

Abstract
Molecular detection via nanopore, achieved by monitoring changes in ionic current arising from analyte interaction with the sensor pore, is a promising technology for multiplex sensing development. Outer Membrane Protein G (OmpG), a monomeric porin possessing seven functionalizable loops, has been reported as an effective sensing platform for selective protein detection. Using flow-cytometry to screen unfavorable constructs, we identified two OmpG nanopores with unique peptide motifs displayed in either loop 3 or 6, which also exhibited distinct analyte signals in single-channel current recordings. We exploited these loops concurrently to facilitate single-molecule multiplex protein detection in a mixture. We additionally report a strategy to increase sensor sensitivity via avidity motif display. These sensing schemes may be expanded to more sophisticated designs utilizing additional loops for increasing multiplicity and sensitivity.
Keywords: Multiplex, Nanopore, Proteins, Sensors, Single-molecule detection
Graphical Abstract
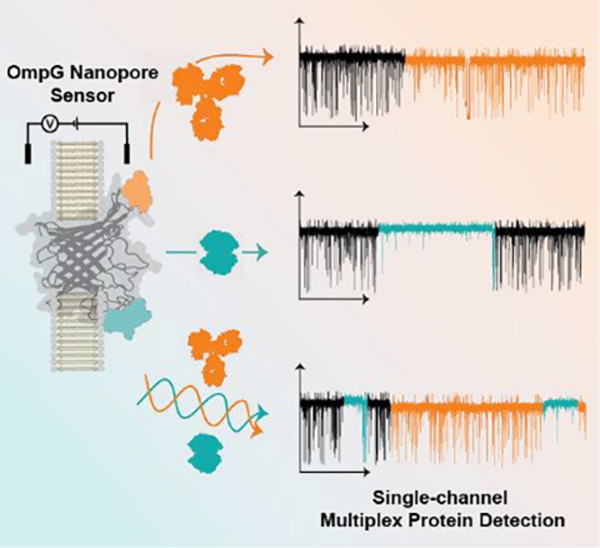
The Outer Membrane Protein G (OmpG) nanopore has been demonstrated as a highly selective platform for protein sensing. By simultaneously functionalizing two of the porin’s seven loops with peptide motifs we achieve single-molecule multiplex protein detection.
Introduction
Rapid and selective detection of biomolecular indicators of disease, referred to as biomarkers, is imperative for accurate diagnostics[1]. However, patient samples are complex and may contain inhibitory or interfering molecules at higher concentrations than the desired biomarker, necessitating sample processing and/or specialized instrumentation and staff for efficient detection[1b, 2]. Therefore, a simple method for selective multiplex detection of independent markers to enable robust and accurate determination of patients’ disease state would be highly desirable[1b]. Nanopore biosensors represent one potential technology capable of fulfilling this role. In nanopore-based sensing ion flow through a biological or synthetic pore is read out as current; when an analyte interacts with the pore or displayed affinity reagent the observed current is perturbed in a characteristic manner, allowing for analyte identification and quantitation[3]. This sensor principle has been successfully applied for genomic DNA sequencing, with ongoing efforts to expand the repertoire of modified nucleobases to enable greater epigenomic analysis[4]. More recently the field advanced toward the detection of peptides and proteins, enabling biomarker detection and even single-molecule protein sequencing[5]. Great effort has been put forth to resolve the twenty proteinogenic amino acids, facilitating the detection and analysis of protein post-translational modifications[6]. Improved resolution of amino acids has thus allowed advances towards protein identification via peptide profiling in a manner analogous to peptide profiling by mass spectrometry[7]. Nanopores have already shown utility in multiplexed detection of peptide(s) or protein(s) with several groups achieving array-based multiplex protein detection using barcoded reporters[8]. As these methods are generally reliant on antibodies to mediate analyte capture, cross-reactivity between the analyte(s) and other structurally similar molecules in complex biological and clinical samples may present a significant challenge to the specificity of multiplex detection[1b, 9].
Outer Membrane Protein G (OmpG) nanopore is a monomeric Escherichia coli (E. coli) porin with a 14-stranded β-barrel possessing seven flexible extracellular loops[10]. The highly dynamic loops regularly undergo conformational changes, altering ion permeability through the pore relative to the conformation of the loop(s) in a phenomenon termed gating[11]. OmpG detection is based on changes to the gating pattern resulting from the interaction of the flexible loops with analytes that were captured by a loop-displayed affinity reagent, rather than the resistive pulse sensing more commonly employed in other nanopore sensors[9a, 12]. This unique sensing scheme is highly selective to the properties of the molecular surface, allowing for target identification and quantitation even among close homologs[13]. We have demonstrated that OmpG is an effective display scaffold as each loop is capable of hosting an affinity reagent for analyte detection, with the most flexible loop 6 demonstrating particular utility in the selective differentiation of protein homologs binding to the same affinity reagent, while remaining insensitive to non-specific interactions in complex solution[9a, 14]. Therefore, we considered OmpG to be a promising scaffold for the generation of a multiplex sensor due to its possession of seven loops amenable as anchor or display sites to host distinct affinity reagents for independent multiplex detection. Herein, we report the development of a simple multiplex OmpG biosensor (Figure 1a) and a general framework from which we can continue the optimization and development of advanced detection platforms for disease biomarkers.
Figure 1.
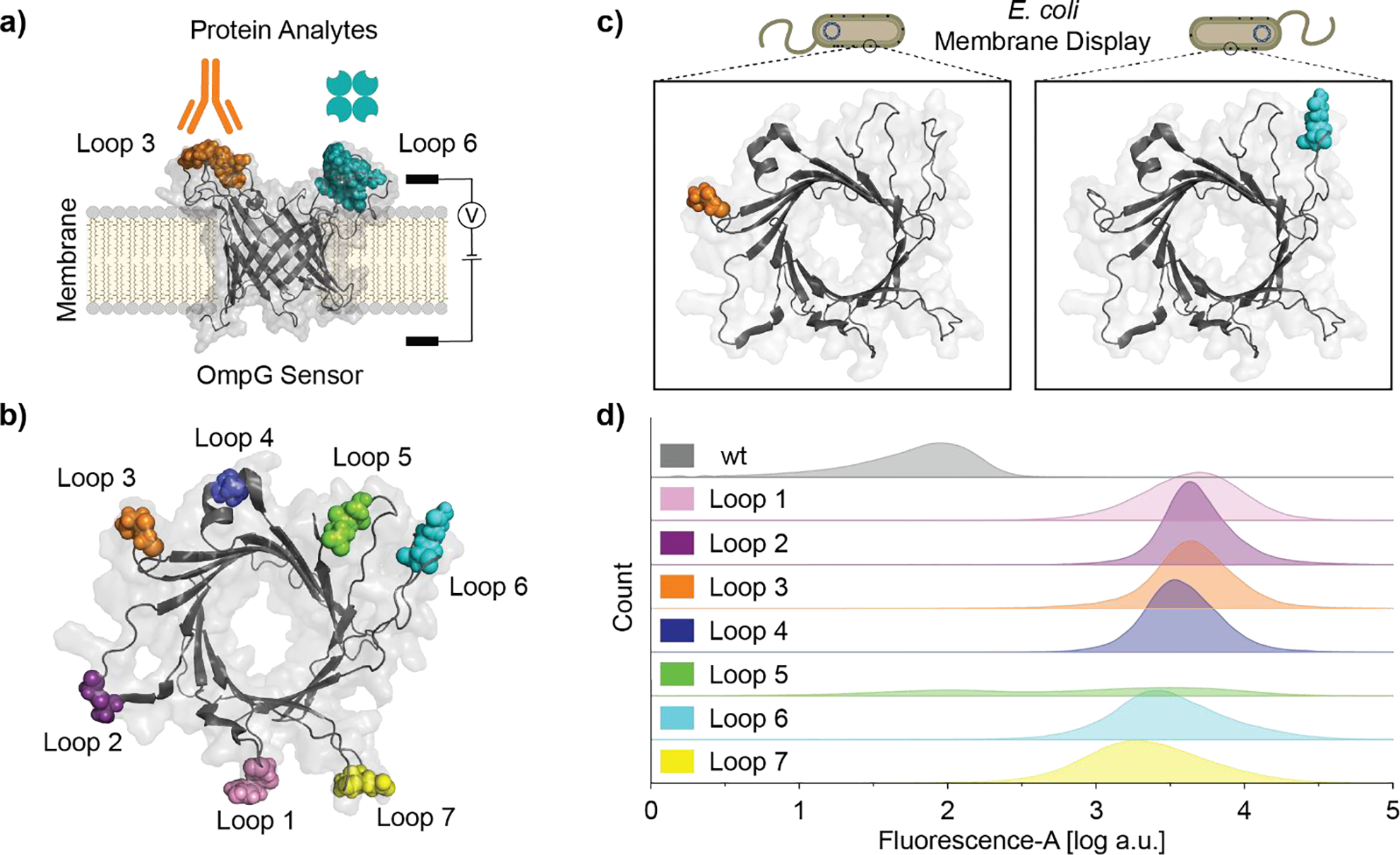
OmpG loops demonstrate variable peptide display efficiency. a) Schematic of OmpG multiplex sensor for single-channel current recording. b) Top view of OmpG sensor, FLAG motif insertion site is indicated by colored spheres (PDB: 5MWV). c) Cartoon schematic of OmpG constructs displayed on E. coli outer membrane. d) Flow cytometry analysis of E. coli cells expressing OmpGLn-FLAG variants, data is presented as kernel density estimation of FITC-channel fluorescence event counts combined from biological triplicates.
