

Abstract
Pseudomonas aeruginosa (PA) provokes a painful, sight-threatening corneal infection. It progresses rapidly and is difficult to treat. In this study, using a mouse model of PA keratitis, we demonstrate the importance of a carbohydrate-binding protein, galectin-8 (Gal-8), in the regulation of innate immune response. First, using two distinct strains of PA, we established that Gal-8−/− mice are resistant to PA keratitis. In contrast, mice deficient in Gal-1, Gal-3 and Gal-9 were fully susceptible. Second, the addition of exogenous rGal-8 to LPS (TLR4 ligand)-stimulated bone marrow-derived macrophages (BMDMs) suppressed: (i) the activation of the NF-κB pathway and (ii) the formation of MD-2/TLR4 complex. Third, the expression level of ROS was substantially higher in infected Gal-8−/− bone marrow-derived neutrophils (BMNs) compared to the Gal-8+/+ BMNs, and the PA killing capacity of Gal-8−/− BMNs was considerably higher compared to that of Gal-8+/+ BMNs. In the bacterial killing assays, the addition of exogenous rGal-8 almost completely rescued the phenotype of Gal-8−/− BMNs. Finally, we demonstrate that a subconjunctival injection of a Gal-8 inhibitor markedly reduces the severity of infection in the mouse model of PA keratitis. These data lead us to conclude that Gal-8 downmodulates innate immune response by suppressing the activation of TLR4 pathway and clearance of PA by neutrophils. These findings have broad implications for developing novel therapeutic strategies for treatment of conditions resulting from hyperactive immune response both in ocular as well as nonocular tissues.
INTRODUCTION
Pseudomonas aeruginosa produces a fulminating, highly destructive, sight-threatening corneal infection in humans (1, 2). Contact lens wear is the prime risk factor (3–5). One of the serious consequences of PA corneal infection is blindness resulting from overactive immune response leading to persistent inflammatory reaction in the stroma, corneal ulceration with subsequent corneal scarring, and in some cases, perforation. The current therapy includes antibiotic treatment, which reduces the bacterial burden; despite this, tissue damage occurs, often rapidly, due to uncontrolled inflammation. While the polymorphonuclear leukocytes (PMN)-predominant innate response is clearly vital in achieving bacterial eradication, it is the persistent dysregulation of innate responses, the primary cause for the acute manifestations of PA keratitis. Because PA keratitis progresses rapidly, the development of effective strategies to control the disease at an early stage to prevent the uncontrolled immune response in the cornea is a high priority. The innate immune system is the first line of defense against pathogens and is initiated by pattern recognition receptors (PRRs), which respond to invading microbes (6–13). The first step in the induction of innate immune response in the context of bacterial infections, is generally achieved by TLR-mediated activation of NF-κB pathway that results in the induction of inflammatory cytokines which are secreted from the cell to mediate downstream inflammatory effects that clear the infection (14–16). TLR4 is critical in host resistance to PA keratitis as its deficiency results in increased PMN infiltration and proinflammatory cytokine production, impaired bacterial killing, and a susceptible phenotype (17)
Galectin-8 (Gal-8) belongs to a tandem repeat-type subfamily of carbohydrate-binding proteins, galectins (18, 19). It contains two distinct carbohydrate recognition domains (CRD), N-terminal CRD (Gal-8N) and C-terminal CRD (Gal-8C), linked by a linker domain. Carbohydrate-binding specificity of the two CRDs is distinct. A distinguishing feature of Gal-8N is its strong preference for α2-3-sialylated glycans. Very high affinity to α2-3-sialylated glycans is a unique feature of Gal-8N that sets Gal-8 apart from all other members of galectin family (20, 21). Gal-8 is widely expressed in many organs and tissues under physiological or pathological conditions. It has been reported that Gal-8 activates antibacterial autophagy to defend cells against bacterial invasion (22). Depending on the context, Gal-8 has been shown to have both pro- as well as anti-inflammatory properties (23). In a number of cell types, including osteoblasts, liver, spleen and lungs, Gal-8 stimulates the secretion of various chemokines and cytokines including TNF-α, IL-1β, MCP-1 and IL-6 and is thus proinflammatory (24). Other published studies using experimental models of autoimmune diseases including rheumatoid arthritis (25), uveitis (26), and encephalomyelitis (27) emphasize that Gal-8 exhibits anti-inflammatory effects. In some scenarios, Gal-8 acts as a proinflammatory molecule in selected resting cells of the immune system but displays anti-inflammatory properties when these cells become activated (23). Therefore, at present, whether Gal-8 restrains an exacerbated response or, to the contrary, fuels ongoing inflammatory response is an open question. In the present study, we demonstrate that: (i) Gal-8−/− mice are resistant to PA keratitis, (ii) Gal-8 exacerbates the PA keratitis pathology by downmodulating TLR4 pathway, and that (iii) Gal-8 downmodulates TLR4 pathway by binding to CD14 and inhibiting the formation of MD2-TLR-4 complex. We further demonstrate that Gal-8 modulated signaling results in repression of ROS expression and reduction in the PA killing capacity of neutrophils. Finally, we explored the potential of pharmacological targeting of Gal-8 for treatment of PA keratitis and demonstrated that subconjunctival injection of Gal-8N inhibitor reduces the severity of PA keratitis in a mouse model.
MATERIALS AND METHODS
Mice, bacterial strains, and recombinant galectin-8 (rGal-8):
Seven to eight weeks old female wild-type (WT, C57BL/6) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The Gal-8−/− mouse strain was created from embryonic stem cell clone (14305A-F8), obtained from the KOMP Repository (www.komp.org) and generated by Regeneron Pharmaceuticals, Inc (28) on C57BL/6N background. The Gal-8 null status of the KO mice was confirmed by Western blotting (29). The Gal-8−/− mice on C57BL/6N background were backcrossed at least 10 generations to C57BL/6J background. Animals were housed in animal facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALC) at Tufts University, and all experimental procedures were in complete agreement with the AAALC guidelines. A cytotoxic strain PA 6077 and a PA invasive strain 6294 were used. Bacteria were cultured overnight on tryptic soy agar plates at 37°C before each infection. Recombinant Gal-8 was produced and purified as previously described (30) with a few modifications. Briefly, lysates of bacteria expressing rGal-8 were chromatographed on a β-lactose-conjugated Sepharose column (EY Labs; 1 mL bed volume). The lectin was eluted from the column with a buffer containing 100 mM β-lactose and dialyzed against PBS containing 2% glycerol and 4 mM β-mercaptoethanol and stored at −80°C. Endotoxin was removed from each preparation of lectin by Detoxin-Gel endotoxin removing gel (Thermo Scientific) and endotoxin levels were detected by ToxinSensor chromogenic LAL endotoxin assay kit (Genscript). Endotoxin levels of all Gal-8 preparations used in this study were <0.1 EU μg −1. Before use, each preparation of Gal-8 was also tested for carbohydrate-binding activity and specificity by the red blood cell (RBC) agglutination assay. To prepare Gal-8 affinity matrix, five mg of rGal-8 was conjugated to 330 mg of Pierce NHS-activated agarose dry resin in accordance with the manufacturer’s instructions (Thermo Scientific, Waltham, MA).
Corneal infection:
Corneas of wild-type (WT) and Gal-8−/− mice were infected under deep anesthesia induced by intraperitoneal injection of ketamine and xylazine. Central corneas of mice were scarified with three parallel 1-mm incisions using a 26-gauge needle, and a 5-μL drop containing 100 CFU of strain PA 6077 or PA6294 was applied to the eye. The eyes were examined on day 1 and day 3 post infection (p.i.) for the development of corneal keratitis and opacity. The degree of opacity was graded from 0–4, as previously described by Berk et al (31): 0, eye macroscopically identical to the uninfected control eye; 1, partial corneal opacity covering the pupil; 2, dense corneal opacity covering the pupil; 3, dense opacity covering the entire anterior segment; and 4, perforation of the cornea, phthisis bulbi (shrinkage of the globe post–inflammatory disease). To enumerate bacterial load, infected corneas of WT and Gal-8−/− mice were harvested, and each cornea was homogenized in 250 μL of tryptic soy broth with 0.05% Tween 20 (TSBT). A 10 μL of the corneal homogenate was serially diluted in TSBT and selected dilutions were plated in triplicate on Pseudomonas isolation agar plates (Becton-Dickinson, Franklin Lakes, NJ). Plates were incubated overnight at 37°C and the number of viable bacteria manually counted. Infiltration of PMN in infected corneas was quantified by myeloperoxidase (MPO) assay (16). Briefly, corneas were harvested at day 1 p.i, homogenized in 1.0 mL of 50 mM phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethyl-ammonium (Sigma, St Louis, MO), subjected to four freeze-thaw cycles, and sonicated in an ice bath (20 sec). After centrifugation, 100 μL of the supernatant was added to 2.9 mL of 50 mM phosphate buffer containing o-dianisidine dihydrochloride (16.7 mg/100mL, Sigma) and H2O2 (0.0005%). The change in absorbance was monitored at 460 nm (5 min at 30 sec intervals), and the numbers of PMNs/cornea were calculated on the standard curve prepared using mouse bone marrow neutrophils (BMNs).
Bacterial growth and adhesion assays:
To determine whether Gal-8 influences bacterial growth, varying concentrations of PA were incubated with rGal-8 (0.75 μM) for 3h at 37°C, and then bacterial counts were enumerated by plating on BBL agar plates as described in the previous section. To determine whether Gal-8 influences the adhesion of PA to corneal surface, corneas of Gal-8−/− and WT mice were scarified and a 5-μl drop of bacteria (100 CFU, PA6077) was applied to the cornea; at 2h and 4h p.i., corneas were harvested and processed for bacterial enumeration.
