

Abstract
Background
Coronary artery disease (CAD) is the leading cause of death worldwide. Recent meta-analyses of genome-wide association studies (GWAS) have identified over 175 loci associated with CAD. The majority of these loci are in non-coding regions and are predicted to regulate gene expression. Given that vascular smooth muscle cells (SMCs) play critical roles in the development and progression of CAD, we aimed to identify the subset of the CAD GWAS risk loci associated with the regulation of transcription in distinct SMC phenotypes.
Methods
Here, we measured gene expression in SMCs isolated from the ascending aortas of 151 heart transplant donors of various genetic ancestries in quiescent or proliferative conditions and calculated the association of their expression and splicing with ~6.3 million imputed single nucleotide polymorphism (SNP) markers across the genome.
Results
We identified 4,910 expression and 4,412 splice quantitative trait loci (sQTL) representing regions of the genome associated with transcript abundance and splicing. 3,660 eQTLs had not been observed in the publicly available Genotype-Tissue Expression dataset. Further, 29 and 880 eQTLs were SMC- and sex-specific, respectively. We made these results available for public query on a user-friendly website. To identify the effector transcript(s) regulated by CAD GWAS loci, we used four distinct colocalization approaches. We identified 84 eQTL and 164 sQTLs that colocalized with CAD loci, highlighting the importance of genetic regulation of mRNA splicing as a molecular mechanism for CAD genetic risk. Notably, 20% and 35% of the eQTLs were unique to quiescent or proliferative SMCs, respectively. One CAD locus colocalized with an SMC sex-specific eQTL (TERF2IP), and another locus colocalized with SMC-specific eQTL (ALKBH8). The most significantly associated CAD locus, 9p21, was an sQTL for the long non-coding RNA CDKN2B-AS1, also known as ANRIL, in proliferative SMCs.
Conclusions
Collectively, our results provide evidence for the molecular mechanisms of genetic susceptibility to CAD in distinct SMC phenotypes.
Keywords: Smooth muscle cell, coronary artery disease, expression and splice quantitative trait loci, Basic Science Research, Vascular biology, Omics, Genetics, Gene Expression and Regulation
Graphical Abstract
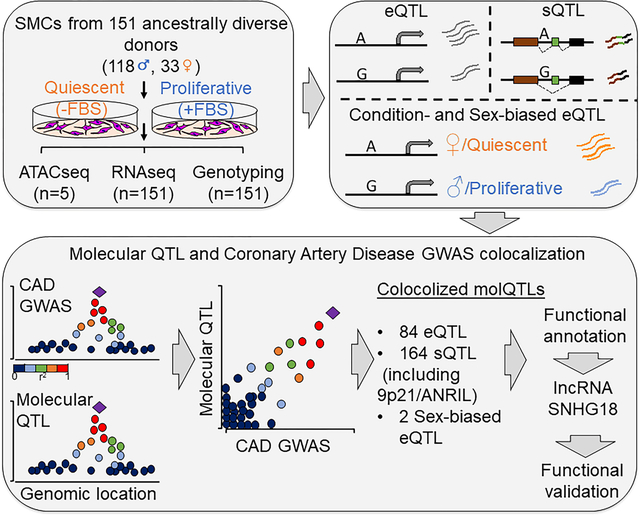
INTRODUCTION
Coronary artery disease (CAD) is the leading cause of death worldwide1. Heritability estimates for CAD vary between 40% to 70%, suggesting a strong genetic contribution to disease pathology2. Genome-wide association studies (GWAS) have identified 175 loci associated with increased risk for CAD3–5. Approximately 40% of the CAD loci are associated with known risk factors, such as blood lipid levels, nitric oxide signaling, and blood pressure3. The remaining 60% have unknown mechanisms, but there is evidence that some of these loci function through the vessel wall where the disease develops6. In addition, 94% of CAD-associated genetic variants are in non-coding regions of the genome7, implying that the disease-causing loci involve regulatory mechanisms that affect the transcription of genes6. Therefore, gene expression studies performed in cells and tissues relevant to CAD in human populations can pinpoint the regulatory mechanisms of disease susceptibility8.
Expression quantitative trait loci (eQTL) and splicing quantitative trait loci (sQTL) analyses are key approaches that link genetic variants with variations in gene expression and splicing patterns, respectively8,9. They have enabled the prioritization of genetic variants within GWAS loci for different traits, and have shown that trait-associated genetic variants often function in a tissue- or cell type-specific manner10–13. Smooth muscle cells (SMCs), which make up the medial layer of arteries, play key roles in the integrity of the vessel wall, regulation of blood pressure and initiation and development of atherosclerosis. Recent studies provided compelling evidence that SMCs can play either beneficial or detrimental roles in lesion pathogenesis depending on the nature of their phenotypic changes, for example from a quiescent to a proliferative phenotype14–17. In addition, SMC phenotypic switching seems to be important in explaining sex differences in atherosclerotic plaque composition18. Thus, identifying the genetic determinants of SMC gene expression is crucial for understanding the biological significance of CAD-associated genetic variants functioning in SMCs. This will also inform the prediction of novel drug candidates targeting the disease processes in the vessel wall.
Despite publicly available population-level gene expression datasets from various tissues and cells, including 54 tissues from the Genotype-Tissue Expression (GTEx) project19 and atherosclerosis-relevant tissues and cell types from the STARNET cohort20, aortic endothelial cells21,22, monocytes23, whole blood24, and coronary artery SMCs25 as well as epigenome profiling from the Roadmap Epigenomics Project26, more than half of the CAD loci are still not functionally annotated. Therefore, in this study, using a multi-omics approach, we identified the genetic variants that are associated with SMC-specific gene expression derived from 151 healthy and ancestrally-diverse heart transplant donors. This allowed us to identify CAD-associated loci, including the most significantly associated CAD locus, 9p21, that perturb SMC transcription, thereby leading to the prediction of candidate effector transcripts and candidate causal variants in these disease-associated loci.
METHODS
A detailed description of the methods and the experimental procedures are provided in the Online Data Supplement. Please see the Major Resources Table in the Supplemental Materials.
The RNAseq and ATACseq data are available at GEO with the accession numbers GSE193817 and GSE198544 respectively. eQTL/sQTL results can be accessed at https://virginia.box.com/s/t5e1tzlaqsf85z13o4ie2f9t1i0zfypd and https://virginia.box.com/s/o81cxrj5xne3xem4au785mupikduuwbu We have created a user-friendly website at http://civeleklab.cphg.virginia.edu to query the dataset published in this paper.
RESULTS
Transcriptional profiling of human aortic SMCs
We performed RNA sequencing of aortic SMCs derived from 151 ancestrally-diverse healthy heart transplant donors (118 males and 33 females) to identify their transcriptional profiles. After quality control filtering, data analyses were performed on 139 and 145 samples cultured in the absence (quiescent) or presence (proliferative) of FBS, respectively (Figure 1). There were 18,637 and 18,116 expressed genes in quiescent and proliferative SMCs, respectively (Table 1). Principal component analysis identified two distinct clusters of samples corresponding to the cells cultured in the two conditions (Supplementary Figure 1A). Further, 2,773 genes were differentially expressed (Padj< 1×10−3), including canonical SMC markers (VCAM1, SMTN, ICAM1, TAGLN, CNN1, ACTA2, SPP1) in agreement with the differences observed in the quiescent and proliferative state of SMCs (Supplementary Figure 1B, Supplementary Table 1). Extracellular matrix organization was among the top biological pathways identified in GO enrichment analysis of upregulated genes in proliferative versus quiescent phenotypes. In contrast, pathways that are associated with DNA replication and proliferation were repressed in the quiescent phenotype (Supplementary Figure 1C).
Figure 1: Study design and overview of analyses.
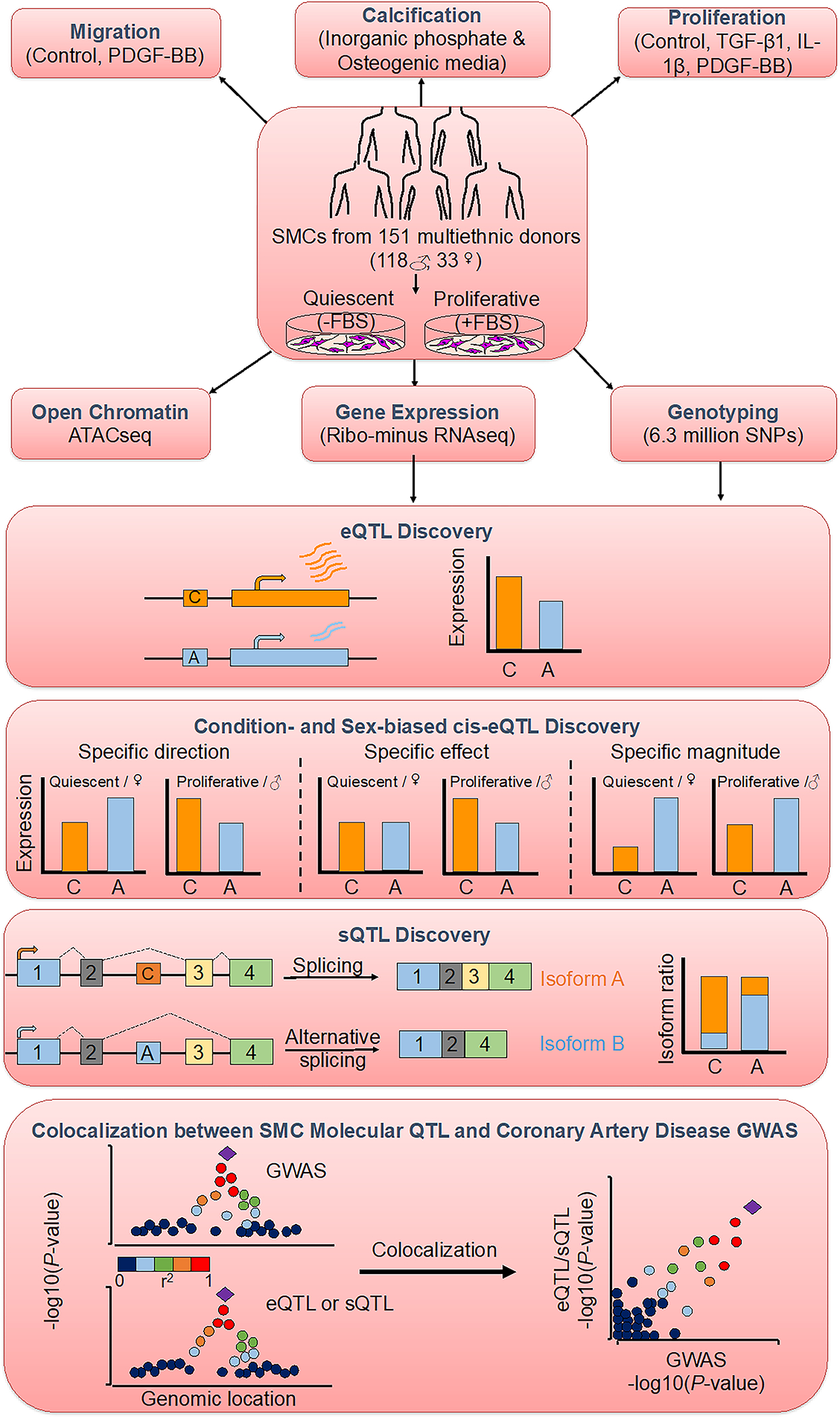
Aortic smooth muscle cells (SMCs) from 151 heart transplant donors of various genetic ancestries were characterized for three atherosclerosis-relevant phenotypes: migration, calcification and proliferation74. To measure gene expression of SMCs, sequencing of ribosomal RNA-depleted total RNA isolated from SMCs cultured in the absence or presence of FBS to simulate quiescent or proliferative phenotypic state was performed. Associations of gene expression and splicing with the genotypes of ~6.3 million imputed SNPs were calculated to discover cis-eQTLs as well as condition-specific and sex-biased eQTLs and sQTLs. Colocalization between molecular QTL and coronary artery disease GWAS associations was identified using four different methods.
