

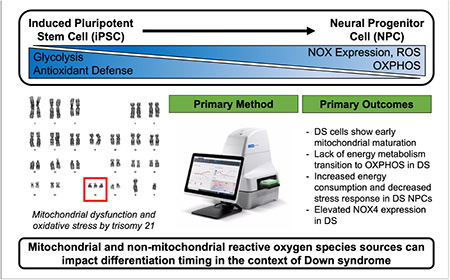
Abstract
Cellular redox state directs differentiation of induced pluripotent stem cells (iPSC) by energy metabolism control and ROS generation. As oxidative stress and mitochondrial dysfunction have been extensively reported in Down syndrome (DS), we evaluated mitochondrial phenotypes and energy metabolism during neural differentiation of DS iPSCs to neural progenitor cells (NPCs). Our results indicate early maturation of mitochondrial networks and elevated NADPH oxidase 4 (NOX4) expression in DS iPSCs. DS cells also fail to transition from glycolysis to oxidative phosphorylation during differentiation. Specifically, DS NPCs show an increased energetic demand that is limited in their mitochondrial and glycolytic response to mitochondrial distress. Additionally, DS iPSC and NPC non-mitochondrial oxygen consumption was significantly impacted by NOX inhibition. Together, these data build upon previous evidence of accelerated neural differentiation in DS that correlates with cellular redox state. We demonstrate the potential for mitochondrial and non-mitochondrial ROS sources to impact differentiation timing in the context of DS, which could contribute to developmental deficits in this condition.
Graphical Abstract
INTRODUCTION
Down syndrome (DS) or trisomy 21 (T21) is an aneuploidy, developmental disorder caused by either partial or complete triplication of chromosome 21 (Hsa21). Overexpression of Hsa21 genes, and their subsequent impact on the entire transcriptome results in intellectual impairment, characteristic facial features, and an increased risk of chronic health conditions including congenital heart defects and early onset Alzheimer’s disease. Mitochondrial dysfunction and elevated oxidative stress have been extensively reported in both in vitro and in vivo models of DS and are commonly described as etiological factors for many DS-related comorbidities [1–8]. Of relevance to this study, T21 is the most common genetic cause of intellectual disability [9]. Assessments of specific triplicated loci on Hsa21 have not yielded the mechanisms behind the ubiquity and interindividual variability of cognitive deficits in DS. Thus, we aim to better understand the impact of T21 gene dosage on mitochondrial phenotypes and related energy metabolism during neural development. Isogenic, induced pluripotent stem cell (iPSC) based modeling utilized here is an optimal approach for interrogating these neurodevelopmental processes. This system provides a reliable and ethical approach to study embryonic developmental processes and ensures that observed differences are specifically due to a third chromosome 21, and not interindividual genetic variation.
Our laboratory and others have shown aberrant mitochondrial structure, function, and energy metabolism, coupled with increased reactive oxygen species (ROS) production, and altered redox homeostasis in DS cells [1–3, 8, 10–13]. The mitochondrial network in DS neuronal cell types (iPSC-derived and in vivo) have been shown to be abnormal. Specifically, decreased mitochondrial inner membrane potential, dysregulated mitochondrial and metabolic gene expression signatures, impaired mitochondrial bioenergetics and biogenesis, and increased mitochondrial fragmentation have been observed [2, 8, 14–19]. Due to the critical role of cellular redox state and energy metabolism during cellular differentiation, these mitochondrial impairments have the potential to affect cell fate specification during neurogenesis. This process is critical for accurate neurodevelopment, and any effect by T21 could contribute to developmental delay and DS-specific neuronal phenotypes.
Based on their critical role in many important metabolic processes including, oxidative phosphorylation (OXPHOS) and lipid metabolism, functional mitochondria networks are essential for accurate cellular differentiation. The balance between stem cell self-renewal and differentiation is regulated by energy metabolism and cellular redox state. Manipulation of cellular metabolism and ROS production has been shown to directly affect stem cell fate decisions [20–22]. ROS production can determine whether a stem cell will self-renew or differentiate (FIG1B). In addition to mitochondrially-derived ROS, a family of enzymes called NADPH oxidases (NOX) are also able to produce ROS ‘on demand’ when these reactive molecules are required for signaling or immune functions [23–28]. Importantly, the ‘glycolysis-to-OXPHOS’ shift that occurs during differentiation precedes genetic changes (FIG1B). Thus, energy metabolism and ROS shifts during specific cellular states or differentiation stages act as signaling molecules, emphasizing the importance of understanding these systems in DS, a genetic disorder with impaired mitochondrial and redox signaling.
Figure 1. Neural differentiation is a redox dependent process.
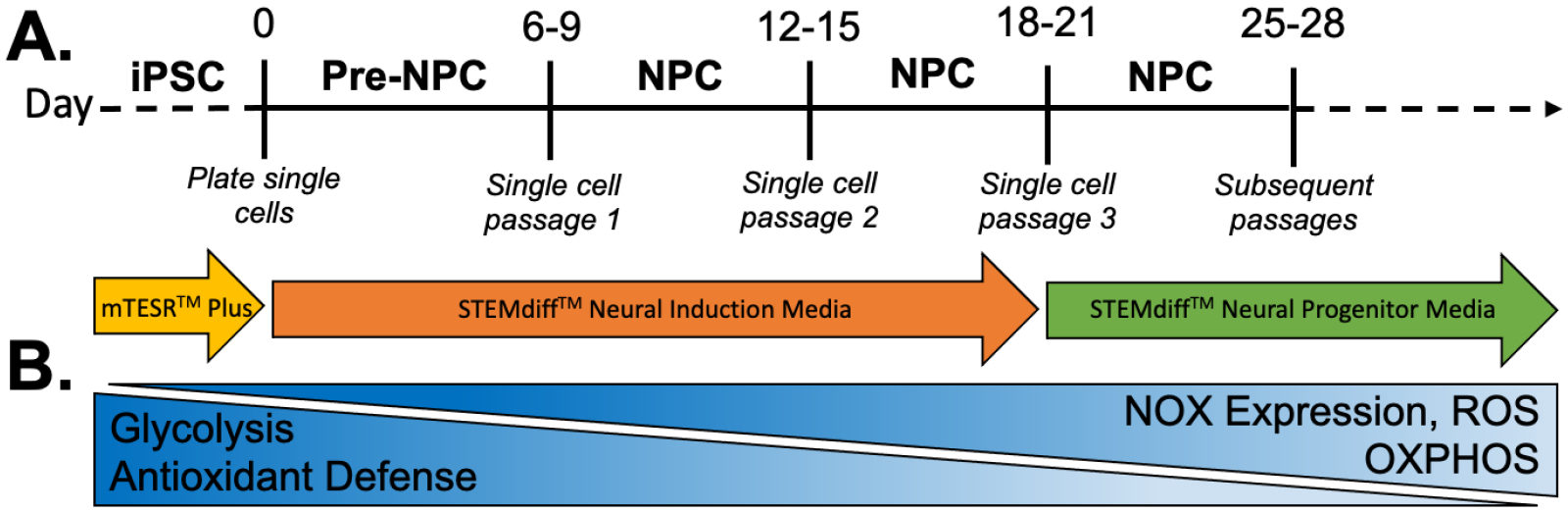
Neural differentiation of iPSCs to NPCs using a two-dimensional, monolayer protocol (A) is redox-regulated (B).
Here, we investigated mitochondrial and metabolic impairments by T21 during two-dimensional, monolayer, neural differentiation of iPSCs. We found that the mitochondrial network of DS iPSCs and NPCs mature earlier compared to isogenic controls. DS cells also showed a lack of metabolic plasticity during differentiation. Euploid and DS cells showed developmental, stage-specific alterations in NOX expression and inhibition response, as well as oxygen consumption rates and extracellular pH. We also show that DS NPCs have a higher level of energy utilization compared to their euploid counterparts. However, DS NPCs show a reduced ability to upregulate glycolysis and mitochondrial respiration despite this ‘energetic’ phenotype. Together, these data demonstrate mitochondrial and metabolic dysfunction in differentiating neural cells by T21. Altered thiol-redox signaling and mitochondrial dysfunction in DS will impact redox-regulated mechanisms of neurogenesis. As DS is characterized by developmental defects and delay, understanding the alterations in redox and mitochondrial driven pathways during neurodevelopment will help to better explain DS-associated neuronal phenotypes.
METHODS
Cell culture
An isogenic pair of human fibroblast-derived induced pluripotent stem cells (iPSCs) were generated by Dr. David Russel’s laboratory (University of Washington) and were provided by Dr. Christopher Link (University of Colorado Boulder) in communication with Gretchen Stein. Both iPSC lines originated from the AG06872 fibroblast cell line through reprogramming. iPSCs were cultured in mTeSR™ Plus (STEMCELL Technologies, 100–0276) on 6-well cell culture dishes coated with Matrigel® hESC-qualified Matrix (Corning, 354277). Quality control of iPSC clones was performed by karyotyping, real-time quantitative PCR (RT-qPCR), and embryoid body (EB) differentiation. Neural progenitor cells (NPCs) derived from iPSCs were cultured on 6-well Matrigel-coated cell culture dishes in STEMdiff™ Neural Progenitor Medium (NPM) (STEMCELL Technologies, 05833).
2-D, monolayer neural differentiation protocol
For differentiation of iPSCs into NPCs, iPSCs were dissociated with ReLeSR™ (STEMCELL Technologies, 100–0484) and resuspended in STEMdiff™ Neural Induction Medium (NIM) (STEMCELL Technologies, 05835), supplemented with 10 μM Y-27632 (STEMCELL Technologies, 72304). 2 × 105 cells/cm2 were plated on Matrigel-coated 6-well plates. Daily medium changes were completed with NIM. On Day 6–7, cells were passaged at 2 × 105 cells/cm2 onto Matrigel-coated 6-well plates. Daily medium changes were completed with NIM. On Day 13–14, cells were passaged again as previously described. After approximately seven days of culture, cells were then passaged as described using STEMdiff™ Neural Progenitor Medium (NPM) (STEMCELL Technologies, 05833). Daily medium changes were completed with NPM.
