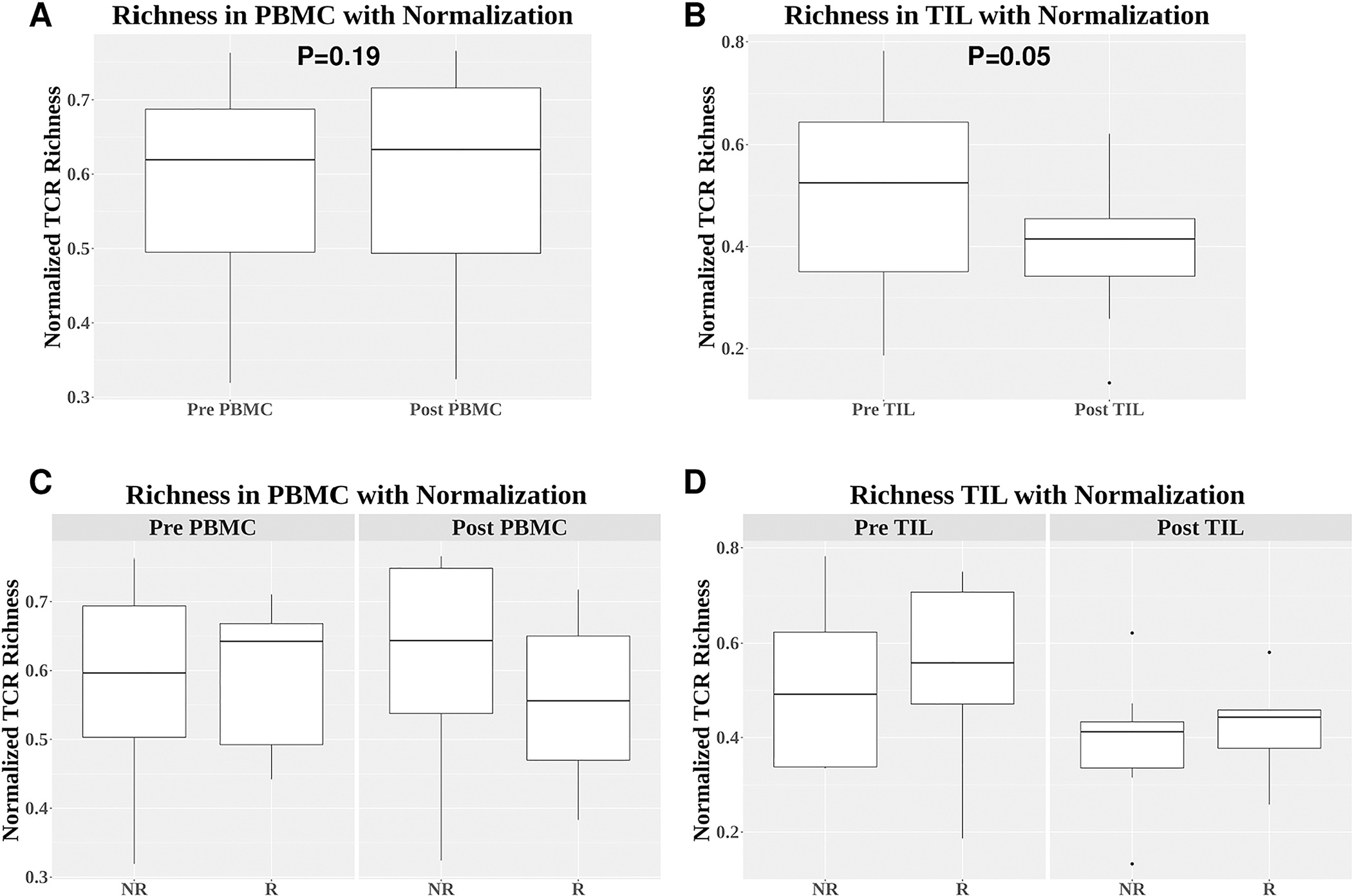
(A-B) Comparison of normalized TCR richness between pre-PBMC and post-PBMC (p=0.19) (A) or between pre-TIL and post-TIL (p=0.05) (B). (C-D) Comparison of normalized TCR richness between responders and non-responders for pre-PBMC (p=1.0), post-PBMC (p=0.44), pre-TIL (p=0.80), and post-TIL (p=0.61). Differences between two independent groups were calculated by Wilcoxon signed rank test.
Characterizing clonal expansion features in pre- vs. post-treatment samples
There are two levels of clonal expansion, and the first one is clonal expansion within a given sample. Each patient has 4 samples: pre-PBMC, post-PBMC, pre-TIL and post-TIL. We can calculate the clonotype frequency for each clone within a given sample, for instance, the clonotype of CASTRTGYPAGPHNEQFF has its frequency as 1.033% in pre-TIL sample and 2.401% in post-TIL sample of patient 16 (Table 1). We can determine the extent of their clonal expansion based on their relative clonal frequency against the frequency of a singleton (see Method) and their ranking in the entire TCR repertoire of each patient’s PBMC or TIL samples. However, the number of total productive TCR sequences of pre-TIL vs. post-TIL of patient 16 varied significantly (Figure 1C), which may influence the baseline of expansion potential within each sample. Hence, we may have to normalize samples when comparing the extent of clonal expansion between different samples.
Table 1.