Table 1:
Expression and Splicing Quantitative Trait Loci
| Gene Expression | Splicing | ||||
|---|---|---|---|---|---|
| Condition | Gene Type | No of genes tested | No of eGenes with at least one eQTL | No of genes tested | No of sGenes with at least one sQTL |
| Quiescent | protein coding | 13,341 | 2,267 | 11,123 | 2,774 |
| lncRNA | 3,183 | 477 | 959 | 244 | |
| pseudogene | 1,484 | 215 | 291 | 129 | |
| other | 629 | 41 | 1 | 0 | |
| Total | 18,637 | 3,000 | 12,374 | 3,147 | |
| Proliferative | protein coding | 13,182 | 3,367 | 11,013 | 3,180 |
| lncRNA | 2,986 | 546 | 996 | 273 | |
| pseudogene | 1366 | 228 | 297 | 125 | |
| other | 582 | 47 | 1 | 0 | |
| Total | 18,116 | 4,188 | 12,307 | 3,578 | |
To confirm that cultured SMCs reflect in vivo physiology, we projected their transcriptomes onto the 49 tissues profiled in GTEx v841 (Supplementary Figure 2). We observed that SMCs formed a distinct cluster and the closest tissue/cell type are fibroblasts, skeletal muscle, blood vessels and heart.
cis-Expression Quantitative Trait Loci in SMCs
We obtained genotype information for ~6.3 million variants with at least 5% minor allele frequency in our population. Clustering of the donor genotypes with the 1000 Genomes reference population samples identified 6, 12, 64, and 69 of the individuals with East Asian, African, Admixed American, and European ancestry, respectively (Supplementary Figure 3).
To identify genetic loci associated with transcript abundance, we performed association mapping with the genotypes of ~6.3 million variants and the expression levels of 18,116 and 18,637 genes in quiescent and proliferative SMCs, respectively using tensorQTL44. We identified 3,000 and 4,188 eGenes with cis-eQTL (< 1Mb from the TSS) at FDR q-value < 0.05 in the quiescent and proliferative phenotypes, respectively (Table 1). We identified that 1,322 and 1,899 eGenes from quiescent and proliferative SMC respectively, were differentially expressed between the two conditions (Padj<0.05) (Supplementary Table 2). Next, we compared our results against the GTEx v8 eQTL dataset composed of 49 different human tissues41 using the QTlizer R package50. We found that 2,818 SMC cis-eQTLs (eSNP and eGene pair) were present in at least one GTEx tissue, whereas 3,660 were unique to our dataset (Figure 2A). Most of the shared SMC eQTLs were enriched in the tissues that are anatomically rich in SMCs (Supplementary Figure 4). Conditioning on the lead SNPs identified 254 and 465 secondary and beyond eQTLs for quiescent and proliferative SMCs, respectively.
Figure 2: Identification of cis-eQTLs, condition-specific and sex-biased eQTLs in aortic smooth muscle cells.
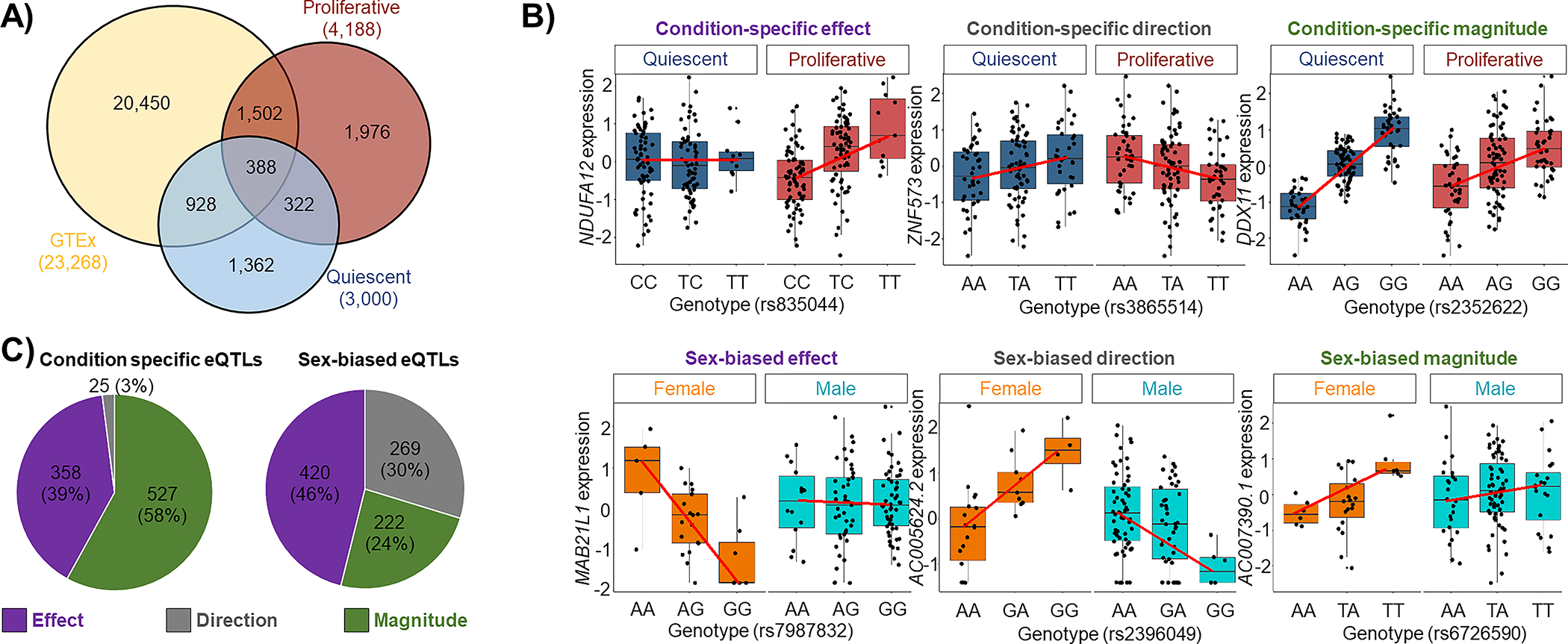
A) Venn diagram comparing eQTL discovered in quiescent (blue) and proliferative (red) conditions versus GTEx tissues (yellow) (FDR q-value<0.05). 3,660 of SMC cis-eQTLs (pair of SNP-gene) were absent or not significant in the GTEx dataset. 1,362 and 1,976 of these novel cis-eQTLs were unique to quiescent and proliferative cells, respectively. B) Condition-specific (top) and sex-biased (bottom) eQTLs were classified into three different categories: condition-specific or sex-biased effect, condition-specific or sex-biased direction, and condition-specific or sex-biased magnitude. C) Quantification of the three different condition-specific (left) and sex-biased (right) eQTL groups.
We identified SMC-specific eQTLs using GTEx tissues as a reference61 with METASOFT50. We identified 29 SMC-specific cis-eQTLs under a stringent criteria of eQTL posterior probability > 0.9 for SMCs and < 0.1 for all the GTEx tissues (Supplementary Table 3, Supplementary Figure 5). To validate these results, we queried these 29 cis-eQTLs in the STARNET dataset of seven cardiometabolic tissues from ~600 donors20. We identified only two of the SMC cis-eQTLs (rs367077-HLA-K and rs4795548-SH3GL1P2) to be present in the STARNET dataset at FDR < 0.05 (Supplementary Table 4). 12 of the 29 SMC-specific cis-eQTLs were present in both quiescent and proliferative SMCs, 9 loci were cis-eQTLs only in quiescent and 8 loci were cis-eQTLs only in proliferative SMCs. Because many tissues in GTEx contain vascular wall cells, we examined if the 29 SMC eQTLs showed associations in monocytes/macrophages23 and aortic endothelial cells21. None of the 29 eQTLs were present in these cells suggesting that the regulatory impact of the variants in these loci are SMC specific.
15% (3,000) and 39% (1,976) of the eGenes were unique to quiescent and proliferative cells, respectively (Figure 2A) (Poverlap <1×10−300, hypergeometric test). Therefore, we determined whether the eQTL effect sizes were statistically different between the two phenotypic states. We compared the regression slopes of an eQTL in quiescent (βnoFBS) vs proliferative (βFBS) phenotypes using a Z-test48. We identified 1,248 eQTLs at FDR q-value < 0.05 with varying effects between the two phenotypic states (Supplementary Table 5). We classified the condition-specific eQTLs into three different categories (Figure 2B). 58% (527) of them showed differences in magnitude of the effect size between the two phenotypes, 39% (358) showed eQTL effect in only one phenotype, and 3% (25) showed differences in the direction of effect between the two phenotypes (Figure 2C). Three examples of condition-specific eQTLs are shown in Figure 2B. These results suggest that regulatory variation impacts SMC gene expression in specific contexts.
Since sex differences in the genetic regulation of gene expression have been observed in many tissues67,68, we separately identified sex-biased cis-eQTLs in quiescent and proliferative SMCs. We identified 457 and 454 sex-biased cis-eQTLs (eigenMT value <0.05) in quiescent and proliferative SMCs, respectively (Supplementary Table 6). 24% of them (combined conditions) showed differences in magnitude of effect size between the two sexes, 46% showed eQTL effect in only one sex, and 30% showed differences in the direction of effect between the two sexes (Figure 2C). Three examples of sex-biased eQTLs are shown in Figure 2B.