Gene expression analyses using qRT-PCR
Total RNA was isolated and extracted using TRIzol™ reagent (ThermoFisher Scientific, 15596026). RNA content was quantified using a NanoDrop200 (Thermo Scientific). RNA (1 μg) was reverse transcribed iScript gDNA Clear cDNA kit (Bio-Rad, 1725035) and T1000 Thermal Cycler (Bio-Rad). Gene expression was measured using 10 ng cDNA, SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, 1725271) and CFX Connect Real Time PCR System (Bio-Rad) as per manufacturer’s protocols. Primer sequences were obtained from the Harvard Primer Bank and purchased from Integrated DNA Technologies (IDT). Forward and reverse sequences of genes utilized in this study can be found in Table 1.
Table 1.
Forward and reverse primer sequences utilized in this study.
| Gene | Sequence (F) | Sequence (R) |
|---|---|---|
| PAX6 | TGGGCAGGTATTACGAGACTG | ACTCCCGCTTATACTGGGCTA |
| OCT4 | CTGGGTTGATCCTCGGACCT | CCATCGGAGTTGCTCTCCA |
| N-Cadherin | AGCCAACCTTAACT GAGGAGT | GGCAAGTTGATTGGAGGGATG |
| NOX4 | TGACGTTGCATGTTT CAGGAG | AGCTGGTTCGGTTAAGACTGAT |
| p22 phox | CCCAGTGGTACTTTGGTGCC | GCGGTCATGTACTTCTGTCCC |
Cell proliferation assay
Approximately 250,000 cells/well (iPSCs) or 150,000 cells/well (NPCs) were plated on a Matrigel-coated, 12-well plate. Cell images were captured at 24hr and 48hr using a 4X objective on the Cytation-1 Cell Imaging Multimode Reader (BioTek). Rate of cell proliferation was calculated as (48hr confluence-24hr confluence)/(24hr confluence).
Immunofluorescence staining of mitochondria
At the appropriate time points, cell cultures on coverslips were fixed using 3.7% formaldehyde in phosphate-buffered saline, 0.1% Tween (PBST), permeabilized using 0.1% Triton X-100, and blocked using PBST, 10% bovine serum albumin (BSA). Primary antibodies were incubated overnight at 4°C in PBST, 0.1% BSA. The primary antibodies used can be found in Table 2. Then, samples were incubated for 1h at room temperature protected from light with an Alexa Fluor 594® (Thermo Scientific) secondary antibody (1:2000) and Hoescht (1:5000) diluted in PBST. Coverslips were washed, dipped in diH2O, inverted, and mounted on a glass microscope slide containing VECTASHIELD Antifade Mounting Media (Vector Laboratories, H-1000–10) and Supermount (BioGenex, HK079). They were allowed to dry overnight at room temperature, protected from light. Cells were visualized on a Nikon Ti2-E microscope equipped with a Nikon A1-plus confocal system with lasers at 405 nm (blue) and 471 nm (red) (Nikon). Analyses of the confocal images were performed using NIS-Elements Viewer (Nikon) and ImageJ.
Table 2.
Antibodies utilized in this study.
| Protein | Supplier | Product # | Host | Clonality | Concentration |
|---|---|---|---|---|---|
| TOM20 | Cell Signaling Tech | 42406 | Rabbit | Mono | 1:100–200 |
Seahorse XFe96 analysis
Live cell analyses of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured with the Seahorse XFe96 system (Agilent). New cell characterization was performed according to the manufacturer’s standard protocol, yielding an optimal cell count and FCCP concentration of 0.5 μM (D 0,3,15) and 1.0 μM (NPC). The day prior to the assay, iPSCs (100,000 cells/well) or NPCs (50,000 cells/well) were plated on a Matrigel coated 96-well Seahorse XFe96 Cell Culture Microplate (Agilent, 101085–004, Agilent). Due to passaging requirements, pre-NPCs (D3) and NPCs (D15) were plated at 20,000 cells per well at Day 0 and 12 respectively and allowed to seed for three days with daily medium changes in a cell culture incubator at 37°C. An XFe96 cartridge was hydrated overnight in a non-CO2 incubator at 37°C prior to all analyses. The XFe96 cartridge was calibrated 30 minutes prior to the assay per manufacturer’s protocol. On the day of the analysis, assay media was prepared similar to culture media. Day 0 assay media contained 1.71 mM glucose, 0.318 mM sodium pyruvate, and 2.92 mM L-glutamine. Day 3 and 15 assay media contained 1.27 mM glucose, 0.24 mM sodium pyruvate, and 2.66 mM L-glutamine. NPC assay media contained 0.89 mM glucose, 0.16 mM sodium pyruvate, and 1.93 mM L-glutamine. The XFe96 cell culture microplate was washed twice with assay media and a final volume of 180 μl assay media was added to cells. Then, the XFe96 cell culture microplate was placed in Cytation-1 Cell Imaging Multimode Reader (BioTek) for cell count prior to assay initiation for normalization. Manufacturer-provided equations were used to calculate metabolic endpoints and can be found in Table 3.
Table 3.
Manufacturer-provided equations to calculate metabolic endpoints, i.e. basal respiration, glycolysis, from each assay trace.
| Assay | Metabolic Endpoint | Equation |
|---|---|---|
| Metabolic Phenotype | Metabolic potential | |
| Cell Mito Stress | Non- mitochondrial oxygen consumption | Minimum rate measurement after rotenone and antimycin injection |
| Basal respiration | (Last rate measurement before first injection) – (Non-mitochondrial respiration) | |
| Maximal respiration | (Maximum rate measurement after oligomycin injection) – (Non-mitochondrial respiration) | |
| ATP production | (Last rate measurement before oligomycin injection) – (Minimum rate measurement after oligomycin injection) | |
| Proton leak | (Minimum rate measurement after oligomycin injection) – (Non-mitochondrial respiration) | |
| Coupling efficiency | ||
| Glycolysis Stress | Glycolysis | (Maximum rate measurement before oligomycin injection) – (Last rate measurement before glucose) |
| Glycolytic capacity | (Maximum rate measurement after oligomycin injection) – (Last rate measurement before glucose injection) | |
| Glycolytic reserve | (Glycolytic Capacity) – (Glycolysis) | |
| Non-Glycolytic acidification | Last rate measurement prior to glucose injection | |
Cell energy phenotype test
Manufacturer’s protocol was followed for the Cell Energy Phenotype Test Kit (Agilent, 103325–100) with port A containing the FCCP/oligomycin stressor mix at 0.5–1.0/2.0 μM (final well concentration).
Cell mito stress test
Manufacturer’s protocol was followed for the Cell Mito Stress Test Kit (Agilent, 103010–100) with port A containing oligomycin (ATP-Synthase inhibitor) at 2.0 μM, port B with 0.5–1.0 μM/ FCCP (mitochondrial membrane depolarizer), and port C with a mixture of 0.5 μM of each rotenone (complex I inhibitor) and antimycin A (complex III inhibitor) (final well concentration).
Glycolysis stress test
Manufacturer’s protocol was followed for the Glycolysis Stress Test Kit with port A containing 100 mM glucose, port B with 2.0 μM oligomycin, and port C with 500 mM 2-deoxyglucose (2-DG) (competitive hexokinase inhibitor) (final well concentration). NOTE: Assay media for this test does not include glucose or sodium pyruvate.
Apocynin
Approximately 30,000 iPSCs were seeded per well on a Matrigel-coated 96-well plate and images were continuously captured using a 10X objective on the Incucyte® S3 Live Cell Analysis System (Sartorius). The following day, apocynin (1–2000 μM; Sigma Aldrich, 178385) was added to each well. Images were analyzed before and after the 24hr apocynin treatment using the vendor’s software to determine optimal apocynin concentration. For the Cell Mito Stress Test, iPSCs (50,000 cells/well) or NPCs (25,000 cells/well) were plated on a Matrigel coated 96-well Seahorse XFe96 Cell Culture Microplate (Agilent, 101085–004) two days prior to the assay. 24 hours before the assay, cells were treated with apocynin (1 or 10 μM). The Cell Mito Stress Test was completed as described above.
Mitochondrial Membrane Potential
Mitochondrial membrane potential was determined using the Mitochondrial Membrane Potential Assay Kit (Cayman Chemical, 10009172) and Cytation-1 Cell Imaging Multimode Reader (BioTek) as per manufacturer’s protocol. Cell count (50,000 iPSCs/well, 25,000 NPCs/well) and TMRE concentration (100 nM) was optimized as per manufacturer’s protocol. Background fluorescence was accounted for in all measurements.
Data analysis
Graphs were generated using GraphPad Prism 9 software and statistical significance was determined via unpaired t-test or 2-way ANOVA with Sidak post-hoc testing. Data were considered statistically significant if p ≤ 0.05. All experiments were done in at least triplicate.
RESULTS
Abnormal mitochondrial networks in DS cells during 2-D neural differentiation
There are currently two different neural induction methods in common usage to differentiate iPSCs to NPCs; three-dimensional (sphere) and two-dimensional (adherent, monolayer). For these studies, we wanted to create a reproducible and robust environment for neural differentiation and eliminate possible artifacts due to physical disruption of three-dimensional spheroids. Therefore, we first optimized a two-dimensional, monolayer neural differentiation protocol shown in FIG1A. The time points utilized in this study were selected based on this protocol – Day 3 (pre-NPC), Day 15 (NPC in neural induction media), and Day 25 (NPC in neural progenitor media) (FIG2A). These three time points allow for characterization of each cell/media condition during differentiation. Implementation of this protocol successfully generated NPCs, as demonstrated by loss of pluripotency gene, OCT4 (FIG2B), and induced expression of neural genes, PAX6 (FIG2C) and CDH2 (N-cadherin) (FIG2D).
