

Abstract
Rationale-
N-myc downstream-regulated gene 1 (NDRG-1) is a member of NDRG family that plays essential roles in cell differentiation, proliferation, and stress responses. Although the expression of NDRG1 is regulated by fluid shear stress, its roles in vascular biology remain poorly understood.
Objective-
The purpose of the study is to determine the functional significance of NDRG1 in vascular inflammation and remodeling.
Methods and Results-
By using quantitative PCR (qPCR), western blot, and immunohistochemistry, we demonstrate that the expression of NDRG1 is markedly increased in cytokine-stimulated endothelial cells (ECs) and in human and mouse atherosclerotic lesions. To determine the role of NDRG1 in endothelial activation, we performed loss-of-function studies using NDRG1 short hairpin RNA (shRNA). Our results demonstrate that NDRG1 knockdown by lentivirus bearing NDRG1 shRNA substantially attenuates both IL-1β and TNF-α-induced expression of cytokines/chemokines and adhesion molecules. Intriguingly, inhibition of NDRG1 also significantly attenuates the expression of procoagulant molecules, such as plasminogen activator inhibitor type 1 (PAI-1) and tissue factor (TF), and increases the expression of thrombomodulin (TM) and tissue-type plasminogen activator (t-PA), thus exerting potent anti-thrombotic effects in ECs. Mechanistically, we showed that NDRG1 interacts with orphan nuclear receptor Nur77 and functionally inhibits the transcriptional activity of Nur77 and nuclear factor Kappa B (NF-κB) in ECs. Moreover, in NDRG1 knockdown cells, both cytokine-induced mitogen-activated protein kinas (MAPK) activation, c-Jun phosphorylation, and activator protein 1 (AP-1) transcriptional activity are substantially inhibited. Neointima and atherosclerosis formation induced by carotid artery ligation and arterial thrombosis were markedly attenuated in EC-specific NDRG1 knockout mice compared with their wild-type littermates.
Conclusions-
Our results for the first time identify NDRG1 as a critical mediator implicated in regulating endothelial inflammation, thrombotic responses, and vascular remodeling, and suggest that inhibition of NDRG1 may represent a novel therapeutic strategy for inflammatory vascular diseases, such as atherothrombosis and restenosis.
Keywords: NDRG1, Endothelial cells, Inflammation, NF-κB, AP-1, Nur77, Basic Science Research, Cell Signaling/Signal Transduction, Vascular Biology
Graphical Abstract

Introduction
Vascular inflammation induces endothelial dysfunction and confers a proadhesive and prothrombotic phenotype of endothelial cells, which contribute significantly to the development and progression of a variety of cardiovascular conditions, most notably atherosclerosis, thrombosis, and congestive heart failure 1 2. In response to inflammatory cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor (TNF-α), endothelial cells (ECs) undergo inflammatory activation, resulting in an increased surface expression of cell adhesion molecules, such as intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, and E-selectin, which contributes to the recruitment of inflammatory cells to arterial wall and their transmigration across the wall 3. Furthermore, vascular inflammation can cause endothelial line of blood vessels from an anticoagulant, profibrinolytic state into a prothromobotic state 4. Indeed, both in vitro and in vivo studies have demonstrated that pro-inflammatory cytokines, such as TNF-α and IL-1β, up-regulate the endothelial production of tissue factor (TF) and fibrinolytic inhibitor plasminogen activator inhibitor-1 (PAI-1) 5 6, while attenuating the expression of thrombomodulin, which consequently leads to enhanced formation of thrombosis and impaired protein C activation 7 8.
Activation of the transcription factor nuclear factor (NF)-κB plays essential roles in the cytokine-induced expression of cell adhesion molecules in vascular ECs 9. The promoter regions of the genes for VCAM-1, ICAM-1, and E-selectin all have binding sites for NF-κB 9. In resting ECs, NF-κB resides inactive in the cytoplasm by forming complexes with its inhibitors, the IκB proteins (IκBs). On stimulation by proinflammatory cytokines, IκBs is phosphorylated by IκB kinase (IKK), ubiquitinated, and proteolytically degraded by 26S proteasomes, resulting in translocation of NF-κB to the nucleus and a subsequent induction of NF-κB target genes, including adhesion molecules, chemokines, and cytokines 10. Recently, in a search for endogenous inhibitors of EC activation, we have identified orphan nuclear receptor Nur77 as a potent negative regulator of NF-κB activation in ECs 11. Our results have demonstrated that overexpression of Nur77 markedly attenuated NF-κB activation and the expression of adhesion molecules through transcriptionally upregulating the IκBα expression in vascular ECs 11. More recently, we further demonstrated that Nur77 exerts potent anti-thrombotic effects in ECs through augmenting the expression of thrombomodulin under inflammatory conditions, further highlighting the functional importance of Nur77 in vascular inflammation and thrombosis 8. However, the molecular mechanism underlying the regulation of Nur77 function in ECs remains poorly understood.
N-myc downstream-regulated gene 1 (NDRG1), together with NDRG2, NDRG3, and NDRG4, are the members of the N-myc downstream-regulated gene family that belongs to the alpha/beta hydrolase superfamily, although it is notable that the NDRG proteins lack a hydrolytic catalytic site and are deficient in hydrolytic enzyme activity 12 13. Depending on the expression patterns of NDRG genes, NDRG family members possess a wide range of important biological functions in cell proliferation, differentiation, development and stress responses 14 12. NDRG1 mRNA is ubiquitously expressed in human tissues 15 and it has been shown to play essential roles in embryogenesis and development 16 17, terminal differentiation 18, lipid biosynthesis 19, 20, stress responses 17 12, and immunity 21. Furthermore, the expression of NDRG1 is often reduced in various types of cancers, and its overexpression has been shown to suppress tumor growth and metastasis 22 13. Interestingly, a recent study suggests that NDRG1 may promote growth of hepatocellular carcinoma cells by directly interacting with glycogen synthase kinase 3β (GSK-3β) and the orphan nuclear receptor 4A1 (NR4A1, Nur77) 23. NDRG1 is a stress response gene and its expression is significantly upregulated by hypoxia, nickel, and homocysteine 24 25 26. Although NDRG1 was initially identified as a responsive gene to homocysteine and flow shear stress in human ECs 24 27, the functional significance of NDRG1 in cardiovascular system is poorly explored. Moreover, NDRG1 is critically involved in the responses of cell stress 17, 24 12, which is a leading risk factor for cardiovascular disease; therefore, it is essential to delineate the functional role of NDRG1 in vascular homeostasis.
In the present study, we found that the expression of NDRG1 is significantly increased in response to inflammatory cytokines in vascular ECs and in human and mouse atherosclerotic lesions. Furthermore, we demonstrated that inhibition of NDRG1 markedly attenuates endothelial activation and vascular remodeling, both in vitro and in vivo, through enhancing Nur77 transcriptional activity and suppressing the NF-κB and AP-1 pathways in vascular ECs.
Methods
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. An expanded Materials and Methods is available in the Expanded Supplemental Materials. All research materials are listed in the Materials and Methods are also included in the Major Resources Table.