To characterize the potential function of the eQTL signals, we evaluated the overlap of the lead variant and LD proxies (r2≥0.8; 1000G EUR) in accessible chromatin regions of SMCs as identified by ATACseq in 5 different donors in quiescent and proliferative phenotypes. 9.86% of the lead variants of the eQTLs were in these accessible chromatin regions. After including LD proxies of these lead variants, 1,386 of 3,000 eQTL signals in quiescent SMCs and 1,945 of 4,188 eQTL signals in proliferative SMCs overlapped accessible chromatin regions, demonstrating the potential regulatory function of these loci. We next sought to identify transcription factor binding sites (TFBSs) overrepresented in ATACseq peaks and overlapping eQTL SNPs in the accessible chromatin regions (Supplementary Figure 6). Motif enrichment analysis showed enrichment of putative binding sites for members of the SP2, SP1, ELK4, and GABPA TF families, some of which are known to play functional roles in SMCs69–71.
Colocalization between eQTLs and CAD GWAS signals
To predict the effector transcripts that are regulated by CAD GWAS loci, we performed colocalization analyses using four distinct approaches. Overall, we had genotypes for 169 of the 175 CAD loci in our dataset. First, to identify the eQTLs that were likely to be driven by the GWAS loci, we assessed if the GWAS and eQTL lead variants were in high linkage disequilibrium (LD) (r2≥0.8) in our population. Using the LD colocalization approach only, we identified 16 and 22 eGenes (FDR q-value < 0.05) in the quiescent and proliferative phenotype that showed an overlap with CAD loci, respectively. We also performed colocalization analysis using three additional methods: SMR52, COLOC54, and eCAVIAR53. We used FDR q-value < 0.05 for SMR and colocalization posterior probability (CLPP) > 0.01 as cutoffs for eCAVIAR and PPH4 > 0.5 for COLOC. Using all four colocalization methods, we identified 84 eGenes that showed statistically significant colocalization. 17 and 30 of them showed an overlap with at least two of the colocalization methods in quiescent and proliferative SMCs, respectively (Figure 3, Supplementary Table 7, Supplementary Figure 7). Some of the eGenes predicted by the four colocalization methods differed; therefore, we visualized the coincidence of the eQTL and GWAS lead SNPs by inspecting the regional colocalization plots using LocusCompare55. This coincidence was also supported by conditional analysis on each lead eSNP and CAD GWAS index SNP.
Figure 3: Summary of the SMC eQTL and CAD GWAS colocalization.
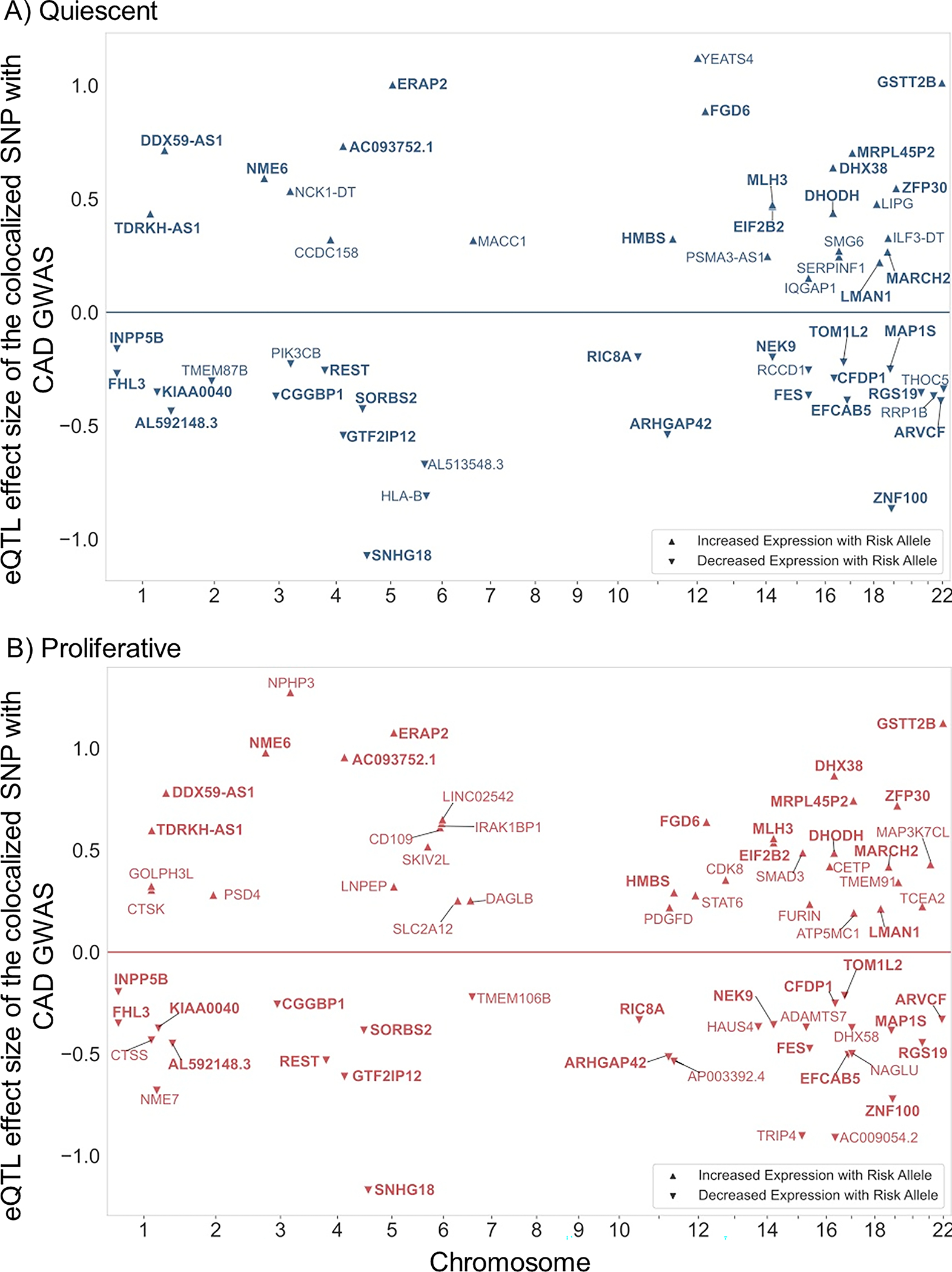
Plots show the colocalization of CAD GWAS and eQTL signals using a combination of four different methods in A) quiescent and B) proliferative SMCs. The X-axis shows the chromosomal position of the colocalized SNP; Y-axis shows the effect size and direction of the eQTL with respect to the risk allele from the coronary artery GWAS4. The effect size value is proportional to gene expression residual after PEER correction43. Risk alleles that are associated with increased gene expression level are shown with up-triangle and risk alleles that are associated with decreased gene expression level are shown with down-triangle. Bolded gene symbols are common between the two phenotypic states.
Next, we assessed the LD between the sex-biased eQTL and CAD GWAS SNPs and identified TERF2IP whose cis-eQTL colocalized with the 16q23.1 CAD locus: (Figure 4). The sex-biased eQTL for TERF2IP was present only in proliferative SMCs. The CAD risk allele of the eSNP rs12929673, T, was associated with lower expression of TERF2IP in females and higher expression in males (Figure 4A). The same locus was also associated with two SMC phenotypes relevant to atherosclerosis in a sex-stratified manner72.The CAD risk allele of the eSNP rs12929673, T, was associated with reduced proliferation response to IL-1β stimulation and lower calcification in females compared to males which showed the opposite effect (Figure 4B). The CAD GWAS signal at this locus was stronger in males (Prs12929673,male=2×10−6; βrs12929673,male=0.28) than in females (Prs12929673,female=1×10−2; βrs2929673,female=0.15) in the UK Biobank cohort73 (Figure 4C, suggesting a protective role for lower TERF2IP expression against atherosclerosis.
Figure 4: Colocalization between TERF2IP sex-biased eQTL and CAD GWAS.
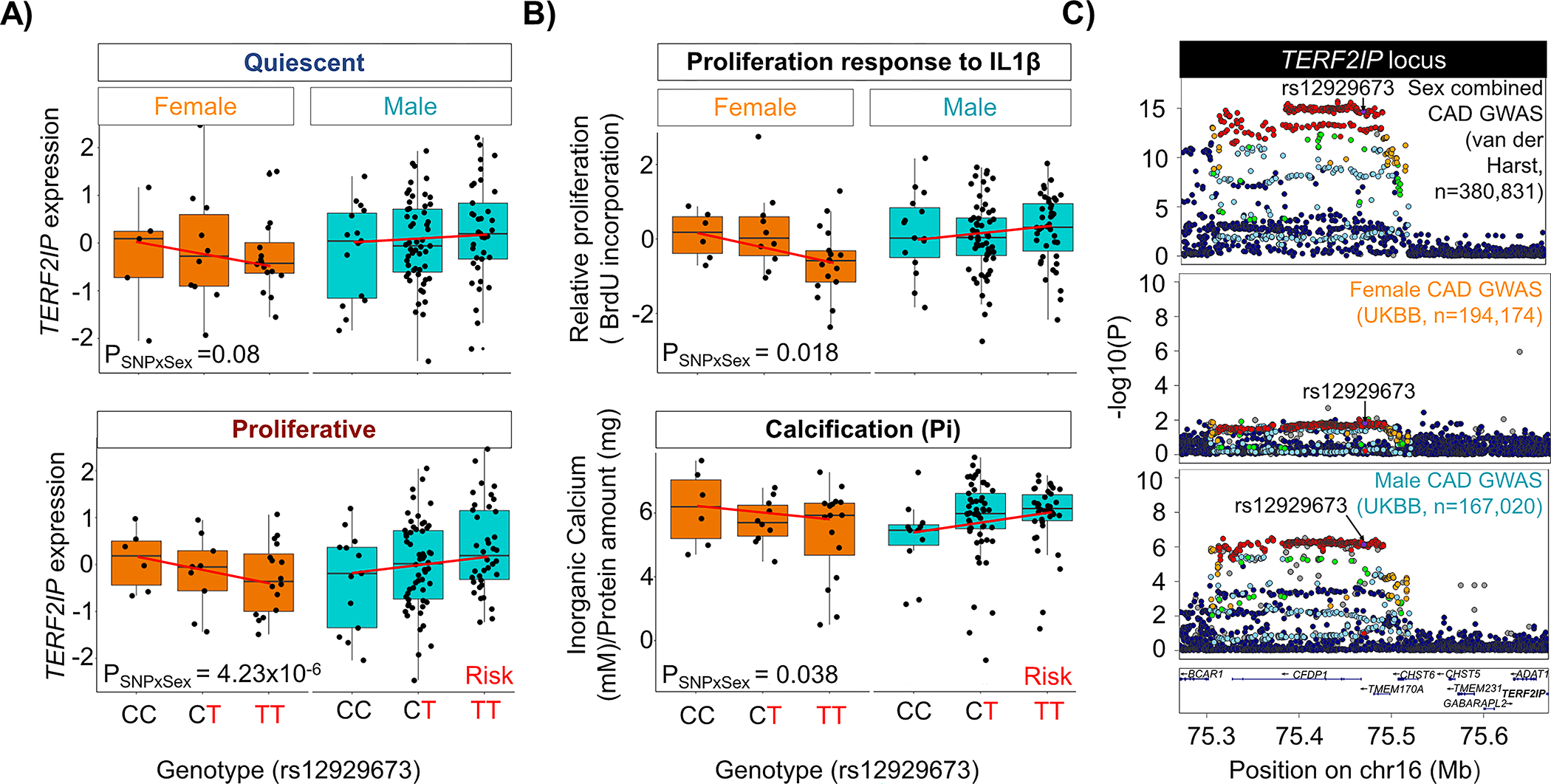
Colocalization based on linkage disequilibrium between sex-biased eQTL SNPs and CAD GWAS identified TERF2IP as a colocalized gene. A) Genotype-gene expression plots for TERF2IP for the colocalized SNP(rs12929673) in quiescent and proliferative SMCs in males and females. B) Sex biased association of rs12929673 with SMC proliferation in response to IL-1β and calcification in response to high inorganic phosphate based on data described in our previous publication74. C) LocusZoom55 plots showing the association signal for sex-combined (Harst and Verweij4) and sex-stratified (UK Biobank73) GWAS for coronary artery disease at the AL160313.1 or TERF2IP locus.
