

Abstract
Osteoarthritis (OA) is a chronic degenerative joint disorder that leads to disability and affects more than 500 million population worldwide. OA was believed to be caused by the wearing and tearing of articular cartilage, but it is now more commonly referred to as a chronic whole-joint disorder that is initiated with biochemical and cellular alterations in the synovial joint tissues, which leads to the histological and structural changes of the joint and ends up with the whole tissue dysfunction. Currently, there is no cure for OA, partly due to a lack of comprehensive understanding of the pathological mechanism of the initiation and progression of the disease. Therefore, a better understanding of pathological signaling pathways and key molecules involved in OA pathogenesis is crucial for therapeutic target design and drug development. In this review, we first summarize the epidemiology of OA, including its prevalence, incidence and burdens, and OA risk factors. We then focus on the roles and regulation of the pathological signaling pathways, such as Wnt/β-catenin, NF-κB, focal adhesion, HIFs, TGFβ/ΒΜP and FGF signaling pathways, and key regulators AMPK, mTOR, and RUNX2 in the onset and development of OA. In addition, the roles of factors associated with OA, including MMPs, ADAMTS/ADAMs, and PRG4, are discussed in detail. Finally, we provide updates on the current clinical therapies and clinical trials of biological treatments and drugs for OA. Research advances in basic knowledge of articular cartilage biology and OA pathogenesis will have a significant impact and translational value in developing OA therapeutic strategies.
Subject terms: Rheumatic diseases, Senescence, Molecular medicine
Introduction
Osteoarthritis (OA) is one of the most common types of arthritis and a chronic degenerative and disabling disease characterized by complex disorders of the whole synovial joint,1 including structural defects of hyaline articular cartilage, loss of intact subchondral bone, tissue hypertrophy and increasing of vascularity in the synovium, and instability of the tendons and ligaments (Fig. 1). In 2021, >22% of adults older than 40 had knee OA, and it is estimated that over 500 million individuals are currently affected by OA worldwide.2 Lacking long-term clinical treatment, OA patients at the end-stage of the disease are ultimately subjected to joint replacement surgery. Joint replacement surgery is growing at a rate of 10% per year globally, and 95% is performed for OA patients.3 However, the lifespan of the artificial joint is limited, and the risk of poor outcomes exists. By 2020, OA is globally estimated to be the fourth leading cause of disability, with a huge amount of medical and healthcare costs and indirect costs caused by loss of jobs and early retirement.
Fig. 1.

Phenotypes of Osteoarthritis (OA). Clinic evidence shows that the majority of OA patients have a diversity of OA phenotypes, including articular cartilage erosion, synovial hyperplasia, abnormal angiogenesis, synovial inflammation, subchondral bone disturbance, ligaments and tendons instability, and joint stiffness. Left-half side shows the structure of the normal synovial joint. Right-half side showed the possible alterations of synovial joint structure and symptoms in osteoarthritis
There is currently no cure for OA. It has been a long time since clinical treatments of OA focused on improving joint pain symptoms rather than on the decline of the disease progression. In recent years, strategies for OA have been shifted to its early prevention and halt or delay OA progression before massive destruction occurs. Therefore, understanding and identifying potential biomarkers and therapeutic targets at different stages of OA are urgent. Scientists and clinicians have devoted great efforts to defining major signaling pathways and molecules that play essential roles in the initiation and development of OA and could finally be developed as potential therapeutical targets to slow down or limit the damage to synovial joints.
Besides the updated epidemiology of OA, including its prevalence, incidence, burden, and risk factors, we have reviewed our current understanding of the pathogenesis in terms of synovial tissue interactions and cellular biology in OA, as well as pathological signaling pathways and essential molecules of OA. We have summarized the roles and functions of those pathological molecular signaling pathways and key molecules in different components of the synovial joints at different stages of OA and their related clinical relevance. We have finally reviewed current clinical therapies applied to OA patients and updated clinical trials of new drugs and biological treatments for future OA treatment.
Prevalence
Osteoarthritis (OA) is among the most prevalent diseases globally, which affects multiple joints, including the hip, knee, ankle, hand, and temporal-mandibular joint (TMJ) and other joints.1,4,5 The knee, hand, and hip joints are most susceptible to developing OA.6 During the past century, the prevalence of OA has grown rapidly in part due to recent increases in lifespan and body weight.7,8 According to a large cohort study in the United States, the prevalence of knee OA has increased by 2.1-fold since the 1950s.9 It is anticipated that, by 2032, the prevalence of OA will rise from 26.6% to 29.5%.10 The prevalence rate of OA can be variable in different studies based on the definitions of OA, e.g., radiographic OA or symptomatic OA. In general, radiographic OA is more prevalent than symptomatic OA.11,12 The Global Burden of Disease (GBD) study showed that the global OA prevalence was about 300 million in 2017 and rapidly increased to 530 million in 2019.13,14 The prevalence rate of OA has elevated by 9.3% from 1990 to 2017 and by 13.25% from 1990 to 2019.13,14 The prevalence standardized by age was estimated at 6,173.38 per 100,000 in 1990 and 6,348.25 per 100,000 in 2019.14 In 2019, China was reported to have the largest number of prevalent OA cases (132.81 million), followed by India (62.36 million) and the U.S. (51.87 million).14 Moreover, the percentages of OA prevalence in China, India, and the United States have dramatically increased by 156.58%, 165.75%, and 79.63%, respectively, from 1990 to 2019.14 The global prevalence of OA in 2019 was significantly higher in females (317.44 million) than in males (210.37 million) and was markedly increased in older people, with a peak level in the 60–64 age group.14 A recent study has developed an algorithm to identify patients with hip OA and conducted a retrospective cohort study using this algorithm together with data from the Integrated Primary Care Information database.15 The results revealed that the prevalence of hip OA increased from 4.03% in 2008 to 7.34% in 2019.15
The prevalence of OA varies in different regions.14,16–18 Epidemiological studies from the 2019 GBD study showed that, after standardization by age, sex, and sociodemographic index, the top three regions of OA prevalence in 2019 were East Asia (137.3 million), South Asia (75.6 million), and Western Europe (57.0 million) (Table 1). In contrast, the prevalence of OA was significantly lower in Oceania (0.4 million (95% UI 0.3–0.4 million)), the Caribbean (3.0 million (95% UI 2.7–3.4 million)) and Central Sub-Saharan Africa (3.2 million (95% UI 2.9–3.6 million)) (Table 1). The standardized prevalence of OA was about 10.7% in the United Kingdom and has gradually increased at an annual rate of 1.4%.19 In China, the prevalence rates of symptomatic knee OA were estimated at 8.1% (5.7% in men and 10.3% in women) in 2012 and 17% (12.3% in men and 22.2% in women) in 2018 based on a population-based meta-analysis.20,21 No marked difference was observed in OA prevalence between the northern and southern areas in China.20 However, the prevalence rates of knee OA were much higher in rural areas of China than in urban regions of China.22 Another study analyzed the overall prevalence of different types of OA in the middle-aged and elder populations in China and reported that lumbar OA was with the highest prevalence (25.03%), followed by knee OA (21.51%) and cervical OA (20.46%).23 Epidemiologists have compared the OA prevalence between China and the U.S. Interestingly, the results showed that both radiographic and symptomatic OAs were more prevalent in women from China than in women from Framingham, Massachusetts.24 In contrast, hip and hand OAs were less prevalent in China than in the United States.25,26
Table 1.