Results
NDRG1 Expression Is Upregulated in Inflamed ECs and Atherosclerotic Lesions
NDRG1 plays an essential role in stress responses 13. Stress, particularly induced by inflammation, has been found to be positively associated with an increased risk of cardiovascular disease 32 1. To elucidate the role of NDRG1 in endothelial dysfunction, we determine the expression of NDRG1 in response to inflammatory cytokines in human umbilical vein endothelial cells (HUVECs). As shown in Figure 1A and 1B, stimulation of ECs with IL-1β, a potent inflammatory cytokine implicated in development of cardiovascular disease 33 34, markedly increased the NDRG1 mRNA levels in HUVECs, in a time and dose dependent manner, as determined by qRT-PCR. Likewise, in a dose and time dependent manner, treatment of HUVECs with IL-1β significantly increased the NDRG1 protein levels by approximately 3.5-fold, as determined by western blot (Figure 1C and 1D). To test the specificity of this response, HUVECs were treated with TNF-α, another potent inflammatory cytokine for EC activation. Similarly, as shown in the supplemental Figure S1, both NDRG1 mRNA and protein levels were significantly augmented in a time and dose dependent manner after TNF-α stimulation.
Figure 1. Expression of NDRG1 is increased in cytokine-stimulated ECs and atherosclerotic lesions.
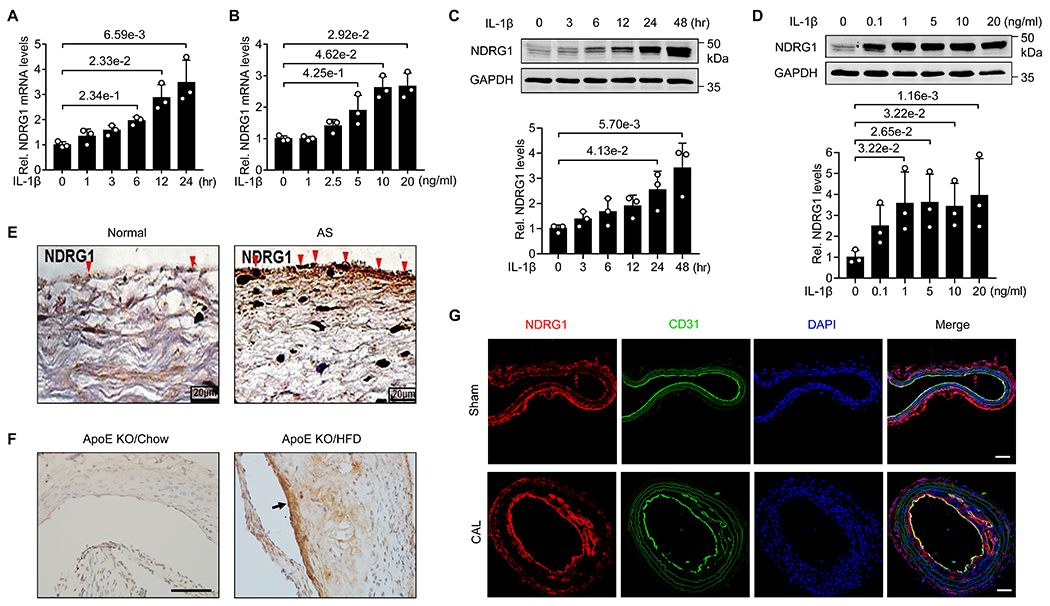
(A) HUVECs were stimulated with 10 ng/mL IL-1β for the different time points as indicated, expression of NDRG1 was determined by qRT-PCR. Data were presented as relative fold change to the 0-hr group (n=3). (B) HUVECs were stimulated with increasing concentrations of IL-1β for 24 hours, expression of NDRG1 was determined by qRT-PCR and data were presented as relative fold change to the 0-ng/ml group (n=3). (C) HUVECs were stimulated with 10 ng/mL IL-1β for the different time points as indicated, expression of NDRG1 was determined by western blot (upper) and quantitated by densitometric analysis (lower). Data were presented as relative fold change to the 0-hr group (n=3). (D) HUVECs were stimulated with increasing concentrations of IL-1β for 24 hours, expression of NDRG1 was then determined by western blot (upper) and quantitated by densitometric analysis (lower). Data were presented as relative fold change to the 0- ng/ml group (n=3). (E) Immunohistochemistry showing expression of NDRG1 in the tissues from normal subjects (NS) and patients with thoracic atherosclerosis (AS). Bars, 20 μm. (F) Immunohistochemistry showing the expression of NDRG1 in the aortic roots from ApoE KO mice fed a high fat diet (HFD) or chow diet. Arrow indicates the expression of NDRG1 in endothelium. Bars, 50 μm. (G) Immunofluorescent staining of NDRG1 in mouse carotid arterial tissues from ApoE KO mice subjected to partial carotid ligation and high-fat diet for 4 weeks. CD31 staining is shown as the endothelial marker. Bars, 50 μm. All data are shown as mean ± SD and analyzed by Kruskal-Wallis followed by Dunn’s test (A-D).
To determine the pathological role of NDRG1 in in vivo vascular inflammation, we examined the expression of NDRG1 in normal thoracic aorta and thoracic atherosclerotic lesions. As shown in Figure 1E, the aortic tissue from atherosclerotic patients displayed significantly increased expression of NDRG1, as compared with normal aortic tissue. Furthermore, we found that the expression of NDRG1 is markedly increased in the aortic roots of ApoE knockout (KO) mice fed a high fat diet (HFD) for 12 weeks, as compared with ApoE KO fed a chow diet (Figure 1F). In a murine accelerated atherosclerotic model induced by partial carotid ligation with a HFD, the expression of NDRG1 was markedly increased in atherosclerotic lesions as compared with sham controls (Figure 1G). Increased NDRG1 expression was predominant localized in mouse vascular endothelium (Figure 1G). Together, these results suggest that NDRG1 may play essential roles in vascular inflammation under both in vitro and in vivo conditions.
Knockdown of NDRG1 Inhibits IL-1β-and TNF-α-induced EC Activation
To investigate the functional significance of NDRG1 in endothelial activation, we performed loss-of-function studies, using lentivirus vector bearing NDRG1 shRNA. As shown in Figure 2A, transduction of HUVECs with lentivirus bearing NDRG1 shRNA markedly inhibited NDRG1 protein expression by approximately 80%, as determined western blot analysis. NDRG1 knockdown did not affect endothelia cell viability, as determined by trypan blue exclusion analysis (data not shown). To investigate whether NDRG1 is involved in EC activation, we determined the expression of cytokines and chemokines in IL-1β and TNF-α-stimulated HUVECs. As shown in Figure 2B, both IL-1β- and TNF-α-induced expression of IL-6, IL-8, and MCP-1, as determined by qRT-PCR, was markedly attenuated in cells transduced with NDRG1 shRNA lentivirus, as compared with the control shRNA lentivirus-transduced cells. Furthermore, NDRG1 knockdown markedly attenuated the IL-1β-induced production of IL-6, IL-8, and MCP-1 in culture supernatants as determined by ELISA (Figure 2C).
Figure 2. NDRG1 knockdown inhibits the expression of inflammatory cytokines in HUVECs.