To identify SMC-specific genetic regulation of CAD risk, we asked if the SMC-specific eQTLs (Supplementary Table 3) colocalized with CAD GWAS loci. We found that cis-eQTL for ALKBH8 colocalized with the 11q22.3 CAD locus (Figure 5A). This SMC-specific eQTL is also colocalized with systolic and diastolic blood pressure (Figure 5B–C), suggesting a role for this gene, which encodes a methyltransferase, in regulating blood pressure and atherosclerosis risk. The risk allele, G, of the eSNP rs7926602 is associated with lower expression of ALKBH8 in quiescent and proliferative SMCs (Figure 5D–E).
Figure 5: Colocalization between SMC-specific eQTL and vascular disease GWAS.
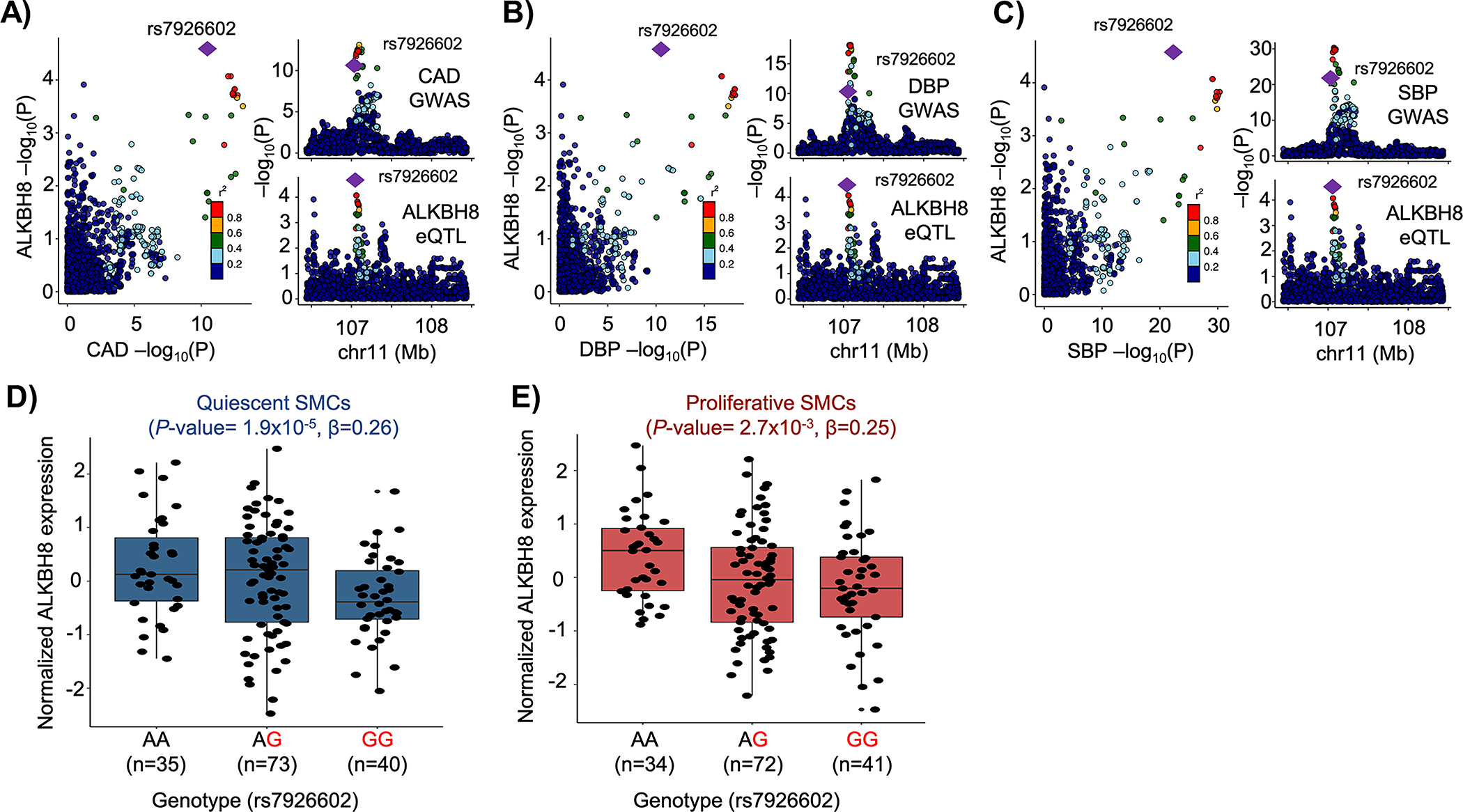
cis-eQTL for ALKBH8 expression colocalized with the 11q22.3 A) Coronary artery disease (CAD), B) Diastolic (DBP) and C) Systolic (SBP) blood pressure GWAS locus. The risk allele (G) of SNP rs7926602 is associated with lower ALKBH8 expression in D) quiescent and E) proliferative SMCs.
Functional annotation of CAD GWAS-colocalized SMC eQTLs
The 84 colocalized eQTLs contain 3,811 SNPs that are in high LD in our study population (r2≥0.8). We overlapped these SNPs with accessible chromatin regions identified in ATACseq experiments performed in quiescent and proliferative SMCs from five donors. We determined that 128 SNPs in 30 eQTL loci in quiescent SMCs and 140 SNPs in 37 eQTL loci in proliferative SMCs were in accessible chromatin peaks (Supplementary Table 8). We predict these SNPs to have a regulatory impact on eQTL gene expression and potentially be causal for SMC gene expression and CAD risk.
We previously characterized the SMC donors for 12 atherosclerosis-relevant phenotypes74. First, we assessed the association of the eSNPs in the CAD loci with these phenotypes. Second, we assessed the correlation between the phenotypes and the colocalized eQTL gene expression. The risk allele of the eSNP rs7195958 at the DHODH locus was associated with increased SMC proliferation compared to the non-risk allele. The risk allele was also associated with decreased expression of DHODH in quiescent SMCs. As would be predicted by these association results, there was a significant positive correlation between DHODH expression and SMC proliferation, suggesting that the 16q22 CAD locus regulates SMC proliferation by perturbing DHODH expression (Supplementary Figure 8A). Similarly, the risk allele of the eSNP rs12817989 at the FGD6 locus had lower association with SMC proliferation compared to the non-risk allele. The risk allele was associated with higher expression of FGD6 in proliferative SMCs. As would be predicted by these association results, there was a significant negative correlation between FGD6 expression and SMC proliferation, suggesting that the 12q22 CAD locus regulates SMC proliferation by perturbing FGD6 expression (Supplementary Figure 8B).
Finally, human single cell RNAseq (scRNAseq) analysis from coronary atherosclerotic plaques confirmed the expression of most of the eQTL genes in SMCs (Supplementary Figure 9)17. We were able to assess the expression of 76 of the 84 eQTL genes in the coronary artery scRNASeq dataset and found that 50 were higher expressed in SMCs, pericytes, and fibroblasts compared to endothelial cells, monocytes, macrophages, and other immune cells. For example, we found that the expression of the long non-coding RNA SNHG18 was regulated by the 5p15 CAD locus in both the quiescent and proliferative SMCs. The risk allele was associated with decreased expression of SNHG18 (Figure 6A). Of the 16 potentially causal SNPs in high LD in this locus, six of them were in accessible chromatin regions in both SMC phenotypic states (Figure 6B, Supplementary Table 8). Nine SNPs also displayed an allelic effect in a massively parallel reporter assay (MPRA) performed in SMCs exposed to cholesterol to induce phenotypic switching to resemble modulated SMCs found in atherosclerotic plaques62 (Figure 6C). SNPs rs1651285, rs1706987, and rs1398337 were in both accessible regions and showed allelic effects in MPRA, identifying them as the potential causal variants at this locus. The expression of SNHG18 was highly enriched in SMC, fibroblast/fibromyocyte, and pericyte clusters in a human carotid artery scRNA-seq dataset17 (Figure 6D). Further, SNHG18 expression was negatively correlated with PDGF-BB-induced proliferation (Figure 6E). When we silenced the SNHG18 expression in immortalized human coronary aortic SMCs (Supplementary Figure 10), we observed increased proliferation (Figure 6F).
Figure 6: Colocalization between SNHG18 eQTL and 5p15.31 CAD GWAS locus.

The risk allele (T) of SNP rs1651285 is associated with lower SNHG18 expression in A) quiescent and proliferative SMCs B) LocusZoom55 plots of the CAD GWAS and SMC cis-eQTL in the 5p15.31 locus. SNPs rs1651285, rs1706987, and rs1398337 are located in an accessible chromatin region identified by ATACseq in SMCs (lower panel). C) Bar plot summarizing the CAD haplotypes in the chr5p15/SNHG18 locus (Hap 1, 2, 3, 4) that demonstrated significant allele-specific enhancer activity in massively parallel reporter assays performed in cholesterol-loaded SMCs (n=3). Significant differences (two-sided T-test) are shown between Hap2/Hap3 vs. Hap1 (reference haplotype). A different color represents each nucleotide. Adenine (A) is indicated by the dark orange color, Cytosine (C) is indicated by the blue color, Guanine (G) is indicated by the green color, and the orange light indicates Thymine (T). D) Uniform manifold approximation and projection plot of single-cell RNA-sequencing data from human coronary atherosclerotic plaques71. E) Negative correlation of SNHG18 expression with PDGF-BB-induced proliferation in SMCs using Spearman correlation. F) Downregulation of SNHG18 in SMCs increased proliferation (n-3). Hap indicates haplotype. G) Representative RNAScope™ Double ISH images (40X) of human aortic SMCs stained for ACTA2, SNHG18, and DAPI. The representative images in H were chosen since they captured the critical cell structures and evident staining. Scale bar represents 20 μm.