Neutrophils sorting from infected corneas:
PA infected corneas were harvested from WT and Gal-8−/− mice on day 1 p.i., pooled group wise (20-30 corneas/group), and digested in 200 μl of DMEM containing collagenase and DNase (100 μg/mL each, Roche Diagnostics, Indianapolis, IN) for 45 min at 37°C in a water bath. After incubation, the corneas were disrupted by grinding with a syringe plunger on a cell strainer, and single-cell suspensions were made in complete DMEM medium. For PMN sorting, cells were blocked with an unconjugated anti-CD32/CD16 mAb (clone 2.4G2, 30 min) in staining buffer (2% FBS, 5 mM EDTA in PBS) and then incubated for 30 min at 4°C with a cocktail of: CD45 (Clone 30-F11), CD11b-PerCP (clone M170), and Ly6G (clone 1A8). All antibodies were purchased from BD Bioscience (San Jose, CA). Then cells were washed three times using PBS and incubated with APC-fixable viability dye (1/1000, 30 min, 4°C), washed again and resuspended for FACS sorting on FACS Aria cell sorter (Becton Dickinson). The yield of neutrophils was 1-1.5 x 105 cells/20 infected corneas.
Gene Expression analyses:
FACS sorted WT and Gal-8−/− neutrophils of infected corneas (1-1.5 x 105 cells) were lysed in buffer RLT (Qiagen) and were processed for transcriptome analysis by RNA-seq. Briefly, total RNA was isolated from cell lysates using RNeasy mini kit (QIAGEN) and quantified using Agilent 5200 fragment analyzer system (Agilent Technologies, Santa Clara, CA). Yield of RNA was 1ng/5000 neutrophils. All RNA samples had 28S/18S ratios between 1.8-2.5. Purified RNA samples (20-40 ng) were sent to Tufts University Genomics Core for library preparation and sequencing. The Ovation® RNA-Seq System (NuGEN Technologies) was used to construct cDNA libraries from total RNA; and cDNA libraries were multiplexed with three samples per lane and loaded onto flow cell lanes. Sequencing-by-synthesis of 51-nucleotide length was performed on Illumina Hiseq 2500 sequencing system (NuGEN). DESEQ2 was used to normalize the expression values of the transcript isoforms. A stringent filtering criterion of raw reads per kilobase of exon model per million mapped (RPKM) value of 50 in at least one sample was used to obtain expressed transcripts. ANOVA was then performed on the log-transformed data to generate fold change between samples collected from different experimental conditions and the P value for each transcript. Differentially expressed mRNA isoforms were filtered cutoff > 50 reads with a fold change > 1.5 and p < 0.05.
Western Blot Analysis:
Protein extracts of infected corneas of WT and Gal-8−/− mice were prepared in a Radioimmunoprecipitation (RIPA) buffer supplemented with a protease inhibitor cocktail (Complete tablets; Roche Applied Science, Mannheim, Germany) and 2% SDS. For preparation of tissue lysates, three to four corneas were pooled and considered one biological replica. Aliquots of lysates containing 10-30 μg of proteins were subjected to electrophoresis in 4% to 15% gradient or 12 % SDS-PAGE gels (Bio-Rad, Hercules, CA, USA), and transferred to a nitrocellulose membrane. The blots were blocked with Odyssey blocking buffer (OBB; Li-Cor Biosciences, Lincoln, NE, USA) and incubated with mouse anti-NLRP3 (clone Cryo-1:1000, Adipogen), anti-caspase-1 (clone Casper-1, 1:1000; AdipoGen, and San Diego) and goat anti-IL-1β (clone AF-401-NA, 1:1000; R&D Systems, Minneapolis, MN, USA) primary antibodies in OBB (4°C, overnight). The secondary antibodies used were anti-goat IgG IRDye 800CW and anti-mouse IgG IRDye 680LT (Li-Cor) diluted in OBB (1:10,000, 25°C, 45 minutes). Blots were then scanned with the Odyssey Infrared Imaging System using Image Studio v3.0 software (Li-Cor). After image acquisition, the blots were stripped using the NewBlot nitrocellulose stripping buffer (Li-Cor) and reprobed with mouse anti-β-actin (clone AC-15, 1:10,000; Santa Cruz Biotechnology, Dallas, TX, USA) as a primary antibody, and anti-mouse IgG IRDye 680LT (Li-Cor) as a secondary antibody.
Preparation of bone marrow-derived macrophages (BMDMs):
BMDMs were prepared by maturing bone marrow cells for 6 to 7 days in DMEM medium containing 10% FBS, 2 mM L-glutamine, 1% penicillin/streptomycin, 1 mM sodium pyruvate, 50 μM β-mercaptoethanol (Life Technologies), and 10% L929 conditioned medium as a source of colony-stimulating factor (32). Over 99.5% of the cells prepared by this procedure were CD11b+F4/80+ macrophages as characterized by flow cytometry analysis.
Experiments to examine the effect of rGal-8 on LPS-induced innate immune response in BMDMs:
To detect the effect of exogenous rGal-8 on the expression levels of the key components of the TLR4 pathway, mature BMDMs in 100 mm petri dishes were serum starved in 5 mL of DMEM for 2-3 hours, rGal-8 (20 μg /mL) was added, after 30 minutes incubation with Gal-8, cells were stimulated with of LPS (100 ng/mL E.coli-LPS, Invitrogen) for 30 min at 37°C. At the end of incubation period, cells were washed, lysed in RIPA buffer and cell lysates were subjected to Western blot analysis to detect the expression levels of phospho-IκBα using mouse anti-phospho-IκBα (Clone 5A5, Cell signaling) as a primary antibody. To determine whether the effect Gal-8 on the innate immune response is carbohydrate-dependent, BMDMs were incubated with Gal-8 in the presence of 0.1 M β-lactose, α2,3-sialyl lactose (3’-SL) or α2,6-sialyl lactose (6’-SL). To detect the effect of exogenous rGal-8 on secretion of IL-6 and TNFα, mature BMDMs were stimulated with LPS in the presence of rGal-8 as described above except that the cells were stimulated with LPS for four hours and nigericin (10μM, Sigma) was present in the media during the last 45 minutes of incubation. At the end of the incubation period, IL-6 and TNFα released into culture supernatant was measured in triplicates by using sandwich ELISA kits (R&D Systems, Minneapolis, MN).
LPS-Galectin binding assay:
To determine whether Gal-8 binds to LPS, wells of 96-well flat bottom plates (Costar) were coated with rGal-8, (2.5 - 20 μg/mL PBS, 2h, room temp), nonspecific binding sites were blocked with 1% BSA in PBS (1h, room temp.). Biotinylated-LPS (Invitrogen, 50 μl, 1 μg/mL PBS containing 1% BSA) was added to the galectin-coated wells, plates were incubated at room temperature for 1h, and then washed with PBS containing 0.05% Tween 20. Streptavidin-HRP (50 μl, 1:40 dilution, R&D Systems) was added to each well, plates were incubated for 20 min. washed, and air dried. Tetramethylbenzidine (TMB) chromogenic substrate (50 μl/well, R&D) was added to each well for HRP detection, reactions were stopped by 1N H2SO4 (50 μl/well), and plates were read in a microplate reader (FilterMax F5 multi-mode, Molecular Devices). Wells coated with mouse CD14 (10 ng/mL, R&D Systems), a well-known LPS-binding protein, served as a positive control. In some experiments, wells were coated with Gal-3 for comparison purposes.
Affinity precipitation assay:
To determine whether CD14 or TLR4 are Gal-8 binding proteins, matured BMDMs in 100 mm petri dishes were stimulated with LPS (E. coli-LPS, Invitrogen; 100 ng/mL DMEM, 2 h), washed with PBS, and then lysed in 0.5 mL of RIPA buffer supplemented with a protease inhibitor cocktail. Protein concentration of the lysates were adjusted to 1 mg/mL, 500μg of protein aliquots were precleared by incubation with 50 μl unconjugated agarose beads (2-4 hours at 4°C), supernatants were collected and incubated overnight with Gal-8-conjugated agarose beads (50 μl at 4°C). Then, beads were washed with PBS three times by centrifugation, bound proteins were eluted by boiling the beads in 30μl Laemmli sample buffer for Western blot analysis using anti-TLR4 (1:100 dilution, Santa Cruz,), and anti-CD14 (1:1000 dilution, clone H-4, Cell signaling) as primary antibodies. Secondary antibodies used were anti-rabbit IgG IRDye 800CW and anti-mouse IgG IRDye 680LT (Li-Cor) diluted in OBB (1:10,000, 25°C, 45 minutes). Controls included incubation of lysates with Gal-8 affinity beads in the presence of β-lactose (100 mM, a pan inhibitor of galectins), or sucrose (100 mM, a noncompeting sugar).
FITC-LPS binding assay to determine the effect of Gal-8 on LPS binding to CD14:
Since CD14 is the key LPS binding protein on BMDMs, to determine the effect of Gal-8 on LPS binding to CD14, serum starved BMDMs in 100 mm petri dishes were treated with rGal8 (20ug/mL in DMEM) for 30 min, FITC-LPS (Sigma, 100 ng/mL in DMEM) was added and plates were incubated for additional 1 hour at 37°C. Plates were then washed 3 times with cold PBS to remove any traces of unbound LPS, stained with a viability dye and were then processed for flow cytometry to detect bound FITC-LPS. Cells were acquired by BD LSRII, and data analyzed by FlowJo.
MD-2-TLR4-complex staining:
BMDMs pretreated with rGal8 (20μg/mL in DMEM) for 30 min as described above were incubated with FITC-LPS (Sigma, 100 ng/mL in DMEM, 1h, 37°C), cells were washed twice with cold PBS, sequentially stained with PE-MD-2/TLR4 complex antibody (dilution1:100, clone MTS510, eBioscience) and a viability dye, and then processed for flow cytometry.