Figure 2. Successful euploid and DS NPC differentiation by 2-D differentiation protocol.
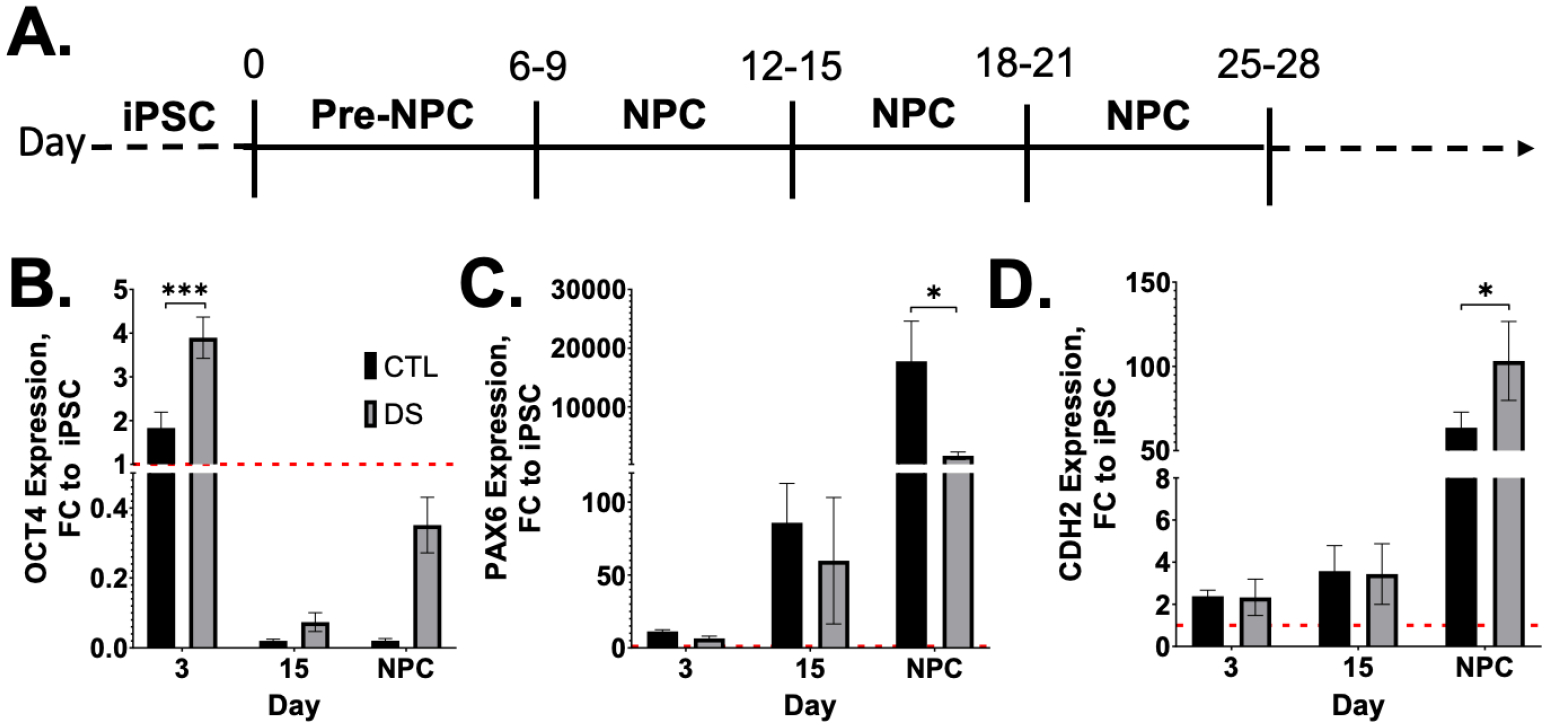
Neural differentiation of iPSCs to NPCs using a two-dimensional, monolayer protocol (A) successfully generated cells with NPC gene signature (B-D). Results are expressed as mean +/− SEM. N = 3–5. * = p ≤ 0.05, *** = p ≤ 0.001.
To investigate mitochondrial morphology in stem cells and multipotent NPCs, we began by staining the outer mitochondrial membrane (TOM20) by immunocytochemistry (ICC) (FIG3A,B). DS iPSCs showed no significant differences in overall TOM20 abundance/fluorescence compared to euploid (FIG3C); however, euploid iPSC mitochondria appeared to be more perinuclear and less distributed throughout the cytoplasm compared to DS (FIG3A). Both DS and euploid NPCs show more dispersed mitochondrial networks compared to their respective iPSCs (FIG3A,B). Further, DS NPCs have a significant increase in TOM20 abundance from DS iPSCs, and a near significant increase in TOM20 abundance from euploid NPCs (FIG3C). Next, we assessed mitochondrial membrane potential in our iPSCs and NPCs by TMRE staining. We found that DS iPSCs and NPCs were hyperpolarized compared to euploid (FIG3D), further defining divergent mitochondrial networks in DS.
Figure 3. Early maturation of mitochondrial networks in DS cells.
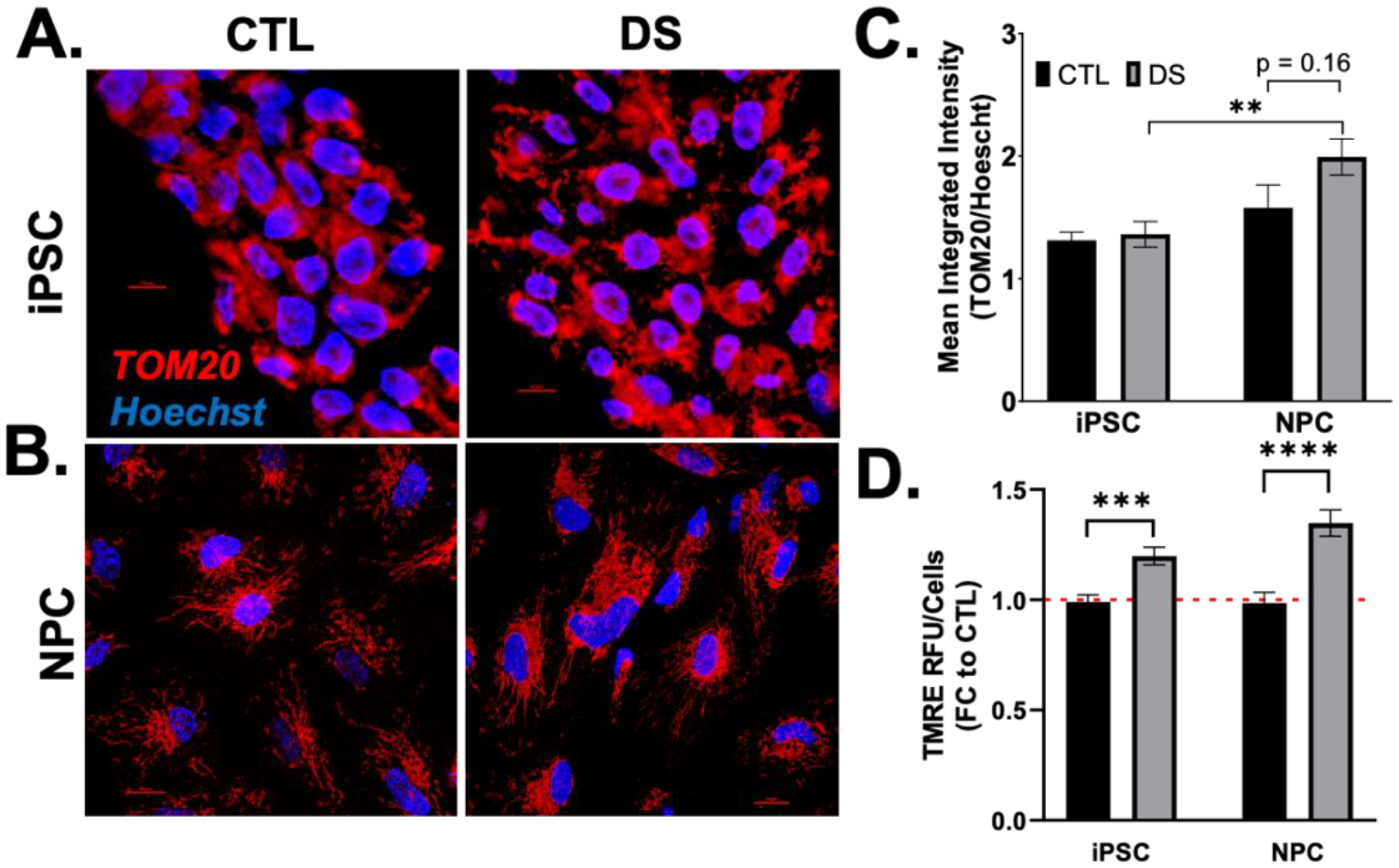
Representative images of mitochondrial shape in DS and euploid iPSCs (A) and NPCs (B) by immunofluorescence detection of TOM20 at 60X magnification show no differences in TOM20 abundance (C). TMRE staining showed increased mitochondrial membrane potential in DS iPSCs and NPCs (D). Nuclei are stained with Hoescht. Bar = 10 μM. Results are expressed as mean +/− SEM. N = 30–40. ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001.