To determine the transcriptional profile of SNHG18 downregulation, we performed transcriptome analysis using RNAseq in human coronary artery SMCs and found 778 genes that were differentially expressed (Padj<0.05) (Supplementary Table 9). Of these, 375 genes showed a log2(fold-change [FC]) above 0.2, and 403 genes were downregulated with log2(FC)<−0.2. Upregulation of MKI67, a cellular marker for proliferation, in response to SNHG18 knockdown confirmed our proliferation results. Furthermore, proliferation/cell cycle, cell migration, cell motility, and blood vessel development/angiogenesis were among the top biological pathways identified in the GO enrichment analysis of downregulated genes Supplementary Table 10). While, pathways that are associated with actin cytoskeleton organization, cell morphogenesis and regulation of cellular component biogenesis were identified in GO enrichment analysis of upregulated genes (Supplementary Table 11).
In contrast, none of the DEGs are cis-regulated suggesting that the effects of SNHG18 are in trans to affect other genes. Next, we assessed SNHG18 mRNA expression in human aortic SMCs from three donors using RNA-scope in situ hybridization and immunofluorescence. Our results confirmed that SNHG18 mRNA localized to the nucleus and cytoplasm and was co-expressed with SMC marker ACTA2 mRNA in all the cells we imaged. In agreement with our results, Zhen et al., demonstrated, using in situ hybridization assays, that SNHG18 is localized to both the nucleus and cytoplasm in different cancer cells and tissues75.
Collectively, these lines of evidence point to three variants in the 5p15 locus as potential causal SNPs regulating the expression of SNHG18 and SMC proliferation, thereby affecting the CAD risk in this locus.
Splicing Quantitative Trait Loci in SMCs
Previous studies showed that RNA splicing explains a large proportion of heritable risk for complex diseases9. To identify genetic loci associated with mRNA splicing, we quantified RNA splicing with LeafCutter46 and performed association mapping with tensorQTL44. We identified 3,147 and 3,578 sGenes with cis-sQTL (< 200 kb from splice sites) in the quiescent and proliferative phenotypes, respectively (FDR q-value <0.05) (Table 1). 1,919 and 1,700 sGenes from quiescent and proliferative SMC respectively, were differentially expressed (Supplementary Table 12). Similar to eQTL findings, the majority of the sGenes were shared (51%) between the two conditions (Poverlap < 1×10−300, hypergeometric test). 19% (834) and 29% (1,265) of the sGenes were unique to quiescent and proliferative conditions, respectively (Supplementary Figure 11A). Conditioning on the lead SNPs identified 144 and 120 secondary and beyond sQTL for quiescent and proliferative conditions, respectively. We also determined the overlap of genes with cis-eQTL or -sQTL. In quiescent SMCs, only 20% (1,008) of the 5,139 genes with a cis-eQTL or -sQTL were genetically regulated both at the mRNA splicing or expression levels (Poverlap = 4.7×10−136, hypergeometric test). Similarly, in proliferative SMCs, only 24% (1,522) of the 6,244 with a cis-eQTL or -sQTL were genetically regulated both at the mRNA splicing or expression levels (Poverlap = 9.1×10−189, hypergeometric test) (Supplementary Figure 11B). This suggests that genetic regulation of mRNA abundance and splicing are largely independent in SMCs, in agreement with studies in other tissues9,76.
Colocalization between sQTLs and CAD GWAS signals
To identify genes whose alternative splicing is associated with genetic risk for CAD, we performed colocalization analyses of splicing QTLs and CAD loci using four distinct approaches similar to the eQTL analysis described above. The intronic excision levels, as measured by LeafCutter46, of 100 and 120 sGenes in the quiescent and proliferative phenotypes were significantly associated with CAD loci, respectively. Colocalization of cis-sQTLs with 44 and 60 genes with CAD loci was unique to quiescent or proliferative SMCs, respectively (Figure 7A; Supplementary Table 13). Significantly more CAD genes were colocalized with sQTLs (164) than eQTLs (84). We examined whether the identified SMC sQTLs that colocalized with CAD could impact the ability of RNA binding proteins (RBPs) playing a role in splicing events. We used RBP-Var77, which provides extensive annotation for the functional variants involved in post-transcriptional interactions. RBP-Var includes collection of RBP binding SNPs which may disrupt the binding of RBPs, derived from crosslinking immunoprecipitation sequencing data sets for 60 RBPs and motif matching sites for 153 RBPs. When considering only 303 sQTL lead variants in 164 loci, 122 (40%) overlapped the binding site of an RBP (Supplementary Table 14).
Figure 7: Summary of the SMC sQTL and CAD GWAS colocalization and the 9p21 locus.
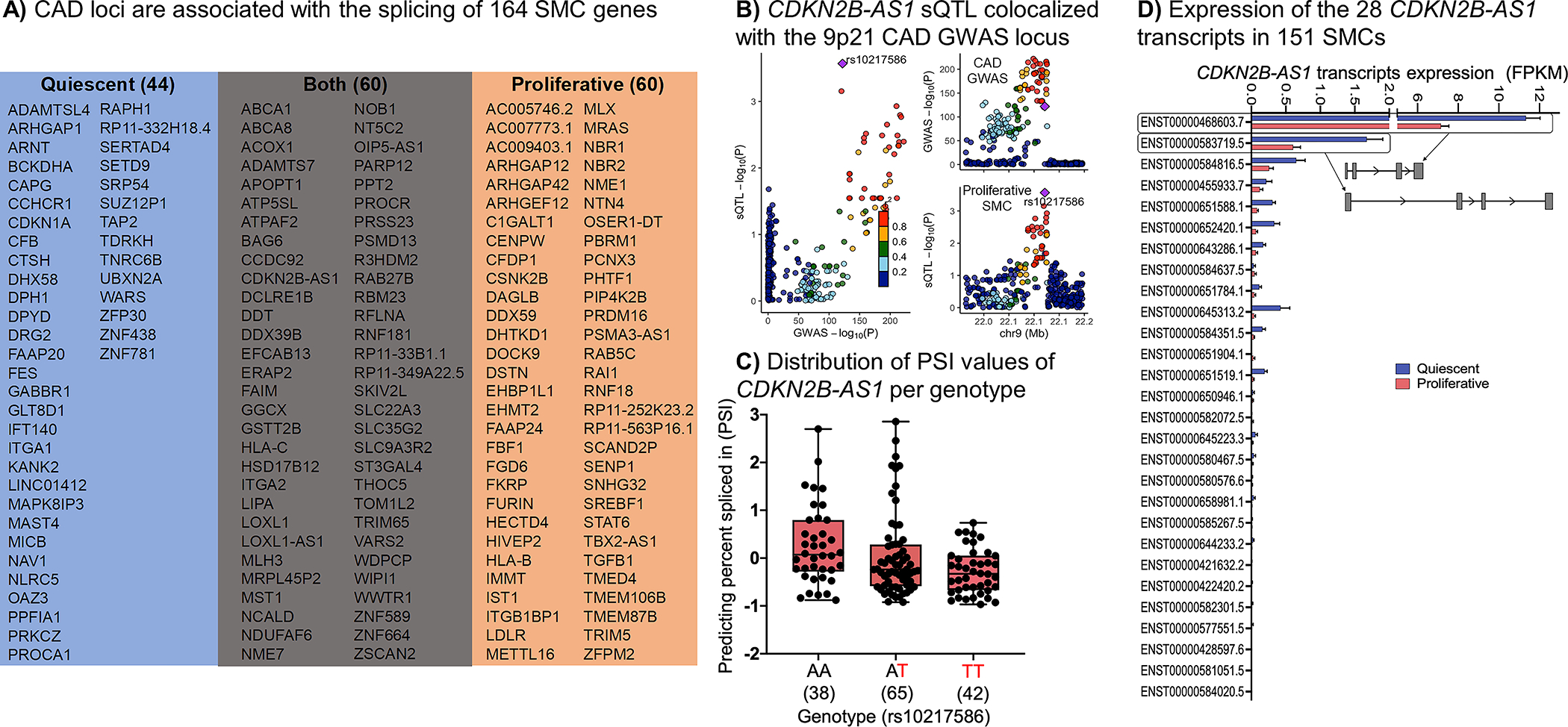
A) Shows the colocalization summary of CAD GWAS and sQTLs signals using four different methods in SMCs. B) CDKN2B-AS1 cis-sQTL signal colocalized with the 9p21 CAD GWAS locus. C) Distribution of the percent spliced in index (PSI) values for CDKN2B-AS1 intron based on the rs10217586 genotype. D) Average expression of the 28 CDKN2B-AS1 transcripts in 151 quiescent and proliferative SMCs. The standard error of the mean is shown. The isoform structures of the two most abundant transcripts are shown. Detailed isoform structures for all transcripts are in Supplementary Figure 14.
We identified 11 genes whose expression and alternative splicing was associated with the same CAD loci (Supplementary Table 15). These results point to the significant role of the genetic regulation of mRNA splicing as a molecular mechanism for CAD genetic risk.
We observed that the 9p21 locus, which has been the most significantly associated CAD locus in many populations, contains an sQTL for CDKN2B-AS1, also known as ANRIL (Figure 7A–C). We detected the expression of 25 of the 28 CDKN2B-AS1 transcripts in SMCs (Figure 7D, Supplementary Figure 12). Our results showed that the most significantly differentially excised intron at 9p21 (chr9:22064018-22096372) was found in CDKN2B-AS1. The frequency of this splicing event found in SMC proliferative phenotype (P = 2.7×10−4; β=−0.28) was colocalized with the genotype of the rs10217586 SNP in the CAD locus (P = 1.9×10−122; β=−0.16). Previous studies conducted in aortic endothelial cells21,22, monocytes23, whole blood24, coronary artery SMCs25, and umbilical artery SMCs78 did not identify an eQTL or sQTL for CDKN2B-AS1 in this locus, suggesting that the genetic variants in the 9p21 locus act in aortic SMCs through splicing.