Eleven Tx-expanded TCR clonotypes shared between PBMC and TIL in top 20 TCR clonotypes of post-PBMC or post-TIL
| PXID | CDR3 region amino_acid | pre_TIL_Freq | pre_TIL_Rank | post_TIL_Freq | post_TIL_Rank | Ratio_TIL | pre_PBMC_Freq | pre_PBMC_Rank | post_PBMC_Freq | post_PBMC_Rank | Ratio_PBMC |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| 6 | CASSPPSRDYVSYEQYF | 1.908% | 3 | 2.263% | 1 | 1.186 | 0.31% | 8 | 0.409% | 8 | 1.321 |
| 15 | CASSHNVYTGELFF | 0.03% | 537 | 0.728% | 5 | 24.571 | 0.807% | 5 | 0.994% | 5 | 1.233 |
| 16 | CASTRTGYPAGPHNEQFF | 1.033% | 4 | 2.401% | 3 | 2.324 | 2.432% | 2 | 3.68% | 2 | 1.513 |
| 16 | CASSLSGGGEQYF | 0.302% | 12 | 0.913% | 8 | 3.027 | 0.194% | 16 | 0.362% | 10 | 1.871 |
| 22 | CASSEAETGGYEQYF | 0.111% | 125 | 1.395% | 6 | 12.628 | 0.618% | 4 | 0.952% | 2 | 1.54 |
| 24 | CASSSLRGPEDYGYTF | 0.802% | 1 | 3.142% | 1 | 3.919 | 0.679% | 2 | 1.222% | 3 | 1.799 |
| 24 | CASSSHYPTDTQYF | 0.345% | 11 | 1.29% | 7 | 3.736 | 0.287% | 6 | 0.52% | 5 | 1.814 |
| 24 | CAISDPRDSYEQYF | 0.274% | 13 | 0.825% | 11 | 3.007 | 0.192% | 11 | 0.224% | 18 | 1.168 |
| 24 | CASSDRGDPSEPQHF | 0.55% | 7 | 0.799% | 12 | 1.452 | 2.832% | 1 | 3.549% | 1 | 1.253 |
| 24 | CASSYSTSGQETQYF | 0.154% | 20 | 0.541% | 17 | 3.507 | 0.184% | 12 | 0.505% | 6 | 2.736 |
| 27 | CASSFHAGAGNTIYF | 0.668% | 5 | 1.306% | 6 | 1.955 | 0.629% | 5 | 0.967% | 6 | 1.537 |
Patient ID indicates the patient who harbors a shared expanded top TCR clonotype.
Patient 6 and 15 are non-responders and patient 16, 22, 24, and 27 are responders (in red fonts).
The second one is clonal expansion between samples by comparing pre- vs. post-treatment, which is likely induced by cetuximab treatment as shown in Table 1. Given the evidence of clonal expansion in TCR repertoire, we next examined how TCR clones underwent expansion by comparing pre-PBMC vs. post-PBMC or pre-TIL vs. post-TIL samples. Expanded clones were defined as a given TCR clone whose clonal frequency was higher in post-treatment than pre-treatment samples. We found that the number of expanded clones was not significantly different between responders and non-responders for both PBMC and TIL samples (Figure 3A, B), likely due to sampling variation.
Figure 3. Differences in the expansion rate of top TCR clonotypes between responders and non-responders.
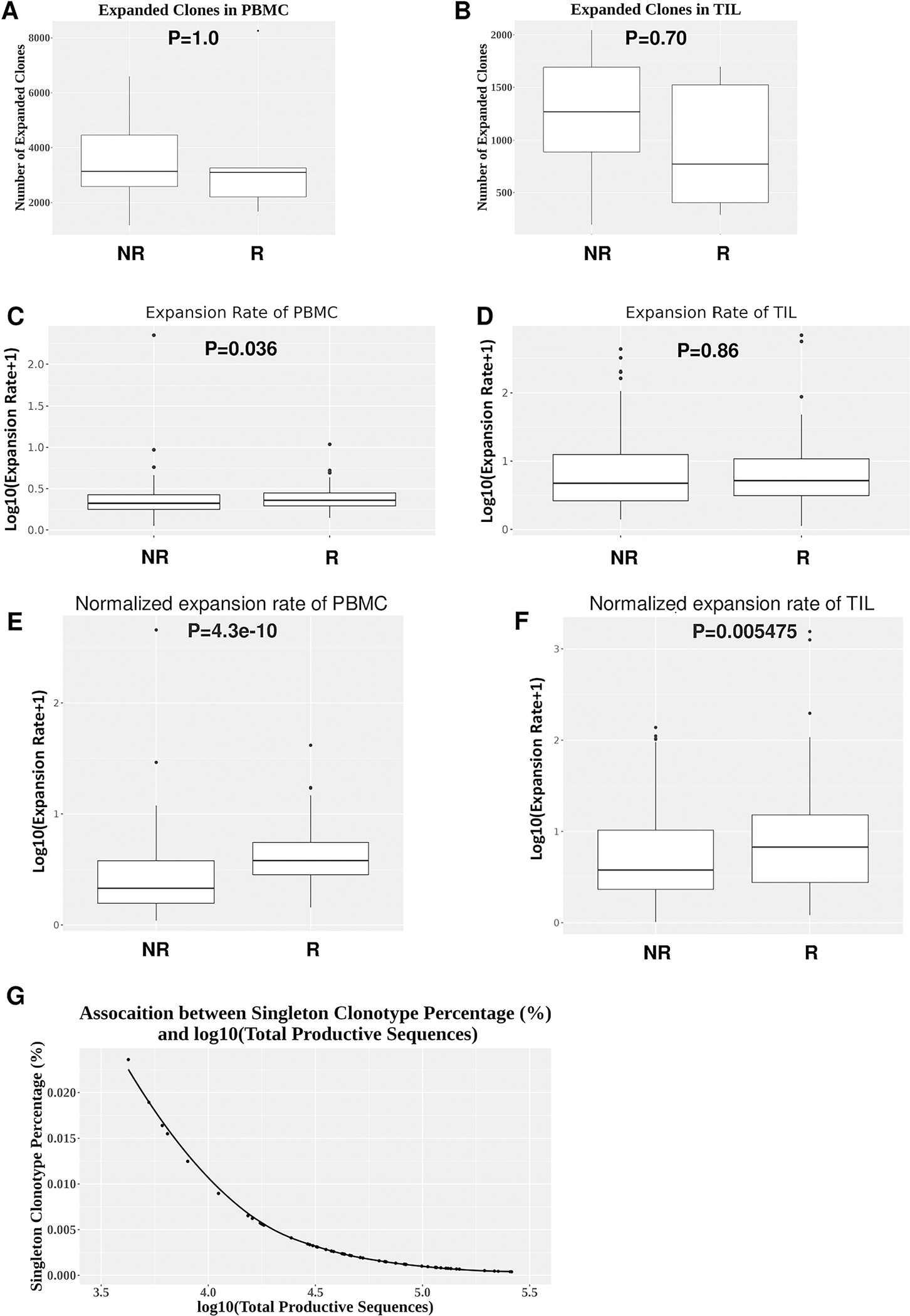
(A-B) Numbers of all expanded clones in PBMC or TIL between responders (n=5) and non-responder (n=9). Expanded clones were calculated by comparing the clonal frequency of a given TCR clone between pre-PBMC vs. post-PBMC or pre-TIL vs. post-TIL. (C-D) Comparison of expansion rate of expanded top 20 TCR clonotypes without normalization between responders (n=5) and non-responders (n=9) in PBMC (p=0.036) (C) or TIL (p=0.86) (D). (E-F) Comparison of expansion rate of expanded top 20 TCR clonotypes between responders (n=5) and non-responders (n=9) in PBMC (p=4.3e-10) (E) or TIL (p=0.005475) (F) normalized by the clonal frequency of a given singleton clonotype. Differences between two independent groups were calculated by Wilcoxon rank sum test. (G) An inverse correlation between the clonal frequency of singleton clonotype and Log10(total productive TCR sequences) (R2=0.73).
