

Abstract
BACKGROUND:
The tyrosine kinase inhibitor (TKI) ponatinib is the only treatment option for chronic myelogenous leukemia (CML) patients with T315I (gatekeeper) mutation. Pharmacovigilance analysis of FDA and WHO datasets have revealed that ponatinib is the most cardiotoxic agent among all FDA-approved TKIs in a real-world scenario. However, the mechanism of ponatinib-induced cardiotoxicity is unknown.
METHODS:
The lack of well-optimized mouse models has hampered the in vivo cardio-oncology studies. Herein, we show that cardiovascular comorbidity mouse models evidence a robust cardiac pathological phenotype upon ponatinib treatment. A combination of multiple in vitro and in vivo models was employed to delineate the underlying molecular mechanisms.
RESULTS:
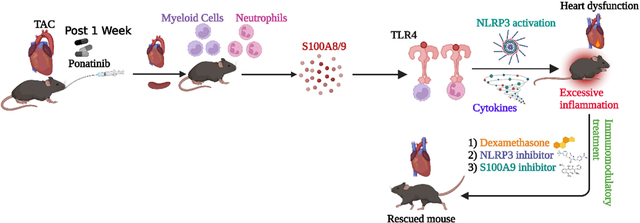
An unbiased RNA-Seq analysis identified the enrichment of dysregulated inflammatory genes, including a multi-fold upregulation of alarmins S100A8/A9, as a top hit in ponatinib-treated hearts. Mechanistically, we demonstrate that ponatinib activates the S100A8/9-TLR4-NLRP3-IL-1β signaling pathway in cardiac and systemic myeloid cells, in vitro and in vivo, thereby leading to excessive myocardial and systemic inflammation. Excessive inflammation was central to the cardiac pathology because interventions with broad-spectrum immunosuppressive glucocorticoid dexamethasone or specific inhibitors of NLRP3 (CY-09) or S100A9 (paquinimod) nearly abolished the ponatinib-induced cardiac dysfunction.
CONCLUSIONS:
Taken together, these findings uncover a novel mechanism of ponatinib-induced cardiac inflammation leading to cardiac dysfunction. From a translational perspective, our results provide critical preclinical data and rationale for a clinical investigation into immunosuppressive interventions for managing ponatinib-induced cardiotoxicity.
Keywords: Cardiotoxicity, Heart Failure, Inflammatory Heart Disease, Remodeling, Cardio-Oncology
Graphical Abstract
Introduction:
Targeted therapies of tyrosine kinase inhibitors (TKIs) have significantly improved the treatment of chronic myeloid leukemia (CML). However, the manifestation of cardiotoxic adverse events has hampered their clinical benefit.1, 2 Among the array of CML-TKIs available, ponatinib is particularly efficacious in patients with the T315I mutation for which no therapy existed previously.3 Based on the PACE trial (with two years of follow-up), heart failure (HF) or left ventricular dysfunction was reported in 8% of patients, including fatal and serious events in 5% of patients.4 Importantly, due to the increased adverse events, further trials were temporarily interrupted and required dose reduction before continuing.5 Furthermore, due to increased cardiovascular (CV) adverse events, ponatinib was temporarily withdrawn from the market in October 2013, with subsequent reintroduction with labeled, boxed warnings of CV adverse effects.6 This was primarily necessitated by the fact that there is no alternative treatment option for CML patients with T315I mutation. Thus, at present, the only option to combat ponatinib cardiotoxicity is to elucidate the underlying molecular mechanisms. A better understanding of the mechanism could guide potential prevention strategies, rescue approaches, and future drug design. Previously, we and others have successfully demonstrated that ponatinib is a potent cardiotoxic TKI, but the precise mechanism of ponatinib cardiotoxicity remains elusive.7–9 Indeed, all previous studies of ponatinib cardiotoxicity are limited by exclusive reliance on cell culture models.
Accumulated data have established that inflammation has a central role in developing heart failure.10 The association of elevated levels of pro-inflammatory cytokines with adverse cardiac outcomes supports the critical involvement of inflammatory mediators in myocardial disease,11–14 and led to the “cytokine hypothesis” as a mechanistic basis for HF progression driven by inflammation.11, 12, 14, 15 Moreover, preclinical studies have repeatedly shown that activation of innate and adaptive immune cells in the heart triggers myocardial inflammation that leads to acute and chronic HF.16–18 The NLRP3 (NLR family, pyrin domain-containing protein 3) inflammasome is a cytosolic multiprotein complex that regulates the inflammatory cytokines IL-1β and IL-18.19 Thus, the NLRP3 inflammasome acts as a crucial pro-inflammatory mediator and may contribute to myocardial inflammation and CV disease pathogenesis.19 The pro-inflammatory alarmins S100A8 and S100A9 are potent activators of the NLRP3 inflammasome.20 Elevated levels of these alarmins are correlated with various inflammatory diseases, including HF.20, 21 S100A8 and its dimerization partner S100A9 are constitutively expressed in immune cells such as monocytes and neutrophils, and their expression is upregulated under inflammatory conditions.22 Extracellular S100A8/9 binds to pattern recognition receptors (PRRs), including toll-like receptors (TLRs) and receptors for advanced glycation end products (RAGE) to promote inflammation.23 S100A8/A9 binding to their respective receptors primes the NLRP3 inflammasome pathway, ultimately leading to IL-1β secretion, and the inflammatory response.20, 21
A major limitation of basic science research in cardio-oncology is the lack of well-optimized animal models mimicking the real-world patient phenotype. For example, ponatinib is clearly cardiotoxic in humans; however, a clinically relevant dose of ponatinib does not lead to cardiac dysfunction in WT mice (C57B6) at baseline.6, 24–26 Thus, there is a clear disconnect from observations in real-world patient populations vs. mice models. Due to this critical limitation, cardio-oncology literature is highly dominated by case reports and meta-analyses. In fact, mechanistic studies with in vivo animal models are scarce. Herein, we report the mouse models of CV comorbidities showing robust cardiac phenotype upon ponatinib treatment. These studies are the first to determine the mechanism of ponatinib-induced cardiotoxicity employing in vivo mouse models. Specifically, studies with high-fat diet (HFD) fed ApoE−/− and pressure overload (PO) murine model suggested the key role of CV-comorbidities in ponatinib-induced cardiomyopathy. Furthermore, we demonstrate that ponatinib causes excessive myocardial inflammation in naïve C57BL/6 mice (no injury), which is a predisposing factor for developing cardiac dysfunction in co-morbid conditions. Additionally, an array of in vivo and ex vivo studies identified the crucial role of the S100A8/9-TLR4-NLRP3-IL1β/IL18 signaling circuit in ponatinib-induced excessive inflammation and cardiac dysfunction. Finally, we demonstrate that ponatinib-induced excessive inflammation is central to cardiac pathology because a broad immunosuppressive agent, dexamethasone, abolished the adverse cardiac remodeling and dysfunction of ponatinib-treated hearts. We also demonstrate that NLRP3-mediated pathways were critical to driving the excessive inflammation and cardiac dysfunction, as CY-09, a direct NLRP3 inhibitor, ameliorated ponatinib-induced detrimental effects. The inflammation hypothesis was further supported by additional in vivo studies with an S100A9 inhibitor, paquinimod, which also mitigated ponatinib-induced inflammation and cardiac dysfunction. In summary, herein, we report the optimization and characterization of CV comorbidity mouse models mimicking the phenotype seen in real-world patient populations. Mechanistically, we discovered that ponatinib exerts its adverse effects and cardiac dysfunction via the proinflammatory S100A8/9-TLR4-NLRP3-IL1β signaling circuit. From a clinical perspective, these findings provide proof of concept for immunosuppressive interventions for managing ponatinib-induced cardiotoxicity.
Methods:
Data Availability.
A detailed description of the Methods is available in the Supplemental Materials section. The data, analytic methods, and study materials are available to other researchers to reproduce the results or replicate the procedures on request.
Statistical Analysis
Analyses were performed by using GraphPad Prism (version 9.3.1). For non-normally distributed data or when N<10, unpaired 2-tailed Mann-Whitney test (for 2 groups) or Kruskal-Wallis test (for 3 or more groups) was performed, followed by Dunn post hoc analysis. For in-vitro cell work, data were analyzed with unpaired t test (2 groups), for multiple group comparisons, 1-way ANOVA followed by Tukey post hoc analysis (for 1 variable) or 2-way ANOVA followed by Sidak’s multiple comparision test (for 2 variables) was conducted. A P of <0.05 was considered statistically significant.
Results:
Ponatinib induces cardiac dysfunction in HFD-fed ApoE−/− mice by promoting myeloid and T cell frequency.
Ponatinib dose selection for in vivo mouse studies was based on multiple preclinical studies, suggesting a dose range of 15 to 30 mg/Kg/day27–29 and our mass spectrometry analysis of plasma samples from ponatinib-treated animals (Figure. S1A). Specifically, our mass spectrometry analysis confirmed that 15mg/Kg was sufficient to produce ponatinib’s blood levels comparable to those seen in patients (50–150ng/ml).30, 31 At 8 weeks of age, wild-type C57Bl/6J mice were treated with 15 mg/kg/day ponatinib or placebo for six weeks.27–31 Surprisingly, serial echocardiography showed no statistically significant difference in cardiac function between ponatinib- and placebo-treated hearts (Fig. 1A–C), despite well-described observations of ponatinib-induced cardiotoxicity in humans.1 This dichotomy underscores the need to identify clinically-relevant preclinical animal models to understand better the “hidden cardiotoxicity” of TKIs and other agents.32 Based on clinical trials and meta-analyses, it has been proposed that patients with CV co-morbidities are at higher risk for development of ponatinib-induced cardiac dysfunction.33, 34 To mimic these real-world scenarios, we employed two co-morbid mouse models: 1) vascular co-morbidity, using HFD-fed ApoE−/− mice, and 2) hypertrophic co-morbidity, using WT C57BL/6J mice with pressure overload. The experimental design for HFD- fed ApoE−/− studies are depicted in Fig. 1D. Briefly, at 8 weeks of age, ApoE−/− mice were fed an HFD diet for 8 weeks, followed by 2 weeks of ponatinib treatment (15 mg/kg/day) while continuing the HFD. Echocardiography revealed a significant decline in cardiac function of ponatinib-treated HFD ApoE−/− mice as compared with placebo-treated HFD-fed animals (Fig. 1E–J). Histological evaluation showed augmented cardiac fibrosis in ponatinib-treated HFD-fed ApoE−/− mice compared to controls (Figure S1B–1C). Collectively, these data indicate that vascular co-morbid conditions predisposed the mice to ponatinib-induced adverse cardiac effects.
Fig. 1|. Ponatinib induces cardiac dysfunction in High Fat Diet (HFD) fed ApoE−/− by promoting myeloid and T cell frequency.
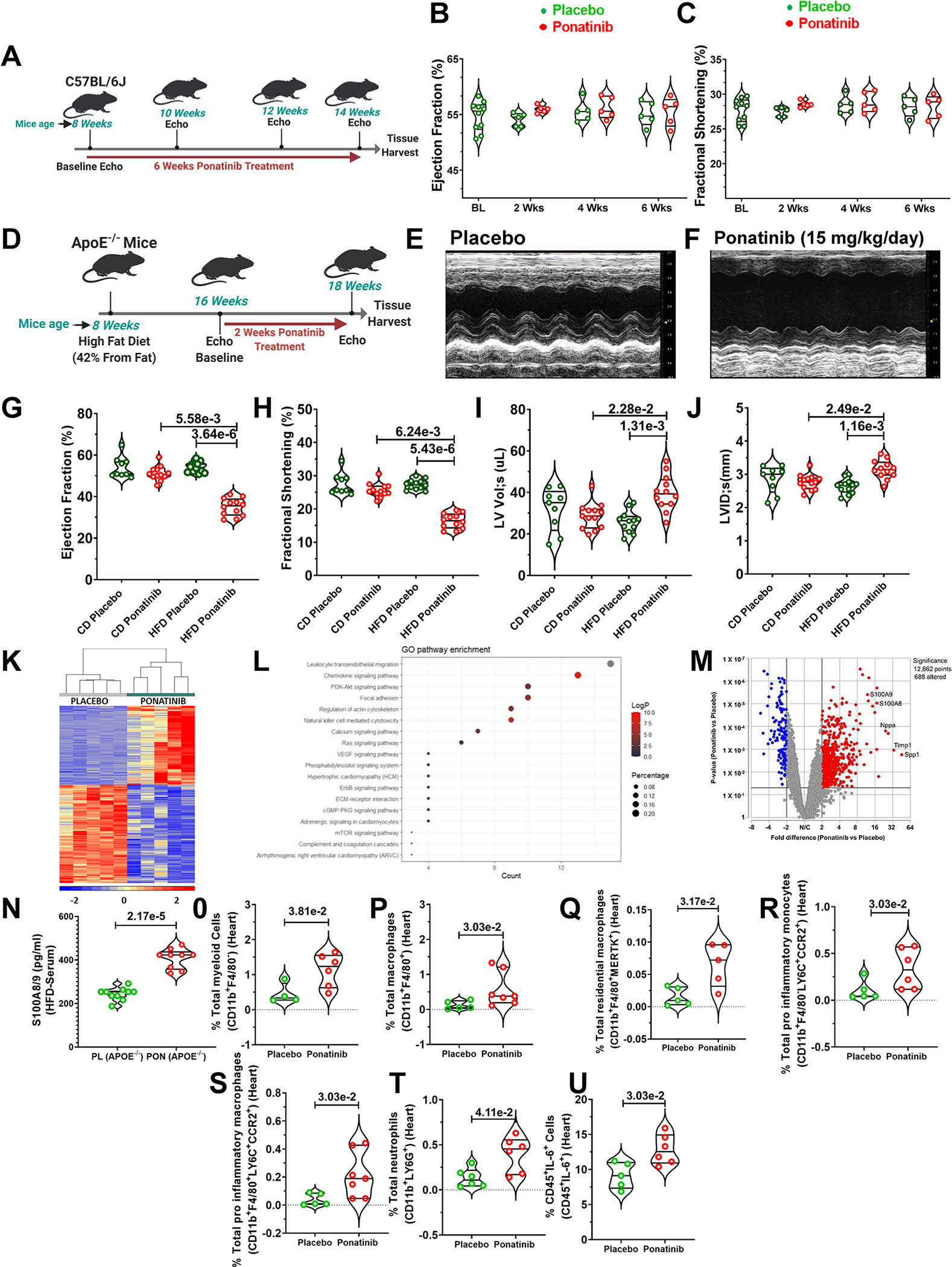
(A) An experimental scheme using 8-week-old male and female C57Bl/6J mice subjected to ponatinib treatment (15 mg/Kg/day) for 4 weeks. (B-C) Ejection Fraction and Fractional Shortening as measured by echocardiography at 2, 4, and 6 weeks. No statistically significant difference was observed in the control and ponatinib-treated group, as measured by the Mann-Whitney U test for each time point and represented as mean±SEM. Basal (BL) (N=10), placebo, and ponatinib (N=5) at 2 weeks, 4 weeks, and 6 weeks. (D) Schematic of experiment performed in which 8-week-old male and female ApoE−/− mice were subjected to high-fat diet (HFD). Chow Diet (CD) animals were used as control. After 8 weeks on HFD, mice were given ponatinib treatment (15mg/Kg/day) for 2 weeks. (E-F) Representative images of echocardiographic measurements in HFD groups. (G-J) Ejection Fraction, Fractional Shortening, Left ventricle volume in systolic (LV Lol;s), and Left ventricle internal diameter in systolic (LVID;s) were measured by echocardiography. Significance was determined by using Kruskal-Wallis followed by Dunn test and represented as mean±SEM CD placebo (N= 9); CD ponatinib, HFD placebo, and HFD ponatinib (N= 12). (K) RNA sequencing (RNA-Seq) analysis of LV samples from HFD placebo and HFD ponatinib groups was performed at 2 weeks after ponatinib treatment by taking LV samples from HFD placebo and HFD ponatinib groups. N=5 per group. The hierarchical clustering of 544 genes was detected as a notably differential between the HFD placebo and HFD ponatinib groups. (L) Dot enrichment plot. (M) Volcano plot of differentially expressed genes. (N) ELISA for quantitative measurement of S100A8/9 from the serum sample of ponatinib treated HFD- fed ApoE−/− mice, placebo (PL) (N=10), and ponatinib (PON) (N=9). (O-U) Quantitation of immune cells as a percentage of total cells (immune and nonimmune) isolated from the digested heart of HFD placebo and HFD ponatinib groups. (O) total percentage of myeloid cells per heart (CD11b+F4/80−), placebo (N=4) and ponatinib (N=6), (P) total percentage of macrophages (CD11b+F4/80+), placebo (N=5) and ponatinib (N=7), (Q) total percentage of residential macrophages (CD11b+F4/80+MERTK+), placebo (N=5) and ponatinib (N=5), (R) total pro-inflammatory monocytes (CD11b+F4/80−LY6C+CCR2+), placebo (N=5) and ponatinib (N=6), (S) total pro-inflammatory macrophages (CD11b+F4/80+LY6C+CCR2+), placebo (N=5) and ponatinib (N=7), (T) total neutrophils (CD11b+LY6G+), placebo (N=6) and ponatinib (N=6), (U) percentage of IL-6 producing leukocytes (CD45+IL-6+), placebo (N=5) and ponatinib (N=6), Data (O-U) were analyzed using the Mann-Whitney U test and represented as mean±SEM.
