

Abstract
Background and Aims.
Visceral hypersensitivity and low grade mucosal inflammation are frequently observed in a subpopulation of irritable bowel syndrome (IBS) patients. The responsible mechanism is unclear. Resolvins are a novel class of anti-inflammatory lipid mediators that regulate resolution of inflammation and pain. We hypothesize that resolvin D1 (RvD1) synthesis is reduced in IBS-D colonic mucosa and contribute to the development of visceral hypersensitivity.
Methods.
We used ELISA and qPCR to quantify the levels of RvD1 synthesis and the gene expression of LOX5 and LOX12 in colonic biopsy samples from healthy individuals (HC) or IBS-D patients. To evaluate the role of gut bacteria in regulating RvD1 synthesis, we colonized germ-free mice with different strains of bacteria, fecal microbiota from HC individuals or IBS-D patients. To evaluate the role of tuft cells in RvD1 synthesis, we examined the effect of fecal supernatant of IBS-D or HC subjects on human colonoids as well as colonoids from mice in which Chat-cre recombinase vector was used to knock-in diphtheria toxin sensitive receptor (DTRf/f) or knock-out the expression of Tlr4f/f or MyD88f/f.
Results.
We report that colonic biopsy samples from IBS-D patients generated significantly lower level of RvD1 and LOX5 mRNA. The conventionalization of germ-free mice with microbiota from IBS-D patients or gram-negative bacteria inhibited RvD1 biosynthesis and caused visceral hypersensitivity and mucosal inflammation. Colonic organoid studies demonstrate that Lps downregulated Lox5 mRNA expression and synthesis of RvD1 via Tlr4-MyD88 receptor signaling pathway in colonic tuft cells.
Conclusions.
Our findings indicate that RvD1 is generated in colonic tuft cells to regulate gut sensitivity to mechanical stimulation. Colonic commensal bacterial composition regulates the synthesis of RvD1 in colonic mucosa which is reduced in IBS-D patients. This appears to be mediated by elevated fecal lipopolysaccharide secondary to gram-negative gut dysbiosis.
Keywords: Endotoxin, brush cells, lypoxygenase 5, visceral hypersensitivity, inflammation'
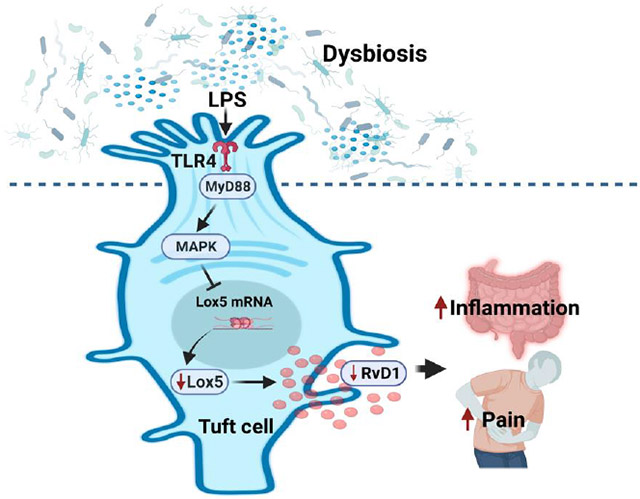
Graphical Abstract
Introduction
Irritable Bowel Syndrome (IBS) is a common disorder affecting 20% of the US population. Although its pathophysiology is unclear, visceral hypersensitivity is frequently observed in IBS patients.1-4 This perceptual abnormality may lead to sensations of pain, gas and bloating. The cause of visceral hypersensitivity is unknown, but several mechanisms have been postulated and includes triggering events like inflammation, psychological or environmental stress and post-injury sensation that may alter peripheral visceral afferent function and/or central processing of afferent information.2,4-6
Recently, low grade colon inflammation has been widely reported in IBS and is common among IBS-D patients.6,7 Of the inflammatory mediators studies, inflammation may trigger the synthesis of resolvins, a novel class of endogenous anti-inflammatory lipid mediators that participate in the resolution of inflammation and pain.8-9 Our recent preclinical studies demonstrate that the activation of mast cells releases mediators that sensitize DRG neurons; it is conceivable that these actions are offset by resolvin D1 (RvD1) which normalize the neuronal excitability.10-11 An imbalance between proinflammatory factors and pro-resolution mediators may contribute to mucosal inflammation and pain in IBS.
Currently the colonic mucosa levels of resolvins in IBS are unknown. Chiang et al12 report that germ-free (GF) mice exhibit abnormally high levels of resolvins in the colonic tissue when compared to wild-type controls suggesting gut microbiota may modulate colonic levels of resolvins. Recent clinical studies suggest that gut dysbiosis is common among IBS patients.13 Hence, it is conceivable that resolvins are reduced in IBS-D colonic mucosa secondary to gut dysbiosis. This failure to counteract the increased primary afferent excitability during inflammation may contribute to VH development.
To investigate this possibility, we obtained biopsy samples from the colon of IBS-D patients and healthy controls (HC) and compared the level of colonic mucosal RvD1. We then examined whether the level of colonic RvD1 can be selectively modulated by gut microbiota and investigated the mechanism by which bacteria modulates RvD1 level. Lastly, we identified the source of colonic RvD1 which is regulated by gut microbiota.
Methods
All human and animal studies were approved by the University of Michigan IRB and IACUC, respectively, in accordance with NIH guidelines.
Patients
Sixteen patients with IBS-D (9 females and 7 males), aged from 28 to 60 years, were recruited from the University of Michigan, Division of Gastroenterology and Hepatology outpatient clinic and primary care clinics. All patients with IBS-D met the Rome IV criteria and were symptomatic at the time of the study. Patients had to be free of other diseases, such as diabetes, celiac disease, and cardiovascular disorders. Biopsies of colonic tissue excluded microscopic colitis. None of study patients with IBS-D used nonsteroidal anti-inflammatory drugs, corticosteroids, histamine antagonists, or mast cell stabilizers in the last 6 months before the study. Sixteen HC (8 females and 8 males, aged 36 to 65 years) served as controls. Consents were obtained from all study subjects and the study was approved by the University of Michigan Human Research Protection Program. Healthy controls were asymptomatic subjects undergoing colonoscopy for colorectal cancer screening. All subjects had a colonoscopy, and 6 mucosal biopsies were obtained from the descending colon of each subject. One biopsy was used for immunohistochemistry to exclude microscopic colitis and 5 biopsies were used for RvD1 extraction.