Next, we focused on the top 20 TCR clones ranked by their individual clonal frequency within each sample, because we envision that these top ranked clones may contribute more to anti-tumor immunity than relatively rare clones. We first compiled top 20 clones in post-treatment samples including post-PBMC and post-TIL from each responder or non-responder and identified the expanded TCR clonotypes by comparing clonal frequency between pre- vs. post-treatment samples, defined as “expanded clonotypes from top 20 clones”, for simplicity, termed “expanded top 20 clonotypes”. Of note, not all of the top 20 ranked TCR clones were expanded between pre- and post-treatment samples (see details below in Figure 5). The expansion rate was calculated to reflect the magnitude of clonal expansion between pre- vs. post-PBMC or between pre- vs. post-TIL for a given TCR clonotype (see details in Methods). We found that the expansion rate of expanded top 20 clonotypes was significantly higher in responder PBMC samples than non-responder ones (Figure 3C), even without baseline normalization for expansion potential. In contrast, we did not detect significant differences in the expansion rate of expanded top 20 clonotypes between responder and non-responder TIL samples without baseline normalization for expansion potential (Figure 3D).
Figure 5. Overlapping expanded top 20 TCR clonotypes between PBMC and TIL.
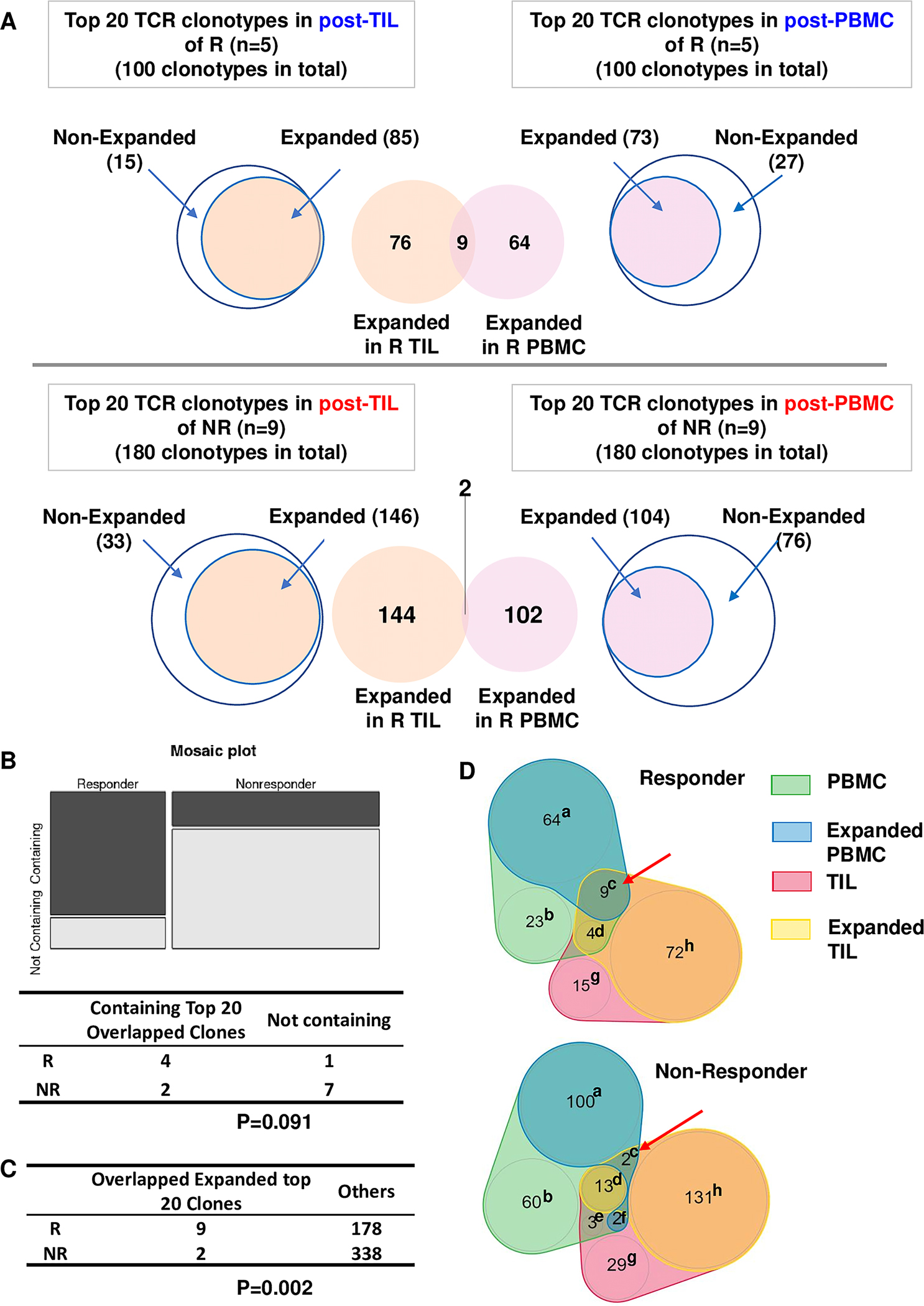
(A) Scheme for identification and comparison of overlapping expanded top 20 TCR clonotypes between PBMC and TIL. (B) Number of responders (n=5) or non-responders (n=9) containing or not containing an overlapping expanded top 20 TCR clonotype. Statistical difference was calculated with Fisher Exact Test (p= 0.091). (C) Number of overlapping expanded vs. others in top 20 TCR clonotypes of responders (n=5) vs. non-responders (n=9). Others are counted as follows: the total number of top 20 TCR clonotypes in PBMC and TIL of responders (n=200) or non-responders (n=360) minus the number of clonotypes in category of c, d, e, and f and the number of clonotypes in category of c. Statistical difference was calculated with Fisher Exact Test (p=0.002). (D) Number of TCR clonotypes distributed into 8 different categories (a-h) for expanding or overlapping conditions between PBMC and TIL. Annotation: (a) PBMC Expanded alone; (b) PBMC Nonexpanded; (c) PBMC Expanded & TIL Expanded; (d) PBMC Nonexpanded & TIL Expanded; (e) PBMC Nonexpanded & TIL Nonexpanded; (f) PBMC Expanded & TIL Nonexpanded; (g) TIL Nonexpanded; (h) TIL Expanded alone.