Failed energy metabolism transition in DS differentiation
Despite reports of mitochondrial dysfunction in DS, few prior studies have assessed the impact of this gene dosage on metabolism during neural differentiation of iPSCs. Thus, we utilized the Seahorse XFe96 Analyzer and coordinated tests to measure mitochondrial function and cellular glycolysis in euploid and DS iPSC-derived cells during in vitro neural differentiation. To begin our investigation, we used the Seahorse XF Cell Energy Phenotype Test. This assay simultaneously measures mitochondrial respiration by oxygen consumption rate (OCR) and glycolysis by extracellular acidification rate (ECAR) under baseline and stressed conditions (FIG4A). An increase in OCR/ECAR ratio during differentiation confirmed the transition from glycolysis to OXPHOS in euploid cells; however, this phenomenon was not observed during DS differentiation (FIG4B) [22, 29]. After baseline measurements, the Cell Energy Phenotype Test then simultaneously adds oligomycin (OLIGO) and FCCP to stress the cells by inhibiting ATP production by the mitochondria and depolarizing the mitochondrial membrane.
Figure 4. DS NPCs are more energetic than euploid with decreased OCR/ECAR ratio.
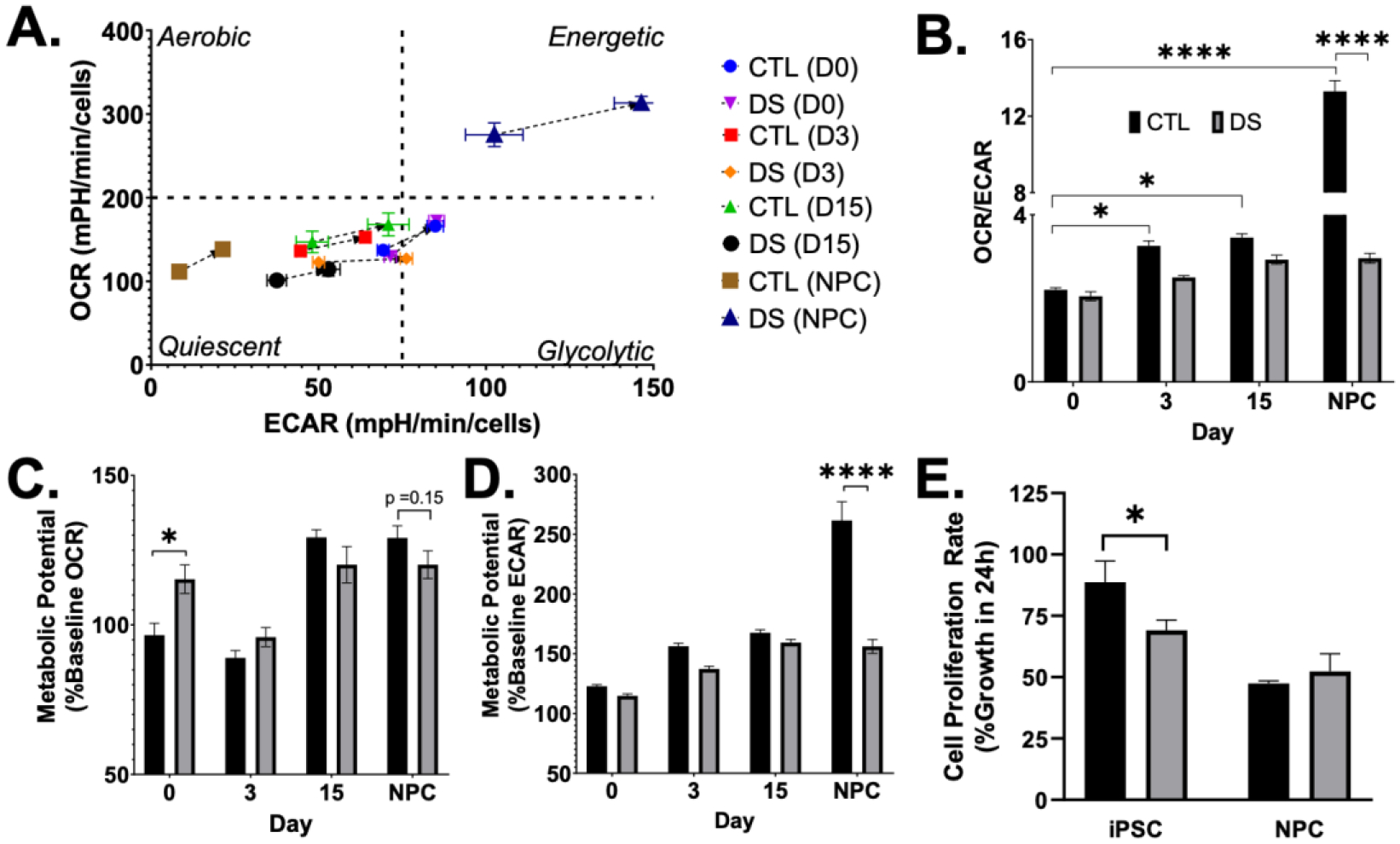
Using the Seahorse XF Cell Energy Phenotype Test (A), DS cells fail to transition from glycolysis to OXPHOS (B). DS iPSCs show an increased ability to respond to stress by respiration (C), and DS NPCs show an energetic phenotype (A) with a reduced ability to meet energy demand via glycolysis under the influence of stressor compounds (D). DS iPSCs proliferate at a slower rate than euploid iPSCs (E). Results are expressed as mean +/− SEM. N = 4–30. * = p ≤ 0.05, **** = p ≤ 0.0001.
We observed significant differences in metabolic potential throughout differentiation between euploid and DS cells (FIG4C,D). Specifically, DS iPSCs showed a significantly increased ability to meet energy demands by respiration, but not glycolysis (FIG4C,D). Additionally, DS NPCs had significantly less metabolic potential via glycolysis and near significant decrease in metabolic potential by respiration under the influence of stressor compounds (FIG4C,D), despite their energetic phenotype (FIG4A). A two-way ANOVA revealed a statistically significant interaction between the effects of cell type (CTL/DS) and day of differentiation on metabolic potential from mitochondrial respiration and glycolysis, indicating that the effect of cell type differed based on the day/stage of differentiation (Table 4). Based on the Cell Energy Phenotype Test results and the use of the OCR/ECAR ratio to detect cellular metabolic and proliferative changes such as the Warburg effect in cancer cells, we completed a cell proliferation assay. DS iPSCs proliferated at a decreased rate when compared to euploid, and NPCs proliferated slower than iPSCs in both cell lines (FIG4E). The observed differences in DS, specifically the pronounced energetic phenotype of NPCs and lack of energy metabolism transition, prompted us to further evaluate mitochondrial function in these cells.
Table 4.
P-values obtained by two-way ANOVA show significant interaction between the effects of cell type and stage of differentiation on mitochondrial respiration and glycolysis potential.
| Source of Variation | % of total variation | P value | P value summary | Sig? | |
|---|---|---|---|---|---|
| Metabolic Potential (%Baseline OCR) | Interaction | 4.988 | 0.0027 | ** | Yes |
| Day of Differentiation | 33.01 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 0.1331 | 0.5325 | ns | No | |
| Metabolic Potential (%Baseline ECAR) | Interaction | 13.33 | <0.0001 | **** | Yes |
| Day of Differentiation | 32.10 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 7.838 | <0.0001 | **** | Yes | |
DS NPCs have increased oxygen consumption
To directly measure mitochondrial function, we employed the Seahorse XF Cell Mito Stress Test. This test measures OCR of cells at baseline and following three serial injections of OLIGO, FCCP, and rotenone/antimycin A (ROT/AA) (FIG5A). DS iPSCs, pre-NPCs (D3), and NPCs (D15) did not show any significant differences compared to euploid in most of the metabolic outputs of mitochondrial respiration (FIG5A–G). However, DS iPSCs had near significant deficit in basal respiration when compared to euploid (FIG5C). Both euploid and DS cells showed similar early time course changes in basal and maximal respiration (FIG5C,D), ATP production (FIG5E), and proton leak (FIG5F). Interestingly, non-mitochondrial oxygen consumption diverged in DS, with DS pre-NPCs (D3) and NPCs showing significantly increased non-mitochondrial oxygen consumption when compared to euploid, and D15 NPCs showing less non-mitochondrial OCR (FIG5B). DS NPCs maintained in neural progenitor media were more energetic compared to euploid counterparts, with significantly more non-mitochondrial oxygen consumption (FIG5B), basal respiration (FIG5C), maximum respiration (FIG5D), ATP production (FIG5E), and proton leak (FIG5F). This also resulted in significantly less coupling efficiency in DS NPCs, which is associated with increased ROS production (FIG5G). All mitochondrial respiration measurements showed a statistically significant interaction effect between cell type (CTL/DS) and day of differentiation (Table 5). Thus, the effect of cell type is dependent on the day of differentiation.
Figure 5. Increased oxygen consumption and decreased coupling efficiency in DS NPCs.