To gain better insight into the molecular processes contributing to ponatinib-induced cardiotoxicity, we performed RNA sequencing (RNA-Seq) with LV tissue of HFD-fed placebo ApoE−/−, and HFD-fed ponatinib ApoE−/− groups at two weeks post-ponatinib treatment. Bioinformatic analyses revealed 244 coding transcripts that were upregulated, and 300 transcripts downregulated in ponatinib-treated HFD ApoE−/− mice (GOTERM Biological Pathway Table, supplementary material) (GEO accession numbers- GSE220121). The two treatment groups were readily distinguishable upon a hierarchical clustering of differentially expressed genes (Fig. 1K–1M). Of note, along with the markers of heart failure, genes related to immune cell activation and inflammation were uniquely enriched. Interestingly, the hallmarks of NLRP3-mediated inflammation, alarmins (S100A8 and S100A9), were remarkably upregulated in ponatinib-treated mice (Fig. 1K–1M). The serum from these mice was collected to perform ELISA for S100A8/9. As expected, the plasma level of S100A8/9 was significantly upregulated in the ponatinib-treated HFD-fed ApoE−/− group (Fig. 1N). To further establish the dynamics of ponatinib-induced immune responses, we performed comprehensive immune profiling by flow cytometry analysis of ponatinib-treated HFD-fed ApoE−/− hearts. Elevation of pro-inflammatory myeloid cells (monocytes, macrophages, neutrophils, and dendritic cells), pro-inflammatory soluble mediators (TNF-α, IL-6, and IL-1β), and pro-inflammatory helper T cells (Th1, Th17, and Th9) are associated with cardiac injury and repair.35–39 Flow cytometry analysis indicated that ponatinib induced the infiltration of myeloid cells (CD11b+F4/80−) and macrophages (CD11b+F4/80+) in HFD fed ApoE−/− hearts compared to placebo-treated groups (Fig. 1O,1P). Additionally, we observed an increased frequency of resident macrophages (CD11b+F4/80+MerTK+) in the ponatinib-treated ApoE−/− group (Fig. 1Q). We also found an increased percentage of pro-inflammatory monocytes (CD11b+F4/80−LY6C+CCR2+) and macrophages (CD11b+F4/80+LY6C+CCR2+) in the ponatinib-treated ApoE−/−hearts (Fig. 1R,1S). Consistent with broad inflammatory repones, ponatinib-treated hearts exhibited increased accumulation of neutrophils (CD11b+LY6G+), IL-6-producing CD45+ leukocytes, and activated CD4+ T cells, including pro-inflammatory IFN-γ producing type I helper T cells (Th1) (Fig. 1T–1U, S1D–S1F). Moreover, ponatinib-induced increased infiltration of immune cells was consistently evident in the histological evaluation of the myocardium (Figure. S1G). The spleen is a secondary lymphoid organ critical for the regulation of systemic immune responses. CD4+IFN-γ+ T cells (Th1), CD4+IL-17+ T cells (Th17), and IL-9 producing CD4+ T cells (Th9) are known to participate in promoting tissue inflammation and myocarditis.40, 41 To examine ponatinib-induced systemic inflammation, splenocytes were isolated from HFD-fed ponatinib- and placebo-treated ApoE−/− mice, and comprehensive immune profiling was performed. Indeed, splenic immunophenotyping corroborated the cardiac immune cell data and clearly indicated that ponatinib exacerbates systemic inflammation by promoting pro-inflammatory myeloid and pro-inflammatory Th1, Th9, and Th17 cell expansion (Figure. S1H–1Q, Figure. S2).
Ponatinib treatment induces cardiac and systemic inflammation in WT naïve mice
HFD-fed ApoE−/− mice are well-known for exhibiting a proinflammatory state. These confounders complicate the interpretation of the ponatinib-mediated cardiac inflammatory phenotype in HFD-fed ApoE−/− mice. To further address ponatinib-mediated effects on cardiac inflammation, naïve C57BL/6J wild-type mice were treated with ponatinib for 2 weeks. Single-cell suspensions of cardiac tissue digests were analyzed for immune cells via flow cytometry. Despite the absence of cardiac dysfunction, ponatinib promoted the expansion of CD45+CD11b+F4/80− myeloid cells and CD45+CD11b+F4/80+ macrophages in the heart (Fig. 2A, 2B). Consistently, the frequency of CCR2+ pro-inflammatory monocytes and macrophages were enhanced in the ponatinib-treated hearts (Fig. 2C, 2D). Furthermore, we found an increased percentage of CD45+CD11C+ and CD45+CD11b+Ly6G+ cells in the ponatinib-treated hearts, suggesting that ponatinib promotes infiltration of dendritic cells (DCs) and neutrophils (Fig. 2E, 2F). IL-17-producing helper T cells (Th17) are closely associated with autoimmunity and myocarditis.42–44 Cardiac Th17 cells were significantly elevated in ponatinib-treated naïve mice compared to placebo (Fig. 2G). To understand the role of ponatinib in systemic inflammation, we immunophenotyped splenocytes isolated from ponatinib and placebo-administered naïve mice. A robust expansion of myeloid cells, including macrophages (CD11b+F480+), MHC-II+ pro-inflammatory M1 macrophages (CD11b+F480+MHC-II+), and neutrophils (CD11b+Ly6G+) were found in the spleens of the ponatinib-treated group (Fig. 2H–2J). Adaptive cell-mediated immunity plays a critical role in the pathogenesis of the inflammatory cardiovascular disease. CD4+ and CD8+ T cells are known to play a crucial role in ventricular remodeling and HF.38, 45 To assess whether ponatinib treatment influences systemic T cells, we analyzed the total percentage of splenic TCRαβ+, CD4+, and CD8+ T cells in ponatinib-treated naïve mice. Ponatinib animals exhibited increased frequency of CD4+, and CD8+ T cells (Fig. 2K–2Q). Furthermore, ponatinib promoted the polarization of pro-inflammatory Th1, Th17, and Th9 subsets of T cells, indicating the potential role of accumulated pro-inflammatory helper T cells in ponatinib-induced myocardial inflammation. Next, we wanted to investigate whether ponatinib-induced inflammation is a transient event or a chronic inflammatory response. The naïve C57BL/6 mice were treated with ponatinib for six weeks, and immune profiling was performed at multiple time points (2wks, 4wks, 6wks). Indeed, ponatinib induced a sustained chronic inflammation in both, the heart and systemically (Figure. S3A–3F). These flow cytometry findings were further confirmed by ELISA assay of multiple inflammatory cytokines in the serum (Figure. S3G–3J). Taken together, these studies suggest that even though ponatinib does not lead to cardiac dysfunction in naïve animals, it induces sustained chronic inflammation.
Fig. 2|. Ponatinib promotes inflammation in naïve wild type C57BL/6J.
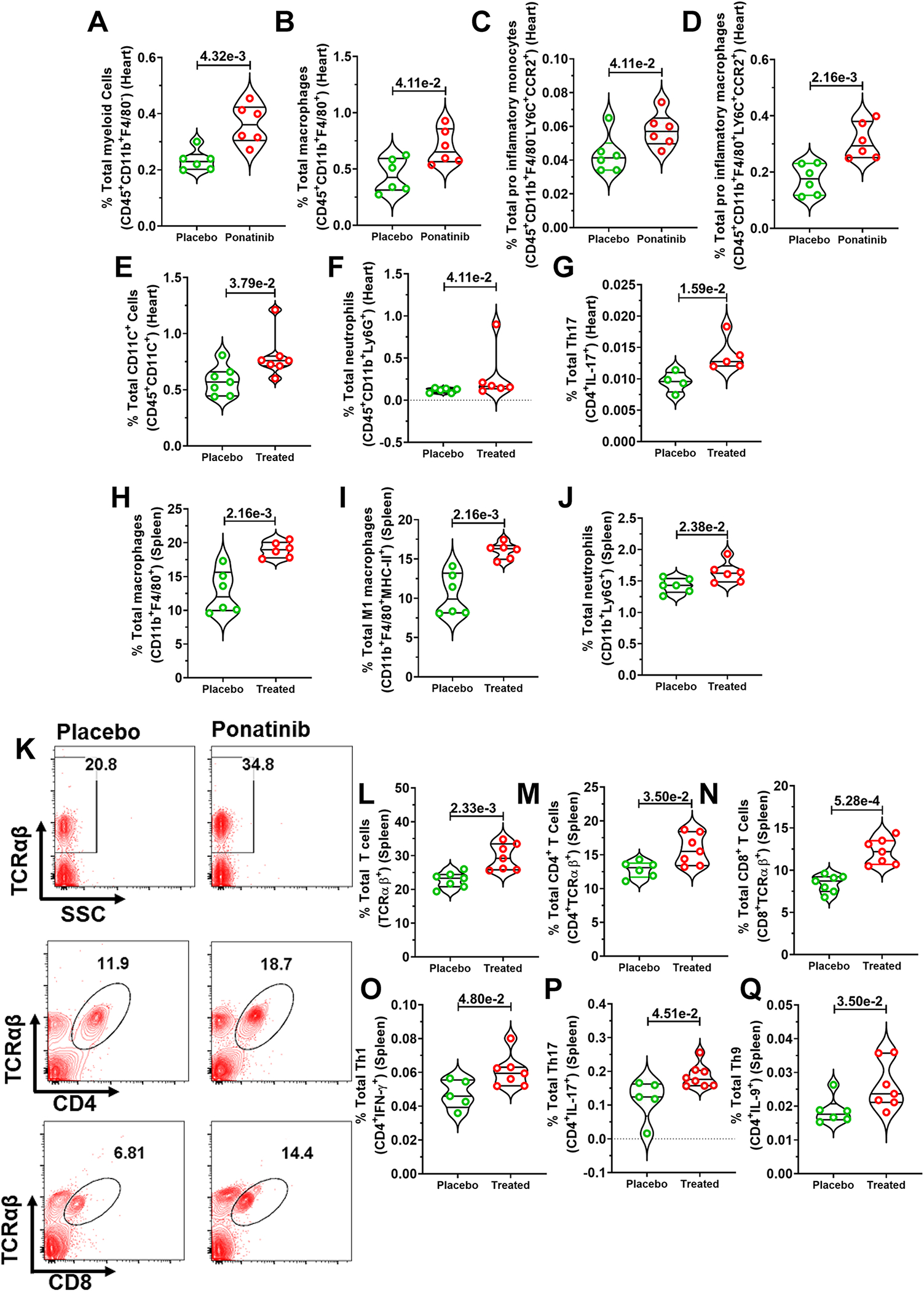
Eight-week-old male and female C57Bl/6J mice were subjected to ponatinib treatment (15 mg/Kg/day) for 2 weeks. After treatment, the whole heart of placebo and ponatinib-treated mice were digested to prepare a single-cell suspension. Cells were then stained with antibodies (CD45, CD11b, F4/80, LY6C, CCR2, LY6G, and IL-17), and flow cytometry was performed. Total percentages of (A) CD45+CD11b+F4/80− myeloid cells, placebo and ponatinib (N=6), (B) CD45+CD11b+F4/80+ macrophages, placebo and ponatinib (N=6), (C) CD45+CD11b+F4/80−LY6C+CCR2+ cells as pro-inflammatory monocytes, placebo and ponatinib (N=6), (D) CD45+CD11b+F4/80+LY6C+CCR2+ cells as pro-inflammatory macrophages, placebo and ponatinib (N=6), (E) CD45+CD11C+ dendritic cells, placebo and ponatinib (N=7), (F) CD45+CD11b+ LY6G+ neutrophils, placebo and ponatinib (N=6), and (G) CD4+IL-17+ Th17 cells, placebo (N=4) and ponatinib (N=5). (H-J) Spleen of placebo and ponatinib treated naïve C57BL/6 wild-type mice were macerated to prepare a single-cell suspension. Cells were then stained with antibodies (CD11b, F4/80, MHC-II, and LY6G) and flow cytometry was performed. Total percentages of (H) CD11b+F4/80+ macrophages, placebo and ponatinib (N=6), (I) CD11b+F4/80+MHC-II+ cells as M1 macrophages, and, placebo and ponatinib (N=6), (J) CD11b+ LY6G+ neutrophils cells, placebo and ponatinib (N=6). (K) Representative figure of flow cytometry showing gating strategy to measure total T cells, CD4+ and CD8+ T cells in the spleen of ponatinib treated and placebo mice post 2 weeks treatment. We analyzed the total percentage of (L) TCRαβ+ T cells, placebo and ponatinib (N=7), (M) TCRαβ+ CD4+ T cells, placebo (N=6) and ponatinib (N=7), (N) TCRαβ+ CD8+ T cells, placebo and ponatinib (N=7),(O) CD4+IFN-γ+ Th1 cells, placebo (N=5) and ponatinib (N=7), (P) CD4+IL-17+ Th17 cells, placebo (N=5) and ponatinib (N=8), and (Q) CD4+IL-9+ Th9 cells, placebo (N=6) and ponatinib (N=7). Data (A-Q) were analyzed using the Mann-Whitney U test and represented as mean±SEM.
