

Abstract
The positive transcription elongation factor b (P-TEFb) is composed of cyclins T1 or T2 and cyclin dependent kinase 9 that regulate the elongation phase of transcription by RNA polymerase II. By antagonizing negative elongation factors and phosphorylating the C-terminal domain of RNA polymerase II, P-TEFb facilitates the elongation and co-transcriptional processing of nascent transcripts. This step is critical for the expression of most eukaryotic genes. In growing cells, P-TEFb is negatively regulated by its reversible associations with HEXIM1/2 in the 7SK snRNP and positively by a number of transcription factors as well as the super elongation complex. In resting cells, P-TEFb falls apart and cyclin T1 is degraded by the proteasome. This complex regulation of P-TEFb has evolved for the precise temporal and spatial regulation of gene expression in the organism. Its dysregulation contributes to inflammatory and neoplastic conditions.
eTOC
P-TEFb promotes transcription elongation by releasing the pause of RNAPII. Research on P-TEFb has expanded our understanding of eukaryotic transcription and diseases such as AIDS and cancer. In this review, Fujinaga et. al. focus on the structure, function and regulation of P-TEFb.
Control of RNAPII elongation.
Eukaryotic transcription of protein-coding genes by RNA polymerase II (RNAPII) is a multi-step process consisting of initiation, promoter clearance, elongation, co-transcriptional processing of nascent transcripts, termination, mRNA cleavage and polyadenylation (Fig.1) 1. Initiation of transcription occurs when RNAPII pre-initiation complex (PIC) is recruited by general transcription factors and the mediator complex at the promoter to the transcription start site. Clearance of RNAPII from the promoter requires the phosphorylation of serines at position 5 (S5) in the tandemly repeated heptapeptide (52 repeats of Y1-S2-P3-T4-S5-P6-S7) of its C-terminal domain (CTD) by CDK7 from the transcription factor-II H (TFIIH) 1. Subsequently, to the 5’-end of each transcript is added 7-methylguanosine cap by the capping enzyme (CE), which protects it from exonuclease digestion 1. Immediately after copying 20–100 nucleotides, RNAPII stalls and remains attached to the DNA template because of negative transcription elongation factors (NTEFs) such as the 5,6-dichloro-1-b-D-ribofuranosylbenzimidazole (DRB) sensitivity-inducing factor (DSIF) and the negative elongation factor (NELF) 2. Although DSIF and NELF have little effect alone, their combination blocks the elongation of RNAPII 3. NELF binds to RNA 4 and a funnel region of RNAPII, which stabilizes the RNA-DNA hybrid in a tilted conformation and immobilizes RNAPII (Fig. 1 bottom left) 5. Interactions between NELF, DSIF and RNAPII also prevent the recruitment of the transcript cleavage factor TFIIS and the elongation promoting polymerase-associated factor (PAF) 6. Since TFIIS can relieve strong pauses and arrests, this inhibitory property stabilizes the confirmation of the paused RNAPII 6. Switching from this paused state to productive transcription elongation is achieved by P-TEFb, which contains cyclins T1 (CycT1), T2a (CycT2a) or T2b (CycT2b) and CDK9 7. Upon recruitment to the paused RNAPII, CDK9 phosphorylates serines at position 2 (S2) in the CTD of RNAPII 8. CDK9 can also phosphorylate serines at positions 5 and 7 (S5 and S7) in the heptapeptide repeats 9,10. Precise roles of these additional phosphorylations during transcription elongation need to be elucidated. NELF-A and E as well as multiple sites in DSIF are also substrates of CDK9 5,11,12. Upon phosphorylation, NELF dissociates from the RNAPII, and is replaced by TFIIS and PAF. SPT6 is recruited to phosphorylated CTD 3,13. DSIF is converted to a positive elongation factor and remains associated with RNAPII 3,13,14. In the RNAPII:PAF structure, the DNA-RNA hybrid is relaxed, which creates a permissive confirmation for RNA synthesis 5 (Fig.1, bottom right). This P-TEFb-dependent release of RNAPII is antagonized by the protein phosphatase (PP) 2A, PP4 and/or a noncanonical PP2A via the integrator complex, which fine-tunes transcription elongation 15–17. The phosphorylated CTD acts as a scaffold for splicing (SR) and polyadenylation (pA) machineries 1. In addition, chromatin-modifying enzymes and other elongation factors travel with the heavily phosphorylated RNAPII 18. Therefore, P-TEFb is the critical factor that releases the pause of RNAPII and promotes transcription elongation.
Figure 1.
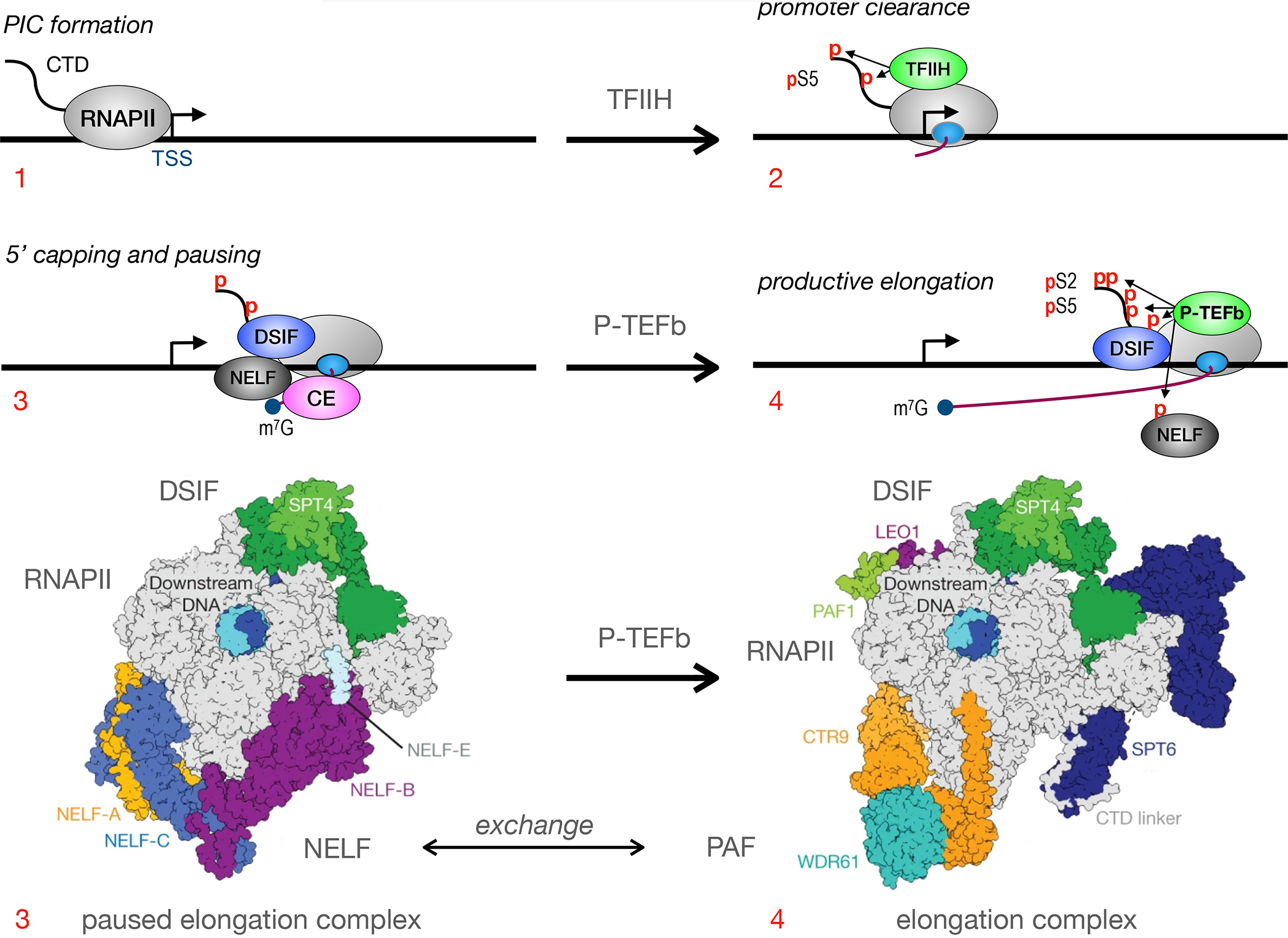
Early steps in RNAPII transcription. Depicted in top panels are: 1. the formation of the pre-initiation complex (PIC); 2. promoter clearance and early phosphorylation of serines at position 5 (pS5) in the C-terminal domain (CTD) of RNAPII by CDK7: 3. RNAPII pausing via DRB sensitivity (DSIF) and negative elongation (NELF) factors and 5’ capping (m7G) of early transcripts by the capping enzyme (CE); 4. recruitment of P-TEFb, the phosphorylation of S2 (pS2) in the CTD, DSIF and NELF by CDK9, followed by transcription elongation. As presented in bottom panels, paused and released elongation complexes now have greater structural understanding. In the paused state, NELF stabilizes the tilted RNA:DNA hybrid state, inhibits the diffusion of nucleotides and obstructs the binding of TFIIS and PAF. For the release of RNAPII to elongation, P-TEFb phosphorylates CTD as well as subunits of NELF and DSIF. NELF dissociates from RNAPII. TFIIS and PAF can now bind to RNAPII. Together with DSIF, they help to relieve the tilt of the RNA-DNA hybrid. They also stabilize the transcription funnel. Co-transcriptional processing of nascent transcripts ensues, i.e. their splicing, termination, polyadenylation and export. TSS is the transcription start site, where the arrow points in the direction of transcription. Other markings: the gray ovals represent RNAPII; its inner smaller oval is where early transcripts emerge; the S shaped line is the CTD with red small ps indicating phosphorylated residues; the blue circle depicts capped mRNA species. Depicted are NELF subunits A, B, C and E, SPT4 and 6, as well as PAF subunits PAF1, LEO1, CTR9 and WDR61. Bottom panels are adapted from Vos et al 5.