Results and Discussion
Development of Flow Cytometry Assay to Assess OmpG Motif Display
Protein nanopore functionalization via chemical labeling can result in population heterogeneity due to incomplete labeling reaction, potentially confounding later analysis, whereas genetic modification assures all constructs harbor the desired affinity ligand with atomic precision. Therefore, we decided to incorporate genetically encoded peptide motifs to build our OmpG nanopore sensor. The FLAG motif (-GDYKDDDDKG-) was selected for this assay due to the availability of monoclonal antibodies and prior use in outer membrane protein display[15]. We additionally selected several peptide sequences which bind streptavidin, as OmpG had previously demonstrated utility in its detection[13a]. We previously observed that the detection sensitivity of biotin-conjugated OmpG constructs was impacted by the loop position where the affinity reagent was attached[13b, 14b]. Thus, we anticipated the site of integration within each loop for display of peptide motifs would also be a factor in the functionality of these constructs. As single-channel current recording in a laboratory setting is a low throughput method for characterizing new nanopore/analyte interactions a more rapid assay was desirable to screen the viability of peptide incorporation sites within OmpG’s loops. Flow cytometry was well suited for screening OmpG nanopore constructs as OmpG is an endogenous E. coli outer membrane protein allowing for efficient display of engineered peptides sequence(s) on the cell surface where they are accessible to protein analytes[10a, 16]. Constructs displaying the FLAG motif in each of OmpG’s seven loops were generated and termed OmpGLn-FLAG (Figure 1b & Figure S1), with n indicating the display loop number.
Each OmpGLn-FLAG construct was separately expressed at the outer membrane of BL21 (DE3) E. coli (Figure 1c). Display of the FLAG motif was then assessed by flow cytometry using a sandwich-style labeling scheme: the FLAG epitope was first recognized by monoclonal mouse anti-FLAG antibody FG4R, which was then bound by goat anti-mouse polyclonal antibody conjugated to fluorescein (FITC) to generate the fluorescence signal. We observed a clear increase in FITC channel emission relative to OmpGwt for most of the constructs (Figure 1d). Constructs OmpGL1/2/3/4-FLAG exhibited mean fluorescence intensity slightly above that of constructs OmpGL6/7-FLAG. Additionally, OmpGL1/6/7-FLAG exhibited a wider distribution in fluorescence intensity, whereas OmpGL2/3/4-FLAG exhibited tighter clustering of their fluorescence signal. Together, the variation in fluorescence intensity and distribution between constructs suggested differences in labeling efficiency as a function of motif location but contributions from differing local environments cannot be fully excluded. Interestingly, OmpGL5-FLAG showed almost no FITC channel emission, while in vitro refolding assessment of OmpGL5-FLAG indicated comparable folding efficiency with other OmpGLn-FLAG constructs (Figure S2a–b), suggesting the low fluorescent signal may be due to inefficiency in the trafficking of the OmpGL5-FLAG construct to the outer cell membrane. Overall, this assay was an effective means to rapidly assess peptide ligand display efficiency, identifying and excluding poorly performing constructs from subsequent studies, with clear trends discernible even from qualitative comparison. Constructs displaying the FLAG motif in loops 2, 3, 4, and 6 were selected for further single-channel analysis because of their strong fluorescence signal. Despite the observation of a comparably strong fluorescence signal in the flow cytometry assay OmpGL1-FLAG was not considered for further analysis as the construct did not appreciably refold in vitro (Figure S2b).
Monoclonal Anti-FLAG Antibody FG4R Detection via Loop 3 Peptide Display
OmpGL2-FLAG, OmpGL3-FLAG, OmpGL4-FLAG, and OmpGL6-FLAG proteins were prepared and characterized by single-channel current measurements. Three proteins, OmpGL2-FLAG, OmpGL3-FLAG, and OmpGL4-FLAG exhibited gating behavior that approximately resembled OmpGwt, as characterized by rapid fluctuations between high and low conductance states, indicating hosting the FLAG motif at these positions did not greatly alter gating dynamics (Figure 2b & S3a–c/e). In contrast, OmpGL6-FLAG exhibited a significant reduction in the intensity and duration of gating events (Figure S3d). Addition of FG4R elicited no change for OmpGwt, OmpGL2-FLAG, OmpGL4-FLAG, and OmpGL6-FLAG constructs (Figure S3a–d) while OmpGL3-FLAG resolved binding events exhibiting a 5.0 ± 1.1% reduction in open pore current (N = 3, n = 9, where N is the number of independent pores and n the number of individual binding events analyzed) with an average duration of minutes (Figure 2a/c–e). MOSAIC was used to collect the intensity (Ires, I/I0) and duration (Dwell, ms) of gating events within OmpGL3-FLAG trace segments covering the basal (I0) and FG4R-bound states, which were then plotted as two-dimensional kernel density plots for comparison[17]. This analysis showed a ~15% reduction in gating frequency during OmpGL3-FLAG/FG4R binding, from a basal (I0) value of 58.0 ± 0.6 Hz to 49.5 ± 1.7 Hz after binding (Figure S3e–f), but with no alteration to the profile of gating events, leaving the reduction in current the most effective parameter for event identification. Due to the excessively long recording time (~hours) necessary to collect enough FG4R binding events with minutes-long duration we did not determine the binding kinetics of FG4R to the OmpGLn-FLAG constructs. This highlights a potential limitation of this detection strategy: analytes with slow off-rate generally exhibit greater pore residence, precluding the detection of additional molecules, allowing for binary detection but not effective quantitation.
Figure 2.