DISCUSSION
GWAS have successfully identified 175 loci associated with CAD risk; however, the genes and mechanisms responsible for many of these loci remain unknown. Majority of the variants are in non-coding regions making the task of identifying causal variants and genes difficult. Systems genetics aims to address this challenge by associating genetic variants with molecular phenotypes to comprehensively uncover the relationship between genotype and phenotype. Colocalized molecular QTL signals enable identification of reasonable candidate genes in disease-relevant cells and tissues. Therefore, we conducted, as far as we know, the largest transcriptome and whole-genome analyses using human aortic SMCs derived from a multiethnic population. We cultured these critical vascular cell types to CAD in two different media formulations to recapitulate the atherosclerosis-relevant quiescent and proliferative state of SMCs. PCA of the transcriptome confirmed the distinctiveness of the two conditions and the comparison with publicly available datasets in GTEx revealed regulatory patterns specific to human SMCs.
Intersection of our SMC eQTL data with the GTEx dataset showed that more than half of SMC eQTLs were not evident in GTEx tissues, indicating genetic regulation of gene expression unique to SMCs. Another study also found about half of the eQTLs from aortic endothelial cells of up to 157 donors were absent in the GTEx dataset22. This is possibly because most GTEx eQTLs have been performed in heterogeneous tissue samples containing various cell types and the genetic effects that are functioning only in rare cell types within a sampled tissue may not be detected. Indeed, most of the SMC eQTLs shared with GTEx samples were in the tissues that are rich in SMCs. Differences in RNA sequencing methods may have also contributed to the differences between GTEx and our study. Cell-type-specific eQTL analysis in disease-relevant tissues will lead to the identification of novel and more precise disease associations that can help elucidate the molecular mechanisms by which the genetic variants affect the disease.
Overlapping TF binding sites with eQTL SNPs identified enrichment of putative binding sites for members of the SP2, SP1, ELK4, and GABPA TF families. While SP2 has an unknown role in SMCs, we predicted that the eQTL SNPs would impact SP2 binding to DNA in both the quiescent and proliferative phenotypes, suggesting an important role for this transcription factor in the regulation of gene expression by genetic variants in SMCs. Chromatin immunoprecipitation followed by sequencing (ChIP–seq) data for the promising TFs, SP2, SP1, ELK4, and GABPA, have yet to be generated in SMCs to support our ATAC-seq analyses. We also predicted the transcription factors KLF5, E2F1, and CTCF to be important for atherosclerosis as their binding sites are enriched in the proliferative SMCs. E2F1 is not only known to regulate cell proliferation79 but it is also upregulated in proliferative SMCs compared to quiescent SMCs suggesting it may be contributing to the phenotypic transition of SMCs from a quiescent to a proliferative state.
Several colocalization approaches have been developed in recent years80. They are sensitive to the parameters, such as thresholds applied to the prior probabilities, and differences in haplotype structures of the populations from which GWAS and molecular QTL data are derived. When the lead variants for the GWAS and eQTL studies are the same or in high LD in both populations, colocalization is straightforward81. Since the donors in our study population had various genetic ancestries and the CAD GWAS participants were of European ancestry, we used four different colocalization methods that may help to account for the differences in the LD structure. While multiple eQTLs and sQTLs had evidence for colocalization with CAD loci with two or more approaches, only seven genes (AL513548.3, EIF2B2, FES, FURIN, MAP3K7CL, SMAD3, REST) had evidence from all four approaches. Further development of colocalization methods is needed to increase the confidence in molecular QTL studies for identifying candidate genes for GWAS loci. For example, analytical approaches by modeling varying LD patterns across datasets with multi-ethnic populations will most likely enhance colocalization discoveries. Notably, most existing molecular QTL and GWAS studies are limited by the cost of the phenotyping, genotyping, and sequencing power and focus on people of European ancestry. In addition, the combination of the improved colocalization methods with data aggregation, e.g., a meta-analysis of molecular QTLs (sQTL, eQTL, pQTL, etc.) and other relevant molecular phenotype studies, such as data from single cells RNAseq, methylation, chromatin accessibility, and histone modification, could significantly enhance both the sensitivity and specificity of the colocalization findings.
In addition, while performing the colocalization analyses using molecular QTL results from a multiethnic population could identify common variants that have disease relevance for all ethnic groups, they could also miss some genetic-ancestry specific colocalizations due to differing haplotype structures between the populations where the molecular QTL studies and CAD GWAS were performed. Therefore, more studies, like the most recent CAD GWAS performed in a multi-ethnic population, are needed82.
A previous study predicted the colocalization of five genes with CAD loci using eQTL data from coronary artery SMCs of 52 donors cultured only under proliferative conditions25. Our study of 151 donors whose SMCs were cultured in two conditions significantly expands these previous findings. Only two of the five predicted causal genes in the coronary SMC study, FES and SMAD3, were replicated in our study. One of the five genes, TCF21, which encodes a transcription factor that inhibits SMC differentiation17,83, is expressed in coronary but not aortic SMCs; therefore, we were not able to test its association with genetic variants. While the other two genes, SIPA1 and PDGFRA, are expressed in aortic SMCs, we did not detect their colocalization with CAD loci. A recent study that was published78 while this manuscript was being revised identified 42,257 eGenes in umbilical cord aortic SMC from 1,486 donors. They identified 85 genes colocalized with CAD GWAS loci, 16 of which overlapped with our study (AL513548.3 (MIA3-AS1), ARHGAP42, CETP, EIF2B2, FES, FGD6, LINC02542, MAP1S, REST, MLH3, NEK9, SKIV2L, SMAD3, SMG6, SNHG18, TDRKH-AS1). The discrepancies between our study and these two studies may be related to differences in methods, such as the P-value thresholds for declaring a cis-eQTL or its colocalization significant in addition to the aortic bed where the SMCs were isolated from. By performing our studies in a large number of donors with deep RNA sequencing, we provide a significant number of the predicted causal CAD genes playing a role in SMCs. The differences between ascending aorta, coronary artery, and umbilical cord aorta SMC eQTLs point to differences in transcriptional regulation among the vascular beds. Thus, our aortic SMC design provides more generalizable knowledge for aortic diseases. Larger numbers of donors or meta-analysis of the SMC eQTL studies should lead to the identification of more causal genes associated with CAD.
Nearby genes to the index SNP in a locus are usually used as signposts for annotation. Previous CAD GWAS identified 366 nearby genes as potentially causal4. We found that only 26 and 33 of these nearby genes matched eQTL and sQTL, respectively. On the other hand, 173 genes were distinct from the initial locus annotations. 58 were derived from eQTL and 127 were derived from sQTL colocalizations. For example, rs11810571 in the 1q21.3 locus is located near TDRKH and RP11-98D18.9. However, our results showed significant colocalizations with the expression level of GOLPH3L, CTSK and CTSS located ~ 1 MB from the variant. For 8 loci, we identified multiple genes associated with the risk variants. For example, 15q21 risk locus was associated with FES and FURIN genes in proliferative state, while in quiescent SMCs it was associated with the expression of FES, RCCD1 and IQGAP1. Finally, previous GWAS identified missense mutations in 20 genes4. For two of the genes, LIPA and TRIM5, we also observed an eQTL effect. For three of the genes, ADAMTS7, DAGLB, DHX58, we also observed both eQTL and sQTL effects. These examples demonstrate the complexity of the molecular mechanisms by which CAD loci affect disease risk.
Long non-coding RNAs (lncRNAs) are typically >200 nucleotides in length and do not contain a functional open reading frame. They can be encoded within protein coding genes or can be encoded in the intergenic regions from the sense or antisense DNA. They are expressed at much lower levels relative to their protein coding counterparts84. By performing library preparation with ribosomal RNA depletion, as opposed to polyA selection, and deep sequencing, we were able to assess the expression of ~3,000 lncRNAs. Recent studies have shown that lncRNA plays an essential role in SMC biology and CAD85,86. We identified that CAD loci were associated with the expression of 12 lncRNAs and with the splicing of 15 lncRNAs. One of the colocalized lncRNA was small nucleolar RNA host gene 18 (SNHG18) which was regulated by the variants in the 5p15 CAD locus in both the quiescent and proliferative SMCs. While the role of SNHG18 in SMC biology and CAD have not been studied, it was observed to be upregulated in glioma and regulate the progression of epithelial-mesenchymal transition and cytoskeleton remodeling of glioma cells87.
Identifying tissue and cell-specific mechanisms of GWAS loci has been challenging with few notable exceptions88–90. We found that SMC-specific eQTL for ALKBH8 colocalized with the 11q22.3 CAD locus, with the risk allele leading to lower ALKBH8 expression. The CAD risk allele is also associated with higher blood pressure73, suggesting a role for this gene, which encodes a tRNA methyltransferase, in regulating the vascular tone. Embryonic fibroblasts isolated from Alkbh8-deficient mice were shown to have increased levels of intracellular reactive oxygen species, lipid peroxidation products and a transcript expression signature indicative of oxidative stress compared to fibroblasts isolated from wild-type littermates91. ALKBH8 agonists have been proposed for treating myocardial infarction injury due to its function on the modulation of autophagy and oxidative stress92.
Significant differences between sexes in the underlying pathology of atherosclerosis and its gene regulation have been described by us and others18,93. We had 118 male and 33 female donors in our population. The sex ratio of the donors is similar to the reported cases over a 20-year period in heart transplant donor registries, where 31.3% of the transplanted hearts are from deceased women94. Despite the imbalance in the numbers of males and females, which may have affected the statistical power of the interaction test, we were able to identify ~1,000 sex-biased eQTLs. One example of sex-biased eQTLs that colocalized with CAD GWAS signal was the TERF2IP gene. Lower TERF2IP leads to telomere elongation95, which is associated with decreased CAD risk96. Risk allele at this locus was associated with higher TERF2IP expression in males and lower expression in females. The same locus had an association with CAD risk only in males, suggesting that the lower TERF2IP expression in females may be playing a protective role against atherosclerosis. These results suggested that considering sex as a biological variable in cardiovascular research is essential to enhance our understanding of sex differences and to inform the development of sex-specific preventions and interventions in multi-ethnic populations.
Identifying genes whose expression is influenced by colocalizing cis-eQTL is just the first step in dissecting SMC and CAD GWAS loci. Discovering the functions of the predicted causal genes in SMC biology and CAD risk is also needed. We had previously shown that CAD loci are associated with atherosclerosis-relevant cellular phenotypes in the same donors74. We combined the two datasets to predict that DHODH and FGD6 are candidate causal genes that regulate SMC proliferation. DHODH encodes dihydroorotate dehydrogenase, which catalyzes the fourth enzymatic step in de novo pyrimidine biosynthesis. Its role in SMCs is not known but pyrimidine nucleotides are involved in the energetics of smooth muscle contracture97. A missense variant in FGD6 has been shown to increase the risk of polypoidal choroidal vasculopathy, which primarily affects the vascular layer of blood vessels in the choroid98.
