

Abstract
Modern mass spectrometry-based workflows employing hybrid instrumentation and orthogonal separations collect multidimensional data, potentially allowing deeper understanding in omics studies through adoption of artificial intelligence methods. However, the large volume of these rich spectra challenges existing data storage and access technologies, therefore precluding informatics advancements. We present MZA (pronounced m-za), the mass-to-charge (m/z) generic data storage and access tool designed to facilitate software development and artificial intelligence research in multidimensional mass spectrometry measurements. Composed by a data conversion tool and a simple file structure based on the HDF5 format, MZA provides easy, cross-platform and cross-programming language access to raw MS-data, enabling fast development of new tools in data science programming languages such as Python and R. The software executable, example MS-data and example Python and R scripts are freely available at https://github.com/PNNL-m-q/mza
Keywords: mass spectrometry, open data format, data conversion, ion mobility spectrometry, data-independent acquisition
Graphical Abstract
In compliance with the guidelines, we confirm that this TOC graphic was originally created by the author Aivett Bilbao and it has not been published elsewhere.
Introduction
Mass spectrometry (MS) is the method of choice for global characterization of molecules in complex samples primarily due to its capability to be coupled to a wide range of separation methods. Untargeted MS-based workflows incorporating orthogonal separations, such as liquid chromatography (LC) and ion mobility spectrometry (IM)1, and collecting comprehensive ion fragmentation data with data-independent acquisition (DIA)2 methods or alternative activation techniques, are providing heterogeneous and multidimensional information which allows deeper understanding in omics studies. However, the large volume and structure of these rich multidimensional spectra challenges current MS-data storage and access technologies.
During LC-MS or LC-IM-MS analyses, the raw spectra is stored in proprietary formats optimized according to the vendor hardware and these files can only be accessed using the vendor’s provided software libraries. The main limitations of vendor libraries are the complex dependencies which are typically Windows-bounded and difficult to maintain and deploy, therefore hindering rapid development of algorithms, prototypes and new software tools in data-science (DS) and artificial intelligence (AI) programming languages such as Python and R, and particularly limiting novice developers and beginners in the MS field. These scripting languages provide considerably easier coding, and faster execution of slow routines has become possible by building the computationally heavy function as C wrapped libraries.
Storing large raw MS-data in text format is impractical due to the resulting enormous file size and slow reading access. Several open formats currently exist, most notably the mzML3 format and the ProteoWizard toolkit4 supporting file conversion from most instrument vendors. These open technologies have been paramount for the advancement of MS-based omics workflows by supporting the development of informatic tools as well as data interchange, and deposition. However, the mzML format is based on the Extensible Markup Language (XML) which is inefficient for storing and accessing large amounts of spectra generated in multidimensional measurements.
Numerous strategies have been developed for either improving over mzML (and its predecessor mzXML5), such as mz56, imML7, OpenMS library8 and pymzML9, or proposing more efficient formats, like mzDB10, Shaduf11, Toffee12, mzMLb13, Aird14 and StackZDPD15. These open solutions offer great file size reduction and fast reading access; however, spectra and metadata are either compressed with customized algorithms, stored in a nested structure of XML tags, or require dedicated software libraries or application programming interfaces (API), all of which complicate data access and utilization. Moreover, some of the tools are specifically designed for a particular instrument or omics application. For instance, the recently reported MS2AI16 pipeline provides automation for large scale machine learning applications, nevertheless, its functionality is exclusive to LC-MS proteomics, uses lossy data representations (i.e., partially discards information) and does not support the IM dimension.
To address these limitations, we developed MZA. Like other recent MS formats, MZA is based on the open, binary, and hierarchical data format, HDF5 (www.hdfgroup.org). but contrary to other solutions, MZA provides a stand-alone and self-contained data conversion tool and a simple MS-data file structure.
Methods
MZA data converter
The MZA data converter was implemented in Python with minimal dependencies: Pythonnet to directly interface with instrument vendor software libraries (.DLL) and read the raw MS-data (currently Agilent MassHunter ‘.d’ and Thermo ‘.raw’ supported), pymzml to read mzML files, h5py and PyTables to write the data as the MZA data structure, and PyInstaller to generate a self-contained software executable. The multiprocessing module was used to parallelize the execution of functions across multiple available computer processors. The MZA executable can be run directly on Windows and Unix-like operating systems require pre-installation of the Wine compatibility layer (www.winehq.org) or a Docker container. Specific instructions and current supported versions can be found in the GitHub page. Raw files from LC-MS and LC-IM-MS experiments, with or without DIA spectra can be converted. Raw files from data-dependent acquisition experiments are also supported. The only required parameter is -file to indicate the file to be converted.