Stool samples
Stool samples were obtained from five HC (two males and three females; 28 to 60 years) and eight patients with IBS-D (three males and five females; 35 to 65 years). The HC had no history of gastrointestinal diseases. The patients were diagnosed with IBS-D according to Rome IV criteria with Bristol stool form scale (≥6), and more than three bowel movements per day, and had symptoms for at least 2 years. Diarrhea was the dominant gastrointestinal symptom in the IBS-D patients included in this study. None of the patients or controls used psychotropic medications, antibiotics, or probiotics for at least 3 months before fecal sample collection. Stool samples were transported anaerobically to the laboratory and frozen at −80°C.
Fecal supernatant extraction
Fecal samples from IBS-D patients and healthy controls were diluted (1 g fecal sample/5 ml PBS), homogenized on ice, and centrifuged (10,000 g, 10 minutes, 4°C). Pellets were discarded, and supernatants were recovered. Bacteria were removed by 0.22-μm syringe filters. Supernatants were kept at −80°C.
Conventionalization of germ-free mice
Diluted stool or bacterial suspension in PBS was gavaged once into 8-12-week-old germ-free mice of either gender. Lactobacillus rhamnosus GG (ATCC 53103) and Proteus mirabilis Hauser (ATCC 43071) (American Type Culture Collection (ATCC), Manassas, VA) were used. Each mouse was gavaged with 0.15 mL of culture grown aerobically overnight at 37°C (~109 CFU grown in Luria-Bertani broth without antibiotics). After 2 weeks, mice visceromotor response to colorectal distention was assessed. Mice were sacrificed thereafter, and tissue samples were collected. No gender effect was observed for any of the independent variables studied.
Collection of mucosal specimens and assays of supernatants
For details regarding ELISA measurements, please see Supplementary Methods.
Animals
Specific pathogen-free C57BL/6 (WT) mice were purchased from Charles Rivers laboratories (Wilmington, MA, USA), Tlr4 KO (C3H/HeJ (TLR4 Lps-d; stock number 000659), Tlr2 KO (B6.129-Tlr2tm1Kir/J, stock number 004650), Tlr4f/f (stock number 024872), MyD88f/f (stock number 008888), ALox12 KO (B6.129S2-Alox15tm1Fun/J; stock number 02778), ALox5 KO (B6.129S2-Alox5tm1Fun/J; stock number 004155), ROSA26iDTR (iDTR or C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J; stock number 007900), Chat-cre (B6;129S6-Chattm2(cre)Lowl/J; stock number 006410), EGFPf/f (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J; stock number 007676) mice were purchased from the Jackson Laboratories (Bar Harbor, ME, USA) and housed in the Animal Facility at the University of Michigan. Germ-free (C57BL/6 background) mice were obtained from the Germ-Free Animal Facility at the University of Michigan. Mice were maintained in a 12-h light/dark cycle and allowed free access to food and water. Germ-free status was checked weekly by aerobic and anaerobic culture. In all animal strains, 8–12-week-old female and male mice were used for the experiments.
Chemicals
Lyposalysacharides (Lps) from E Coli and diphtheria toxin were purchased from Sigma-Aldrich (St Luis, MO, USA), LTA (InvivoGen, San Diego, CA, USA). RVD1, resolvin E1, NFkB inhibitory peptide (SN50) and MK886 were purchased from Cayman chemical (Ann Arbor MI, USA), WRW4 and U0126 from Tocris (Minneapolis, MN, USA). ELISA for RvD1 (#500380; Cayman Chemical, Ann Arbor, MI, USA), IL6 (#583371, Cayman Chemical), TNFa (#500850, Cayman Chemical), and IL1b (#DY401, Tocris) were performed according to the instructions provided by the manufacturer. siRNAs for NFKB p65 (sc-29411), Erk1 (sc-29038), and Erk2 (sc-35336) and a siRNA-A unrelated to these genes to be used as the control (sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and diluted in Opti-MEM (Thermo Fisher Scientific, Waltham, MA, USA).
VMR to Colorectal Distension and Recording
The visceromotor responses (VMR) were recorded by quantifying reflex contractions of the abdominal musculature induced by colorectal distention (CRD). Please see Supplementary Methods.
Lps removal
For details regarding endotoxin (Lps) removal, please see Supplementary Methods.
Histology and immunohistochemistry
For details regarding immunohistochemistry, please see Supplementary Methods.
Colon organoid preparation
Human colonic biopsies and mouse colonic tissue was washed with PBS containing 1% penicillin-streptomycin (Thermo Fisher Scientific). After 3-5 washes, tissue was incubated in cold PBS containing 20 mM EDTA with DTT (Sigma-Aldrich, St Louis, MO, USA) for 5 minutes to remove mucus and in cold PBS containing 20 mM EDTA for 30 minutes for further digestion. Crypts were released by rhythmic shaking in PBS containing 0.1% BSA for 1–2 minutes. Isolated crypts were centrifuged at 300 g for 2 minutes. Colonic crypts were collected and plated in 24-well plates in a 5% CO2 humidified incubator at 37°C in DMEM supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Transfection with silencing RNA in colonoid cultures
For details regarding transfection and silencing RNA, please see Supplementary Methods.
Reverse transcriptase–PCR studies
For details regarding primers and reverse transcription quantitative real-time polymerase chain reaction (qPCR) analysis, please see Supplementary Methods and Supplementary Table 1.
