

Abstract
Background & Aims
Elimination of chronic HBV/HDV infection remains a major global health challenge. Targeting excessive hepatitis B surface antigen (HBsAg) release may provide an interesting window of opportunity to break immune tolerance and to achieve a functional cure using additional antivirals.
Methods
We evaluated a HBsAg-specific human monoclonal antibody, as part of either a prophylactic or therapeutic strategy, against HBV/HDV infection in cell culture models and in human-liver chimeric mice. To assess prophylactic efficacy, mice were passively immunized prior to infection with HBV or HBV/HDV (coinfection and superinfection setting). Therapeutic efficacy was assessed in HBV and HBV/HDV-coinfected mice receiving 4 weeks of treatment. Viral parameters (HBV DNA, HDV RNA and HBsAg) were assessed in mouse plasma.
Results
The antibody could effectively prevent HBV/HDV infection in a dose-dependent manner with IC50 values of ∼3.5 ng/ml. Passive immunization showed complete protection of mice from both HBV and HBV/HDV coinfection. Moreover, HDV superinfection was either completely prevented or at least attenuated in HBV-infected mice. Finally, antibody treatment in mice with established HBV/HDV infection resulted in a significant decline in viremia and a concomitant drop in on-treatment HBsAg, with a moderate viral rebound following treatment cessation.
Conclusion
We present data on a valuable antibody candidate that could complement other antivirals in strategies aimed at achieving functional cure of chronic HBV and HDV infection.
Impact and implications
Patients chronically infected with HBV may eventually develop liver cancer and are at great risk of being superinfected with HDV, which worsens and accelerates disease progression. Unfortunately, current treatments can rarely eliminate both viruses from chronically infected patients. In this study, we present data on a novel antibody that is able to prevent chronic HBV/HDV infection in a mouse model with a humanized liver. Moreover, antibody treatment of HBV/HDV-infected mice strongly diminishes viral loads during therapy. This antibody is a valuable candidate for further clinical development.
Keywords: Viral hepatitis, hepatitis B, hepatitis D, human monoclonal antibody, hepatitis B surface antigen, prevention, neutralization, human-liver chimeric mouse model
Abbreviations: hu-mAb, human monoclonal antibody; HBsAg, hepatitis B surface antigen; IC50, half maximal inhibitory concentration; IP, intraperitoneally; LLOQ, lower limit of quantification; LOD, limit of detection; NTCP, sodium taurocholate co-transporting polypeptide
Graphical abstract
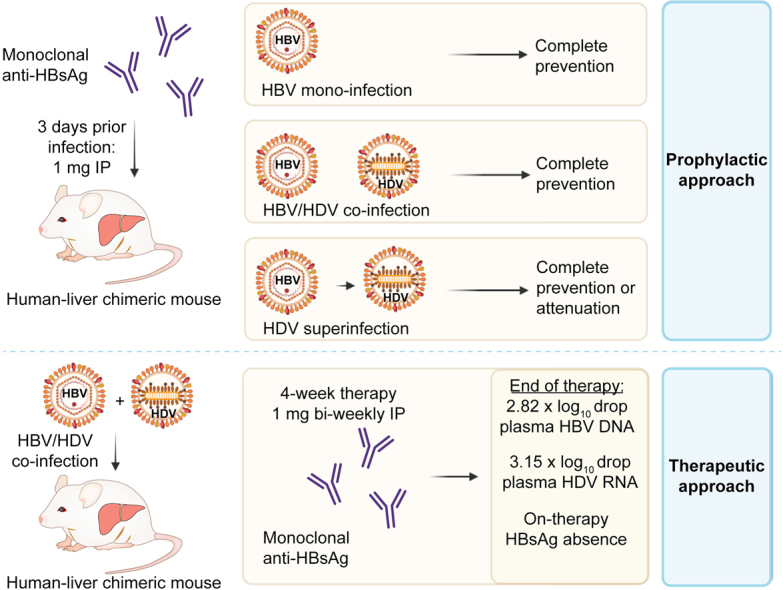
Highlights
-
•
A human mAb that targets HBsAg was developed.
-
•
This human mAb prevents HBV and HDV infection in permissive cell lines and in human-liver chimeric mice.
-
•
The human mAb prevents, or at least attenuates, HDV superinfection in vivo.
-
•
This mAb also showed therapeutic potential in vivo.
Introduction
Despite the availability of safe and efficacious HBV vaccines, approximately 300 million people worldwide are currently chronic carriers of hepatitis B surface antigen (HBsAg).1 Chronic HBV infection may eventually lead to cirrhosis and hepatocellular carcinoma (HCC) development. HBV is responsible for a heavy disease burden and an estimated 820,000 liver-related deaths annually.2,3 Moreover, at least 12 million and up to 60 million patients with chronic HBV are estimated to suffer from a concomitant HDV infection.[4], [5], [6], [7] Since HDV infection greatly exacerbates disease progression by rapidly inducing cirrhosis, liver dysfunction and HCC, it is considered the most severe form of viral hepatitis.5 Patients coinfected with HBV and HDV usually recover spontaneously through immune-mediated viral elimination. However, in 80% of cases, patients with chronic HBV who become superinfected with HDV progress to a chronic disease state, resulting in rapid deterioration of the pre-existing HBV-related liver damage and very high mortality rates.8
Current lifelong HBV therapy with nucleos(t)ide analogues (NAs) suppresses viral replication, but only about 10% of all treated HBeAg-positive patients (and 1% in HBeAg-negative patients) may achieve a functional cure, i.e. complete HBsAg loss with/without seroconversion after a 5-year follow-up.[9], [10], [11] Therefore, NAs will not eliminate the risk of cancer development. Regarding HDV therapy, this virus does not code for polymerases or proteases that could be therapeutically targeted as is the case for HBV or HCV, but relies on both the host replicative machinery and on the helper function of HBV to complete its life cycle.12 Therefore, the only recommended clinical regimen remains pegylated-IFNα, a therapy showing poor efficacy and infrequent long-term responses.13 Specifically, only 28% of treated patients achieved clearance of serum HDV 6 months after cessation of a 48-week regimen and 50% of responders experienced late HDV RNA relapse during longer follow-up.[14], [15], [16] While phase III results are still pending, in 2020 the European Medicines Agency conditionally approved the entry-inhibitor Myrcludex B (bulevirtide), a preS1-derived peptide able to block the HBV/HDV host receptor sodium taurocholate co-transporting polypeptide (NTCP), for the treatment of patients with compensated chronic HDV infection.[17], [18], [19], [20] Several other HDV treatment options are currently being assessed in clinical trials.21 However, it is widely accepted that a combination of antiviral compounds targeting different steps in the viral life cycle will be required to eliminate both viruses.
In this study, we evaluated the capacity of a previously generated HBsAg-specific human monoclonal antibody (hu-mAb) to prevent and treat HBV and HDV infection both in vitro and in vivo.22 Targeting the entry-step in combination with diminishing circulating HBsAg would simultaneously block viral spread and potentially provide a window of opportunity for other antivirals to break immune tolerance in the context of chronic infection.
Materials and methods
More details can be found in the supplementary materials and methods.
Human monoclonal anti-HBsAg production and purification
The human monoclonal anti-HBsAg-specific antibody was produced using classical hybridoma technology, more specifically by fusion of SCID-engrafted human peripheral blood lymphocytes isolated from a vaccinated individual (6,981 mIU/ml anti-HBsAg at time of blood donation) with K6H6/B5 heteromyeloma cells as previously described.22 Fused cells were then seeded in culture medium supplemented with hybridoma growth factors and the selection drugs hypoxanthine-aminopterin-thymidine and ouabain. Anti-HBs-positive cultures were sequentially cloned and several monoclonal hybridoma lines were isolated and confirmed via commercial anti-HBs ELISA (DiaSorin, Italy). The hu-mAb was then purified from collected supernatant using conventional Protein G columns (Hitrap Protein G HP, Sigma Aldrich, USA). After evaluation by serum protein electrophoresis (SAS-MX Serum Protein-10, Sysmex, Japan), the antibody was concentrated (Amicon Ultra-15 50k, Sigma Aldrich, USA) and its concentration was determined using spectrophotometry at 280 nm.