Ponatinib promotes excessive inflammation in pressure-overload WT mice
To further test our hypothesis that ponatinib-mediated inflammation is a predisposing factor for cardiac dysfunction, we used the transverse aortic constriction (TAC) pressure-overload mouse model of heart failure. The experimental timeline and treatment plan are depicted in the schematic diagram (Fig. 3A). As anticipated, ponatinib treatment exacerbated the marked cardiac dysfunction in TAC mice as reflected by significantly reduced ejection fraction, fractional shortening, and increased LV systolic volume (Fig. 3B, 3C). These functional deteriorations were associated with adverse cardiac remodeling as reflected by significantly increased LV internal diameter in ponatinib-administered mice (Fig. 3D). Pathological adverse cardiac remodeling was also evident by elevated myocardial fibrosis in the ponatinib-treated TAC group (Figure. S4A–B). Consistent with our findings with HFD-fed ApoE−/− model, ponatinib-induced cardiac dysfunction in TAC animals was associated with increased immune cell infiltration as assessed by H&E staining (Figure. S4C). Using immunostaining, we consistently observed elevated numbers of CD45+ leukocytes and CD3+ T cells in the cardiac tissue of ponatinib-treated mice (Fig. 3E, 3F). These results support the role of ponatinib-induced expansion of myeloid and T cells in producing cardiotoxicity. Comprehensive immune profiling of ponatinib vs. placebo-treated hearts was performed to better understand ponatinib-mediated immunological responses. The flow cytometry data validated the IHC results and showed the recruitment and proliferation of CD45+ leukocytes in ponatinib-treated hearts (Fig. 3G, 3H). Subsequently, we examined myeloid cells in the heart. Augmentation of ventricular myeloid cells (CD45+CD11b+F4/80−), macrophages (CD45+CD11b+F4/80+), resident macrophages (CD45+CD11b+F4/80+MERTK+), M1 macrophages (CD45+CD11b+F4/80+MHC-II+), DCs (CD45+CD11c+) and neutrophils (CD45+CD11b+Ly6G+) were observed in the ponatinib-treated group versus placebo (Fig. 3I–3N). Interestingly, CXCL9-producing CD45+ cells were significantly higher in the ventricular tissue of ponatinib-treated TAC mice (Fig. 3O). We also observed a strong increase in Th1 cytokine (TNF-α, IL-6, and IL-1β) producing CD45+ leukocytes in the ponatinib-treated hearts. (Fig. 3O–3R). Consistently, TNF-α, IL-6, and IL-1β were increased in the circulation of ponatinib-treated mice (Fig. 3S–3U). Thus, ponatinib promotes myeloid cell proliferation and induces pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α), leading to myocardial inflammation. A network of upregulated chemokines and cytokines facilitates T-cell recruitment to the heart and plays a critical role in inflammation-induced cardiac remodeling and dysfunction.45 To determine the dynamics of T cells in cardiac tissue, we analyzed the total frequency of TCRαβ+, CD4+, and CD8+ T cells. Indeed, ponatinib-induced myocardial inflammation was associated with T-cell infiltration in cardiac tissue (Fig. S5A–C). It is well established that the release of danger signals such as alarmins and IL-1β in heart failure stimulates hematopoiesis and medullary monocytopoiesis in the bone marrow.46 These monocytes and myeloid progenitor cells then migrate toward the spleen for extramedullary monocytopoiesis.47 Later, this pool of pro-inflammatory immune cells, mainly monocytes from the spleen, migrate towards the heart.48, 49 Apart from chemokines, intracellular cell adhesion molecule 1 (ICAM-1) expressed on both blood monocytes, and cardiac endothelial cells regulate cardiac inflammation by mediating left ventricular leukocyte recruitment.45 Interestingly, enhanced expression of ICAM-1 was demonstrated in splenic and blood myeloid cells post-ponatinib treatment, suggesting ponatinib facilitates the recruitment of leukocytes (Figure. S5F–J). Further analysis of T cell subtypes revealed an elevated frequency of pro-inflammatory Th1 and Th17 helper T cells (Fig. S5D, S5E). Enhanced levels of serum cytokines in the ponatinib group suggested ponatinib-induced systemic inflammation (Fig. 3S–3U). Consistently, we demonstrated increased proliferation (Ki67+) of splenocytes, enhanced frequency of DC, M1 macrophages, IL-6, and TNF-α producing cells in the spleen of ponatinib-treated animals. (Figure. S5K–O). Excessive systemic inflammation was further supported by the upregulation of pro-inflammatory Th1 and Th17 T cells in the ponatinib group (Figure. S5P–Q). Ponatinib–induced M1 macrophage polarization was further confirmed by utilizing additional M1 marker CD86 (Figure. S6A–D). Taken together, the cardiac function of placebo and ponatinib-treated animals was not significantly different in naïve or sham groups; in contrast, ponatinib led to a robust cardiac phenotype in HFD-fed ApoE−/− and TAC mice, confirming the critical role of CVD comorbidities for the development of ponatinib mediated cardiotoxicity. These findings support the notion that subclinical inflammation (without cardiac pathologies) predisposes the heart to accelerated dysfunction and remodeling in the presence of CVD comorbidities. These findings implicate excessive inflammation as a critical determinant of ponatinib-induced pathologies in failing hearts.
Fig. 3|. Ponatinib-induced inflammation promotes cardiac dysfunction in pressure overload WT mice.
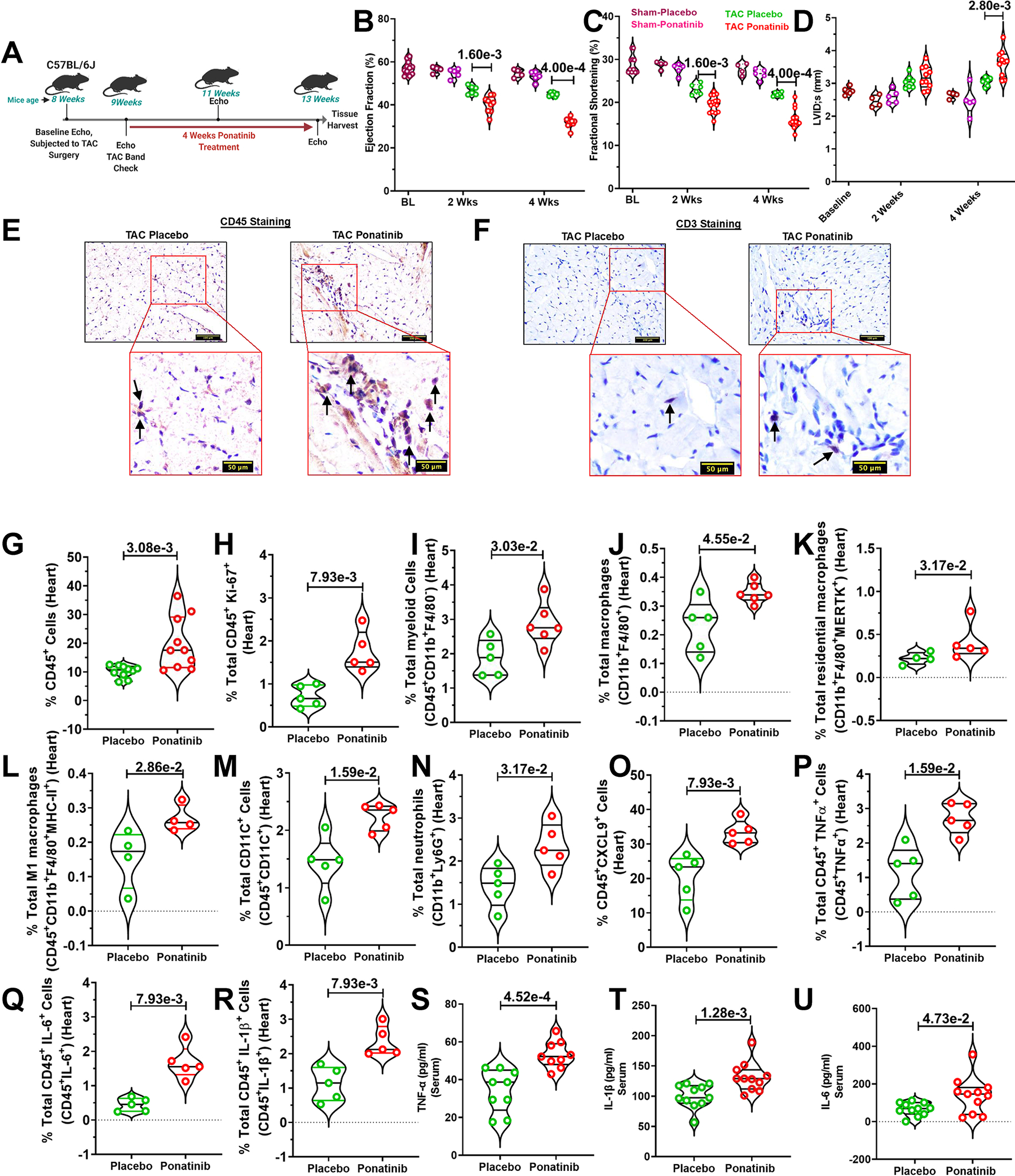
(A) Schematic outline of the experiment performed. Eight-week-old male and female C57Bl/6J mice were subjected to TAC surgery followed by ponatinib treatment (15 mg/Kg/day) for 4 weeks. (B-C) Ejection Fraction and Fractional Shortening and Left ventricle internal diameter in systolic (LVID; s) as measured by echocardiography at 2 and 4 weeks, basal (BL) (N=11); At 2 and 4 weeks, sham placebo (N=4), sham ponatinib (N=5), TAC placebo (N=6), and TAC ponatinib (N=9); At 4 weeks, sham placebo (N=4), sham ponatinib (N=5), TAC placebo (N=6), and TAC ponatinib (N=9). (D) Left ventricle internal diameter in systolic (LVID; s), Basal (BL) (N=6), At 2 weeks, sham placebo (N=4), sham ponatinib (N=5), TAC placebo (N=7), and TAC ponatinib (N=10); At 4 weeks, sham placebo (N=4), sham ponatinib (N=5), TAC placebo (N=6), and TAC ponatinib (N=10). Significance was compared between TAC Placebo vs TAC Ponatinib for 2 weeks and 4 weeks separately by using the Mann-Whitney U test and represented as mean±SEM. (E) CD45-positive leukocytes and (F) CD3-positive T cells in the heart of ponatinib-treated TAC mice and control. A black arrow in the inset image indicates a positive cell. The scale bar indicates 50 μm. (G-R) Quantitation of immune cells as a percentage of total cells isolated from the heart of TAC-placebo and TAC-ponatinib groups. Data represents quantitation of percent of (G) total leukocytes (CD45+), placebo and ponatinib (N=10),(H) total proliferative leukocytes (CD45+Ki67+), placebo and ponatinib (N=5), (I) total myeloid (CD45+CD11b+F4/80−), placebo (N=5) and ponatinib (N=6), (J) total macrophages (CD11b+F4/80+), placebo (N=5) and ponatinib (N=6), (K) total residential macrophages (CD11b+F4/80+MERTK+), placebo (N=4) and ponatinib (N=5), (L) total M1 macrophages (CD11b+F4/80+MHC-II+), placebo and ponatinib (N=4), (M) total dendritic cells (CD45+CD11C+), placebo and ponatinib (N=5),(N) total neutrophils (CD11b+LY6G+), placebo and ponatinib (N=5), (O) total CXCL9 producing leukocytes (CD45+CXCL9+), placebo and ponatinib (N=5), (P) total TNF-α producing cells (CD45+TNF-α+), placebo and ponatinib (N=5),(Q), total IL-6 producing cells (CD45+IL-6+), placebo and ponatinib (N=5), (R) total IL-1β producing cells (CD45+IL-1β+), placebo and ponatinib (N=5). ELISA was performed from the serum obtained from the TAC Placebo and TAC Ponatinib animals after 4 weeks of ponatinib treatment. Data showing serum levels of (S) TNF-α, placebo and ponatinib (N=9), (T) IL-1β, placebo and ponatinib (N=11) (U) IL-6, placebo and ponatinib (N=11). Data (G-U) were analyzed using the Mann-Whitney U test and represented as mean±SEM.
Fibroblast activation or myocardial fibrosis is not the driver of ponatinib cardiotoxicity
Fibroblast activation and fibrosis are critical to adverse cardiac remodeling and cardiac dysfunction. As discussed above, we observed a significantly increased myocardial fibrosis in ponatinib-treated hearts at 4 weeks post-TAC (Figure. S4A–B). This prompted us systematically investigate the role of fibroblast activation and fibrosis in ponatinib-induced cardiac dysfunction. Therefore, to investigate the potential role of myocardial fibrosis in ponatinib’s cardiotoxicity, both in vivo and in vitro experimental strategies were employed. To determine the direct effect of ponatinib on fibroblasts activation, isolated cardiac fibroblasts were treated with relevant ponatinib concentrations (100nM, 250nM) in the presence of fibrosis agonist TGF-β1 (10ng/ml, 1h), and the critical readout SMAD3 activation (P-SMAD3Ser423/425) was determined by western blotting. To our complete surprise, TGF-β1 mediated activation of SMAD3 was not statistically significant between TGF-β1-alone vs TGF-β1 and ponatinib groups (Figure. S7A–B). This data suggest that ponatinib does not directly activate cardiac fibroblast activation. Since increased fibroblast number (proliferation) is a crucial marker of fibroblast activation/fibrotic remodeling, we quantified the total fibroblast percentage in ponatinib-treated hearts. Indeed, ponatinib-induced total fibroblast percentages were not statistically significant at 2 weeks, and a significant increase was only noted starting from a 4 weeks time point (Figure. S7C). Consistently, analysis of trichrome-stained heart sections at 2 weeks post-TAC showed a statistically insignificant level of fibrosis in ponatinib vs. placebo-treated hearts (Figure. S7D–E). Indeed, the ponatinib-induced cardiac dysfunction phenotype was obvious and significant at 2 weeks post-TAC. Thus, the excessive inflammation and cardiac dysfunction phenotype (obvious at 2 wk post-TAC) preceded the onset of fibrotic remodeling. Taken together, these data exclude fibrotic remodeling as the primary driver of ponatinib-induced cardiac dysfunction. Furthermore, it suggests that observed excessive fibrosis at the later time point (4 weeks, post-TAC) is secondary to ponatinib-induced cardiac inflammation and dysfunction.
Ponatinib promotes inflammation through the NLRP3 pathway.
Our unbiased RNA-Seq analysis revealed a multi-fold (10+) upregulation of alarmins S100A8/A9 expression in ponatinib-treated hearts (Fig. 1K–1N). As anticipated, elevated serum S100A8/A9 levels in ponatinib-treated WT naïve mice corroborated our RNA seq data (Figure. S3J). Consistently, flow cytometry analysis revealed robust activation of the innate and adaptive immune response in ponatinib-treated hearts (Fig. 3G–3U, S5A–E). In vivo inflammation studies are confounded by systemic effects; therefore, an isolated splenocyte model was employed to test whether ponatinib directly activates immune cells. Ex vivo cultures of whole splenocytes from naïve WT mice were treated with either vehicle (DMSO) or ponatinib (100 nM and 500 nM, 72h). Increased proliferation of myeloid cells and T cells (CD4+ and CD8+) suggested a potential direct immune activation effect of ponatinib (Fig. 4A–4E). Furthermore, enhanced M1/M2 ratio, and Th1, Th17 frequency was determined in ponatinib-treated samples (Fig. 4F–4H). ELISA analysis revealed elevated levels of inflammatory cytokines (TNF-α and IL-1β) in the media supernatant of ponatinib-treated samples (Fig. 4I–4J). To gain further mechanistic insight, we determined if ponatinib directly promotes S100A8/A9 expression in immune cells. Indeed, ponatinib promotes the frequency of S100A8/A9-producing myeloid cells and neutrophils in vitro (Fig. 4K, 4L). Furthermore, ponatinib induced robust activation of the S100A8/A9-NLRP3-IL-1β signaling axis in ex-vivo experimental settings as described above (Fig. 4M–4R). Advanced image stream single-cell analysis further confirmed ponatinib-induced NLRP3-IL1β expression in myeloid cells in vitro (Fig. 4S, 4T). We performed similar experiments with human peripheral blood mononuclear cells (PBMCs) (iXCells Biotechnology). As anticipated, ponatinib promoted proliferation and NLRP3 expression in human PBMCs (Fig. S8A–D). To gain insight into signaling mechanisms responsible for this immunoinflammatory response in vivo, we analyzed whether ponatinib alters the S100A8/A9-TLR4-NLRP3-IL-1β axis in vivo (Ponatinib treated TAC animals). Indeed, ponatinib promotes S100A8/A9/TLR4/NLRP3/IL-1β expression in cardiac and systemic myeloid cells (Fig. 5A–5L) (Figure. S9, 10, 11). The S100A8/9-NLRP3 signaling axis is known to upregulate chemotaxis-related molecules (CCR2, CXCR6) to promote immune cell proliferation.50 To determine if ponatinib-induced inflammatory signaling is transient or chronic activation, naïve C57BL/6 mice were treated with ponatinib for up to 6 weeks and analyzed at multiple time points (2wks, 4wks, and 6 wks). Indeed, these studies suggested a chronic activation of the S100A8/A9-TLR4-NLRP3-IL-1β pathway (Figure. S12). Furthermore, these animals also exhibit the proliferation of myeloid cells and neutrophils. We also observed proliferation of T cells and increased frequency of Th1, Th9, and Th17 cells in these mice (Figure. S12). Based on these robust findings, we hypothesized that the S100A8/A9-NLRP3-IL-1β signaling axis is critical for ponatinib-mediated immune cell proliferation. To test this hypothesis, isolated splenocytes from WT and NLRP3 KO were treated with vehicle (DMSO) or ponatinib, and proliferation was determined. Indeed, ponatinib-induced proliferation and pro-inflammatory polarization of immune cells were markedly reduced in the NLRP3 KO compared to controls. (Fig. 5M–5T). To further consolidate the signaling circuit of the ponatinib-induced inflammatory response, pure myeloid cells were flow-sorted, and experiments were performed with various agonists and antagonists of the NLRP3 signaling cascade. These include TLR4 agonist (LPS), TLR4 inhibitor (TLR-IN-C34), NLRP3 inhibitor (CY09), and alarmin inhibitor (paquinimod). As expected, LPS Induced myeloid cell proliferation by several folds (Fig. 6A–C). Indeed, ponatinib-induced myeloid cell proliferation was nearly abolished by inhibitors of TLR4, NLRP3, and alarmins (Fig. 6A–C). These data are consistent with in vivo findings with NLRP3 KO animals and support our overall hypothesis that ponatinib drives excessive inflammation through the alarmins-TLR4-NLRP3 signaling axis. Consistently, ponatinib-mediated myeloid cell proliferation was abolished by alarmins inhibitor (Fig. 6A–C). These findings confirm the critical role of NLRP3 signaling in ponatinib-induced immune cell activation and proliferation, leading to excessive inflammation.