Prevalent cases, age-standardized rate and percent for osteoarthritis in 2019 for both sexes, and percentage change of age-standardized rates by Global Burden of Disease regions
| Counts millions (2019) | Percent % (2019) | Age-standardized estimates (2019) | Annual rate of change in age-standardized rates between 1990 and 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Women | Men | Total | Women | Men | Both | Women | Men | Both | ||
| Global |
317.4 (350.3–288.5) |
210.4 (190.2–233.7) |
527.8 (478.7–584.8) |
8.46 (7.69–9.34) |
5.71 (5.16–6.32) |
7.09 (6.43–7.85) |
7278.0 (6613.6–8038.6) |
5324.0 (4827.0–5885.2) |
6348.3 (5776.3–7023.0) |
3.9 (2.8–5.1) |
| Andean Latin America |
2.1 (2.3–1.9) |
1.4 (1.2–1.5) |
3.5 (3.2–3.8) |
6.87 (6.22–7.54) |
4.63 (4.18–5.13) |
5.77 (5.23–6.36) |
7139.4 (6472.6–7810.8) |
5048.7 (4565.3–5561.9) |
6130.8 (5571.7–6733.0) |
12.4 (9.8–15.3) |
| Australasia |
2.4 (2.7–2.2) |
1.4 (1.3–1.5) |
3.8 (3.4–4.2) |
17.26 (15.45–19.28) |
10.51 (9.53–11.58) |
13.99 (12.67–15.52) |
10148.5 (9090.2–11415.8) |
6369.5 (5779.7–7041.4) |
8335.2 (7537.9–9261.7) |
10.2 (7.6–12.7) |
| Caribbean |
1.8 (2.0–1.6) |
1.3 (1.1–1.4) |
3.0 (2.7–3.4) |
7.57 (6.82–8.43) |
5.66 (5.08–6.32) |
6.64 (5.99–7.37) |
6421.6 (5784.5–7143.6) |
5131.9 (4601.8–5708.3) |
5808.8 (5244.4–6439.2) |
7.9 (6.2–9.6) |
| Central Asia |
2.7 (3.0–2.4) |
1.6 (1.5–1.8) |
4.3 (3.8–4.9) |
5.87 (5.2–6.66) |
3.82 (3.41–4.29) |
4.87 (4.35–5.51) |
6129.4 (5462.4–6943.4) |
4722.3 (4262.2–5263.1) |
5518.5 (4957.3–6181.7) |
4.9 (3.7–6.1) |
| Central Europe |
6.1 (6.7–5.5) |
4.4 (4.0–4.8) |
10.4 (9.4–11.5) |
10.67 (9.61–11.79) |
8.36 (7.56–9.25) |
9.56 (8.64–10.55) |
5374.2 (4854.1–5977.7) |
4910.2 (4436.2–5416.3) |
5165.5 (4661.3–5715.9) |
7.7 (6.6–8.7) |
| Central Latin America |
9.1 (10.1–8.2) |
6.7 (6.0–7.4) |
15.8 (14.2–17.5) |
7.37 (6.66–8.2) |
5.87 (5.28–6.52) |
6.65 (5.99–7.39) |
7037.6 (6346.2–7809.2) |
5958.1 (5380.1–6605.2) |
6536.8 (5908.8–7244.4) |
11.1 (10.0–12.5) |
| Central Sub-Saharan Africa |
1.8 (2.1–1.6) |
1.4 (1.2–1.5) |
3.2 (2.9–3.6) |
2.82 (2.51–3.16) |
2.13 (1.91–2.41) |
2.48 (2.21–2.8) |
5998.9 (5355.4–6713.3) |
5071.1 (4576.9–5660.5) |
5580.1 (5021.9–6250.8) |
3.7 (1.8–5.5) |
| East Asia |
81.7 (91.8–72.8) |
55.5 (49.3–62.5) |
137.3 (122.0–154.1) |
11.65 (10.36–13.07) |
7.84 (6.96–8.82) |
9.73 (8.65–10.92) |
7357.8 (6566.5–8236.3) |
5239.7 (4678.5–5891.6) |
6324.6 (5651.0–7078.9) |
10.6 (9.3–11.8) |
| Eastern Europe |
17.0 (19.4–15.0) |
9.7 (8.7–10.9) |
26.7 (23.7–30.3) |
15.65 (13.84–17.85) |
10.61 (9.49–11.97) |
13.35 (11.86–15.15) |
8397.1 (7409.8–9606.2) |
7283.7 (6516.6–8207.7) |
7951.4 (7061.0–9031.1) |
2.4 (0.7–4.2) |
| Eastern Sub-Saharan Africa |
5.4 (6.1–4.8) |
4.3 (3.9–4.9) |
9.7 (8.7–10.9) |
2.66 (2.37–2.99) |
2.21 (1.99–2.48) |
2.44 (2.19–2.73) |
5994.9 (5390.6–6709.0) |
5184.0 (4684.9–5771.8) |
5608.4 (5056.0–6260.2) |
5.8 (5.0–6.7) |
| High-income Asia Pacific |
22.1 (24.4–20.1) |
10.1 (9.1–11.1) |
32.2 (29.4–35.4) |
23.97 (21.75–26.39) |
11.61 (10.47–12.82) |
17.97 (16.37–19.71) |
10912.3 (9818.5–12134.5) |
5639.9 (5084.2–6229.0) |
8369.7 (7626.9–9263.7) |
8.0 (6.6–9.5) |
| High-income North America |
34.9 (38.9–31.4) |
21.8 (19.6–24.4) |
56.7 (51.0–63.4) |
19.56 (17.6–21.82) |
13.04 (11.7–14.62) |
16.41 (14.74–18.34) |
11264.2 (10111.1–12613.5) |
7983.3 (7219.5–8914.5) |
9704.7 (8749.7–10856.4) |
4.1 (0.7–7.6) |
| North Africa and Middle East |
13.3 (14.8–11.9) |
11.3 (10.2–12.7) |
24.6 (22.1–27.3) |
4.68 (4.2–5.23) |
3.81 (3.41–4.26) |
4.24 (3.81–4.71) |
5925.2 (5326.4–6569.0) |
4786.2 (4300.3–5304.4) |
5342.8 (4815.9–5907.8) |
10.4 (9.2–11.6) |
| Oceania |
0.2 (0.2–0.2) |
0.2 (0.1–0.2) |
0.4 (0.3–0.4) |
3.4 (3.01–3.82) |
2.53 (2.24–2.86) |
2.96 (2.61–3.32) |
5697.4 (5083.0–6347.3) |
4159.9 (3701.9–4635.1) |
4907.2 (4386.3–5466.5) |
7.0 (4.4–9.7) |
| South Asia |
43.9 (49.0–39.6) |
31.7 (28.4–35.4) |
75.6 (68.1–84.5) |
5.06 (4.56–5.66) |
3.57 (3.2–3.98) |
4.31 (3.88–4.81) |
5988.7 (5408.6–6664.5) |
4435.0 (3990.0–4914.0) |
5219.0 (4711.2–5790.9) |
10.4 (9.4–11.2) |
| Southeast Asia |
15.9 (17.8–14.1) |
10.8 (9.5–12.1) |
26.7 (23.6–29.9) |
4.84 (4.29–5.42) |
3.36 (2.96–3.77) |
4.11 (3.63–4.6) |
4633.5 (4117.8–5161.8) |
3548.0 (3157.6–3959.6) |
4128.9 (3676.7–4603.3) |
13.0 (11.6–14.4) |
| Southern Latin America |
4.3 (4.8–3.8) |
2.3 (2.0–2.5) |
6.5 (5.9–7.3) |
12.98 (11.67–14.61) |
7.55 (6.83–8.36) |
10.38 (9.39–11.57) |
9592.2 (8604.2–10784.8) |
6156.8 (5563.7–6806.5) |
8013.5 (7246.6–8953.5) |
9.3 (7.5–11.5) |
| Southern Sub-Saharan Africa |
2.1 (2.4–1.9) |
1.7 (1.5–1.9) |
3.8 (3.5–4.3) |
5.48 (4.9–6.15) |
4.68 (4.24–5.21) |
5.1 (4.59–5.69) |
6437.4 (5777.2–7194.6) |
6676.9 (6058.9–7380.3) |
6542.3 (5904.3–7279.5) |
6.7 (5.6–7.7) |
| Tropical Latin America |
8.6 (9.5–7.7) |
5.9 (5.3–6.5) |
14.5 (13.0–16.1) |
7.71 (6.92–8.56) |
5.71 (5.12–6.32) |
6.75 (6.07–7.49) |
6403.5 (5754.3–7090.1) |
5237.9 (4702.7–5776.5) |
5869.8 (5280.9–6498.3) |
9.7 (8.5–11.1) |
| Western Europe |
35.3 (38.9–32.2) |
21.8 (19.7–23.9) |
57.0 (51.9–62.9) |
16.46 (14.98–18.15) |
10.92 (9.9–12.03) |
13.79 (12.58–15.21) |
8166.6 (7396.8–9070.3) |
5828.6 (5278.1–6422.3) |
7056.9 (6394.9–7801.7) |
6.3 (4.8–9.3) |
| Western Sub-Saharan Africa |
6.8 (7.6–6.1) |
5.2 (4.7–5.8) |
12.0 (10.7–13.4) |
2.97 (2.65–3.34) |
2.4 (2.15–2.68) |
2.69 (2.41–3.02) |
6422.2 (5788.3–7164.8) |
5374.8 (4850.5–5984.6) |
5922.7 (5346.0–6596.3) |
5.7 (5.0–6.4) |
All data in this table were extracted from the following website http://ghdx.healthdata.org/gbd-results-tool
Incidence
According to the 2017 GBD study, about 14.9 million incident cases of OA were reported globally with an age-standardized incidence rate (ASIR) of 181.2 per 100,000 person-years, an increase of 8.2% from 1990 to 2017.13 The ASIR of OA was highest in high-income regions, such as North America (306.6), Australasia (236), and Asia Pacific (233.4), while it was lowest in Eastern Sub-Saharan Africa (133), Central Sub-Saharan Africa (135.9) and East Asia (136.2).13 At the regional levels, the ASIR of OA in 2017 was highest in the U.S. (316.9), Kuwait (260.7), and Qatar (258), whereas it was lowest in Taiwan (99.5), North Korea (105.7), and Madagascar (122.3).13 Moreover, the U.S., Oman, and Equatorial Guinea were the top three countries with the greatest growth in ASIR of OA from 1990 to 2017.13 The Central African Republic and DR Congo were the only two of the 195 countries that exhibited a trend toward decline in the OA incidence.13 In a Chinese longitudinal cohort study, the 10-year average ASIR of knee OA was 25.2 per 1000 person-years.27 Data from a population-based healthcare database in England reported that the annual consultation incidence of OA was 8.6/1000 persons ≥15 years of age, 6.3 in males, and 10.8 in females.28 Another study estimated that the ASIR (per 100,000 person-years) was 240 for knee OA, 88 for hip OA, and 100 for hand OA.29 The incidence of hand, hip, and knee OA increased with age, reached the peak level at 55–59 years old, and gradually declined in the older groups.13,29 Women have a higher incidence of hand, foot, and knee OA, while men have a higher incidence of shoulder and cervical OA.29,30