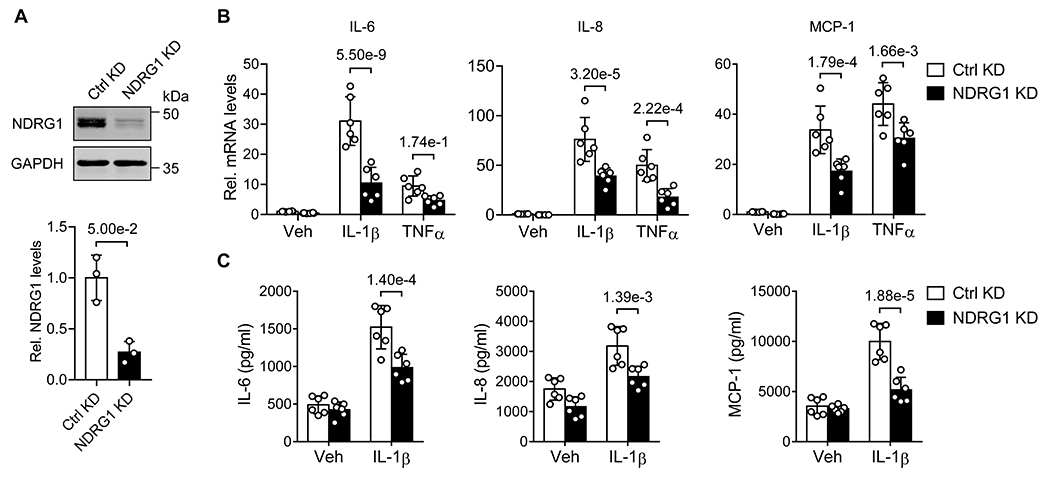
(A) HUVECs were transduced with lentivirus bearing control shRNA (Ctrl KD) or shNDRG1 (NDRG1 KD) at a moi of 5. 72 hours later, the expression of NDRG1 was determined by western blot (upper) and quantitated by densitometric analysis (lower). Data were presented as relative fold change to Ctrl KD (n=3) and analyzed by Mann-Whitney U coupled with Dunn’s test. (B) HUVECs were transduced with indicated shRNA lentivirus for 72 hours (moi 5), and then stimulated with vehicle (Veh, PBS), IL-1β (10 ng/mL) or TNF-α (20 ng/mL) for 6 hours. The expression of IL-6, IL-8 and MCP-1 were determined by qRT-PCR. Data were presented as relative fold change to Ctrl KD/Veh (n=6). Two-way ANOVA and tukey’s post hoc test. (C) The levels of IL-6, IL-8, and MCP-1 in the culture supernatants were determined by ELISA. Data were analyzed by two-way ANOVA and tukey’s post hoc test, n=6. All results are exhibited as mean ± SD.
Under inflammatory conditions, increased expression of adhesion molecules, such as VCAM-1 and ICAM-1, mediates the adhesion of monocytes to inflamed ECs, which is an important event in the initiation and development of vascular diseases, such as atherosclerosis and restenosis 1. Therefore, we examined the effects of NDRG1 knockdown on the cytokine-induced VCAM-1 and ICAM-1 expression in HUVECs. As shown in Figure 3A, NDRG1 knockdown markedly attenuated both TNF-α– and IL-1β–induced expression of VCAM-1 and ICAM-1, as determined by qRT-PCR, and IL-1β–induced protein expression of VCAM-1 and ICAM-1 as determined by western blot (Figure 3B). Furthermore, we assessed the effect of NDRG1 knockdown on U937 cell adhesion to the activated HUVECs. Stimulation of HUVECs with IL-1β (10 ng/mL) increased the adhesion of U937 cells, which was suppressed by approximately 70% in NDRG1 depleted HUVECs (Figure 3C). Taken together, these results identify NDRG1 as an essential regulator of the cytokine-induced inflammatory responses in ECs.
Figure 3. NDRG1 knockdown attenuates the expression of adhesion molecules.

(A) HUVECs were transduced with lentivirus bearing control shRNA (Ctrl KD) or shNDRG1 (NDRG1 KD) for 72 hours (moi 5), and then stimulated with vehicle (Veh), IL-1β (10 ng/mL) or TNF-α (20 ng/mL) for 12 hours. Expression of VCAM-1 and ICAM-1 were determined by qRT-PCR and data were presented as relative fold change to Ctrl KD (n=6). (B) HUVECs were transduced with indicated shRNA lentivirus for 72 hours followed by treatment of vehicle (Veh) or IL-1β for 24 hours. Expression of VCAM-1 and ICAM-1 were determined by western blot (left) and quantitated by densitometric analysis (right). Data were presented as relative fold change to Ctrl KD/Veh, n=6. (C) HUVECs were transduced with indicated shRNA lentivirus for 72 hours before starvation overnight and then were treated with or without IL-1β (10 ng/mL) for 8 hours. HUVECs were then incubated with calcein-labeled U937 cells for another 1 hour. Following washing, attached U937 cells were visualized on an inverted fluorescent microscopy and fluorescent intensity was then determined by Synergy 2 Multi-Mode Microplate Reader and data were presented as relative fold change to Ctrl KD/Veh (n=6). Scale bars, 50 μm. All results are expressed as mean ± SD and analyzed by two-way ANOVA coupled with tukey’s post hoc test (A-C).
NDRG1 Knockdown Attenuates Cytokine-induced Procoagulant Properties in HUVECs
Proinflammatory stimuli promote procoagulant properties in endothelial cells 4. For instance, cytokines, such as IL-1β and TNFα, have been shown to induce expression of procoagulant molecules, such as TF and PAI-1, and inhibit expression of thrombomodulin (TM), a key regulator implicated in the thrombin-mediated activation of the natural anticoagulant activated protein C (APC) 8. To determine whether NDRG1 is involved in promoting procoagulant states in ECs, we assessed the effects of NDRG1 knockdown on the expression of several key factors involved in blood coagulation. As shown in Figure 4, under basal conditions, knockdown NDRG1 strongly inhibited the expression of PAI-1, TF, and von Willebrand factor (vWF) (Figure 4A), and upregulated the expression of TM and t-PA. Furthermore, the cytokine-mediated induction of PAI-1 and TF was potently attenuated, whereas the cytokine-mediated inhibition of TM was prevented in NDRG1 knockdown cells. To further determine the functional consequence of NDRG1 knockdown in ECs, we performed an in vitro blood clotting assays 8. As shown in Figure 4B, exposure of recalcified human plasma to HUVECs transduced with NDRG1 shRNA lentivirus significantly prolonged the clotting time, when compared with that in control shRNA transduced cells. Moreover, TNF-α stimulation significantly shortened the clotting times, and this prothrombotic effect was markedly prevented in NDRG1-depleted HUVECs. Together, these results suggest that NDRG1 is critically involved in modulating thrombotic function in ECs under both basal and inflammatory conditions.
Figure 4. NDRG1 knockdown inhibits cytokine-induced pro-coagulant states in ECs.