Preparation of bone marrow neutrophils (BMNs):
Bone marrow neutrophils were isolated as described by Swamydas et al (33). Briefly, femurs and tibias of WT and Gal-8−/− mice were collected; bone marrow was flushed over a cell strainer (70μm). Cells were washed and resuspended in 3 mL of DMEM and loaded slowly over two layers of histopaque 1119 and 1077 (Sigma), respectively, and centrifuged at 2500 rpm (25 min, room temperature). Neutrophils were collected from the interface of the Histopaque 1119 and Histopaque 1077 layers. Over 95% of the cells collected from the interface were CD11b+Ly6G+ neutrophils as characterized by flow cytometry analysis.
Reactive Oxygen Species (ROS) detection:
To determine whether Gal-8 modulates ROS expression in BMNs, WT and Gal-8−/− BMNs (2x105 cells) were incubated in 100 μl of 1X ROS Label in the ROS Assay Buffer (BioVision, Milpitas, CA; 30 min, 37°C, protected from light), and were then co-cultured with PA 6294 (PA:BMNs ratio 1:1, 1:5 and 1:10). After 2-hour coculture period, fluorescence intensity of ROS was measured by flow cytometry. Cells were acquired by BD LSRII and data analyzed by FlowJo. BMNs incubated with 1X ROS inducer (BioVision, Milpitas, CA) for 1 hour before flow cytometry served as a positive control. For rescue experiments, the assays were performed using Gal-8−/− BMNs in the presence of rGal-8 (5μg-20μg /ml Gal-8).
Neutrophil bacterial killing assay:
To assess the role of Gal-8 in the modulation of bacterial killing capacity of neutrophils, BMNs prepared from WT and Gal-8−/− mice were seeded in triplicates in a 48 well-plate (5 x 104 cells /well in 250 μl of DMEM +10% of FBS without antibiotics). Cells were allowed to adhere to wells (1h at 37 °C in a CO2 incubator) and were then co-cultured with PA 6294 at a BMN:PA ratio of 1:1, 1:5 and 1:10 (in 250 μl, 4h). At the end of the incubation period, cells were homogenized in the same media, and homogenates were processed for bacterial enumeration as described above. For rescue experiments, Gal-8−/− BMNs were preincubated with an ascending concentration of exogenous rGal-8 (10-20 μg/mL, 30 min, 37°C) and coculture incubations were carried out in the presence of rGal-8.
Experiments to determine the effect of Gal-8 inhibition on the course of keratitis:
To examine the effect of Gal-8 inhibition on the outcome of keratitis, a Gal-8 inhibitor {3,4-dichlorophenyl 3-O-(5-carboxy-1-methyl-1H-benzo[d]imidazole-2-ylmethyl)-1-thio-α-D-galactopyranoside}, referred to here after as 19a, was used (34). This inhibitor is a benzimidazole-derivatized galactoside designed and synthesized based on the crystal structure of Gal-8, selectively inhibits Gal-8 N-terminal domain (Kd 1.8 μM), with a weaker affinity for Gal-3 (Kd 5 μM) and little affinity for Gal-1, −2, −4, −7 and −9 (34). To examine the effect of Gal-8 inhibition, corneas of two groups of WT mice were infected with PA (5 corneas/group). The control group received a subconjunctival injection of 10 μL of vehicle (PBS+23% DMSO) and the experimental group received a 10 μL injection of a Gal-8 inhibitor, 19a (5 mg/mL in vehicle), immediately prior to PA infection. The eyes were examined on day 1 p.i., degree of keratitis was recorded, and the corneas were harvested: (i) individually for bacterial enumeration by plating the homogenate on agar plates and PMN enumeration using the myeloperoxidase assay as described earlier or (ii) pooled groupwise (5 corneas /group) and processed for quantification of IL1-β. Briefly, corneas were homogenized in 500 μl PBS containing 1% BSA and a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) and centrifuged at 12,000xg for five minutes. A 100 microliter aliquot of supernatant was assayed in triplicate by ELISA to quantify IL-1β, using a Duoset kit (R&D Systems, Minneapolis, MN).
Statistical analysis:
Differences between two groups were compared using two-tailed unpaired t tests using GraphPad Prism software (version 6.05). Data in graphs represent mean ± SEM. P-values less than 0.05 were considered significant.
RESULTS
Gal-8−/− mice are resistant to PA keratitis:
To determine whether Gal-8 has an impact on the course of PA keratitis, central corneas of Gal-8−/− and WT (C57BL/6) mice were infected with PA, and at post infection (p.i.) day 1, the severity of bacterial keratitis was graded by slit lamp and then corneas were harvested for bacterial and PMN enumeration. As expected, WT mouse corneas developed severe keratitis (Figure 1Ai–ii); the degree of keratitis was reduced in Gal-8−/− mice as evident from a substantial decrease in opacity score (Figure 1Ai–ii), bacterial load (Figure 1B) and PMN infiltration (Figure 1C). Similar results were obtained on day 3 p.i. (supplementary Figure S1). Also, similar results were obtained regardless of whether the corneas were infected with the cytotoxic strain PA 6077 (Figure 1) or an invasive strain PA 6294 (Supplementary Figure S2) in four independent experiments. Additional studies revealed that rGal-8 did not influence bacterial growth (Supplementary Figure S3A), or adhesion of bacteria to the abraded corneas of WT and Gal-8−/−mice (Supplementary Figure S3B). Thus, the resistance of Gal-8−/− mice to PA keratitis is not due to the ability of Gal-8 to facilitate bacterial growth or bacterial adhesion; nor is it likely to be due to the ability of Gal-8 to facilitate bacterial entry into the cells because similar results were obtained regardless of whether the infection was induced with the extracellular cytotoxic strain PA 6077 or an invasive strain PA 6294.
Figure 1: Galectin-8 KO mice are resistant to PA keratitis:
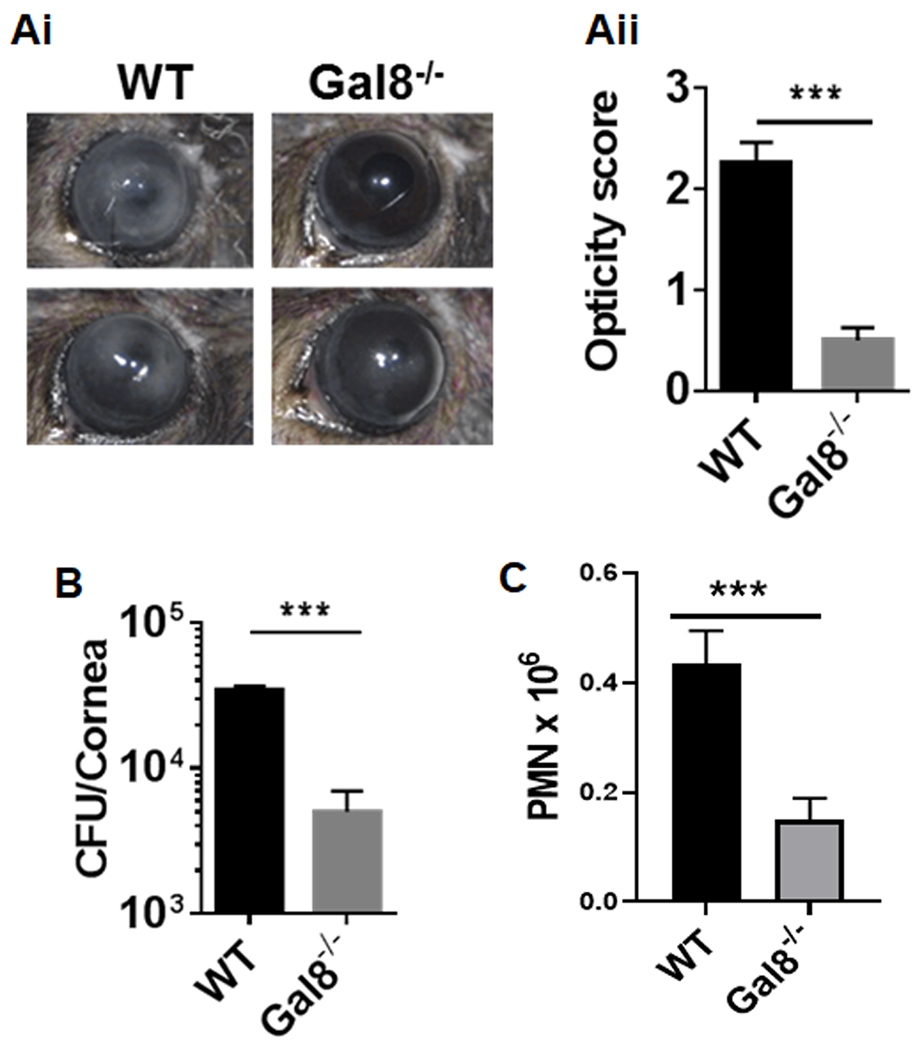
Central corneas of Gal-8−/− and WT (C57BL/6) mice were challenged with PA and the severity of bacterial keratitis was graded on day 1 post-infection (p.i.) using scoring system ranging from 0 to 4 to grade the degree of keratitis as described in Methods, and then corneas were harvested for bacterial and PMN enumeration. (Ai) Representative eye images comparing the disease severity of WT and Gal-8−/− corneas; (Aii) Corneal opacity score of Gal-8−/− and WT mice on day 1 p.i.; (B) Bacterial load and (C) PMN counts in Gal-8−/− and WT mice at day1 p.i. Data are plotted as Mean±SEM. Panels Aii and B: Combined results of 3 independent experiments are shown, N=18; Panel C: Combined results of two independent experiments are shown. N=20. Statistical levels of significance were analyzed by the Student’s t-test. ***P<0.001 vs WT.