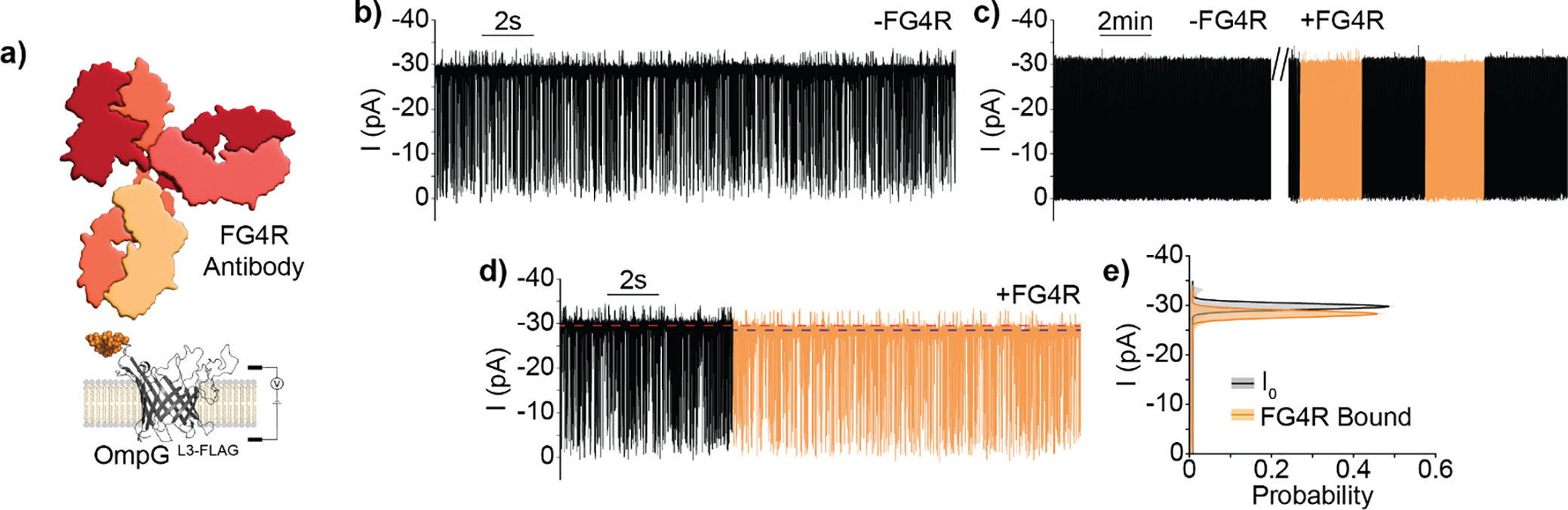
Detection of FG4R via OmpGL3-FLAG nanopore. a) Schematic model of OmpGL3-FLAG sensor and FG4R depicted to relative scale. b,c,d) Representative current traces of OmpGL3-FLAG in the b) absence or c-d) presence of FG4R (20 nM). Regions of the trace showing FG4R binding are colored in orange. The red and blue dotted lines represent I0 and FG4R-bound current respectively. e) All-points histogram of 30 second segments of OmpGL3-FLAG during I0 and FG4R bound states. Experiments were performed in 50 mM Na2HPO4 pH 6.0 buffer containing 300 mM KCl at ±50 mV. Traces were filtered using a 500 Hz lowpass digital gaussian filter.
From this study we observed antibody detection at nanomolar concentration within tens of minutes via direct interaction of the analyte with a loop-displayed peptide motif. Interestingly, while the majority of OmpGLn-FLAG constructs demonstrated effective interaction with FG4R by flow cytometry assay, only OmpGL3-FLAG successfully translated the molecular interaction to a discernible current signal. This observation suggests flow cytometry measurements are not sufficient to predict the detection ability of OmpG nanopore constructs, and as such additional optimization of ligand presentation position within the display loop may be required to allow the analyte/motif interactions to induce resolvable changes in the sensor’s loop gating dynamic readout.
Streptavidin Detection via Loop 6 Peptide Display
We have previously demonstrated the detection of streptavidin using an OmpG nanopore with biotin conjugated to loop 6[13a], here we attempt to detect the same target by utilizing our motif display strategy. We initially selected four streptavidin-binding motifs, including Nano-tag9 (-GDVEAWLGARG-, Kd 17 ± 4 nM) derived from a ribosome-display library, and a series of SA sequences (SA consensus: ICMNVC; SA-3: TVLICMNICWTGETQ; SA-3 truncate: TVLICMNICWT; and SA-1 truncate: LEICQNVCYY) derived from an OmpA-scaffold bacterial display library with the unaltered motif’s reported Kd ranging from 4 nM to 10 nM[18]. Truncation of SA-3 and SA-1, as well as the streptavidin-binding consensus sequence reported from Bessette et al. were considered (Figure S4a) in an attempt to bring the bound analyte closer to the pore entrance to facilitate recognition[13b]. Additionally, to enhance the fluorescence signal, a glycine was added to either side of the SA-1 and SA-3 truncation sequence to generate SA1.1 and SA3.1 (Table S1). Following motif insertion into OmpG loop 6 we utilized flow cytometry to screen these constructs for their ability to bind streptavidin conjugated to the Alexa-647 fluorophore. Four constructs SA-3 truncate, SA3.1, SA-1 truncate, and SA1.1 exhibited an increase in Alexa647 fluorescence, to different extents (Figure S4b), suggesting motif composition/length and the local loop environment influence the efficiency of molecular recognition. Notably, the displayed motifs with the lowest reported Kd values did not exhibit the strongest fluorescence signal in our flow cytometry assay (Figure S4b), showing further considerations of the local motif environment must be made in sensor design, highlighting the utility of the moderate throughput flow cytometry screen.
Among these constructs, SA1.1 was the best candidate for streptavidin detection as the flow cytometry signal showed a single peak distribution with the highest average intensity (Figure S4b) and the construct refolded well in vitro (Figure S2c). Single-channel current measurements of OmpGL6-SA1.1 revealed ‘quiet’ pore behavior with a reduction in gating frequency relative to wildtype (Figure 3b & S5a–b). Addition of streptavidin (strp) resulted in a heterogenous alteration of the gating signature which was not observed following streptavidin dosing to OmpGwt (Figure 3a/c and S5c). Altered pore gating signals could be parameterized into three main subtypes. However, there was considerable variation observed between events categorized to each subtype, indicating this subtyping was not exhaustive, but sufficient for our purpose of representing observed signals. Subtype I was dominated by a substantial reduction of gating frequency during the binding event, with a concurrent 5.30 ± 0.44 % increase (N =3, n = 15) in the open pore current (Figure 3d). Spontaneous gating was not completely precluded during this state but was greatly reduced in frequency, with a stark reduction in the average blockage depth as well. Subtype II was characterized by high frequency transient gating to ~50–75% open pore current but did not demonstrate altered open pore current during the bound state (Figure 3e). Subtype III was also characterized by an increased frequency of gating; however, the frequency and current occlusion was less than that observed in subtype II. Additionally, subtype III exhibited a 4.83 ± 1.40 % increase in the open pore current (N = 3, n = 15) (Figure 3f). The overall prevalence of binding subtypes was III>II≈I. The reduction in gating intensity and/or frequency upon target interaction likely stems from reduced loop 6 mobility due to the bound analyte, with the other observed ‘spikes’ potentially contributed by the motion of other loops sampling the bound analyte near the pore interface[13a, 19]. Interestingly, the single analyte resulted in heterogenous yet motif-specific binding signals (Figure 3d–f). We propose this could be due to different interaction interfaces formed between the OmpG nanopore and the surface of the streptavidin captured by SA1.1 motif.
Figure 3.