(A) HUVECs were transduced with control shRNA (Ctrl KD) or shNDRG1 (NDRG1 KD) lentivirus for 72 hours (moi 5), and then stimulated with vehicle (Veh, PBS), IL-1β (10 ng/mL) or TNF-α (20 ng/mL) for 12 hours. The expression of PAI-1, TF, vWF, TM and t-PA were determined by qPCR and data were presented as relative fold change to Ctrl KD (n=6). (B) Knockdown NDRG1 inhibits blood clotting in vitro. HUVECs were transduced with indicated shRNA lentivirus for 72 hours and then treated with vehicle (PBS) or TNF-α (20 ng/mL) for 5 hours. After washing, decalcified plasma was added and clotting kinetics was measured by monitoring the absorbance at 405 nm. Analysis of kinetic profiles was performed to determine time to reach the half-maximal absorbance (T1/2 max), n=9. All results are shown as mean ± SD, analyzed by two-way ANOVA coupled with tukey’s post hoc test (A, B).
NDRG1 Promotes NF-κB Activation through Functional Interaction with Nur77
The activation of the transcription factor NF-κB has been well characterized to induce endothelial inflammation and pro-coagulation under inflammatory conditions 11. To examine whether NDRG1 regulates the expression of cytokines and adhesion molecules by interfering with NF-κB pathway, we investigated the effect of NDRG1 knockdown on NF-κB activity by electrophoretic mobility shift assay (EMSA). As shown in Figure 5A, we found that knockdown of NDRG1 significantly attenuated the DNA binding activity of NF-κB. Likewise, the nuclear translocation of NF-κB subunit p65 induced by IL-1β in HUVECs was inhibited in NDRG1 knockdown cells, as determined by western blot and immunofluorescent staining (Figure 5B and 5C).
Figure 5. NDRG1 knockdown attenuates NF-κB activation.

(A) HUVECs were transduced with control shRNA (Ctrl KD) or shNDRG1 (NDRG1 KD) lentivirus for 72 hours (moi 5), and then stimulated with either vehicle or IL-1β (10 ng/mL) for 30 minutes. Nuclear fraction was extracted and NF-κB DNA-binding activity was determined by EMSA (upper) and then quantitated by densitometric analysis (lower). The competitor was 50-fold unlabeled probe. Data were presented as relative fold change to Ctrl KD (n=6). (B) HUVECs were transduced with indicated shRNA lentivirus for 72 hours, and then stimulated with IL-1β (10 ng/mL) for 30 minutes. NF-κB p65 in the cytoplasmic and nuclear fractions were determined by western blot (upper) and quantitated by densitometric analysis (lower). Lamin A/C and GAPDH were used as nuclear and cytoplasmic markers. Data were presented as relative fold change to unstimulated Ctrl KD (n=6). (C) HUVECs were transduced with indicated shRNA lentivirus for 72 hours, followed by IL-1β stimulation for 30 minutes. Localization of p65 was determined by immunofluorescent staining. Nuclei were stained with DAPI. Scale bars, 20 μm. All results are displayed as mean ± SD, two-way ANOVA and tukey’s post hoc test (A, B).
NDRG1 has recently been shown to interact with orphan nuclear receptor 4A1 (Nur77) in hepatocellular carcinoma cells 23. We and other have shown that in ECs, Nur77 is essential for controlling NF-κB activation under both basal and inflammatory conditions 11 35. We hypothesized that NDRG1 might affect NF-κB activation through a functional interaction with Nur77 in ECs. To this end, we first performed immunoprecipitation to examine the interaction of NDRG1 with Nur77 in mouse aorta. As shown in Figure 6A, lysates from mouse aortic tissues were immunoprecipitated with anti-NDRG1 antibody and then analyzed by western blot. We found that NDRG1 physically interacts with Nur77. Confocal immunofluorescent staining demonstrated a strong localization of Nur77 and NDRG1 in HUVECs (Figure 6B). Furthermore, we performed an immunoprecipitation in HEK293T cells transfected with myc-tagged NDRG1 and Flag-tagged Nur77 expression vectors using anti-Flag antibody. We found that NDRG1 bound to the full length Nur77 and Nur77 mutants bearing the DNA binding domain (DBD). Deletion of DBD caused a loss of the interaction of NDRG1 with Nur77, indicating that the DBD of Nur77 is responsible for the binding of NDRG1 in Nur77 (Figure 6C). Since the DBD is responsible for the transcriptional activity of Nur77 11 36, we then determined whether NDRG1 affects the transcriptional activity of Nur77. As shown in Figure 6D, overexpression of NDRG1 dose-dependently inhibited the Nur77 response element (NBRE, AAAGGTCA)-driven luciferase activity in ECs. Further, co-transfection of NDRG1 and Nur77 markedly inhibited the binding of Nur77 to NBRE (Figure 6E), while knockdown of NDRG1 significantly increased the Nur77 activity in HUVECs, as indicated by increased Nur77 binding to the NBRE in EMSA (Figure 6F). Importantly, we found that the effects of NDRG1 knockdown on IL-1β-induced vascular inflammation and thrombotic responses were significantly prevented in Nur77 knockdown ECs (Figure 6G–6H), further implicating a functional interaction of NDRG1 with Nur77 in ECs.
Figure 6. Functional interaction of NDRG1 with Nur77 in ECs.

(A) Immunoprecipitation of NDRG1 followed by western blot detection of NDRG1 and Nur77 in lysates isolated from the wild-type C57BL/6 mouse aorta. (B) Immunofluorescent confocal staining of NDRG1 and Nur77 in HUVECs. Bars, 20 μm. (C) HEK-293T cells were transfected with Myc-tagged NDRG1 and Flag-tagged full length Nur77 and its deletion mutants. 48 hours after co-transfection, cell lysates were obtained and precipitated by anti-Flag antibody. Immunocomplexes were separated by 12% SDS-PAGE. The transferred membrane was immunoblotted with either anti-Flag or anti-Myc antibody. (D) EA.hy926 cells were transfected with NF-κB reporter together with indicated plasmids. Cells were harvested 48 hours later to determine luciferase activity. Results were presented as relative fold change to the non-overexpression group (n=3). (E) EA.hy926 cells were transfected with NDRG1 and Nur77 expression plasmids as indicated for 48 hours and nuclear protein was extracted for EMSA to measure Nur77 DNA-binding activity. Competitor was 50-fold unlabeled probe. (F) HUVECs were transfected with control (Ctrl KD) or anti-NDRG1 siRNA lentivirus (NDRG1 KD) for 72 hours and the nuclear protein was extracted for EMSA with Nur77 DNA binding probes. (G, H) HUVECs were transduced with lentivirus bearing control shRNA (Ctrl KD), NDRG1 shRNA (NDRG1 KD) or Nur77 shRNA (Nur77 KD) at a moi of 5. 72 hours after transduction, cells were then stimulated with or without IL-1β (10 ng/mL) for 12 hours. The expression of indicated genes was determined by qRT-PCR. Results were presented as relative fold change to Ctrl KD (n=6). All results are expressed as mean ± SD, analyzed by Kruskal-Wallis corrected with Dunn’s test (D) two-way ANOVA and tukey’s post hoc test (G) or one-way ANOVA and tukey’s post hoc test (H).