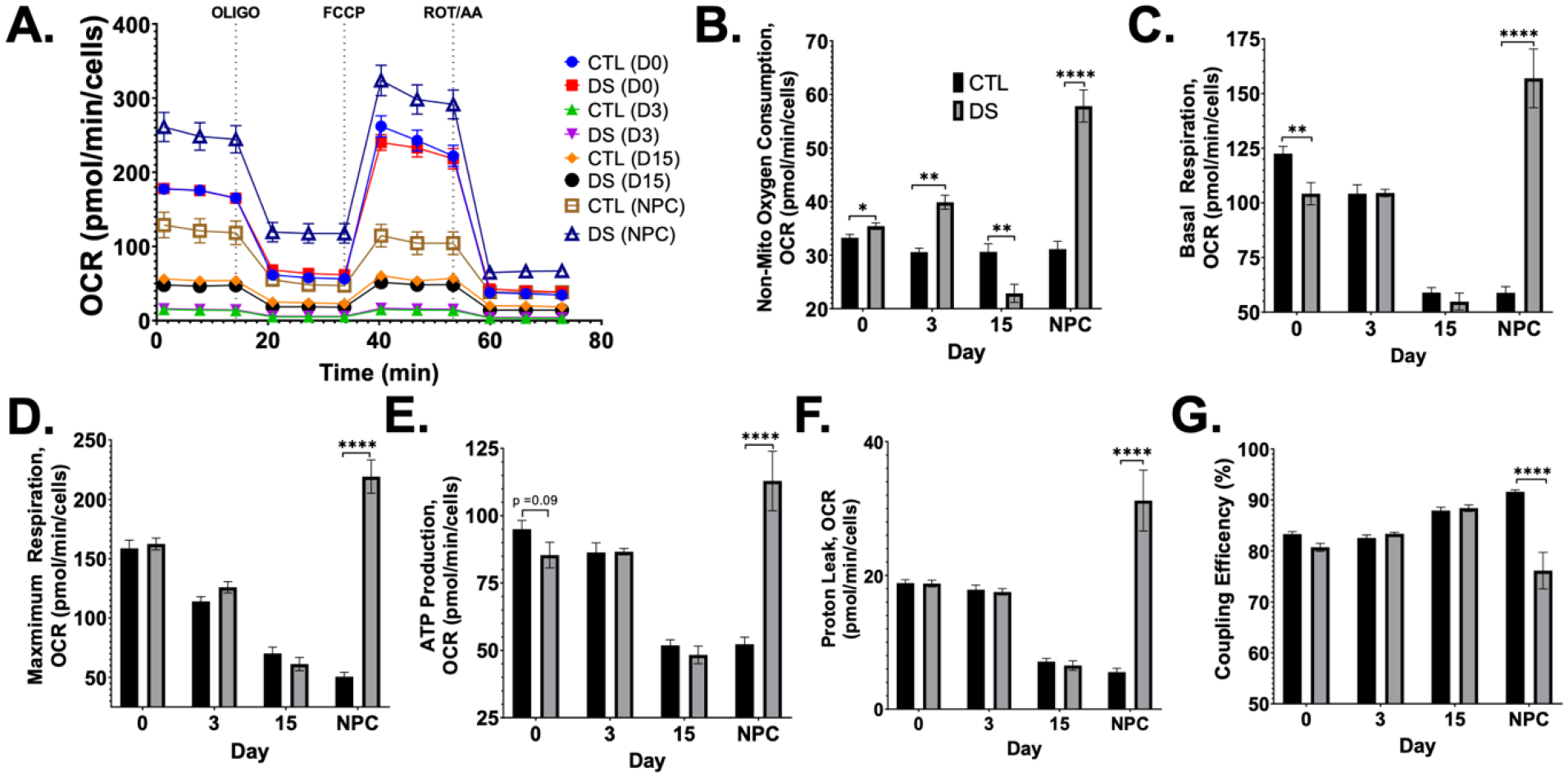
Mitochondrial function was measured using the Seahorse XF Cell Mito Stress Test (A) which showed significantly increased oxygen respiration (A-E) with more proton leakage (F) and reduced coupling efficiency (G) in DS NPCs. Results are expressed as mean +/− SEM. N = 15–32. *** = p ≤ 0.001, **** = p < 0.0001.
Table 5.
P-values obtained by two-way ANOVA show significant interaction effect from cell type and stage of differentiation on all metabolic endpoints of the Cell Mito Stress Test.
| Source of Variation | % of total variation | P value | P value summary | Sig? | |
|---|---|---|---|---|---|
| Non-Mito Oxygen Consumption | Interaction | 16.23 | <0.0001 | **** | Yes |
| Day of Differentiation | 15.84 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 4.623 | <0.0001 | **** | Yes | |
| Basal Respiration | Interaction | 18.88 | <0.0001 | **** | Yes |
| Day of Differentiation | 9.269 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 1.910 | 0.0063 | ** | Yes | |
| Maximum Respiration | Interaction | 19.48 | <0.0001 | **** | Yes |
| Day of Differentiation | 12.73 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 5.426 | <0.0001 | **** | Yes | |
| ATP Production | Interaction | 11.58 | <0.0001 | **** | Yes |
| Day of Differentiation | 7.506 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 1.139 | 0.0469 | * | Yes | |
| Proton Leak | Interaction | 11.19 | <0.0001 | **** | Yes |
| Day of Differentiation | 4.412 | 0.0034 | ** | Yes | |
| Cell Type (CTL/DS) | 2.617 | 0.0043 | ** | Yes | |
| Coupling Efficiency | Interaction | 8.684 | <0.0001 | **** | Yes |
| Day of Differentiation | 2.226 | 0.0656 | ns | No | |
| Cell Type (CTL/DS) | 2.485 | 0.0047 | ** | Yes | |
NOX inhibition impacts T21 non-mitochondrial oxygen consumption
The inconsistent pattern in DS non-mitochondrial oxygen consumption during differentiation was intriguing and led to further investigation. Non-mitochondrial oxygen consuming systems include NOX and cytochrome p450, and both have a role in cellular proliferation and differentiation. To better understand non-mitochondrial oxygen consumption in our system, we first measured NOX4 and p22phox expression in iPSCs and NPCs. DS iPSCs were observed to have increased NOX4 expression and no difference in p22phox when compared to euploid (FIG6A,B). DS NPCs showed no difference in NOX4 or p22phox expression from euploid cells (FIG6A,B). These data indicate that the NOX4 system is possibly responsible for enhanced non-mitochondrial OCR in DS.
Figure 6. iPSCs have more NOX4 expression and are impacted by NOX inhibition.
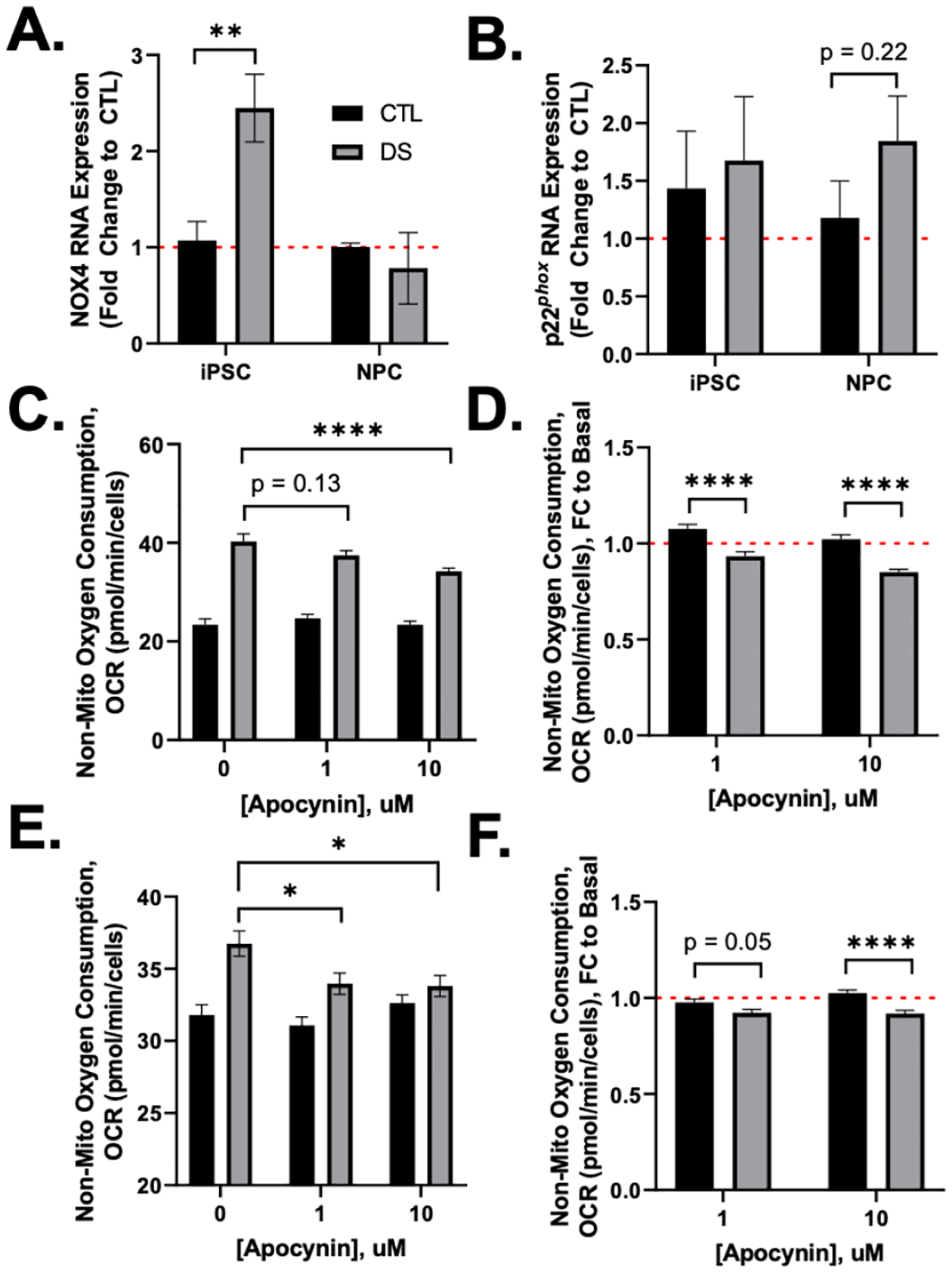
DS iPSCs have increased NOX4 expression with no difference in p22phox (A,B). Mitochondrial function was measured in iPSCs (C,D) and NPCs (E,F) after 24hr treatment with NOX inhibitor, apocynin (APO). DS iPSCs and NPCs showed significant decreases in non-mitochondrial oxygen consumption after APO treatment (D,F). Results are expressed as mean +/− SEM. N = 3–35. * = p ≤ 0.05, ** = p ≤ 0.01, **** = p ≤ 0.0001.