Results
Synthesis of RvD1 in colonic mucosa of IBS-D patients
As shown in Fig 1A, RvD1 is derived from docosahaxenoic acid (DHA;28:6n-3) via 17-hydroxydocosahexaenoic acid (17-HpDHA).8,9,14 This is mediated by LOX12 which converts DHA to 17-HpDHA, and LOX5 which transforms 17-HpDHA to RvD1. RvD1 acts via two GPCRs, the formyl peptide receptor 2 (FPR2) and or orphan receptor GPR32 on human leukocytes.8,15 We measured the levels of RvD1 in the biopsy samples from IBS-D patients and HC using ELISA. Our data show that biopsy samples from IBS-D patients contained significantly lower levels of RvD1 when compared to HC (mean 3.5±0.55 vs 6.05±0.88 pg/ml respectively n=16 each group, *P<0.01 Fig 1B). Furthermore, we investigated the levels of key molecules involved in RvD1 synthesis and signaling using qPCR (Fig 1). Our data show that colonic biopsy samples from IBS-D patients expressed significantly lower levels of LOX5 mRNA when compared to HC but not LOX12, FPR2 or GPR32 mRNA (Fig 1 C, D, Supplementary figure 1). These data suggest that IBS-D patients exhibit impaired synthesis of RvD1 in the colonic mucosa.
Fig 1.

Role of RvD1 in colon. (A) Schematic pathway of RvD1 synthesis. (B) ELISA data show the level of RvD1 in colonic biopsy samples from healthy control individuals (HC, n=16) and IBS-D patients (n=16). *, P<0.01. qPCR data showing relative expression of LOX5 mRNA (C), LOX12 mRNA (D) in colonic biopsy samples from HC or IBS-D patients. Data are expressed as means with SEM. *, P<0.05 from HC; unpaired t-test. (E) Representative recordings of VMR to colorectal distention after administration of fecal supernatant from HC (HC-FS, left panel) or fecal supernatant from IBS-D patient (IBS-FS, middle panel), and 30 minutes after administration of RvD1 (100ng/mouse ip, right panel). (F) Summary graph of VMR to colorectal distention shows that intracolonic administration of IBS-FS (n = 13) cause visceral hypersensitivity when compared to VMR generated in response to intracolonic administration of HC-FS (n=8). The enhanced VMR was abolished by i.p. administration of RvD1 (n = 7) but not RvE1 (1000 ng/mouse, n=6). The effect of RvD1 was inhibited by FPR2 receptor blocking peptide WRW4 (n = 6). (G) qPCR data show relative expression of mRNA for IL1b, IL-6, TNFα in colonic tissue from mice pretreated with HC-FS, IBS-FS, IBS-FS+RvD1 and IBS-FS+RvD1+WRW4. Each dot represents an individual mouse. Results are expressed as mean ± SEM; *, P<0.05 ANOVA with Bonferoni post-hock test. (H) ELISA data show relative expression of IL1b, IL-6, TNFα in supernatants from colonic tissue pretreated with HC-FS, IBS-FS, IBS-FS+RvD1 and IBS-FS+RvD1+WRW4. Each dot represents an individual mouse. Results are expressed as mean ± SEM; *, P<0.05 ANOVA with Bonferoni post-hock test.
RvD1 modulates mucosal inflammation and pain response in IBS-D.
It has been reported that resolvins exert analgesic actions and have anti-inflammatory properties in pain models11,14 raising the possibility that reduced level of colonic RvD1 may contribute to IBS symptoms including allodynia and mucosal inflammation. We next evaluated the ability of RvD1 in the modulation of pain and inflammation using an IBS mouse model in which visceral hypersensitivity and intestinal inflammation were induced by intracolonic administration of fecal supernatant from IBS-D patient (IBS-FS).16 Our pain behavior studies show that administration of IBS-FS caused hypersensitivity to colonic distention when compared to VMR to colonic distention in mice that received HC-FS (Fig 1 E, F). Intraperitoneal administration of RvD1 (100ng/mouse) but not RvE1 (1000ng/mouse) abolished visceral hypersensitivity, suggesting specific analgesic action of RvD1 (Fig 1 E and F). We further show that the analgesic action of RvD1 was blocked using WRW4, a FPR2 receptor antagonist,17 suggesting that the action RvD1 is mediated via FPR2 receptor (Fig 1F). Our qPCR data also show that i.p. administration of RvD1 significantly reduced the levels of mRNAs and the release of proinflammatory mediators in the colonic tissue (TNFα, IL1β, IL6) induced by intracolonic administration of IBS-FS (Fig 1 G H). The anti-inflammatory effect of RvD1 was blocked using WRW4 (Fig 1 G H).
Gut microbiota modulates the synthesis of RvD1 in colonic mucosa
In separate studies we show that colonic tissue from germ-free (GF) mice contained abnormally high levels of RvD1 when compared to wild-type (WT) mice (10.84±26 vs 2.6±.86 pg/ml/μg respectively, **,p<0.01, Fig 2 A and B). Furthermore, pain behavior studies show that GF mice were resistant to IBS-FS which induced visceral hypersensitivity in WT mice suggesting that gut microbiota may regulate visceral hypersensitivity via modulation of the level of RvD1 in the gut (Fig 2 C). To address this hypothesis, we conventionalized germ-free mice with (i)fecal material from wild-type (WTm) mice or (ii)monocolonized with Proteus mirabililis (PMm, a strain of gram-negative bacteria that is increased in IBS patient fecal samples);6,13 (ii)with lactobacillus rhamnosus GG (LGGm, a strain of gram-positive bacteria that show analgesic properties).18 Conventionalization of germ-free mice with the fecal material from wild-type mouse (WTm) or Proteus mirabilis (PMm) significantly reduced the level of RvD1 in the colonic tissue (Fig 2B) and regained the exaggerated pain response to intracolonic infusion of IBS-FS (Figure 2C). Interestingly, conventionalization with the Lactobacillus rhamnosus (LGGm) did not reduce the level of RvD1 when compared to the germ-free mice (Fig 2B) and the mice did not develop visceral hypersensitivity in response to intracolonic administration of IBS-FS (Fig 2C). These observations support the hypothesis that RvD1 in the colonic tissue modulates visceral pain.
Fig 2.