Fig. 4|. Ponatinib promotes inflammation through the NLRP3 pathway.
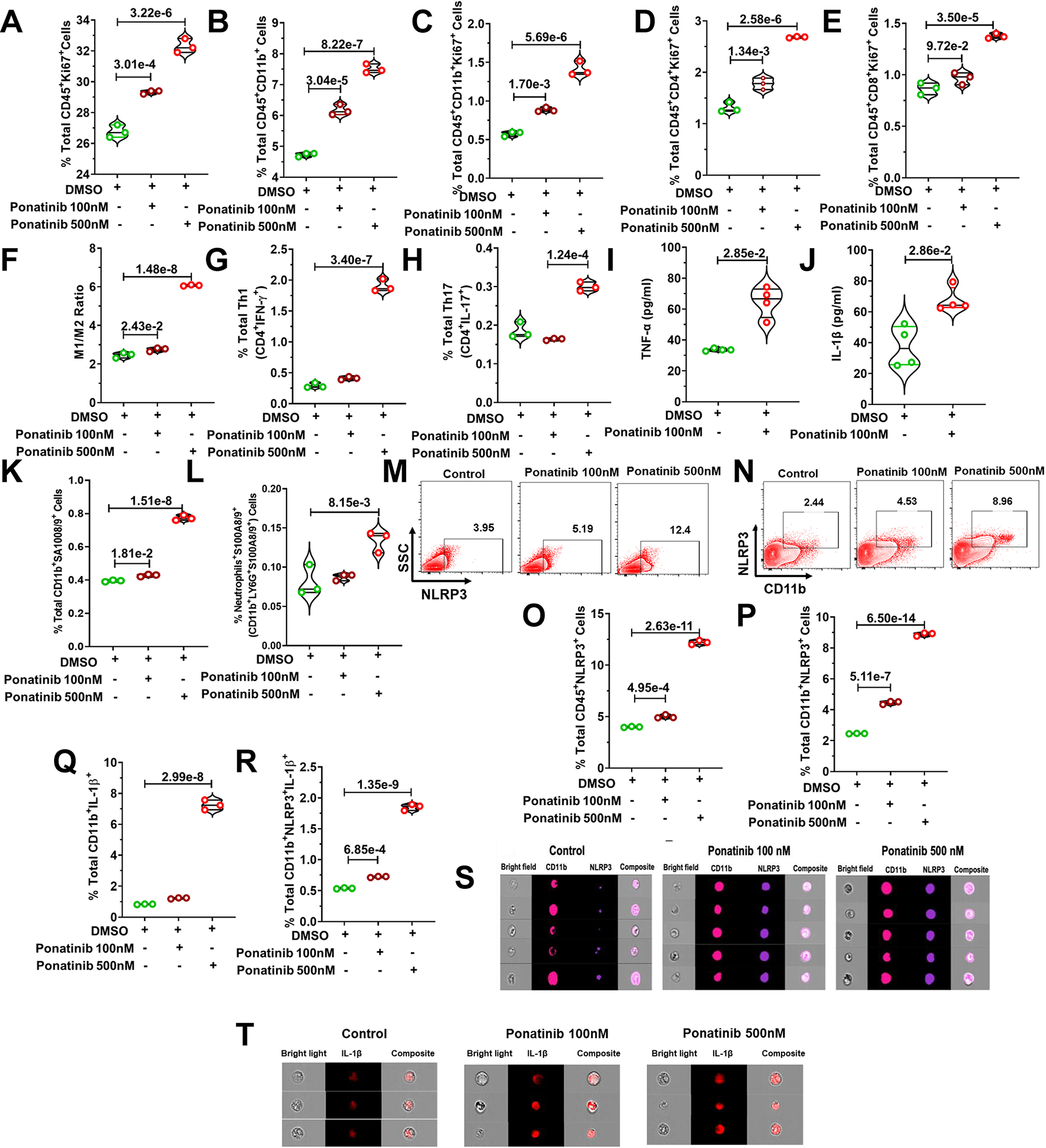
Whole splenocytes of 8-week-old C57BL/6J mice, disaggregated and seeded with vehicle control (DMSO) and ponatinib (100 nM and 500 nM, 72h), and flow cytometry was performed. (A) Increased proliferation of leukocytes (CD45+Ki67+). (B) total % of myeloid cells (CD45+CD11b+) and proliferation (C) (CD45+CD11b+Ki67+). Increased frequency of (D) CD4+ T cell proliferation (CD45+CD4+Ki67+), (E) CD8+ T cell proliferation (CD45+CD8+Ki67+), (F) M1/M2 ratio, (G) total Th1 cells (CD4+IFN-γ+), and (H) Th17 cells (CD4+IL-17+). ELISA was performed for in vitro assessment of the concentration of (I) TNF-α and (J) IL-1β in media supernatant of ponatinib-treated splenocytes. (K) Total percentage of S100A8/9 producing myeloid (CD11b+S100A8/9+). (L) Total percentage of S100A8/9 producing neutrophils (CD11b+LY6G+S100A8/9+). (M, N) Flow cytometry analysis of NLRP3+ cells in ponatinib-treated splenocytes. The total percentage of (O) CD45+NLRP3+cells (P) CD11b+NLRP3+ cells (Q) IL-1β producing CD11B+ cells (CD11b+IL-1β+) and (R) NLRP3 expressing myeloid secreting IL-1β (CD11b+NLRP3+IL-1β+). Experiments repeated 3 times (N= 3). Experiment was repeated three times (N= 3), and Data (A-L, O-R) were analyzed by using ordinary one way ANOVA followed by Tukey’s multiple comparisions test and represented as mean±SEM. (S, T) Splenocytes were obtained from mouse and treated with ponatinib for 72 hours. Cells were then stained with antibodies (CD11b, NLRP3 and IL-1β) and ImageStream analysis was performed. Representative ImageStream pictures of single cell staining showing ponatinib treatment upregulate NLRP3 expression in, (S) CD11b+ myeloid cells, and (T) IL-1β producing cells.
Fig. 5|. Ponatinib activates the formation of NLRP3 inflammasome.
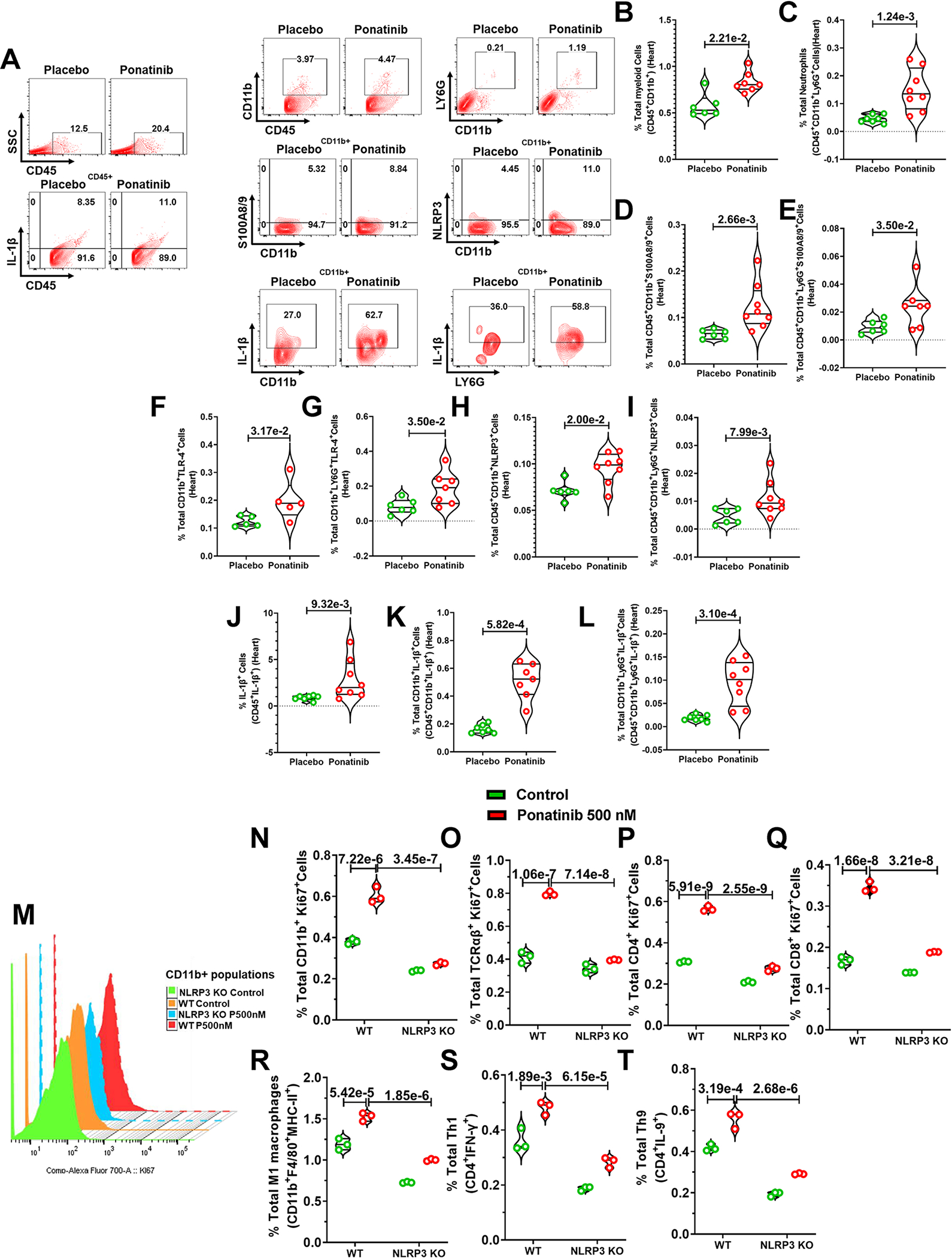
(A-L) Eight-week-old male and female C57Bl/6J mice were subjected to TAC surgery followed by ponatinib treatment (15 mg/Kg/day) for 4 weeks. Immune cells were isolated from the heart of TAC-placebo and TAC-ponatinib animal groups by enzymatic digestion after ponatinib treatment for 4 weeks. Next, cells were stained, and flow cytometry was performed. (A) Representative figure of flow cytometry showing gating strategy to measure IL-1β, CD11b, S100A8/9, and NLRP3 expressing immune cells. Data represents quantitation of percent of (B) total myeloid cells (CD45+CD11b+-), placebo (N=6) and ponatinib (N=7), (C) total neutrophils (CD45+CD11b+LY6G+), placebo (N=7) and ponatinib (N=8), (D) total S100A8/9 producing myeloid cells (CD45+CD11b+S100A8/9+), placebo (N=6) and ponatinib (N=8),(E) total S100A8/9 producing neutrophils (CD45+CD11b+LY6G+S100A8/9+), placebo (N=6) and ponatinib (N=7), (F) total TLR-4 expressing myeloid cells (CD11b+TLR-4+), placebo and ponatinib (N=5), (G) total TLR-4 expressing neutrophils (CD11b+LY6G+TLR-4+), placebo (N=6) and ponatinib (N=7), (H) total NLRP3+ myeloid cells (CD45+CD11b+NLRP3+),(I) total NLRP3+ neutrophils (CD45+CD11b+LY6G+NLRP3+), placebo (N=6) and ponatinib (N=8), (J) total IL-1β producing leukocytes (CD45+IL-1β+), placebo (N=6) and ponatinib (N=8), (K) total IL-1β producing myeloid cells (CD45+CD11b+IL-1β+), placebo and ponatinib (N=7), (L) total IL-1β producing neutrophils (CD45+CD11b+LY6G+IL-1β+), placebo (N=7) and ponatinib (N=8). Data (B-L) were analyzed using the Mann-Whitney U test and represented as mean±SEM. (M-T) Immune cells from NLRP3 KOs are resistant to ponatinib-induced proliferation. Splenocytes from NLRP3 KO and WT mice were treated with control (DMSO) and ponatinib (500 nM, 72hrs). Flow cytometry analyzed for the proliferation of myeloid cells and T cells with the staining of Ki67. (M) Staggered overlay diagram showing reduced proliferation in KO myeloid cells with ponatinib treatment. (N) Diminished ex vivo proliferation of CD11b+ myeloid cells in KO compared to WT. (O) Diminished ex vivo proliferation of TCRαβ+ T cell in KO compared to WT. (P) Diminished ex vivo proliferation of CD4+ T cell in KO compared to WT. (Q) Diminished ex vivo proliferation of CD8+ T cell in KO compared to WT. Decreased frequency of total (R) M1 macrophages, (S) Th1 cells, and (T) Th9 cells KO compared to WT. Experiment was repeated three times (N= 3). Data were analyzed by using ordinary two way ANOVA followed by Sidak’s multiple comparision test and represented as mean±SEM.
Fig. 6|. Ponatinib-induced myeloid cell proliferation is S100A8/9, TLR-4, and NLRP3 dependent, while Ponatinib concurrently activates human PBMCs with anticancer efficacy.
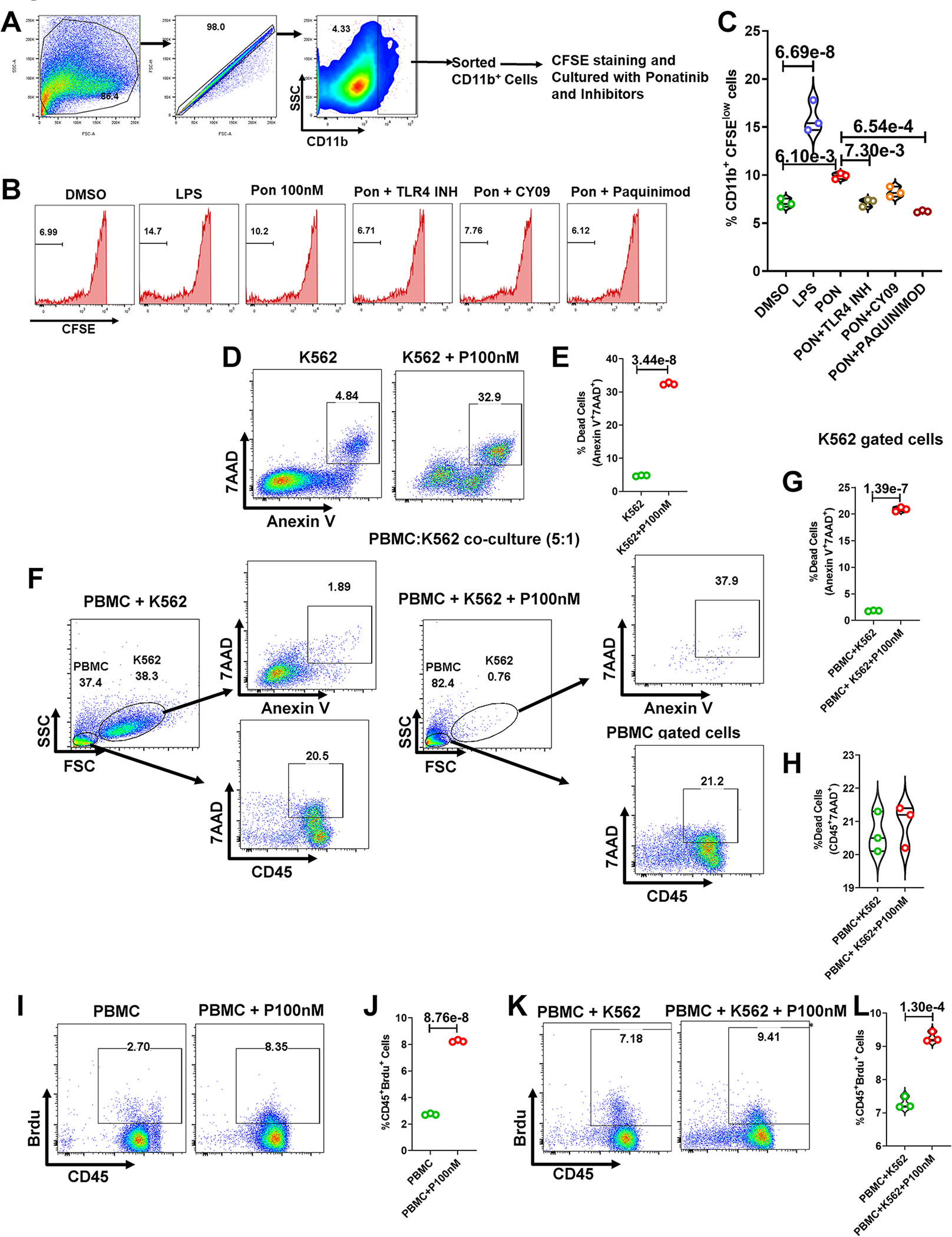
(A) Gating strategy for sorting CD11b+ cells from mouse splenocytes. (B) CFSE stained CD11b+ cells were cultured and treated with combination of antagonists and ponatinib for 72 hours (DMSO as control, LPS, Ponatinib 100nM, Ponatinib 100nM+ TLR4 inhibitor, Ponatinib 100nM+ CY09, Ponatinib 100nM+ Paquinimod). Histograms representing the percentage of diluted CFSE-stained CD11b+ cells. (C) Quantification of CD11b+ proliferation (percentage of CFSE low CD11b+ cells). Experiment was repeated three times (N= 3). Data were analyzed by using ordinary one way ANOVA followed by Tukey’s multiple comparisions test and represented as mean±SEM. (D) Human CML cells K562 were treated with ponatinib (100nM) for 72 hours. Next, cells were stained with Annexin V and 7AAD, and flow cytometry was performed. (E) Violin plot showing the percentage of dead K562 cells after ponatinib treatment. (F) Human K562 CML cells and PBMCs were co-cultured (1:5) and treated with ponatinib. Representative flow cytometry figures show gating strategy to measure dead cells in co-culture. (G-H) Data represents the quantitation of the percent of K562 dead cells and PBMCs. (I) Human PBMCs were treated with ponatinib (100nM) for 72 hours and BrdU was incorporated to culture 24 hours prior to flow cytometry analysis. (J) Violin plot shows the percent of proliferating immune cells (CD45+Brdu+). (K-L) Data represents a proliferation of immune cells in co-culture of human PBMCs and K562 cells post ponatinib treatment (100nM, 72 hours). Experiment was repeated three times (N= 3). Data (E, G, H, J, and L) were analyzed using the unpaired t test and represented as mean±SEM.