Since the baseline of expansion potential varied markedly in different groups, we first normalized the expansion rate with the clonal frequency of a given singleton clonotype (see details in Method). After applying this normalization approach, we found that the normalized expansion rate was significantly higher in both PBMC and TIL samples in responders than non-responders (Figure 3E, F). However, we noticed that the clonal frequency of a given singleton clonotype reversely correlated to Log10(the number of total productive TCR sequences) (Figure 3G). For instance, if a sample has 5000 productive TCR sequences, the singleton frequency would be 1/5000=0.02%; if a sample has 10,000 productive TCR sequences, the singleton frequency would be 1/10,000=0.01%. Thus, this normalization approach will again be influenced by the number of total productive TCR sequences, namely, sampling variation.
To better quantify the expansion potential of expanded top 20 clonotypes, we divided the clonal frequency of each expanded top 20 clonotype with the sum of a singleton clonotype frequency within a given sample, then calculated the normalized expansion rate between pre- and post-treatment samples (see details in Method). We chose the sum of a singleton clonotype frequency to calibrate expansion potential between different samples due to the following reasons: (1) we found that this parameter did not correlate with the number of total productive TCR sequences (Figure 4A), thus, was not affected by sampling variation; (2) we did not detect any significant differences in the sum of a singleton clonotype frequency between responders and non-responders for all groups including pre-PBMC, post-PBMC, pre-TIL and post-TIL (Figure 4B). Hence, this parameter was not affected by sample size or sample type; (3) in a certain sense, the sum of a singleton clonotype frequency sets the baseline for expansion potential. Basically, it represents the percentage of all nonexpanded clones. After applying this normalization approach, we found that the expansion rate of expanded top 20 clonotypes was still significantly higher in responder PBMC (Figure 4C) and, especially, in responder TIL samples (Figure 4D) than non-responder ones. Taken together, we conclude that the expansion rate of expanded top 20 clonotypes was significantly higher in responder PBMC regardless of normalization and remarkably higher in responder TIL upon normalization.
Figure 4. Differences in the normalized expansion rate of expanded top 20 TCR clonotypes between responder and non-responder TILs.
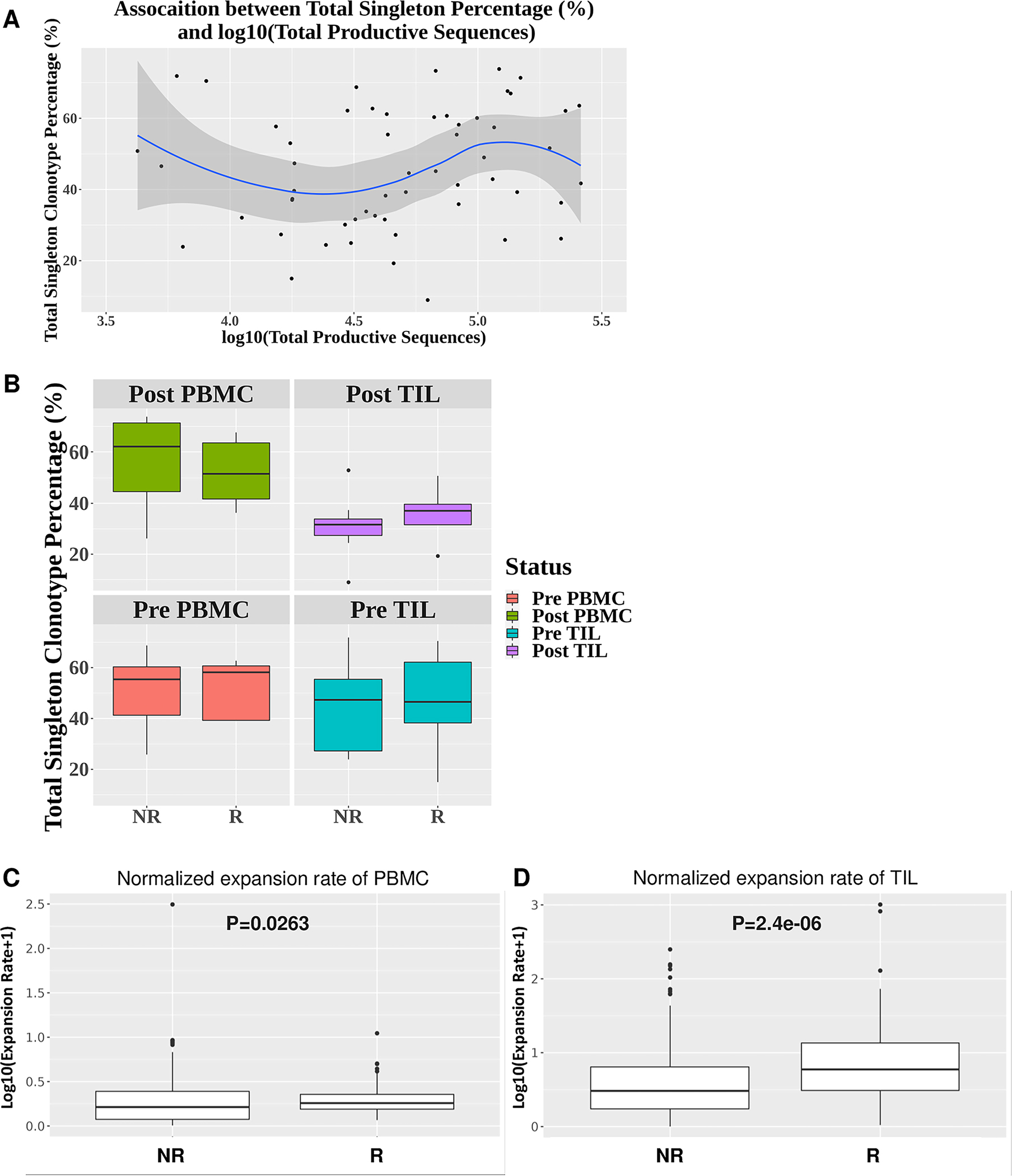
(A) Little association between the sum of frequency of singleton clonotypes and the number of total productive TCR sequences (n=56) (R2=0.035). (B) No significant differences in the sum of frequency of singleton clonotypes between responders (n=5) and non-responders (n=9) for post-PBMC (p=0.44), post-TIL (p=0.52), pre-PBMC (p=0.80), and pre-TIL (p=0.90). (C-D) Significant differences in the normalized expansion rate of expanded top 20 TCR clonotypes between responders (n=5) and non-responders (n=9) for PBMC (p=0.0263) or TIL (p=2.4e-06). Statistical difference was calculated by Wilcoxon rank sum test.
We also tested whether HPV status affected the extent of TCR clonotype expansion. We failed to detect any significant correlation between HPV status and the number of all expanded clones or the expansion rate of top 20 TCR clonotypes (Supplemental Figure 1A–C). However, the sample size is rather small, and additional studies are needed to validate the findings.