Burden
OA has been a leading cause of disability globally, which leads to enormous healthcare and socioeconomic burdens.31 In 2010, hip and knee OAs were among the top 20 causes of disability globally and the top 40 highest in the disability-adjusted life-years (DALYs).32 In 2017, OA caused nearly 9.6 million years lived with disability (YLDs), with an age-standardized rate (ASR) of 118.8 YLD per 100,000.13 Moreover, the age-standardized YLD was elevated by 9.6% from 1990 to 2017.13 Data from the 2019 GBD study showed that OA caused 18.9 million YLDs with an ASR of 244.9 (95% UI 123.7–486.7) YLDs per 100,000. The percentage changes in age-standardized YLD rates between 1990 and 2019 were 6.45% (95% UI 4.84% to 7.96%). OA is estimated to contribute $460 billion in all-cause medical costs in 2019 in the United States.33 It was also reported that OA patients had four times excess total medical costs than people without OA ($14,521 vs. $3629 per year per person).34 The 2015 GBD study analyzed 315 causes of diseases and their burdens. OA ranked as the 15th leading cause of YLDs, contributing a total of 52,661 (UI 34,056–77,499) YLDs in the Nordic region in 2015.35 In China, the YLDs caused by knee OA were 4,149,628, and the YLDs per 100,000 was 968. Moreover, the total YLDs caused by OA was 1.97 million in 2017, accounting for 1.08% of all YLDs.36 Currently, the burden of OA is still continuously increasing in most countries.16,37 Risk factors in OA etiology, such as female gender and advanced age, are correlated with high YLD rates.38
Risk factors
Risk factors of OA include obesity, female gender, aging, knee injuries, and high-impact sports, such as marathons, speed skating, and weightlifting. Although aging and OA could be totally independent processes, they are closely associated from a statistical point of view.39–41 Aging has been accepted as the most prominent risk factor for OA. Except for increasing exposure to other risk factors with age, aging-related biological and molecular signaling alterations significantly contribute to the disorganization of the joint structure. Several potential pathological mechanisms of how aging contributes to OA have been proposed recently.41,42 The cellular senescence-associated secretory phenotype (SASP) has been detected in the degradative articular cartilage and synovial joint tissue of developing OA.43–45 In addition, aging-related mitochondrial dysfunction that induces oxidative stress, characterized by excessive accumulation of the reactive oxygen species (ROS) with the imbalance of energy metabolism of articular chondrocytes, is believed to promote articular chondrocyte apoptosis and articular cartilage destruction.46–48 Furthermore, age-related inflammation in the synovial joint, which is also associated with SASP, leads to destructive changes in the extracellular matrix (ECM) of the articular cartilage and promotes OA.49
Obesity is another major risk factor that leads to a higher incidence of hip and knee OA. Obesity is one of the most significant risk factors for knee OA partly because the excessive weight of obese patients leads to an abnormal increase in mechanical loading on knee joints, which results in the wearing and tearing of articular cartilage accompanied by ligament destruction and eventually leads to the occurrence of OA. Surprisingly, obese patients also have a higher incidence of OA in the hands that do not usually bear the weight.50,51 This leads to the general belief that it is the systemic factor(s) released by other tissues that induce OA in obese patients. In obese patients, cytokines released by adipocytes, also known as “adipokines”, such as resistin, visfatin, leptin, omentin, adiponectin, retinol-binding protein 4 (RBP4), and other factors, were reported to be associated with promoting the initiation and progression of OA.52–56 Furthermore, cytokines, such as TNF-α, IL-1, IL-6, and IL-8, were shown to trigger joint inflammation, which leads to ECM breakdown and cartilage degeneration.
OA affects more than 500 million populations worldwide, with a higher prevalence in the female gender than in males. Women are known to be more susceptible to OA onset and development than men are.6,57,58 Several studies showed that OA development could be triggered by the plunge in sex hormone levels in menopausal women.59,60 Besides, compared to male OA patients, female patients were reported to have higher levels of joint inflammation and clinical pain, thinner articular cartilage, and severe physical joint mobilities.57,60,61 The potential contributing factors for this gender difference in OA are not fully understood and need further attention in the OA research community.
Knee injury is another major risk factor for knee OA. Post-traumatic OA is one of the OA subtypes that occurs in those joints that have been injured. Current studies have shown that joints that have been traumatized are five times more likely to develop OA than joints that have never been damaged.62 U.S. clinical statistics predict that post-traumatic OA accounts for 12–42% of OA (the proportion varies by age), and the actual proportion could be higher.3 Trauma in the joints has been demonstrated to induce massive gene expression alterations in different compartments of knee joints. Besides injury, sports-related excessive joint loading also increases the chance of OA development. Professional athletes of high-impact sports present a higher prevalence of early knee OA than non-professional athletes and the universal population.63 The new technology of instrument innovations has been developed and utilized for investigating the role of joint mechanical stress in OA pathogenesis, which could study the pattern, force, and duration of mechanical stress on joint loading.64,65 Our advanced knowledge of how mechanical loading contributes to OA onset and progression is just beginning to be used in the applications of biomechanics assessment for guiding the clinical physical therapy of OA patients.66 However, the molecular mechanisms of how mechanical stress contributes to OA onset and development need to be investigated in great detail.