The 175 CAD loci contain >6,000 SNPs and identifying which of these alter transcriptional activity in SMCs is a necessary step to dissect the molecular mechanism of the loci. We overlaid the genomic locations of eQTL SNPs with accessible chromatin regions to predict that 194 SNPs may alter gene expression, thereby significantly reducing the number of predicted causal variants in CAD loci. Our studies show that integrating across multiple scales, from genotype to cellular phenotypes, allows us to focus on a few plausible hypotheses to test in subsequent in vitro and in vivo studies to identify the molecular and cellular genetic mechanisms of CAD loci.
The landscape of CAD-relevant RNA splicing events are mostly unknown. We observed that significantly more CAD loci were associated with splicing than expression, suggesting that the majority of the genetic risk for CAD acts through regulating transcript splicing in SMCs rather than transcript abundance. This observation is in agreement with a previous study that showed that sQTLs are more likely to be enriched for Alzheimer’s disease GWAS SNPs than eQTLs76. Further, we observed that sQTLs that colocalized with CAD loci were associated with a distinct group of genes than eQTLs, indicating that our results can explain additional factors of the genetic architecture of CAD.
Of significant note, we identified the colocalization of CDKN2B-AS1 (ANRIL) sQTL with the 9p21 locus. This locus has been under intense scrutiny because it is the most significantly associated CAD locus and has been replicated in many populations with diverse ancestries99. Since there is no association with traditional risk factors such as dyslipidemia, diabetes mellitus, age, and sex, previous studies focused on identifying the effects at the vessel wall. eQTL studies in endothelial cells did not identify an impact of the genetic variants in this locus on gene expression22; however, they have been shown to regulate adhesion, contraction, and proliferation in SMCs derived from induced pluripotent stem cells100. Aortic SMCs isolated from mice with a knock-out of the homologous region showed excessive proliferation and diminished senescence101. When these mice were bred to an atheroprone background, they developed larger atherosclerotic plaques with no changes in blood pressure, lipid levels, body weight, or fasting glucose102. Primary SMCs were prone to dedifferentiation and had accelerated calcification, reflective of the susceptibility mechanisms of the humans carrying the risk allele. This region contains five tightly clustered genes, which partly overlap. CDKN2B-AS1 overlaps in antisense the full length of the CDKN2B gene body while sharing a bidirectional promoter with CDKN2A. There are 28 linear and multiple circular isoforms of ANRIL. We detected the expression of 25 of the 28 linear isoforms in SMCs. Previous studies showed associations of linear ANRIL isoforms, as well as CDKN2A and CDKN2B with the variants in the 9p21 locus in whole blood, peripheral blood monocytes, peripheral blood T lymphocytes, lymphoblastoid cells lines, vascular tissues such as carotid atherosclerotic plaque samples, aorta, mammary artery, as well as subcutaneous or omental adipose tissue99,103. Our study shows these variants affect linear ANRIL splicing in SMCs; however, the associations of these variants with circular forms of ANRIL remain to be determined. The mechanism by which ANRIL isoforms affect SMC functions such as proliferation, migration, and calcification also needs to be explored.
Collectively, our results predicted candidate causal genes playing a role in SMCs that modulate the genetic risk for CAD. Some of the loci act differentially in quiescent and proliferative SMC phenotypes emulating different stages of atherosclerosis. They also have distinct effects in males and females, and some are SMC-specific. Taken together, our results provide evidence for the complexity of the molecular mechanisms of CAD loci. We expect that our findings will provide a rich catalog of molecular QTLs to the cardiovascular community and candidates for future preclinical development.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Genome-wide association studies (GWAS) identified 175 loci associated with coronary artery disease (CAD).
Most of these loci are in non-coding regions, have unknown mechanisms, but are predicted to regulate gene expression.
The most significantly associated CAD GWAS locus, 9p21, remains a mystery after many years of studies.
Vascular smooth muscle cells (SMCs) play critical roles in the development and progression of CAD.
What New Information Does This Article Contribute?
We identify 84 genes whose expression and 164 genes whose splicing are regulated by loci associated with increased risk for CAD.
We discover distinct genetic architectures of gene expression in quiescent and proliferative SMC phenotypes.
We predict that long non-coding RNA SNHG18 is a likely causal transcript for CAD and affects SMC proliferation.
We identify the colocalization of TERF2IP with a sex-biased CAD locus.
We show that the 9p21, the most significantly associated CAD locus, affects the splicing of the long non-coding RNA CDKN2B-AS1, also known as ANRIL, in SMCs.
CAD is the leading cause of death worldwide. Recent meta-analyses of GWAS have identified 175 loci associated with CAD. Given that vascular SMCs play critical roles in the development and progression of CAD, we hypothesized that a subset of the CAD GWAS risk loci are associated with the regulation of transcription in distinct SMC phenotypes. We identified 4,910 eQTL and 4,412 sQTL that represent regions of the genome associated with transcript abundance and splicing. 3,660 of the eQTLs had not been observed in the publicly available GTEx dataset. We identified 84 eQTL and 164 sQTLs that colocalized with CAD loci. Notably, 20% and 35% of the eQTLs were unique to quiescent or proliferative SMCs, respectively. A CAD locus colocalized with a sex-specific eQTL, TERF2IP, and another locus colocalized with SMC-specific eQTL, ALKBH8. The most significantly associated CAD locus, 9p21, was an sQTL for the long non-coding RNA CDKN2B-AS1, also known as ANRIL, in proliferative SMCs. We created a user-friendly website (http://civeleklab.cphg.virginia.edu) for cardiovascular researchers to query our dataset. Our study provided evidence for the molecular mechanisms of genetic susceptibility to CAD in distinct SMC phenotypes.
Sources of Funding
This work was supported by an American Heart Association Postdoctoral Fellowship 18POST33990046 (to R.A.), Transformational Project Award 19TPA34910021 (to M.C.), National Institutes of Health Grants: R21HL135230 (to M.C.); R01HL148239 (to C.L.M); F31HL156463 (to D.W.), Academy of Finland (Grant No’s 287478 and 319324 to M.U.K), European Research Council Horizon 2020 Research and Innovation Programme (Grant No. 802825 to M.U.K), the Finnish Foundation for Cardiovascular Research (to M.U.K), and Transatlantic Network of Excellence Awards (12CVD02, 18CVD02) from Foundation Leducq (to M.C., J.B, H.dR., C.L.M) and its Junior Investigator Award (to R.A.)
Non-Standard Abbreviations and Acronyms
- ATACseq
Assay for Transposase-Accessible Chromatin with high-throughput sequencing
- CAD
Coronary artery disease
- CLPP
Colocalization posterior probability
- eQTL
Expression quantitative trait locus
- FBS
Fetal bovine serum
- GTEx
Genotype-Tissue Expression
- GWAS
Genome-wide association studies
- LD
linkage disequilibrium
- RBP
RNA binding protein
- SMC
Smooth muscle cells
- SNP
Single nucleotide polymorphism
- sQTL
Splicing quantitative trait locus
- STARNET
Stockholm-Tartu Atherosclerosis Reverse Networks Engineering Task
Footnotes
Disclosures
Johan Bjorkegren is a shareholder in Clinical Gene Network AB that has an invested interest in STARNET. The remaining authors have nothing to disclose
REFERENCES
- 1.Tsao CW, Aday AW, Almarzooq ZI, et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation. 2022;145:e153–e639. [DOI] [PubMed] [Google Scholar]
- 2.McPherson R, Tybjaerg-Hansen A. Genetics of Coronary Artery Disease. Circ Res. 2016;118:564–78. [DOI] [PubMed] [Google Scholar]
- 3.Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114:1241–1257. [DOI] [PubMed] [Google Scholar]
- 4.Harst P van der, Verweij N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease Novelty and Significance. Circ Res. 2018;122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishigaki K, Akiyama M, Kanai M, et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat Genet. 2020;52:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18:331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braenne I, Civelek M, Vilne B, et al. Prediction of Causal Candidate Genes in Coronary Artery Disease Loci. Arter Thromb Vasc Biol. 2015;35:2207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16:197–212. [DOI] [PubMed] [Google Scholar]
- 9.Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK. RNA splicing is a primary link between genetic variation and disease. Science. 2016;352:600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown CD, Mangravite LM, Engelhardt BE. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet. 2013;9:e1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fairfax BP, Makino S, Radhakrishnan J, Plant K, Leslie S, Dilthey A, Ellis P, Langford C, Vannberg FO, Knight JC. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat Genet. 2012;44:502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu J, Wolfs MG, Deelen P, et al. Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet. 2012;8:e1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hormozdiari F, Gazal S, van de Geijn B, et al. Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat Genet. 2018;50:1041–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cherepanova OA, Gomez D, Shankman LS, et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med. 2016;22:657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shankman LS, Gomez D, Cherepanova OA, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huize Pan, Chenyi Xue, Auerbach Benjamin J, et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation. 2020;142:2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wirka RC, Wagh D, Paik DT, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019;25:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartman RJG, Owsiany K, Ma L, et al. Sex-Stratified Gene Regulatory Networks Reveal Female Key Driver Genes of Atherosclerosis Involved in Smooth Muscle Cell Phenotype Switching. Circulation. 2021;143:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gamazon ER, Segrè AV, Bunt M van de, et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet. 2018;50:956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franzen O, Ermel R, Cohain A, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016;353:827–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erbilgin A, Civelek M, Romanoski CE, Pan C, Hagopian R, Berliner JA, Lusis AJ. Identification of CAD candidate genes in GWAS loci and their expression in vascular cells. J Lipid Res. 2013;54:1894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stolze LK, Conklin AC, Whalen MB, et al. Systems Genetics in Human Endothelial Cells Identifies Non-coding Variants Modifying Enhancers, Expression, and Complex Disease Traits. Am J Hum Genet. 2020;106:748–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeller T, Wild P, Szymczak S, et al. Genetics and beyond--the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu B, Pjanic M, Wang T, et al. Genetic Regulatory Mechanisms of Smooth Muscle Cells Map to Coronary Artery Disease Risk Loci. Am J Hum Genet. 2018;103:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roadmap Epigenomics C, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navab M, Imes SS, Hama SY, Hough GP, Ross LA, Bork RW, Valente AJ, Berliner JA, Drinkwater DC, Laks H. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest. 1991;88:2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmermann O, Zwaka TP, Marx N, Torzewski M, Bucher A, Guilliard P, Hannekum A, Hombach V, Torzewski J. Serum starvation and growth factor receptor expression in vascular smooth muscle cells. J Vasc Res. 2006;43:157–165. [DOI] [PubMed] [Google Scholar]