Overlapping probability of expanded top TCR clonotypes between PBMC and TILs
Since we identified expanded TCR clonotypes in PBMC or TIL samples, we next asked whether the expanded top TCR clonotypes were shared between PBMC and TIL samples in a given patient. We first compiled the top 20 TCR clonotypes of post-PBMC or post-TIL from responders (n=5) (Figure 5A, top) or non-responders (n=9) (Figure 5A, bottom), identified the expanded clones in PBMC or TIL, then overlapped the two groups of the expanded clones to find the shared expanded TCR clonotypes between PBMC and TIL samples. To define an overlapping expanded TCR clonotype, we calculated the ratio of (Freq in post-PBMC)/(Freq in pre-PBMC) and the ratio of (Freq in post-TIL)/(Freq in pre-TIL), both of which had to be more than 1 (Table 1). Using the TCR clonotype of CASSLSGGGEQYF as an example, the ratio of post-TIL/pre-TIL is 3.027 and the ratio of post-PBMC/pre-PBMC is 1.871 (Table 1), showing that this particular TCR clonotype was expanded in both PBMC and TIL after cetuximab treatment. Overall, we identified 11 TCR clonotypes shared between PBMC and TIL samples (Table 1); furthermore, 9 out of 11 clonotypes were found in responders (Figure 5A, Table 1). Notably, 4 out of 5 responders were found to harbor 9 shared TCR clonotypes, whereas only 2 out of 9 non-responders contained 2 shared TCR clonotypes (Figure 5A, B, Table 1). Clinical responders showed a significantly higher chance to harbor a shared expanded top 20 TCR clonotype between PBMC and TIL than non-responders (Figure 5C). Of note, the TCR clonotypes can distribute into 8 different categories based on their expansion and overlapping status in PBMC and/or TIL (Figure 5D, see details in figure legend for category a-h). The only category that had a higher number of clonotypes in responders is category c (PBMC Expanded & TIL Expanded), while all others showed higher numbers in non-responders likely due to random chance since non-responders had a higher number of total clones (n=180 for non-responders vs. n=100 for responders).
Next, we extended our analysis to the top 50 TCR clonotypes identified in post-treatment samples (Figure 6A). We found that all responders harbored shared expanded top TCR clonotypes, while 6 out of 9 non-responders harbored such TCR clonotypes (Figure 6B). Similar to what we observed for top 20 TCR clonotypes, the responders still showed a significantly higher chance to harbor a shared, expanded top TCR clonotype between PBMC and TIL than did non-responders (Figure 6C). Again, we observed that only category c showed a higher number of TCR clonotypes in responders than non-responders (21 vs. 13), whereas all other categories showed higher numbers in non-responders (Figure 6D), likely due to larger sample size (n=450 for non-responders vs. n=250 for responders). These data strongly suggest that the shared expanded top TCR clonotypes occurred much more frequently in responders against random chance. When all expanded TCR clonotypes were included, we did not find the overlapping expanded TCR clonotypes occurred more frequently in responders, instead, they occurred more frequently in non-responders (Supplemental Figure 2), likely due to random chance since non-responder samples contained more productive TCR clonotypes. Thus, we conclude that the expanded top TCR clonotypes can be shared between PBMC and TIL samples, which occurred much more frequently in responders than non-responders.
Figure 6. Overlapping expanded top 50 TCR clonotypes between PBMC and TIL.