In comparison, few genetic mutations have been confirmed to be linked to human OA before. It is until recently that genetics has been discovered as a risk factor in 11 types of OA, including OAs in the hand, hip, and spine.67 A recently reported genome-wide association study (GWAS) meta-analysis of more than 820,000 East Asian and European individuals from 13 international cohorts of 9 populations, including over 170,000 OA patients, identified around 10,000 significantly associated single-nucleotide variants (SNVs), in which 100 were unique and showed independent genetic correlation with OA phenotypes and symptoms.67 Among these identified 100 SNVs, 60 were genome-wide significantly associated with more than one type of OA, and 77 potential effector genes were identified. Though genetic studies identified risk variants associated with new molecular signals and already reported effector genes contributing to the OA development,68–71 these genetic risk data need to be further verified and investigated to reveal options for the translational intervention of OA.
Clinical symptoms
Clinical symptoms of OA include joint functional limitations, stiffness, pain, disability of walking or running, and probably other symptoms.1,72,73 Bony enlargement and swollen and inflamed joints could be found in OA patients in physical examination. Clinical radiographic examination, such as MRI (magnetic resonance imaging), is able to visualize marginal osteophytes, joint space narrowing, structural changes of osteochondral tissue, and other OA lesions. Pain is one of the most distinctive symptoms and the main reason OA patients seek medical help,72 but the underlining mechanisms of OA pain are still poorly understood. Pain is a clinical indicator of tissue damage, inflammations, or disorders of the nervous system.74,75 Articular cartilage is an avascular tissue without any nerve invasion, and OA pain can happen both before and after an articular cartilage lesion detected by the imaging system. Therefore, it is unlikely that the destruction of articular cartilage directly causes OA pain. OA pain has been reported to be associated with synovitis and bone marrow lesion,76,77 as well as alterations in subchondral bone, osteophyte formation, abnormalities of infrapatellar fat pads, and lesion of ligaments, in which tissues have highly distributed sensory nerves.78 Molecular mechanisms underlying OA pain have been comprehensively updated and discussed in two recent review articles.79,80
Pathogenesis
During the past decades, the pathogenesis of OA has been extensively studied.81 Although its risk factors were characterized, and the structural changes of the synovial joint in OA are well understood, the complex pathological mechanisms of the onset and development of OA remain elusive. We summarize our current understanding of OA pathogenesis from the perspective of tissue interactions, changes in cellular biology, and pathogenic signaling pathways and molecules.
Tissue interactions in OA
Numerous studies reported that subchondral bone sclerosis could be one of the major reasons that cause aging-related OA and that the abnormal bone remodeling related to dysregulation of osteoblasts and osteoclasts plays key roles in the OA initiation and development.82–87 Increased subchondral bone porosity and remodeling, reduced bone density, and bone mineralization with irregular matrix organization, which were believed to be stimulated by bone-cartilage crosstalk through subchondral pores and vascular invasion, were observed in the early stage of OA.88–91 These changes in subchondral bone were found to be happening at the same time with or earlier than the early destruction of articular cartilage.92–95 On the other hand, the late stage of OA showed architectural alterations of the subchondral bone characterized by a reduction of bone remodeling and enhanced subchondral bone densification leading to sclerosis.96–98 However, the mechanism of how articular cartilage and joint issues crosstalk with subchondral bone leading to the initiation and development of OA is incompletely understood and needs further investigation.
Besides the subchondral bone dysregulation, the synovium is another most-related tissue that showed significant changes at the early stage of OA, even before cartilage degradation occurs.91,99 The contribution of synovium to the initiation and development of OA has been investigated in the past 20 years. At the early stage of OA, histological changes of the synovium include synovial lining hypertrophy and hyperplasia, increased angiogenesis, a low level of synovial inflammation, and synovial fibrosis observed.100–103 Synovitis with a high level of macrophages could be found at the end stage of the OA.104 Synovitis scores used as one of the OA assessments are based on these histological features.99,105,106 Low-grade synovial inflammation can be detected in >50% of OA patients at the early and late stages of the disease.107,108 Therefore, among the synovial features, synovial inflammation has received the most attention from the OA research community. It is widely believed that pro-inflammatory factors that are released by synovial tissue induce the ECM destruction of the articular cartilage. However, the interactions of different cell types in the synovial joint and features of the synovium at different stages of OA need to be extensively investigated.
Obesity acts as an OA risk factor not solely through loading excessive body weight onto knee joints,109 the pathogenesis involves a complex network of tissue and cellular interactions. As mentioned above, adipokines released by adipose tissue that interact with different tissues are believed to be critically involved in OA pathogenesis.110–112 In addition, the local adipose tissue, such as the infrapatellar fat pad (IPFP), also cross-talks with other synovial tissues to affect OA development.111,113 Recently, a completely fat-free transgenic lipodystrophy (LD) mouse model, which has a comparable body weight to wild-type controls, was used in an OA study to demonstrate the contribution of adipose tissues to OA onset and development. LD mice with a total lack of adipose tissue are resistant to DMM-induced or spontaneous OA. The susceptibility to posttraumatic OA was restored after mature fat depots were implanted in the LD mice, suggesting the notion that the adipose tissue and factors released by it can promote OA lesions.114 On the other hand, adipose tissue could be used as a good source of heterogeneous cell populations, including lymphoid, adipose-derived stromal/stem cells, and myeloid cells and scaffolds, for OA treatment.115
Cell biology of the articular cartilage
Cellular changes of different cell types of synovial joint tissue in OA have been discussed in several excellent review articles.1,116 Although tissue interactions are important in the onset and progression of OA, the destruction of articular cartilage is a hallmark of the majority of OA cases, especially knee OA. Chondrocytes are the only cell type in articular cartilage. Chondrocytes in the degradative articular cartilage undergo more active cell death than those in normal articular cartilage. Apoptosis, a highly regulated programmed cell death, is the earliest reported cell death form.117 While it is known that a low level of apoptosis detected by TUNEL assay exists in normal articular cartilage, the number of apoptotic cells is dramatically increased in the early and late stages of OA articular chondrocytes. Several studies have shown that death receptors (e.g., FAS, TNFR),118,119 cytokines (e.g., TNF-α, IL-1β),120–122 and abnormal mechanical stress and physicochemical factors, including ultraviolet radiation and ionizing radiation, can cause chondrocyte apoptosis.123 In addition, three signaling pathways have been shown to induce apoptosis of articular chondrocytes in OA: the mitochondrial-mediated caspase-dependent pathways, the death receptor pathway, and the endoplasmic reticulum stress (ER stress)-induced unfolded protein response (UPR) pathway. However, whether apoptosis is the cause or consequence of OA remains to be determined. Besides apoptosis, autophagy has been considered to be involved in combination with apoptosis in contributing to OA development.124 While autophagy is activated at the early stage of OA to promote cell survival, it facilitates cell death at the end stage of OA. Ferroptosis is a newly discovered type of cell death driven by iron-dependent lipid peroxidation and characterized by excessive accumulation of lipid ROS.81,125 Pyroptosis, which can be triggered by proinflammatory factors, was recently shown to promote chondrocyte death and OA.126
The superficial layer accounts for 10–20% of the total thickness of the articular cartilage, and chondrocytes in this layer are flat and have a relatively high-density,127 while the chondrocytes in the middle and deep layers are relatively sparse. Chondrocytes, which are large and round in shape in the deeper layer, are divided and differentiated by chondrocytes from the superficial layer.128 OA is characterized by the increasing production of ECM-degrading enzymes and the removal of articular cartilage, which is very similar to the process of endochondral ossification, and abnormally induced articular chondrocyte hypertrophic differentiation is one of the key factors that promote the onset and progression of OA. Thus, factors that accelerate this process are probably important risk factors for inducing OA. However, the mechanisms that regulate articular chondrocyte differentiation are poorly defined. It is known that the ultimate differentiation process and cell function of chondrocyte hypertrophy is regulated by a variety of in vivo systemic factors and local signaling factors, including growth hormone, insulin-like growth factor, parathyroid hormone, parathyroid hormone-associated protein, Indian hedgehog, fibroblast growth factor, classical and non-classical Wnt signaling pathways, transforming growth factor families, RUNX2 (Runt-related transcription factor 2) and MEF2 (myocyte enhancer factor 2) transcription factors, etc.129–131
In the past decades, more and more evidence suggests that metabolism plays a crucial role in maintaining the energy homeostasis of articular chondrocytes and that abnormalities in chondrocyte metabolism can lead to the onset and progression of OA.132,133 Abnormally accelerated catabolism of articular chondrocytes, which promotes ECM degradation and overtakes ECM synthesis, is a major feature of the OA cartilage.132 With the low-grade inflammation in the synovial membrane and abnormal blood vessel invasion through the subchondral bone, the cellular hypoxic stress, oxidative stress, and autophagy level of synovial membrane cells and articular chondrocytes are constantly changing and interplaying with each other, thus contributing to the pathogenesis of OA (Fig. 2).