In early days, the control of transcription elongation was studied in specific transcription units such as c-myc, human immunodeficiency virus type 1 (HIV), and heat shock protein (HSP) genes 8. However, more recent genome-wide studies revealed that promoters of most inactive and inducible genes are already engaged by stalled RNAPII 19,20. Therefore, the elongation step of transcription, which is regulated by P-TEFb, is the main gate keeper of eukaryotic gene expression. While early transcription elongation is mediated largely by P-TEFb, later stages are also impacted by other CDKs that also phosphorylate S2 in the CTD. For example, CycK:CDK12 are recruited to the phosphorylated CTD by SR proteins and/or PAF1C and mediate the cleavage and polyadenylation (CPA) of some genes 21,22. On other genes, CycL:CDK11 are recruited by the TREX/THO (transcription/export // transcriptional defects of Hpr1Δ by over-expression) complex or FLASH (Fas-via death domain-associated-like ICE-associated huge protein) to facilitate their CPA and mRNA export 23,24. Since P-TEFb is also involved in splicing and 3’ end formation 25,26, it remains to be investigated how all these different CDKs coordinate their effects during transcription.
P-TEFb was discovered in early 1990’s as a nuclear factor that stimulates transcription elongation 27. First, the ATP analog DRB was demonstrated to inhibit greatly overall transcription with the recovery of only short, capped transcripts. In vitro transcription elongation was inhibited as well. Next, the use of a fly in vitro transcription system demonstrated that DRB did not inhibit RNAPII but a factor required for its elongation, most likely a kinase 27. Named P-TEFb, it was subsequently purified as a heterodimer, where its smaller subunit (~42K) was identified as CDK9 28. Next, its cyclin subunit, CycT1, was co-purified with the transactivator of transcription (Tat) from HIV 29. Thus, P-TEFb became central to our understanding of viral replication. CycT2a and CycT2b were cloned soon thereafter 30. Subsequently, many cellular transcription factors, chromatin remodeling machineries, and other proteins were demonstrated to interact with P-TEFb, leading to the current model of transcription elongation by RNAPII.
The structure of P-TEFb
In human cells, different P-TEFb complexes exist 8. They contain CycT1, CycT2a or CycT2b and shorter or longer forms of CDK9 (42 kDa and 55 kDa) 8. The two isoforms of CDK9 are produced from different transcription start sites in the first exon of the CDK9 gene 31. For purposes of this review, only CycT1 and the shorter CDK9 are diagramed in Fig. 2 (left). The N-terminal lobe (residues from positions 16 to 108), and C-terminal lobe (residues from positions 109–330) of CDK9 form a cleft which is highly conserved among CDKs (Fig. 2, right). CycT1 binds to CDK9 at the N-terminal lobe. The N-terminal lobe also contains the G-loop domain required for ATP-binding 9. The aspartate at position 104 (D104) and cysteine at position 106 (C106) interact directly with ATP. The adjacent phenylalanine at position 103 (F103) determines the specificity of nucleotide binding and is therefore called the gatekeeper. A mutation of this residue (F103A) sensitizes CDK9 for bulkier ATP analog inhibitors, which do not inhibit other cellular kinases. Such an analog-sensitive CDK9 protein was utilized to identify additional substrates of CDK9 32,33. The PITALRE motif which is unique for each CDK is also located within the N-terminal lobe. The substrate-binding site is located in the cleft formed by N- and C-terminal lobes. It targets consensus CDK substrate motifs consisting of serine/threonine-proline-X-arginine/lysine (S/T-P-X-K/R) residues 9. Next is the DFG (aspartate, phenylalanine, glycine) motif that binds to Mg++. When CycT1 is absent, the cleft is masked by the self-inhibitory domain called the T-loop. The kinase activity of CDK9 requires that the threonine at position 186 (T186) in the T-loop be phosphorylated. CDK7 and CDK9 are candidates for the responsible CDK-activating kinase (CAK) 9,34. The serine at position 175 (S175) is also phosphorylated, which can affect the function of P-TEFb 35,36 (Fig. 2, right). Additional phosphorylation of the C-terminus of CDK9 improves interactions between P-TEFb, Tat and its target, the transactivation response (TAR) RNA 37.
Figure. 2.
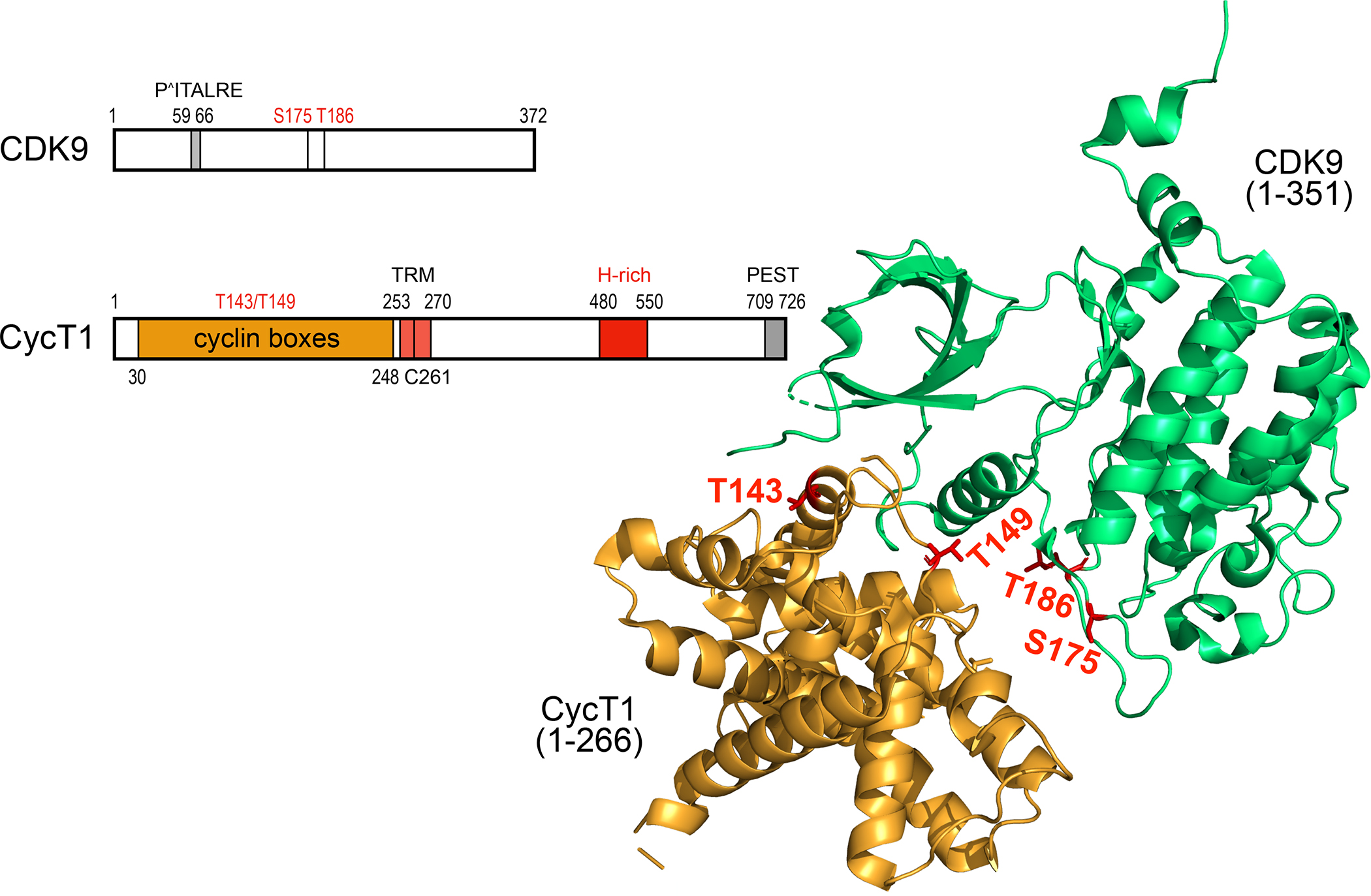
Schematic representation and the 3-D structure of human CDK9 and CycT1 proteins. CDK9 contains 372 residues. From N- to C-termini, important elements contain the PITALRE sequence. serine at positions 175 (S175) and threonine at position 186 (T186, T-loop). S175 and T186 are phosphorylated in the active P-TEFb. CycT1 contains 726 residues. From the N-terminus, there are 2 cyclin boxes (yellow), the Tat-TAR recognition motif (TRM), the histidine-(H)-rich region (red) and the putative PEST sequence (gray). Threonines at positions 143 and 149 (T143. T149) are targets of PKC and facilitate the binding between CDK9 and CycT1. In the unbound CycT1, the degron, where Siah1/2 bind, is located between position 210 and 260. The TRM is the binding site for the HIV transcriptional transactivator Tat. The H-rich sequence binds to targets of CDK9, i.e. the CTD of RNAPII, DSIF and NELF, among others. The function of the PEST sequence remains enigmatic. Some of these features are presented on the 3-D structure of P-TEFb 45,46. The ordered structure (ribbons) contains 266 residues from CycT1 and 351 residues from CDK9. T143 and T149 in CycT1 form intra- and inter-molecular contacts with CycT1 and CDK9, respectively. T186 (T-loop) and S175 in CDK9 are phosphorylated in the active P-TEFb.