Knockdown of NDRG1 Attenuates Cytokine-induced AP-1 Activation
The activating protein 1 (AP-1) pathway is crucial for a potent transcriptional response to cytokine stimulation in vascular ECs 37. To test whether NDRG1 knockdown attenuates AP-1 activation in HUVECs, we performed EMSA. As shown in Figure 7A, NDRG1 knockdown markedly attenuated the AP-1 DNA-binding activity under IL-1β-stimulated conditions. To determine the molecular mechanism involved, we assessed the phosphorylation of c-Jun, which is a major component of AP-1 transcriptional complex 38, in HUVECs transduced with either control or NDRG1 shRNA lentivirus. As shown in Figure 7B, NDRG1 knockdown markedly attenuated IL-1β-induced phosphorylation of c-Jun in HUVECs. To further examine the molecular mechanism involved in AP-1 activation, we determined the effect of NDRG1 on the activation of MAPK pathway, which is a major kinase pathway responsible for the activation of the AP-1 in ECs 38. As shown in Figure 7B, NDRG1 knockdown significantly attenuated the IL-1β-stimulated phosphorylation of ERK1/2, JNK, and p38 in HUVECs. Taken together, these results suggest that NDRG1 regulates the AP-1 activation through modulating the MAPK signaling pathway in ECs.
Figure 7. NDRG1 knockdown inhibits MAPK phosphorylation and AP-1 activity.

(A) HUVECs were transduced with control shRNA (Ctrl KD) or shNDRG1 (NDRG1 KD) lentivirus for 72 hours (moi 5), and then stimulated with IL-1β (10 ng/mL) for 30 minutes. Nuclear fraction was extracted and AP-1 DNA-binding activity was determined by EMSA (upper) and then quantitated by densitometric analysis (lower). Competitor was 50-fold unlabeled probe. Results were presented as relative fold change to the unstimulated Ctrl KD (n=6). (B) HUVECs were transduced with indicated shRNA lentivirus for 72 hours before starvation overnight, and then were treated with or without IL-1β (10 ng/mL) for indicated time points. Total proteins were extracted and western blot was performed using anti-ERK1/2, anti-phospho-ERK1/2 (Thr202/Tyr204), anti-p38 MAPK, anti-phospho-p38 MAPK (Thr180/Tyr182), anti-JNK, and anti-phospho-JNK (Thr183/Tyr185) antibodies (left). The levels of phosphorylation related to the total expression levels were quantitated by densitometric analysis (right). Results were presented as relative fold change to the unstimulated Ctrl KD (n=6). All Results are displayed as mean ± SD, two-way ANOVA and tukey’s post hoc test.
Endothelial-specific NDRG1 Deletion Attenuates Neointima and Atherosclerosis Formation Following Carotid Artery Injury in Mice
To investigate the effect of endothelial-specific deletion of NDRG1 on vascular lesion formation, we used a murine carotid artery ligation as a model for non–endothelium-denuding neointimal formation 39. The endothelial activation and increased coagulation have been shown to play a significant role in the development of vascular lesions in this model 39–41. Through breeding NDRG1 floxed mice (NDRG1F/F) with Tie2-Cre transgenic mice, we acquired endothelial specific NDRG1 knockout mice (NDRG1eKO), and the endothelial specific deletion of NDRG1 was confirmed by detection of NDRG1 expression in primary EC cultures isolated from NDRG1eKO mice by both qPCR and western blot (Figure 8A and 8B). 4 weeks after carotid artery ligation, neointimal formation was significantly decreased in NDRG1eKO mice as compared with NDRG1F/F mice, as determined by neointima/media ratio (Figures 8C). This is accompanied by a substantial inhibition of the expression of adhesion molecules, such as VCAM-1 and ICAM-1, in NDRG1eKO mice, as determined by immunohistochemical staining (Figure 8D). Endothelial deletion of NDRG1 significantly prevented vascular injury-induced expression of VCAM-1, ICAM-1, IL-1β, vWF, and TF, as determined by qPCR (Figure 8E). Expression of endothelin-1 (ET-1), which was previously identified as a downstream target that was negatively regulated by Nur77 in ECs 28, was inhibited in the vasculature of NDRG1eKO mice as compared with NDRG1F/F mice (Figure 8E), indicating an increased Nur77 activity in NDRG1 deficient ECs. Furthermore, we found that the carotid atherosclerosis formation, as induced by a partial carotid ligation with a high-fat diet 29, 30, was markedly inhibited in NDRG1eKO as compared with NDRG1F/F mice (Figure 8F and 8G). Consistent with our previous observation that Nur77 suppresses arterial thrombosis in vivo 8, we found that arterial thrombus formation was also significantly protected in NDRG1eKO mice (Figure 8H), while the tail bleeding time was not affected in NDRG1eKO mice (Figure 8I). Together, these results indicate that endothelial NDRG1 is an essential regulator of neointimal formation and vascular remodeling in vivo.
Figure 8. Endothelial specific deletion of NDRG1 attenuates vascular remodeling and atherothrombosis formation in mice.

(A) Expression of NDRG1 was determined by qRT-PCR in primary aortic ECs isolated from NDRG1 WT and EC specific KO mice. Gene expression was presented as relative fold change to the NDRG1F/F group (n=3). (B) Expression of NDRG1 was determined by western blot in primary aortic ECs isolated from NDRG1 WT and EC specific KO mice. (C) H&E staining photomicrographs of carotid arteries 4 weeks after ligation of WT and EC specific NDRG1 KO mice (left), with quantitative analysis of vascular lesions in ligated carotid arteries (right), n=15 (NDRG1F/F) and 13 (NDRG1eKO). Bars, 50 μm. (D) Immunostaining of the vascular wall for the expression of ICAM-1 and VCAM-1 in NDRG1 WT and EC-specific KO mice after carotid artery ligation (left) with quantification result (right), n=9. Bars, 10 μm. (E) Expression of VCAM1, ICAM1, IL-1β, vWF, TF, and ET-1 was determined by qRT-PCR in right (sham) and left (ligated) carotid arterial tissues from NDRG1F/F and NDRG1eKO mice (2 weeks after ligation). Data were presented as relative fold change to the NDRG1F/F/sham group (n=7). (F) NDRG1F/F and NDRG1eKO mice were injected with AAV-PCSK9 and then subjected to partial carotid ligation and high-fat diet. 3 weeks later, carotid arterial tissues were dissected out for gross plaque imaging using a dissection microscopy and atherosclerotic plaques were then quantitated, n=7. Bars, 1 mm. (G) Carotid arterial tissue sections from indicated mice were stained with Oil Red-O, followed by hematoxylin counterstaining. Atherosclerotic plaques were shown by representative images (left) with the quantitative results (right), n=7. Bars, 50 μm. (H) Results of carotid artery thrombus formation time of NDRG1F/F and NDRG1eKO mice, n=7. (I) Results of tail bleeding time of NDRG1 WT and EC-specific KO mice, n=8. All results are shown as mean ± SD and analyzed by Mann-Whitney U and Dunn’s test (A), two tailed student’s t-test (C, D, F-I) or two-way ANOVA and tukey’s post hoc test (E).