MZA file format
The MZA file structure is simple: a metadata table and two groups of 1D arrays stored individually as HDF5 datasets. Each row in the metadata table represents a spectrum and the columns represent the properties of the spectrum such as scan number (unique to each spectrum), MS level, activation (i.e., ion fragmentation type), retention time, ion mobility arrival time, etc. Most of the column names follow the standardized vocabulary established for mzML by the Proteomics Standards Initiative. Spectra are stored omitting zero-intensity values and as two jagged arrays, one for m/z values and one for corresponding intensity values. The data structure is mass analyzer agnostic, e.g., time-of-flight (TOF) and Fourier transform-based MS are supported. For IM spectra, the m/z dimension is stored as indexes of a single full m/z array common for all spectra in the file.
Raw MS-data and conversion to open formats
Three representative raw files (available at ftp://massive.ucsd.edu/MSV000090269) were used: a 30 min LC-Drift Tube (DT) IM-MS lipidomics run (MassHunter ‘.d’ format) acquired from a standard human plasma on a Agilent 6560 Ion Mobility quadrupole TOF MS system (Agilent Technologies, Santa Clara, CA) in the DIA All Ions mode for a previous study17, a 60 min LC-MS DIA proteomics run (.raw format) acquired from our bacterium (Shewanella oneidensis) quality control sample on an Orbitrap Fusion Lumos Tribrid MS (Thermo Scientific) with 30 precursor isolation windows, and a 60 min LC-Structures for Lossless Ion Manipulations (SLIM) IM-MS proteomics run (MassHunter ‘.d’ format) acquired from our mammalian (Mus musculus) quality control sample on an in-house SLIM-IM-TOF MS system. Raw files were converted with ProteoWizard (version v 3.0.22139.aa8be89 for mzXML, mzML and mz5, and version 3_0_20059_33c55d87 for mzMLb) using a Windows 10 64-bit computer (Intel Core i9-10920X CPU, 3.5 GHz). For each format, spectra were converted in profile mode with lossless compression (--zlib) and consecutive zero intensity peaks removed (--filter "zeroSamples removeExtra”).
Results and Discussion
MZA design and purpose
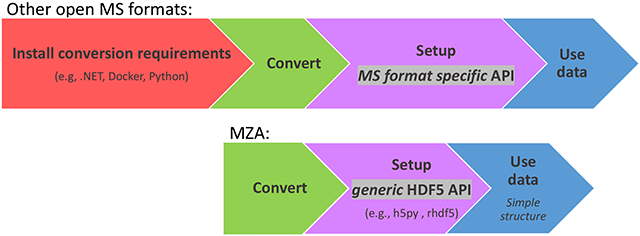
MZA has two components, a data conversion tool and a simple MS-data file structure, which provide open, easy, and efficient programmatic access to raw MS-data from multiple instrument vendors and mass analyzers (Fig. 1). Rather than replace the utilization of other formats in the existing tools, the focus of MZA is to facilitate access of multidimensional MS-data and enable fast development of prototypes and new tools in DS programming languages (e.g., Python and R). MZA is based on the HDF5 format, which was specifically designed to store and organize large amounts of data, while supporting efficient reading with fast random access.
Figure 1. MZA converter and file structure.

The MZA converter is self-contained (i.e., it is fully portable, it does not require installation of dependencies, not even a Python installation is required) and in Windows it can be immediately run to convert raw MS-data files from mzML or proprietary instrument formats supported. The loose coupling model enables development of software tools that are independent of MS format specific or instrument APIs, thus facilitating deployment. For AI tools, the stand-alone executable can be called from a script or notebook. The generated MZA file can be read using generic HDF5 libraries natively available in the language preferred by the developer. Each spectrum is stored as two HDF5 datasets (m/z and intensity 1D arrays) in two HDF5 groups. All metadata is easily accessible from a single table.