Intestinal bacteria modulate the level of colonic RvD1 (A) Schematic overview of experimental setup to assess the effect of gut bacteria in mice. (B) ELISA data show the levels of RvD1 in colonic tissue from wild-type mice (WT), germ-free mice (GF), GF mice conventionalized with fecal microbiota from WT mice (GF+WTm), GF mice conventionalized with Proteus mirablis (GF+PMm) or Lactobacillus rhamnosus GG bacteria (GF+LGGm). Each dot represents an individual mouse. *P<0.05, ** P<0.01 from WT (C) VMR to colorectal distention in mice shows that intracolonic administration of HC-FS (n=6) or IBS-FS (n=6) did not induce visceral hypersensitivity in germ-free mice (GF+HC-FS or GF+IBS-FS). Intracolonic administration of IBS-FS supernatant to germ-free mice that were conventionalized with fecal microbiota from wild-type mice (GF-WTm+IBS-FS, n=7) or PM bacteria (GF-PMm+IBS-FS, n=6) produced visceral hypersensitivity to mechanical distention, but not in germ-free mice conventionalized with LGG bacteria (GF-LGGm+IBS-FS, n=9). Results are expressed as mean ± SEM, *, P<0.05 from GF+IBS-FS. (D). Representative recordings of VMR to colorectal distention in GF mice conventionalized with fecal microbiota from HC individuals (HCm) or IBS-D patients (IBSm). (E) Summary graph of VMR to colorectal distention in germ-free mice conventionalized with fecal microbiota from healthy individuals (HCm, n=8) or IBS-D patients (IBSm, n=8). Results are expressed as mean ± SEM, ***, P<0.001 from GF-HCm. (F) ELISA data showing the level of RvD1 in colonic tissue from GF mice conventionalized with fecal microbiota from HC or IBS-D patient. Results are expressed as mean ± SEM, *, P<0.05. (G) qPCR data of Lox5 gene expression showing conventionalization of GF mice with the fecal material from healthy subjects (GF+HCm, n=4) or IBS-D patients (GF+IBSm, n=4) inhibited the level of Lox5 mRNA in the colonic tissues when compared to germ-free mice (n=4). **P<0.01 or ***, P<0.001 from GF. Results are expressed as mean ± SEM, ANOVA with Bonferoni post-hock test. (H) qPCR data showing the levels of mRNA for IL1b. IL6, TNFα in germ-free, germ-free mice conventionalized with microbiota from HC subjects (GF+HCm) or IBS-D patients (GF+IBSm). ***, P<0.001. ANOVA with Bonferoni post-hock test.
To evaluate the role of commensal bacteria from human on RvD1 synthesis, we colonized germ-free mice with the fecal material from healthy control individuals (HCm) or IBS-D patients with diarrhea (IBSm), and measured visceral sensitivity, inflammation markers and the level of RvD1 synthesis in the colonic tissue. VMR to colorectal distention study demonstrated that conventionalization of germ-free mice with the fecal material from IBS-D patients (n=8, IBSm material from 4 different patients) generated heightened VMR to colorectal distention when compared to the pain response to colorectal distention generated in germ-free mice inoculated with fecal material from HC (n=6, HCm fecal material from 3 different subjects, P<0.001) (Fig 2 D, E). This was associated with significantly lower level of RvD1 (Fig 2 F) and reduced gene expression of Lox5 mRNA (Fig 2 G) in the colonic tissue of germ-free mice conventionalized IBS-D fecal material. In addition, our qPCR data show significantly higher levels of mRNA for IL1β, IL6 and TNFα in the colonic tissue of germ-free mice inoculated with fecal material from IBS-D patients when compared germ-free mice inoculated with fecal material from HC subjects (Fig 2 H). These data demonstrate that the gut microbiota regulates colonic level of RvD1 and that dysregulation in the production of this mediator may lead to visceral hypersensitivity and mucosal inflammation.
Synthesis of RvD1 in the colonic mucosa is Tlr4-Lox5 pathway dependent
The Lox5 pathway is the only known biosynthetic route for the formation of RvD1 (Fig 1A).19,20 We compared the level of RvD1 in the colonic tissue from WT(C57BL/6), Lox12 KO and Lox5 KO mice and found a low level of RvD1 in the colonic tissue of Lox12 KO and Lox5 KO mice (Fig 3 A). Furthermore, we show that pretreatment of WT mice with MK886 (1mg/kg), an inhibitor of 5-lipoxygenase-activating protein (FLAP) markedly reduced the levels of the RvD1 in the colonic tissue when compared to sham/controls (Fig 3 A). Importantly, pain behavior studies show that Lox12 KO and Lox5 KO mice or WT mice pretreated with MK886 exhibited visceral hypersensitivity to colorectal distention (Fig 3 B).
Fig.3.

Modulation of RvD1 synthesis via Tlr4-Lox5 pathway in colonic mucosa. (A) ELISA data show the level of RvD1 in the colonic tissue from wild-type (WT, n=8), Lox12 KO (Lox 12, n=4), Lox5 KO (Lox 5, n=4), WT mice pretreated with MK886 (1 mg/kg, n=6), Tlr2 KO (n=4) and Tlr4 KO mice (n=13). *, P<0.05 ANOVA with Bonferoni post-hock test. (B) VMR to colorectal distention data show that Lox12 KO (n=4), Lox5 KO (n=4) and wild-type pretreated with MK886 mice (n=6) exhibited hypersensitivity when compared to wild-type (n=6). Results are expressed as mean ± SEM, ***, P<0.001 and **, P<0.01 from WT. (C) VMR to colorectal distention shows that intracolonic administration of IBS-FS failed to generate visceral hypersensitivity in Tlr4 KO mice (n=5), when compared to wild-type (n=8). Results are expressed as mean ± SEM, ***, P<0.001; **, P<0.01 and *, P<0.05 from WT+HC-FS.
Next, we investigated the mechanisms by which gut microbiota modulate the production of RvD1 in the colon. We tested the hypothesis that microbial product(s) act via TLRs to regulate the expression of Lox5 mRNA levels and RvD1 production. Under basal conditions, Tlr4 KO mice generated significantly higher levels of RvD1 when compared to WT mice or Tlr2 KO (Fig 3 A) suggesting that microbial products such as Lps act via Tlr4 receptor to inhibit the synthesis of RvD1. Interestingly, Tlr4 KO mice but not Tlr2 KO mice were resistant to development of visceral hypersensitivity induced by intracolonic administration of IBS-FS (Fig 3 C).