In vitro prevention of HBV and HDV infection
For in vitro prevention of HBV infection, anti-HBsAg was applied to HepG2.hNTCP cells in duplicate at 5-fold serial dilutions ranging from 10 μg/ml to 0.128 ng/ml. Two hours later, cells were infected with HBV (4,990 IU/cell; genotype D) and after 1 week, infection was assessed using immunofluorescent (IF) staining of hepatitis B core antigen-positive cells. Images were captured by automated spinning disk microscopy using a 40x objective (CSU-X1, Nikon). Per condition, a 20 x 10 field was captured (in duplicate) and positive cells were automatically counted using ImageJ software v1.53c. In vitro prevention of HDV infection (genotype 1) was examined in the same manner, but in three different cell lines (HepG2.hNTCP, Huh7.5.hNTCP and NEB2.7). Imaging was performed using the Leica TCS-SPE microscope with a 20x objective. Per condition, three random pictures were taken (in duplicate) and HDV-positive cells were automatically counted using ImageJ software v1.53c. More details are provided in the supplementary materials and methods.
Mice
All mice were bred under sterile conditions and all experiments were approved by the Animal Ethics Committee of the Faculty of Medicine and Health Sciences of Ghent University. Human-liver chimeric mice were generated by transplanting 106 primary human hepatocytes (donor C342, C399 and HH223 from Corning, the Netherlands; and donor L191501 from Lonza, Switzerland) into homozygous uPA+/+-SCID mice as previously described.23,24 Human albumin was quantified in mouse plasma to evaluate successful humanization of the mouse liver using conventional ELISA (Bethyl Laboratories, USA). Mice with albumin levels ranging between 2-10 mg/ml were selected for this study and groups were randomized, taking into consideration human albumin levels, infection levels and general condition (e.g. based on body weight). Mice were 8- to 9-weeks-old at the start of infection.
In vivo prevention of HBV, HBV/HDV and HDV superinfection
For in vivo prevention of HBV, human-liver chimeric mice (n = 6) repopulated with human hepatocytes from two different donors (n = 3 for C342 and n = 3 for HH223) were passively immunized through intraperitoneal (IP) administration of 1 mg hu-mAb 3 days before challenge with HBV (patient serum, 106 IU/mouse). As controls, six additional mice were infected without prior passive immunization. For prevention of HBV/HDV coinfection, humanized mice (n = 4) engrafted with hepatocytes from donor C342 were passively immunized (1 mg hu-mAb, IP) 3 days prior to HBV/HDV coinfection with cell culture-derived virus (5 x 106 IU HBV DNA/mouse and 2.3 x 106 IU HDV RNA/mouse). As controls, three additional mice were coinfected with HBV/HDV without prior passive immunization. Mice (co-)infected with HBV or HBV/HDV were monitored until week 12-13 post-inoculation. Finally, for prevention of HDV superinfection, humanized mice were first infected with either patient-derived HBV (106 IU/mouse, n = 7, mice engrafted with donor L191501 hepatocytes) or cell culture-derived HBV (5 x 106 IU per mouse, n = 14, i.e. five donor C342 and nine C399 engrafted mice). Mice were superinfected with patient or cell culture-derived HDV (both 2.55 x 105 IU/mouse) 6 or 8 weeks later, respectively. Three days prior to superinfection, 5/7 mice infected with patient-derived HBV and 9/14 mice infected with cell culture-derived HBV were passively immunized with 1 mg of hu-mAb IP. Mice superinfected with HDV were monitored for 9-10 weeks following HDV inoculation. Blood plasma was collected every 1-2 weeks (depending on the condition of the mouse and the timing post-infection) and HBV DNA and HDV RNA levels were quantified via commercial HBV/HDV RealStar® (reverse transcription-)qPCR (Altona Diagnostics, Germany) or the RealTime m2000 HBV assay (Abbot, USA). Anti-HBsAg ELISA was performed using a commercially available quantitative kit (Beijing Wantai Biological, China).
Treatment of HBV and HBV/HDV (co-)infected mice
Human-liver chimeric mice (n = 7) transplanted with donor C342 hepatocytes were chronically infected with cell culture-derived HBV (5 x 106 IU/mouse). Four out of seven mice received 1 mg hu-mAb (IP) twice a week for 4 consecutive weeks (eight doses in total), starting at week 11 post viral inoculation. The remaining three infected mice were left untreated as controls. In a second treatment experiment, 13 human-liver chimeric mice transplanted with donor L191501 hepatocytes were first infected with cell culture-derived HBV (5 x 106 IU/mouse) before being superinfected with cell culture-derived HDV (2.55 x 105 IU/mouse) 6 weeks later. At week 10, 7/13 mice received 1 mg hu-mAb (IP) twice a week for 4 consecutive weeks (nine doses in total). The six remaining coinfected mice did not receive any treatment (control). Blood plasma was collected every 1 to 2 weeks (depending on the condition of the mouse and the timing post-infection) and the levels of HBV DNA, HDV RNA and hu-mAb were determined as described above. Plasma samples were analyzed by SDS-PAGE and western blot was used to assess HBsAg levels.
SDS-PAGE and western blot analysis
Exactly 0.5 μl of mouse plasma was used for sample preparation and denaturation (70 °C, 10 min), and subjected to SDS-polyacrylamide gel (12%) electrophoresis, with proteins transferred to a PVDF membrane. Membranes were blocked (5% skim milk in TBS-T) and HBsAg was detected by a primary goat anti-HBsAg antibody (70-HG15; Fitzgerald industries; 1/1,000), followed by a horse radish peroxidase-conjugated rabbit anti-goat antibody (31402; Thermo Fisher Scientific; 1/20,000). Immunoblots were developed using the SuperSignal™ West Femto Maximum Sensitivity Substrate kit (Thermo Fisher Scientific, USA) and exposed to the ImageQuant LAS4000 chemiluminescent imaging system (GE Healthcare, Diegem, Belgium). More details are provided in the supplementary materials and methods.
Statistical analysis
GraphPad Prism v9.3.1 was used for graph visualization and statistical analyses. IC50 values were calculated using non-linear regression (curve fit). Normality tests and Mann-Whitney U tests enabled comparisons between normalized log mean virus levels of treated vs. non-treated mice. Data were considered statistically significant if p <0.05.
Results
In vitro prevention of HBV infection
The HBV-permissive cell line HepG2.hNTCP was (pre-)incubated with 5-fold serial antibody dilutions before, during and after viral inoculation. Infected cells were visualized via immunofluorescent staining (hepatitis B core antigen) after 1 week. Near complete abrogation of infection was shown when at least 80 ng/ml hu-mAb was applied, while an overall dose-dependent inhibitory effect was observed with an IC50 value of 3.531 ng/ml (Fig. 1).
Fig. 1.

In vitro prevention of HBV infection.
(A) A 5-fold serial dilution of hu-mAb was evaluated for the prevention of HBV infection in HepG2.hNTCP cells. Infectivity was assessed using hepatitis B core antigen immunofluorescence staining 1 week after infection. Relative infection (%): total number of infected cells relative to the control condition where no antibody was applied. (B) The IC50 value was calculated from the antibody dose-response curve by non-linear regression (curve fit). All conditions were performed in duplicate. All data represented as mean ± SD (error bars). hu-mAb, human monoclonal antibody; IC50, half maximal inhibitory concentration.