Gal-8 dampens the innate immune response:
Next, we investigated whether Gal-8 has the capacity to influence the immune response. Since TLR4 plays a critical role in host resistance to PA keratitis (17), we first examined whether the expression of genes related to TLR4 pathway is altered in Gal-8−/− neutrophils of infected corneas. Briefly, FACS sorted WT and Gal-8−/− neutrophils of infected corneas were processed for RNA-seq and pathway analysis by Ingenuity Pathway Analysis software (Qiagen). Analysis of the data revealed that many genes of the innate immune pathway including NLRP3, NFκB, TNF and CD14 were upregulated in Gal-8−/− neutrophils (Figure 2A). Next, we performed studies to determine whether the expression levels of key components of TLR4 pathway are upregulated in infected corneas of Gal-8−/− mice. Western blot analysis of infected corneas of Gal-8−/− and WT mice, harvested on day 1 p.i., revealed that the expression levels of various components known to be upregulated upon activation of TLR4 pathway including pro-IL-1β, NLRP3 and procaspase-1 are substantially higher in infected Gal-8−/− corneas compared to corresponding wild-type mouse corneas (Supplementary Figure S4). These data suggest that activation of TLR pathway is augmented in infected Gal-8−/− corneas and that Gal-8 dampens the innate immune response. Next, to further confirm the role of Gal-8 in down modulation of innate immune response, mature BMDMs were stimulated with 100 ng/mL of LPS (TLR4 ligand) in the presence and the absence of exogenous rGal-8 (20μg/mL). We found that exogenous rGal-8 downregulated phosphorylation of IκBα (Figure 2B), a key readout to assess the activation of NFκB pathway. A critical outcome of TLR4 pathway activation is the secretion of inflammatory cytokines. Specifically, TNFα and IL-6 expression are key readouts of the activation of TLR4 pathway. To conclusively establish the suppressive role of Gal-8 on the activation of TLR4 pathway, next set of studies were designed to determine whether treatment with exogenous rGal-8 will diminish secretion of TNFα and IL-6 in BMDMs. In this study, mature BMDMs were serum starved overnight and were then stimulated with 100 ng/mL of LPS (TLR4 ligand) in the presence and the absence of exogenous rGal-8 (4 hours, 37°C). At the end of incubation period, levels of key inflammatory cytokines released into culture supernatants were measured by ELISA. Indeed, the addition of exogenous rGal-8 markedly reduced the secretion of TNFα and IL-6 (Figure 2C). The inhibitory effect of Gal-8 on LPS-induced activation of TLR pathway was specifically counteracted by α2-3-sialyl lactose, a Gal-8N competing saccharide, but not by α2-6-sialyl lactose, a Gal-8 noncompeting saccharide (Figure 2B and 2C) suggesting that the carbohydrate-dependent function of Gal-8N is directly involved in the inhibitory effect of Gal-8 on the activation of TLR4 pathway.
Figure 2: Galectin 8 dampens innate immune response:
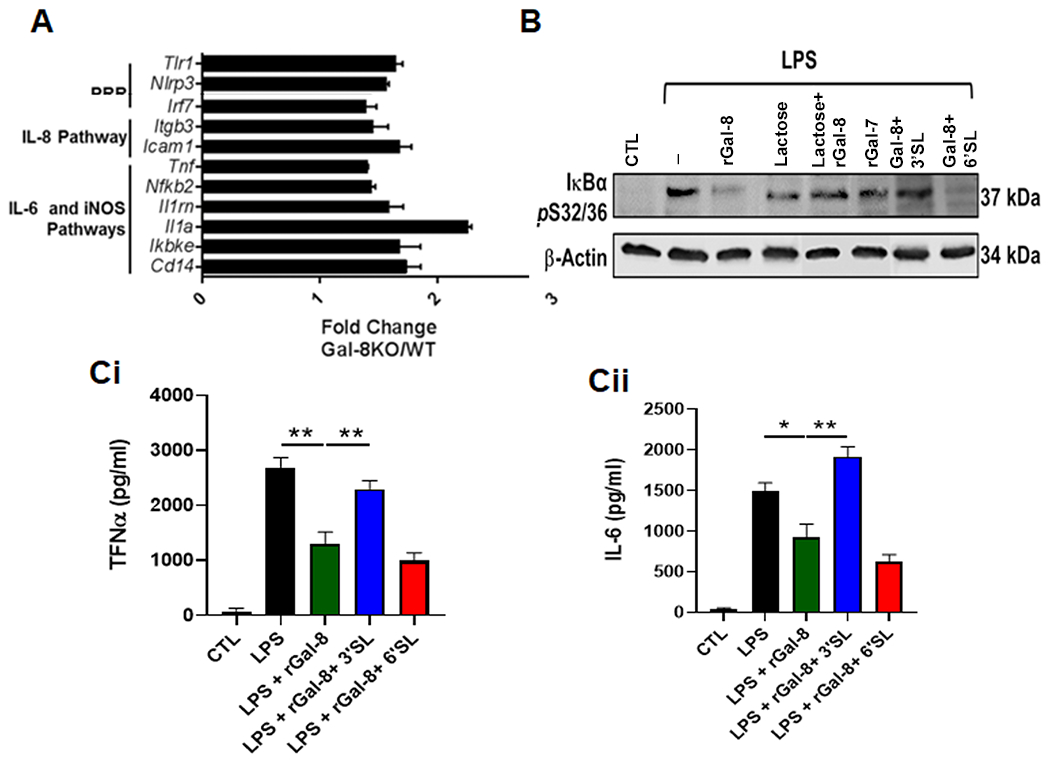
(A) The expression of genes related to innate immune pathway are upregulated in Gal-8−/− neutrophils of infected corneas. FACS sorted WT and Gal-8−/− neutrophils of infected corneas were processed for RNA-seq and pathway analysis by Ingenuity Pathway Analysis software (Qiagen). Histogram depicts upregulated expression of genes related to innate immune response. Combined results of two independent experiments are shown (20 corneas/group). (B) Gal-8 negatively regulates TLR4 signaling: Mature BMDMs were serum starved overnight and were then stimulated with 100 ng/mL of LPS (TLR4 ligand) in the presence and the absence of exogenous rGal-8 (20μg/mL) or rGal-8+control saccharides for 30 min at 37°C. Electrophoresis blots of cell lysates were processed for Western blot analysis using antibodies to phospho-IκBα. A representative blot of three independent experiments done in duplicates is shown. (C) Exogenous rGal-8 diminishes secretion of TNFα and IL-6 in BMDMs: Mature BMDMs were serum starved overnight and were then stimulated with 100 ng/mL of LPS in the presence and the absence of exogenous rGal-8 (20μg/mL). At the end of incubation period, TNFα and IL-6 released into culture supernatants were measured by ELISA (N=4). Data are plotted as Mean±SEM. Statistical levels of significance were analyzed by the Student’s t-test. *P<0.05 **P<0.01.
Together, our findings that TLR4 pathway is augmented in the infected Gal-8−/− corneas and that exogenous rGal-8 reduces the TLR4 pathway activation in the LPS-treated BMDMs lead us to conclude that Gal-8 down modulates TLR4 pathway, and, thereby, plays a critical role in the regulation of innate immune response.
Gal-8 downmodulates innate immune response by inhibiting the formation of MD-2/TLR4 Complex:
We next sought to define the mechanism by which Gal-8 represses the TLR4 pathway. The first step in the activation of TLR4 signaling is the delivery of LPS to CD14 that is present on macrophage cell surface or in soluble form. LPS is then transferred from CD14 to the myeloid differentiation (MD-2) protein. The LPS–MD2 subsequently interacts with TLR4 resulting in assembly of TLR4–MD2–CD14 complex that triggers the signaling pathways including the activation NFκB pathway and secretion of inflammatory cytokines. To define the mechanism by which Gal-8 downmodulates innate immune response, we first conducted studies to establish whether Gal-8 binds to LPS, TLR4 or CD14. To determine whether Gal-8 is an LPS-binding protein, Gal-8-coated wells were sequentially incubated with: (i) biotinylated-LPS, (ii) Streptavidin-HRP, and (iii) TMB chromogenic substrate (R&D) for HRP detection. This study revealed that unlike Gal-3 and CD14, Gal-8 is not an LPS-binding protein (Figure 3A). To determine whether Gal-8 binds to TLR4 or CD14, BMDM lysates were incubated overnight with Gal-8-conjugated agarose beads and bound proteins were examined along with total cell lysates (input) by Western blot using anti-TLR4 and anti-CD-14. TLR4 did not bind to Gal-8 beads (Figure 3B). In contrast, CD14 bound to Gal-8 beads (Figure 3B, lane media). Binding of CD14 to Gal-8 was inhibited by a competing saccharide, lactose, whereas, a noncompeting sugar, sucrose had no effect (Figure 3B) suggesting that CD14 binds to Gal-8 in a carbohydrate-dependent manner. Subsequently, we conducted studies to determine whether Gal-8 hinders TLR4 signaling by: (i) interfering with the uptake of LPS by CD14, or (ii) by deterring delivery of LPS from CD14 to MD2 and formation of TLR4-MD2-CD14 complex. To determine whether Gal-8 interferes with the binding of LPS to CD14, BMDMs pretreated with rGal8 (30 min) were incubated with FITC-LPS (1h) and were then processed for flow cytometry to detect cell bound FITC-LPS. These experiments revealed that Gal-8 does not interfere with binding of CD-14 to LPS as indicated by complete overlap of the FITC-LPS plot regardless of whether the cells were pretreated with rGal-8 (Figure 3C). Next, to determine whether Gal-8 prevents formation of MD-2/TLR4 complex, BMDMs pretreated with rGal8 were stimulated with FITC-LPS (30 mins), stained with MD-2/TLR4 complex antibody and then processed for flow cytometry. As expected, stimulation with LPS, resulted in the formation the MD2/TLR4 complex (Figure 3D). Treatment with rGal-8 inhibited the formation of MD-2/TLR4 complex. The inhibitory effect of Gal-8 on the formation of MD-2/TLR4 complex was abrogated by α2-3-sialyl lactose (3’SL), a Gal-8 competing saccharide, but not by α2-6-sialyl lactose (6’SL), a noncompeting saccharide (Figure 3D). These data lead us to conclude that Gal-8 binds to CD14 and down modulates the TLR4 pathway by inhibiting the formation of MD-2/TLR4 complex in a carbohydrate-dependent manner.