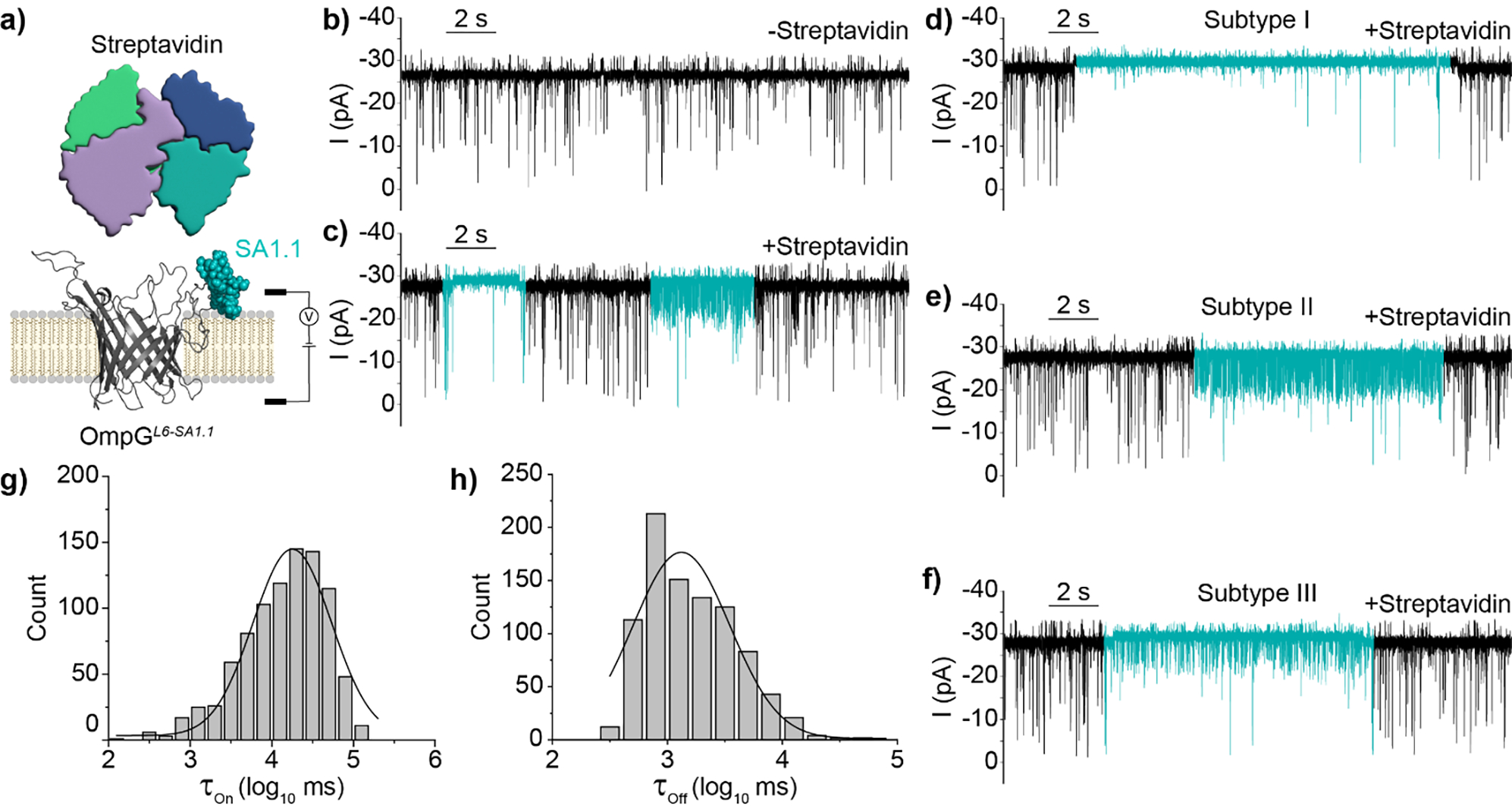
Detection of Streptavidin by OmpGL6-SA1.1 nanopore. a) Schematic view of OmpGL6-SA1.1 sensor and analyte streptavidin. b-c) Representative traces of OmpGL6-SA1.1 in the absence and presence of streptavidin (800 nM). The regions of the trace demonstrating streptavidin binding signals are colored in blue. d-f) Representative traces of streptavidin binding Subtypes I-III. g-h) Black line shows gaussian fit of τon and τoff from log transformed millisecond values (N = 3, n = 903). Experiments were performed in 50 mM Na2HPO4 pH 6.0 buffer containing 300 mM KCl at an applied potential of ±50 mV. Traces were filtered using a 500 Hz lowpass digital gaussian filter.
Following binding signal parameterization, the interevent (τOn) and dwell (τOff) of streptavidin binding at 800 nM was calculated by accounting for all binding subtypes (N = 3, n = 903) resulting in a τOn of 18.43 ± 1.01 s and τOff of 1.27 ± 0.02 s determined from the average of three independent gaussian fittings (Figure 3g–h). From these values the average association rate constant (kOn = 1/[C × τOn], mean ± SD) and dissociation rate constant (kOff = 1/τOff, mean ± SD) of streptavidin were calculated to be 7.76 ± 1.37 × 104 M−1*s−1 and 78.0 ± 5.0 × 10−2 s−1 respectively. Thus, the apparent dissociation constant (Kd = kOff/kOn) was calculated to be 10.05 ± 1.37 μM. This value was over two orders of magnitude higher than was reported for the original un-truncated motif (Kd = 10 nM) suggesting a substantial loss in affinity stemming from the truncation of the original sequence. However, despite this apparent loss in affinity our study was able to demonstrate effective detection of nanomolar streptavidin within minutes. In summary, we have now demonstrated the display of two motifs, FLAG and SA1.1, within two distinct OmpG loops can generate unique, analyte-specific, and reversible gating signatures arising from direct target interaction.
Multiplex Detection of FG4R and Streptavidin in a Simple Mixture
With two motifs (FLAG and SA1.1) exhibiting unique gating signatures arising from target interaction with two different loops, we sought to generate a construct for the simultaneous display of both motifs (Figure 4a). This new sensor, termed OmpGL3-FLAG/L6-SA1.1, contains the FLAG motif within loop 3 and SA1.1 within loop 6 and its gating behavior was assessed via single-channel current measurements as before. In the absence of target analytes, the gating profile resembled that observed for OmpGL6-SA1.1, suggesting the motif incorporated into loop 6 dominates the gating behavior (Figure 4b & Figure S6b–d). Addition of FG4R resulted in recapitulation of the minutes long, reversible reduction in open pore current observed with OmpGL3-FLAG sensor (Figure 4c). Similarly, addition of streptavidin recapitulated the heterogenous signal observed with OmpGL6-SA1.1 (Figure 4d). Analyte dosing to OmpGwt or pores otherwise lacking the corresponding affinity motif did not evoke any change in current or gating behavior (Figure S7a–d). We next tested if the analyte-specific signals could be reproduced when both streptavidin and FG4R were present. We observed that gating signature unique to each analyte could be observed individually (FG4R shown in Figure 4e, streptavidin shown in in Figure 4f). Surprisingly, we also noticed that the two analyte-binding signatures could occur concurrently (Figure 4g), indicating both analytes could bind their respective motif without one analyte’s binding precluding the binding of any further analytes due to steric occlusion. Interestingly, concurrent occupation of the sensor pore by both analytes appears to alter the distribution of streptavidin binding subtypes via complete ablation of Subtype II (Figure S8a–c).
Figure 4.