In vitro prevention of HDV infection
The HDV inoculum was pre-incubated with 5-fold serial hu-mAb dilutions and three cell lines were subjected to this virus-antibody mixture. Antibodies were maintained for 1 week until visualization of infected cells by IF. As shown in Fig. 2, infection was nearly completely abrogated using at least 16 ng/ml hu-mAb for all three cell lines tested. A clear dose-dependent inhibitory effect was observed with IC50 values of 3.275 ng/ml, 4.393 ng/ml and 3.080 ng/ml for HepNB2.7, HepG2.hNTCP and Huh7.5.hNTCP cells, respectively.
Fig. 2.

In vitro prevention of HDV infection.
(A) A 5-fold serial dilution of hu-mAb was evaluated for the prevention of HDV infection in HepNB2.7 (grey), HepG2.hNTCP (purple) and Huh7.5.hNTCP (dark purple) cells. The hu-mAb was pre-incubated with the HDV inoculum before challenge and the antibody was maintained for 7 days until HDAg immunofluorescence staining. Relative infection (%): total number of infected cells relative to the control condition where no antibody was applied. (B) The IC50 values were calculated from the antibody dose-response curves by non-linear regression (curve fits). Baseline for background immunofluorescence is set at 0.04% (not depicted). All conditions were performed in duplicate. All data represented as mean ± SD (error bars). hu-mAb, human monoclonal antibody; IC50, half maximal inhibitory concentration.
In vivo prevention of HBV infection
Six human-liver chimeric mice (repopulated with hepatocytes from two distinct donors) were passively immunized (1 mg hu-mAb/mouse) 3 days prior to HBV inoculation. As controls, six animals were injected with the virus without prior passive immunization. In donor C342 engrafted mice, no virus was detected in the three passively immunized mice until at least week 6 (on which two were found dead) and one mouse experienced HBV DNA breakthrough at week 10, while the controls showed strongly increasing levels from week 4 onwards (Fig. 3A). This was also confirmed in mice repopulated with donor HH223 hepatocytes: two out of three passively immunized mice remained HBV DNA negative until at least week 12 post-infection (end of the observation period), one mouse only showed signs of infection at week 12 and all three control mice demonstrated increasing HBV DNA levels from week 3 onwards (Fig. 3B). In the two passively immunized mice that rebounded at week 10 and 12 post-infection, circulating plasma anti-HBsAg was no longer detectable as of week 6 (Fig. S1).
Fig. 3.

In vivo prevention of HBV infection.
Mice transplanted with hepatocytes from donor C342 (A) or donor HH223 (B) were passively immunized with 1 mg of hu-mAb (IP) 3 days prior to HBV challenge (each n = 3). Control mice (each n = 3) were infected with the same viral inoculum (patient serum, 106 IU/mouse), but without prior passive immunization. HBV DNA was quantified using either the RealStar® qPCR (Altona Diagnostics, Germany; LOD = 3,750 IU/ml) (A) or the RealTime m2000 HBV assay (Abbot, US; LOD = 300 IU/ml) (B). †: mouse found dead. hu-mAb, human monoclonal antibody; IP, intraperitoneally; LOD, limit of detection.
In vivo prevention of HBV/HDV coinfection
Four human-liver chimeric mice were injected with 1 mg of hu-mAb (IP), 3 days prior to inoculation of a viral preparation containing 5 x 106 IU HBV and 2.28 x 106 IU HDV (both cell culture produced). Control mice (n = 3), injected with the same virus prep without prior passive immunization, experienced increasing plasma levels of HBV DNA (Fig. 4A) and HDV RNA (Fig. 4B) from week 4 and 8 onwards, respectively. Interestingly, all passively immunized mice did not show any signs of infection until 13 weeks post-infection when the experiment was terminated. Anti-HBs antibody levels exceeded 1,000 mIU/ml during the first 2-4 weeks following passive immunization, gradually declined over time and became undetectable in our assay at around 8-10 weeks post antibody injection (Fig. S2).
Fig. 4.

In vivo prevention of HBV/HDV coinfection.
Passive immunization using 1 mg of hu-mAb (IP) was performed 3 days prior to HBV/HDV inoculation (n = 4) in mice engrafted with donor C342 hepatocytes. Control mice (n = 3) were accordingly infected with cell culture-derived virus (5 x 106 IU HBV and 2.28 x 106 IU HDV per mouse). HBV DNA (A) and HDV RNA (B) was analyzed in mouse plasma via RealStar® (reverse transcription-)qPCR (Altona Diagnostics, Germany; LOD HBV = 3,750 IU/ml; LOD HDV = 187.5 IU/ml). †: mouse found dead. hu-mAb, human monoclonal antibody; IP, intraperitoneally; LOD, limit of detection.
In vivo prevention of HDV superinfection
To evaluate whether a single antibody injection could avert an HDV infection in mice with pre-existing HBV infection, human-liver chimeric mice engrafted with donor L191501 hepatocytes (n = 7) were first infected with HBV and 6 weeks later, five out of seven mice were passively immunized with the antibody 3 days prior to HDV superinfection (Fig. 5A-B). At the moment of HDV inoculation (week 6), the HBV DNA plateau had been reached in all mice (Fig. 5A). In both control mice, HDV RNA levels could already be detected 1 week after HDV inoculation and these increased steeply to 108 IU/ml at week 10. In most passively immunized mice, HDV RNA was only detected at week 10 with a delayed peak at week 14 (Fig. 5B). Accordingly, circulating anti-HBs was lost between week 8 and 14 for all passively immunized mice (Fig. S3A). This experimental setup was repeated in 14 mice engrafted with either donor C342 or C399 hepatocytes, but here, passive immunization (in 9/14 mice) and HDV superinfection was performed at week 8 (Fig. 5C-D). Interestingly, eight mice were completely protected from HDV superinfection and the five animals that reached the endpoint of the observation period (week 16-17) were still HDV RNA negative (Fig. 5D). The single passively immunized mouse that became HDV RNA positive at week 12 also lost its circulating anti-HBs at week 9 (Fig. S3B).
Fig. 5.

In vivo prevention of HDV superinfection.
(A-B) Mice were engrafted with human hepatocytes from donor L191501 and infected at week 0 with patient-derived HBV (106 IU/mouse, n = 7). Three days prior to HDV superinfection with patient-derived inoculum (2.55 x 105 IU/mouse) at week 6, passive immunization in 5/7 mice was performed with 1 mg of hu-mAb (IP). (C-D) Mice were engrafted with human hepatocytes from donor C342 (n = 5) or C399 (n = 9) as depicted and infected at week 0 with cell culture-derived HBV (5 x 106 IU/mouse). Three days prior to HDV superinfection with either cell culture-derived inoculum or patient-derived inoculum (∗) at week 8 (both using 2.55 x 105 IU/mouse), passive immunization in 9 (n = 3 for C342 mice and n = 6 for C399 mice) out of 14 mice was performed with 1 mg of hu-mAb (IP). HBV DNA (A, C) and HDV RNA (B, D) was quantified by RealStar® (reverse transcription-)qPCR (Altona Diagnostics, Germany; LOD HBV = 3,750 IU/ml; LOD HDV = 187.5 IU/ml). †: mouse found dead. hu-mAb, human monoclonal antibody; IP, intraperitoneally; LOD, limit of detection.