It has been suggested that macrophages and other immunocytes are major source of specialized pro-resolving mediators (SPM).21 When exposed to pathogenic bacteria, human macrophages increase SPM synthesis.22 On the other hand colonic tissue from GF mice which exhibit low numbers of macrophages and other immune cells,23 synthesize high levels of RvD1 when compared to WT controls (Fig 2B). This suggests that immunocytes are not the major source of RvD1 in the gut. To demonstrate that gut epithelial cells produce RvD1, we show that isolated colonoids devoid of immunocytes synthesize high level of RvD1 which was inhibited by IBS-FS (Fig 4A). The inhibitory effect of IBS-FS was abolished by pretreating the IBS-FS with endotoxin scavenger to remove Lps (Fig 4A). Furthermore, we show that RvD1 levels were dose-dependently inhibited by gram-negative bacteria-derived endotoxin Lps (Tlr4 receptor agonist) but not by the gram-positive bacteria-derived lipoteichoic acid (LTA, Tlr2 receptor agonist) (Fig 4A). Importantly, our data show that pretreatment of human colonoids generated from colonic biopsy samples from healthy controls with IBS-FS for 24 h significantly inhibited RvD1 and LOX5 mRNA expression but LOX12 or DCLK mRNA expression (Supplementary figure 2). Furthermore, we show that IBS-FS inhibited RvD1 synthesis in colonoids from wild-type and Tlr2 KO mice but not from Tlr4 KO mice (Fig 4B). These findings suggest that gram-negative bacteria act by Tlr4 receptor to inhibit RvD1 synthesis in the colonic epithelial cells.
Fig 4.

Commensal bacteria derived Lps modulates RvD1 synthesis in colonic epithelial cells. (A) ELISA data show that the inhibitory action of IBS-FS on RvD1 biosynthesis in colonoids was abolished by removal of endotoxin from the IBS fecal supernatant (IBS-Lps). Administration of Lps but not LTA dose dependency inhibited colonic RvD1 levels. *, P<0.05 or **, P<0.01. Results are expressed as mean ± SEM, ANOVA with Bonferoni post-hock test. (B) Summary bar graph shows that IBS-FS (10 μl/ml/24h) inhibited RvD1 synthesis in colonoids from Tlr2 KO and WT mice but not from Tlr4 KO mice when compared to pretreatment with HC-FS (10 μl/ml/24h). *, P<0.05 or ***, P<0.001 compared to HC-FS. Results are expressed as mean ± SEM, ANOVA with Bonferoni post-hock test. (C-D) ELISA measurement of RvD1 synthesis in colonoids shows that the inhibitory effect of IBS-FS (10 μgr/ml/24h) was abolished with U0126 (10 μM) or silencing Erk1/2 using siRNA but not with SN50 (100 μgr/ml) or NFκB p65 siRNA. Results are expressed as mean ± SEM, ANOVA with Bonferoni post-hock test.
Downstream events in the activation of Tlr4 receptor involve the activation of NF-κB and/or MAPK pathways.24 Our data show that inhibition of Mek 1/2 kinase activity (U0126, 10 μM) or silencing Erk1 and Erk2 genes using siRNAs abolished the inhibitory effect of IBS-FS on RvD1 synthesis in colonoids (Figure 4 C, D). qPCR data confirmed successful silencing of Erk1 and Erk2 (Supplementary figure 3). On the other hand, inhibition of NF-κB using SN50 peptide25 or silencing NF-κB failed to affect the inhibitory actions of IBS-FS. qPCR studies confirmed successful silencing of NF-κB. (Supplementary Figure 2). These findings suggest that Lps in IBS-FS acts via MAPK pathways independent of NF-κB activation to modulate RvD1 synthesis.
Colonic tuft cells generate RvD1
The colonic mucosa epithelial cell layer consists of at least five different cell linages. We next investigated whether a specific epithelial cell type is responsible for RvD1 generation in the colonic mucosa. Analysis of recently published single-cell RNA-sequencing data26 and our immunohistochemical data (Supplementary figure 4) show that colonic mucosa tuft cells (also called intestinal brush cells) exclusively express Lox5, suggesting that these cells may be responsible for RvD1 synthesis in colonic mucosa. In addition to Lox5, tuft cells express a number of specific endogenous markers including choline acetyltransferase (Chat) and doublecortin-like kinase 1 (Dclk).26-31 To explore the hypothesis that tuft cells synthesize RvD1, we generated mouse in which Chat-cre recombinase vector was used to activate simian diphtheria toxin receptor (DTR; from simian Hbegf) inserted into the Gt(ROSA)26S locus in which Cre-mediated excision of a STOP cassette renders cells sensitive to diphtheria toxin (DTX)32 or expressing enhanced green fluorescent protein (EGFP) under the control of the Chat-cre promoter. We first demonstrate that colonic tissue and colonoids from EGFP-Chat-cre mice expressed GFP positive cells, indicating successful activation of the insert and showed a predicted distribution profile consistent with the distribution of tuft cells (Fig 5 A).33 Our data also show that that Chat immunoreactivity completely overlapped with Dclk1imunoreactivity in epithelial cells (Supplementary figure , 5). Next, we studied the role of Chat expressing mucosal epithelial cells in RvD1 synthesis in colonoids from DTR-Chat-cre mouse. Pretreatment of colonoids with diphtheria toxin (100 ng/ml/24h) significantly reduced the level of expression of Chat and Lox5 mRNA (75% and 55% respectively) (Fig 5B) and reduced RvD1 synthesis by >50% when compared to sham-treated colonoids (Fig 5 C). To demonstrate that Tlr4 receptor on tuft cells modulates RvD1 synthesis we crossed Tlr4 floxed mouse (Tlr4f/f) or MyD88 (MyD88f/f) with Chat-cre mouse. Our data show that targeted ablation of Tlr4 or MyD88 genes in tuft cells abolished the inhibitory effect of IBS-FS on RvD1 synthesis in colonoids when compared to treatment with HC-FS (Figure 5 D). These findings indicate that tuft cells constitutively express Lox5 and generate RvD1 in the colonic epithelium.
Fig. 5.