The N-termini of CycT1, CycT2a and CycT2b consist of two highly conserved cyclin boxes (residues from positions 30 to 248), which are essential for CDK9 binding (Fig. 2) 30. Phosphorylation at the two evolutionally conserved threonine residues at positions 143 (T143) and 149 (T149) by the protein kinase C (PKC) potentiates interactions between CDK9 and CycT1 (P-TEFb assembly) 38. In the absence of CDK9, cyclin boxes bind to ubiquitin E3 ligases Siah (seven in absentia homolog)1/2, which mediate the polyubiquitination of CycT1 and lead to its rapid degradation 39. In CycT1, these cyclin boxes also serve as a platform for binding to Tat and other transcription factors 8. A short stretch of lysines and arginines adjacent to these cyclin boxes serves as the nuclear localization signal (NLS) 29. This region also includes the Tat-TAR recognition motif (TRM) (residues from positions 253 to 270). Interestingly, the unique cysteine at position 261 (C261) in the TRM of the human CycT1 is critical for its ternary complex with Tat and TAR 40. Thus, CycT2a and CycT2b as well as the mouse CycT1 fail to mediate Tat transactivation due to the absence of this C261. Next, there is a histidine-rich (H-rich) region from positions 480 to 550 that mediates direct interactions between P-TEFb and the CTD of RNAPII 41. Recent studies also revealed that it is required for the tight association between P-TEFb and chromatin and for creating functional phase-separated compartments with RNAPII in nuclear speckles. There, kinases and substrates are densely packed, providing an optimal environment for transcription elongation 42. Interestingly, poly ADP-ribosylation of the H-rich region in CycT1 upon DNA-damage prevents the transition of P-TEFb into these phase-separated compartments, which can shut down transcription globally 43. In contrast to consensus N-termini, C-termini of CycT proteins are less well conserved. The unique C-terminus of CycT1 from positions 709 to 726 resembles the canonical PEST (proline, glutamate, serine, threonine) motif (Fig. 2) 29. It can be targeted by Skp2 (S-phase kinase associated protein 2), which mediates the subsequent ubiquitination of CDK9 44. However, the removal of this PEST motif does not affect the degradation of P-TEFb in cells 39.
Several crystal structures of P-TEFb have been reported 45,46. They contain only the N-terminal 266 residues of CycT1 and 351 residues of CDK9 (Fig. 2, right). These structures confirm previously mapped surfaces between CycT1 and CDK9 and conform to those found in other CDKs. Tat was also co-crystalized with P-TEFb. Its residues from positions 1 to 49 bind tightly to CycT1 and also to sequences near the T-loop in CDK9. Finally, subunits of the super elongation complex (SEC) were found to interact with P-TEFb, especially AFF1 and AFF4 (AF4/FMR2 family members 1 and 4) 47. From the N- to the C-terminus, different surfaces on AFF1/4 bind to CycT1, ELL2 (elongation factor for RNAPII 2) and ENL/AF9 (eleven-nineteen-leukemia protein/ALL1-fused gene from chromosome 9). In these structures, Tat and SEC bind to non-overlapping surfaces on P-TEFb and create a more stable multi protein complex for transcription 47. These structures also suggest surfaces, where small molecules could interfere with Tat transactivation and HIV transcription 48.
The function of P-TEFb
P-TEFb is recruited to the paused RNAPII by various mechanisms including (1) DNA-tethering, (2) RNA-tethering, and (3) sequence-independent mechanisms (Fig. 3 top) 8. Many DNA-bound activators mediate their enhancer function via P-TEFb. They include NF-κB, c-Myc, the Signal Transducer and Activator of Transcription (STAT) 3, the Myoblast Determination protein (MyoD), Myocyte Enhancer Factor-2 (MEF2), as well as hormone receptors (estrogen and androgen receptors) 49. This P-TEFb-dependent transcription activation can be recapitulated by artificial DNA-tethering systems. For example, CycT1 fused to the yeast Gal4 DNA binding domain activates transcription of reporter genes containing its target sequences upstream or downstream of promoters and coding sequences 41. This activity requires the H-rich region that binds to the CTD of RNAPII and NTEFs. Therefore, interactions between distal sites, CycT1 (also CycT2a and CycT2b) and the CTD enable P-TEFb to act as a co-activator of transcription and P-TEFb can link activators at enhancers and PIC at promoters, which results in transcription elongation (Fig. 3, top) 41.
Figure 3.
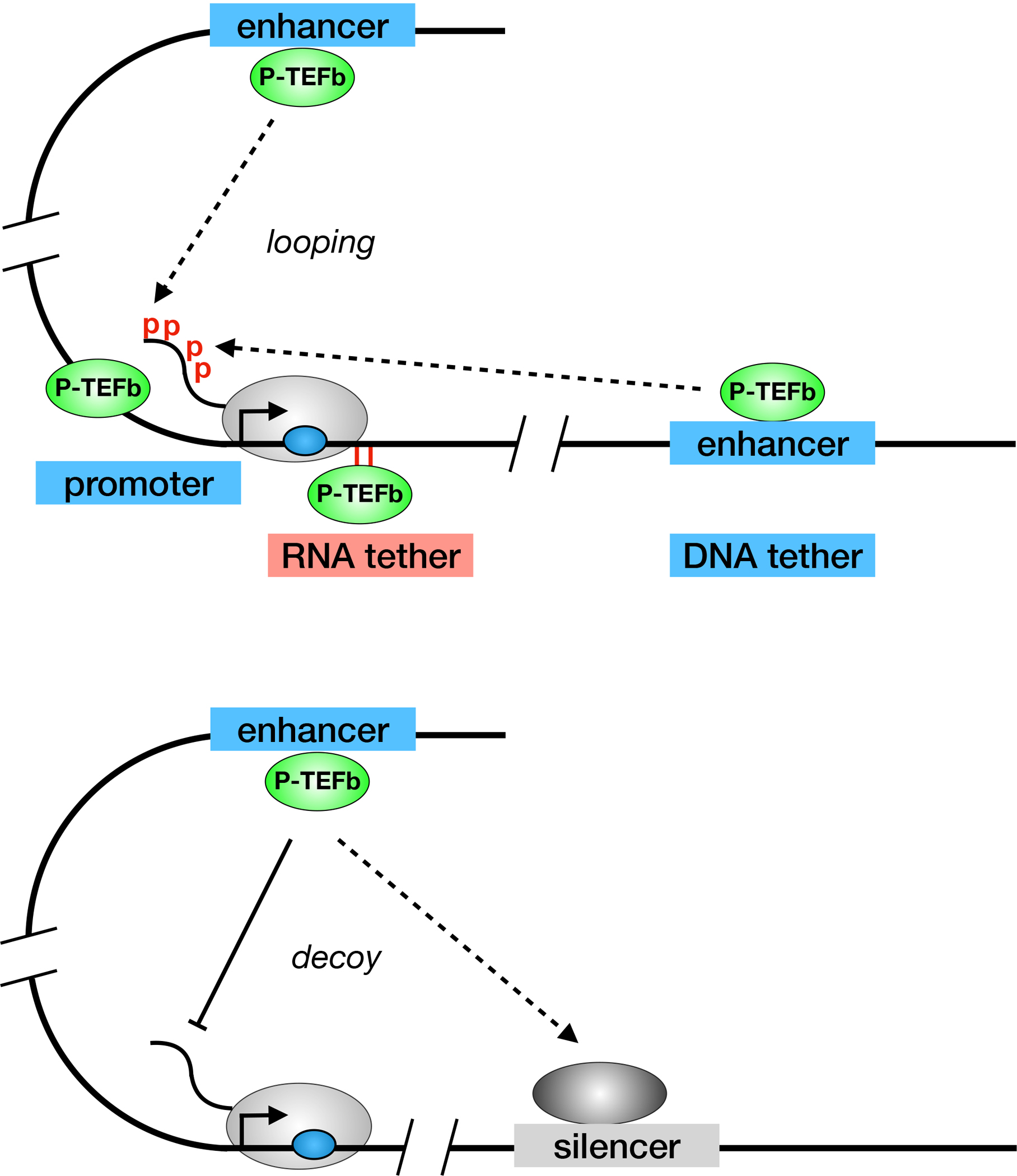
P-TEFb is critical for transcription elongation of most eukaryotic genes. Top panel depicts how P-TEFb can be recruited to RNAPII from promoters, 5’ and 3’ enhancers (via looping), and even RNA, as is the case with Tat from HIV. Bottom panel depicts how some silencers and/or repressors of transcription decoy P-TEFb away from RNAPII. Such is the case with global transcriptional repression by PIE1 and temporary silencing of the CD4 gene by RunX1. The other markings are as in Fig. 1.
P-TEFb can also be recruited to RNAPII via RNA. The most prominent example is Tat from HIV, which brings P-TEFb to TAR RNA 40. Such recruitment of transcription factors via RNA had not been observed previously in eukaryotic systems. Again, this RNA-enhancer function can be recapitulated by artificial RNA-tethering systems, including the coat protein of the bacteriophage MS2/R17, which binds to its operator RNA to block translation in bacteria, or to the regulator of viral gene expression (Rev), which binds to the Rev response element (RRE) RNA in the env gene of HIV 50,51. CycT1 or CDK9 fused to Rev activate transcription from an RRE’s Stem-Loop IIB (SLIIB)-containing reporter gene to levels that are similar to Tat-transactivation 52. This finding implies that interactions between P-TEFb and nascent RNA are sufficient for the stimulation of transcription elongation (Fig. 3, top). Since RNA passes the CTD co-transcriptionally, a shorter CycT1 (from positions 1 to 280) is equally effective as its wild type counterpart 53. Importantly, studies indicate that only proteins that interact with P-TEFb can activate transcription in this assay 54. Thus, RNA-tethering represents a genetic assay for activators that recruit P-TEFb for their effects.