Fig. 2.

Histological characteristics of OA articular cartilage. Articular cartilage is composed of a dense extracellular matrix (ECM) and sparse distribution of chondrocytes. A cross-sectional diagram of articular cartilage can be divided into the superficial zone, middle zone, and deep zone (the lower part is calcified). Chondrocytes and ECM in the OA cartilage are extensively changed. Cell apoptosis and necrosis are increased in OA chondrocytes with inhibited anabolism and boosted catabolism leading to suppressed ECM synthesis and enhanced ECM degradation. Inflammatory factors, such as TNFα secreted by chondrocytes and synovial membrane fibroblasts, stimulate inflammation in the whole joint. Synovial hyperplasia and fibrosis, and abnormal blood vessel invasion from subchondral bone is observed in OA synovial joint. Increased chondrocyte hypertrophy and oxidative stress with excessive ROS production are reported
Pathogenic pathways and molecules
Wnt signaling
Wingless-like (Wnt) glycoproteins are a family of more than 19 secreted proteins, which are poorly soluble molecules due to their lipid sidechains. The reduction of solubility of Wnt ligands strongly limits their physical signaling transduction capacity in different tissues and typically generates concentration gradients impacting adjacent cell behavior.134,135 With both paracrine and autocrine effects, Wnt signaling was demonstrated to play an essential role in skeletal development in terms of mesenchymal stem cell condensation, chondrogenic differentiation, chondrocyte hypertrophy, growth plate chondrocyte organization, and osteoblast differentiation and maturation.
Wnt pathways are classified into two types by their dependency on β-catenin (Fig. 3): the canonical pathway that is β-catenin-dependent and the non-canonical pathway that is independent of the β-catenin signaling.136 β-catenin is a subunit of the cadherin protein complex and the co-activator to promote its downstream target gene transcription.137 In the cytoplasm, β-catenin binds to cadherin protein on the cell membrane attaching to cytoskeleton protein actin and participating in cell adhesion.138 After being phosphorylated by GSK3β, β-catenin forms a complex with GSK3β and is degraded through ubiquitination.138 In the canonical pathway, Wnt ligands bind to Frizzled and co-receptor LRP5/6 (low-density receptor-related protein 5 or 6), which then recruits protein complex to inhibit the proteasomal degradation and promote nucleus translocation of β-catenin to transcribe Wnt target genes.139–141
Fig. 3.
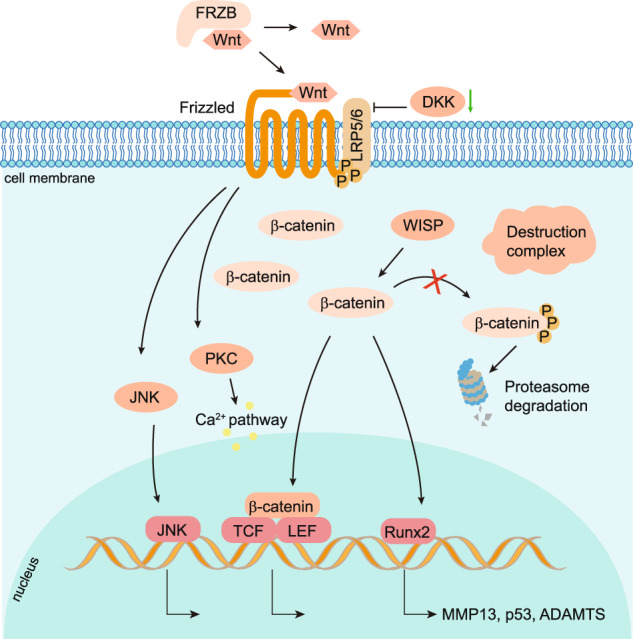
Activated Wnt canonical and non-canonical signaling pathways in OA. In healthy articular cartilage, the Wnt signaling pathway in articular cartilage and synovium is profoundly inhibited by the absence of Wnt-Wnt receptor interactions and β-catenin degradation through the proteasome pathway. In OA chondrocytes and synovial membrane cells, Wnt ligands bind to Frizzled/LRP5/6 receptors to activate the canonical pathway, in which the destruction complex moves to the cell membrane and releases β-catenin to translocate to the nucleus and binding with TCF/LEF family to control target gene transcription. For the non-canonical Wnt pathway, the protein kinase C (PKC) and c-Jun N-terminal Kinase (JNK) are activated to upregulate downstream target genes and pathways, including the calcium pathway
Modest Wnt canonical signaling activation is necessary for maintaining healthy articular cartilage. Inhibiting the activity of β-catenin in articular chondrocytes to inactivate the canonical Wnt pathway by overexpressing a small molecule ICAT (Inhibitor of β-catenin and T cell factor) in Col2a1-ICAT transgenic mice, caused articular cartilage degradation with enhanced chondrocyte apoptosis.142 In addition, mice with global knockout of Wnt16, which is dramatically upregulated in articular cartilage of OA patients and mouse models, developed a more severe articular cartilage degradation than wild-type mice with increased chondrocyte apoptosis and reduction of lubricin expression after induced destabilization of the medial meniscus (DMM).143 Wnt16 alleviates the progression of OA by reducing cartilage catabolism through the planar cell polarity (PCP)/JNK-mTORC1-PTHRP cascade and inhibiting the chondrocyte hypertrophy.144 Most Wnt ligands play non-redundant crucial roles during mouse embryogenesis.145 Currently, there are few genetic mouse models with Wnt genes knocked out in articular chondrocytes that develop spontaneous OA. Of note, the excessive Wnt signaling might promote the development of OA. β-catenin is significantly upregulated in the OA mouse superficial articular chondrocytes and increased in the cartilage of OA patients. The polymorphisms in the frizzled-related protein (FRZB) gene, which is a secreted Wnt antagonist, showed an association with hip OA in females.146 Though they do not develop spontaneous OA nor stabilize more β-catenin in the articular chondrocyte, Frzb global knock-out mice exhibited increased cartilage loss with upregulation of MMP3 in the mouse arthritis models induced by instability and inflammation.147 Activating the Wnt signaling by mutating the three GSK3β phosphorylation sites within the β-catenin molecule in chondrocytes using the Col2a1-Cre transgenic mice led to the embryonic lethetic phenotype.148–150 However, inducible activation of the β-catenin in chondrocytes of adult mice using the Col2a1-CreERT2 transgenic mice caused dramatic destruction of the articular cartilage and other OA-like phenotypes in the knee, hip, and TMJ joints.5,151–154 Moreover, a recent study showed that activating β-catenin by injection of LiCl in the synovial joint of rabbits promoted cartilage degeneration, and the increase of MMP-13 and P53, leading to cartilage degradation and OA development.155 All these lines of evidence indicate that the overactivation of β-catenin in articular chondrocytes promotes the initiation and progress of OA. Interestingly, activating β-catenin signaling by expressing a constitutively active form of β-catenin under the control of the cartilage-specific Col11a1 promoter in chondrocytes in adult mice induces early closure of the growth plate and leads to significant increases in thickness, cell proliferation rate, and cell density, especially in the superficial layer of the articular cartilage.156,157
To extensively understand the fundamental role of Wnt signaling in the articular cartilage and the development of OA, more animal models with genetic manipulation of Frizzled (Fz) receptor proteins, single Wnt ligands, and β-catenin in the articular cartilage should be studied. Wnt signaling pathway-related genes are more extensively dysregulated in the synovium than in the articular cartilage in the STR/Ort mice and collagenase-induced OA mouse models, especially at the early stage of OA.158 In the articular cartilage, the expression of Frizzled receptors generally, especially Fz6, responded to OA initiation much earlier than detectable changes of Wnt ligands in mouse OA models,158 suggesting that Wnt signaling might initially have the paracrine effects in the early stage of mouse OA. Dickkopf-1 (DKK1, Wnt antagonist) levels in plasma and synovial fluid of OA patients were significantly reduced,159 and the level of DKK1 were negatively associated with the severity of OA.160 Overexpression of Dkk1 but not Dkk2 using Col2a1-Cre in chondrocytes inhibited the articular cartilage damage induced by DMM surgery in mice.161 Overexpression of Dkk1 in bone cells using Col1a1-Cre induced fewer cartilage lesions in OA mouse models.161,162 The expression of Dkk3 was significantly increased in the human OA cartilage, synovial tissue, and synovial fluid.163,164 However, the role of Dkk3 and Dkk4 in the onset and progression of OA needs to be determined. In the synovium of OA patients, multiple Wnt family proteins are overexpressed165; however, their potential mechanisms in synovial inflammation are still unclear. WISP-1 (WNT1-inducible-signaling pathway protein 1) significantly increased in the synovium and articular cartilage in human OA and experimental mice OA. Overexpressing recombinant WISP-1 in synovium and articular chondrocyte stimulated macrophages and chondrocytes, causing interleukin-1 to independently induce several aggrecanase and matrix metalloproteinases (MMPs), and the overexpression of MMPs and aggrecanase in joints caused articular cartilage damage.158,160 In the non-canonical pathway, Wnt ligands bind to frizzled, and the co-receptors, PKC (the protein kinase C) and JNK (c-Jun N-terminal Kinase) are then activated to upregulate downstream target genes and pathways, including calcium pathway. These two pathways are both involved in the onset and development of the OA.141,166 Wnt 5a and Wnt11 are the two ligands that are associated with the non-canonical pathway, and they are both detected in the human OA cartilage.167 The role of the Wnt non-canonical pathway in OA development remains to be identified.