Treatment of chronic HBV infection
Humanized mice with an established HBV infection were treated twice a week for 4 consecutive weeks with 1 mg of hu-mAb (IP; n = 4). While non-treated HBV-infected controls (n = 3) demonstrated quite stable or moderately increasing plasma HBV DNA levels, all antibody-treated mice showed strongly, although not statistically significantly, decreasing viral levels on therapy (Fig. 6A-B and Table S1). Specifically, already after 1 week of therapy, mean plasma HBV DNA levels were reduced by 1.78 x log10 compared to non-treated mice. HBV DNA even became undetectable in our assay in one mouse (M240L). On week 2, 3 and 4 (end) of treatment, we observed a 1.55 x log10, 2.09 x log10 and 2.02 x log10 fold reduction, respectively, compared to the controls. Importantly, only one (M257RR) out of four mice rebounded after treatment cessation and this coincided with a complete loss of circulating anti-HBs antibodies (Fig. 6C). Three weeks post-therapy, a mean difference of 2.03 x log10 was still present between the two groups.
Fig. 6.

Treatment of chronic HBV infection.
Human-liver chimeric mice (engrafted with donor C342 hepatocytes) were infected with HBV for 11 weeks and then treated with hu-mAb for 4 consecutive weeks: 2 IP injections of 1 mg per week (n = 4). Control mice were accordingly infected with cell culture-derived virus (5 x 106 IU HBV/mouse) but were not treated (n = 3). (A) Plasma HBV DNA. LOD (HBV) = 3,750 IU/ml. (B) Log10 reduction of plasma HBV DNA, calculated towards levels observed 1 day prior to first antibody injection (week 11). (C) Anti-HBsAg determined in mouse plasma via quantitative ELISA (Wantai, China). LLOQ (anti-HBsAg) = 100 mIU/ml and ULOQ (anti-HBsAg) = 1,600 mIU/ml. †: mouse found dead. hu-mAb, human monoclonal antibody; IP, intraperitoneally; LLOQ, lower limit of quantification; LOD, limit of detection; ULOQ, upper limit of quantification.
Treatment of HBV/HDV-coinfected mice
Six weeks post HBV infection, 13 humanized mice were inoculated with HDV to obtain HBV/HDV-coinfected mice. Four weeks later, the animals were divided in two groups: the first group (n = 7) was treated with hu-mAb for 4 weeks, while the second group (n = 6) served as untreated controls. At the start of therapy, plasma HBV DNA levels ranged between 106 and 109 IU/ml, while HDV RNA levels were more diverse, varying from just above the limit of detection (LOD) to almost 108 IU/ml, with one mouse (M264RL) having undetectable HDV RNA in our assay (Fig. 7A,C). Two weeks following the onset of therapy, mean HBV DNA and HDV RNA levels were significantly reduced compared to controls: 1.96 x log10 (p <0.01) and 2.82 x log10 (p <0.05) difference, respectively (Fig. 7A-D and Table S2). Even more pronounced mean differences could be observed after 4 weeks (end of therapy): -2.82 x log10 and -3.15 x log10 (both p <0.01) for HBV DNA and HDV RNA, respectively, while stable or even increasing viral loads were measured in the controls. Two and 4 weeks after treatment cessation, mean differences of -1.97 x log10 and -1.69 x log10 for HBV DNA, respectively, and -3.20 x log10 and -3.10 x log10 for HDV RNA, respectively, were observed; these differences were accompanied by hu-mAb plasma levels exceeding 1,000 mIU/ml in treated animals. However, two mice (M241R and M298R) demonstrated HBV/HDV rebound at week 2-4 post-therapy, which coincided with hu-mAb loss at these time points (Fig. 7E). Remarkably, HBsAg was observed at the onset of therapy in the treated mice (week 10) but was undetectable on therapy (based on western blot analysis) (Fig. 7F). For the two mice showing strongly diminished or even absent viral loads post-therapy (M296 and M296R), HBsAg was still absent. On the other hand, the post-therapy viral rebound observed in M241R (at week 16) and M298R (at week 18) was accompanied by recurrence of HBsAg.
Fig. 7.

Treatment of chronic HBV/HDV infection.
Human-liver chimeric mice engrafted with donor L191501 hepatocytes were infected with HBV and superinfected with HDV at week 6. From week 10 onwards, mice were treated with hu-mAb for 4 consecutive weeks: 2 IP injections of 1 mg per week (n = 7). Control mice were accordingly infected with cell culture-derived virus (5 x 106 IU HBV and 2.55 x 105 IU HDV per mouse) but were not treated (n = 6). (A) Plasma HBV DNA. LOD (HBV) = 3,750 IU/ml. (B) Log10 reduction of plasma HBV DNA, calculated towards levels observed 1 day prior to first antibody injection (week 10). (C) Plasma HDV RNA. LOD (HDV) = 187.5 IU/ml. (D) Log10 reduction of plasma HDV RNA, calculated towards levels observed 1 day prior to first antibody injection (week 10). Statistical analyses performed using Mann-Whitney U test: ∗p value <0.05; ∗∗p value <0.01. (E) Anti-HBsAg determined in mouse plasma via quantitative ELISA. LLOQ (anti-HBsAg) = 100 mIU/ml and ULOQ (anti-HBsAg) = 1,600 mIU/ml. (F) HBsAg Western blot analysis. †: mouse found dead. (g)p, (glycosylated) protein; hu-mAb, human monoclonal antibody; IP, intraperitoneally; LLOQ, lower limit of quantification; LOD, limit of detection; ULOQ, upper limit of quantification.
Discussion
The excessive HBsAg release in patients with chronic HBV tolerizes antibody- and cell-mediated immune responses, representing a major obstacle to viral eradication with current treatments.25,26 Consequently, it is of utmost importance to find ways to break immune tolerance, enabling the host to mount effective immune responses to clear the virus and to prevent HDV superinfection. Herein, we demonstrate the prophylactic and therapeutic efficacy of a hu-mAb targeting the HBV envelope in the context of HBV and HDV infection in vitro and in human-liver chimeric mice.
The hu-mAb neutralized HBV and HDV in vitro with respective IC50 values of 3.53 ng/ml (∼0.23 nM) and 3.58 ng/ml (∼0.24 nM) and can be considered as highly potent, since Hehle et al.27 evaluated 170 different hu-mAbs isolated from natural HBV controllers or vaccinated individuals and showed that only 35% of antibodies harbored IC50 values <50 ng/ml.
Next, we demonstrated efficient prevention of HBV mono- and HBV/HDV coinfection in human-liver chimeric mice. Conceivably, once a chronic HBV infection is fully established, preventing HDV superinfection may be even more challenging due to the presence of large quantities of HBsAg. However, depending on the hepatocyte donor, we showed either near complete prevention (donor C342 and C399) or at least delayed HDV RNA levels (donor L191501). Notably, transplantation of mice with donor L191501 human hepatocytes rendered the animals more prone to hepatitis infection compared to other donors tested (data not shown), which may explain viral breakthrough. Overall, our HDV superinfection prevention data is quite impressive since only a single injection of 1 mg hu-mAb was sufficient to overcome the high load of circulating HBsAg and to (partially) neutralize the high-titer HDV inoculum. Furthermore, since HDV can silently persist and replicate in human hepatocytes for up to 6 weeks, the hu-mAb effectively prohibits entry of HDV virions into hepatocytes.28,29
Notably, antibody half-life varies throughout our experiments, i.e. the antibody is still present in circulation 3-4 weeks or longer after a 1 mg injection. This may potentially be explained by variability in body weight (and hence distribution volume) since we injected 1 mg of antibody irrespective of mouse body weight. Furthermore, antibody clearance might be greatly accelerated by high replication of the virus (high circulating viral load). The source of viral inoculum also appears to influence the success of prevention and lack of viral rebound. For example, in the prophylactic HBV/HDV coinfection experiment, cell culture-derived virus was used and no viral rebound was observed, while in the prophylactic HBV mono-infection experiment, patient-derived virus was applied and HBV DNA rebound was observed. Additionally, more animals were protected from HDV superinfection when infected with cell culture-derived viruses. Therefore, it seems that primary patient-derived virus preparations are intrinsically less sensitive to antibody neutralization, or may contain variants that are.