Finally, there are mechanisms of P-TEFb recruitment independent of DNA- or RNA-sequences. They include co-activators such as the autoimmune regulator (AIRE) and the class II transactivator (CIITA) that require P-TEFb for their activities 8. Other factors directly associated with RNAPII, such as components of the mediator complex (Med26) also interact with P-TEFb and recruit it to the paused RNAPII 55. Components of the SEC, such as AFF1 and AFF4, also bind to P-TEFb and form protein complexes containing multiple transcription factors 56. Finally, P-TEFb can be recruited to chromatin by the bromodomain containing protein 4 (BRD4), whose bromodomain binds to the acetylated histone H3 (H3K27Ac) 57. Although BRD4 interacts with the active histone code (H3K27Ac), the precise role of this recruitment of P-TEFb for the global or gene-specific transcription needs to be elucidated. Of note, the pharmacological degradation of BRD4 by a small molecule degrader of BET family proteins (dBET1) results in global inhibition of transcription elongation without affecting the chromatin localization of CDK9, suggesting that P-TEFb recruitment can occur independently of BRD4 58. Moreover, BRD4 is dispensable for effects of P-TEFb on NF-κB and AIRE 59 and some genes require other factors, such as JMJD6 (Jumonji Domain Containing 6) for their transcription 60. Nevertheless, interactions between BRD4 and P-TEFb were reported to act as the bookmark of late mitosis, especially for the expression of genes required for the progression of the cell cycle 61.
Given that the recruitment of P-TEFb to paused RNAPII results in transcription elongation of target genes, active repression can be achieved simply by inhibiting the substrate recognition of CDK9 (Fig. 3, bottom). The best candidates contain sequences that resemble the CTD but cannot be phosphorylated. Indeed, when S2 and S5 in the heptad repeats are changed to alanines, this unmodifiable pseudosubstrate blocks the activity of P-TEFb. PIE-1 from C. elegans contains such a changed heptapeptide repeat and inhibits P-TEFb globally 62. Other repressors, such as Runx1, which binds to the CD4 silencer and represses the expression of the CD4 gene during T cell development in the thymus, also contains such an inhibitory domain. Importantly, this Runx1-dependent inhibition occurs despite the engagement of RNAPII on the CD4 promoter (Fig. 3B) 63. Another mechanism of antagonizing P-TEFb during transcription elongation is to recruit specific protein phosphatases, which include canonical PP2A, PP4, and/or atypical PP2A that dephosphorylate P-TEFb substrates, to RNAPII 15–17.
P-TEFb not only phosphorylates the CTD in RNAPII, but has additional protein targets. Many of them are involved in transcription, e.g. RNA processing and 3’ end formation. For example, by phosphorylating and activating the exoribonucease Xrn2, P-TEFb affects CPA 33. P-TEFb also phosphorylates and inhibits PP1, which stabilizes DSIF and RNAPII during transcription elongation. In addition, P-TEFb phosphorylates and activates UBE2A histone ubiquitin E2 ligase 64. The ubiquinated histone H2B (H2BUb), regulates gene expression and DNA repair. Additional protein targets were revealed using an analogue sensitive CDK9 and mass spectrometry 32,33. They are involved in chromatin remodeling, RNA splicing, RNA turnover, cell cycle progression and apoptosis 32,33. Further validation and analysis of these substrates will reveal new cellular processes that are impacted P-TEFb.
The regulation of P-TEFb
7SK snRNP
In proliferating cells, there exists an equilibrium between the active (free) P-TEFb and its inactive (sequestered) form in the 7SK small nuclear ribonucleoprotein (7SK snRNP) complex (Fig. 4) 65,66. This small ribonuclear complex forms on the 7SK snRNA, which contains 332 nucleotides and is transcribed by RNAPIII 8. It forms the scaffold for multiple protein:RNA and protein:protein interactions. The 5’ end of 7SK snRNA is targeted by the methyl phosphate capping enzyme (MePCE), which adds a γ-monomethyl cap to the 5’ end of this RNA. Via its N-terminal Lupus antigen (La) module, the La related protein 7 (LARP7) binds to the 3’ terminal U residues in the 7SK snRNA 67. Its C-terminal xRRM motif binds to the 3’ stem loop of 7SK snRNA. Both proteins stabilize 7SK snRNA 68. Without them, 7SK snRNA is rapidly degraded. Next, hexamethylene bisacetamide inducible mRNAs 1 and 2 (HEXIM1 and HEXIM2) proteins, also known as cardiac lineage protein (CLP) 1 69, estrogen-downregulated gene (EDG) 1 70, or menage a quatre (MAQ) 1 71, bind to the GAUC bulge in the 5’ stem loop of 7SK snRNA via their KHRR (lysine, histidine, arginine, arginine) repeats 71. P-TEFb binds to the central loop of this hairpin structure 72. Upon binding to RNA, conformational changes in HEXIM1/2 expose its PYNT (proline, tyrosine, arginine, threonine) motif that targets the ATP pocket in CDK9 and inhibits its kinase activity 71. The inducible HEXIM1 is more abundant in most cells. These interactions resemble those between Tat, P-TEFb and TAR RNA 8. Thus, it is not surprising that Tat can compete for this binding and release P-TEFb from the 7SK snRNP 73,74.
Figure 4.
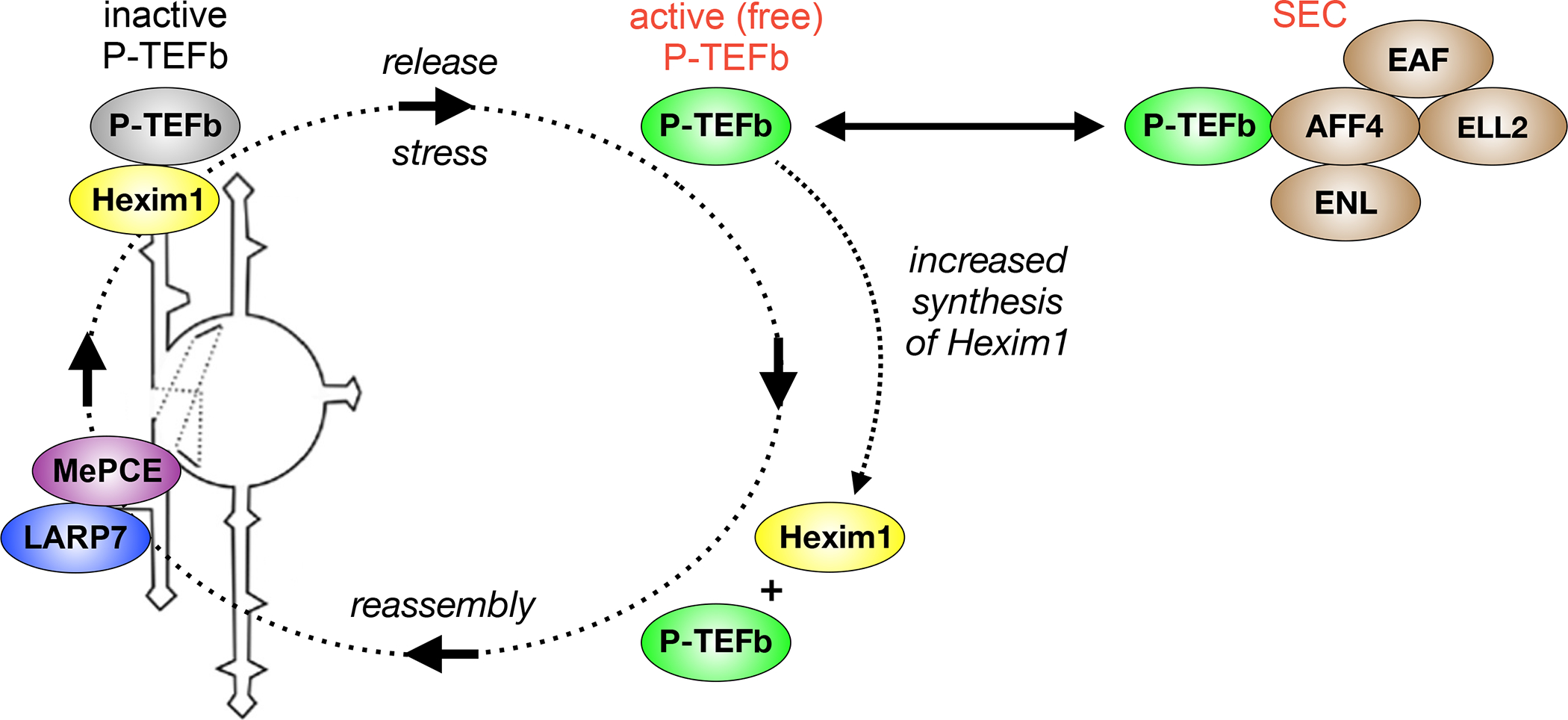
The regulation of P-TEFb in growing cells. In growing, dividing cells, P-TEFb partitions between the active (free) state and the inactive (sequestered) form. In the former, it binds other transcription factors (TFs) or is incorporated into the super elongation complex (SEC). In the latter, it is incorporated into the 7SK small nuclear ribonucleoprotein (7SK snRNP) complex by 7SK snRNA and HEXIM1. 7SK snRNA is stabilized by the La related protein 7 (LARP7) and methyl phosphate capping enzyme (MePCE) at its 3’ and 5’ ends, respectively. P-TEFb is released by stress from the 7SK snRNP, which increases the synthesis of HEXIM1. Thus, 7SK snRNP reassembles quickly. Stress can come in many forms, from cellular signaling pathways, certain signaling molecules (cytokines and lymphokines), DNA damage (e.g. UV light) to chromatin stress (HDAC and BET bromodomain protein inhibition), differentiation agents (HMBA) and viral infections. Of interest, HEXIM1 and P-TEFb bind to the 5’ bulge and central loop in stem loop 1 of 7SK snRNA, which is analogous to how Tat interacts with TAR RNA. Indeed, Tat can displace P-TEFb from the 7SK snRNP.