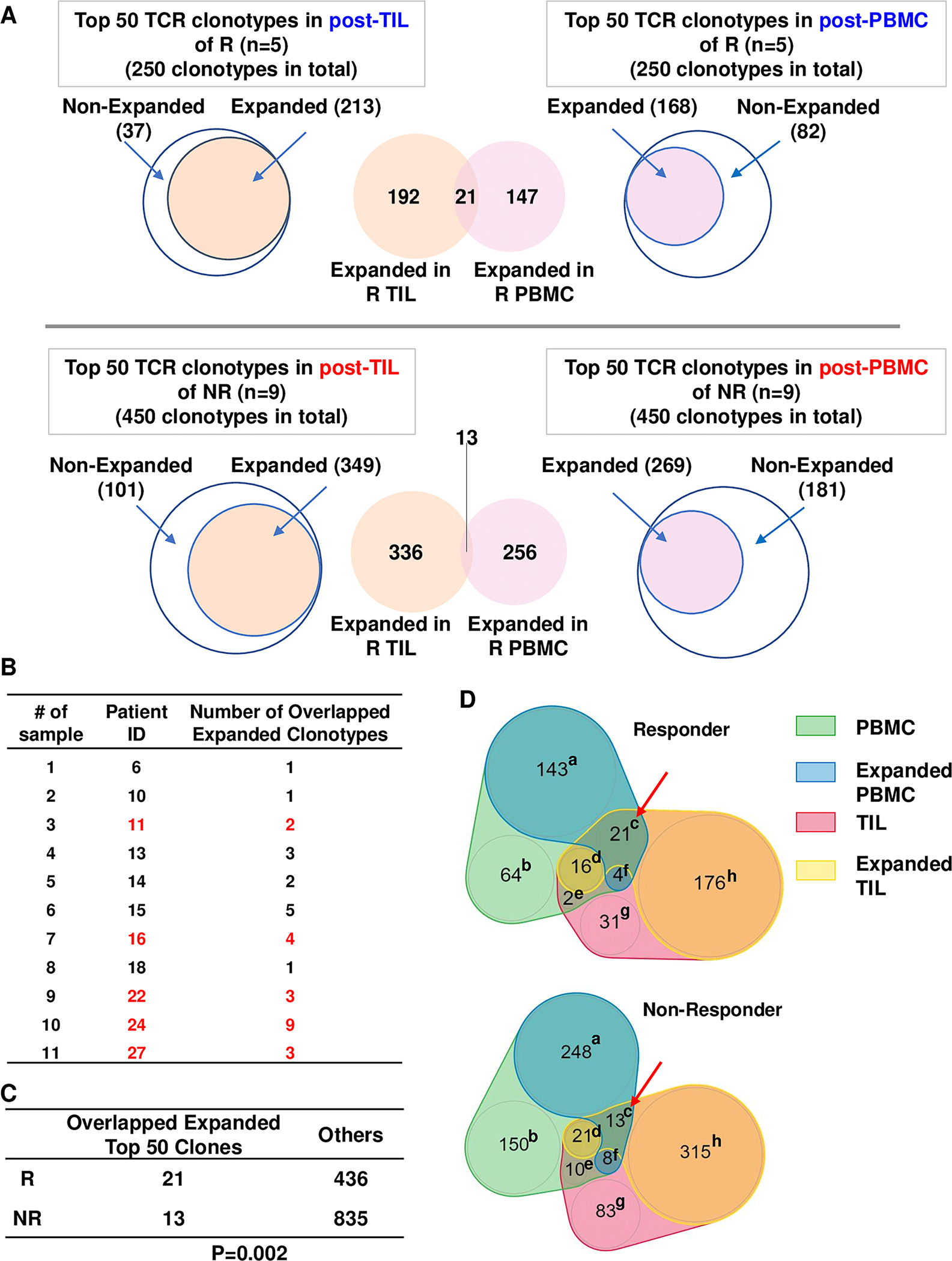
(A) Scheme for identification and comparison of overlapping expanded top 50 TCR clonotypes between PBMC and TIL. (B) Number of overlapping expanded top 50 TCR clonotypes in each HNSCC patient. Responder IDs are denoted in red fonts. (C) Number of overlapping expanded vs. others in top 50 TCR clonotypes of responders (n=5) vs. non-responders (n=9). Others are counted as follows: the total number of top 50 TCR clonotypes in PBMC and TIL of responders (n=500) or non-responders (n=900) minus the number of clonotypes in category of c, d, e, and f and the number of clonotypes in category of c. Statistical difference was calculated with Chi-square test (p=0.002). (D) Number of TCR clonotypes distributed into 8 different categories (a-h) for expanding or overlapping conditions between PBMC and TIL. Annotation: (a) PBMC Expanded alone; (b) PBMC Nonexpanded; (c) PBMC Expanded & TIL Expanded; (d) PBMC Nonexpanded & TIL Expanded; (e) PBMC Nonexpanded & TIL Nonexpanded; (f) PBMC Expanded & TIL Nonexpanded; (g) TIL Nonexpanded; (h) TIL Expanded alone.