Discussion
NDRG1 is a stress responsive gene that is regulated by various patho-physiological stimuli, including hypoxia, cellular iron levels, DNA damage, and homocysteine 12. Although NDRG1 was identified as a responsive gene to homocystenine and flow sheer stress in ECs24, 42 27, the functional role of NDRG1 in endothelial biology remains unknown. In the present study, we demonstrate that the expression of NDRG1 is markedly upregulated in cytokine-stimulated ECs as well as in the inflammatory aortic tissues from atherosclerotic patients. Furthermore, we have shown that specific knockdown of NDRG1 expression in ECs substantially inhibits the expression of cytokines and adhesion molecules, and prevents the cytokine-induced prothrombotic function in ECs. Furthermore, we found that endothelial-specific deletion of NDRG1 attenuates the expression of adhesion molecules and neointima and athrothrombosis formation carotid artery injury in mice. Mechanistically, we found that NDRG1 functionally interacts with Nur77 and inhibits its transcriptional activity, which may be accountable for the inhibition of NF-κB and AP-1 activation in ECs 11 28.
Proinflammatory cytokines such as TNF-α and IL-1β have been shown to induce NF-κB activation and influence atherosclerosis by inducing cellular adhesion molecules and promoting recruitment of leukocytes to the vessel wall 1 33. Indeed, recent genetic studies in atherosusceptible strains of mice have demonstrated that disruption of the cytokine-induced NF-κB signaling pathway suppresses endothelial activation and lesion formation in animal disease models 43 10. In the present study, we identified NDRG1 as a key player in endothelial inflammation through an inhibitory interaction with Nur77, which has emerged as an essential regulator for controlling the NF-κB activation through an inhibitory interaction with p65 or upregulating the IκBα expression in vascular ECs 11 44.. In response to the cytokine stimulation, Nur77 expression is rapidly induced, which subsequently inhibits NF-κB activation through induction of the IκBα expression, thus constituting a potent negative feedback mechanism in inhibiting NF-κB activation in ECs 11. Moreover, we found that the DNA binding domain (DBD) of Nur77 is critically important for the anti-inflammatory effects of Nur77 in ECs, since it mediates the binding of Nur77 to the promoter region of the IκBα gene and transcriptional activation of IκBα expression 11. In accordance with this observation, we show that NDRG1 functions as a repressor of Nur77 through its functional interaction with the DBD of Nur77, which may directly inhibit the binding of Nur77 to its consensus DNA binding element and/or affect the nuclear translocation of Nur77 in vascular ECs. Furthermore, we demonstrate that overexpression of NDRG1 markedly inhibits both Nur77 dependent DNA binding activity and transcriptional activation, while depletion of NDRG1 in ECs significantly increases the Nur77 transcriptional activity, which may contribute significantly to the inhibition of NF-κB activation under inflammatory conditions. In this regard, our results for the first time identify NDRG1 as a novel regulator of Nur77 and endothelial activation, and shed new light on the molecular mechanism underlying the regulation of the Nur77/NF-κB axis in vascular inflammation.
In addition to inhibiting the NF-κB pathway, we found that depletion of NDRG1 in ECs potently inhibits the cytokine-induced activation of AP-1 pathway, which is a critical mediator for the expression of inflammatory cytokines, adhesion molecules, and pro-coagulant factors in vascular inflammation 45. In this regard, our results are consistent with a previous notion showing that NDRG4, which belongs to the same NDRG family, affects the AP-1 activity in PC12 cells 46. Furthermore, we demonstrate that inhibition of NDRG1 markedly attenuates IL-1β-induced MAPK activation, which has been implicated in the phosphorylation of c-Jun and the activation of the AP-1 transcriptional pathway 47. The molecular mechanism underlying regulating MAPK activation by NDRG1 remains elusive. NDRG1 has been shown to be involved in actin-filament polymerization and stress fiber assembly 22, which is essential for the cytokine-induced activation of MAPK in ECs 48 49. Therefore, we speculate that inhibition of NDRG1 may affect the activation of MAPK pathway through its potential effects on the cytokine-induced actin-filament polymerization and stress fiber formation in ECs. This hypothesis is under active investigation.
Inflammation has been shown to induce prothrombotic effects in vascular endothelium 4. Inflammatory cytokines, including TNF-α and IL-1β, have been shown to induce the expression of procoagulant molecules, including TF and PAI-1, through activating the NF-κB and AP-1 pathways 20. In consistent with the potential effects on the regulation of the NF-κB and AP-1 transcriptional pathways, we found that inhibition of NDRG1 expression markedly inhibited the expression of both PAI-1 and TF under both basal and cytokine-stimulated conditions. Furthermore, we demonstrate that the expression of anti-thrombotic molecules, such as TM and t-PA, was substantially increased. Accordingly, the clotting time was significantly prolonged in NDRG1-depleted ECs under both basal and TNF-α-stimulated conditions. Interestingly, we found that the expression of vWF was substantially inhibited (>90%) in NDRG1-depleted cells. vWF is a blood glycoprotein that is required for normal hemostasis, and its levels have been associated with the risk of cardiovascular diseases, such as atherosclerosis, acute coronary syndrome, thrombosis, and pulmonary hypertension 50. Several transcriptional factors, including GATA, YY1, and Ets transcription factors, have been shown to function as activators, whereas Oct1 and nuclear factor (NF)-I function as repressors of vWF promoter activity 51 52. Furthermore, in the carotid artery ligation model, the elevated von Willebrand factor plasma levels and TF deposition in endothelium have been shown to play critical roles in neointimal formation, therefore, interruption of inflammation and the coagulation efficiency reduced fibrin deposition and ultimately diminished neointima formation 41. In the present study, we show that NDRG1 is critically involved in regulating both endothelial inflammation and coagulation. Accordingly, endothelial specific deletion of NDRG1 markedly attenuated neointimal formation, atherosclerosis, and arterial thrombosis formation after injury. Moreover, in pulmonary ECs, hypoxia has been shown to potently induce vWF expression through increasing YY1 transcriptional activity 53. Since hypoxia is one of the potent stimuli for induction of NDRG1 expression 17, and our results suggest the essential role of NGRG1 in regulating vWF expression, it would be very interesting to investigate whether NDRG1 is involved in the regulation of vWF expression under hypoxic conditions.
In conclusion, we identified NDRG1 as a critical mediator implicated in regulating endothelial inflammation and vascular remodeling through regulating Nur77/NF-κB and AP1 transcriptional pathways. As NDRG1 was initially identified in vascular ECs in response to the treatment of homocysteine and endoplasmic-reticulum (ER) stress, both of which have been shown to promote the inflammatory and thrombotic states in cardiovascular system, our studies may provide a novel therapeutic target for the treatment of inflammatory cardiovascular diseases, such as atherosclerosis and thrombotic disorders.
Supplementary Material
Novelty and Significance.
What Is Known?
Vascular inflammation induces endothelial dysfunction and confers a proadhesive and prothrombotic phenotype of endothelial cells, which contribute significantly to the development and progression of a variety of cardiovascular conditions.
N-myc downstream-regulated gene 1 (NDRG-1) is a member of NDRG family that plays essential roles in cell differentiation, proliferation, and stress responses.
Stress responses such as acute inflammation, hypoxia, laminar shear stress, and vascular injury are implicated in the development of cardiovascular disease.
What New Information Does This Article Contribute?
Expression of NDRG1 is significantly increased in cytokine-induced endothelial activation and atherosclerotic lesions.
Inhibition of NDRG1 markedly inhibited inflammatory responses and pro-coagulant activity in endothelial cells.