Figure 3: Galectin 8 dampens TLR4 signaling by inhibiting MD-2 complex formation in a carbohydrate-dependent manner:
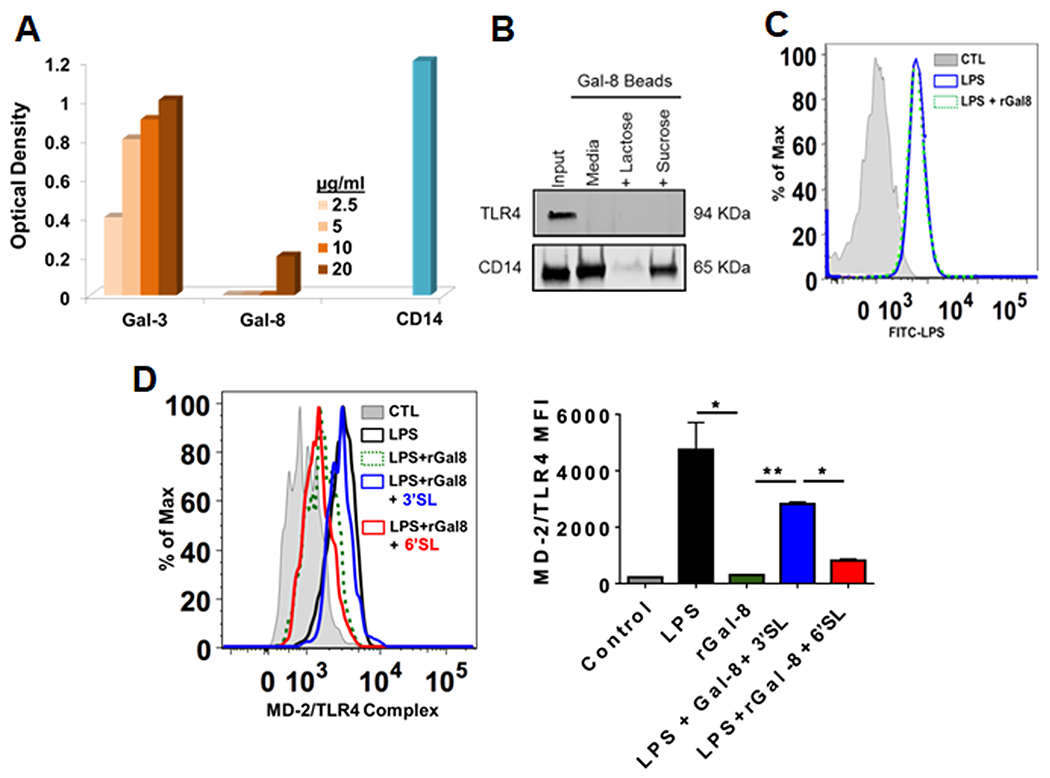
(A) Unlike Gal-3, Gal-8 is not an LPS-binding protein: Biotinylated-LPS was added to the Gal-8-coated wells, and the plates were sequentially incubated with Streptavidin-HRP and then TMB chromogenic substrate. Wells coated with known LPS-binding proteins, Gal-3 and CD14, served as positive controls. Combined results of three independent experiments in triplicates are shown (n=9). (B) Gal-8 binds to CD14 but not TLR4: BMDM lysates were incubated overnight with Gal-8-conjugated agarose beads in the presence or absence of lactose or sucrose (100 mM), bound proteins were examined along with total cell lysates (input, 30 μg protein) by Western blot using anti-TLR4 and anti-CD-14. A representative blot of three independent experiments is shown. (C) Gal-8 does not interfere with the binding of LPS to CD14: BMDMs pretreated with rGal8 were incubated with FITC-LPS, washed, and were then processed for Flow cytometry to detect bound FITC-LPS. Data are representative of three independent experiments in duplicate. (D) Gal-8 prevents delivery of LPS from CD14 to MD-2, and there by, prevents formation of MD-2/TLR4 complex: BMDMs pretreated with rGal8 (0.75 uM) in the absence and presence of control sugars were stimulated with 100 ng/mL FITC-LPS (30 mins), washed, stained with MD-2/TLR4 complex antibody (MTS510, eBioscience) and then processed for flow cytometry. Left: a representative FACS plot of three independent experiments in duplicate is shown (n=6); Right: combined results of 2-3 independent experiments in duplicate are shown (n=6 for LPS and rGal-8 groups; n=4 for 3’SL and 6’SL groups). Data are plotted as Mean±SEM. Statistical levels of significance were analyzed by the Student’s t-test. *P<0.05, **P<0.01.
Gal-8 reduces bacterial killing capacity of neutrophils via ROS pathway:
As described earlier, innate immune response results is the recruitment of neutrophils at the site of infection. Production of ROS by neutrophils is a vital component of innate immune response that enables killing and clearance of pathogens. Therefore, next we investigated the role of Gal-8 in the modulation of ROS pathway and the bacterial killing capacity of neutrophils. In this study, we first determined whether the expression of genes related to ROS pathway is altered in Gal-8−/− neutrophils. FACS sorted WT and Gal-8−/− neutrophils of infected corneas were processed for RNA-seq and pathway analyses by Ingenuity Pathway Analysis software (Qiagen). Analysis of the data revealed that many genes of the ROS pathway were upregulated in Gal-8−/− BMNs (Figure 4A). To determine whether expression of ROS is upregulated in infected Gal-8−/− BMNs, WT and Gal-8−/− BMNs co-cultured with PA 6294 (BMNs:PA ratio 1:10, 2h), were incubated with ROS Label for 30 min, and then fluorescence intensity of ROS was measured by flow cytometry. Infected WT BMNs expressed low levels of ROS (Figure 4B, red). Expression levels of ROS was substantially higher in corresponding Gal-8−/− BMNs (Figure 4B, green). To determine whether Gal-8 modulates the bacterial killing capacity of neutrophils, freshly isolated WT or Gal-8−/− BMNs were incubated with an ascending dose of PA (4 h), homogenized and processed for bacterial enumeration. Number of PA colonies were substantially higher in the WT BMNs compared to the Gal-8−/− BMNs (Figure 5A) indicating that the bacterial killing capacity of Gal-8−/− BMNs is considerably higher compared to that of Gal-8+/+ BMNs. To determine whether the exogenous rGal-8 rescues the altered bacterial killing capacity of Gal-8−/− neutrophils, Gal-8−/− BMNs were preincubated with ascending concentration of rGal-8 (30 mins), co-cultured with PA 6294 (2 h), and then processed for bacterial enumeration. In these experiments, exogenous rGal-8 reduced the killing capacity of Gal-8−/− BMNs in a dose dependent manner resulting in a complete rescue of the phenotype at rGal-8 concentration of 10 μg/mL or higher (Figure 5B). To determine whether the bacterial killing capacity of BMNs is totally dependent ROS, BMNs were pre-incubated with a ROS inhibitor (DPI, 2h) prior to co-culture with PA. ROS inhibitor almost completely abolished the bacterial killing capacity of BMNs (Figure 5C).