We and others have reported the ponatinib direct effect on cardiomyocyte death.7, 8 Therefore, it is conceivable that ponatinib treatment may lead to cardiomyocyte death or cardiac endothelial cell injury, which may attract immune cells or act as DAMPs to produce alarmins. To determine the effect of ponatinib on cardiomyocyte death, we performed TUNEL assay on cardiac tissue sections from ponatinib- and placebo-treated animals (2 weeks & 4 weeks post-TAC). Indeed, we did not observe measurable cardiomyocyte death at any of these time points (Figure. S13A–C). We also investigated the potential apoptotic death of leukocytes (CD45+tive) and other cardiac cells (CD45-tive cells) by performing Annexin/7AAD staining and flow cytometry analysis of single-cell preparation from myocardial tissue. Indeed, the pre-apoptotic and apoptotic cells were not significantly different in the ponatinib and placebo-treated hearts (Figure. S13D–G). Next, we turned our focus to endothelial cell injury, which could account for ponatinib-induced inflammation. The heart sections of ponatinib-treated TAC animals were stained with CD31 to quantify vessel densities in an in vivo setting. Interestingly, there was no difference in the vessel densities of control vs. ponatinib-treated hearts (Figure. S14A–C). Furthermore, the total endothelial cell numbers and death in the myocardium of ponatinib-treated hearts from naïve C57BL/6 animals were not statistically significant (Figure. S15A–C). Indeed, these findings were also consistent in the TAC studies (Figure. S15D–G). Taken together, these findings suggest that adverse effect on vasculature plays a minimal role in developing ponatinib-induced cardiac dysfunction. That said, further experimentation and functional analysis are required to fully establish the ponatinib effects on endothelial cell function and its potential role in cardiotoxicity. To further delineate the primary source of ponatinib-induced alarmins and inflammatory signaling, S100A8/9 and NLRP3-producing endothelial cells and fibroblasts were analyzed in the myocardium of ponatinib treated TAC mice. Indeed, there was no difference in S100A8/9 and NLRP3 expressing endothelial cells and fibroblasts (Figure. S16A–D and Fig. 17A–D). In our continued effort to investigate the primary source of ponatinib-induced alarmins, an ELISA assay was performed to quantify the S100A8/9 levels in the culture medium of ponatinib-treated cardiomyocytes. The S100A8/9 levels in ponatinib and vehicle-treated culture medium were not statistically different (Figure. S17E). These data collectively exclude the possibility of endothelial cells, fibroblasts, and cardiomyocytes as primary sources of alarmins and NLRP3 in ponatinib-treated animals.
Our unbiased RNAseq analysis indicated altered reactive oxygen species (ROS) pathways in ponatinib-treated hearts (Figure 1K–1N). Therefore, we next planned to determine mitochondrial ROS production in leukocytes, and non-leukocytes cell populations in ponatinib-treated TAC hearts. Notably, ponatinib-induced increased ROS production was limited to leukocytes, suggesting immune cell-specific ROS production (Figure. S18A–C). Additionally, ponatinib did not affect the ROS level in cardiomyocytes (Figure. S18D). Indeed, activated immune cells are known for increased ROS production.51, 52 Taken together, these findings exclude the potential involvement of cardiomyocytes and reinstate that immune cells mediated mechanism as the primary driver of ponatinib-mediated cardiotoxicity.
Ponatinib concurrently inhibits cancer cells but activates immune cells
We next sought to investigate ponatinib’s anticancer efficacy and its potential to induce immune activation in a co-culture model of cells of human origin. We co-cultured human PBMCs and human K562 CML cells (5:1) and treated them with ponatinib (100nM, 72 hrs) followed by cell death analysis using Annexin-7AAD flow cytometry analysis. Indeed, ponatinib was killing the cancer cells as evidenced by a dramatic increase in K562 cell apoptosis (AnnexinV+7AAD+), leading to a sharp decrease in the K562 gated population (Fig. 6D–G). On the other hand, as identified by increased BrdU incorporated CD45+ PBMCs, ponatinib induced the proliferation of immune cells in the same culture (Fig. 6H–L). These studies suggest that our mouse model’s findings are consistent with the human cell-based co-culture model. Furthermore, these findings indicate that two independent mechanisms potentially mediate the ponatinib’s ability to kill cancer cells and induce immune cells. Therefore, interventions to modulate the excessive immune response should not interfere with the ponatinib’s cancer efficacy. Having that said, future studies with humanized CML models are warranted to achieve both these readouts simultaneously, namely cancer efficacy and cardiotoxicity.
Dexamethasone rescued ponatinib-induced cardiotoxicities by suppressing excessive inflammation.
Our robust findings with multiple models strongly implicate excessive inflammation and cytokine elaboration as critical factors in ponatinib-induced cardiac dysfunction. Therefore, we hypothesized that preventing inflammation would mitigate ponatinib-mediated cardiotoxicity. Corticosteroids remain the cornerstone in the current management of immune-related adverse events due to the fast onset of action and high efficacy.53, 54 The corticosteroid dexamethasone has been used in leukemia patients for its anti-cancerous and immunosuppressive properties.55, 56 However, it has never been assessed in the context of its immunosuppressive action to mitigate cardiac adverse events in these patients. Therefore, for proof of concept, we tested whether dexamethasone would rescue ponatinib-induced cardiac inflammation and dysfunction. As shown in Fig. 7A, we treated TAC mice with dexamethasone (2 mg/kg i.p.) three times a week, in parallel with ponatinib (15 mg/kg/day) treatment. As reflected by the relative preservation of cardiac function, dexamethasone prevented ponatinib-induced cardiac dysfunction. (Fig. 7B–C). To our complete surprise, dexamethasone alone did not show a significant anti-inflammatory effect compared to the placebo group. However, animals treated with both dexamethasone and ponatinib exhibited a dramatically diminished frequency of IL6 producing CD45+ and CD11b+ cells in the heart (Figure. S19A–B). Moreover, dexamethasone treatment inhibited the ponatinib-induced TLR4-NLRP3-IL1β response in cardiac myeloid cells (Figure. S19C–E). These findings suggest that dexamethasone alleviates ponatinib-induced inflammation by inhibiting the NLRP3 inflammasome signaling. Furthermore, dexamethasone dramatically protected against ponatinib-induced myocardial T-cell activation (Figure. S19F–G). Consistently, the animals treated with both dexamethasone and ponatinib exhibited drastically decreased systemic myeloid cells, macrophages, pro-inflammatory M1 macrophages, and TNF-α producing myeloid cells compared to animals that only received ponatinib (Figure. S19H–K). Next, we sought to examine the effect of dexamethasone treatment on ponatinib-mediated NLRP3 expression. Indeed, dexamethasone attenuated the ponatinib-induced NLRPR3 expression (Figure. S19L). Consistently, dexamethasone also significantly diminished ponatinib-induced splenic T-cell expansion (Figure. S19M). Altogether, these findings indicate that dexamethasone prevents ponatinib-induced excessive inflammation, and thereby protects against ponatinib-induced cardiotoxicity.
Fig. 7|. Dexamethasone and direct NLRP3 inflammasome inhibitor CY-09 rescued ponatinib-induced cardiotoxicities by suppressing excessive inflammation.
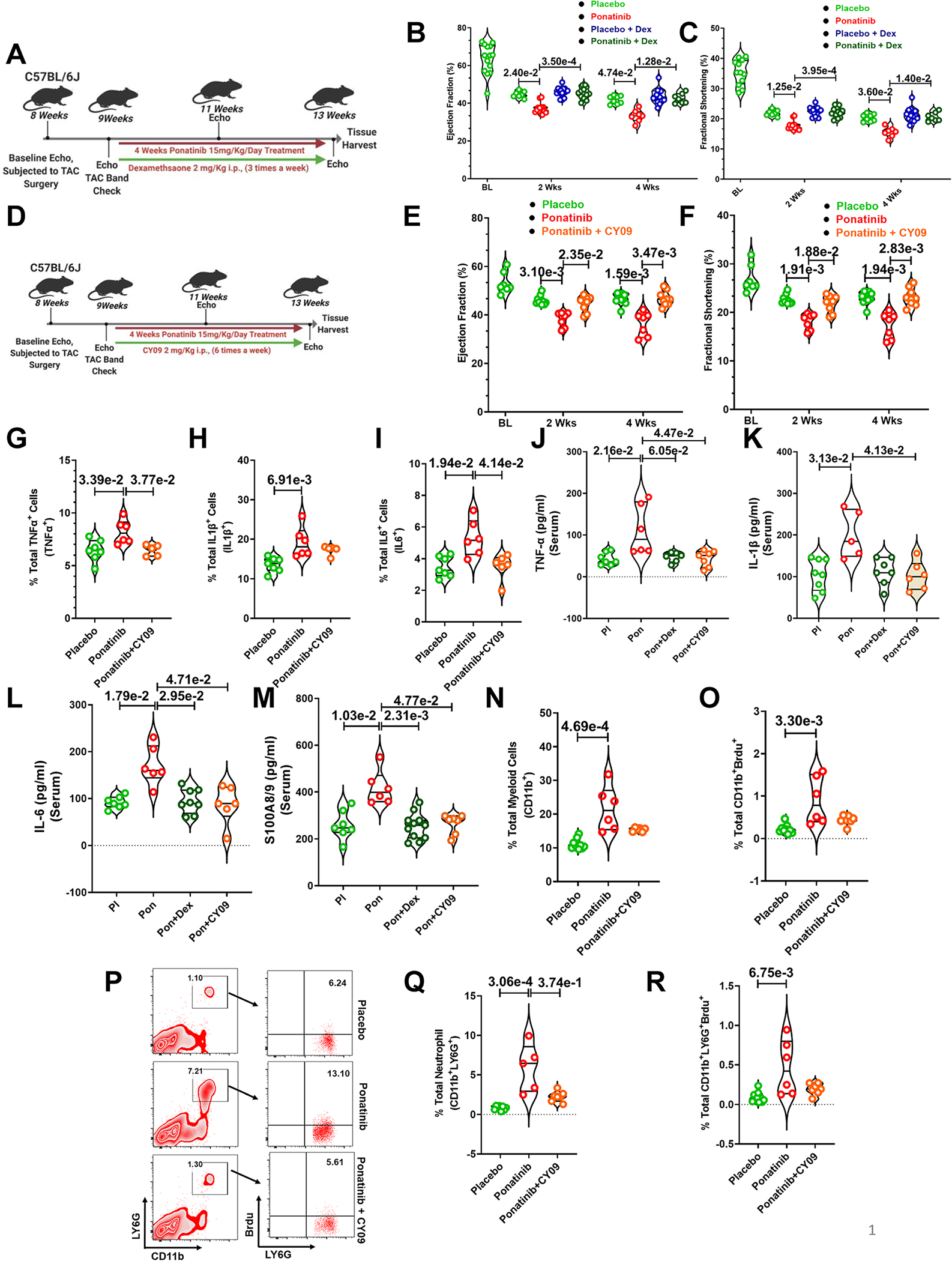
(A) Schematic outline of experiments performed in this figure. Eight-week-old C57BL6 wild-type (WT) mice were subjected to TAC surgery followed by ponatinib treatment (15 mg/Kg/day) after 1 week. Mice were given dexamethasone (2 mg/Kg i.p.) three times a week and concurrently ponatinib for four weeks. Only TAC groups have been taken for this study. (BC) Ejection Fraction and Fractional Shortening as measured by echocardiography at 2 and 4 weeks. (B) Ejection Fraction, Basal (BL) (N=14); At 2 weeks, placebo (N=7), ponatinib (N=11), placebo Dex (N=14), and ponatinib+ Dex (N=13); At 4 weeks, placebo (N=8), ponatinib (N=7), placebo Dex (N=14) and ponatinib+ Dex (N=7) (C) Fractional Shortening, Basal (BL) (N=12); At 2 weeks, placebo (N=7), ponatinib (N=13), placebo Dex (N=14) and ponatinib+ Dex (N=13); At 4 weeks, placebo (N=8), ponatinib (N=7), placebo Dex (N=14) and ponatinib+ Dex (N=7). Significance was compared between TAC placebo vs TAC ponatinib and TAC ponatinib vs TAC ponatinib + Dex for 2 weeks and 4 weeks separately by using Kruskal-Wallis followed by Dunn test and represented as mean±SEM. (D) Experimental overview. Eight-week-old C57BL6 wild-type (WT) mice were subjected to TAC surgery. After 1 week, mice were given CY-09 (2 mg/Kg i.p.) six times a week and concurrently ponatinib for four weeks (15 mg/Kg/day). Only TAC groups have been taken for this study. (E-F) Ejection Fraction and Fractional Shortening as measured by echocardiography at 2 and 4 weeks. Basal (BL) (N=7); At 2 weeks, placebo (N=10), ponatinib (N=7) and ponatinib+CY09 (N=8); At 4 weeks, placebo (N=10), ponatinib (N=7), and ponatinib+CY09 (N=9). Significance was compared between TAC placebo vs TAC ponatinib and TAC ponatinib vs TAC ponatinib + CY09 for 2 weeks and 4 weeks separately by using Kruskal-Wallis followed by Dunn test and represented as mean±SEM. (G-I) Quantitation of immune cells as a percentage of total cells isolated from the heart of animal groups mentioned in the figures. Data represents quantitation of percent of the total (G) TNF-α producing leukocytes (CD45+TNF-α+); placebo (N=7), ponatinib (N=6), and ponatinib+CY09 (N=5), (H) IL-1β producing leukocytes (CD45+IL-1β+); placebo (N=7), ponatinib (N=6), and ponatinib+CY09 (N=5), (I) IL-6 producing leukocytes (CD4+IL6+); placebo (N=7), ponatinib (N=6), and ponatinib+CY09 (N=6). (J-M) ELISA was performed from the serum obtained from animal groups. Data showing serum levels of (J) TNF-α; placebo (N=7), ponatinib (N=6), ponatinib + Dex (N=6) and ponatinib+CY09 (N=7), (K) IL-1β; placebo (N=8), ponatinib (N=5), ponatinib + Dex (N=7) and ponatinib+CY09 (N=6),(L) IL-6; placebo (N=7), ponatinib (N=6), ponatinib + Dex (N=8) and ponatinib+CY09 (N=6), (M) S100A8/9; placebo (N=7), ponatinib (N=6), ponatinib + Dex (N=11) and ponatinib+CY09 (N=7). (N-R) Quantitation of immune cells as a percentage of total cells isolated from spleen of animal groups mentioned in the figures. Data represents % total of (N) myeloid cells (CD11b+); placebo (N=10), ponatinib (N=6), and ponatinib+CY09 (N=6), (O) proliferating myeloid cells (CD11b+Brdu+); placebo (N=9), ponatinib (N=6), and ponatinib+CY09 (N=7), (P) Flow cytometry plots representing neutrophil populations in spleen and a total percentage of (Q) neutrophils (CD11b+LY6G+); placebo (N=8), ponatinib (N=5), and ponatinib+CY09 (N=7), and (R) proliferating neutrophils (CD11b+LY6G+Brdu+); placebo (N=10), ponatinib (N=6), and ponatinib+CY09 (N=7). Data (G-I) and (Q-R) were analyzed by using Kruskal-Wallis followed by Dunn test and represented as mean±SEM.