Benchmarking
To evaluate the performance of MZA in terms of file size and conversion time, we converted raw MS-data from three example files in vendor format: a 60 min LC-DIA-MS proteomics run with 68,850 spectra, a 30 min LC-DTIM-MS lipidomics run with 480,299 spectra and a 60 min LC-SLIM-MS proteomics run with 5,288,068 spectra. Files in the most commonly used and instrument-independent open formats available through ProteoWizard were generated: mz5, mzXML, mzML and mzMLb. As Fig. 2 illustrates, the IM dimension considerably increases the file size compared to a DIA analysis alone. When coupled with LC, an IM separation is performed at each LC time point, and thus, the number of spectra is significantly increased. In terms of file size, MZA files were larger than mz5 and mzMLb, and smaller than mzXML and mzML, most notably for the LC-SLIM-MS file, which is the most challenging one due to the larger number of spectra produced by the SLIM separation. The mz5 format provided the smallest output for all files due to its compression approach. However, mz5 has a custom compression method and it cannot be easily accessed with generic HDF5 libraries. Most formats offer several options for compression, in both lossless and lossy forms. Lossy compression methods typically provide smaller file size at the expense of reducing the data fidelity or precision of the floating-point numerical values, which normally add very small parts-per-million error that does not alter results in downstream data analyses13. Yet, the optimal compression method may be different depending on the instrument type and analysis setup, and thus, finding the best compression method may become a burden to the user in practice. For a more equitable comparison, we have used a lossless compression method for the other formats. However, it is worth noting that MZA does not apply any compression method to the spectra. The only compression applied is to the metadata table, which is transparent to the user as it is automatically decompressed by the generic HDF5 libraries when it is read. Our strategy to reduce the file size is to ignore the zero intensity peaks (i.e., by default only peaks with intensity higher than zero are included), which was also applied in the conversion to the other formats benchmarked. Like most formats, filtering the spectra in MZA can be applied using a minimum intensity threshold characteristic to the instrument and analysis setup (i.e., optional parameter -intensityThreshold). In terms of conversion speed, MZA was consistently faster than mzMLb, but resulted in longer times compared to the optimized parallel processing available in ProteoWizard for mzXML, mzML and mz5. However, the conversion is performed only once by a computer, while the simplicity of the usage of MZA will translate into much fewer human hours needed for software development (e.g., researchers can easily execute the converter to generate the MZA file and quickly start using the data, without worrying about installation or complicated formats and APIs).
Figure 2. MZA comparison to other MS formats.

Three representative experiments were compared: a 60 min LC-DIA-MS proteomics run, a 30 min LC-DTIM-DIA-MS lipidomics run, and a 60 min LC-SLIM-MS proteomics run. Top row: MZA vs. text formats. Bottom row: MZA vs. binary formats. Left panels: file size. Right panels: conversion time. MZA produced smaller files than mzXML and mzML, and required less execution time than mzMLb, most notably for the LC-SLIM-IM-MS run which is the most challenging to process due to the larger number of spectra collected with high IM resolving power.
To illustrate the usage of MZA in Python, we extracted the signals of an example peptide, VLEEANQAINPK (M+2H)2+, from the LC-SLIM-MS data (Fig. 3). The generated figures displayed the same information as the vendor software, exemplifying the multidimensional data fidelity in MZA compared to the instrument format (i.e., MZA can be used to develop software that properly utilizes MS data containing multiple front-end separations: LC and IM). The extracted ion chromatograms (XICs) of the 4 isotopic peaks across the complete file took 2.14 min to generate, compared to 13.24 s using the vendor software. However, the ion mobility heatmap view from the subset of spectra for RT 52-52.5 min was generated in 3 s compared to 8.89 s with the vendor software. These times suggest that using MZA in Python could be slower than the vendor software for generating full XICs (likely due to the ~3x increase in file size of MZA compared to the instrument format), but more efficient for accessing subsets of spectra.
Figure 3. Example of raw MS-data visualization from an MZA file containing liquid chromatography and ion mobility spectrometry separations.

Peptide VLEEANQAINPK (M+2H)2+ m/z 663.3567 in the LC-SLIM-MS run. Top panel: XICs of 4 isotopic peaks. Bottom panel: heatmap of arrival time vs. m/z for RT 52-52.5 min. Figure generated from a Python script using the packages h5py, hdf5plugin, numpy and matplotlib.
MZA advantages
Compared to other MS-data conversion and access technologies, MZA provides several advantages for both developers and end users. First, data in MZA files is read using generic HDF5 libraries. Once converted, the MS-data in the MZA file is explicit and can be easily and efficiently accessed from any programming language and operating system using common HDF5 libraries available (e.g., h5py and rhdf5). Different programming languages could be used depending on the project needs or developer skills and developers do not need to deal with complex dependencies of MS APIs or have specialized instrument knowledge to support them. Moreover, rather than having a dedicated API supported by the smaller computational MS community, the growing DS and AI communities across multiple scientific fields and industries support the generic HDF5 libraries (a large ecosystem with 700+ GitHub projects). Importantly, due to the HDF5 design, parallel file processing can support massively parallel high performance computing environments.