To further assess the roll of NOX, we inhibited NOX using a 24hr treatment with the commonly utilized NOX inhibitor, apocynin (APO). iPSCs and NPCs were treated with 1 and 10 μM APO (FIGS1) for 24hr and then subjected to the Cell Mito Stress Test. DS iPSCs and NPCs displayed significant decreases in non-mitochondrial oxygen consumption after 1 and 10 μM APO treatment, while euploid non-mitochondrial oxygen consumption was not impacted by APO (FIG6C,E). When OCR was visualized as fold change to basal (0 μM APO), we observed a significant decrease in the impact of 1 and 10 μM APO on non-mitochondrial oxygen consumption in DS iPSCs and NPCs when compared to euploid (FIG6D,F). All other metabolic endpoints from Cell Mito Stress Test in iPSCs besides coupling efficiency were not significantly affected by APO in both euploid and DS iPSCs (FIGS2). However, 10 μM APO treatment significantly decreased maximum respiration and proton leak in DS NPCs, and increased ATP production and coupling efficiency in DS (FIGS3). Together, these data demonstrate impaired NOX systems in DS which can lead to impaired redox homeostasis impacting energy metabolism and differentiation.
Lower extracellular pH in NPCs by T21
To complete our investigation of energy metabolism during neural differentiation, we assessed glycolytic function using the Seahorse XF Glycolysis Stress Test. Glycolysis provides a rapid source of ATP, intermediate metabolites for anabolic processes, and flux through this pathway decreases as stem cells undergo neural induction and differentiation (FIG1A). DS iPSCs had no significant differences in glycolytic outputs (FIG7A–E). At D3, DS pre-NPCs had increased glycolytic outputs that were not sustained at D15 (FIG7B–E). DS NPCs reciprocated D3 results, with significantly more glycolysis (FIG7B), non-glycolytic acidification (FIG7E), glycolytic capacity (FIG7C), and glycolytic reserve (FIG7D). Euploid cells showed a decrease in glycolysis throughout the differentiation, as expected from iPSC to NPCs; a pattern that was not shown in our DS cells (FIG7B). All glycolytic measurements showed a statistically significant interaction effect between cell type (CTL/DS) and day of differentiation, emphasizing the importance of timing and developmental stage specific alterations in understanding DS differences in glycolysis (Table 6).
Figure 7. Lower extracellular pH in NPCs by T21.
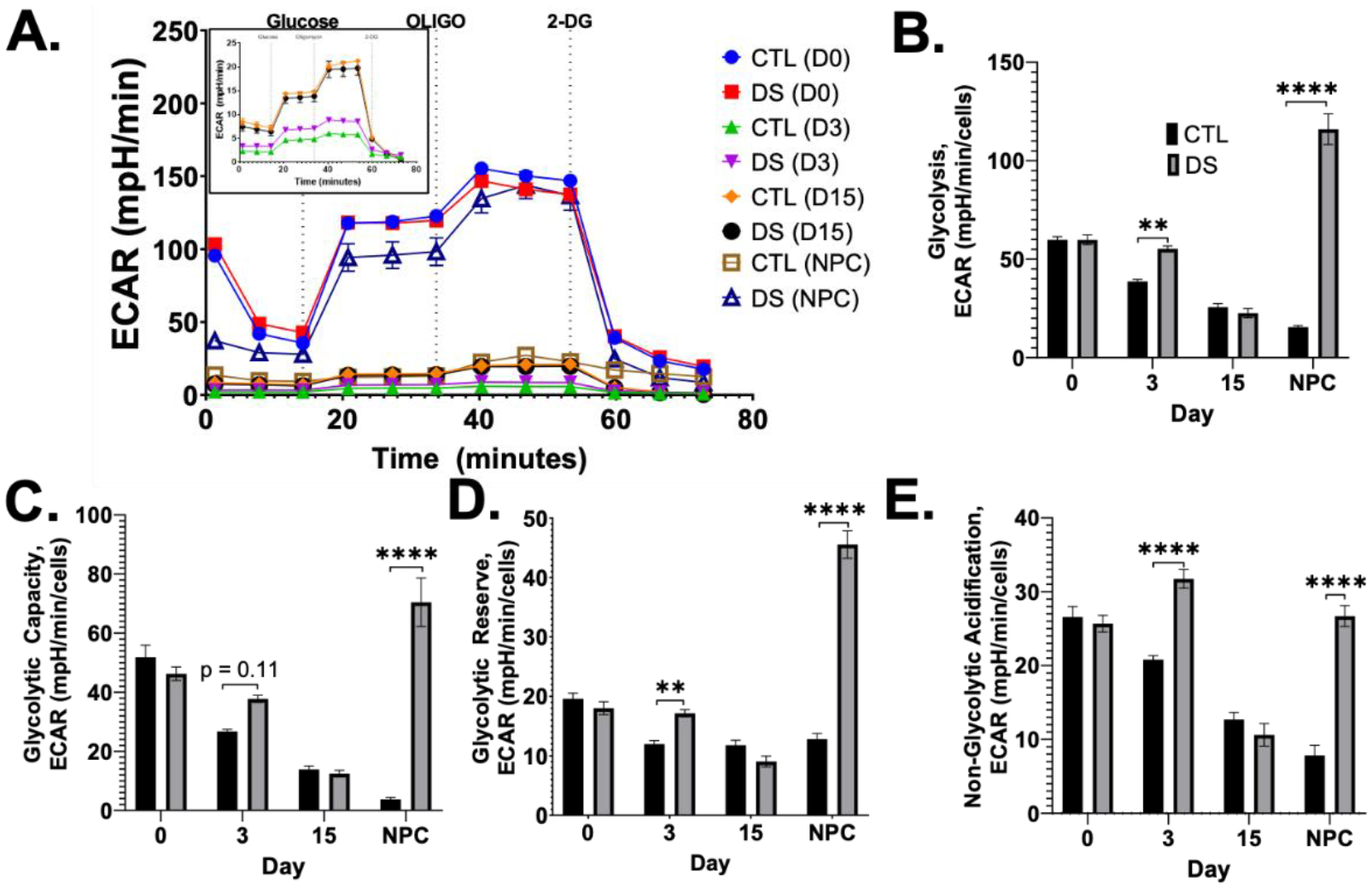
Glycolysis was measured using the Seahorse XF Glycolysis Stress Test (A) which showed significantly increased glycolysis in DS pre-NPCs at Day 3 and DS NPCs (A-E). Intermediate differentiation stages (Day 3, 15) showed reduced glycolysis compared to iPSCs and DS NPCS (A, inset). Results are expressed as mean +/− SEM. N = 15–30. ** = p ≤ 0.01, **** = p ≤ 0.0001.
Table 6.
P-values obtained by two-way ANOVA show significant interaction effect from cell type and stage of differentiation on all metabolic endpoints of the Glycolysis Stress Test.
| Source of Variation | % of total variation | P value | P value summary | Sig? | |
|---|---|---|---|---|---|
| Glycolysis | Interaction | 35.40 | <0.0001 | **** | Yes |
| Day of Differentiation | 15.14 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 13.80 | <0.0001 | **** | Yes | |
| Glycolytic Capacity | Interaction | 26.85 | <0.0001 | **** | Yes |
| Day of Differentiation | 13.91 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 8.380 | <0.0001 | **** | Yes | |
| Glycolytic Reserve | Interaction | 31.03 | <0.0001 | **** | Yes |
| Day of Differentiation | 28.25 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 8.996 | 0.0001 | **** | Yes | |
| Non-Glycolytic Acidification | Interaction | 17.06 | <0.0001 | **** | Yes |
| Day of Differentiation | 31.50 | <0.0001 | **** | Yes | |
| Cell Type (CTL/DS) | 9.877 | <0.0001 | **** | Yes | |
DISCUSSION
Many reports have attributed DS phenotypes, such as intellectual disability and early Alzheimer’s pathology, to the established mitochondrial defects and dysfunction present in various tissues and cell types in both human and animal models of DS [1–7, 30]. To better understand the mitochondrial role in DS development, specifically its impact on neurogenesis, we employed an isogenic, iPSC system to model DS neurogenesis. Research using iPSC-derived NPCs and neurons report that these cells resemble those from fetal brains, encouraging use of this system in lieu of less than optimal T21 animal models, and the paucity of embryonic and fetal brain tissue from people with DS [1, 31]. In addition, multiple large-scale gene expression analyses in DS, specifically DS neural cells, have shown that many of the genes involved in oxidative stress, energy metabolism (i.e. OXPHOS), and mitochondrial function are dysregulated [19]. Utilization of iPSC-based models to better understand DS development and pathology requires proper characterization of critical intermediate stages of neural induction in vitro. Here, we analyzed the transition from iPSC to NPC. A functional NPC population is critical for proper neural development because of their ability to differentiate into most neuronal cell types and glia. The research presented here allows for future research focused on DS iPSC-derived neuronal and glial cells by our laboratory and others.
The iPSC-derived neural cell types investigated showed gene expression and mitochondrial organizational patterns consistent with neural differentiation using an optimized two-dimensional, monolayer differentiation protocol. The characteristic, perinuclear, and immature mitochondrial networks of iPSCs was observed in our study in euploid and DS iPSCs, when compared to the more organized, filamentous networks in NPCs [22, 32–36]. However, DS iPSCs displayed more dispersed cytoplasmic distribution of mitochondria and increased mitochondrial membrane potential when compared to euploid. Importantly, cells with less perinuclear mitochondria have been shown to have a propensity for spontaneous differentiation, which has been observed in DS [11, 37]. By NPC differentiation, DS cells show an increase in TOM20 abundance and mitochondrial membrane potential. Mitochondrial membrane potential is associated with cellular differentiation, and several studies have demonstrated increased mitochondrial membrane potential with differentiation [38–40]. These data also agree with previously published reports showing accelerated differentiation in DS and extensive reports of mitochondrial dysfunction in DS, specifically in DS astrocytes and neurons [11, 18]. The mitochondrial maturation process during differentiation has been previously described, and coordinates with bioenergetic demands by energy metabolism transitioning from anaerobic glycolysis to OXPHOS, increasing ROS production [22, 32, 34, 35]. Here, we found a general lack of ‘energy metabolism switch’ in DS cells with no difference in NPC proliferation rate. This energy metabolism transition is intimately involved with redox homeostasis and is essential for differentiation into many different cellular lineages [41–43]. Dysfunction in this process by T21 gene dosage could have detrimental impacts on not only neural differentiation but development of other tissues and organs, which must be further explored in DS.