In different proliferating cells, 50% to 90% of P-TEFb is found within 7SK snRNP 8. Only the active (free) P-TEFb turns on target genes, many of which are required for cell activation, growth and proliferation. Thus, a precise P-TEFb equilibrium is required for the proper homeostasis of cells and tissues in the organism 75. When P-TEFb is not incorporated into the 7SK snRNP, 7SK snRNA also interacts with heterologous nuclear RNP Q and/or R 76,77, or the high mobility group A1 (HMGA1) proteins 78. 7SK snRNA undergoes large-scale conformational changes between these two states 79,80. When P-TEFb is released from the 7SK snRNP, the hexim1 gene is one of its earliest targets. Newly synthesized HEXIM1 proteins then reassemble the 7SK snRNP (Fig. 4) 81. This negative feedback loop ensures that the P-TEFb equilibrium is restored 82. HEXIM1 and 7SK snRNP have also been observed at different transcription units, possibly for their repression or a more rapid deployment of P-TEFb to target genes following its release 83. Nevertheless, this association cannot be very tight, as 7SK snRNP elutes at low salt concentrations from the nucleus in cell fractionation experiments. In contrast, active P-TEFb requires very high salt concentrations for its removal from chromatin 8.
P-TEFb releasing factors
Release of P-TEFb from the 7SK snRNP occurs in various ways 84. Most of them represent some form of stress to the cell 7. They include strong intracellular signals, UV light, other forms of DNA damage and chromatin perturbations 8. For example, TCR-mediated signaling can release P-TEFb from the 7SK snRNP 85. PKC agonists can mimic this effect 85. Other stimuli include small compounds such as histone deacetylase inhibitors (HDACis), bromodomain and extra-terminal (BET) bromodomain inhibitors (BETis) and hexamethylene bisacetamide (HMBA) 8. For HMBA, this release is mediated by the phosphorylation of HEXIM1 by Akt 86. For others, different protein phosphatases (PP1a and PP2B) are involved. They dephosphorylate T186 in the T-loop of CDK9 87. Although T186 has to be rapidly re-phosphorylated for activity, S175 remains modified in the released P-TEFb 35. Other physiological stress-induced proteins include RNA-binding motif protein (RMB)7 and serine-arginine rich splicing factor (SRSF) 2 88,89. They can bind to the 7SK snRNP and displace P-TEFb. Released P-TEFb is then recruited to the paused RNAPII. The stronger the perturbation, the faster the release from and subsequent reassembly of the 7SK snRNP. Thus, increased levels of HEXIM1 commonly result in a P-TEFb disequilibrium that inhibits transcription 82.
Degradation
P-TEFb is also regulated by levels of its subunits. This total amount is correlated to the growth and proliferation of cells. For example, levels of P-TEFb, especially of CycT1, are low to absent in most quiescent cells, such as resting, memory and naive T cells as well as terminally differentiated macrophages 90. Despite the loss of CycT1, levels of CDK9 remain high due to its binding to HSP70 and HSP90 in these cells 91. Importantly, mRNA levels of CycT1 and CDK9 are not diminished 90. This finding implies that CycT1 is regulated post-transcriptionally in quiescent cells 90. Various mechanisms have been proposed, including micro (miRNA)-mediated inhibition of CycT1 translation 90. In addition, proteasomal inhibitors reverse the depletion of CycT1 in terminally differentiated macrophages, which implicates the proteasome 92. This degradation of CycT1 is due to the dissociation of P-TEFb by PP1 (Fig. 5) 38. It removes phosphate groups from threonines at positions 143 and 149 in CycT1 (T143 and T149), which are required for optimal interactions between CycT1 and CDK9. The E3 ligases Siah1/2 then target the unbound CycT1 for degradation 39. The degron and ubiquitination sites were mapped to residues from positions 210 to 280 in the unbound CycT1 39. PKCα and PKCβ phosphorylate T143 and T149, which explains how P-TEFb is reassembled in growing cells 39. In contrast, when PKC is inactive in resting cells or degraded following strong activation signals or by the chronic administration of phorbol esters, P-TEFb falls apart in anergic, exhausted or terminally differentiated lymphocytes 38. However, to maintain cellular homeostasis, the extent of this degradation should vary between tissue resident, circulating and ex vivo cultured cells.
Figure 5.
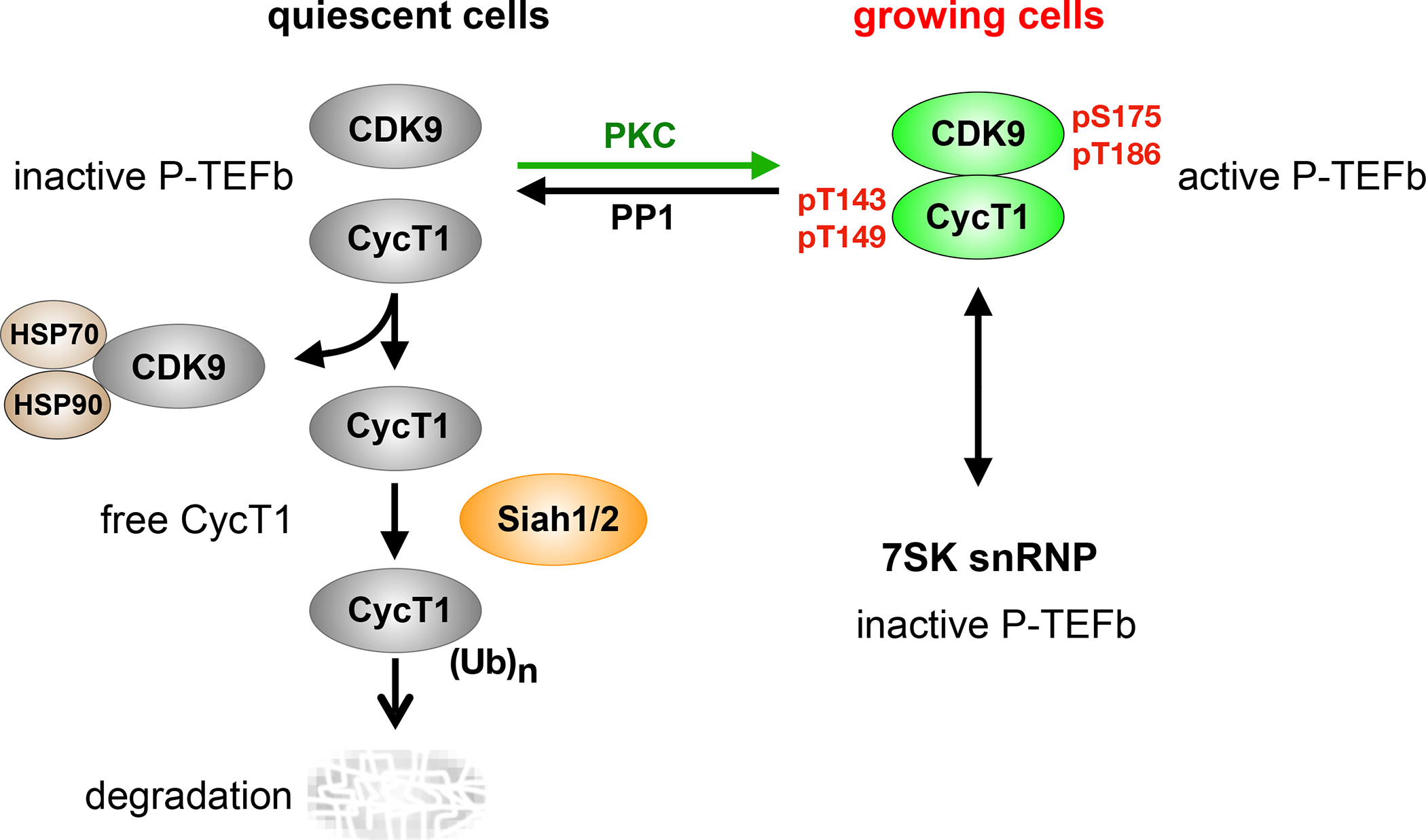
The regulation of P-TEFb in quiescent cells. In contrast to growing cells, P-TEFb is degraded in quiescent cells. In these cells, the lack of active PKC allows protein phosphatase 1 (PP1) to dephosphorylate threonines at positions 143 and 149 (T143 and T149) in CycT1, thus dissociating it from CDK9. HSP 70 and HSP90 bind and stabilize the free CDK9. Siah1/2 bind to and ubiquitinate the unbound CycT1, which is quickly degraded. CDK9 and CycT1 transcripts remain high in these cells. Upon cell activation, PKC phosphorylates T143 and T149 and reverses this process. Arrows point in the direction of the unbound CycT1 degradation. However, upon PKC activation, the first step is reversed and P-TEFb is stabilized. Phosphorylated serine and threonine at positions 175 and 186 (pS175 and pT186) in CDK9 and phosphorylated T143 and T149 (pT143 and pT149) are found in CycT1 in the active P-TEFb.
P-TEFb in HIV transcription and latency
Since P-TEFb is the cellular co-activator of HIV transcription, it plays a central role in the pathogenesis of AIDS. Indeed, viral replication was demonstrated early to be regulated by transcription elongation 93. Without Tat, RNAPII copies only short transcripts that form the TAR RNA structure. Tat then recruits P-TEFb to RNAPII via TAR. In addition, following T cell activation, NF-κB also binds to the HIV LTR 94. Therefore, P-TEFb is recruited for Tat-independent (basal, NF-κB) and Tat-dependent HIV transcription. In HIV-positive patients who receive antiretroviral therapy (ART), levels of HIV are kept low to undetectable in the peripheral blood. Although the majority of HIV-infected cells encode replication-defective HIV proviruses, a small population contain the intact HIV. Upon the cessation of ART, fully infectious progeny virions are produced from this reservoir, leading to the rebound of the disease. Several possible mechanisms of establishment and maintenance of HIV latency include integration into inactive heterochromatin, epigenetic silencing (i.e. DNA methylation, histone modifications), absence of required transcription factors (e.g. NF-κB, Tat), transcriptional interference (i.e. integration into actively transcribed host cell genes), and increased levels of host restriction factors 40.