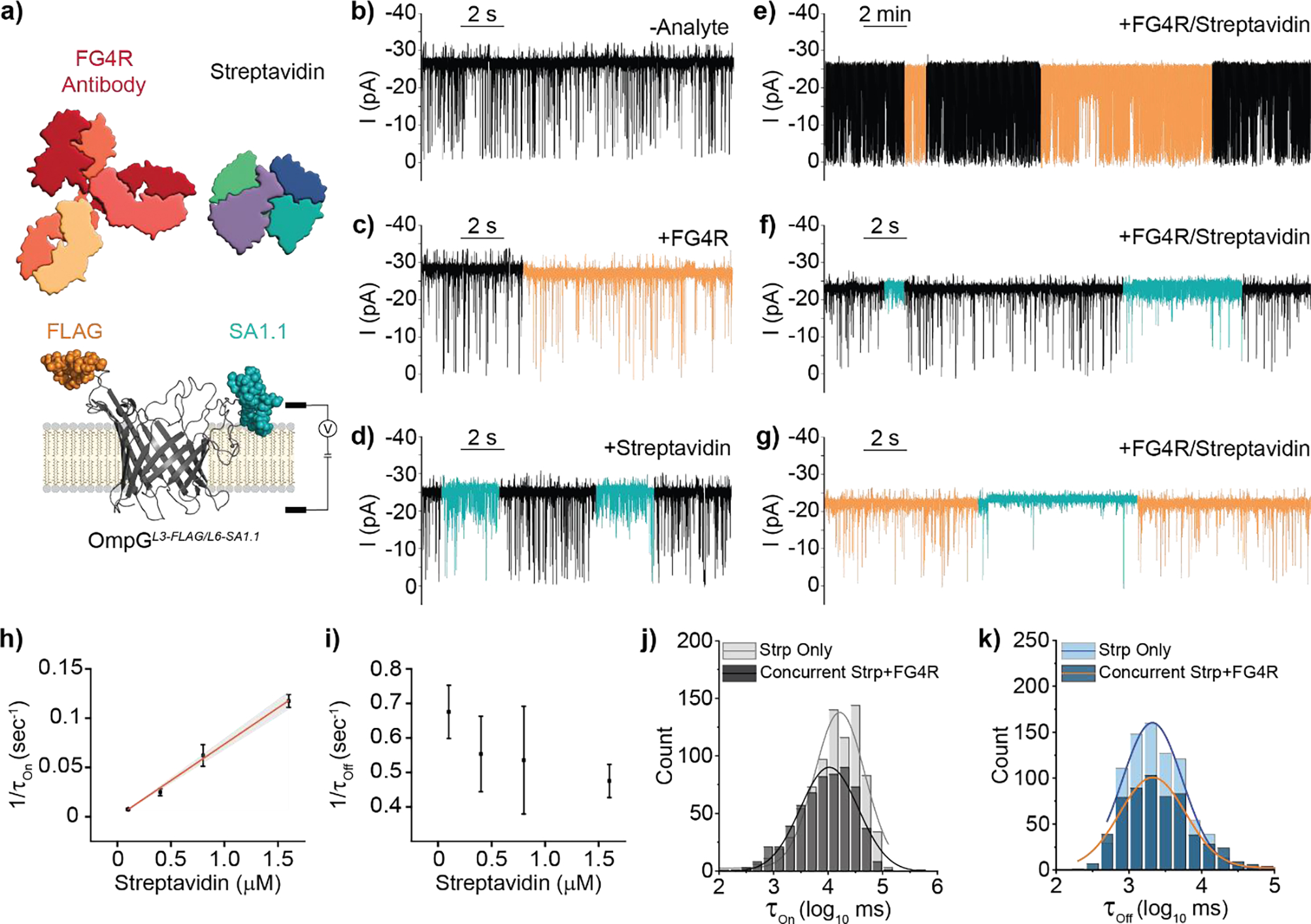
Multiplex detection of two protein analytes in a simple mixture by OmpGL3-FLAG/L6-SA1.1 nanopore a) Schematic view of OmpGL3-FLAG/L6-SA1.1 sensor. b-d) Representative single-channel current recordings of OmpGL3-FLAG/L6-SA1.1 b) basal behavior and recapitulation of c) FG4R (20 nM) and d) streptavidin (800 nM) binding signals. e-f) Representative single-channel current recording traces showing recapitulation of individual analyte signals when analytes are present in a simple mixture g) Concurrent observation of both binding signals. h-i) Concentration dependence of streptavidin 1/τOn and 1/τOff. Error bars represent standard deviation from three independent pores. j-k) Gaussian fit of τOn and τOff values from log transformed millisecond values representing quantification of streptavidin binding. Light gray and blue represent when only streptavidin is present (N = 3, n = 813), dark gray and blue represent concurrent streptavidin and FG4R quantification (N = 3, n = 594). Experiments were performed in 50 mM Na2HPO4 pH 6.0 buffer containing 300 mM KCl at an applied potential of ±50 mV. Traces were filtered using a 500 Hz lowpass digital gaussian filter.
We then determined the binding kinetics of streptavidin with the OmpGL3-FLAG/L6-SA1.1sensor when only streptavidin was present for direct comparison with the binding kinetics of streptavidin with OmpGL6-SA1.1. The average dissociation rate constant, kOff, was 55.0 ± 3.0 × 10−2 s−1 (N = 12, n = 2846) and was independent of streptavidin concentration (Figure 4i & S9b/d). The 1/τOn values increased linearly with streptavidin concentration (Figure 4h & Figure S9a/c) resulting in a calculated association rate constant, kOn, of 7.22 ± 1.0 × 104 M−1s-1. These values result in a calculated Kd of streptavidin for the SA1.1 multiplex sensor of 7.51 ± 0.26 μM, in close agreement with the ~10 μM value calculated for OmpGL6-SA1.1, suggesting the presence of the FLAG motif on loop 3 did not perturb the streptavidin/loop 6 interaction.
Subsequently, we determined if concurrent analyte occupancy of the pore altered this kinetic. We initially calculated τOn and τOff of streptavidin at 800 nM when it was the only analyte present (Strp Only) to be 16.46 ± 3.16 s and 1.97 ± 0.55 s (N = 3, n = 813) respectively. Then, to determine the influence of concurrent binding, a recording was performed where both streptavidin (800 nM) and FG4R (20 nM) were present. Due to the slow kOff of FG4R, and the more rapid association kinetics of streptavidin, we were able to observe numerous streptavidin binding events within the FG4R bound state, despite the latter’s relatively infrequent occurrence. For the concurrent binding analysis, we only considered regions of the trace where both analytes were bound (Concurrent Strp+FG4R) and calculated the τOn and τOff of concurrently bound streptavidin to be 11.70 ± 5.49 s and 2.11 ± 0.35 s respectively (N = 3, n = 594). Overall, we concluded there was no significant interference of association (Student’s T-test p-value 0.26) or dissociation kinetics (Student’s T-test p-value 0.73) when both analytes were bound to the sensor scaffold, a significant finding as this is the first demonstration of multiplex detection allowing dual occupancy of a nanopore scaffold by two bulky protein analytes. Additionally, the observation that the occupation of loop 3 by FG4R changes the distribution of streptavidin binding signal characteristics but not their overall frequency suggests that some streptavidin/OmpG sampling interface were prohibited by the bound FG4R.
Next, we tested the performance of our OmpG sensor in the presence of 150 μM bovine serum albumin (BSA) (Figure S10a), an abundant protein in serum with non-specific protein binding propensity. Addition of BSA to the recording chamber caused a general increase in OmpGL3-FLAG/L6-SA1.1 gating ‘noise’ a (Figure S10b/c) but without any discrete binding signal changes associated with an affinity-mediated interaction. After confirming BSA influence on the sensor we then dosed streptavidin (1.6 μM) to determine if the previously reported binding subtypes could be recapitulated (Figure S10d). Despite the increased gating noise the binding signals were still well resolved, allowing for determination of streptavidin τOn and τOff values (Figure S10e/f). τOn of streptavidin at 1.6 μM was calculated as 18.72 ± 3.53 s by accounting for all binding subtypes, showing an increase of ~2.2 fold (Student’s T-test p-value 0.007) in comparison with 8.53 ± 0.48 s without BSA (Figure S10e). τOff was determined to be 2.72 ± 0.67 s and 2.12 ± 0.21 s with and without BSA respectively (Figure S10f), exhibiting no significant difference between conditions (Student’s T-test p-value 0.21). We concluded high concentration of BSA appeared to reduce analyte capture efficiency, potentially through one or more of the following mechanisms: 1) BSA acts a molecular crowding reagent slowing down the diffusion rate of streptavidin 2) BSA transiently occupies the affinity sites on either the sensor or streptavidin therefore impeding the analyte binding to OmpG nanopore. Despite the reduction in the association rate streptavidin detection was not substantially impacted, suggesting this sensor strategy is viable for biomarker detection in complex solution.