Inhibition of NDRG1 attenuated multiple inflammatory signal pathways, including NF-κB and AP-1 activation, through a functional interaction with orphan nuclear receptor Nur77.
Endothelial specific deficiency of NDRG1 inhibits neointima and atherothrombosis formation after carotid artery injury in mice.
Stress responses such as acute inflammation, hypoxia, flow shear stress, and vascular injury are essentially implicated in the development of cardiovascular disease. N-myc downstream-regulated gene 1 (NDRG-1) is a member of NDRG family and its expression is significantly regulated by cell stress responses such as laminar shear stress and homocysteinemia in vascular cells. By knocking down NDRG1 in endothelial cells, we found that endothelial inflammation and pro-coagulant activity were markedly suppressed through inhibiting NF-κB activation and AP-1 transcriptional activity in NDRG1 knockdown cells. Specific deletion of NDRG1 in vascular endothelium prevents the development of neointimal and atherothrombosis in mice after vascular injury. Our studies unveil a novel mechanism in controlling endothelial activation and may provide an effective therapeutic target for treating inflammatory vascular diseases.
Sources of Funding
This work was funded by National Heart, Lung, and Blood Institute grant R01HL159168 and R01HL152703 to JS.
Nonstandard Abbreviations and Acronyms
- NDRG1
N-myc downstream-regulated gene 1
- MAPK
Mitogen-activated protein kinase
- NF-κB
Nuclear Factor Kappa B
- Activating protein-1
AP-1
- NR4A1
Nuclear receptor subfamily 4, group A, member 1
- IL-1β
Interleukin-1 beta
- TNF-α
Tumor necrosis factor-alpha
- HUVECs
human umbilical vein endothelial cells
- ELISA
Enzyme-linked immunosorbent assay
- EMSA
Electrophoretic mobility shift assay
- qRT-PCR
Quantitative real time PCR
Footnotes
Disclosures
None.
References
- 1.Libby P Inflammation in atherosclerosis. Nature. 2002;420:868–74. [DOI] [PubMed] [Google Scholar]
- 2.Glezeva N, Gilmer JF, Watson CJ and Ledwidge M. A Central Role for Monocyte-Platelet Interactions in Heart Failure. J Cardiovasc Pharmacol Ther. 2015. [DOI] [PubMed] [Google Scholar]
- 3.Libby P, Sukhova G, Lee RT and Galis ZS. Cytokines regulate vascular functions related to stability of the atherosclerotic plaque. J Cardiovasc Pharmacol. 1995;25 Suppl 2:S9–12. [DOI] [PubMed] [Google Scholar]
- 4.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM and Stern DM. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–61. [PubMed] [Google Scholar]
- 5.Sitter T, Toet K, Fricke H, Schiffl H, Held E and Kooistra T. Modulation of procoagulant and fibrinolytic system components of mesothelial cells by inflammatory mediators. Am J Physiol. 1996;271:R1256–63. [DOI] [PubMed] [Google Scholar]
- 6.Hezi-Yamit A, Wong PW, Bien-Ly N, Komuves LG, Prasad KS, Phillips DR and Sinha U. Synergistic induction of tissue factor by coagulation factor Xa and TNF: evidence for involvement of negative regulatory signaling cascades. Proc Natl Acad Sci U S A. 2005;102:12077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sohn RH, Deming CB, Johns DC, Champion HC, Bian C, Gardner K and Rade JJ. Regulation of endothelial thrombomodulin expression by inflammatory cytokines is mediated by activation of nuclear factor-kappa B. Blood. 2005;105:3910–7. [DOI] [PubMed] [Google Scholar]
- 8.Yang P, Wei X, Zhang J, Yi B, Zhang GX, Yin L, Yang XF and Sun J. Antithrombotic Effects of Nur77 and Nor1 Are Mediated Through Upregulating Thrombomodulin Expression in Endothelial Cells. Arterioscler Thromb Vasc Biol. 2016;36:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D and Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 10.May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS and Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–4. [DOI] [PubMed] [Google Scholar]
- 11.You B, Jiang YY, Chen S, Yan G and Sun J. The orphan nuclear receptor Nur77 suppresses endothelial cell activation through induction of IkappaBalpha expression. Circ Res. 2009;104:742–9. [DOI] [PubMed] [Google Scholar]
- 12.Melotte V, Qu X, Ongenaert M, van Criekinge W, de Bruine AP, Baldwin HS and van Engeland M. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB J. 2010;24:4153–66. [DOI] [PubMed] [Google Scholar]
- 13.Fang BA, Kovacevic Z, Park KC, Kalinowski DS, Jansson PJ, Lane DJ, Sahni S and Richardson DR. Molecular functions of the iron-regulated metastasis suppressor, NDRG1, and its potential as a molecular target for cancer therapy. Biochim Biophys Acta. 2014;1845:1–19. [DOI] [PubMed] [Google Scholar]
- 14.Kovacevic Z and Richardson DR. The metastasis suppressor, Ndrg-1: a new ally in the fight against cancer. Carcinogenesis. 2006;27:2355–66. [DOI] [PubMed] [Google Scholar]
- 15.Lachat P, Shaw P, Gebhard S, van Belzen N, Chaubert P and Bosman FT. Expression of NDRG1, a differentiation-related gene, in human tissues. Histochem Cell Biol. 2002;118:399–408. [DOI] [PubMed] [Google Scholar]
- 16.Kyuno J, Fukui A, Michiue T and Asashima M. Identification and characterization of Xenopus NDRG1. Biochem Biophys Res Commun. 2003;309:52–7. [DOI] [PubMed] [Google Scholar]
- 17.Chen B, Nelson DM and Sadovsky Y. N-myc down-regulated gene 1 modulates the response of term human trophoblasts to hypoxic injury. J Biol Chem. 2006;281:2764–72. [DOI] [PubMed] [Google Scholar]
- 18.van Belzen N, Dinjens WN, Eussen BH and Bosman FT. Expression of differentiation-related genes in colorectal cancer: possible implications for prognosis. Histol Histopathol. 1998;13:1233–42. [DOI] [PubMed] [Google Scholar]
- 19.Hunter M, Angelicheva D, Tournev I, Ingley E, Chan DC, Watts GF, Kremensky I and Kalaydjieva L. NDRG1 interacts with APO A-I and A-II and is a functional candidate for the HDL-C QTL on 8q24. Biochem Biophys Res Commun. 2005;332:982–92. [DOI] [PubMed] [Google Scholar]
- 20.Peiretti F, Alessi MC, Henry M, Anfosso F, Juhan-Vague I and Nalbone G. Intracellular calcium mobilization suppresses the TNF-alpha-stimulated synthesis of PAI-1 in human endothelial cells. Indications that calcium acts at a translational level. Arterioscler Thromb Vasc Biol. 1997;17:1550–60. [DOI] [PubMed] [Google Scholar]
- 21.Schmid E, Xuan NT, Zahir N, Russo A, Yang W, Kuhl D, Faggio C, Shumilina E and Lang F. Serum- and glucocorticoid-inducible kinase 1 sensitive NF-kappaB signaling in dendritic cells. Cell Physiol Biochem. 2014;34:943–54. [DOI] [PubMed] [Google Scholar]
- 22.Sun J, Zhang D, Bae DH, Sahni S, Jansson P, Zheng Y, Zhao Q, Yue F, Zheng M, Kovacevic Z and Richardson DR. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis. 2013;34:1943–54. [DOI] [PubMed] [Google Scholar]