Importantly, levels of P-TEFb are low in resting CD4+ T cells and their memory subsets harboring silent HIV proviruses, which represents a major factor that maintains the latent viral reservoir 90. To achieve a functional cure of AIDS, attempts have been made to maintain proviral latency (“block and lock” strategy) or to reverse it by reactivating HIV and remove infected cells by host immune responses or via direct viral cytotoxic effects (“shock and kill” strategy) 95. To achieve this “shock and kill” of HIV, levels of active P-TEFb must be increased as well as those of NF-κB and Tat. Proteasomal inhibitors, T cell receptor (TCR) or PKC agonists, HDACis and BETis alone or in combination have all been studied for HIV reactivation from latency 95. In this context, bortezomib induces the reactivation of latent HIV without global T cell activation 96. Of interest, even when 7SK snRNP has reassembled and the expression of most host cell genes is inhibited, HIV transcription continues because Tat competes with HEXIM1 for P-TEFb 73,74. The sequence of these events can be very rapid, resulting in waves of HIV transcription. Interestingly, since other viruses, such as HTLV-1, KSHV, Influenza virus, EBV, CMV, HSV as well as SIV, EIAV, and BIV also utilize P-TEFb for their replication, manipulating levels and activity of P-TEFb could also reactivate them from their reservoirs 97.
P-TEFb in health and disease
P-TEFb plays a critical role in many human diseases and conditions. In some, P-TEFb is not regulated by 7SK snRNP. In others, it is aberrantly recruited to specific loci via genetic rearrangements. One early example is cardiac hypertrophy, where treatment of cardiomyocytes with hypertrophic stimuli such as endothelin-1 (ET1) or mechanical stresses releases P-TEFb from the 7SK snRNP 98. In addition, high levels of P-TEFb or the genetic inactivation of HEXIM1 leads to cardiac hypertrophy and heart failure in mice 69.
High levels of active P-TEFb have also been implicated in autoimmune diseases, where TNFα signaling releases it from the 7SK snRNP. NF-κB and P-TEFb, or STAT3 and P-TEFb then increase the expression of their target genes, leading to inflammation 49. An important target are joints, e.g. rheumatoid arthritis, which is a condition ameliorated by TNFα inhibition. In addition, specific CDK9 inhibitors have beneficial effects in mouse models of the disease 99.
The P-TEFb equilibrium is also shifted in various solid tumors and hematopoietic malignancies. In particular, mutations in LARP7 have been observed in gastric, breast and ovarian cancers 100. In these malignant cells, 7SK snRNA is degraded and 7SK snRNP cannot form 101. Thus, all P-TEFb is in the active form. An example of cancer where P-TEFb is recruited to specific loci is mixed-lineage leukemia (MLL) 56. Here, the translocations and genetic rearrangements occur between the MLL gene and components of the SEC, which then recruits P-TEFb 56. A majority of MLL rearrangements (~50% of infant cases and ~75% of adult cases of acute lymphoblastic leukemia) result in in-frame fusion proteins between MLL and AFF4. MLL rearrangements also occur with ENL and AF9 genes 56. These MLL rearrangements lead to aberrant transcription elongation by P-TEFb. A different example is c-Myc, whose levels are increased in many cancers. The expression and activity of c-Myc are regulated by P-TEFb and BRD4 102,103, and the removal of BRD4 from the c-myc locus by BETi results in potent anti-tumor effects 104. In addition, P-TEFb binds to and regulates the activities of many other transcription factors involved in cancer, which includes estrogen and androgen receptors 49. For the treatment of autoimmune diseases and cancer, many chemical CDK9 inhibitors have been developed with varying degrees of success in clinical trials. Their use has been limited primarily because of off target effects (inhibiting other CDKs) or severe side effects of inhibiting P-TEFb globally. However, their development, uses and clinical trials go beyond the scope of this manuscript and have been covered extensively in other reviews 49.
The opposite situation, where P-TEFb is absent, pertains to anergic, exhausted and terminally differentiated lymphocytes. T cell exhaustion impairs greatly the effectiveness of cancer immunotherapy, including the check point blockade with antibodies that inhibit negative lymphocyte signaling and the chimeric antigen receptor (CAR)-T cell therapy 105. Exhausted T cells occur with chronic infections or tumors where constant antigen stimulation via TCR depletes PKC and other signaling molecules, leading to the degradation of CycT1 39. Since P-TEFb is required for the expression of most activated genes, which include those coding for cytokines and lymphokines, its absence in exhausted T cells could contribute greatly to their unresponsiveness 106. Increasing levels of active P-TEFb in these cells could potentially mitigate T cell exhaustion and improve the effectiveness of these therapies.
Future Directions
Although P-TEFb has been studied extensively, we still do not know many aspects of its structure and function. For example, we have no structure of the full length CycT1 and CDK9 or the inhibitory 7SK snRNP complex. What roles do different P-TEFb components (such as CycT2a and CycT2b as well as CDK9-55) play in development and tissues? How many transcription factors interact with P-TEFb directly or via the SEC? What roles do interactions between BRD4 and P-TEFb play in transcription elongation? Indeed, how well does chromatin bound BRD4 interact with P-TEFb? Finally, how are three different Cyclin/CDK complexes that phosphorylate S2 of RNAPII CTD (P-TEFb, CycK:CDK12 and CycL:CDK11) co-ordinated for the precise temporal and spatial regulation of specific genes, for elongation, RNA processing and 3’ end formation?
Besides targeted questions concerning the structure and function of P-TEFb, what do low levels of P-TEFb mean for quiescent, anergic and exhausted T cells? Indeed, how are they controlled between different differentiated cells in the organism? How much P-TEFb is required for their simple housekeeping, vegetative functions? If levels of P-TEFb could be manipulated artificially, how would that affect cellular functions and the establishment or reversal of proviral latency?
The other side of the coin addresses the inhibition of P-TEFb in inflammation, autoimmunity and cancer. Since many cancers have mutations in LARP7, could the introduction of the WT protein act as a tumor suppressor? Alternatively, would specific inhibition or targeted degradation of P-TEFb decrease inflammation, ameliorate autoimmunity and arrest the growth of malignant cells. Possibly, new, more specific CDK9 inhibitors could be used in smaller, less toxic doses. Alternatively, one could target surfaces on P-TEFb that interact with specific transcription factors or use pseudo-substrate decoys that would inhibit CDK9 only for the transcription of specific genes? Thus, the field of transcription elongation and P-TEFb in particular remains a very fruitful area of future research, where most answers will benefit not only our knowledge of gene expression but of growth, development and disease.
Acknowledgements
We thank Drs. David Price, Matjaz Barboric, and Matthias Geyer for carefully reading the manuscript and providing insightful suggestions. We apologize for not able to cite many interesting and important work due to limitations in the number of citation. KF is supported by research grants from NIH (1R21AI127274, 1R56AI136683, R01AI167778, 1R21AI170195) and Nora Eccles Treadwell Foundation. BMP is supported by research grants from NIH (R01 AI049104, P50AI150476).