Next, we examined whether the top 20 TCR clonotypes of pre-treatment samples at baseline also expanded in post-treatment samples, namely, whether the information of pre-existing TCR clonotypes can help to predict the dynamic changes of TCR clonotypes upon cetuximab treatment. We first compiled the top 20 TCR clonotypes of pre-PBMC or pre-TIL from responders or non-responders, identified the clones that expanded upon treatment in PBMC and TIL, then overlapped the two groups of the expanded clones to find the shared expanded TCR clonotypes between pre-PBMC and pre-TIL samples (Supplemental Figure 3A). We noticed that the percentage of expanded TCR clonotypes in pre-treatment samples appeared to be less than that in corresponding post-treatment samples, for example, by comparing top 20 TCRs in pre-TIL of responders vs. post-TIL of responders (35 expanded out of 100 in Supplemental Figure 3A vs. 85 expanded out of 100 in Figure 5A). These data suggest that if a given TCR ranked among top 20 in post-TIL samples, it appeared to have a higher chance to be a treatment-expanded TCR clone. We found that 3 out of 5 responders harbored shared expanded top TCR clonotypes, while only 1 out of 9 non-responders did so (Supplemental Figure 3B, Supplemental Table 1). Similar to what we observed for post-treatment samples, the responders still showed a significantly higher chance to harbor a shared, expanded top TCR clonotype between pre-PBMC and pre-TIL than did non-responders (Supplemental Figure 3C). A higher number of TCR clonotypes in category c was detected in responders than non-responders (9 vs. 1) (Supplemental Figure 3D). Of note, 8 out of 9 shared expanded top TCR clonotypes identified between pre-PBMC and pre-TIL (Supplemental Table 1) were identical to their counterparts identified between post-PBMC and post-TIL (Table 1), suggesting that the shared expanded top TCR clonotypes existed prior to treatment. Next, we extended our analysis to the top 50 TCR clonotypes identified in pre-PBMC and pre-TIL samples (Supplemental Figure 4A–D). We observed similar findings when top 50 TCR clonotypes were analyzed, although the level of significance was less (Supplemental Figure 4C). These data strongly suggest that the shared expanded top TCR clonotypes occurred much more frequently in responders against random chance.
Discussion
We applied new analytic methods to the TCR sequencing dataset containing 56 samples from 14 cetuximab-treated HNSCC patients comparing pre versus post treatment as well as responder versus non-responders. Using this highly unique data set, for each patient, we sequenced 4 types of samples: pre-PBMC, post-PBMC, pre-TIL and post-TIL. By examining the differences between pre- and post-treatment samples of HNSCC patients, we uncovered dynamic changes of the TCR repertoires that correlated with cetuximab clinical responses (tumor shrinkage), including the expansion rate of top TCR clonotypes in PBMC or TIL and the overlapping probability of expanded top TCR clonotypes between PBMC and TIL. Our current analysis revealed that TCR richness strongly associated with the number of total productive TCR sequences, with the latter varying significantly in different samples. After normalizing TCR richness with the number of total productive TCR sequences, we did not detect differences in normalized TCR richness in paired comparison among all samples except between pre- and post-TIL. While these observations may be distinct from our prior study (11) but not inconsistent, this is because different analytic methods were employed taking into account or not the influence of the number of total productive TCR sequences.
Prior studies characterized the TCRβ clonotypes in locoregionally advanced HNSCC patient samples (12) or compared HPV+ vs. HPV− HNSCC samples (13), without linking TCR clonotypes with treatment outcomes. Our data showed that the expansion rate in top TCR clonotypes was significantly higher in responder PBMC than non-responder even without normalization. These results suggest that the expansion rate between pre- and post-PBMC samples may serve as a minimally invasive and readily accessible marker predicting clinical response to cetuximab. In line with our observation, a recent study also showed that peripheral TCR repertoire changes correlated with response to anti-PD1 treatment in renal cell carcinoma (14), in which the ratio of post-ICI/pre-ICI TCR diversity index and the number of expanded TCR clones were used for comparison between responders and non-responders. However, it is unclear whether these parameters are influenced by sampling variation and whether they have been normalized to reduce the influence of such variation. Nevertheless, we did not observe significant differences in the number of expanded TCR clones between responders and non-responders (Figure 3A) or in the ratio of post-/pre-treatment TCR diversity index (data not shown). Taken together, our current study highlights the importance of data normalization for TCR repertoire analysis.
After testing and applying different normalization methods, the expansion rate of responder TIL became remarkably higher than non-responder for the top TCR clonotypes. Assuming the expanded top TCRs are tumor-reactive, at least some of them, these results suggest that cetuximab may function as an immunotherapy and promote a much stronger T cell-mediated anti-tumor immune response in responding HNSCC patients. These results also suggest that combining cetuximab and anti-PD1 may synergize in a fraction of patients whose anti-tumor immunity can be promoted via TCR expansion, thereby broadening the spectrum of anti-PD1 responding patients. Consistent with this notion, a recent trial reported that pembrolizumab combined with cetuximab showed promising clinical activity for R/M HNSCCs with the overall response rate as 45% by 6 months (15). In contrast, such a combination of anti-PD1/cetuximab may not be effective in the patients who do not contain useful TCRs (16) that can be expanded significantly upon treatment, similar to what we present here for cetuximab non-responders. Another study examined the peripheral blood TCR repertoires of HNSCC patients treated with concurrent cetuximab and nivolumab and reported that the peripheral TCR repertoires from pre-PBMC might be developed as biomarkers for predicting outcome of combined treatment of cetuximab and nivolumab (17).
We found that the expanded top TCR clonotypes overlapped between PBMC and TIL which occurred much more frequently in responders than non-responders. These data suggest that TIL clones may migrate into peripheral blood after their expansion in the tumor microenvironment (TME), thus, can be detected in the post-PBMC samples. However, another possibility is that certain TCR clones expanded in post-PBMC, then migrated into the TME, similar to the “clonal replacement” concept proposed in a prior study (18). It is also possible that these clones are already prominent before treatment and their shared expansion after treatment is the result of homeostasis between peripheral blood and tumor tissue. Notably, the frequency of overlapping expanded TCR clonotypes is significantly higher in responders than non-responders only when we count the top-20 or top-50 clonotypes. If we included all expanded TCR clonotypes, we do not detect a preferential association between response and overlapping frequency. These data suggest that the dynamic changes in the top ranking TCR clonotypes might be more relevant to therapeutic responses, whereas other clonotypes with limited expansion do not contribute substantially.
A limitation of our current study is that paired pre- and post-treatment samples were only available from 14 patients, with 5 responders vs. 9 non-responders. Nevertheless, we were able to perform analyses on paired peripheral blood and TIL samples, which identified potentially novel biomarkers of response including the expansion rate of top TCR clonotypes in PBMC or TILs and their overlapping probability between PBMC and TIL. Future studies are needed to confirm our initial findings. Our study also highlights the importance of data normalization for TCR repertoire analysis given that the number of total productive TCR sequences varies remarkably in different patient samples, based on specimen size and degree of T cell infiltration. We propose that future TCR repertoire analysis needs to take into consideration of sampling variation.