Second, the MZA data and metadata structure is simple and instrument agnostic. Like mzML, jagged arrays are used to directly store m/z and intensities for each spectrum separately and independently of instrument types. A single metadata table facilitates access to multidimensional information combining various analytical methods and hybrid instrumentation. Since most of the storage is occupied by the spectra instead of the metadata, this design is feasible and favors simplicity and usability rather than minimizing redundancy. All metadata can be read into memory at once (i.e., as a data frame in Python or R) in a very fast operation which requires minimal memory and avoids the need of iterating the complete file to access the metadata. Any spectrum can be read into memory as needed with random access and in a very fast operation (independently of the position or the total number of scans in the file). This avoids the need for loading all the spectra into memory and allows implementation of tools with low memory consumption. The needed subset of spectra can be identified from the metadata table (e.g., MS1, MS2, or by activation type in lipidomics or glycoproteomics analyses employing more than one ion fragmentation technique, including collision-induced dissociation, higher-energy dissociation, electron transfer dissociation or ultraviolet photodissociation) to only read that subset of spectra. In addition, the metadata table can be easily updated or extended to facilitate evaluations for instrument development.
Third, the self-contained converter minimizes installation of dependencies and can be effortlessly deployed, scaled, and used (i.e., installation of Python and MS instrument software libraries are not required). It can be used in different computer systems, from laptops to desktops and high-performance computers. Using a loose-coupling model, processing software, scripts or notebooks can simply call the MZA executable to convert the MS-data.
Conclusions
The large volume of multidimensional spectra collected by advanced and hybrid MS instrumentation is challenging current data storage and access technologies. We have described MZA as an alternative to address those challenges by simplifying programmatic access to raw MS-data from multiple instrument vendors and mass analyzers. While improvements in speed and file size are still needed, MZA provides a practical interface between researchers and MS instrument vendors. With the loose-coupling model in MZA researchers can readily utilize the raw MS-data while saving time and effort. At the same time, MS instrument vendors can protect the specifications of their optimized proprietary formats while avoiding maintenance of data-access APIs.
Since the self-contained MZA converter renders complicated API installation and documents unnecessary, we expect that by using MZA, software prototypes and tools can be developed in a faster way, and easy deployment can be provided to end users.
Direct and easy access to the raw MS-data is critical to perform tasks like feature engineering and data transformations needed for developing machine learning algorithms. Using our MZA tool, we are currently developing AI-based algorithms for different omics studies.
Ultimately, we anticipate that the advantages that MZA provides will accelerate the adoption of AI for processing raw MS-data, since developers can focus on the scientific problem and algorithm to solve it. Similar to many other research areas, advanced MS-tools based on AI may achieve human-level or super-human AI systems18 and have the potential to exploit the rich multidimensional MS-data to derive new molecular knowledge from complex biological and environmental samples, and to replace pipelines of traditional tools that heavily require intervention from human experts.
Acknowledgements
We thank Dr. John C. Fjeldsted and Dr. Sarah M. Stow for their assistance in accessing the MassHunter format. This work was supported by the Pacific Northwest National Laboratory (PNNL) Laboratory Directed Research and Development Program, and is a contribution of the m/q Initiative. Portions of this research were performed under the National Institute of General Medical Sciences grant P41 GM103493 and supported by the Environmental Molecular Sciences Laboratory (EMSL), a DOE Office of Science User Facility sponsored by the Biological and Environmental Research program under Contract No. DE-AC05-76RL01830.
Abbreviations
- AI
artificial intelligence
- AT
arrival time
- DIA
data independent acquisition
- DT
drift tube
- DS
data science
- IM
ion mobility spectrometry
- LC
liquid chromatography
- MS
mass spectrometry
- RT
retention time
- SLIM
structures for lossless ion manipulations
- XIC
extracted ion chromatogram
Footnotes
The authors declare no competing financial interest.