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cramer P (2019). Eukaryotic Transcription Turns 50. Cell 179, 808–812. 10.1016/j.cell.2019.09.018. [DOI] [PubMed] [Google Scholar]
- 2.Yamaguchi Y, Inukai N, Narita T, Wada T, and Handa H (2002). Evidence that negative elongation factor represses transcription elongation through binding to a DRB sensitivity-inducing factor/RNA polymerase II complex and RNA. Mol Cell Biol 22, 2918–2927. 10.1128/MCB.22.9.2918-2927.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adelman K, and Henriques T (2018). Transcriptional speed bumps revealed in high resolution. Nature 560, 560–561. 10.1038/d41586-018-05971-8. [DOI] [PubMed] [Google Scholar]
- 4.Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J, and Handa H (1999). NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97, 41–51. 10.1016/s0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]
- 5.Vos SM, Farnung L, Urlaub H, and Cramer P (2018). Structure of paused transcription complex Pol II-DSIF-NELF. Nature 560, 601–606. 10.1038/s41586-018-0442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, and Cramer P (2018). Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature 560, 607–612. 10.1038/s41586-018-0440-4. [DOI] [PubMed] [Google Scholar]
- 7.Egloff S (2021). CDK9 keeps RNA polymerase II on track. Cell Mol Life Sci 78, 5543–5567. 10.1007/s00018-021-03878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peterlin BM, and Price DH (2006). Controlling the elongation phase of transcription with P-TEFb. Mol Cell 23, 297–305. 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 9.Anshabo AT, Milne R, Wang S, and Albrecht H (2021). CDK9: A Comprehensive Review of Its Biology, and Its Role as a Potential Target for Anti-Cancer Agents. Front Oncol 11, 678559. 10.3389/fonc.2021.678559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eick D, and Geyer M (2013). The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev 113, 8456–8490. 10.1021/cr400071f. [DOI] [PubMed] [Google Scholar]
- 11.Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, and Peterlin BM (2004). Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol 24, 787–795. 10.1128/mcb.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivanov D, Kwak YT, Guo J, and Gaynor RB (2000). Domains in the SPT5 protein that modulate its transcriptional regulatory properties. Mol Cell Biol 20, 2970–2983. 10.1128/mcb.20.9.2970-2983.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohamed AA, Vazquez Nunez R, and Vos SM (2022). Structural advances in transcription elongation. Curr Opin Struct Biol 75, 102422. 10.1016/j.sbi.2022.102422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Yamaguchi Y, Tsugeno Y, Yamamoto J, Yamada T, Nakamura M, Hisatake K, and Handa H (2009). DSIF, the Paf1 complex, and Tat-SF1 have nonredundant, cooperative roles in RNA polymerase II elongation. Genes Dev 23, 2765–2777. 10.1101/gad.1834709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang KL, Jee D, Stein CB, Elrod ND, Henriques T, Mascibroda LG, Baillat D, Russell WK, Adelman K, and Wagner EJ (2020). Integrator Recruits Protein Phosphatase 2A to Prevent Pause Release and Facilitate Transcription Termination. Mol Cell 80, 345–358 e349. 10.1016/j.molcel.2020.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parua PK, Booth GT, Sanso M, Benjamin B, Tanny JC, Lis JT, and Fisher RP (2018). A Cdk9-PP1 switch regulates the elongation-termination transition of RNA polymerase II. Nature 558, 460–464. 10.1038/s41586-018-0214-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vervoort SJ, Welsh SA, Devlin JR, Barbieri E, Knight DA, Offley S, Bjelosevic S, Costacurta M, Todorovski I, Kearney CJ, et al. (2021). The PP2A-Integrator-CDK9 axis fine-tunes transcription and can be targeted therapeutically in cancer. Cell 184, 3143–3162 e3132. 10.1016/j.cell.2021.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morillon A, Karabetsou N, O’Sullivan J, Kent N, Proudfoot N, and Mellor J (2003). Isw1 chromatin remodeling ATPase coordinates transcription elongation and termination by RNA polymerase II. Cell 115, 425–435. 10.1016/s0092-8674(03)00880-8. [DOI] [PubMed] [Google Scholar]
- 19.Guenther MG, Levine SS, Boyer LA, Jaenisch R, and Young RA (2007). A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88. 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, and Adelman K (2007). RNA polymerase is poised for activation across the genome. Nat Genet 39, 1507–1511. 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eifler TT, Shao W, Bartholomeeusen K, Fujinaga K, Jager S, Johnson JR, Luo Z, Krogan NJ, and Peterlin BM (2015). Cyclin-dependent kinase 12 increases 3’ end processing of growth factor-induced c-FOS transcripts. Mol Cell Biol 35, 468–478. 10.1128/MCB.01157-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, and Roeder RG (2015). RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science 350, 1383–1386. 10.1126/science.aad2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gajduskova P, Ruiz de Los Mozos I, Rajecky M, Hluchy M, Ule J, and Blazek D (2020). CDK11 is required for transcription of replication-dependent histone genes. Nat Struct Mol Biol 27, 500–510. 10.1038/s41594-020-0406-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pak V, Eifler TT, Jager S, Krogan NJ, Fujinaga K, and Peterlin BM (2015). CDK11 in TREX/THOC Regulates HIV mRNA 3’ End Processing. Cell Host Microbe 18, 560–570. 10.1016/j.chom.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barboric M, Lenasi T, Chen H, Johansen EB, Guo S, and Peterlin BM (2009). 7SK snRNP/P-TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc Natl Acad Sci U S A 106, 7798–7803. 10.1073/pnas.0903188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laitem C, Zaborowska J, Isa NF, Kufs J, Dienstbier M, and Murphy S (2015). CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat Struct Mol Biol 22, 396–403. 10.1038/nsmb.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marshall NF, and Price DH (1992). Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol 12, 2078–2090. 10.1128/mcb.12.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grana X, De Luca A, Sang N, Fu Y, Claudio PP, Rosenblatt J, Morgan DO, and Giordano A (1994). PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retinoblastoma protein in vitro. Proc Natl Acad Sci U S A 91, 3834–3838. 10.1073/pnas.91.9.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei P, Garber ME, Fang SM, Fischer WH, and Jones KA (1998). A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462. 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 30.Peng J, Zhu Y, Milton JT, and Price DH (1998). Identification of multiple cyclin subunits of human P-TEFb. Genes Dev 12, 755–762. 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shore SM, Byers SA, Dent P, and Price DH (2005). Characterization of Cdk9(55) and differential regulation of two Cdk9 isoforms. Gene 350, 51–58. 10.1016/j.gene.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Decker TM, Forne I, Straub T, Elsaman H, Ma G, Shah N, Imhof A, and Eick D (2019). Analog-sensitive cell line identifies cellular substrates of CDK9. Oncotarget 10, 6934–6943. 10.18632/oncotarget.27334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanso M, Levin RS, Lipp JJ, Wang VY, Greifenberg AK, Quezada EM, Ali A, Ghosh A, Larochelle S, Rana TM, et al. (2016). P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev 30, 117–131. 10.1101/gad.269589.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, and Fisher RP (2012). Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol 19, 1108–1115. 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mbonye UR, Gokulrangan G, Datt M, Dobrowolski C, Cooper M, Chance MR, and Karn J (2013). Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog 9, e1003338. 10.1371/journal.ppat.1003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nekhai S, Petukhov M, and Breuer D (2014). Regulation of CDK9 activity by phosphorylation and dephosphorylation. Biomed Res Int 2014, 964964. 10.1155/2014/964964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garber ME, Mayall TP, Suess EM, Meisenhelder J, Thompson NE, and Jones KA (2000). CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 tat-P-TEFb complex to TAR RNA. Mol Cell Biol 20, 6958–6969. 10.1128/MCB.20.18.6958-6969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang F, Nguyen TT, Echeverria I, Rakesh R, Cary DC, Paculova H, Sali A, Weiss A, Peterlin BM, and Fujinaga K (2021). Reversible phosphorylation of cyclin T1 promotes assembly and stability of P-TEFb. Elife 10. 10.7554/eLife.68473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang F, Feng Y, Peterlin BM, and Fujinaga K (2022). P-TEFb is degraded by Siah1/2 in quiescent cells. Nucleic Acids Res 50, 5000–5013. 10.1093/nar/gkac291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cary DC, Fujinaga K, and Peterlin BM (2016). Molecular mechanisms of HIV latency. J Clin Invest 126, 448–454. 10.1172/JCI80565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taube R, Lin X, Irwin D, Fujinaga K, and Peterlin BM (2002). Interaction between P-TEFb and the C-terminal domain of RNA polymerase II activates transcriptional elongation from sites upstream or downstream of target genes. Mol Cell Biol 22, 321–331. 10.1128/mcb.22.1.321-331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, and Zhou Q (2018). Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323. 10.1038/s41586-018-0174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu H, Liu R, Jia Z, Li R, Zhu F, Zhu W, Shao Y, Jin Y, Xue Y, Huang J, et al. (2022). Poly(ADP-ribosylation) of P-TEFb by PARP1 disrupts phase separation to inhibit global transcription after DNA damage. Nat Cell Biol 24, 513–525. 10.1038/s41556-022-00872-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiernan RE, Emiliani S, Nakayama K, Castro A, Labbe JC, Lorca T, Nakayama Ki K, and Benkirane M (2001). Interaction between cyclin T1 and SCF(SKP2) targets CDK9 for ubiquitination and degradation by the proteasome. Mol Cell Biol 21, 7956–7970. 10.1128/MCB.21.23.7956-7970.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baumli S, Lolli G, Lowe ED, Troiani S, Rusconi L, Bullock AN, Debreczeni JE, Knapp S, and Johnson LN (2008). The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J 27, 1907–1918. 10.1038/emboj.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, and Price DH (2010). Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 465, 747–751. 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schulze-Gahmen U, Upton H, Birnberg A, Bao K, Chou S, Krogan NJ, Zhou Q, and Alber T (2013). The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. Elife 2. ARTN e00327. 10.7554/eLife.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schulze-Gahmen U, Lu HS, Zhou Q, and Alber T (2014). AFF4 binding to Tat-P-TEFb indirectly stimulates TAR recognition of super elongation complexes at the HIV promoter. Elife 3. ARTN e02375. 10.7554/eLife.02375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujinaga K (2020). P-TEFb as A Promising Therapeutic Target. Molecules 25. 10.3390/molecules25040838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Selby MJ, and Peterlin BM (1990). Trans-activation by HIV-1 Tat via a heterologous RNA binding protein. Cell 62, 769–776. 10.1016/0092-8674(90)90121-t. [DOI] [PubMed] [Google Scholar]
- 51.Tiley LS, Madore SJ, Malim MH, and Cullen BR (1992). The VP16 transcription activation domain is functional when targeted to a promoter-proximal RNA sequence. Genes Dev 6, 2077–2087. 10.1101/gad.6.11.2077. [DOI] [PubMed] [Google Scholar]