The Cell Energy Phenotype Test allows for further investigation into metabolic potential and transitioning as they differentiate from pluripotent stem cell to multipotent NPC. This is accomplished by stressing the cell through ATP synthase inhibition and mitochondrial membrane depolarization, causing compensatory glycolytic and OXPHOS changes to meet energy demands. The output of ‘metabolic potential’ reveals how each ATP-producing pathway responds to the stressor. Increased OXPHOS potential in DS iPSCs correlates with the more mature mitochondrial networks and reports of spontaneous differentiation in these cells [11]. Further, euploid cells during differentiation show a gradual transition from an energetic iPSC to a more quiescent NPC. In contrast, DS cells show a similar transition as euploid from Day 0–15, but this pattern substantially diverges when DS NPCs switch from neural induction media to neural progenitor media. Then, DS NPCs show a more energetic phenotype that exceeds euploid and DS iPSCs. Despite this energetic phenotype, DS NPCs have a decreased ability to respond to stressors by both metabolic pathways. We hypothesize that this is in part due to mitochondrial dysfunction resulting in elevated basal energy metabolism by both systems, as evidenced by Cell Mito and Glycolysis Tests, and increased mitochondrial superoxide production in DS NPCs [11]. This may result in an inability to augment energy production further when stressed, skewing metabolic potential outputs in NPCs. Thus, this energetic phenotype may be indicative of dysregulated differentiation and mitochondrial defects powering increased anaerobic and aerobic energy production.
The energetic phenotype of DS NPCs is persistent in the Cell Mito Stress Test, where DS NPCs show significant increases in oxygen consumption. Highly proliferative cells tend to rely on glycolysis, and energy-demanding, differentiated cells prefer to utilize OXPHOS. For example, metabolic analyses of neural stem cells and their differentiated neuronal lineages have shown that stem cells rely predominantly on glycolysis, and differentiated neurons prefer mitochondrial respiration [44–49]. Thus, this OCR increase may be due to more mature neurons and early differentiation occurring in our DS NPCs, as reported previously [11]. Interestingly, we observed a significant increase in proton leakage and a significant decrease in coupling efficiency in DS NPCs. Uncoupling can indicate lower mitochondrial respiratory rate or ROS production by proton leakage, which has been previously demonstrated in DS cells [10, 11]. Here, we observe elevated proton leakage with no changes in ATP production providing evidence for mitochondrial dysfunction and ROS production driving this process.
While mitochondrial generated ROS are a byproduct of respiration, NOXs directly produce ROS by transporting electrons to oxygen producing superoxide radical. Thus, NOX is critically involved in stem cell fate and function by non-mitochondrial oxygen consumption and ROS production [26, 28, 50–52]. A limitation of our study pertains to the use of APO as a chemical tool. While APO has been traditionally described as a NOX inhibitor, several studies have shown that it acts as an antioxidant [53–56]. Here we show APO acting on NOX, based on its significant impact on non-mitochondrial oxygen consumption without interfering on mitochondrial OCR. In our models, low dose APO significantly impacted non-mitochondrial oxygen consumption in DS iPSCs and NPCs, with no impact on euploid cells. These data are also strengthened by the expression of NOX4 and p22phox. Together, these data suggest that dysregulated DS non-mitochondrial oxygen consumption is related to NOX-dependent oxygen consumption, generating superoxide radicals at an increased rate when compared to euploid. NOX4 is unique in that it is constitutively active and is widely expressed in many cell types, in contrast to more specialized cellular localization and activation processes in other NOX enzyme systems, like NOX2 [28]. Thus, the elevated NOX4 expression in DS iPSCs can directly contribute to ROS generation and stimulate proliferation and differentiation. Importantly, depletion of the subunit p22phox has been shown to inhibit NOX4 activity [57]. Here, DS and euploid cells show no differences in p22phox expression leading to reliance on NOX4 expression to better understand NOX generated ROS. Based on the lack of NOX research in DS, this finding provokes further investigation into this system during neuronal differentiation, but also its role in more generalized metabolic regulation to better understand reported redox phenotypes.
The mitochondrial processes explored here are intimately involved in the tricarboxylic acid cycle (TCA), with constant feedback between these systems being critical for cell health and function. Glycolysis and TCA generate reducing equivalents (NAD(P)H) for cellular redox maintenance and ATP production. Increased delivery of reducing equivalents into the mitochondrial matrix can lead to increased electron flux and leakage from the respiratory chain resulting in ROS production. Previous published studies have demonstrated alterations in glycolytic and TCA metabolites in DS samples [58–65]. Here, we directly measured ECAR using the Glycolysis Stress Test. The process of breaking down glucose by glycolysis results in secretion of protons and lactate, acidifying the media. While glycolysis is the major ECAR driver, there are other sources that impact ECAR. Studies have demonstrated substantial ECAR contribution from carbon dioxide production from the TCA [66]. Additionally, other carboxylic acids, besides lactate can increase extracellular acidification, particularly by mitochondrial dysfunction [65]. These additional contributors are usually described as ‘non-glycolytic acidification rate’. However, this acidification may carry over to succeeding measurements. Non-glycolytic acidification rate progressively decreases during differentiation in euploid cells; a pattern not observed in DS differentiation. Thus, DS cells have additional ECAR contribution from the TCA and proton leakage by mitochondrial dysfunction as described above, impacting cellular differentiation and function.
Our laboratory has previously demonstrated accelerated neural differentiation in DS [11]. Here, we build upon our previous findings and show a deficient ‘glycolysis to OXPHOS’ transition in DS that leads to an increased energetic demand in differentiated NPCs. Neoplastic and stem cells preferentially utilize glycolysis-based metabolism, despite normal oxygen levels, allowing for protection against oxidative stress and NADPH generation [67–69]. As cellular metabolism must continually adapt and respond to constantly changing stimuli and ATP demands, cells are not typically operating at their maximum potential. However, these data suggest that DS NPCs are limited in their energetic response to mitochondrial stress, in part due to an elevated basal metabolic rate that corresponds with a more differentiated cellular phenotype. We present a potential mechanism for accelerated neural differentiation by T21 that builds upon altered redox homeostasis in DS and emphasizes mitochondrial and non-mitochondrial ROS sources. However, it is important to note that ROS is not the only way to alter thiol redox status, and other factors, such as reducing equivalents, can also alter this mechanism.
DS cognition has been extensively linked to the early stages of neurodevelopment, e.g. neurogenesis, synaptic connectivity, and myelination [70–72]. These processes drive the anatomy and physiology of the developed brain. Of interest, the developing brain is susceptible to oxidative stress. Enhanced oxidative stress and a lack of metabolic plasticity in DS will interfere with these crucial developmental stages. This could lead to anatomical and pathological phenotypes seen in DS, which are associated with developmental delay and intellectual disability. The results shown here emphasize the importance of timing during neural differentiation and underlines the sensitivity of this process to external and intrinsic redox stressors which must be further explored to better understand vulnerabilities that lead to DS phenotypes and comorbidities.