Figure 4: Gal-8 downmodulates ROS expression in neutrophils:
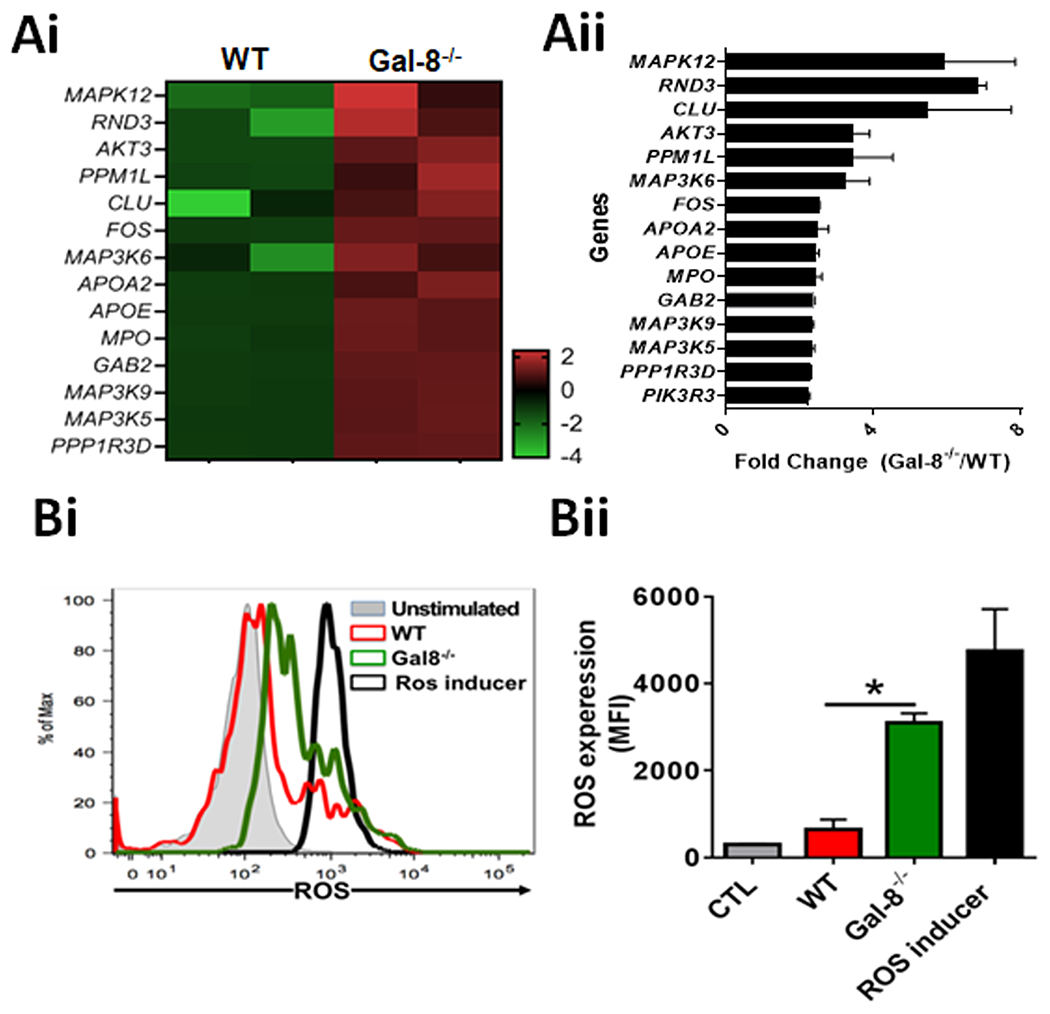
(A) Genes related to ROS pathway are upregulated in Gal-8−/− neutrophils of infected corneas: FACS sorted WT and Gal-8−/− neutrophils of infected corneas were processed for next-generation transcriptomic RNA sequencing (RNA-seq) and pathway analyses. (Ai) A representative heat map of three independent experiments is shown. All signals are compared with a mean value and change from the mean is visually represented by a color assignment; (Aii) Combined results of three independent experiments in triplicates are shown (n=9). (B) ROS expression is upregulated in Gal-8−/− neutrophils: WT and Gal-8−/− BMNs were preincubated with 1x ROS label for 30 min at 37°C prior to co-culture with PA 6294 (2h), and then fluorescence intensity of ROS was quantified by flow cytometry. BMNs incubated with a ROS inducer (BioVision) served as a positive control. (Bi) A representative FACS plot; (Bii) quantification of ROS expressed as mean fluorescence intensity (MFI)± SEM. Combined results of two independent experiments in duplicate are shown. *P<0.05
Figure 5: Gal-8 reduces bacterial killing capacity of neutrophils via ROS pathway:
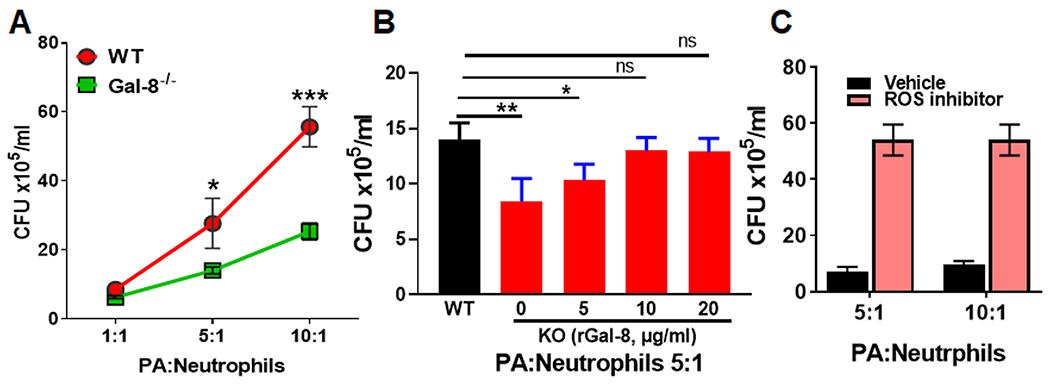
(A) Bacterial killing capacity of Gal-8−/− neutrophils is enhanced: Freshly isolated BMNs of WT or Gal-8−/− mice were incubated with an ascending dose of PA 6294 for 4 hours, homogenized, 10 μL aliquots of homogenates were plated on BBL blood agar overnight, for bacterial enumeration. Note that the number of PA colonies are substantially higher in the WT BMNs compared to the Gal-8−/− BMNs. (B) Exogenous rGal-8 rescues the altered bacterial killing capacity of Gal-8−/− neutrophils: BMNs were preincubated with ascending concentration of rGal-8 (5-20 μg/mL) for 30 min at 37°C prior to co-culture with PA and then processed for bacterial enumeration. (C) Bacterial killing capacity of neutrophils is dependent on ROS pathway: BMNs were preincubated with ROS inhibitor (Diphenyleneiodonium,10μM) prior to co-culture with PA and then processed for bacterial enumeration. Combined results of three independent experiments in duplicate are shown (N=6). Data are plotted as Mean±SEM. Statistical levels of significance were analyzed by the Student’s t-test. *p<0.05; **p<0.01, ***p<0.001.
Galectin-8 inhibition reduces the severity of PA keratitis:
Our findings that Gal-8−/− mice are resistant to PA keratitis suggest that inhibiting Gal-8 may improve the outcome of the disease. To explore the potential of pharmacological targeting of Gal-8 for the treatment of PA keratitis, two groups of mice were infected with PA. Prior to challenge with PA, one group of mice received a subconjunctival injection of a Gal-8N inhibitor (19a), and the other group of mice was treated with vehicle only. The severity of bacterial keratitis was graded on day 1 p.i. and then corneas were harvested for bacterial and neutrophil enumeration and for quantification of IL-1β. Treatment with Gal-8 inhibitor substantially reduced PA keratitis as detected by reduction in corneal opacity (Figure 6A), bacterial load (Figure 6B), PMN infiltration (Figure 6C) and expression level of IL1β (Figure 6D). These data lead us to conclude that targeting Gal-8 is an attractive strategy for the development of novel treatment for blinding immunopathology resulting from bacterial keratitis and possibly other disorders resulting from chronic inflammation.
Figure 6: Galectin-8 inhibition reduces the severity of PA keratitis:
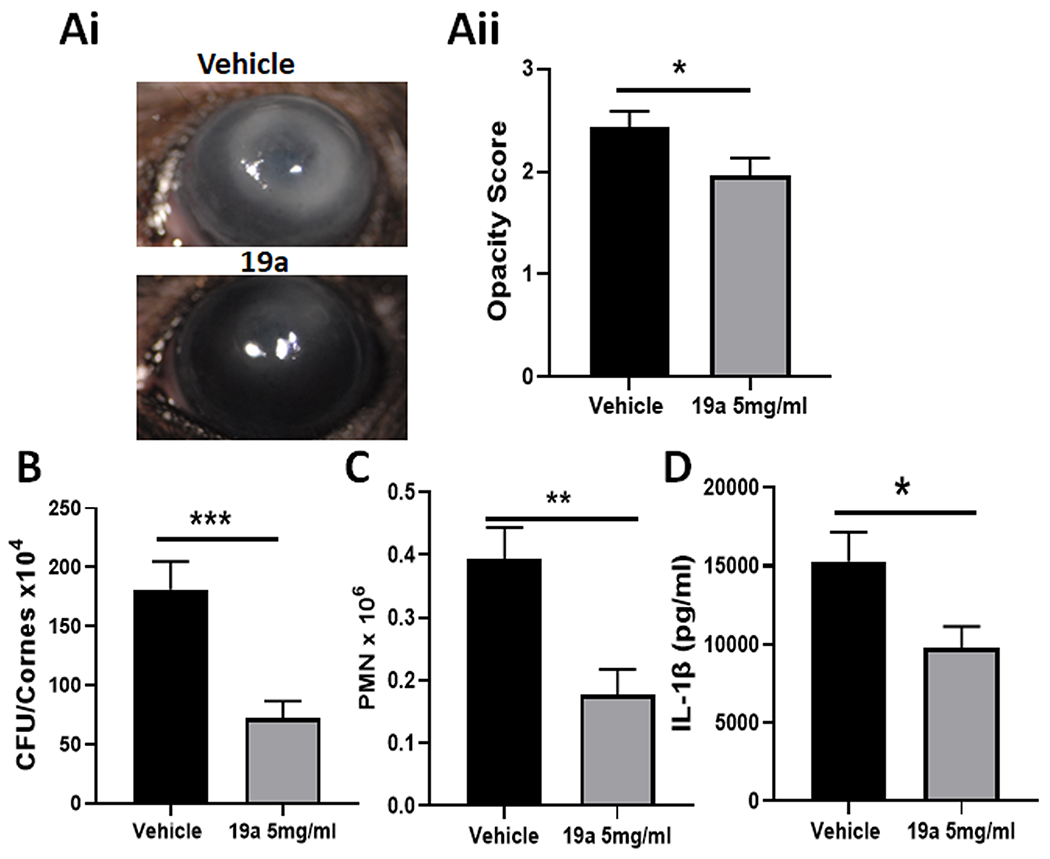
Corneas of WT mice were infected using PA strain 6077. Immediately, prior to infection, control group mice received a subconjunctival injection of vehicle (10μl PBS+23% DMSO) and experimental group mice received a 10 μL injection of Gal-8N inhibitor,19a (5 mg/mL in vehicle). On day 1 p.i., the eyes were examined to assess the severity of corneal keratitis and then corneas were harvested for bacterial and PMN enumeration and quantification of IL-1β. A: (Ai) Photographs of vehicle and Gal-8N inhibitor-treated corneas; (Aii): Quantification of corneal opacity score (n=30); B: Bacterial load (N=32); C: Infiltration of PMN (N=19). D: Quantification of total IL-1β by ELISA (N=6). Data are plotted as Mean±SEM. Statistical levels of significance were analyzed by the Student’s t-test. *p<0.05; **p<0.01, ***p<0.001.