NLRP3 inflammasome drives ponatinib-induced cardiotoxicity.
The rescue studies with broad immunosuppressive dexamethasone clearly established the crucial role of excessive inflammation in ponatinib-induced cardiac dysfunction. Consistently, our mechanistic studies indicated the critical involvement of NLRP3 inflammasome in ponatinib-mediated cardiotoxicity. Based on these observations, we hypothesize that NLRP3 inflammasome drives ponatinib-induced cardiotoxicity. To test this hypothesis, we planned to perform two independent in vivo rescue experiments, one with CY-09, a direct inhibitor of NLRP3 57, and another with paquinimod, a specific inhibitor of alarmins (S100A9). The experimental design of CY-09 studies is depicted in Fig. 7D. In brief, we treated TAC mice with CY-09 (2 mg/kg i.p.) six times a week along with ponatinib. Indeed, intervention with CY-09 nearly abolished the detrimental cardiac effect of ponatinib (Fig. 7E–F). As anticipated, CY-09 treatment reduced the frequency of TNF-α, IL-1β and IL-6-producing cells in the myocardium (Fig. 7G–I). Consistently, ponatinib-induced plasma TNF-α, IL-1β, IL-6, and S100A8/9 levels were significantly reduced by CY-09 treatment (Fig. 7J–M). As expected, CY-09 treatment also reduced the proliferation of myeloid cells and neutrophils in spleens, demonstrating reduced systemic inflammation in CY-09-treated animals (Fig. 7N–R). Furthermore, CY-09 treatment alleviated the ponatinib-induced macrophage induction, T cell proliferation, and inflammatory monocytes (Figure. S20).
Considering the high translational potential of the current studies, we also investigated the potential metabolic consequences of proposed interventions, specifically dexamethasone and CY-09, in the setting of ponatinib treatment. Notably, ponatinib itself did not have any effect on plasma glucose and cholesterol levels, suggesting ponatinib exerts its cardiotoxic effects independent of these metabolic changes (Figure. S20). As steroids are known for their adverse effect on cholesterol levels, dexamethasone intervention on ponatinib-treated animals showed elevated plasma cholesterol levels compared to ponatinib and placebo groups (Figure. S21). Remarkably, analysis of serum glucose and cholesterol levels revealed a favorable metabolic effect of CY-09 intervention. The improved plasma glucose and cholesterol by CY-09 were not too surprising as it’s reported that CY-09 reverses metabolic disorders in diabetic animals.57 Thus, CY-09 treatment successfully mitigated the ponatinib-induced adverse cardiac effects without unwanted metabolic complications, as seen with dexamethasone.
In the rescue experiment with S100A9 inhibitor paquinimod, we used a very similar experimental design as CY-09 studies. TAC animals were treated with paquinimod ad libitum in drinking water daily58 along with ponatinib (Fig. 8A). As expected, concurrent treatment of paquinimod along with ponatinib preserved the cardiac dysfunction caused by ponatinib (Fig. 8B–C). We next assessed serum levels of S100A8/9 and found that paquinimod treatment significantly reduced the concentration of S100A8/9 in the ponatinib+paquinimod-treated group (Fig. 8D). Consistently, paquinimod reduced the frequency and proliferation of immune cells in the heart (Fig. 8E–O) and spleen (Fig. 8P–W, Figure. S22, 23).
Fig. 8|. Inhibition of S100A8/9 is critical to protect against ponatinib-induced cardiotoxicity.
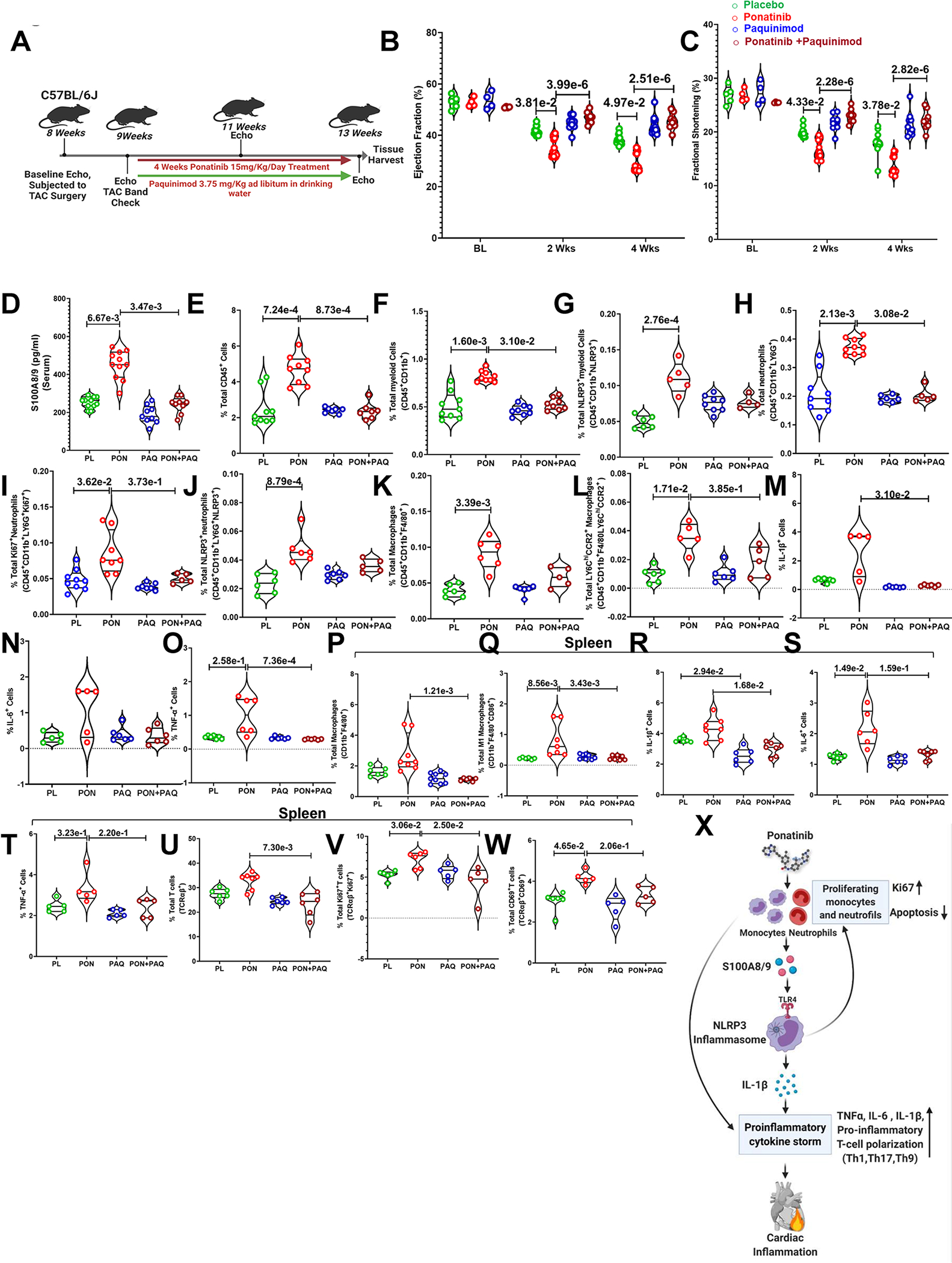
(A) Experimental overview. Eight-week-old C57BL6 wild-type (WT) mice were subjected to TAC surgery. After 1 week, mice were given paquinimod (3.75 mg/Kg weight ad libitum in drinking water) and concurrently ponatinib for four weeks (15 mg/Kg/day). Only TAC groups have been taken for this study. (B-C) Ejection Fraction and Fractional Shortening as measured by echocardiography at 2 and 4 weeks. (B) Ejection Fraction - At Base line (BL), placebo (N=5), ponatinib (N=4), paquinimod (N=4) and ponatinib+ paquinimod (N=4); At 2 weeks, placebo (N=13), ponatinib (N=12), paquinimod (N=9) and ponatinib+ paquinimod (N=8); At 4 weeks, placebo (N=13), ponatinib (N=12), paquinimod (N=10) and ponatinib+ paquinimod (N=8). (C) Fractional Shortening; At Basal (BL), placebo (N=5), ponatinib (N=4), paquinimod (N=4) and ponatinib+ paquinimod (N=4); At 2 weeks, placebo (N=13), ponatinib (N=13), paquinimod (N=9) and ponatinib+ paquinimod (N=8); At 4 weeks, placebo (N=13), ponatinib (N=12), paquinimod (N=10) and ponatinib+ paquinimod (N=8). Significance was compared between TAC placebo vs TAC ponatinib and TAC ponatinib vs TAC ponatinib + paquinimod for 2 weeks and 4 weeks separately by using Kruskal-Wallis followed by Dunn test and represented as mean±SEM. (D) Data showing the concentration of S100A8/9 in serum placebo (N=5), ponatinib (N=4), paquinimod (N=4), and ponatinib + paquinimod (N=4) placebo (N=11), ponatinib (N=11), paquinimod (N=9), and ponatinib + paquinimod (N=9). (E-O) The figures mention quantitation of immune cells as a percentage of total cells isolated from the heart of animal groups. Data represents % total of (E) total leukocytes (CD45+); placebo (N=9), ponatinib (N=9), paquinimod (N=7), and ponatinib + paquinimod (N=7), (F) total myeloid cells (CD45+CD11b+); placebo (N=9), ponatinib (N=9), paquinimod (N=8), and ponatinib+ paquinimod (N=8), (G) total NLRP3+ myeloid cells (CD45+CD11b+NLRP3+); placebo (N=6), ponatinib (N=5), paquinimod (N=8), and ponatinib + paquinimod (N=4), (H) total neutrophils (CD45+CD11b+LY6G+); placebo (N=9), ponatinib (N=9), paquinimod (N=6), and ponatinib + paquinimod (N=5) (I) total proliferating neutrophils (CD45+CD11b+LY6G+Ki67+); placebo (N=9), ponatinib (N=5), paquinimod (N=7) and ponatinib+ paquinimod (N=5), (J) total NLRP3+ neutrophils (CD45+CD11b+LY6G+NLRP3+); placebo (N=6), ponatinib (N=6), paquinimod (N=7) and ponatinib+ paquinimod (N=5) (K) total macrophages (CD45+CD11b+F4/80+); placebo (N=6), ponatinib (N=6), paquinimod (N=6) and ponatinib + paquinimod (N=5), (L) total CCR2+ macrophages (CD45+CD11b+F4/80+Ly6Chi+CCR2+); placebo (N=6), ponatinib (N=5), paquinimod (N=6) and ponatinib+ paquinimod (N=5), (M) total IL-1β producing cells (IL-1β+); placebo (N=7), ponatinib (N=5), paquinimod (N=6) and ponatinib+ paquinimod (N=6), (N) total IL-6 producing cells (IL-6+); placebo (N=5), ponatinib (N=5), paquinimod (N=6), and ponatinib+ paquinimod (N=6), and (O) total TNF-α producing cells (TNF-α+); placebo (N=7), ponatinib (N=6) paquinimod (N=6), and ponatinib+ paquinimod (N=6). (P-W) Quantitation of immune cells as a percentage of total cells isolated from the spleen of animal groups mentioned in the figures. Data represents quantitation of percent of (P) total macrophages (CD11b+F4/80+); placebo (N=6), ponatinib (N=8), paquinimod (N=9), and ponatinib + paquinimod (N=6), (Q) total M1 macrophages (CD11b+F4/80+CD86+); placebo (N=6), ponatinib (N=7), paquinimod (N=8), and ponatinib + paquinimod (N=7) (R) total IL-1β producing cells (IL-1β+); placebo (N=6), ponatinib (N=7), paquinimod (N=6), and ponatinib + paquinimod (N=7). (S) total IL-6 producing cells (IL-6+); placebo (N=7), ponatinib (N=6), paquinimod (N=6), and ponatinib+ paquinimod (N=7), and (T) total TNF-α producing cells (TNF-α+); placebo (N=5), ponatinib (N=5), paquinimod (N=5), and ponatinib+ paquinimod (N=5), (U) total T-cells (TCRαβ+); placebo (N=6), ponatinib (N=7), paquinimod (N=6), and ponatinib+ paquinimod (N=6), (V) total proliferating T-cells (TCRαβ+Ki67+); placebo (N=6), ponatinib (N=6), paquinimod (N=5), and ponatinib+ paquinimod (N=5), and (W) total activated T-cells (TCRαβ+CD69+); placebo (N=6), ponatinib (N=5), paquinimod (N=5), and ponatinib+ paquinimod (N=5). Data (D-W) were analyzed by using Kruskal-Wallis followed by Dunn test and represented as mean±SEM. (X) Schematic depicting findings of the study.
Finally, we investigated if the proposed pharmacological agents to mitigate cardiotoxicity interfere with ponatinib’s anti-cancerous efficacy. To address this concern, we treated human CML K562 cells with CY-09, ponatinib, and paquinimod, along with control groups. Indeed, CY-09 or paquinimod treatment did not affect the ponatinib-induced apoptosis of CML K562 cells. These data strongly advocate that NLRP3 or alarmin inhibitors do not interfere with ponatinib efficacy (Fig. S24).
In summary, our findings confirmed the driving role of the ponatinib-induced S100A8/9-NLRP3 pro-inflammatory axis in ponatinib-mediated cardiotoxicity; therefore, strategies to target this signaling axis could be novel therapeutic targets to prevent ponatinib-associated adverse cardiac events.
Discussion:
This study identified a novel mechanism of ponatinib-mediated cardiotoxicity, the only treatment option for CML patients with the T315I mutation. Specifically, we demonstrate that ponatinib-induced excessive inflammation is crucial to driving the adverse cardiotoxic effects. Mechanistically, we identified the vital role of the S100A8/A9-TLR4-NLRP3-IL-1β signaling circuit in ponatinib-mediated inflammation and cardiac dysfunction. As depicted in the schematic figure (Fig. 8X), ponatinib induces S100A8/A9 production, which primes the NLRP3 inflammasome, and stimulates the release of proinflammatory IL-1β. The inflammation hypothesis is strongly supported by three independent rescue studies with the immunosuppressive corticosteroid, dexamethasone, CY-09, a direct inhibitor of NLRP3 inflammasome, and paquinimod, an inhibitor of S100A9. Thus, we have discovered a previously unknown effect of ponatinib on immune cell activation and inflammation, which predisposes the development of ponatinib-induced cardiac dysfunction.