Here we have shown that the binding kinetics of an analyte with its corresponding loop-displayed motif are not significantly impacted by the scaffold hosting other loop-displayed motifs, suggesting this approach is feasible to pursue with other analytes. Overall, we have demonstrated that OmpG is a promising malleable scaffold for the development of a nanopore multiplex protein biosensor, but specific construct optimization may be necessary to resolve the binding of certain target analytes. This study represents the first multi-functionalization of a protein nanopore for the direct single-molecule multiplex detection of protein analytes. Prior reports of nanopore multiplex protein detection have been accomplished either by 1) target-specific functionalization of many pores in an array or 2) use of DNA barcoding to detect and quantify the corresponding protein biomarker[8a, 8c, 8h–j, 20]. The methods utilizing DNA consistently achieve lower limit of detection due to the intrinsic capture efficiency of DNA, and channel functionalization is expandable based on the repertoire of available affinity ligands. However, these methods lack the molecular recognition resolution of OmpG sensing which allowed for discrimination among closely related targets that bind to the same affinity reagent, allowing for molecular subtyping not available in other systems.
Avidity Display of SA1.1 Significantly Impacts Binding Kinetics
OmpG sensing is stochastic and diffusion mediated[21]. At low analyte concentrations (≤nM), the detection time becomes inordinately long compared to systems where analytes can be driven to the pore[3, 22]. We attempted to address the diffusion limitations of OmpG sensing by enhancing the avidity through the display of multiple copies of the same motif on OmpG nanopore. We elected to utilize the streptavidin binding motif SA1.1 as it showed moderate affinity and faster dissociation kinetics relative to FG4R. A new construct, OmpGL3/L6-SA1.1 was generated displaying the SA1.1 motif in both loops 3 and 6 (Figure 5a).
Figure 5.
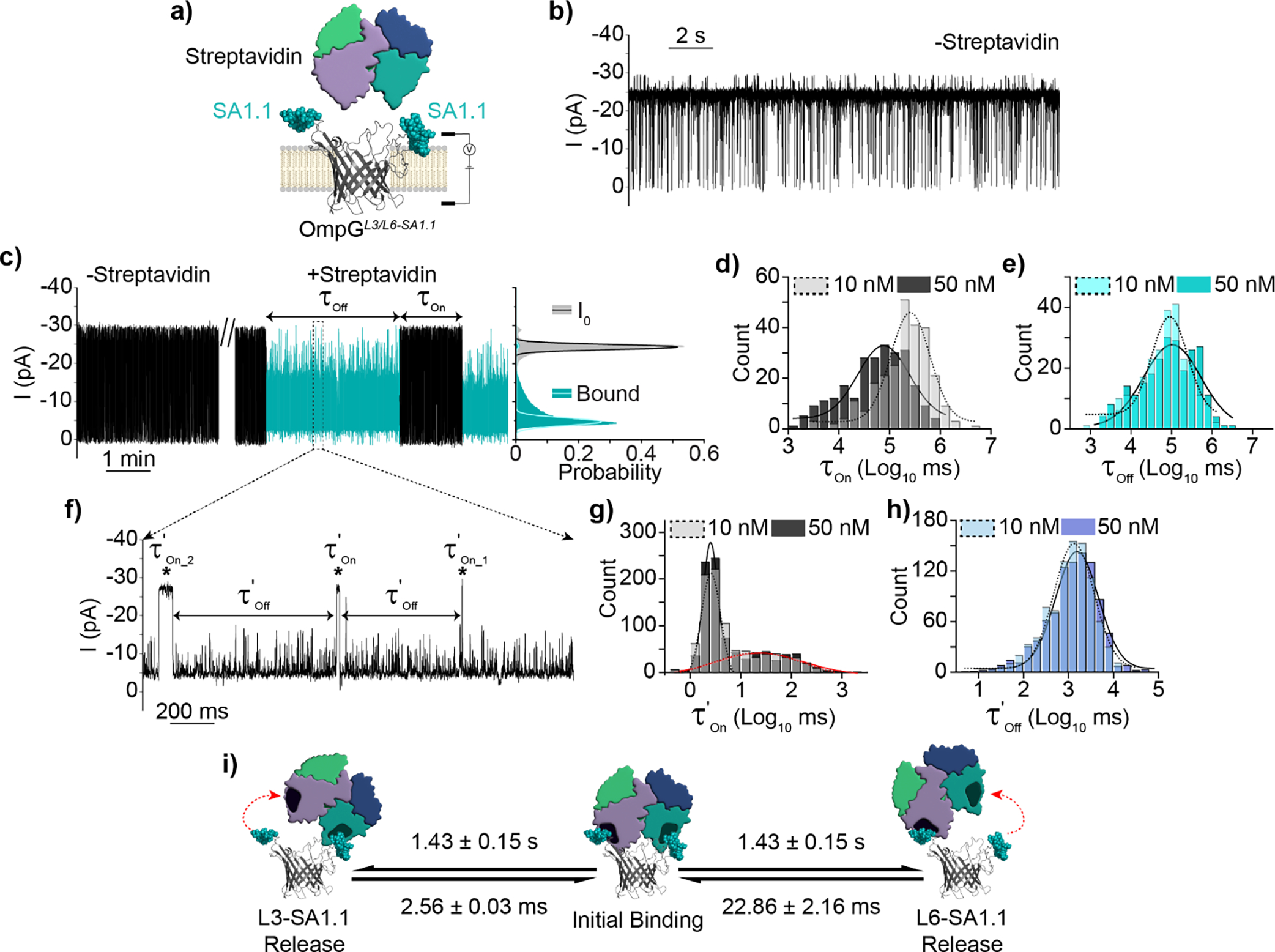
Detection of streptavidin by OmpGL3/L6-SA1.1 avidity nanopore sensor. a) Schematic view of OmpGL3/L6-SA1.1 avidity sensor. b) Representative current trace in the absence of streptavidin c) Representative current trace and all-points histogram in the presence of streptavidin, discrete binding signal(s) indicated in blue. All-points histogram generated from 30 second segments of indicated state. d-e) Gaussian fit of τOn and τOff values derived from discrete streptavidin binding events at either 10 nM (N = 7, n = 258) or 50 nM (N = 5, n = 247). f) Zoomed in view of discrete bound state highlighting streptavidin rebinding sub-states within the discrete signal. g-h) Gaussian fit of kinetic parameters τ’On_1&2 and τ’Off of rebinding events at either 10 nM (N = 3, n = 902) or 50 nM (N = 3, n = 896). i) Proposed model of streptavidin interaction with OmpGL3/L6-SA1.1. Experiments were performed in 50 mM Na2HPO4 pH 6.0 buffer containing 300 mM KCl at an applied potential of ±50 mV. Traces were filtered using a 500 Hz lowpass digital gaussian filter.
In the absence of target OmpGL3/L6-SA1.1 basal gating resembled that of OmpGL6-SA1.1 and OmpGL3-FLAG/L6-SA1.1 (Figure 5b & S6c/d/e). Addition of streptavidin resulted in a strikingly different signal from that observed in OmpGL6-SA1.1. This new signal (Figure 5c) consisted of minute(s)-long regions with a ~72.8 ± 6.3% reduction in current (N = 5, n = 25), and transient high-frequency fluctuations at sub-conductance states between 20 and 75% open pore current. Streptavidin binding kinetics were starkly different for OmpGL3/L6-SA1.1 relative to OmpGL6-SA1.1. Streptavidin bound to OmpGL3/L6-SA1.1 exhibited an average kOff of 1.0 ± 0.1 × 10−2 s−1 (10 nM N = 7, n = 258 and 50 nM N = 5, n = 247, average of gaussian fit from each concentration N = 12, n = 505) compared to that of OmpGL6-SA1.1 at 78.0 ± 5.0 × 10−2 s−1, exhibiting an increase of analyte residence by ~78 fold (Figures 4i & 5e). The calculated kon of OmpGL6-SA1.1 was 7.76 ± 1.37 × 104 M−1*s−1 compared to 3.26 ± 0.90 × 105 M−1s−1 for OmpGL3/L6-SA1.1, an increase of ~4.2 fold (Figures 4h & 5d). As with OmpGL6-SA1.1 1/τOn increased with analyte concentration (Figure 5d) while τOff was concentration independent (Figure 5e) indicating the observed signals represented unique and analyte-specific interactions with the pore. From these kinetic values we calculated the Kd of streptavidin for OmpGL3/L6SA1.1 to be ~30 nM, an increase of ~335 fold compared to OmpGL6-SA1.1.