The therapeutic potential of the hu-mAb was furthermore demonstrated by (significantly) reduced mean viral plasma levels at the end of a 4-week treatment in HBV-infected (2.02 x log10 drop) and HBV/HDV-coinfected mice (2.82 x log10 and 3.50 x log10 drops for HBV and HDV, respectively). Since the repeated injections of antibody result in a clear drop in HBV DNA (and HDV RNA levels) in this therapeutic setup, we assume that the single dose regimen in the HDV superinfection prevention experiment was insufficient to show an antiviral effect on the already established HBV infection (i.e. no effect on HBV DNA). Interestingly, our 4-week treatment data are in line with the most potent hu-mAb (namely Bc1.187), which was tested by Hehle et al.27 in HUHEP mice (weekly ∼1 mg (50 mg/kg) hu-mAb IP for 3 weeks), and induced a 1.76 x log10 decrease in serum HBV DNA at the end of therapy. Notably, circulating HBsAg levels dropped and remained undetectable for 2 weeks following the last injection in 60% of treated animals. Importantly, these mice had lower initial HBV DNA levels (<106 IU/ml) compared to mice in our study. Another hu-mAb (HBC34) was evaluated at a very comparable schedule, but at lower dose (a 4-week treatment with 2 IP injections per week of 1 mg/kg) in HBV/HDV-coinfected USB/USG mice.30,31 They revealed a similar reduction in HBV DNA and HDV RNA levels (mean 1 to 2.7 x log10 and 2.4 to 2.5 x log10 reduction, respectively, in both models) and a concomitant 2.7 to 2.8 x log10 reduction of HBsAg levels after 4 weeks. Finally, we also show on-treatment HBsAg loss with rebound after treatment cessation, a phenomenon that is frequently noted upon withdrawal of mAb therapy.27,32 Interestingly, the HBsag loss might provide a therapeutic window for viral eradication using NAs, possibly in combination with other antivirals, that could take advantage of a (partially) reconstituted adaptive immune response.23,24 Interestingly, although no effect on HBsAg was seen when bulevirtide was used as monotherapy, HBsAg decreases could be observed when combined with peg-IFNα.17,20 Notably, decreasing HBsAg might be a valuable therapeutic strategy, but currently no direct evidence is available to support this hypothesis and therefore, additional studies addressing the relationship between circulating HBsAg levels and HBV-specific immune responses are required to provide further insights on the immunobiology of HBV; presumably, additional immune activation would be necessary to control the virus.33 Nowadays, polyclonal antibodies derived from pooled plasma from recovered or vaccinated individuals (hepatitis B immunoglobulin) are successfully and routinely used in clinical practice as post-exposure prophylaxis, to avoid HBV recurrence after liver-transplantation or therapeutic immunosuppression, or to prevent perinatal transmission.[34], [35], [36] However, only a small antibody proportion in hepatitis B immunoglobulin preparations is actually HBV-specific and neutralizing, and blood products always remain subject to bio-safety concerns, especially for immunosuppressed patients.37,38 Consequently, monoclonal antibodies may be an attractive alternative since they represent a stable, reproducible source for prolonged immunotherapy. Hu-mAbs have already been successfully explored in many therapeutic fields such as oncology,39, auto-immune diseases40 and recently also for infectious diseases such as SARS-CoV-2.41,42 Our research group also demonstrated successful hu-mAb use in the context of HCV infection.[43], [44], [45] Regarding other HBV-specific hu-mAbs, previous studies have similarly shown efficient prevention, delayed infection, or therapeutic efficacy in cell culture,27,[46], [47], [48], chimpanzees[49], [50], [51], [52], [53], [54] and various mouse models.27,38,[54], [55], [56], [57], [58] Some have reached clinical stages as well, such as lenvervimab59 and VIR-3434, the Fc-engineered version of antibody HBC34, with extended serum half-life; NCT04423393 and NCT04856085.60 Hu-mAb studies involving HDV prophylaxis or therapy are currently very limited: Li et al.40 demonstrated prevention in a mouse model harboring human NTCP receptors in the absence of HBV infection and, moreover, on a mouse background; and HBC34 led to a potent decrease in HBV/HDV viremia in HBV/HDV-coinfected USB/USG mice.30,31
Currently evaluated or approved antibody dosages vary tremendously in human applications, even beyond the field of infectious diseases. For example, omalizumab, a humanized recombinant monoclonal anti-IgE antibody approved for the treatment of allergic asthma is recommended at 75 to 600 mg every 2 weeks.61 Rituximab, a genetically engineered chimeric mouse/human mAb approved for rheumatoid arthritis is recommended at two infusions of 1,000 mg IV every 2 weeks.62 Eculizumab, a humanized mAb approved for myasthenia gravis, is administered at 600 mg IV weekly for 4 weeks, followed by 900 mg IV in the 5th week and then, 900 mg every 2 weeks as maintenance therapy.63 Finally, the FDA recently authorized two mAbs (casirivimab and imdevimab) for the treatment of COVID-19 disease at dosages as high as 1,400 mg IV.64,65 These examples illustrate the high variability of applied doses in various applications, but also that much higher dosages are achievable and tolerated compared to our regimen. Assuming an average mouse body weight of 15 g, we applied about 66.7 mg/kg per injection. In the treatment regimen, we hence administered a total dose of 533 mg over a 4-week period; clearly in range of what is feasible in humans. However, dosages used in mice cannot simply be translated into human doses. More specifically, the common perception of scaling of dose based on body weight (mg/kg) alone is not entirely correct. This is primarily because the functional systems in species vary which, in turn, alters pharmacokinetics.66
The main putative mechanism of action of the HBsAg-targeting antibody in the applied mouse model is functional neutralization of the viral envelope, which directly blocks entry of HBV and HDV, and consequently prevents spread of infection to naïve hepatocytes. Furthermore, the antibody also promotes HBsAg reduction (or clearance) in the circulation. It has also been shown that HBsAg-specific antibodies could be internalized into infected cells and therefore partially inhibit HBsAg secretion from these cells, implicating intracellular blocking of virion and protein release as an additional mechanism of action.67,68 Importantly, in the context of infected patients, additional intrinsic effector functions could be induced through the formation of immune complexes. More specifically, clearance of circulating virions, antigens or infected cells could be promoted via antibody-dependent cell-mediated cytotoxicity and/or other Fc-mediated effector responses such as complement lysis or phagocytosis.55,56,[69], [70], [71], [72], [73] Accordingly, many therapeutic advantages could be proposed in a clinical setting: 1) reduction of the hepatic accumulation of covalently closed circular DNA, one of the main causes of HBV persistence;74,75 2) mediation of serum HBV/HDV and HBsAg clearance, which may ultimately counteract the challenging T-cell exhaustion mechanisms that are characteristic in patients with chronic HBV infection;76 3) overall risk reduction of viral reactivation (following liver-transplantation);77 and finally 4) potential allowance of safe withdrawal of antiviral drugs.78
In conclusion, we show the added value of an entry-inhibiting hu-mAb in the context of HBV and HDV (super)infection prophylaxis and its potential use as treatment for chronic HBV or HBV/HDV (co-)infection. For the experiments presented herein, we used either patient-derived virus or cell culture-derived virus (genotype D for HBV and genotype 1 for HDV) in human-liver chimeric mice that were transplanted with hepatocytes from four different donors. Further confirmation using additional viral genotypes and various hepatocyte donors with distinct genetic backgrounds is warranted. Overall, our data suggest that this hu-mAb is an interesting candidate to complement current therapies or antivirals (under development) to eradicate both HBV and HDV infections. Decreasing or even eliminating the high levels of circulating HBsAg in patients with chronic infection may provide an opportunistic window for other antiviral therapies targeting later steps in the viral life cycle or the immune system to eventually cure chronic infection.