- 23.Lu WJ, Chua MS, Wei W and So SK. NDRG1 promotes growth of hepatocellular carcinoma cells by directly interacting with GSK-3beta and Nur77 to prevent beta-catenin degradation. Oncotarget. 2015;6:29847–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kokame K, Kato H and Miyata T. Homocysteine-respondent genes in vascular endothelial cells identified by differential display analysis. GRP78/BiP and novel genes. J Biol Chem. 1996;271:29659–65. [DOI] [PubMed] [Google Scholar]
- 25.Zhang P, Tchou-Wong KM and Costa M. Egr-1 mediates hypoxia-inducible transcription of the NDRG1 gene through an overlapping Egr-1/Sp1 binding site in the promoter. Cancer Res. 2007;67:9125–33. [DOI] [PubMed] [Google Scholar]
- 26.Hickok JR, Sahni S, Mikhed Y, Bonini MG and Thomas DD. Nitric oxide suppresses tumor cell migration through N-Myc downstream-regulated gene-1 (NDRG1) expression: role of chelatable iron. J Biol Chem. 2011;286:41413–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beguin EP, Janssen EFJ, Hoogenboezem M, Meijer AB, Hoogendijk AJ and van den Biggelaar M. Flow-induced Reorganization of Laminin-integrin Networks Within the Endothelial Basement Membrane Uncovered by Proteomics. Mol Cell Proteomics. 2020;19:1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin Q, Chen M, Yi B, You X, Yang P and Sun J. Orphan nuclear receptor Nur77 is a novel negative regulator of endothelin-1 expression in vascular endothelial cells. J Mol Cell Cardiol. 2014;77:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D and Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol. 2009;297:H1535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar S, Kang DW, Rezvan A and Jo H. Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab Invest. 2017;97:935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li P, Zhu N, Yi B, Wang N, Chen M, You X, Zhao X, Solomides CC, Qin Y and Sun J. MicroRNA-663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circ Res. 2013;113:1117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glass CK and Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–16. [DOI] [PubMed] [Google Scholar]
- 33.McCarty S and Frishman W. Interleukin 1beta: a proinflammatory target for preventing atherosclerotic heart disease. Cardiol Rev. 2014;22:176–81. [DOI] [PubMed] [Google Scholar]
- 34.Ridker PM and Luscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J. 2014;35:1782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurakula K, Vos M, Logiantara A, Roelofs JJ, Nieuwenhuis MA, Koppelman GH, Postma DS, van Rijt LS and de Vries CJ. Nuclear Receptor Nur77 Attenuates Airway Inflammation in Mice by Suppressing NF-kappaB Activity in Lung Epithelial Cells. J Immunol. 2015;195:1388–98. [DOI] [PubMed] [Google Scholar]
- 36.van Tiel CM and de Vries CJ. NR4All in the vessel wall. The Journal of steroid biochemistry and molecular biology. 2012;130:186–93. [DOI] [PubMed] [Google Scholar]
- 37.Van-Assche T, Huygelen V, Crabtree MJ and Antoniades C. Gene therapy targeting inflammation in atherosclerosis. Curr Pharm Des. 2011;17:4210–23. [DOI] [PubMed] [Google Scholar]
- 38.Huang H, Petkova SB, Cohen AW, Bouzahzah B, Chan J, Zhou JN, Factor SM, Weiss LM, Krishnamachary M, Mukherjee S, Wittner M, Kitsis RN, Pestell RG, Lisanti MP, Albanese C and Tanowitz HB. Activation of transcription factors AP-1 and NF-kappa B in murine Chagasic myocarditis. Infect Immun. 2003;71:2859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar A and Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–44. [DOI] [PubMed] [Google Scholar]
- 40.Kawashima S, Yamashita T, Ozaki M, Ohashi Y, Azumi H, Inoue N, Hirata K, Hayashi Y, Itoh H and Yokoyama M. Endothelial NO synthase overexpression inhibits lesion formation in mouse model of vascular remodeling. Arterioscler Thromb Vasc Biol. 2001;21:201–7. [DOI] [PubMed] [Google Scholar]
- 41.Kawasaki T, Dewerchin M, Lijnen HR, Vreys I, Vermylen J and Hoylaerts MF. Mouse carotid artery ligation induces platelet-leukocyte-dependent luminal fibrin, required for neointima development. Circ Res. 2001;88:159–66. [DOI] [PubMed] [Google Scholar]
- 42.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H and Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–63. [DOI] [PubMed] [Google Scholar]
- 43.Boyle EM, Jr., Kovacich JC, Canty TG, Jr., Morgan EN, Chi E, Verrier ED and Pohlman TH. Inhibition of nuclear factor-kappa B nuclear localization reduces human E-selectin expression and the systemic inflammatory response. Circulation. 1998;98:II282–8. [PubMed] [Google Scholar]
- 44.Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K and Hedrick CC. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110:416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M, Kluger MS, D’Alessio A, Garcia-Cardena G and Pober JS. Regulation of arterial-venous differences in tumor necrosis factor responsiveness of endothelial cells by anatomic context. Am J Pathol. 2008;172:1088–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohki T, Hongo S, Nakada N, Maeda A and Takeda M. Inhibition of neurite outgrowth by reduced level of NDRG4 protein in antisense transfected PC12 cells. Brain Res Dev Brain Res. 2002;135:55–63. [DOI] [PubMed] [Google Scholar]
- 47.Mossman BT, Lounsbury KM and Reddy SP. Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am J Respir Cell Mol Biol. 2006;34:666–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsu HJ, Lee CF, Locke A, Vanderzyl SQ and Kaunas R. Stretch-induced stress fiber remodeling and the activations of JNK and ERK depend on mechanical strain rate, but not FAK. PLoS One. 2010;5:e12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boon RA, Leyen TA, Fontijn RD, Fledderus JO, Baggen JM, Volger OL, van Nieuw Amerongen GP and Horrevoets AJ. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood. 2010;115:2533–42. [DOI] [PubMed] [Google Scholar]
- 50.Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost. 2006;4:1186–93. [DOI] [PubMed] [Google Scholar]
- 51.Schwachtgen JL, Remacle JE, Janel N, Brys R, Huylebroeck D, Meyer D and Kerbiriou-Nabias D. Oct-1 is involved in the transcriptional repression of the von willebrand factor gene promoter. Blood. 1998;92:1247–58. [PubMed] [Google Scholar]
- 52.Peng Y and Jahroudi N. The NFY transcription factor functions as a repressor and activator of the von Willebrand factor promoter. Blood. 2002;99:2408–17. [DOI] [PubMed] [Google Scholar]
- 53.Mojiri A, Nakhaii-Nejad M, Phan WL, Kulak S, Radziwon-Balicka A, Jurasz P, Michelakis E and Jahroudi N. Hypoxia results in upregulation and de novo activation of von Willebrand factor expression in lung endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:1329–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. An expanded Materials and Methods is available in the Expanded Supplemental Materials. All research materials are listed in the Materials and Methods are also included in the Major Resources Table.