References
- (1).Zheng X; Wojcik R; Zhang X; Ibrahim YM; Burnum-Johnson KE; Orton DJ; Monroe ME; Moore RJ; Smith RD; Baker ES Coupling Front-End Separations, Ion Mobility Spectrometry, and Mass Spectrometry For Enhanced Multidimensional Biological and Environmental Analyses. Annu Rev Anal Chem (Palo Alto Calif) 2017, 10 (1), 71–92. DOI: 10.1146/annurev-anchem-061516-045212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bilbao A; Varesio E; Luban J; Strambio-De-Castillia C; Hopfgartner G; Muller M; Lisacek F Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics 2015, 15 (5-6), 964–980. DOI: 10.1002/pmic.201400323. [DOI] [PubMed] [Google Scholar]
- (3).Deutsch E mzML: a single, unifying data format for mass spectrometer output. Proteomics 2008, 8 (14), 2776–2777. DOI: 10.1002/pmic.200890049. [DOI] [PubMed] [Google Scholar]
- (4).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 2012, 30 (10), 918–920. DOI: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pedrioli PG; Eng JK; Hubley R; Vogelzang M; Deutsch EW; Raught B; Pratt B; Nilsson E; Angeletti RH; Apweiler R; et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol 2004, 22 (11), 1459–1466. DOI: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- (6).Wilhelm M; Kirchner M; Steen JAJ; Steen H mz5: space- and time-efficient storage of mass spectrometry data sets. Mol Cell Proteomics 2012, 11 (1), O111 011379. DOI: 10.1074/mcp.O111.011379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Schramm T; Hester Z; Klinkert I; Both JP; Heeren RMA; Brunelle A; Laprevote O; Desbenoit N; Robbe MF; Stoeckli M; et al. imzML--a common data format for the flexible exchange and processing of mass spectrometry imaging data. J Proteomics 2012, 75 (16), 5106–5110. DOI: 10.1016/j.jprot.2012.07.026. [DOI] [PubMed] [Google Scholar]
- (8).Rost HL; Schmitt U; Aebersold R; Malmstrom L Fast and Efficient XML Data Access for Next-Generation Mass Spectrometry. PLoS One 2015, 10 (4), e0125108. DOI: 10.1371/journal.pone.0125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kosters M; Leufken J; Schulze S; Sugimoto K; Klein J; Zahedi RP; Hippler M; Leidel SA; Fufezan C pymzML v2.0: introducing a highly compressed and seekable gzip format. Bioinformatics 2018, 34 (14), 2513–2514. DOI: 10.1093/bioinformatics/bty046. [DOI] [PubMed] [Google Scholar]
- (10).Bouyssie D; Dubois M; Nasso S; Gonzalez de Peredo A; Burlet-Schiltz O; Aebersold R; Monsarrat B mzDB: a file format using multiple indexing strategies for the efficient analysis of large LC-MS/MS and SWATH-MS data sets. Mol Cell Proteomics 2015, 14 (3), 771–781. DOI: 10.1074/mcp.O114.039115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Askenazi M; Ben Hamidane H; Graumann J The arc of Mass Spectrometry Exchange Formats is long, but it bends toward HDF5. Mass Spectrom Rev 2017, 36 (5), 668–673. DOI: 10.1002/mas.21522 From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tully B Toffee - a highly efficient, lossless file format for DIA-MS. Sci Rep 2020, 10 (1), 8939. DOI: 10.1038/s41598-020-65015-y From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bhamber RS; Jankevics A; Deutsch EW; Jones AR; Dowsey AW mzMLb: A Future-Proof Raw Mass Spectrometry Data Format Based on Standards-Compliant mzML and Optimized for Speed and Storage Requirements. J Proteome Res 2021, 20 (1), 172–183. DOI: 10.1021/acs.jproteome.0c00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lu M; An S; Wang R; Wang J; Yu C Aird: a computation-oriented mass spectrometry data format enables a higher compression ratio and less decoding time. BMC Bioinformatics 2022, 23 (1), 35. DOI: 10.1186/s12859-021-04490-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang J; Lu M; Wang R; An S; Xie C; Yu C StackZDPD: a novel encoding scheme for mass spectrometry data optimized for speed and compression ratio. Sci Rep 2022, 12 (1), 5384. DOI: 10.1038/s41598-022-09432-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rehfeldt TG; Krawczyk K; Bogebjerg M; Schwammle V; Rottger R MS2AI: Automated repurposing of public peptide LC-MS data for machine learning applications. Bioinformatics 2021. DOI: 10.1093/bioinformatics/btab701 From NLM Publisher. [DOI] [PubMed] [Google Scholar]
- (17).Bilbao A; Gibbons BC; Stow SM; Kyle JE; Bloodsworth KJ; Payne SH; Smith RD; Ibrahim YM; Baker ES; Fjeldsted JC A Preprocessing Tool for Enhanced Ion Mobility-Mass Spectrometry-Based Omics Workflows. J Proteome Res 2022, 21 (3), 798–807. DOI: 10.1021/acs.jproteome.1c00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Matsuo Y; LeCun Y; Sahani M; Precup D; Silver D; Sugiyama M; Uchibe E; Morimoto J Deep learning, reinforcement learning, and world models. Neural Networks 2022. DOI: 10.1016/j.neunet.2022.03.037. [DOI] [PubMed] [Google Scholar]