Interestingly, upon closer inspection of each minute(s) long binding event we observed that each of these streptavidin binding signals constituted numerous short duration sub-events separated by current signals transiently returning to the fully open pore level (Figure 5f). We calculated the k’Off of the sub-events to be 70.0 ± 8.0 × 10−2 s−1, close to that of OmpGL6-SA1.1 78.0 ± 5.0 × 10−2 s−1 (Figure 4i & 5h). The inter-event durations were fitted with bimodal Gaussian distribution which resulted in two average duration times τ’On_1 and τ’On_2 as 2.56 ± 0.03 ms (black lines) and 22.86 ± 2.16 ms (red lines), corresponding to a k’On_1 and k’On_2 of 3.9 × 1010 M−1s−1 and 4.4 × 109 M−1s−1 respectively (10 nM N = 3, n = 902 and 50nM N = 3, n = 896, average of gaussian fit from each concentration N = 6, n = 1798). Importantly, increasing the streptavidin concentration showed no effect on the on and off rates of these sub-events (Figure 5g–h).
Based on the current signal and kinetic analysis, we propose that each of the minutes long streptavidin signals represents the binding of a unique streptavidin molecule, and as such we refer to these events as ‘discrete’ binding. As streptavidin is a homotetramer it has a valency of four potential SA1.1 binding sites. When a single subunit is bound by a SA1.1 copy, the other three subunits are available for interaction with the second loop-displayed SA1.1. Thus, the sub-event binding signal observed within discrete binding events likely reflects the concurrent docking of two SA1.1 motifs to the tetramer, bridging the streptavidin molecule over the pore’s luminal entrance, which resulted in the obstruction of ionic flow as indicated by the substantial reduction in current compared to streptavidin binding to the single SA1.1 site on OmpGL6-SA1.1. The sub-events within each “discrete” streptavidin binding can be explained by a “partial-release and re-bridging” model: when dissociation of streptavidin from one of the SA1.1 sites on OmpGL3/L6-SA1.1 occurred, the current signals returned to the open-pore current as the streptavidin is no longer held over the pore’s luminal entrance. This explains why the dwell time (τ’Off) of OmpGL3/L6-SA1.1 sub-events was identical to that of streptavidin/OmpGL6-SA1.1 complex as the duration of both scenarios was controlled by the interaction between a single subunit of streptavidin and a SA1.1 motif, while the unoccupied SA1.1 motif could quickly re-bind to streptavidin and resume the bridge mode. Notably, the “re-bridging” on-rates of sub-events exhibited a bimodal gaussian distribution, corresponding to a more rapid process (2.56 ± 0.03 ms) and a slower one (22.86 ± 2.16 ms). We propose the ten-times variation in the two rates may reflect the different efficiency of analyte re-binding to the SA 1.1 motif displayed at loop 3 or loop 6. For example, re-binding to a loop 3-displayed SA1.1 motif may occur more efficiently than to a loop 6-displayed one as loop 6 is longer and more dynamic. Another explanation is that the analyte may dissociate from both motifs causing it temporarily disengage from the pore surface, while remaining in the local area where it could then be recaptured by OmpGL3/L6-SA1.1. The full “release and recapture” could account for the slower k’On_2 while the fast k’On_1 originates when one subunit dissociates from a SA1.1 and then resumes the “bridge” binding mode. This is less likely as we did not observe a bimodal distribution of the τOn for the streptavidin interaction with OmpGL6-SA1.1 and OmpGL3-Flag/L6-SA1.1. Should the recapture of a fully released streptavidin by OmpGL3/L6-SA1.1 occur at the rate ~4 × 109 M−1s−1, such “recapture” events would also take place with OmpGL6-SA1.1 and OmpGL3-Flag/L6-SA1.1 with expected lower efficiency. However, we only observed a single population of slow association events at a rate of ~7 × 104 M−1s-1. More importantly, the ratio of the total number of two types of events, calculated as the areas of two gaussian peaks, were close to 1:1 at both concentrations (10 nM and 50 nM) indicating that the two types of events occurred at almost equal frequency. If the “release and recapture” phenomena occurred, the slower recapture process would appear at greatly reduced frequency relative to the faster recapture processes, as the full release of the molecule from both motifs would occur less often than release from a single motif. Furthermore, the on-rates of sub-events were independent of the analyte concentration and exceeded the diffusion-controlled process, again supporting that these sub-events originate from the same streptavidin molecule alternating its binding-modes on OmpGL3/L6-SA1.1.
Thus, we attributed the enhanced avidity of streptavidin binding with OmpGL3/L6-SA1.1 predominantly to the extended dwell time (τOff), which was enabled by the formation of dual interaction interfaces and multiple cycles of the release and re-formation of either interaction (Figure 5i). Such unique binding kinetics likely reflected the architecture of dual binding motifs presented by the OmpG scaffold and streptavidin’s multivalency. Further study is needed to determine if, and to what extent, a monovalent protein analyte may also achieve prolonged binding time. Based on our study, one could also envisage a strategy to enhance the detection sensitivity by placing two weak affinity motifs that recognize separate regions of a protein into the OmpG nanopore for analyte capture when high affinity motifs are not available. However, we only observed a moderately additive effect in the association rate for “discrete” binding, which suggested the multivalent motif strategy would not offer significant improvement to the sensor’s limit of detection.
Conclusion
In summary, we demonstrated that peptide motif display via OmpG nanopore scaffold expressed at the bacterial outer membrane allows for medium throughput screening of sensor constructs via flow cytometry. The OmpG nanopore sensor hosting duo binding motifs can be produced as a single polypeptide chain without multiple chemical labeling processes. Furthermore, the OmpG nanopore can integrate two distinct binding motifs that enable multiplex detection of protein analytes in a mixture with high specificity and accuracy. Our data also provided a mechanistic explanation for avidity binding at single molecule level.