Since overactivation of Wnt signaling is associated with OA development, the Lorecivivint (SM04690),168 the first inhibitor of the β-catenin-related pathway, was subjected to the phase I clinical trial as a disease-modifying osteoarthritis drug (DMOAD) in 2015,169,170 and has recently entered phase III clinical trial (NCT05603754) in Nov. 2022 as an OA-modifying agent. SM04690 was shown to induce the bone-marrow-derived human mesenchymal stem cells to chondrocyte differentiation and promote regeneration and protection of articular cartilage in vivo and in vitro through inhibiting CLK2 and DYRK1A, thus leading to the inactivation of β-catenin associated pathway.168,169,171 Intra-articular injection of other small-molecule inhibitors of Wnt/β-catenin, such as XAV-939 and C113, alleviated OA phenotypes in terms of decreased articular cartilage degradation and synovitis in vivo.172 Several other Wnt/β-catenin inhibitors have been investigated in cultured cells and animal models.173–176 However, when the Wnt/β-catenin pathway was excessively inhibited, proliferation and hypertrophy of growth plate chondrocyte were disrupted, the formation of secondary ossification center was delayed in long bone, and articular cartilage destruction occurred.177 Therefore, other β-catenin-independent Wnt pathway-related strategies need to be investigated and developed. Injecting Ad-Wnt16 into the joint to activate the Wnt16 pathway significantly reduced the symptoms of OA.144 This may provide new ideas for the design of DMOAD.
NF-κB pathway
Nuclear factor-κB (NF-κB) constitutes a group of transcription factors in which the pathways can be activated by different kinds of pro-inflammatory cytokines.178 NF-κB plays a critical role in the inflammation, differentiation, proliferation, and survival of mammalian cells.178,179 The NF-κB family contains five members: RelA/p65, RelB, c-Rel, NF-κB1/p50(p105), and NF-κB2/p52(p100). These five proteins all contain an N-terminal Ref-1-homology domain, which is evolutionally conserved and contributes to the dimerization of protein complexes, nuclear localization, DNA banding, and interaction with NF-κB inhibitors.180 The NF-κB is a homo- or hetero-dimeric complex which can be divided into two categories according to its subunit combination.181 One category includes p50 and/or p52 which are processed from their precursor p105 (NF-κB1) and p100 (NF-κB2),182 respectfully. The second category includes c-Rel, p65, and Rel B, which contain a transcriptional trans-activation domain (TAD).182 Lacking TAD, the dimers formed only by p50 and p52, either homodimers or heterodimers, could not drive the transcription.181 In contrast, the heterodimers of c-Rel:p50, p65:p50, and RelB:p52 could function as transcriptional activators.181 In eukaryotes, two classic dimers (p65:p50 and RelB:p52) are the most common NF-κB complexes that work as transcriptional activators.181 Inhibitors of NF-κB (IκB) proteins, including IκBα, IκBβ, IκBγ, IκBε, IκBζ, and Bcl-3, bind to NF-κB family members in the cytoplasm to block phosphorylation and activation of NF-κB.183,184 A variety of inflammatory signals, such as TNFα, can activate the IκB kinases (IKKs), and the activated IKKs lead to IκB degradation by regulating its phosphorylation.183 Then, the NF-κB complex translocates to the nucleus, where it triggers the transcriptions of downstream target genes.185
The expression of NF-κB proteins varies in different stages of cartilage development.186 p65 is expressed in the entire growth plate, especially highly expressed in hypertrophic chondrocytes in vivo and in vitro, suggesting that the NF-κB pathway participates in the endochondral ossification.186,187 Over-expression of IκBα blocks NF-κB activation, thereby inhibiting the Rel/NF-κB protein expression and leading to abnormal development of limb bud in chicken embryos.188 In turn, activation of the NF-κB pathway promotes the proliferation and maturation of chondrocytes by treating with IGF-1 in vitro.189 Deleting p100 in mice resulted in dwarfism, primarily caused by impaired chondrocyte hypertrophy. However, these defects were partially rescued when the RelB gene was knocked down in p100−/− mice.190 Using tissue-specific knockout mice, Kobayashi, H. et al. found that Rela/p65 deletion in embryonic limb cartilage promoted apoptosis of chondrocytes leading to impaired skeletal growth, while homozygous knockout of Rela in articular cartilage at seven weeks resulted in accelerated progression of arthritis, but heterozygotes knockout led to delayed OA development by inhibiting the expression of catabolic genes.191
NF-κB signaling is extensively involved in OA pathology through a variety of patterns (Fig. 4). Mechanoreceptors, cytokine receptors, TNFR, and TLR are located on the surfaces of the articular chondrocyte cellular membrane. Activation of these receptors by pro-inflammatory mediators (such as TNFα or IL-1β), fibronectin fragments, and mechanical stress induces the NF-κB signaling192,193 and cross-talk with BMP (bone morphogenetic protein), Wnt, and other signaling cascades as well.192 NF-κB signaling induces the secretion of degrading enzymes, such as matrix metalloproteinases (MMP) (MMP1, MMP2, MMP3, MMP7, MMP8, MMP9, MMP13), ADAMTS4 and ADAMTS5, leading to degradation of articular cartilage.192 Additionally, numerous NF-κB-mediated cytokines and chemokines, including receptor activators (RANKL) of TNF-α, IL-1β, IL-6, and NF-κB (RANK) ligands expressed in OA articular chondrocytes could upregulate MMP production, reduce the cellular synthesis of collagen and proteoglycan, and act in a positive feedback loop to enhance NF-κB signaling.194 NF-κB can enhance joint injury by inducing PGE2 (prostaglandin E2), NOS (nitric oxide synthase), NO (nitric oxide), and COX2 (cyclooxygenase-2), thereby promoting tissue inflammation, synthesis of catabolic factors, and apoptosis of articular chondrocytes.192 NF-κB promotes the activation of other transcription factors, such as hypoxia-inducible factor 2α (HIF-2α), E74-like factor 3 (ELF3), and RUNX2.195–198 These transcription factors trigger the expression of ADAMTS5 and MMP13 proteases, which promotes the differentiation of pre-hypertrophic articular chondrocytes to terminal differentiated chondrocytes.199 In addition, Deng and coworkers found that inflammatory cytokines regulated TAK1-mediate-phosphorylation to promote proteasomal degradation of YAP (Yes-associated protein), which is a downstream regulating factor of the Hippo pathway.200 Activation of YAP in the articular chondrocytes inhibited substrate accessibility of TAK1, thus attenuating the NF-κB signaling.200 Therefore, the interactions of the Hippo-YAP pathway and NF-κB signaling regulate ECM degradative enzymes to control articular cartilage homeostasis.200
Fig. 4.