Financial support
PM was supported by the Special Research Fund of Ghent University (BOFEXP2017001002), the Research Foundation-Flanders (FWO-Vlaanderen; projects G089515N, G047417N and Excellence of Science projects VirEOS and VirEOS2.0) and the European Union (FP7, HepaMab).
Authors’ contributions
PM devised the study. General experimental setup was designed by PM and RB. RB and FVH performed in vitro neutralization studies. Animal work was conducted by RB, LV, AAM and PM. Viral parameters were monitored in mouse plasma by RB and FVH. GLR provided the antibody clone. SC, HW and PR delivered patient-derived viruses that were essential for the execution of the study. Data analyses and writing were performed by RB and PM. All authors approved the final version of the manuscript.
Data availability statement
The data that supports the findings of this study is available upon request. Please contact the corresponding author.
Conflict of interest
Nothing to declare related to the content of this manuscript.
Please refer to the accompanying ICMJE disclosure forms for further details.
Acknowledgments
Cell lines HepNB2.7, HepG2.hNTCP and Huh7.5.hNTCP were kindly provided by Prof. Dr. Stephan Urban (Heidelberg University, Germany); HepAD38 cells by Dr. Christoph Seeger (Fox Chase Cancer Center, Philadelphia, USA), and Huh7.5 cells by Prof. Dr. Charles M. Rice (The Rockefeller University, USA). Dr. John Taylor and Prof. Dr. Camille Sureau provided plasmids pSVL(D)3 and pT7HB2.7. Recombinant HBsAg (HEF002) was kindly provided by GlaxoSmithKline Biochemicals (Rixensart, Belgium). We thank Sophie Vermaut for antibody purification and technical assistance. We acknowledge the Centre for Advanced Light Microscopy at Ghent University (Belgium) for the use and support on the Nikon spinning disk CSU-X1 microscope. The first draft of the graphical abstract was created with Biorender.com.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhepr.2022.100646.
Supplementary data
The following are the supplementary data to this article:
References
- 1.Razavi-Shearer D.G.I., Gamkrelidze I., Nguyen M.H., Chen D.S., Van Damme P., Abbas Z., et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol. 2018;3:383–403. doi: 10.1016/S2468-1253(18)30056-6. [DOI] [PubMed] [Google Scholar]
- 2.Suk-Fong Lok A. Hepatitis B Treatment: What we know now and what remains to Be researched. Hepatol Commun. 2019;3:8–19. doi: 10.1002/hep4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO hepatitis B fact sheet; 2022. [cited; Available from: Hepatitis B (who.int)
- 4.Stockdale A.J., Kreuels B., Henrion M.Y.R., Giorgi E., Kyomuhangi I., de Martel C., et al. The global prevalence of hepatitis D virus infection: systematic review and meta-analysis. J Hepatol. 2020;73:523–532. doi: 10.1016/j.jhep.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes S.A., Wedemeyer H., Harrison P.M. Hepatitis delta virus. Lancet. 2011;378:73–85. doi: 10.1016/S0140-6736(10)61931-9. [DOI] [PubMed] [Google Scholar]
- 6.Miao Z., Zhang S., Ou X., Li S., Ma Z., Wang W., et al. Estimating the global prevalence, disease progression and clinical outcome of hepatitis delta virus infection. J Infect Dis. 2020;221:1677–1687. doi: 10.1093/infdis/jiz633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H.Y., Shen D.T., Ji D.Z., Han P.C., Zhang W.M., Ma J.F., et al. Prevalence and burden of hepatitis D virus infection in the global population: a systematic review and meta-analysis. Gut. 2019;68:512–521. doi: 10.1136/gutjnl-2018-316601. [DOI] [PubMed] [Google Scholar]
- 8.Negro F. Hepatitis D virus coinfection and superinfection. Cold Spring Harb Perspect Med. 2014;4:a021550. doi: 10.1101/cshperspect.a021550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Locarnini S., Hatzakis A., Chen D.S., Lok A. Strategies to control hepatitis B: public policy, epidemiology, vaccine and drugs. J Hepatol. 2015;62:S76–S86. doi: 10.1016/j.jhep.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 10.Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 11.European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67:370–398. doi: 10.1016/j.jhep.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 12.Caviglia G.P., Rizzetto M. Treatment of hepatitis D, an unmet medical need. Clin Microbiol Infect. 2020;26:824–827. doi: 10.1016/j.cmi.2020.02.031. [DOI] [PubMed] [Google Scholar]
- 13.Abbas Z., Memon M.S., Mithani H., Jafri W., Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real-world experience. Antivir Ther. 2014;19:463–468. doi: 10.3851/IMP2728. [DOI] [PubMed] [Google Scholar]
- 14.Wedemeyer H., Yurdaydin C., Hardtke S., Caruntu F.A., Curescu M.G., Yalcin K., et al. Peginterferon alfa-2a plus tenofovir disoproxil fumarate for hepatitis D (HIDIT-II): a randomised, placebo controlled, phase 2 trial. Lancet Infect Dis. 2019;19:275–286. doi: 10.1016/S1473-3099(18)30663-7. [DOI] [PubMed] [Google Scholar]
- 15.Wedemeyer H., Yurdaydin C., Dalekos G.N., Erhardt A., Cakaloglu Y., Degertekin H., et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. New Engl J Med. 2011;364:322–331. doi: 10.1056/NEJMoa0912696. [DOI] [PubMed] [Google Scholar]
- 16.Heidrich B., Yurdaydin C., Kabacam G., Ratsch B.A., Zachou K., Bremer B., et al. Late HDV RNA relapse after peginterferon alpha-based therapy of chronic hepatitis delta. Hepatology (Baltimore, Md) 2014;60:87–97. doi: 10.1002/hep.27102. [DOI] [PubMed] [Google Scholar]
- 17.Wedemeyer H., Schöneweis K., Bogomolov P.O., Chulanov V., Stepanova T., Viacheslav M., et al. 48 weeks of high dose (10 mg) bulevirtide as monotherapy or with peginterferon alfa-2a in patients with chronic HBV/HDV co-infection. J Hepatol. 2020;73:S52–S53. [Google Scholar]
- 18.Wedemeyer H., Schöneweis K., Bogomolov P.O., Voronkova N., Chulanov V., Stepanova T. Interim results of a multicentre, open-label phase 2 clinical trial (MYR203) to assess safety and efficacy of Myrcludex B in combination with Peg-Interferon alpha 2a in patients with chronic HBV/HDV co-infection. J Hepatol. 2019;70:E81. [Google Scholar]
- 19.Wedemeyer H., Bogomolov P., Blank A., Allweiss L., Dandri-Petersen M., Bremer B., et al. Final results of a multicenter, open-label phase 2b clinical trial to assess safety and efficacy of Myrcludex B in combination with Tenofovir in patients with chronic HBV/HDV co-infection. J Hepatol. 2018;68:S3. [Google Scholar]
- 20.Degasperi E., Anolli M.P., Lampertico P. Bulevirtide for patients with compensated chronic hepatitis delta: a review. Liver Int. 2022 doi: 10.1111/liv.15389. PMID: 35942695. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 21.Sandmann L., Wedemeyer H. New treatments for chronic hepatitis B virus/hepatitis D virus infection. Clin Liver Dis. 2021;25:831–839. doi: 10.1016/j.cld.2021.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Depraetere S., Verhoye L., Leclercq G., Leroux-Roels G. Human B cell growth and differentiation in the spleen of immunodeficient mice. J Immunol (Baltimore, Md: 1950) 2001;166:2929–2936. doi: 10.4049/jimmunol.166.5.2929. [DOI] [PubMed] [Google Scholar]