Supplementary Material
Acknowledgements
J.C.F. was supported in part by a fellowship from the University of Massachusetts Amherst as part of the Chemistry-Biology Interface (CBI) training grant (National Institutes of Health TM32-GM008515 & TM32-GM139789). This research was supported by the US National Institutes of Health grant R01-GM115442 (to M.C.) and US Department of Agriculture National Institute of Food and Agriculture grant AFRI 2018–07590 (to M.D.M.). We would like to thank Dr. AS Burnside for assistance with flow cytometry data acquisition.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Califf RM, Exp Biol Med 2018, 243, 213–221; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cohen L, Walt DR, Chem Rev 2019, 119, 293–321; [DOI] [PubMed] [Google Scholar]; c) Nimse SB, Sonawane MD, Song KS, Kim T, Analyst 2016, 141, 740–755. [DOI] [PubMed] [Google Scholar]
- [2].Leva-Bueno J, Peyman SA, Millner PA, Med Microbiol Immunol 2020, 209, 343–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Reiner JE, Balijepalli A, Robertson JW, Campbell J, Suehle J, Kasianowicz JJ, Chem Rev 2012, 112, 6431–6451. [DOI] [PubMed] [Google Scholar]
- [4].a) Wang Y, Zhao Y, Bollas A, Wang Y, Au KF, Nat Biotechnol 2021, 39, 1348–1365; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tabatabaei SK, Pham B, Pan C, Liu J, Chandak S, Shorkey SA, Hernandez AG, Aksimentiev A, Chen M, Schroeder CM, Milenkovic O, Nano Lett 2022, 22, 1905–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Huang G, Willems K, Soskine M, Wloka C, Maglia G, Nat Commun 2017, 8, 935; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hwang HJ, Kim JS, Lee J, Min JS, Jeong KB, Kim E, Lee MK, Chi SW, Anal Chem 2022, 94, 7449–7454; [DOI] [PubMed] [Google Scholar]; c) Yan S, Zhang J, Wang Y, Guo W, Zhang S, Liu Y, Cao J, Wang Y, Wang L, Ma F, Zhang P, Chen HY, Huang S, Nano Lett 2021, 21, 6703–6710; [DOI] [PubMed] [Google Scholar]; d) Brinkerhoff H, Kang ASW, Liu J, Aksimentiev A, Dekker C, Science 2021, 374, 1509–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Ouldali H, Sarthak K, Ensslen T, Piguet F, Manivet P, Pelta J, Behrends JC, Aksimentiev A, Oukhaled A, Nat Biotechnol 2020, 38, 176–181; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Restrepo-Pérez L, Huang G, Bohländer PR, Worp N, Eelkema R, Maglia G, Joo C, Dekker C, ACS nano 2019, 13, 13668–13676; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Restrepo-Pérez L, Wong CH, Maglia G, Dekker C, Joo C, Nano letters 2019, 19, 7957–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Lucas FLR, Versloot RCA, Yakovlieva L, Walvoort MTC, Maglia G, Nat Commun 2021, 12, 5795; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang S, Huang G, Versloot RCA, Bruininks BMH, de Souza PCT, Marrink SJ, Maglia G, Nat Chem 2021, 13, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Duan L, Yobas L, ACS Nano 2018, 12, 7892–7900; [DOI] [PubMed] [Google Scholar]; b) He L, Karau P, Tabard-Cossa V, Nanoscale 2019, 11, 16342–16350; [DOI] [PubMed] [Google Scholar]; c) Zhang Z, Wang X, Wei X, Zheng SW, Lenhart BJ, Xu P, Li J, Pan J, Albrecht H, Liu C, Biosens Bioelectron 2021, 181, 113134; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wei S, Weiss ZR, Williams Z, G3 2018, 8, 1649–1657; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Huang S, Romero-Ruiz M, Castell OK, Bayley H, Wallace MI, Nat Nanotechnol 2015, 10, 986–991; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Cardozo N, Zhang K, Doroschak K, Nguyen A, Siddiqui Z, Bogard N, Strauss K, Ceze L, Nivala J, Nat Biotechnol 2022, 40, 42–46; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Palla M, Punthambaker S, Stranges B, Vigneault F, Nivala J, Wiegand D, Ayer A, Craig T, Gremyachinskiy D, Franklin H, Sun S, Pollard J, Trans A, Arnold C, Schwab C, McGaw C, Sarvabhowman P, Dalal D, Thai E, Amato E, Lederman I, Taing M, Kelley S, Qwan A, Fuller CW, Roever S, Church GM, ACS Nano 2021, 15, 489–502; [DOI] [PubMed] [Google Scholar]; h) Bell NA, Keyser UF, Nat Nanotechnol 2016, 11, 645–651; [DOI] [PubMed] [Google Scholar]; i) Keyser UF, Nat Nanotechnol 2016, 11, 106–108; [DOI] [PubMed] [Google Scholar]; j) Liu L, Li T, Zhang S, Song P, Guo B, Zhao Y, Wu HC, Angew Chem Int Ed 2018, 57, 11882–11887. [DOI] [PubMed] [Google Scholar]
- [9].a) Fahie MA, Yang B, Mullis M, Holden MA, Chen M, Anal Chem 2015, 87, 11143–11149; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chuah K, Wu Y, Vivekchand SRC, Gaus K, Reece PJ, Micolich AP, Gooding JJ, Nat Commun 2019, 10, 2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Conlan S, Zhang Y, Cheley S, Bayley H, Biochemistry 2000, 39, 11845–11854; [DOI] [PubMed] [Google Scholar]; b) Liang B, Tamm LK, Proc Natl Acad Sci U S A 2007, 104, 16140–16145; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Retel JS, Nieuwkoop AJ, Hiller M, Higman VA, Barbet-Massin E, Stanek J, Andreas LB, Franks WT, van Rossum BJ, Vinothkumar KR, Handel L, de Palma GG, Bardiaux B, Pintacuda G, Emsley L, Kühlbrandt W, Oschkinat H, Nat Commun 2017, 8, 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Bainbridge G, Gokce I, Lakey JH, FEBS Lett 1998, 431, 305–308; [DOI] [PubMed] [Google Scholar]; b) Beckstein O, Biggin PC, Bond P, Bright JN, Domene C, Grottesi A, Holyoake J, Sansom MS, FEBS Lett 2003, 555, 85–90; [DOI] [PubMed] [Google Scholar]; c) Yildiz O, Vinothkumar KR, Goswami P, Kühlbrandt W, Embo j 2006, 25, 3702–3713; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhuang T, Chisholm C, Chen M, Tamm LK, J Am Chem Soc 2013, 135, 15101–15113; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhuang T, Tamm LK, Angew Chem Int Ed Engl 2014, 53, 5897–5902; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Perez-Rathke A, Fahie MA, Chisholm C, Liang J, Chen M, J Am Chem Soc 2018, 140, 1105–1115; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Pham B, Chisholm CM, Foster J, Friis E, Fahie MA, Chen M, Biochim Biophys Acta Biomembr 2021, 1863, 183485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Zernia S, van der Heide NJ, Galenkamp NS, Gouridis G, Maglia G, ACS Nano 2020, 14, 2296–2307; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kozak D, Anderson W, Vogel R, Trau M, Nano Today 2011, 6, 531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Fahie MA, Chen M, J Phys Chem B 2015, 119, 10198–10206; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fahie MA, Yang B, Pham B, Chen M, ACS Sens 2016, 1, 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Pham B, Eron SJ, Hill ME, Li X, Fahie MA, Hardy JA, Chen M, Biophys J 2019, 117, 844–855; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fahie MA, Candido J, Andree G, Chen M, ACS Sens 2021, 6, 1286–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Visudtiphole V, Chalton DA, Hong Q, Lakey JH, Biochem Biophys Res Commun 2006, 351, 113–117; [DOI] [PubMed] [Google Scholar]; b) Verhoeven GS, Alexeeva S, Dogterom M, den Blaauwen T, PLoS One 2009, 4, e6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rollauer SE, Sooreshjani MA, Noinaj N, Buchanan SK, Philos Trans R Soc Lond B Biol Sci 2015, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Balijepalli A, Ettedgui J, Cornio AT, Robertson JW, Cheung KP, Kasianowicz JJ, Vaz C, ACS Nano 2014, 8, 1547–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Lamla T, Erdmann VA, Protein Expr Purif 2004, 33, 39–47; [DOI] [PubMed] [Google Scholar]; b) Bessette PH, Rice JJ, Daugherty PS, Protein Eng Des Sel 2004, 17, 731–739. [DOI] [PubMed] [Google Scholar]
- [19].Chen M, Khalid S, Sansom MS, Bayley H, Proc Natl Acad Sci U S A 2008, 105, 6272–6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kong J, Bell NA, Keyser UF, Nano Lett 2016, 16, 3557–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ayub M, Bayley H, Curr Opin Chem Biol 2016, 34, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang G, Wang L, Han Y, Zhou S, Guan X, Accounts of chemical research 2013, 46, 2867–2877. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.