- 52.Bieniasz PD, Grdina TA, Bogerd HP, and Cullen BR (1999). Recruitment of cyclin T1/P-TEFb to an HIV type 1 long terminal repeat promoter proximal RNA target is both necessary and sufficient for full activation of transcription. Proc Natl Acad Sci U S A 96, 7791–7796. 10.1073/pnas.96.14.7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fujinaga K, Taube R, Wimmer J, Cujec TP, and Peterlin BM (1999). Interactions between human cyclin T, Tat, and the transactivation response element (TAR) are disrupted by a cysteine to tyrosine substitution found in mouse cyclin T. Proc Natl Acad Sci U S A 96, 1285–1290. 10.1073/pnas.96.4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kurosu T, and Peterlin BM (2004). VP16 and ubiquitin; binding of P-TEFb via its activation domain and ubiquitin facilitates elongation of transcription of target genes. Curr Biol 14, 1112–1116. 10.1016/j.cub.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi H, Parmely TJ, Sato S, Tomomori-Sato C, Banks CA, Kong SE, Szutorisz H, Swanson SK, Martin-Brown S, Washburn MP, et al. (2011). Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 146, 92–104. 10.1016/j.cell.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, and Shilatifard A (2010). AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37, 429–437. 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, and Zhou Q (2005). Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 19, 535–545. 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 58.Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell 67, 5–18 e19. 10.1016/j.molcel.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang F, Shao W, Fujinaga K, and Peterlin BM (2018). Bromodomain-containing protein 4-independent transcriptional activation by autoimmune regulator (AIRE) and NF-kappaB. J Biol Chem 293, 4993–5004. 10.1074/jbc.RA117.001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal A, and Rosenfeld MG (2013). Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 155, 1581–1595. 10.1016/j.cell.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang Z, He N, and Zhou Q (2008). Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol Cell Biol 28, 967–976. 10.1128/MCB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang F, Barboric M, Blackwell TK, and Peterlin BM (2003). A model of repression: CTD analogs and PIE-1 inhibit transcriptional elongation by P-TEFb. Genes Dev 17, 748–758. 10.1101/gad.1068203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang H, Zhang F, Kurosu T, and Peterlin BM (2005). Runx1 binds positive transcription elongation factor b and represses transcriptional elongation by RNA polymerase II: possible mechanism of CD4 silencing. Mol Cell Biol 25, 10675–10683. 10.1128/MCB.25.24.10675-10683.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shchebet A, Karpiuk O, Kremmer E, Eick D, and Johnsen SA (2012). Phosphorylation by cyclin-dependent kinase-9 controls ubiquitin-conjugating enzyme-2A function. Cell Cycle 11, 2122–2127. 10.4161/cc.20548. [DOI] [PubMed] [Google Scholar]
- 65.Nguyen VT, Kiss T, Michels AA, and Bensaude O (2001). 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414, 322–325. 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- 66.Yang Z, Zhu Q, Luo K, and Zhou Q (2001). The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414, 317–322. 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- 67.Markert A, Grimm M, Martinez J, Wiesner J, Meyerhans A, Meyuhas O, Sickmann A, and Fischer U (2008). The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep 9, 569–575. 10.1038/embor.2008.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xue Y, Yang Z, Chen R, and Zhou Q (2010). A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res 38, 360–369. 10.1093/nar/gkp977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Espinoza-Derout J, Wagner M, Shahmiri K, Mascareno E, Chaqour B, and Siddiqui MA (2007). Pivotal role of cardiac lineage protein-1 (CLP-1) in transcriptional elongation factor P-TEFb complex formation in cardiac hypertrophy. Cardiovasc Res 75, 129–138. 10.1016/j.cardiores.2007.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wittmann BM, Fujinaga K, Deng H, Ogba N, and Montano MM (2005). The breast cell growth inhibitor, estrogen down regulated gene 1, modulates a novel functional interaction between estrogen receptor alpha and transcriptional elongation factor cyclin T1. Oncogene 24, 5576–5588. 10.1038/sj.onc.1208728. [DOI] [PubMed] [Google Scholar]
- 71.Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, Lania L, and Bensaude O (2004). Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J 23, 2608–2619. 10.1038/sj.emboj.7600275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujinaga K, Luo Z, and Peterlin BM (2014). Genetic analysis of the structure and function of 7SK small nuclear ribonucleoprotein (snRNP) in cells. J Biol Chem 289, 21181–21190. 10.1074/jbc.M114.557751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, Geyer M, Matija Peterlin B, and Zhou Q (2007). Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res 35, 2003–2012. 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sedore SC, Byers SA, Biglione S, Price JP, Maury WJ, and Price DH (2007). Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res 35, 4347–4358. 10.1093/nar/gkm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He N, Pezda AC, and Zhou Q (2006). Modulation of a P-TEFb functional equilibrium for the global control of cell growth and differentiation. Mol Cell Biol 26, 7068–7076. 10.1128/MCB.00778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barrandon C, Bonnet F, Nguyen VT, Labas V, and Bensaude O (2007). The transcription-dependent dissociation of P-TEFb-HEXIM1–7SK RNA relies upon formation of hnRNP-7SK RNA complexes. Mol Cell Biol 27, 6996–7006. 10.1128/MCB.00975-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ji C, Deng C, Antor K, Bischler T, Schneider C, Fischer U, Sendtner M, and Briese M (2022). hnRNP R negatively regulates transcription by modulating the association of P-TEFb with 7SK and BRD4. EMBO Rep, e55432. 10.15252/embr.202255432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eilebrecht S, Benecke BJ, and Benecke A (2011). 7SK snRNA-mediated, gene-specific cooperativity of HMGA1 and P-TEFb. RNA Biol 8, 1084–1093. 10.4161/rna.8.6.17015. [DOI] [PubMed] [Google Scholar]
- 79.Olson SW, Turner AW, Arney JW, Saleem I, Weidmann CA, Margolis DM, Weeks KM, and Mustoe AM (2022). Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Mol Cell 82, 1708–1723 e1710. 10.1016/j.molcel.2022.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang Y, Liu S, Egloff S, Eichhorn CD, Hadjian T, Zhen J, Kiss T, Zhou ZH, and Feigon J (2022). Structural basis of RNA conformational switching in the transcriptional regulator 7SK RNP. Mol Cell 82, 1724–1736 e1727. 10.1016/j.molcel.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu P, Xiang Y, Fujinaga K, Bartholomeeusen K, Nilson KA, Price DH, and Peterlin BM (2014). Release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein (snRNP) activates hexamethylene bisacetamide-inducible protein (HEXIM1) transcription. J Biol Chem 289, 9918–9925. 10.1074/jbc.M113.539015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tan JL, Fogley RD, Flynn RA, Ablain J, Yang S, Saint-Andre V, Fan ZP, Do BT, Laga AC, Fujinaga K, et al. (2016). Stress from Nucleotide Depletion Activates the Transcriptional Regulator HEXIM1 to Suppress Melanoma. Mol Cell 62, 34–46. 10.1016/j.molcel.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McNamara RP, Guzman C, Reeder JE, and D’Orso I (2016). Genome-wide analysis of KAP1, the 7SK snRNP complex, and RNA polymerase II. Genom Data 7, 250–255. 10.1016/j.gdata.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.AJ CQ, Bugai A, and Barboric M (2016). Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res 44, 7527–7539. 10.1093/nar/gkw585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim YK, Mbonye U, Hokello J, and Karn J (2011). T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J Mol Biol 410, 896–916. 10.1016/j.jmb.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Contreras X, Barboric M, Lenasi T, and Peterlin BM (2007). HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog 3, 1459–1469. 10.1371/journal.ppat.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen R, Liu M, Li H, Xue Y, Ramey WN, He N, Ai N, Luo H, Zhu Y, Zhou N, and Zhou Q (2008). PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev 22, 1356–1368. 10.1101/gad.1636008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bugai A, Quaresma AJC, Friedel CC, Lenasi T, Duster R, Sibley CR, Fujinaga K, Kukanja P, Hennig T, Blasius M, et al. (2019). P-TEFb Activation by RBM7 Shapes a Pro-survival Transcriptional Response to Genotoxic Stress. Mol Cell 74, 254–267 e210. 10.1016/j.molcel.2019.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, and Fu XD (2013). SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 153, 855–868. 10.1016/j.cell.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ramakrishnan R, Chiang K, Liu H, Budhiraja S, Donahue H, and Rice AP (2012). Making a Short Story Long: Regulation of P-TEFb and HIV-1 Transcriptional Elongation in CD4+ T Lymphocytes and Macrophages. Biology (Basel) 1, 94–115. 10.3390/biology1010094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.O’Keeffe B, Fong Y, Chen D, Zhou S, and Zhou Q (2000). Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. J Biol Chem 275, 279–287. 10.1074/jbc.275.1.279. [DOI] [PubMed] [Google Scholar]
- 92.Liou LY, Haaland RE, Herrmann CH, and Rice AP (2006). Cyclin T1 but not cyclin T2a is induced by a post-transcriptional mechanism in PAMP-activated monocyte-derived macrophages. J Leukoc Biol 79, 388–396. 10.1189/jlb.0805429. [DOI] [PubMed] [Google Scholar]
- 93.Kao SY, Calman AF, Luciw PA, and Peterlin BM (1987). Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 330, 489–493. 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- 94.Nabel G, and Baltimore D (1987). An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326, 711–713. 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 95.Darcis G, Van Driessche B, and Van Lint C (2017). HIV Latency: Should We Shock or Lock? Trends Immunol 38, 217–228. 10.1016/j.it.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 96.Cary DC, and Peterlin BM (2020). Proteasomal Inhibition Potentiates Latent HIV Reactivation. AIDS Res Hum Retroviruses 36, 800–807. 10.1089/AID.2020.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zaborowska J, Isa NF, and Murphy S (2016). P-TEFb goes viral. Inside Cell 1, 106–116. 10.1002/icl3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, DeMayo FJ, and Schneider MD (2002). Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med 8, 1310–1317. 10.1038/nm778. [DOI] [PubMed] [Google Scholar]
- 99.Sekine C, Sugihara T, Miyake S, Hirai H, Yoshida M, Miyasaka N, and Kohsaka H (2008). Successful treatment of animal models of rheumatoid arthritis with small-molecule cyclin-dependent kinase inhibitors. J Immunol 180, 1954–1961. 10.4049/jimmunol.180.3.1954. [DOI] [PubMed] [Google Scholar]
- 100.Stavraka C, and Blagden S (2015). The La-Related Proteins, a Family with Connections to Cancer. Biomolecules 5, 2701–2722. 10.3390/biom5042701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ji X, Lu H, Zhou Q, and Luo K (2014). LARP7 suppresses P-TEFb activity to inhibit breast cancer progression and metastasis. Elife 3, e02907. 10.7554/eLife.02907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eberhardy SR, and Farnham PJ (2001). c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem 276, 48562–48571. 10.1074/jbc.M109014200. [DOI] [PubMed] [Google Scholar]
- 103.Kanazawa S, Soucek L, Evan G, Okamoto T, and Peterlin BM (2003). c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene 22, 5707–5711. 10.1038/sj.onc.1206800. [DOI] [PubMed] [Google Scholar]
- 104.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gumber D, and Wang LD (2022). Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine 77, 103941. 10.1016/j.ebiom.2022.103941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hargreaves DC, Horng T, and Medzhitov R (2009). Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145. 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]