- 23.Meuleman P., Libbrecht L., De Vos R., de Hemptinne B., Gevaert K., Vandekerckhove J., et al. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology (Baltimore, Md) 2005;41:847–856. doi: 10.1002/hep.20657. [DOI] [PubMed] [Google Scholar]
- 24.Meuleman P., Vanlandschoot P., Leroux-Roels G. A simple and rapid method to determine the zygosity of uPA-transgenic SCID mice. Biochem Biophys Res Commun. 2003;308:375–378. doi: 10.1016/s0006-291x(03)01388-3. [DOI] [PubMed] [Google Scholar]
- 25.Kuipery A., Gehring A.J., Isogawa M. Mechanisms of HBV immune evasion. Antivir Res. 2020;179 doi: 10.1016/j.antiviral.2020.104816. [DOI] [PubMed] [Google Scholar]
- 26.Bertoletti A., Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut. 2012;61:1754–1764. doi: 10.1136/gutjnl-2011-301073. [DOI] [PubMed] [Google Scholar]
- 27.Hehle V., Beretta M., Bourgine M., Ait-Goughoulte M., Planchais C., Morisse S., et al. Potent human broadly neutralizing antibodies to hepatitis B virus from natural controllers. J Exp Med. 2020:217. doi: 10.1084/jem.20200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giersch K., Helbig M., Volz T., Allweiss L., Mancke L.V., Lohse A.W., et al. Persistent hepatitis D virus mono-infection in humanized mice is efficiently converted by hepatitis B virus to a productive co-infection. J Hepatol. 2014;60:538–544. doi: 10.1016/j.jhep.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Giersch K., Hermanussen L., Volz T., Kah J., Allweiss L., Casey J., et al. Murine hepatocytes do not support persistence of Hepatitis D virus mono-infection in vivo. Liver Int. 2021;41:410–419. doi: 10.1111/liv.14677. [DOI] [PubMed] [Google Scholar]
- 30.Corti D., Volz T., Giersch K., Agatic G., Cameroni E., Kah J., et al. Prevention of viral spread, viremia reduction and HBsAg clearance in hepatitis B and D coinfected humanized mice by a human neutralizing monoclonal antibody. J Hepatol. 2016;2:S510. [Google Scholar]
- 31.Lempp F.A., volz T., cameroni E., Benigni F., Zhou J., Rosen L.E., et al. The potent broadly neutralizing antibody VIR-3434 controls Hepatitis B and D Virus infection and reduces HBsAg in humanized mice. bioRxiv. 2022 doi: 10.1016/j.jhep.2023.07.003. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 32.Galun E., Eren R., Safadi R., Ashour Y., Terrault N., Keeffe E.B., et al. Clinical evaluation (phase I) of a combination of two human monoclonal antibodies to HBV: safety and antiviral properties. Hepatology (Baltimore, Md) 2002;35:673–679. doi: 10.1053/jhep.2002.31867. [DOI] [PubMed] [Google Scholar]
- 33.Kim J.H., Ghosh A., Ayithan N., Romani S., Khanam A., Park J.J., et al. Circulating serum HBsAg level is a biomarker for HBV-specific T and B cell responses in chronic hepatitis B patients. Sci Rep. 2020;10:1835. doi: 10.1038/s41598-020-58870-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerino A., Mantovani S., Mele D., Oliviero B., Varchetta S., Mondelli M.U. Human monoclonal antibodies as adjuvant treatment of chronic hepatitis B virus infection. Front Immunol. 2019;10:2290. doi: 10.3389/fimmu.2019.02290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fox A.N., Terrault N.A. The option of HBIG-free prophylaxis against recurrent HBV. J Hepatol. 2012;56:1189–1197. doi: 10.1016/j.jhep.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 36.Shouval D., Samuel D. Hepatitis B immune globulin to prevent hepatitis B virus graft reinfection following liver transplantation: a concise review. Hepatology (Baltimore, Md) 2000;32:1189–1195. doi: 10.1053/jhep.2000.19789. [DOI] [PubMed] [Google Scholar]
- 37.Cerino A., Bremer C.M., Glebe D., Mondelli M.U. A human monoclonal antibody against hepatitis B surface antigen with potent neutralizing activity. PLoS One. 2015;10 doi: 10.1371/journal.pone.0125704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang W., Sun L., Li T., Ma Y., Li J., Liu Y., et al. A human monoclonal antibody against small envelope protein of hepatitis B virus with potent neutralization effect. MAbs. 2016;8:468–477. doi: 10.1080/19420862.2015.1134409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimiz-Gebologlu I., Gulce-Iz S., Biray-Avci C. Monoclonal antibodies in cancer immunotherapy. Mol Biol Rep. 2018;45:2935–2940. doi: 10.1007/s11033-018-4427-x. [DOI] [PubMed] [Google Scholar]
- 40.Barnas J.L., Looney R.J., Anolik J.H. B cell targeted therapies in autoimmune disease. Curr Opin Immunol. 2019;61:92–99. doi: 10.1016/j.coi.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinreich D.M., Sivapalasingam S., Norton T., Ali S., Gao H., Bhore R., et al. REGEN-COV antibody combination and outcomes in outpatients with covid-19. New Engl J Med. 2021;385:e81. doi: 10.1056/NEJMoa2108163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.ACTIV-3/Therapeutics for Inpatients with COVID-19 (TICO) Study Group. Efficacy and safety of two neutralising monoclonal antibody therapies, sotrovimab and BRII-196 plus BRII-198, for adults hospitalised with COVID-19 (TICO): a randomised controlled trial. Lancet Infect Dis. 2022;22:622–635. doi: 10.1016/S1473-3099(21)00751-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desombere I., Mesalam A.A., Urbanowicz R.A., Van Houtte F., Verhoye L., Keck Z.Y., et al. A novel neutralizing human monoclonal antibody broadly abrogates hepatitis C virus infection in vitro and in vivo. Antivir Res. 2017;148:53–64. doi: 10.1016/j.antiviral.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mesalam A.A., Desombere I., Farhoudi A., Van Houtte F., Verhoye L., Ball J., et al. Development and characterization of a human monoclonal antibody targeting the N-terminal region of hepatitis C virus envelope glycoprotein E1. Virology. 2018;514:30–41. doi: 10.1016/j.virol.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vercauteren K., Van Den Eede N., Mesalam A.A., Belouzard S., Catanese M.T., Bankwitz D., et al. Successful anti-scavenger receptor class B type I (SR-BI) monoclonal antibody therapy in humanized mice after challenge with HCV variants with in vitro resistance to SR-BI-targeting agents. Hepatology (Baltimore, Md) 2014;60:1508–1518. doi: 10.1002/hep.27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jo G., Jeong M.S., Wi J., Kim D.H., Kim S., Kim D., et al. Generation and characterization of a neutralizing human monoclonal antibody to hepatitis B virus PreS1 from a phage-displayed human synthetic Fab library. J Microbiol Biotechnol. 2018;28:1376–1383. doi: 10.4014/jmb.1803.03056. [DOI] [PubMed] [Google Scholar]