DISCUSSION
The present study was designed to investigate the immunomodulatory role of Gal-8 in PA keratitis. We demonstrate here that Gal-8, which is known to be highly upregulated in pathological corneas, including PA infected corneas (35), plays a critical role in the pathobiology of PA keratitis. First, we established that Gal-8−/− mice are resistant to PA keratitis as demonstrated by marked reduction in bacterial load and opacity in infected Gal-8−/− corneas compared to the corresponding WT corneas. Clinical isolates of PA fall into one of two categories; invasive strains and cytotoxic strains. Both strain types cause human corneal disease (36) but the disease they cause has been shown to involve different pathological processes in an animal model (37). Our findings that Gal-8−/− mice are resistant to PA keratitis regardless of whether the corneas were infected with the cytotoxic strain PA 6077 or an invasive strain PA 6294, suggest that the observed phenotype of Gal-8−/− mice is unrelated to the invasiveness and cytotoxicity virulence determinants. Interestingly, a published study has shown that Gal-8−/− animals are also partially resistant to the growth and development of primary tumors and metastatic lesions (38). In contrast, in Trypanosoma cruzi chronic infection, Gal-8 protects from the disease as shown by a generalized increase in heart, skeletal muscle, and liver inflammation and fibrosis in Gal-8−/− mice compared to the WT mice (39). Other published studies using experimental models of autoimmune diseases including rheumatoid arthritis (25), uveitis (26), and encephalomyelitis (27) emphasize that Gal-8 exhibits anti-inflammatory effects, mostly by modulating adaptive immune response. Dual behavior of Gal-8, acting as both pro- as well as anti-inflammatory molecule based on the context and microenvironment of tissue can easily be explained by the differential glycosylation profile exhibited by the cells of the innate and adaptive immune system as well as naïve vs activated immune cells and the expression of other galectins in the milieu. In this respect, Gal-1 has also been shown to have a range of pro- and -anti-inflammatory functions dependent upon its expression and cellular localization (40, 41).
Various members of galectin family have been shown to bind to bacteria and kill the bound bacteria. For example, human Gal-3 binds to and induces death of Candida albicans (42), Gal-2 and Gal-3 kill H. pylori (43, 44) and fish Gal-8 has been shown to exert bactericidal activity against some gram negative bacterial pathogens (45). Also, Stowell et al (46) have demonstrated that rGal-8 is able to selectively kill bacteria expressing human blood group antigens, both in vivo and in vitro. In addition, a number of studies have shown that that intracellular Gal-8 targets invading microbes for their destruction by autophagy (22). None of these mechanisms of bacterial killing are likely to be directly relevant to the reduced severity of PA keratitis we observed in the Gal-8−/− mice since: (i) we found that Gal-8−/− mice were resistant to PA keratitis regardless of whether the corneas were infected with the extracellular cytotoxic strain PA 6077, or an invasive strain PA 6294 and that (ii) Gal-8 does not influence PA growth. Regarding the mechanism by which Gal-8−/− mice resist the PA keratitis, we demonstrated that Gal-8 downmodulates the innate immune response. The first step in the induction of innate immune response in the context of bacterial infections, is generally achieved by TLR-mediated activation of NF-κB pathway that results in the induction of: (i) proinflammatory cytokines, (ii) Rho pathway (iii) assembly of inflammasomes and (iv) recruitment of neutrophils that ultimately clear the infection. That Gal-8 dampens the innate immune response is demonstrated by our: (i) RNA-seq data showing that many genes of the innate immune pathway including NLRP3, NFκB, TNF and CD14 are upregulated in Gal-8−/− neutrophils compared to that of Gal-8+/+ neutrophils of PA-infected mouse corneas; (ii) western blot data showing that expression levels of proteins known to be upregulated upon activation of TLR4 pathway including pro-IL-1β, NLRP3 and procaspase-1 were substantially higher in infected Gal-8−/− corneas compared to corresponding wild-type mouse corneas; (iii) findings that BMDMs stimulated with LPS in the presence of exogenous rGal-8 exhibited reduction in the activation of the NF-κB pathway as indicated by the expression of markedly reduced level of phosphorylated iκBα and proinflammatory cytokines, TNFα and IL-6. Our striking finding that unlike Gal-8−/− mice, mice deficient in several other members of galectin family including Gal-1, Gal-3 and Gal-9 were fully susceptible to PA keratitis suggests that Gal-8-mediated resistance to PA keratitis involves the affinity of N-CRD of Gal-8 for 3’-sialylated galactosides that is unique among animal galectins (20, 21). In support of this notion, specific inhibition of N-CRD of Gal-8 by 3’-SL abrogated the effect of exogenous rGal-8 on suppression of TLR4 pathway in BMDMs stimulated with LPS. In contrast, control saccharide, 6’-SL, which lacks the affinity for the N-CRD (Gal-8N) of Gal-8 had no effect. Together, these data suggest that carbohydrate-dependent function of Gal-8N is directly involved in the inhibitory effect of Gal-8 on the activation of TLR4 pathway.
That Gal-8 dampens the innate immune response by suppressing the formation of MD-2/TLR4 complex via interacting with CD14 in a carbohydrate-dependent manner is demonstrated by our robust mechanistic studies showing that: (i) CD14 is a Gal-8 binding protein; unlike Gal-3, Gal-8 is not an LPS-binding protein; Gal-8 is also not a TLR4 binding; (ii) Gal-8 does not interfere with the delivery of LPS to CD14, and (iii) Gal-8 prevents delivery of LPS from CD14 to MD-2, and there by, averts the formation of MD-2/TLR4 complex. Again, our findings that the inhibitory effect of Gal-8 on the formation of MD-2/TLR4 complex was abrogated by α2-3-sialyl lactose (3’SL), a Gal-8 competing saccharide, but not by α2-6-sialyl lactose (6’SL), a noncompeting saccharide, suggests that Gal-8-mediated suppression of the formation of TLR4 complex involves the affinity of N-CRD of Gal-8 for 3’-sialylated galactosides.
In most cases, killing and clearance of pathogens is dependent on ROS production by neutrophils at the site of infection. ROS deficiency in human results in recurrent and severe bacterial infections, while their unregulated release leads to pathology from excessive inflammation. That Gal-8 downmodulates the neutrophil-mediated killing of bacteria in a ROS-dependent manner is suggested by our findings that: (i) many genes of the ROS pathway were upregulated in Gal-8−/− BMNs, (ii) expression levels of ROS are substantially higher in infected Gal-8−/− BMNs compared to the Gal-8+/+ neutrophils, and (iii) ROS inhibitor almost completely abolished the PA killing capacity of BMNs. Our findings that the bacterial killing capacity of Gal-8−/− BMNs is considerably higher compared to that of Gal-8+/+ BMNs, and that exogenous rGal-8 completely rescues the enhanced bacterial killing phenotype of Gal-8−/− neutrophils, leads us to conclude with a high level of confidence that Gal-8 is a key player in the modulation of bacterial killing capacity of neutrophils.
Regardless of the mechanisms involved, our findings that treatment with a Gal-8 inhibitor markedly reduces the severity of PA keratitis has broad implications for developing novel therapeutic strategies for treatment of conditions resulting from hyperactive immune response both in ocular as well as nonocular tissues. At present, treatment of PA keratitis is a major clinical problem. As described earlier, PA keratitis is notorious for causing rapidly fulminant disease often associated with corneal melting and permanent vision loss. While the current therapy including broad-spectrum topical antibiotics ultimately eliminates the bacteria, it fails to salvage vision in ~30% of patients who develop long-term moderate-to-severe vision loss (1). Unfortunately, strategies to treat PA keratitis has not changed in the last 30 years or so and there is a tremendous need of developing evidence based therapy to downmodulate unwanted, excessive immune response. Implications of developing novel treatment for unwarranted, out of control, immune response extend far beyond the diseases of ocular tissues. Indeed, excessive inflammation due to an unchecked inflammatory cytokine storm is responsible for pathogenesis of many cytokines storm-associated diseases (47, 48) including sepsis, cancers, autoimmune diseases, infections and other inflammatory diseases, where TLR-signaling plays a significant role. Again, targeting Gal-8 may help downmodulate unwanted immune response in such cases.
Supplementary Material
KEY POINTS:
Galectin-8−/− mice are resistant to Pseudomonas keratitis
Galectin-8 downmodulates innate immune response
Treatment with Galectin-8 inhibitor markedly reduces the severity of PA keratitis
ACKNOWLEDGEMENTS
We thank Dr. Albert Tai (Tufts Genomic Core) for his expert help and guidance in RNA Seq analyses. We also thank Drs. Tanveer Zaidi and Pablo Argueso for helpful discussions.
Support:
This work was Supported by National Eye Institute Grant R01EY028570, Massachusetts Lions Eye Research Fund, New England Corneal Transplant Fund, an unrestricted challenge award from Research to Prevent Blindness, and Galecto Biotech.