Supplementary Material
Statement of translational relevance.
We uncovered dynamic changes in the T cell receptor (TCR) repertoires of cetuximab-treated HNSCC patients that correlated with therapeutic responses. Our data indicate that response to cetuximab may be mediated by enhanced T cell-mediated anti-tumor immunity in responders. We propose that evaluating the expansion rate of top TCR clonotypes in PBMC samples may serve as a minimally invasive marker to predict clinical response to cetuximab treatment. Further characterizing the expansion rate of top TCR clonotypes in TIL and their overlapping probability between PBMC and TIL may improve the prognostic accuracy of biomarkers for cetuximab treatment. Our study also highlights the importance of data normalization for TCR repertoire analysis given that the number of total productive TCR sequences varies remarkably in different patient samples, based on specimen size and degree of T cell infiltration.
Acknowledgments
This work was supported by UPMC Hillman Cancer Center startup fund to J.H.W., NIH R01-DE027329 and R01-DE028420 to J.H.W., R01-DE031947 to R.L.F. and J.H.W., P30-CA047904, P50-CA097190, and R01-CA206517 to R.L.F. We thank Dr. Lazar Vujanovic for helpful comments on the manuscript. This research was partially supported by the University of Pittsburgh Center for Research Computing (CRC) through the resources provided. We apologize to those whose work was not cited due to length restrictions.
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interests to the current study.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68(6):394–424 doi 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Cramer JD, Burtness B, Le QT, Ferris RL. The changing therapeutic landscape of head and neck cancer. Nat Rev Clin Oncol 2019;16(11):669–83 doi 10.1038/s41571-019-0227-z. [DOI] [PubMed] [Google Scholar]
- 3.Du E, Mazul AL, Farquhar D, Brennan P, Anantharaman D, Abedi-Ardekani B, et al. Long-term Survival in Head and Neck Cancer: Impact of Site, Stage, Smoking, and Human Papillomavirus Status. Laryngoscope 2019;129(11):2506–13 doi 10.1002/lary.27807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers 2020;6(1):92- doi 10.1038/s41572-020-00224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boeckx C, Baay M, Wouters A, Specenier P, Vermorken JB, Peeters M, et al. Anti-epidermal growth factor receptor therapy in head and neck squamous cell carcinoma: focus on potential molecular mechanisms of drug resistance. Oncologist 2013;18(7):850–64 doi 10.1634/theoncologist.2013-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell 2002;109 Suppl:S45–55. [DOI] [PubMed] [Google Scholar]
- 7.Jung D, Alt FW. Unraveling V(D)J recombination; insights into gene regulation. Cell 2004;116(2):299–311. [DOI] [PubMed] [Google Scholar]
- 8.Calis JJ, Rosenberg BR. Characterizing immune repertoires by high throughput sequencing: strategies and applications. Trends Immunol 2014;35(12):581–90 doi 10.1016/j.it.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, Franke A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol 2017;17(1):61 doi 10.1186/s12896-017-0379-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ming Lim C, Stephenson R, Salazar AM, Ferris RL. TLR3 agonists improve the immunostimulatory potential of cetuximab against EGFR(+) head and neck cancer cells. Oncoimmunology 2013;2(6):e24677 doi 10.4161/onci.24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kansy BA, Shayan G, Jie HB, Gibson SP, Lei YL, Brandau S, et al. T cell receptor richness in peripheral blood increases after cetuximab therapy and correlates with therapeutic response. Oncoimmunology 2018;7(11):e1494112 doi 10.1080/2162402X.2018.1494112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saloura V, Fatima A, Zewde M, Kiyotani K, Brisson R, Park JH, et al. Characterization of the T-Cell Receptor Repertoire and Immune Microenvironment in Patients with Locoregionally Advanced Squamous Cell Carcinoma of the Head and Neck. Clin Cancer Res 2017;23(16):4897–907 doi 10.1158/1078-0432.CCR-17-0103. [DOI] [PubMed] [Google Scholar]
- 13.Poropatich K, Fontanarosa J, Swaminathan S, Dittmann D, Chen S, Samant S, et al. Comprehensive T-cell immunophenotyping and next-generation sequencing of human papillomavirus (HPV)-positive and HPV-negative head and neck squamous cell carcinomas. J Pathol 2017;243(3):354–65 doi 10.1002/path.4953. [DOI] [PubMed] [Google Scholar]
- 14.Kato T, Kiyotani K, Tomiyama E, Koh Y, Matsushita M, Hayashi Y, et al. Peripheral T cell receptor repertoire features predict durable responses to anti-PD-1 inhibitor monotherapy in advanced renal cell carcinoma. Oncoimmunology 2021;10(1):1862948 doi 10.1080/2162402X.2020.1862948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sacco AG, Chen R, Worden FP, Wong DJL, Adkins D, Swiecicki P, et al. Pembrolizumab plus cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: an open-label, multi-arm, non-randomised, multicentre, phase 2 trial. The Lancet Oncology 2021;22(6):883–92 doi 10.1016/S1470-2045(21)00136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JH. Why the Outcome of Anti-Tumor Immune Responses is Heterogeneous: A Novel Idea in the Context of Immunological Heterogeneity in Cancers. Bioessays 2020;42(10):e2000024 doi 10.1002/bies.202000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Muzaffar J, Kirtane K, Song F, Johnson M, Schell MJ, et al. T cell repertoire in peripheral blood as a potential biomarker for predicting response to concurrent cetuximab and nivolumab in head and neck squamous cell carcinoma. Journal for immunotherapy of cancer 2022;10(6) doi 10.1136/jitc-2022-004512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nature medicine 2019;25(8):1251–9 doi 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Adaptive ImmunoSEQ sequencing data are available via clients.adaptivebiotech.com/pub/ge-2022-ccr. The immuneACCESS DOI is https://doi.org/10.21417/HG2022CCR. Additional analysis information is available upon request to Dr. Jing H. Wang at jhw51@pitt.edu