Supplementary Material
Highlights:
Isogenic iPSCs from euploid and trisomy 21 individuals were studied for alterations in energy metabolism during neural differentiation
Differentiation of trisomy 21 iPSCs resulted in early maturation of mitochondrial networks
Trisomy 21 resulted in elevated levels of NOX4
Trisomy 21 neural progenitor cells display increased metabolic demand in response to mitochondrial stress
ACKNOWLEDGEMENTS
This work was supported by funds provided by the National Institutes of Health (R01ES027593 to JRR; T32ES02974 to KMP) and the Linda Crnic Institute for Down Syndrome (KNM). K.N.M gratefully acknowledges financial support from the William R. Hummel Homocystinuria Research Fund and holds the Ehst-Hummel-Kaufmann Family Endowed Chair in Inherited Metabolic Disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Busciglio J and Yankner BA, Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature, 1995. 378(6559): p. 776–9. [DOI] [PubMed] [Google Scholar]
- 2.Helguera P, et al. , Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab, 2013. 17(1): p. 132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piccoli C, et al. , Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Hum Mol Genet, 2013. 22(6): p. 1218–32. [DOI] [PubMed] [Google Scholar]
- 4.Pastore A, et al. , Glutathione metabolism and antioxidant enzymes in children with Down syndrome. J Pediatr, 2003. 142(5): p. 583–5. [DOI] [PubMed] [Google Scholar]
- 5.Carratelli M, et al. , Reactive oxygen metabolites and prooxidant status in children with Down’s syndrome. Int J Clin Pharmacol Res, 2001. 21(2): p. 79–84. [PubMed] [Google Scholar]
- 6.Jovanovic SV, Clements D, and MacLeod K, Biomarkers of oxidative stress are significantly elevated in Down syndrome. Free Radic Biol Med, 1998. 25(9): p. 1044–8. [DOI] [PubMed] [Google Scholar]
- 7.Perrone S, et al. , Early oxidative stress in amniotic fluid of pregnancies with Down syndrome. Clin Biochem, 2007. 40(3–4): p. 177–80. [DOI] [PubMed] [Google Scholar]
- 8.Izzo A, et al. , Mitochondrial dysfunction in down syndrome: molecular mechanisms and therapeutic targets. Mol Med, 2018. 24(1): p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiseman FK, et al. , A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci, 2015. 16(9): p. 564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson CC, et al. , Trisomy 21 results in modest impacts on mitochondrial function and central carbon metabolism. Free Radic Biol Med, 2021. 172: p. 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prutton KM, et al. , Oxidative stress as a candidate mechanism for accelerated neuroectodermal differentiation due to trisomy 21. Free Radic Biol Med, 2022. 186: p. 32–42. [DOI] [PubMed] [Google Scholar]
- 12.Zamponi E and Helguera PR, The Shape of Mitochondrial Dysfunction in Down Syndrome. Dev Neurobiol, 2019. 79(7): p. 613–621. [DOI] [PubMed] [Google Scholar]
- 13.Aivazidis S, et al. , The burden of trisomy 21 disrupts the proteostasis network in Down syndrome. PLoS One, 2017. 12(4): p. e0176307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mollo N, et al. , Targeting Mitochondrial Network Architecture in Down Syndrome and Aging. Int J Mol Sci, 2020. 21(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim SH, et al. , The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer’s disease. Life Sci, 2001. 68(24): p. 274150. [DOI] [PubMed] [Google Scholar]
- 16.Coskun PE and Busciglio J, Oxidative Stress and Mitochondrial Dysfunction in Down’s Syndrome: Relevance to Aging and Dementia. Curr Gerontol Geriatr Res, 2012. 2012: p. 383170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arbuzova S, Hutchin T, and Cuckle H, Mitochondrial dysfunction and Down’s syndrome. Bioessays, 2002. 24(8): p. 681–4. [DOI] [PubMed] [Google Scholar]
- 18.Busciglio J, et al. , Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron, 2002. 33(5): p. 677–88. [DOI] [PubMed] [Google Scholar]
- 19.Weick JP, et al. , Deficits in human trisomy 21 iPSCs and neurons. Proc Natl Acad Sci U S A, 2013. 110(24): p. 9962–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takubo K, et al. , Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell, 2013. 12(1): p. 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu WM, et al. , Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell, 2013. 12(1): p. 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, et al. , UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. Embo j, 2016. 35(8): p. 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sart S, Song L, and Li Y, Controlling Redox Status for Stem Cell Survival, Expansion, and Differentiation. Oxid Med Cell Longev, 2015. 2015: p. 105135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Funato Y, et al. , The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol, 2006. 8(5): p. 501–8. [DOI] [PubMed] [Google Scholar]
- 25.Shin SY, et al. , Hydrogen peroxide negatively modulates Wnt signaling through downregulation of beta-catenin. Cancer Lett, 2004. 212(2): p. 225–31. [DOI] [PubMed] [Google Scholar]
- 26.Bigarella CL, Liang R, and Ghaffari S, Stem cells and the impact of ROS signaling. Development, 2014. 141(22): p. 4206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saretzki G, et al. , Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells, 2008. 26(2): p. 455–64. [DOI] [PubMed] [Google Scholar]
- 28.Maraldi T, et al. , NADPH Oxidases: Redox Regulators of Stem Cell Fate and Function. Antioxidants (Basel), 2021. 10(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, et al. , Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat Protoc, 2012. 7(6): p. 1068–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pallardó FV, et al. , Mitochondrial dysfunction in some oxidative stress-related genetic diseases: Ataxia-Telangiectasia, Down Syndrome, Fanconi Anaemia and Werner Syndrome. Biogerontology, 2010. 11(4): p. 401–19. [DOI] [PubMed] [Google Scholar]
- 31.Chambers SM, et al. , Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol, 2009. 27(3): p. 275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.St John JC, et al. , The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells, 2005. 7(3): p. 141–53. [DOI] [PubMed] [Google Scholar]
- 33.St John JC, et al. , The analysis of mitochondria and mitochondrial DNA in human embryonic stem cells. Methods Mol Biol, 2006. 331: p. 347–74. [DOI] [PubMed] [Google Scholar]
- 34.Suhr ST, et al. , Mitochondrial rejuvenation after induced pluripotency. PLoS One, 2010. 5(11): p. e14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Folmes CD, et al. , Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab, 2011. 14(2): p. 264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prigione A, et al. , The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells, 2010. 28(4): p. 721–33. [DOI] [PubMed] [Google Scholar]
- 37.Lonergan T, Brenner C, and Bavister B, Differentiation-related changes in mitochondrial properties as indicators of stem cell competence. J Cell Physiol, 2006. 208(1): p. 149–53. [DOI] [PubMed] [Google Scholar]
- 38.Vega-Naredo I, et al. , Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells. Cell Death Differ, 2014. 21(10): p. 1560–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voccoli V and Colombaioni L, Mitochondrial remodeling in differentiating neuroblasts. Brain Res, 2009. 1252: p. 15–29. [DOI] [PubMed] [Google Scholar]
- 40.Dai DF, et al. , Mitochondrial Maturation in Human Pluripotent Stem Cell Derived Cardiomyocytes. Stem Cells Int, 2017. 2017: p. 5153625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crespo FL, et al. , Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells, 2010. 28(7): p. 1132–42. [DOI] [PubMed] [Google Scholar]
- 42.Ji AR, et al. , Reactive oxygen species enhance differentiation of human embryonic stem cells into mesendodermal lineage. Exp Mol Med, 2010. 42(3): p. 175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin CY, et al. , Exacerbation of oxidative stress-induced cell death and differentiation in induced pluripotent stem cells lacking heme oxygenase-1. Stem Cells Dev, 2012. 21(10): p. 1675–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khacho M, et al. , Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell, 2016. 19(2): p. 232–247. [DOI] [PubMed] [Google Scholar]
- 45.Agostini M, et al. , Metabolic reprogramming during neuronal differentiation. Cell Death Differ, 2016. 23(9): p. 1502–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Brien LC, Keeney PM, and Bennett JP Jr., Differentiation of Human Neural Stem Cells into Motor Neurons Stimulates Mitochondrial Biogenesis and Decreases Glycolytic Flux. Stem Cells Dev, 2015. 24(17): p. 1984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng X, et al. , Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorenz C, et al. , Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell, 2017. 20(5): p. 659–674.e9. [DOI] [PubMed] [Google Scholar]
- 49.Homem CCF, et al. , Ecdysone and mediator change energy metabolism to terminate proliferation in Drosophila neural stem cells. Cell, 2014. 158(4): p. 874–888. [DOI] [PubMed] [Google Scholar]
- 50.Pervaiz S, Taneja R, and Ghaffari S, Oxidative stress regulation of stem and progenitor cells. Antioxid Redox Signal, 2009. 11(11): p. 2777–89. [DOI] [PubMed] [Google Scholar]
- 51.Liang R and Ghaffari S, Stem Cells Seen Through the FOXO Lens: An Evolving Paradigm. Curr Top Dev Biol, 2018. 127: p. 23–47. [DOI] [PubMed] [Google Scholar]
- 52.Kobayashi CI and Suda T, Regulation of reactive oxygen species in stem cells and cancer stem cells. J Cell Physiol, 2012. 227(2): p. 421–30. [DOI] [PubMed] [Google Scholar]
- 53.Augsburger F, et al. , Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol, 2019. 26: p. 101272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbieri SS, et al. , Apocynin prevents cyclooxygenase 2 expression in human monocytes through NADPH oxidase and glutathione redox-dependent mechanisms. Free Radic Biol Med, 2004. 37(2): p. 156–65. [DOI] [PubMed] [Google Scholar]
- 55.Heumüller S, et al. , Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension, 2008. 51(2): p. 211–7. [DOI] [PubMed] [Google Scholar]
- 56.Stefanska J and Pawliczak R, Apocynin: molecular aptitudes. Mediators Inflamm, 2008. 2008: p. 106507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawahara T, et al. , Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem, 2005. 280(36): p. 31859–69. [DOI] [PubMed] [Google Scholar]
- 58.Mapstone M, et al. , Metabolic correlates of prevalent mild cognitive impairment and Alzheimer’s disease in adults with Down syndrome. Alzheimers Dement (Amst), 2020. 12(1): p. e12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gross TJ, et al. , Plasma metabolites related to cellular energy metabolism are altered in adults with Down syndrome and Alzheimer’s disease. Dev Neurobiol, 2019. 79(7): p. 622–638. [DOI] [PubMed] [Google Scholar]
- 60.Sotelo-Orozco J, et al. , Association Between Plasma Metabolites and Psychometric Scores Among Children With Developmental Disabilities: Investigating Sex-Differences. Front Psychiatry, 2020. 11: p. 579538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Orozco JS, et al. , Metabolomics analysis of children with autism, idiopathic-developmental delays, and Down syndrome. Transl Psychiatry, 2019. 9(1): p. 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pecze L, Randi EB, and Szabo C, Meta-analysis of metabolites involved in bioenergetic pathways reveals a pseudohypoxic state in Down syndrome. Mol Med, 2020. 26(1): p. 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caracausi M, et al. , Plasma and urinary metabolomic profiles of Down syndrome correlate with alteration of mitochondrial metabolism. Sci Rep, 2018. 8(1): p. 2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Culp-Hill R, et al. , Red blood cell metabolism in Down syndrome: hints on metabolic derangements in aging. Blood Adv, 2017. 1(27): p. 2776–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Powers RK, et al. , Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat Commun, 2019. 10(1): p. 4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mookerjee SA, et al. , The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta, 2015. 1847(2): p. 171–181. [DOI] [PubMed] [Google Scholar]
- 67.Ma T, et al. , Progress in the reprogramming of somatic cells. Circ Res, 2013. 112(3): p. 562–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varum S, et al. , Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One, 2011. 6(6): p. e20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vander Heiden MG, Cantley LC, and Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 2009. 324(5930): p. 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Becker L, et al. , Growth and development of the brain in Down syndrome. Prog Clin Biol Res, 1991. 373: p. 133–52. [PubMed] [Google Scholar]
- 71.Becker LE, Armstrong DL, and Chan F, Dendritic atrophy in children with Down’s syndrome. Ann Neurol, 1986. 20(4): p. 520–6. [DOI] [PubMed] [Google Scholar]
- 72.Takashima S, et al. , Dendrites, dementia and the Down syndrome. Brain Dev, 1989. 11(2): p. 131–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.