Footnotes
The RNA-seq data presented in this article have been submitted to the ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) under accession number E-MTAB-12376
REFERENCES
- 1.Ung L, and Chodosh J. 2021. Foundational concepts in the biology of bacterial keratitis. Exp Eye Res 209: 108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lakhundi S, Siddiqui R, and Khan NA. 2017. Pathogenesis of microbial keratitis. Microb Pathog 104: 97–109. [DOI] [PubMed] [Google Scholar]
- 3.Ormerod LD, and Smith RE. 1986. Contact lens-associated microbial keratitis. Arch Ophthalmol 104: 79–83. [DOI] [PubMed] [Google Scholar]
- 4.Pachigolla G, Blomquist P, and Cavanagh HD. 2007. Microbial keratitis pathogens and antibiotic susceptibilities: a 5-year review of cases at an urban county hospital in north Texas. Eye Contact Lens 33: 45–49. [DOI] [PubMed] [Google Scholar]
- 5.Schein OD, Ormerod LD, Barraquer E, Alfonso E, Egan KM, Paton BG, and Kenyon KR. 1989. Microbiology of contact lens-related keratitis. Cornea 8: 281–285. [PubMed] [Google Scholar]
- 6.Akira S, and Takeda K. 2004. Toll-like receptor signalling. Nat Rev Immunol 4: 499–511. [DOI] [PubMed] [Google Scholar]
- 7.Pandey RK, Yu FS, and Kumar A. 2013. Targeting toll-like receptor signaling as a novel approach to prevent ocular infectious diseases. Indian J Med Res 138: 609–619. [PMC free article] [PubMed] [Google Scholar]
- 8.Pearlman E, Sun Y, Roy S, Karmakar M, Hise AG, Szczotka-Flynn L, Ghannoum M, Chinnery HR, McMenamin PG, and Rietsch A. 2013. Host defense at the ocular surface. Int Rev Immunol 32: 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathinam VA, and Fitzgerald KA. 2016. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 165: 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahoo M, Ceballos-Olvera I, del Barrio L, and Re F. 2011. Role of the inflammasome, IL-1beta, and IL-18 in bacterial infections. ScientificWorldJournal 11: 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Storek KM, and Monack DM. 2015. Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev 265: 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobden JA, Masinick SA, Barrett RP, and Hazlett LD. 1997. Proinflammatory cytokine deficiency and pathogenesis of Pseudomonas aeruginosa keratitis in aged mice. Infect Immun 65: 2754–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Church LD, Cook GP, and McDermott MF. 2008. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol 4: 34–42. [DOI] [PubMed] [Google Scholar]
- 14.Karmakar M, Sun Y, Hise AG, Rietsch A, and Pearlman E. 2012. Cutting edge: IL-1beta processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1. J Immunol 189: 4231–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dinarello CA 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27: 519–550. [DOI] [PubMed] [Google Scholar]
- 16.Davis BK, Wen H, and Ting JP. 2011. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29: 707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X, Du W, McClellan SA, Barrett RP, and Hazlett LD. 2006. TLR4 is required for host resistance in Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci 47: 4910–4916. [DOI] [PubMed] [Google Scholar]
- 18.Bidon-Wagner N, and Le Pennec JP. 2002. Human galectin-8 isoforms and cancer. Glycoconj J 19: 557–563. [DOI] [PubMed] [Google Scholar]
- 19.Zick Y, Eisenstein M, Goren RA, Hadari YR, Levy Y, and Ronen D. 2002. Role of galectin-8 as a modulator of cell adhesion and cell growth. Glycoconj J 19: 517–526. [DOI] [PubMed] [Google Scholar]
- 20.Ideo H, Matsuzaka T, Nonaka T, Seko A, and Yamashita K. 2011. Galectin-8-N-domain recognition mechanism for sialylated and sulfated glycans. J Biol Chem 286: 11346–11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlsson S, Oberg CT, Carlsson MC, Sundin A, Nilsson UJ, Smith D, Cummings RD, Almkvist J, Karlsson A, and Leffler H. 2007. Affinity of galectin-8 and its carbohydrate recognition domains for ligands in solution and at the cell surface. Glycobiology 17: 663–676. [DOI] [PubMed] [Google Scholar]
- 22.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, and Randow F. 2012. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482: 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tribulatti MV, Carabelli J, Prato CA, and Campetella O. 2020. Galectin-8 in the onset of the immune response and inflammation. Glycobiology 30: 134–142. [DOI] [PubMed] [Google Scholar]
- 24.Zick Y 2022. Galectin-8, cytokines, and the storm. Biochem Soc Trans 50: 135–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eshkar Sebban L, Ronen D, Levartovsky D, Elkayam O, Caspi D, Aamar S, Amital H, Rubinow A, Golan I, Naor D, Zick Y, and Golan I. 2007. The involvement of CD44 and its novel ligand galectin-8 in apoptotic regulation of autoimmune inflammation. J Immunol 179: 1225–1235. [DOI] [PubMed] [Google Scholar]
- 26.Sampson JF, Hasegawa E, Mulki L, Suryawanshi A, Jiang S, Chen WS, Rabinovich GA, Connor KM, and Panjwani N. 2015. Galectin-8 Ameliorates Murine Autoimmune Ocular Pathology and Promotes a Regulatory T Cell Response. PLoS One 10: e0130772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pardo E, Carcamo C, Uribe-San Martin R, Ciampi E, Segovia-Miranda F, Curkovic-Pena C, Montecino F, Holmes C, Tichauer JE, Acuna E, Osorio-Barrios F, Castro M, Cortes P, Oyanadel C, Valenzuela DM, Pacheco R, Naves R, Soza A, and Gonzalez A. 2017. Galectin-8 as an immunosuppressor in experimental autoimmune encephalomyelitis and a target of human early prognostic antibodies in multiple sclerosis. PLoS One 12: e0177472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas J, Yasenchak J, Chernomorsky R, Boucher M, Elsasser AL, Esau L, Zheng J, Griffiths JA, Wang X, Su H, Xue Y, Dominguez MG, Noguera I, Torres R, Macdonald LE, Stewart AF, DeChiara TM, and Yancopoulos GD. 2003. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol 21: 652–659. [DOI] [PubMed] [Google Scholar]
- 29.Chen WS, Cao Z, Sugaya S, Lopez MJ, Sendra VG, Laver N, Leffler H, Nilsson UJ, Fu J, Song J, Xia L, Hamrah P, and Panjwani N. 2016. Pathological lymphangiogenesis is modulated by galectin-8-dependent crosstalk between podoplanin and integrin-associated VEGFR-3. Nat Commun 7: 11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poland PA, Kinlough CL, and Hughey RP. 2015. Cloning, expression, and purification of galectins for in vitro studies. Methods Mol Biol 1207: 37–49. [DOI] [PubMed] [Google Scholar]
- 31.Berk RS, Hazlett LD, and Beisel KW. 1987. Genetic studies on resistant and susceptibility genes controlling the mouse cornea to infection with Pseudomonas aeruginosa. Antibiot Chemother (1971) 39: 83–91. [DOI] [PubMed] [Google Scholar]
- 32.Broz P, and Monack DM. 2013. Measuring inflammasome activation in response to bacterial infection. Methods Mol Biol 1040: 65–84. [DOI] [PubMed] [Google Scholar]
- 33.Swamydas M, and Lionakis MS. 2013. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp: e50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassan M, van Klaveren S, Hakansson M, Diehl C, Kovacic R, Baussiere F, Sundin AP, Dernovsek J, Walse B, Zetterberg F, Leffler H, Anderluh M, Tomasic T, Jakopin Z, and Nilsson UJ. 2021. Benzimidazole-galactosides bind selectively to the Galectin-8 N-Terminal domain: Structure-based design and optimisation. Eur J Med Chem 223: 113664. [DOI] [PubMed] [Google Scholar]
- 35.Chen WS, Cao Z, Truong L, Sugaya S, and Panjwani N. 2015. Fingerprinting of galectins in normal, P. aeruginosa-infected, and chemically burned mouse corneas. Invest Ophthalmol Vis Sci 56: 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fleiszig SM, Zaidi TS, Preston MJ, Grout M, Evans DJ, and Pier GB. 1996. Relationship between cytotoxicity and corneal epithelial cell invasion by clinical isolates of Pseudomonas aeruginosa. Infect Immun 64: 2288–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cole N, Willcox MD, Fleiszig SM, Stapleton F, Bao B, Tout S, and Husband A. 1998. Different strains of Pseudomonas aeruginosa isolated from ocular infections or inflammation display distinct corneal pathologies in an animal model. Curr Eye Res 17: 730–735. [PubMed] [Google Scholar]
- 38.Shatz-Azoulay H, Vinik Y, Isaac R, Kohler U, Lev S, and Zick Y. 2020. The Animal Lectin Galectin-8 Promotes Cytokine Expression and Metastatic Tumor Growth in Mice. Sci Rep 10: 7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bertelli A, Sanmarco LM, Pascuale CA, Postan M, Aoki MP, and Leguizamon MS. 2020. Anti-inflammatory Role of Galectin-8 During Trypanosoma cruzi Chronic Infection. Front Cell Infect Microbiol 10: 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sundblad V, Morosi LG, Geffner JR, and Rabinovich GA. 2017. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J Immunol 199: 3721–3730. [DOI] [PubMed] [Google Scholar]
- 41.Lei T, Moos S, Klug J, Aslani F, Bhushan S, Wahle E, Frohlich S, Meinhardt A, and Fijak M. 2018. Galectin-1 enhances TNFalpha-induced inflammatory responses in Sertoli cells through activation of MAPK signalling. Sci Rep 8: 3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohatsu L, Hsu DK, Jegalian AG, Liu FT, and Baum LG. 2006. Galectin-3 induces death of Candida species expressing specific beta-1,2-linked mannans. J Immunol 177: 4718–4726. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki T, Saito R, Oyama M, Takeuchi T, Tanaka T, Natsume H, Tamura M, Arata Y, and Hatanaka T. 2020. Galectin-2 Has Bactericidal Effects against Helicobacter pylori in a beta-galactoside-Dependent Manner. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park AM, Hagiwara S, Hsu DK, Liu FT, and Yoshie O. 2016. Galectin-3 Plays an Important Role in Innate Immunity to Gastric Infection by Helicobacter pylori. Infect Immun 84: 1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang T, Jiang S, and Sun L. 2020. A Fish Galectin-8 Possesses Direct Bactericidal Activity. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stowell SR, Arthur CM, Dias-Baruffi M, Rodrigues LC, Gourdine JP, Heimburg-Molinaro J, Ju T, Molinaro RJ, Rivera-Marrero C, Xia B, Smith DF, and Cummings RD. 2010. Innate immune lectins kill bacteria expressing blood group antigen. Nat Med 16: 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan L, Karimi N, Morovati S, Alizadeh K, Kakish JE, Vanderkamp S, Fazel F, Napoleoni C, Alizadeh K, Mehrani Y, Minott JA, Bridle BW, and Karimi K. 2021. The Roles of Neutrophils in Cytokine Storms. Viruses 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar V 2020. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int Immunopharmacol 89: 107087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.