Recent clinical reports and meta-analysis studies of patients treated with TKIs indicate that those with CVD comorbidities are highly susceptible to TKI-induced cardiac dysfunction.32 Keeping this in mind, we used cardiovascular comorbidity mouse models, which allowed us to expose the “hidden cardiotoxicity” of ponatinib. Historically, the cardiac safety data of TKIs from the clinical development stage is always under-reported.59–61 This is primarily due to most clinical trials done prior to FDA approval excluding patients with comorbidities or pre-existing cardiovascular disease (CVD). However, such patients are frequently treated post-FDA approval.62 In this regard, very few cases of cardiotoxicity were reported in the early clinical trials of ponatinib. However, the additional follow-up has revealed a higher frequency of serious adverse cardiac events.2, 6, 63–66 Moreover, pharmacovigilance analysis of adverse reports from the real-world patient population has revealed that ponatinib is the most cardiotoxic agent among all FDA-approved TKIs.67 Thus, the CV comorbidity mouse models employed herein more closely mimic the clinical situation of cancer patients with co-existing cardiovascular disease. We strongly advocate using similar CV comorbidity models in future studies to expose the hidden cardiotoxicity of other TKIs and agents. Our findings are also consistent with the multi-hit hypotheses68 that naïve mice treated with ponatinib showed no evidence of cardiotoxicity despite a robust subclinical inflammation (without any cardiac pathologies). However, this subclinical inflammation predisposes these animals to develop accelerated heart failure due to CVD comorbidities. Hence, the likelihood of developing ponatinib-induced cardiotoxicity is substantially higher in patients with cardiovascular comorbidities, and this group should be more closely monitored for potential adverse cardiac effects. Of note, the vascular effects of ponatinib are known and well-studied. Herein, we have focused on its adverse cardiac effects. Notably, the cardiac vascular density and endothelial cells were comparable in ponatinib and placebo groups in the current experimental setting. Thus, the ponatinib cardiotoxicity was independent of the potential effect on cardiac vasculature. However, it is conceivable that ponatinib’s extra-cardiac vascular effects might be playing a part in the development of cardiac dysfunction. Nonetheless, we cannot stress enough that the real-world patient population is often complicated by multiple comorbidities; thus, the in vivo models employed herein are more representative of the actual clinical scenarios. This is further supported by the fact that cancer and CVD share several common risk factors.69, 70
The lack of effective treatments to overcome ponatinib-induced cardiac adverse events stem from an incomplete understanding of the underlying mechanisms. All previous studies focusing on identifying mechanisms for ponatinib cardiotoxicity were limited by exclusive reliance on cell culture models.7–9 Therefore, identifying a mechanism that involves crosstalk of multiple systems and systemic effects was not possible. Herein, our in vivo setting with multiple CV comorbidity models allowed us to discover the driving role of inflammation in ponatinib-mediated cardiotoxicity. Unbiased RNA-Seq analysis initially indicated the involvement of inflammation in ponatinib-mediated cardiotoxicity. Consistently, ponatinib induced cardiac infiltration and proliferation of multiple immune cells, including myeloid cells, neutrophils, and T cells (Th1, Th17, and Th9). Of note, the role of Th9 cells and their primary cytokine IL-9 in cardiac remodeling is understudied and needs further investigation. In stark contrast to our findings, Chen et al.71 employed the influenza A/PR8 virus infection model to demonstrate that ponatinib protects from influenza infection by suppressing cytokine storms. However, it’s important to note that Chen et al. used the BALB/c animals for an infection-induced inflammation model, and herein, we employed the C57B6 mouse for a sterile inflammation model. Thus, the two studies’ mouse backgrounds and experimental settings are quite different; therefore, the findings are not directly comparable. In our model, the inflammation hypothesis of ponatinib cardiotoxicity is further supported by numerous findings, including differentiation of monocytes to proinflammatory monocytes and macrophages, increased production of monocytes and macrophages in BM and spleen, increased expression of ICAM-1 in splenic monocytes and neutrophils, and T cell polarization into pro-inflammatory T cells. Taken together, these findings support the notion that ponatinib promotes excessive inflammation by activating both innate and adaptive immune responses. In future studies, it is attractive to employ one of the upcoming technologies to directly image the inflamed myocardium without any downstream processing.72
RNA-Seq analysis, combined with bulk flow cytometry and single-cell image stream validation revealed the multiple-fold induction of the proinflammatory alarmins S100A8 and S100A9 in ponatinib-treated mice. In fact, along with known cardiac remodeling markers (ANP, TIMP, etc.), S100A8 and S100A9 were the top hits (>10-fold upregulation) in the RNA-Seq dataset. The S100A8/9 heterodimers are more prevalent and known to activate the NLRP3 inflammasome pathway. Indeed, alarmin-primed NLRP3 inflammasome activation is a critical mediator of myocardial and systemic inflammation.23 Our findings using multiple experimental approaches support the hypothesis that ponatinib directly activates myeloid cells and neutrophils, leading to enhanced secretion of the alarmins S100A8/9, which is vital for ponatinib-mediated myocardial and systemic inflammation. S100A8 and S100A9 activate the pattern recognition receptor TLR-4 that further inducing sterile inflammatory signals.73 Increased expression of TLR4 over myeloid cells and neutrophils further supports the S100A8/9-TLR4 mediated excessive inflammatory response in ponatinib-treated animals. Mechanistic studies with multiple agonists or antagonists were employed to establish the critical role of this signaling circuit in ponatinib-mediated excessive inflammation. Furthermore, the ponatinib-mediated inflammatory response was remarkably attenuated in the NLRP3 deficient cells, confirming the crucial role of this signaling axis in ponatinib-mediated excessive inflammation. Therefore, it’s not surprising that multiple studies have proposed targeting this pathway as a novel therapeutic approach for preventing heart failure.18, 19, 44 Additionally, this signaling axis is critical for promoting chemotaxis and immune cell proliferation. However, future studies with approaches to precisely ablate the specific immunocyte (e.g., neutrophils or macrophages) are warranted to establish the cause-and-effect relationship of specific immune cell population recruitment in the heart tissue.
After establishing the ponatinib-mediated cardiac inflammation, we rescued this adverse effect using immunosuppressive strategies. The corticosteroid dexamethasone has been used previously with CML tyrosine kinase inhibitors and in patients who have undergone allogeneic stem cell transplantation.56 This prompted us to use dexamethasone as a concurrent treatment during ponatinib administration. Indeed, dexamethasone suppressed the ponatinib-induced excessive immune response and rescued cardiac dysfunction. Thus, it is reasonable to conclude that strategies for managing excessive inflammation will potentially protect the heart from ponatinib-induced cardiotoxicity. Encouragingly, as per the ongoing clinical trial (ClinicalTrials.gov Identifier: NCT03576547), a combination of ponatinib with dexamethasone and other chemotherapy agents appears safe.74 As this trial is still recruiting, the final outcome will be critical to establishing the safety and efficacy of this combination. Moreover, dexamethasone was sufficient to manage the “cytokine release syndrome (CRS)” in a patient treated with a combination therapy of ponatinib and blinatumomab.75 Of course, we are well aware of the concerns associated with long-term corticosteroid treatment. Remarkably, additional recuse studies with NLRP3 inhibitor CY-09 or alarmin inhibitor paquinimod revealed that it could successfully mitigate the ponatinib-induced adverse cardiac effects without unwanted metabolic complications seen with dexamethasone. These intriguing findings warrant further investigation to employ immunomodulatory strategies to combat ponatinib cardiotoxicity. Of note, Paquinimod was well tolerated in humans with only mild to moderate adverse effects.76, 77 Having that said, it’s important to note that the precise mechanism of how inflammation leads to cardiac dysfunction is not entirely known and is currently under intense investigation by multiple groups.
In conclusion, we have identified a novel and central role for excessive inflammation in regulating adverse remodeling and dysfunction in ponatinib-treated hearts. Ponatinib activates the myeloid cell inflammasome pathway by increasing the production of alarmins S100A8/9, leading to the activation of the proinflammatory S100A8/A9-TLR4-NLRP3-IL-1β signaling axis. We clearly demonstrate that ponatinib-induced excessive inflammation is the primary mechanism leading to adverse cardiac remodeling and ventricular dysfunction. This conclusion is strongly supported by our studies with the broad-spectrum immunosuppressive glucocorticoid dexamethasone and direct NLRP3 inhibitor, which rescued the detrimental phenotype observed in ponatinib-treated hearts. Our findings suggest that patients on ponatinib should be followed closely for excessive inflammation, cytokine elevation, and cardiac dysfunction. Therefore, immunosuppressive medication could be further optimized and tested to manage ponatinib-mediated cardiac complications in CML patients.
Supplementary Material
Novelty and Significance.
What Is Known?
Targeted therapies of tyrosine kinase inhibitors (TKIs) have significantly improved cancer treatment.
The adverse cardiotoxic effects of TKIs are a serious clinical concern.
Ponatinib is the most cardiotoxic among all FDA-approved TKIs in a real-world patient scenario. However, the mechanism of ponatinib-induced cardiotoxicity is unknown.
What New Information Does This Article Contribute?
Preexisting cardiovascular (CV) comorbidities are critical to developing ponatinib-induced cardiotoxicity.
S100A8/A9-NLRP3-IL-1β signaling circuit drives the ponatinib-induced excessive inflammation and cardiotoxicity.
Immunomodulatory interventions with dexamethasone or specific inhibitors of NLRP3 (CY-09) or S100A9 (paquinimod) rescued the ponatinib-induced cardiotoxicity.
These preclinical data provide the rationale for a clinical investigation into immunosuppressive interventions for ponatinib-induced cardiotoxicity.
Pharmacovigilance analysis of FDA and WHO datasets have established that ponatinib is the most cardiotoxic agent among all FDA-approved TKIs in a real-world patient scenario. However, the mechanism of ponatinib-induced cardiotoxicity is unknown, primarily because of the lack of well-optimized mouse models for preclinical cardio-oncology research. Herein, we have established mouse models of CV comorbidities showing robust cardiac phenotype. These newly optimized models will be critical to progressing the mechanistic preclinical research of additional TKIs-mediated cardiotoxicity. To this end, these CV comorbidity models facilitated us to discover S100A8/A9-NLRP3-IL-1β signaling axis as the primary driver of ponatinib-induced excessive inflammation and cardiotoxicity. Multiple rescue experiments with various immunosuppressive interventions confirmed the central role of excessive inflammation in ponatinib’s cardiotoxicity. Our findings provide the rationale for a clinical investigation into immunosuppressive interventions for managing ponatinib-induced cardiotoxicity.
Sources of funding
This work was supported by research grants to HL from the NHLBI (R01HL143074-01A1, R01HL133290), PU was supported by American Heart Association (19POST34460025).
Non-standard Abbreviations and Acronyms
- BrdU
Bromodeoxyuridine
- CFSE
Carboxyfluorescein Succinimidyl Ester
- CML
Chronic Myelogenous Leukemia
- CV
cardiovascular
- CVD
Cardiovascular Disease
- DC
Dendritic Cells
- DMSO
Dimethyl Sulfoxide
- ELISA
Enzyme-linked immunoassay
- EF
Ejection Fraction
- FBS
Fetal Bovine Serum
- FDA
Food and Drug Administration
- FS
Fractional Shortening
- HF
Heart Failure
- HFD
High-Fat Diet
- ICAM-1
Intracellular Cell Adhesion Molecule 1
- LPS
Lipopolysaccharides
- LV
Left Ventricle
- NLRP3
NLR Family Pyrin Domain Containing 3
- NRVMs
Neonatal Rat Ventricular Cardiomyocytes
- PBMCs
Peripheral Blood mononuclear cells
- RAGE
Receptors for Advanced Glycation End Products
- RNA-Seq
RNA Sequencing
- TAC
Transverse Aortic Constriction
- TKI
Tyrosine Kinase Inhibitor
- TLRs
Toll-like Receptors
- WHO
World Health Organization
Footnotes
Disclosures
None
Supplemental Materials:
References:
- 1.Douxfils J, Haguet H, Mullier F, Chatelain C, Graux C and Dogne JM. Association Between BCR-ABL Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia and Cardiovascular Events, Major Molecular Response, and Overall Survival: A Systematic Review and Meta-analysis. JAMA Oncol. 2016;2:625–632. [DOI] [PubMed] [Google Scholar]
- 2.Moslehi JJ and Deininger M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J Clin Oncol. 2015;33:4210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA 3rd, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MW, Druker BJ and Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoy SM. Ponatinib: a review of its use in adults with chronic myeloid leukaemia or Philadelphia chromosome-positive acute lymphoblastic leukaemia. Drugs. 2014;74:793–806. [DOI] [PubMed] [Google Scholar]
- 5.Dorer DJ, Knickerbocker RK, Baccarani M, Cortes JE, Hochhaus A, Talpaz M and Haluska FG. Impact of dose intensity of ponatinib on selected adverse events: Multivariate analyses from a pooled population of clinical trial patients. Leuk Res. 2016;48:84–91. [DOI] [PubMed] [Google Scholar]
- 6.Gainor JF and Chabner BA. Ponatinib: Accelerated Disapproval. Oncologist. 2015;20:847–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, Churko JM, Kitani T, Wu H, Holmstrom A, Matsa E, Zhang Y, Kumar A, Fan AC, Del Alamo JC, Wu SM, Moslehi JJ, Mercola M and Wu JC. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh AP, Glennon MS, Umbarkar P, Gupte M, Galindo CL, Zhang Q, Force T, Becker JR and Lal H. Ponatinib-induced cardiotoxicity: delineating the signalling mechanisms and potential rescue strategies. Cardiovasc Res. 2019;115:966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talbert DR, Doherty KR, Trusk PB, Moran DM, Shell SA and Bacus S. A multi-parameter in vitro screen in human stem cell-derived cardiomyocytes identifies ponatinib-induced structural and functional cardiac toxicity. Toxicol Sci. 2015;143:147–55. [DOI] [PubMed] [Google Scholar]
- 10.Adamo L, Rocha-Resende C, Prabhu SD and Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. 2020;17:269–285. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann U and Frantz S. How can we cure a heart “in flame”? A translational view on inflammation in heart failure. Basic Res Cardiol. 2013;108:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Menyar AA. Cytokines and myocardial dysfunction: state of the art. J Card Fail. 2008;14:61–74. [DOI] [PubMed] [Google Scholar]
- 13.Anand IS, Latini R, Florea VG, Kuskowski MA, Rector T, Masson S, Signorini S, Mocarelli P, Hester A, Glazer R, Cohn JN and Val-He FTI. C-reactive protein in heart failure: prognostic value and the effect of valsartan. Circulation. 2005;112:1428–34. [DOI] [PubMed] [Google Scholar]
- 14.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT and Anti TNFTACHFI. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–40. [DOI] [PubMed] [Google Scholar]
- 15.Ueland T, Gullestad L, Nymo SH, Yndestad A, Aukrust P and Askevold ET. Inflammatory cytokines as biomarkers in heart failure. Clin Chim Acta. 2015;443:71–7. [DOI] [PubMed] [Google Scholar]
- 16.Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N and Teichmann SA. Cells of the adult human heart. Nature. 2020;588:466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Bauersachs J and Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail. 2017;19:1379–1389. [DOI] [PubMed] [Google Scholar]
- 18.Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116:1254–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–17. [DOI] [PubMed] [Google Scholar]
- 20.Simard JC, Cesaro A, Chapeton-Montes J, Tardif M, Antoine F, Girard D and Tessier PA. S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-dependent activation of NF-kappaB(1.). PLoS One. 2013;8:e72138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, Quaife-Ryan GA, Al-Sharea A, Pernes G, Dragoljevic D, Lal H, Schroder K, Hanaoka BY, Raman C, Grant MB, Hudson JE, Smyth SS, Porrello ER, Murphy AJ and Nagareddy PR. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation. 2020;141:1080–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryckman C, Vandal K, Rouleau P, Talbot M and Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003;170:3233–42. [DOI] [PubMed] [Google Scholar]