- 47.Zankharia U.S., Kudchodkar S., Khoshnejad M., Perales-Puchalt A., Choi H., Ho M., et al. Neutralization of hepatitis B virus by a novel DNA-encoded monoclonal antibody. Hum Vaccin Immunother. 2020;16:2156–2164. doi: 10.1080/21645515.2020.1763686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corti D., Benigni F., Shouval D. Viral envelope-specific antibodies in chronic hepatitis B virus infection. Curr Opin Virol. 2018;30:48–57. doi: 10.1016/j.coviro.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Chi S.W., Maeng C.Y., Kim S.J., Oh M.S., Ryu C.J., Kim S.J., et al. Broadly neutralizing anti-hepatitis B virus antibody reveals a complementarity determining region H3 lid-opening mechanism. Proc Natl Acad Sci United States America. 2007;104:9230–9235. doi: 10.1073/pnas.0701279104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang P., Yu M.Y., Venable R., Alter H.J., Shih J.W. Neutralization epitope responsible for the hepatitis B virus subtype-specific protection in chimpanzees. Proc Natl Acad Sci United States America. 2006;103:9214–9219. doi: 10.1073/pnas.0603316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong H.J., Ryu C.J., Hur H., Kim S., Oh H.K., Oh M.S., et al. In vivo neutralization of hepatitis B virus infection by an anti-preS1 humanized antibody in chimpanzees. Virology. 2004;318:134–141. doi: 10.1016/j.virol.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 52.Ogata N., Ostberg L., Ehrlich P.H., Wong D.C., Miller R.H., Purcell R.H. Markedly prolonged incubation period of hepatitis B in a chimpanzee passively immunized with a human monoclonal antibody to the a determinant of hepatitis B surface antigen. Proc Nat Acade Sci USA. 1993;90:3014–3018. doi: 10.1073/pnas.90.7.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim S.H., Shin Y.W., Hong K.W., Chang K.H., Ryoo K.H., Paik S.H., et al. Neutralization of hepatitis B virus (HBV) by human monoclonal antibody against HBV surface antigen (HBsAg) in chimpanzees. Antivir Res. 2008;79:188–191. doi: 10.1016/j.antiviral.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 54.Eren R., Ilan E., Nussbaum O., Lubin I., Terkieltaub D., Arazi Y., et al. Preclinical evaluation of two human anti-hepatitis B virus (HBV) monoclonal antibodies in the HBV-trimera mouse model and in HBV chronic carrier chimpanzees. Hepatology (Baltimore, Md) 2000;32:588–596. doi: 10.1053/jhep.2000.9632. [DOI] [PubMed] [Google Scholar]
- 55.Li D., He W., Liu X., Zheng S., Qi Y., Li H., et al. A potent human neutralizing antibody Fc-dependently reduces established HBV infections. Elife. 2017;6 doi: 10.7554/eLife.26738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang T.Y., Yuan Q., Zhao J.H., Zhang Y.L., Yuan L.Z., Lan Y., et al. Prolonged suppression of HBV in mice by a novel antibody that targets a unique epitope on hepatitis B surface antigen. Gut. 2016;65:658–671. doi: 10.1136/gutjnl-2014-308964. [DOI] [PubMed] [Google Scholar]
- 57.Wang Q., Michailidis E., Yu Y., Wang Z., Hurley A.M., Oren D.A., et al. A combination of human broadly neutralizing antibodies against hepatitis B virus HBsAg with distinct epitopes suppresses escape mutations. Cell Host Microbe. 2020;28:335–349.e336. doi: 10.1016/j.chom.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim J.-H., Kim H., Kim T.-H., Kim W., Jung J., Lee A. PS-077-Lenvervimab, a monoclonal antibody against HBsAg, can induce sustained HBsAg loss in a chronic hepatitis B mouse model. J Hepatol. 2019;70:e50. [Google Scholar]
- 59.Lee H.W., Park J.Y., Hong T., Park M.S., Ahn S.H. Efficacy of lenvervimab, a recombinant human immunoglobulin, in treatment of chronic hepatitis B virus infection. Clin Gastroenterol Hepatol. 2020;18:3043–3045.e3041. doi: 10.1016/j.cgh.2019.09.038. [DOI] [PubMed] [Google Scholar]
- 60.Beretta M., Mouquet H. Advances in human monoclonal antibody therapy for HBV infection. Curr Opin Virol. 2022;53 doi: 10.1016/j.coviro.2022.101205. [DOI] [PubMed] [Google Scholar]
- 61.EMA Xolair product information. [cited; Available from: https://www.ema.europa.eu/en/documents/product-information/xolair-epar-product-information_en.pdf.
- 62.EMA MabThera product information. [cited; Available from: https://www.ema.europa.eu/en/documents/product-information/mabthera-epar-product-information_en.pdf.
- 63.Xiao H., Wu K., Liang X., Li R., Lai K.P. Clinical efficacy and safety of eculizumab for treating myasthenia gravis. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.715036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Razonable R.R., Pawlowski C., O'Horo J.C., Arndt L.L., Arndt R., Bierle D.M., et al. Casirivimab-Imdevimab treatment is associated with reduced rates of hospitalization among high-risk patients with mild to moderate coronavirus disease-19. EClinicalMedicine. 2021;40 doi: 10.1016/j.eclinm.2021.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Casirivimab . National Library of Medicine (US); Bethesda (MD): 2006. Drugs and lactation database (LactMed) [Google Scholar]
- 66.Nair A.B., Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schilling R., Ijaz S., Davidoff M., Lee J.Y., Locarnini S., Williams R., et al. Endocytosis of hepatitis B immune globulin into hepatocytes inhibits the secretion of hepatitis B virus surface antigen and virions. J Virol. 2003;77:8882–8892. doi: 10.1128/JVI.77.16.8882-8892.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neumann A.U., Phillips S., Levine I., Ijaz S., Dahari H., Eren R., et al. Novel mechanism of antibodies to hepatitis B virus in blocking viral particle release from cells. Hepatology (Baltimore, Md) 2010;52:875–885. doi: 10.1002/hep.23778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marasco W.A., Sui J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol. 2007;25:1421–1434. doi: 10.1038/nbt1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Imai K., Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- 71.Halper-Stromberg A., Lu C.L., Klein F., Horwitz J.A., Bournazos S., Nogueira L., et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell. 2014;158:989–999. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wen Y.M., Mu L., Shi Y. Immunoregulatory functions of immune complexes in vaccine and therapy. EMBO Mol Med. 2016;8:1120–1133. doi: 10.15252/emmm.201606593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Celis E., Chang T.W. Antibodies to hepatitis B surface antigen potentiate the response of human T lymphocyte clones to the same antigen. Science. 1984;224:297–299. doi: 10.1126/science.6231724. [DOI] [PubMed] [Google Scholar]
- 74.Allweiss L., Volz T., Giersch K., Kah J., Raffa G., Petersen J., et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut. 2018;67:542–552. doi: 10.1136/gutjnl-2016-312162. [DOI] [PubMed] [Google Scholar]
- 75.Volz T., Allweiss L., Ben M.M., Warlich M., Lohse A.W., Pollok J.M., et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol. 2013;58:861–867. doi: 10.1016/j.jhep.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 76.Bachmann M.F., Hunziker L., Zinkernagel R.M., Storni T., Kopf M. Maintenance of memory CTL responses by T helper cells and CD40-CD40 ligand: antibodies provide the key. Eur J Immunol. 2004;34:317–326. doi: 10.1002/eji.200324717. [DOI] [PubMed] [Google Scholar]
- 77.Paul S., Dickstein A., Saxena A., Terrin N., Viveiros K., Balk E.M., et al. Role of surface antibody in hepatitis B reactivation in patients with resolved infection and hematologic malignancy: a meta-analysis. Hepatology (Baltimore, Md) 2017;66:379–388. doi: 10.1002/hep.29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Whitacre D.C., Peters C.J., Sureau C., Nio K., Li F., Su L., et al. Designing a therapeutic hepatitis B vaccine to circumvent immune tolerance. Hum Vaccin Immunother. 2020;16:251–268. doi: 10.1080/21645515.2019.1689745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study is available upon request. Please contact the corresponding author.