Tuft cells biosynthesize RvD1 in colonic mucosa. (A) The micrograph showing the distribution of EGFP positive cells (arrows) in the colonic tissue from Chat-cre-EGFP mouse. (B) qPCR data of Chat and Lox5 mRNA in colonoids with sham (DTR) or DTx (100ng/ml 24h) pretreatment. Results are expressed as mean ± SEM; ***, P<0.001 from DTR. (C) Pretreatment of colonoids from DTRf/f-Chat-cre mice with DTx (100ng/ml 24h) significantly reduced the level of RvD1 biosynthesis. *, P<0.05 compared to DTR (D) ELISA demonstration that targeted gene knock-out of Tlr4 (Tlr4f/f-Chat-cre) and MyD88 (MyD88f/f-Chat-cre) abolished the inhibitory effect of IBS-FS on RvD1 biosynthesis observed in colonoids from WT mice. Results are expressed as mean ± SEM; **, P<0.01 from HC-FS.
Several recent studies show that environmental factors can modulate the expansion of tuft cell numbers in the GI system.31,33 Our qPCR and immunostaining data show that the level of expression of Dclk1 and IL25 mRNA or the number of Dclk1 immunoreactive cells in the colonic mucosa was significantly higher in germ-free mice when compared to germ-free mice conventionalized with single strand bacteria or microbiota from IBS-D patients or HC individuals (Fig 6 A, C). There was however no difference in number of the cells and the level of Dclk1 and IL25 mRNA among the conventionalized mice with different bacteria (Fig 6 A, B). Additionally, our qPCR data show that the mRNA levels of DCLK1, IL25 and TRPM5 (another biomarker of Tuft cell)33,34 in the colonic biopsies from healthy control subjects and IBS patients were not significantly different (Supplementary figure 6). The increased number of Tuft cells in germ-free mice corelate with the abnormally high levels of RvD1 in the colonic mucosa. On the other hand, when germ-free mice conventionalized with different microbial contents (gram-positive (LGGm), gram negative bacteria (PMm), WTm, HCm, IBSm) they showed similar numbers of Tuft cell markers but different levels of RvD1 (Figure 2 B, E). Hence it appears that gut microbiota modulates colonic RvD1 by at least two distinct mechanisms: it can regulate the number of Tuft cells such as in germ-free mice or alternatively gut microbiota can directly modulate Tuft cell synthesis of RvD1 as in gut dysbiosis.
Fig 6.

Microbial composition does not modulate the number of the tuft cells. (A) qPCR data showing the level of Dclk1 mRNA in colonic tissue from GF mice or GF conventionalized with Lactobacillus rhamnosus GG (+LGGm), Proteus mlrabllls (+PMm) or fecal microbiota from WT mice (+WTm) or HC subjects (+HCm) or IBS-D patients (+IBSm). Conventionalization with microbial agents inhibited expression of Dclk1 gene expression. **, P<0.01 from WT. (B) qPCR data showing the level of IL25 mRNA in colonic tissue from GF mice or GF conventionalized with fecal microbiota from HC subjects (+HCm) or IBS-D patients (+IBSm). Conventionalization with microbial agents inhibited expression of Dclk1 gene expression. *, P<0.05. (C) Summary bar graph showing the number of Dclk1-immunoreactive cells in cross sections of distal colon from GF mice and GF mice conventionalized with fecal microbiota from HC subjects (GF+HCm) or IBS-D patients (GF+IBSm). *, P<0.05 from GF. Results are expressed as mean ± SEM, ANOVA with Bonferoni post-hock test. (D) Immunohistochemical demonstration of Dclk1 immunoreactivities (red) and Dapi staining (blue) in cross sections of distal colonic mucosa from GF mouse, GF mouse conventionalized with fecal microbiota from HC subjects (GF+HCm) or IBS-D patients (GF+IBSm) at low power magnification (x4, left colon) and high-power insets (right colon). Bar = 600μm.
Discussion
This paper contains several groundbreaking concepts in the physiology of pro-resolving lipid mediators. Current leading concept is that pro-resolving mediators are generated by immune cells (predominantly macrophages) in response to inflammatory insults.8-9 However, our study for the first time shows that RvD1 is generated in colonic tuft cells and plays an important role maintaining homeostasis of the colon. Until now, tuft cells in the small intestine have been shown to mediate type 2 immunity in response to parasite infection.33, 35-37 Here we uncovered a new unique role for RvD1 synthesis in colonic tuft cell to regulate gut sensitivity to mechanical stimulation. We show that colonic commensal bacterial composition regulates the level of RvD1 synthesis in colonic mucosa and this in turn regulates visceral sensitivity and inflammation. For the first time, we show that colonic biopsy samples from IBS-D patients display a low level of RvD1 when compared to healthy controls. Hence it appears that in response to low grade inflammation induced by gut dysbiosis, colonic RvD1 synthesis is inhibited in IBS-D patients resulting in visceral hypersensitivity. This may contribute to the exaggerated pain response to mechanical stimulation commonly observed in IBS patients.
Resolvins have been shown to dampen inflammatory and postoperative pain.38-40 More recent studies show that resolvin D2 normalize histamine/IBS supernatant-induced sensitization of TRPV1 in dorsal root ganglion neurons. This effect is mediated by G protein receptor 18 (GPR 18).41 In our current study we show that in germ-free mice the colonic RvD1 is >400% higher compared to wild-type. These mice are resistant to developing visceral hypersensitivity evoked by intracolonic infusion of IBS-FS. One may argue that these two events may not be causally related. However, we show that by introducing different strains or groups of gut microbiota, we were able to generate different levels of colonic RvD1 levels (Fig 2). For example, conventionalization of germ-free mice with fecal materials from wild-type mice or from healthy individuals decreased colonic RvD1, whereas conventionalization with Lactobacillus strains had no effects on colonic RvD1 levels when compared to germ-free. On the other hand, conventionalization with fecal material from IBS-D patients markedly reduced colonic RvD1. This provides us with an excellent model to demonstrate a good functional relationship between levels of RvD1 and their abilities to combat VH induced by intracolonic infusion of IBS-D FS (Fig 2).
Until now the source of resolvins in the gut is believed to originate from immunocytes such as the macrophages. When exposed to pathogenic E. coli or S. aureus, each stimulates proinflammatory Lox5 pathway to generate specialized pro-resolving mediators in M1 macrophages.12, 22 In this study we uncovered an alternative source of RvD1. We show that colonoids which consist of epithelial cells devoid of immunocytes generate high levels of RvD1 and that the level of RvD1 synthesis is subjected to regulation by Lps. This new source of RvD1 which responds to commensal bacteria may play a more important role in various physiological conditions.