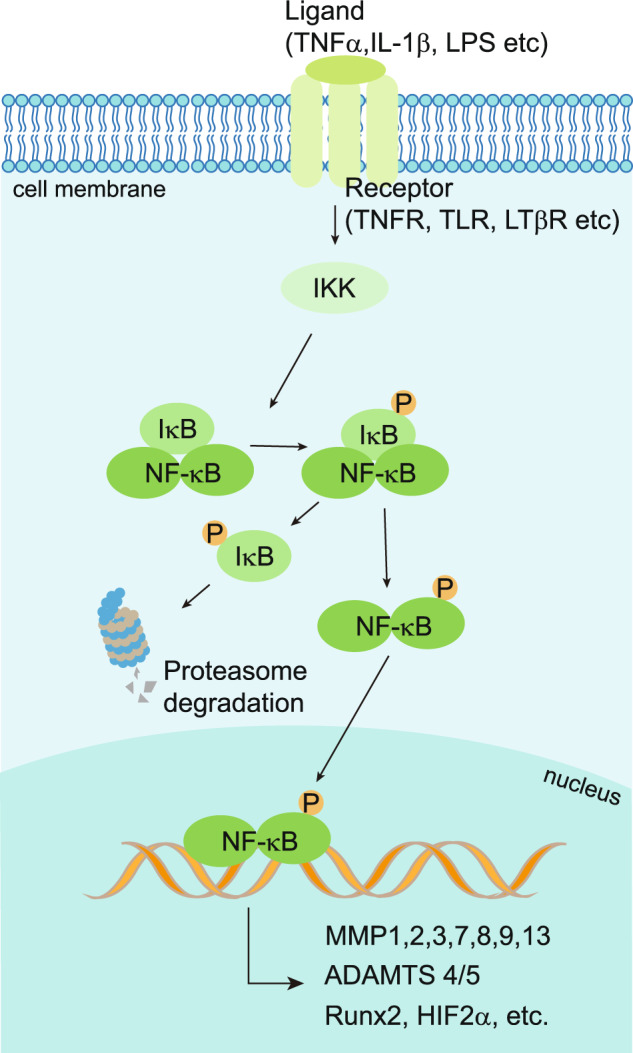
The activity of NF-κB signaling in OA chondrocytes. The NF-κB signaling is activated by a variety of ligands, including TNFα, IL-1β, and LPS, leading to phosphorylation (P) and proteasome degradation of IκB. Receptors of NF-κB signaling include TNFR, TLR, LTβR and etc. Activated IKKs lead to IκB degradation by regulating its phosphorylation. Thus, the translocation of NF-κB to the nucleus results in the transcription of the downstream target genes encoding RUNX2, MMPs, ADAMTS, and HIF2α
NF-κB signaling has been well accepted as a therapeutic target of OA.201,202 Thus, a series of drugs aiming at inhibiting the NF-κB signaling pathway have been developed. For example, the COX-2 isomer is regarded as a proinflammatory enzyme induced by inflammatory stimulation, and COX-2 is responsible for the production of pro-inflammatory PGE2.201 Glucocorticoids are also used to treat inflammatory diseases. It has been suggested that glucocorticoid treatment can not only up‐regulate the NF‐κB inhibiting protein like I‐κB but also prevent DNA binding and transcriptional activation of NF‐κB.203 Furthermore, receptor antagonists, such as TNF receptor inhibitors, IL‐1β receptor antagonists, IKK inhibitors, and inhibitors of NF‐κB nuclear translocation also considered novel therapeutics for the treatment of OA. The TNF inhibition strategy has recently been tested in two clinical trials. The results showed that anti-TNF antibody adalimumab and etanercept (TNF inhibitor) treatment for 12 weeks and 24 weeks, respectively, showed no differences in pain control in hand OA.204,205 The results of clinical trials for IL-1 inhibitors seem to be controversial. Both AMG108, a monoclonal antibody that blocks IL-1α and IL-1β, and ABT-981, an immunoglobulin targeting both IL-1α and IL-1β, displayed negative results in amelioration of the OA symptoms in patients,204,206 but IL-1 inhibitor canakinumab, which is used to treat atherosclerotic disease to reduce cardiovascular risk, showed a reduced rate of joint replacement cases in the treatment group.77 Biological treatment of OA by knee joint intra-articular injection of the anti-inflammatory drug canakinumab has been under the clinical trial phase II since Nov 2022 (NCT04814368).
AMPK pathway
Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a heterotrimeric complex that contains one catalytic subunit α and two regulatory subunits β and γ,207 and each subunit is encoded by two or three different genes leading to 12 different α/β/γ combinations of distinct AMPK complexes (Fig. 5). It senses low cellular ATP levels and responds to the decreasing of ATP to AMP ratio by regulating metabolic enzymes to boost ATP generation and suppress ATP consumption, which is an essential regulator for energy homeostasis. When cellular energy is low, Thr-172 in the α subunit is phosphorylated, leading to the activation of the AMPK complex. The major kinases that catalyze this reaction are the LKB1 (liver kinase B1) and calcium-sensitive kinase CAMKK2 (also known as CAMKKβ) which respond to calcium flux. AMPK expression and potent phosphorylation of AMPKα are detected in normal articular chondrocytes. Compared with normal joint tissues, the articular cartilage of OA patients and joint tissues of aging-related and surgically-induced OA mice have significantly reduced AMPKα phosphorylation at T172.208 Several studies have demonstrated that the expression of AMPK is significantly reduced in mouse OA articular chondrocytes and human knee OA chondrocytes.209,210 AMPKα phosphorylation was also significantly reduced in a DMM-induced mouse model of OA and acute gouty arthritis (AGA) rats.208,211 In line with these findings, deletion of AMPKα1, AMPKα2, or both in mouse cartilage caused more severe knee injury 2–4 weeks after DMM surgery,212 suggesting AMPK activation is a protective factor for articular cartilage. However, how the expression and activation of AMPK are reduced in OA chondrocytes is still unknown. Reduced LKB1 activity could be one of the reasons why the phosphorylated AMPK is decreased in human and mouse OA chondrocytes since downregulation of LKB1 and AMPK activity has been found in biomechanically induced OA to modulate matrix catabolism,208 but genetic studies on the role of LKB1 in the onset and development of OA is still missing. Interestingly, CaMKK2, another kinase that induces AMPK phosphorylation, has been reported to be elevated in DMM-induced OA cartilage and IL-1β-treated articular chondrocytes. Suppression or loss of CaMKK2-protected DMM-induced cartilage destruction.213 This discrepancy might be due to the other potential functions of CaMKK2 that are independent of AMPK or CaMKK2 not being the major enzyme fulfilling the AMPK phosphorylation in the articular cartilage context.