- 23.Wang S, Song R, Wang Z, Jing Z, Wang S and Ma J. S100A8/A9 in Inflammation. Front Immunol. 2018;9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seruga B, Sterling L, Wang L and Tannock IF. Reporting of serious adverse drug reactions of targeted anticancer agents in pivotal phase III clinical trials. J Clin Oncol. 2011;29:174–85. [DOI] [PubMed] [Google Scholar]
- 25.Cross MJ, Berridge BR, Clements PJ, Cove-Smith L, Force TL, Hoffmann P, Holbrook M, Lyon AR, Mellor HR, Norris AA, Pirmohamed M, Tugwood JD, Sidaway JE and Park BK. Physiological, pharmacological and toxicological considerations of drug-induced structural cardiac injury. Br J Pharmacol. 2015;172:957–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richey EA, Lyons EA, Nebeker JR, Shankaran V, McKoy JM, Luu TH, Nonzee N, Trifilio S, Sartor O, Benson AB 3rd, Carson KR, Edwards BJ, Gilchrist-Scott D, Kuzel TM, Raisch DW, Tallman MS, West DP, Hirschfeld S, Grillo-Lopez AJ and Bennett CL. Accelerated approval of cancer drugs: improved access to therapeutic breakthroughs or early release of unsafe and ineffective drugs? J Clin Oncol. 2009;27:4398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madonna R, Moscato S, Polizzi E, Pieragostino D, Cufaro MC, Del Boccio P, Bianchi F, De Caterina R and Mattii L. Connexin 43 and Connexin 26 Involvement in the Ponatinib-Induced Cardiomyopathy: Sex-Related Differences in a Murine Model. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eide CA, Zabriskie MS, Savage Stevens SL, Antelope O, Vellore NA, Than H, Schultz AR, Clair P, Bowler AD, Pomicter AD, Yan D, Senina AV, Qiang W, Kelley TW, Szankasi P, Heinrich MC, Tyner JW, Rea D, Cayuela JM, Kim DW, Tognon CE, O’Hare T, Druker BJ and Deininger MW. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell. 2019;36:431–443 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanova ES, Tatarskiy VV, Yastrebova MA, Khamidullina AI, Shunaev AV, Kalinina AA, Zeifman AA, Novikov FN, Dutikova YV, Chilov GG and Shtil AA. PF114, a novel selective inhibitor of BCRABL tyrosine kinase, is a potent inducer of apoptosis in chronic myelogenous leukemia cells. Int J Oncol. 2019;55:289–297. [DOI] [PubMed] [Google Scholar]
- 30.Castagnetti F, Pane F, Rosti G, Saglio G and Breccia M. Dosing Strategies for Improving the Risk-Benefit Profile of Ponatinib in Patients With Chronic Myeloid Leukemia in Chronic Phase. Front Oncol. 2021;11:642005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molica M, Scalzulli E, Colafigli G, Foa R and Breccia M. Insights into the optimal use of ponatinib in patients with chronic phase chronic myeloid leukaemia. Ther Adv Hematol. 2019;10:2040620719826444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferdinandy P, Baczko I, Bencsik P, Giricz Z, Gorbe A, Pacher P, Varga ZV, Varro A and Schulz R. Definition of hidden drug cardiotoxicity: paradigm change in cardiac safety testing and its clinical implications. Eur Heart J. 2019;40:1771–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saussele S, Haverkamp W, Lang F, Koschmieder S, Kiani A, Jentsch-Ullrich K, Stegelmann F, Pfeifer H, La Rosee P, Goekbuget N, Rieger C, Waller CF, Franke GN, le Coutre P, Kirchmair R and Junghanss C. Ponatinib in the Treatment of Chronic Myeloid Leukemia and Philadelphia Chromosome-Positive Acute Leukemia: Recommendations of a German Expert Consensus Panel with Focus on Cardiovascular Management. Acta Haematol. 2020;143:217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casavecchia G, Galderisi M, Novo G, Gravina M, Santoro C, Agricola E, Capalbo S, Zicchino S, Cameli M, De Gennaro L, Righini FM, Monte I, Tocchetti CG, Brunetti ND, Cadeddu C and Mercuro G. Early diagnosis, clinical management, and follow-up of cardiovascular events with ponatinib. Heart Fail Rev. 2020;25:447–456. [DOI] [PubMed] [Google Scholar]
- 35.Dick SA and Epelman S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ Res. 2016;119:159–76. [DOI] [PubMed] [Google Scholar]
- 36.Vaduganathan M, Greene SJ, Butler J, Sabbah HN, Shantsila E, Lip GY and Gheorghiade M. The immunological axis in heart failure: importance of the leukocyte differential. Heart Fail Rev. 2013;18:835–45. [DOI] [PubMed] [Google Scholar]
- 37.Swirski FK and Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18:733–744. [DOI] [PubMed] [Google Scholar]
- 38.Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G and Prabhu SD. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ Heart Fail. 2017;10:e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A and Pizzinat N. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–24. [DOI] [PubMed] [Google Scholar]
- 40.Qing K, Weifeng W, Fan Y, Yuluan Y, Yu P and Yanlan H. Distinct different expression of Th17 and Th9 cells in coxsackie virus B3-induced mice viral myocarditis. Virol J. 2011;8:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Q, Ming T, Wang Y, Ding S, Hu C, Zhang C, Cao Q and Wang Y. Increased Th9 cells and IL-9 levels accelerate disease progression in experimental atherosclerosis. Am J Transl Res. 2017;9:1335–1343. [PMC free article] [PubMed] [Google Scholar]
- 42.Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, Fairweather D, Stoner JA, Cox CJ and Cunningham MW. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM and Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steinman L A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–45. [DOI] [PubMed] [Google Scholar]
- 45.Blanton RM, Carrillo-Salinas FJ and Alcaide P. T-cell recruitment to the heart: friendly guests or unwelcome visitors? Am J Physiol Heart Circ Physiol. 2019;317:H124–H140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Libby P, Nahrendorf M and Swirski FK. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum”. J Am Coll Cardiol. 2016;67:1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, Weissleder R and Nahrendorf M. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR and Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R and Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sreejit G, Abdel Latif A, Murphy AJ and Nagareddy PR. Emerging roles of neutrophil-borne S100A8/A9 in cardiovascular inflammation. Pharmacol Res. 2020;161:105212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muri J and Kopf M. Redox regulation of immunometabolism. Nat Rev Immunol. 2021;21:363–381. [DOI] [PubMed] [Google Scholar]
- 52.Rashida Gnanaprakasam JN, Wu R and Wang R. Metabolic Reprogramming in Modulating T Cell Reactive Oxygen Species Generation and Antioxidant Capacity. Front Immunol. 2018;9:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Agarwal K, Yousaf N and Morganstein D. Glucocorticoid use and complications following immune checkpoint inhibitor use in melanoma. Clin Med (Lond). 2020;20:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, Chau I, Ernstoff MS, Gardner JM, Ginex P, Hallmeyer S, Holter Chakrabarty J, Leighl NB, Mammen JS, McDermott DF, Naing A, Nastoupil LJ, Phillips T, Porter LD, Puzanov I, Reichner CA, Santomasso BD, Seigel C, Spira A, Suarez-Almazor ME, Wang Y, Weber JS, Wolchok JD, Thompson JA and National Comprehensive Cancer N. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018;36:1714–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jabbour E, Short NJ, Ravandi F, Huang X, Daver N, DiNardo CD, Konopleva M, Pemmaraju N, Wierda W, Garcia-Manero G, Sasaki K, Cortes J, Garris R, Khoury JD, Jorgensen J, Jain N, Alvarez J, O’Brien S and Kantarjian H. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: long-term follow-up of a single-centre, phase 2 study. Lancet Haematol. 2018;5:e618–e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rea D, Legros L, Raffoux E, Thomas X, Turlure P, Maury S, Dupriez B, Pigneux A, Choufi B, Reman O, Stephane D, Royer B, Vigier M, Ojeda-Uribe M, Recher C, Dombret H, Huguet F, Rousselot P, Intergroupe Francais des Leucemies Myeloides C and Group for Research in Adult Acute Lymphoblastic L. High-dose imatinib mesylate combined with vincristine and dexamethasone (DIV regimen) as induction therapy in patients with resistant Philadelphia-positive acute lymphoblastic leukemia and lymphoid blast crisis of chronic myeloid leukemia. Leukemia. 2006;20:400–3. [DOI] [PubMed] [Google Scholar]
- 57.Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, Wang A, Tao J, Wang C, Liu Q, Jin T, Jiang W, Deng X and Zhou R. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214:3219–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schelbergen RF, Geven EJ, van den Bosch MH, Eriksson H, Leanderson T, Vogl T, Roth J, van de Loo FA, Koenders MI, van der Kraan PM, van den Berg WB, Blom AB and van Lent PL. Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase-induced osteoarthritis. Ann Rheum Dis. 2015;74:2254–8. [DOI] [PubMed] [Google Scholar]
- 59.Rhea IB and Oliveira GH. Cardiotoxicity of Novel Targeted Chemotherapeutic Agents. Curr Treat Options Cardiovasc Med. 2018;20:53. [DOI] [PubMed] [Google Scholar]
- 60.Sheng CC, Amiri-Kordestani L, Palmby T, Force T, Hong CC, Wu JC, Croce K, Kim G and Moslehi J. 21st Century Cardio-Oncology: Identifying Cardiac Safety Signals in the Era of Personalized Medicine. JACC Basic Transl Sci. 2016;1:386–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Damrongwatanasuk R and Fradley MG. Cardiovascular Complications of Targeted Therapies for Chronic Myeloid Leukemia. Curr Treat Options Cardiovasc Med. 2017;19:24. [DOI] [PubMed] [Google Scholar]
- 62.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S and Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. [DOI] [PubMed] [Google Scholar]
- 63.Santoro M, Accurso V, Mancuso S, Contrino AD, Sardo M, Novo G, Di Piazza F, Perez A, Russo A and Siragusa S. Management of Ponatinib in Patients with Chronic Myeloid Leukemia with Cardiovascular Risk Factors. Chemotherapy. 2019;64:205–209. [DOI] [PubMed] [Google Scholar]
- 64.Garcia-Gutierrez V and Hernandez-Boluda JC. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front Oncol. 2019;9:603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Massaro F, Molica M and Breccia M. Ponatinib: A Review of Efficacy and Safety. Curr Cancer Drug Targets. 2018;18:847–856. [DOI] [PubMed] [Google Scholar]
- 66.Prasad V and Mailankody S. The accelerated approval of oncologic drugs: lessons from ponatinib. JAMA. 2014;311:353–4. [DOI] [PubMed] [Google Scholar]
- 67.van Hasselt JGC, Rahman R, Hansen J, Stern A, Shim JV, Xiong Y, Pickard A, Jayaraman G, Hu B, Mahajan M, Gallo JM, Goldfarb J, Sobie EA, Birtwistle MR, Schlessinger A, Azeloglu EU and Iyengar R. Transcriptomic profiling of human cardiac cells predicts protein kinase inhibitor-associated cardiotoxicity. Nat Commun. 2020;11:4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schoormans D, Pedersen SS, Dalton S, Rottmann N and van de Poll-Franse L. Cardiovascular co-morbidity in cancer patients: the role of psychological distress. Cardiooncology. 2016;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alexandre J, Cautela J, Ederhy S, Damaj GL, Salem JE, Barlesi F, Farnault L, Charbonnier A, Mirabel M, Champiat S, Cohen-Solal A, Cohen A, Dolladille C and Thuny F. Cardiovascular Toxicity Related to Cancer Treatment: A Pragmatic Approach to the American and European Cardio-Oncology Guidelines. J Am Heart Assoc. 2020;9:e018403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Campia U, Moslehi JJ, Amiri-Kordestani L, Barac A, Beckman JA, Chism DD, Cohen P, Groarke JD, Herrmann J, Reilly CM and Weintraub NL. Cardio-Oncology: Vascular and Metabolic Perspectives: A Scientific Statement From the American Heart Association. Circulation. 2019;139:e579–e602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen S, Liu G, Chen J, Hu A, Zhang L, Sun W, Tang W, Liu C, Zhang H, Ke C, Wu J and Chen X. Ponatinib Protects Mice From Lethal Influenza Infection by Suppressing Cytokine Storm. Front Immunol. 2019;10:1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sagoo P, Garcia Z, Breart B, Lemaitre F, Michonneau D, Albert ML, Levy Y and Bousso P. In vivo imaging of inflammasome activation reveals a subcapsular macrophage burst response that mobilizes innate and adaptive immunity. Nat Med. 2016;22:64–71. [DOI] [PubMed] [Google Scholar]
- 73.Schiopu A and Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. 2013;2013:828354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Short NJ, Konopleva M, Jabbour E, Kadia TM, Daver N, Cook R, Jain N and Ravandi F. Interim Results of the Phase I/II Study of the Ponatinib, Venetoclax and Dexamethasone for Patients with Relapsed or Refractory Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood. 2020;136:11–12.32276273 [Google Scholar]
- 75.Yuda J, Yamauchi N, Kuzume A, Guo YM, Sato N and Minami Y. Molecular remission after combination therapy with blinatumomab and ponatinib with relapsed/refractory Philadelphia chromosome-positive acute lymphocytic leukemia: two case reports. J Med Case Rep. 2021;15:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hesselstrand R, Distler JHW, Riemekasten G, Wuttge DM, Torngren M, Nyhlen HC, Andersson F, Eriksson H, Sparre B, Tuvesson H and Distler O. An open-label study to evaluate biomarkers and safety in systemic sclerosis patients treated with paquinimod. Arthritis Res Ther. 2021;23:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bengtsson AA, Sturfelt G, Lood C, Ronnblom L, van Vollenhoven RF, Axelsson B, Sparre B, Tuvesson H, Ohman MW and Leanderson T. Pharmacokinetics, tolerability, and preliminary efficacy of paquinimod (ABR-215757), a new quinoline-3-carboxamide derivative: studies in lupus-prone mice and a multicenter, randomized, double-blind, placebo-controlled, repeat-dose, dose-ranging study in patients with systemic lupus erythematosus. Arthritis Rheum. 2012;64:1579–88. [DOI] [PubMed] [Google Scholar]
References for Expanded Materials and Methods:
- 1.Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, Quaife-Ryan GA, Al-Sharea A, Pernes G, Dragoljevic D, Lal H, Schroder K, Hanaoka BY, Raman C, Grant MB, Hudson JE, Smyth SS, Porrello ER, Murphy AJ and Nagareddy PR. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation. 2020;141:1080–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schelbergen RF, Geven EJ, van den Bosch MH, Eriksson H, Leanderson T, Vogl T, Roth J, van de Loo FA, Koenders MI, van der Kraan PM, van den Berg WB, Blom AB and van Lent PL. Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase-induced osteoarthritis. Ann Rheum Dis. 2015;74:2254–8. [DOI] [PubMed] [Google Scholar]
- 3.Ren M, Qin H, Ren R and Cowell JK. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia. 2013;27:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Clackson T and Rivera VM. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther. 2012;11:690–9. [DOI] [PubMed] [Google Scholar]
- 5.Singh AP, Glennon MS, Umbarkar P, Gupte M, Galindo CL, Zhang Q, Force T, Becker JR and Lal H. Ponatinib-induced cardiotoxicity: delineating the signalling mechanisms and potential rescue strategies. Cardiovasc Res. 2019;115:966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmad F, Singh AP, Tomar D, Rahmani M, Zhang Q, Woodgett JR, Tilley DG, Lal H and Force T. Cardiomyocyte-GSK-3alpha promotes mPTP opening and heart failure in mice with chronic pressure overload. J Mol Cell Cardiol. 2019;130:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou J, Lal H, Chen X, Shang X, Song J, Li Y, Kerkela R, Doble BW, MacAulay K, DeCaul M, Koch WJ, Farber J, Woodgett J, Gao E and Force T. GSK-3alpha directly regulates beta-adrenergic signaling and the response of the heart to hemodynamic stress in mice. J Clin Invest. 2010;120:2280–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umbarkar P, Singh AP, Gupte M, Verma VK, Galindo CL, Guo Y, Zhang Q, McNamara JW, Force T and Lal H. Cardiomyocyte SMAD4-Dependent TGF-beta Signaling is Essential to Maintain Adult Heart Homeostasis. JACC Basic Transl Sci. 2019;4:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woudstra L, Biesbroek PS, Emmens RW, Heymans S, Juffermans LJ, van der Wal AC, van Rossum AC, Niessen HWM and Krijnen PAJ. CD45 is a more sensitive marker than CD3 to diagnose lymphocytic myocarditis in the endomyocardium. Hum Pathol. 2017;62:83–90. [DOI] [PubMed] [Google Scholar]
- 10.Zhou J, Ahmad F, Parikh S, Hoffman NE, Rajan S, Verma VK, Song J, Yuan A, Shanmughapriya S, Guo Y, Gao E, Koch W, Woodgett JR, Madesh M, Kishore R, Lal H and Force T. Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy. Circ Res. 2016;118:1208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lal H, Zhou J, Ahmad F, Zaka R, Vagnozzi RJ, Decaul M, Woodgett J, Gao E and Force T. Glycogen synthase kinase-3alpha limits ischemic injury, cardiac rupture, post-myocardial infarction remodeling and death. Circulation. 2012;125:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E and Force T. Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation. 2014;130:419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A detailed description of the Methods is available in the Supplemental Materials section. The data, analytic methods, and study materials are available to other researchers to reproduce the results or replicate the procedures on request.