The human gut is believed to contain up to 1g of Lps and fecal Lps level is significantly higher in IBS patients than in healthy subjects.16, 42 Tlr4 receptor specifically recognizes and binds Lps and its expression varies by the regions of the gastrointestinal tract and is determined largely by the bacterial composition of that region.43,44 Our studies show that in contrast to wild-type and Tlr2 KO mice intracolonic infusion of IBS-FS failed to inhibit colonic RvD1 levels in Tlr4 KO mice. These mice were also resistance to develop VH in response to IBS-FS (Fig 3 C). These findings not only demonstrate that colonic sensitivity varies according to the levels of RvD1 but also confirm that importance of Tlr4 in mediating the inhibitory action of gram-negative gut dysbiosis. Importantly or data also show that the inhibitory effect of IBS-FS on RvD1 synthesis and LOX5 mRNA expression can be replicated in colonoids derived from human colonic mucosa.
Our studies also reveal a new function of colonic tuft cells which are rare primary solitary cells of epithelial lineage distributed in hollow organs (GI tract and respiratory tract) with distinct morphologic characteristics – bottle-shaped intraepithelial cells with apical microvilli.45 Intestinal tuft cells are positioned to detect information from luminal content including bacterial products and parasites and have the capacity to produce a unique spectrum of biological effector molecules, including IL-25, eicosanoids and the neurotransmitters acetylcholine.31,33,35,36 GPCR signaling is a dominant mechanism whereby tuft cells receive signals from the environment. Succinate acting via GPR91/SUCNR1 has been identified as a potent activator of tuft cells in IL-25-, and POU2F3-dependent manner to promote small intestinal ILC2 proliferation and IL-13 expression and subsequent tuft cell expansion.31,34,35,46 GPR91/SUCNR1 receptor enables sensing succinate-producing luminal organisms, including the intestinal protist Tritrichomonas and certain bacteria. 31,34,35,46 Tuft cells also express several GPCRs involved in taste sensing (T1R2, T1R1 and T1R3 receptors).47 Activation of these receptors leads to stimulation of the canonical taste transduction cascade – which involves α-gustducin (GNAT3), phospholipase Cβ2 (PLCβ2), calcium mobilization, and activation of TRPM5 that leads to depolarization of the cell. Here, we uncovered an unexpected role of tuft cells in the colonic mucosa to detect Lps from gram negative commensal bacteria which in turn inhibits the synthesis of RvD1. This mechanism may be responsible for mucosal inflammation and visceral pain in the presence of gut dysbiosis.
Supplementary Material
BACKGROUND AND CONTEXT
Resolvins mediate resolution of inflammation and pain. We hypothesize that resolvin D1 synthesis is reduced in IBS-D colonic mucosa secondary to gut dysbiosis and this may contribute to visceral hypersensitivity.
NEW FINDINGS
Resolvin D1 is generated in colonic tuft cells to regulate gut sensitivity. Commensal bacterial composition regulates of resolvin D1 synthesis in colonic mucosa, which is reduced in IBS-D patients.
LIMITATIONS
Clinical studies in IBS-D patients are needed to demonstrate the correlation of colonic resolvin D1 levels and degree of visceral hypersensitivity.
IMPACT
Identify a new role for colonic tuft cells in synthesizing resolvin D1 and provide a possible explanation for the development of visceral hypersensitivity in IBS-D patients.
LAY SUMMARY
Gut commensal bacteria modulate synthesis of resolvin D1 in colonic tuft cells. Decreased synthesis of colonic resolvin D1 may contribute to the development of pain in IBS-D patients.
Support:
The studies were supported by NIH grants: R01DK110436 and R01DK122350.
Abbreviations:
- cDNA
complementary DNA
- Chat
choline acetyltransferase
- CRD
colorectal distention
- Dclk1
serine/threonine-protein kinase DCLK1
- DTR
diphtheria toxin receptor
- DTX
diphtheria toxin
- FLAP
5-lipoxygenase-activating protein
- FPR2
formyl peptide receptor 2
- FS
fecal supernatant
- GF
germ-free
- GPR32
G protein coupled receptor 32
- HC
healthy control
- IBS
irritable bowel syndrome
- KO
knockout
- Lps
lipopolysaccharide
- LTA
lipoteichoic acid
- Lox5
lipoxygenase 5
- MAPK
mitogen-activated protein kinase
- MyD88
myeloid differentiation primary response 88
- mRNA
messenger RNA
- NFkB
nuclear factor kappa-light-chain-enhancer of activated B cells
- RvD1
resolvin D1
- RvE1
resolving E1
- siRNA
silencing RNA
- SPF
specific pathogen-free
- SPM
specialized pro-resolving mediators
- TRPM5
transient receptor potential cation channel subfamily M member 5
- Tlr2 and 4
toll-like receptor subtype 2 or 4
- VMR
visceromotor response
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
The authors have declared that no conflict of interest exists.