- 29.Han M, Wen J-K, Zheng B, Cheng Y, Zhang C. Serum deprivation results in redifferentiation of human umbilical vascular smooth muscle cells. Am J Physiol Cell Physiol. 2006;291:C50–58. [DOI] [PubMed] [Google Scholar]
- 30.1000Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. Robust relationship inference in genome-wide association studies. Bioinforma Oxf Engl. 2010;26:2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire M-D, Williams C, Reich M, Winckler W, Getz G. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung A, Stapleton K, Natarajan R. Functional Long Non-coding RNAs in Vascular Smooth Muscle Cells [Internet]. In: Morris KV, editor. Long Non-coding RNAs in Human Disease. Cham: Springer International Publishing; 2016. [cited 2020 Feb 16]. p. 127–141.Available from: 10.1007/82_2015_441 [DOI] [PubMed] [Google Scholar]
- 36.Lee S, Lee S, Ouellette S, Park W-Y, Lee EA, Park PJ. NGSCheckMate: software for validating sample identity in next-generation sequencing studies within and across data types. Nucleic Acids Res. 2017;45:e103–e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jun G, Flickinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, Kang HM. Detecting and Estimating Contamination of Human DNA Samples in Sequencing and Array-Based Genotype Data. Am J Hum Genet. 2012;91:839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinforma Oxf Engl. 2012;28:882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tarazona S, Furió-Tarí P, Turrà D, Pietro AD, Nueda MJ, Ferrer A, Conesa A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43:e140–e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012;7:500–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor-Weiner A, Aguet F, Haradhvala NJ, Gosai S, Anand S, Kim J, Ardlie K, Van Allen EM, Getz G. Scaling computational genomics to millions of individuals with GPUs. Genome Biol. 2019;20:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U A. 2003;100:9440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, Pritchard JK. Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet. 2018;50:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim-Hellmuth S, Bechheim M, Pütz B, et al. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat Commun. 2017;8:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis JR, Fresard L, Knowles DA, Pala M, Bustamante CD, Battle A, Montgomery SB. An Efficient Multiple-Testing Adjustment for eQTL Studies that Accounts for Linkage Disequilibrium between Variants. Am J Hum Genet. 2016;98:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Munz M, Wohlers I, Simon E, Reinberger T, Busch H, Schaefer AS, Erdmann J. Qtlizer: comprehensive QTL annotation of GWAS results. Sci Rep. 2020;10:20417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han B, Eskin E. Interpreting Meta-Analyses of Genome-Wide Association Studies. PLOS Genet. 2012;8:e1002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7. [DOI] [PubMed] [Google Scholar]
- 53.Hormozdiari F, van de Bunt M, Segrè AV, Li X, Joo JWJ, Bilow M, Sul JH, Sankararaman S, Pasaniuc B, Eskin E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am J Hum Genet. 2016;99:1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wallace C. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLOS Genet. 2020;16:e1008720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB. Abundant associations with gene expression complicate GWAS follow-up. Nat Genet. 2019;51:768–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, Liu T, Meyer CA, et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar S, Ambrosini G, Bucher P. SNP2TFBS – a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017;45:D139–D144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Örd T, Õunap K, Stolze LK, et al. Single-Cell Epigenomics and Functional Fine-Mapping of Atherosclerosis GWAS Loci. Circ Res. 2021;129:240–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020;38:276–278. [DOI] [PubMed] [Google Scholar]
- 64.ShinyGO: a graphical gene-set enrichment tool for animals and plants | Bioinformatics | Oxford Academic [Internet]. [cited 2022 Oct 6];Available from: https://academic.oup.com/bioinformatics/article/36/8/2628/5688742?login=true [DOI] [PMC free article] [PubMed]
- 65.Barry DM, Liu X-T, Liu B, et al. Exploration of sensory and spinal neurons expressing gastrin-releasing peptide in itch and pain related behaviors. Nat Commun. 2020;11:1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oliva M, Muñoz-Aguirre M, Kim-Hellmuth S, et al. The impact of sex on gene expression across human tissues. Science. 2020;369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anderson WD, Soh JY, Innis SE, et al. Sex differences in human adipose tissue gene expression and genetic regulation involve adipogenesis. Genome Res. 2020;30:1379–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crook MF, Olive M, Xue H-H, Langenickel TH, Boehm M, Leonard WJ, Nabel EG. GA-binding protein regulates KIS gene expression, cell migration, and cell cycle progression. FASEB J. 2008;22:225–235. [DOI] [PubMed] [Google Scholar]
- 70.Kimura TE, Duggirala A, Hindmarch CCT, Hewer RC, Cui M-Z, Newby AC, Bond M. Inhibition of Egr1 expression underlies the anti-mitogenic effects of cAMP in vascular smooth muscle cells. J Mol Cell Cardiol. 2014;72:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang X, Li R, Qin X, Wang L, Xiao J, Song Y, Sheng X, Guo M, Ji X. Sp1 Plays an Important Role in Vascular Calcification Both In Vivo and In Vitro. J Am Heart Assoc. 7:e007555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aherrahrou R, Guo L, Nagraj VP, et al. Genetic Regulation of Atherosclerosis-Relevant Phenotypes in Human Vascular Smooth Muscle Cells. Circ Res. 2020;127:1552–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Redouane Aherrahrou, Liang Guo, Peter Nagraj V., et al. Genetic Regulation of Atherosclerosis-Relevant Phenotypes in Human Vascular Smooth Muscle Cells. Circ Res. 2020;127:1552–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng R, Yao Q, Ren C, Liu Y, Yang H, Xie G, Du S, Yang K, Yuan Y. Upregulation of Long Noncoding RNA Small Nucleolar RNA Host Gene 18 Promotes Radioresistance of Glioma by Repressing Semaphorin 5A. Int J Radiat Oncol. 2016;96:877–887. [DOI] [PubMed] [Google Scholar]
- 76.Raj T, Li YI, Wong G, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet. 2018;50:1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mao F, Xiao L, Li X, Liang J, Teng H, Cai W, Sun ZS. RBP-Var: a database of functional variants involved in regulation mediated by RNA-binding proteins. Nucleic Acids Res. 2016;44:D154–D163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Solomon CU, McVey DG, Andreadi C, et al. Effects of Coronary Artery Disease–Associated Variants on Vascular Smooth Muscle Cells. Circulation. 2022;146:917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dubrez L Regulation of E2F1 Transcription Factor by Ubiquitin Conjugation. Int J Mol Sci. 2017;18:2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wainberg M, Sinnott-Armstrong N, Mancuso N, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51:592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu Y, Broadaway KA, Raulerson CK, et al. Colocalization of GWAS and eQTL signals at loci with multiple signals identifies additional candidate genes for body fat distribution. Hum Mol Genet. 2019;28:4161–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tcheandjieu C, Zhu X, Hilliard AT, et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat Med. 2022;28:1679–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nagao M, Lyu Q, Zhao Q, et al. Coronary Disease-Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ Res. 2020;126:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Turner AW, Wong D, Khan MD, Dreisbach CN, Palmore M, Miller CL. Multi-Omics Approaches to Study Long Non-coding RNA Function in Atherosclerosis. Front Cardiovasc Med. 2019;6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fasolo F, Jin H, Winski G, et al. Long Noncoding RNA MIAT Controls Advanced Atherosclerotic Lesion Formation and Plaque Destabilization. Circulation. 2021;144:1567–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jaé N, Dimmeler S. Noncoding RNAs in Vascular Diseases. Circ Res. 2020;126:1127–1145. [DOI] [PubMed] [Google Scholar]
- 87.Zheng R, Yao Q, Li X, Xu B. Long Noncoding Ribonucleic Acid SNHG18 Promotes Glioma Cell Motility via Disruption of α-Enolase Nucleocytoplasmic Transport. Front Genet. 2019;10:1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Small KS, Todorčević M, Civelek M, et al. Regulatory variants at KLF14 influence type 2 diabetes risk via a female-specific effect on adipocyte size and body composition. Nat Genet. 2018;50:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krause MD, Huang R-T, Wu D, et al. Genetic variant at coronary artery disease and ischemic stroke locus 1p32.2 regulates endothelial responses to hemodynamics. Proc Natl Acad Sci U S A. 2018;115:E11349–E11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Endres L, Begley U, Clark R, Gu C, Dziergowska A, Małkiewicz A, Melendez JA, Dedon PC, Begley TJ. Alkbh8 Regulates Selenocysteine-Protein Expression to Protect against Reactive Oxygen Species Damage. PLOS ONE. 2015;10:e0131335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xiao M-Z, Liu J-M, Xian C-L, Chen K-Y, Liu Z-Q, Cheng Y-Y. Therapeutic potential of ALKB homologs for cardiovascular disease. Biomed Pharmacother Biomedecine Pharmacother. 2020;131:110645. [DOI] [PubMed] [Google Scholar]
- 93.Yahagi K, Davis HR, Arbustini E, Virmani R. Sex differences in coronary artery disease: Pathological observations. Atherosclerosis. 2015;239:260–267. [DOI] [PubMed] [Google Scholar]
- 94.Khush KK, Kubo JT, Desai M. Influence of donor and recipient sex mismatch on heart transplant outcomes: Analysis of the International Society for Heart and Lung Transplantation Registry. J Heart Lung Transplant. 2012;31:459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cai Y, Kandula V, Kosuru R, Ye X, Irwin MG, Xia Z. Decoding telomere protein Rap1: Its telomeric and nontelomeric functions and potential implications in diabetic cardiomyopathy. Cell Cycle Georget Tex. 2017;16:1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Codd V, Nelson CP, Albrecht E, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45:422–7, 427e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.James SG, Appleby GJ, Miller KA, Steen JT, Colquhoun EQ, Clark MG. Purine and pyrimidine nucleotide metabolism of vascular smooth muscle cells in culture. Gen Pharmacol Vasc Syst. 1996;27:837–844. [DOI] [PubMed] [Google Scholar]
- 98.Huang L, Zhang H, Cheng C-Y, et al. A missense variant in FGD6 confers increased risk of polypoidal choroidal vasculopathy. Nat Genet. 2016;48:640–647. [DOI] [PubMed] [Google Scholar]
- 99.Holdt LM, Teupser D. Long Noncoding RNA ANRIL: Lnc-ing Genetic Variation at the Chromosome 9p21 Locus to Molecular Mechanisms of Atherosclerosis. Front Cardiovasc Med. 2018;5:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lo Sardo V, Chubukov P, Ferguson W, et al. Unveiling the Role of the Most Impactful Cardiovascular Risk Locus through Haplotype Editing. Cell. 2018;175:1796–1810.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, Blow MJ, Cohen JC, Rubin EM, Pennacchio LA. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kojima Y, Ye J, Nanda V, et al. Knockout of the Murine Ortholog to the Human 9p21 Coronary Artery Disease Locus Leads to Smooth Muscle Cell Proliferation, Vascular Calcification, and Advanced Atherosclerosis. Circulation. 2020;141:1274–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Civelek M, Wu Y, Pan C, et al. Genetic Regulation of Adipose Gene Expression and Cardio-Metabolic Traits. Am J Hum Genet. 2017;100:428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.