References
- 1.Mertz H, Naliboff B, Munakata J, et al. Altered rectal perception is a biological marker of patients with irritable bowel symptom. Gastroenterology 1995;109: 40–52. [DOI] [PubMed] [Google Scholar]
- 2.Camilleri M, Lasch K, Zhou K. Irritable bowel syndrome: methods, mechanisms, and pathophysiology. The confluence of increased permeability, inflammation, and pain in irritable bowel syndrome. Am. J. Physiol. Gastrointest. Liver. Physiol 2012;303:G775–G785. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein CN, Niazi N, Robert M, et al. Rectal afferent function in patients with inflammatory and functional intestinal disorders. Pain 1996;66:151–61. [DOI] [PubMed] [Google Scholar]
- 4.Mearin F, Lacy BE, Chang L, et al. Bowel Disorders. Gastroenterology. 2016:S0016-5085(16)00222-5. [DOI] [PubMed] [Google Scholar]
- 5.Hughes PA, Zola H, Penttila IA, et al. Immune activation in irritable bowel syndrome: can neuroimmune interactions explain symptoms? Am. J. Gastroenterol 2013;108:1066–1074. [DOI] [PubMed] [Google Scholar]
- 6.Enck P, Aziz Q, Barbara G, et al. Irritable bowel syndrome. Nat Rev Dis Primers 2016;2:16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohman L, Simrén M. Pathogenesis of IBS: role of inflammation, immunity and neuroimmune interactions. Nat Rev Gastroenterol Hepatol. 2010;7(3):163–73. [DOI] [PubMed] [Google Scholar]
- 8.Ji RR, Xu ZZ, Strichartz G, Serhan CN. Emerging Roles of Resolvins in the Resolution of Inflammation and Pain. Trends Neuroscie 2011;34:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 2008;8:349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grabauskas g, Wu X, Owyang C. Resolvin D1 Acting on G1 Coupled Fpr2 Receptor Inhibits Mast Cell Degranulation and Prevents Development of Visceral Hypersensitivity in lbs. Gastroenterology 2018;154 (Supl 6);S–214 [Google Scholar]
- 11.Grabauskas G, Wu X, Gao J, et al. Prostaglandin E2, Produced by Mast Cells in Colon Tissues From Patients With Irritable Bowel Syndrome, Contributes to Visceral Hypersensitivity in Mice. Gastroenterology 2020;158:2195–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang N, Fredman G, Bäckhed F, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012;484:524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pittayanon R, Lau JT, Yuan Y, et al. Gut microbiota in patients with irritable bowel syndrome—a systematic review. Gastroenterology 2019;157: 97–108. [DOI] [PubMed] [Google Scholar]
- 14.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014;510(7503):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnamoorthy S, Recchiuti A, Chiang N, et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci U S A. 2010;107(4):1660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou SY, Gillilland M 3rd, Wu X, et al. FODMAP diet modulates visceral nociception by lipopolysaccharide-mediated intestinal inflammation and barrier dysfunction. J Clin Invest. 2018;128:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bae YS, Lee HY, Jo EJ, et al. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J Immunol. 2004;173:607–14. [DOI] [PubMed] [Google Scholar]
- 18.Vanderhoof JA, Whitney DB, Antonson DL, et al. Lactobacillus GG in the prevention of antibiotic-associated diarrhea in children. J Pediatr 1999;135:564–568. [DOI] [PubMed] [Google Scholar]
- 19.Sun YP, Oh SF, Uddin J, et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem 2007;282:9323–34. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann C, Homann J, Ball AK, et al. Lipoxin and resolvin biosynthesis is dependent on 5-lipoxygenase activating protein. FASEB J 2015;29:5029–43. [DOI] [PubMed] [Google Scholar]
- 21.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197 [DOI] [PubMed] [Google Scholar]
- 22.Werz O, Gerstmeier J, Libreros S, et al. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun 2018;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Round J, Mazmanian S. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009;9:313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine 2008;42:145–151. [DOI] [PubMed] [Google Scholar]
- 25.Lin YZ, Tao S, Veach RA, et al. Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 1995;270:14255–14258. [DOI] [PubMed] [Google Scholar]
- 26.Schaum N, Karkanias J, Neff NF, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westphalen CB, Asfaha S, Hayakawa Y, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest. 2014;124:1283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schütz B, Jurastow I, Bader S et al. Chemical coding and chemosensory properties of cholinergic brush cells in the mouse gastrointestinal and biliary tract. Front Physiol 2015;6:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schütz B, Ruppert AL., Strobel O. et al. Distribution pattern and molecular signature of cholinergic tuft cells in human gastro-intestinal and pancreatic-biliary tract. Sci Rep 2010. 9;9:17466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Middelhoff M, Nienhüser H, Valenti G et al. Prox1-positive cells monitor and sustain the murine intestinal epithelial cholinergic niche. Nat Commun 2020;11:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerjee A, Herring CA, Chen C, et al. Succinate Produced by Intestinal Microbes Promotes Specification of Tuft Cells to Suppress Ileal Inflammation. Gastroenterology 2020;159:2101–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buch T, Heppner FL, Tertilt C, et al. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods 205;2:419–426. [DOI] [PubMed] [Google Scholar]
- 33.Howitt MR, Lavoie S, Monia Michaud M, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016;351(6279):1329–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lei W, Ren W, Ohmoto M, et al. Activation of intestinal tuft cell-expressed Sucnr1 triggers type 2 immunity in the mouse small intestine. Proc Natl Acad Sci U S A 2018;115:5552–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35,Schneider C, O'Leary CE, von Moltke J, et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell 2018;174:271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Moltke J, Ji M, Liang HE, et al. Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 2016;529: 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerbe F, Sidot E, Smyth DJ, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016;529,226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu ZZ, Zhang L, Liu T, Park JY, et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat Med. 2010;16:592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang J, Burston JJ, Li L, et al. Targeting the D-series resolving receptor system for the treatment of osteoarthritic pain. Arthritis Rheumatol 2016;69:996–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meesawatsom P, Burston J, et al. Inhibitory effects of aspirin-triggered resolvin D1 on spinal nociceptive processing in rat pain models. J Neuroinflammation 2016;13:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perna E, Aguilera-Lizarraga J, Florens MV et al. Effect of resolvins on sensitisation of TRPV1 and visceral hypersensitivity in IBS. Gut 2021;70(7):1275–1286. [DOI] [PubMed] [Google Scholar]
- 42.d'Hennezel E, Abubucker S, Murphy LO, et al. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems 2017;2:e00046–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ortega-Cava CF, Ishihara S, Rumi MAK, et al. Strategic Compartmentalization of Toll-Like Receptor 4 in the Mouse Gut. J Immunol 2003;170:3977–3985; [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Devkota S, Musch MW, et al. Regional mucosa-associated microbiota determine physiological expression of TLR2 and TLR4 in murine colon. PLoS One 2010;5(10):e13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato A. Tuft cells. Anatomical science International 2007;82:187–199. [DOI] [PubMed] [Google Scholar]
- 46.Nadjsombati MS, McGinty JW, Lyons-Cohen MR, et al. Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 2018:49:33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sukumaran SK, Lewandowski BC, Qin Y, et al. Whole transcriptome profiling of taste bud cells. Scl Rep 2017;7:7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.