Fig. 5.
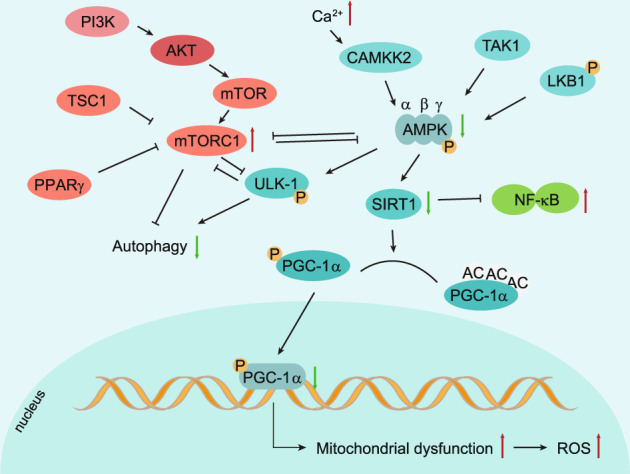
Regulation of chondrocyte autophagy and metabolism by AMPK and mTOR signaling pathways. Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a heterotrimeric complex that contains subunits α, β, and γ. LKB1 (liver kinase B1), CAMKK2 (calcium-sensitive kinase) and TAK1 (transforming growth factor-β-activated protein kinase-1) are three major enzymes that can introduce AMPK phosphorylation. In OA chondrocytes, inhibited AMPK phosphorylation decreases ULK1 expression, which is an essential initiator of autophagy. Decreased phosphorylation of AMPK also downregulates the expression of PGC-1α through SIRT-1 in articular chondrocytes, leading to mitochondrial dysfunction
TAK1 (transforming growth factor-β-activated protein kinase-1) was another enzyme that can introduce AMPK phosphorylation in certain cellular contexts in vitro.214,215 Over-expressing Tak1 by intra-articular injection of adenovirus expressing TAK1 induced cartilage destruction in rats, and inhibiting TAK1 by small molecule 5Z-7 protected against DMM-induced cartilage degradation.216 However, this study did not examine the expression level and activity of AMPK. Therefore, whether TAK1 is one of the enzymes that phosphorylate and activate the AMPK pathway and the effect of AMPK overactivation in the articular cartilage context in vivo remains to be determined.
Dysfunction of mitochondria and impaired mitochondrial biogenesis in OA chondrocytes have been determined to be related to abnormal AMPK activity during OA development.208,217 AMPK activator, A-769662, which induces the upregulation of peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC1α) could reverse the impaired mitochondria biogenesis in OA chondrocytes through regulating SIRT1 signaling.209,218 Impairment of AMPK activity further induced mitochondrial dysfunction resulting in insufficient energy supply and excessive ROS production.208 Furthermore, it was reported that NF-κB signaling was directly or indirectly regulated by AMPK as an important inflammatory response pathway. AMPK deacetylated p65 through SIRT1, induced proteasomal degradation of p65, and ultimately inactivated the NF-κB signaling.219 Activation of AMPK with a glycolysis inhibitor 2-deoxyglucose (2-DG)) reduces synovial inflammation and joint damage primarily by inhibiting the NF-κB pathway in adjuvant arthritis (AA) rats.220 AMPK phosphorylated and activated the ULK1(Unc-51-like kinase 1), which is a key initiator of the autophagy.221 AMPK also plays a key role in the regulation of ER stress. CHOP (C/EBP homologous protein), one of the UPR components, inhibited AMPK activity via IL-1β in OA cartilage where UPR activation is triggered, and activation of AMPK by AICAR (5-Aminoimidazole-4-carboxamide ribonucleotide) attenuated the enhanced expression of CHOP in biomechanical injured articular cartilage.222 Quercetin was reported to suppress ER stress by activating the SIRT1/AMPK signaling pathway.223 AMPK is involved not only in the degradation of articular cartilage but also in the pathological changes of the synovium. AMPK is involved in the anti-inflammatory response of the synovium. For example, transforming growth factor-β1 (TGF-β1) activates AMPK to promote FOXO3 expression, which inhibits the synthesis of inflammatory factors in the synovial fibroblasts of human knee OA.224
Restoring AMPK activity may be able to reduce inflammation and prevent OA progression. For example, the catabolic response of chondrocytes to inflammatory cytokines was significantly inhibited by the selective AMPK agonist AICAR.209 Metformin was reported to limit OA by upregulating the expression of total and phosphorylated AMPK in articular cartilage tissue in mice and rats.225–227 Further experiments demonstrated that metformin could upregulate AMPKα1 expression and delay the development of OA.225–227 In addition, metformin could also relieve OA pain by upregulating the expression of total AMPKα1 in DRG cells and inhibiting the pain sensitivity caused by DMM surgery.225,228,229 In addition, active vitamin D reduced inflammation by triggering the AMPK-mTOR pathway in mouse OA.230 Chitosan oligosaccharide (COS) has also been reported to inhibit synovial inflammation in vivo by activating AMPK at the early-stage OA.231 Administration of rapamycin, which inhibits mTOR, reduced the severity of experimental OA in mice.232 In addition, the safflower yellow,233 estrogen,210 puerarin,234 and bilobalide235 were reported to attenuate OA by promoting AMPK activation. They could be some novel therapeutic options for OA treatment.
Metformin is the first-line clinical drug in the treatment of type 2 diabetes.236 Currently, more and more studies have proved that it has not only hypoglycemic properties but also anti-aging and anti-inflammation properties.237 In recent years, studies of metformin in OA treatment have attracted extensive attention. Metformin, as an anti-diabetic drug, could indirectly ameliorate the negative impact of the obesity factor on OA disorder.238 Clinical studies have also shown that metformin had beneficial effects on long-term knee prognosis in obese patients.239 In addition, metformin could act as an effective activator of AMPK.240 Because of the important role of AMPK in pain signal transduction and metabolism of chondrocytes, studies have found that metformin also plays important roles in OA treatment by suppressing inflammation, regulating autophagy, reducing oxidative stress, and relieving pain.227,241–244 Inflammation can upregulate the expression of catabolic enzymes, such as ADAM, ADAMTS, and MMPs family, resulting in cartilage degeneration.245 Metformin acts directly or indirectly on a variety of signaling pathways, including mTOR/STAT,246 sirt1/NF-κB,247 Dicer/microRNA-34a-5p, and miR125b-5p,248 and HDAC5/KLF2,249 reduces the release of early inflammatory mediators and inhibits the inflammatory response.244 Autophagy is associated with maintaining cartilage homeostasis during OA development.250 AMPK can activate key kinases that induce autophagy.251,252 It has been reported that metformin treatment can enhance AMPK in a dose-dependent manner to inhibit mTORC1 and upregulate the expression of LC3, an autophagy marker, in chondrocytes.226 Metformin also enhanced autophagy in chondrocytes via the AMPKα2-SIRT1 signaling pathway, and the expression of autophagy-associated proteins was decreased by silencing AMPKα2 but not AMPKα1.227 Metformin also plays a protective role in cartilage by upregulating SIRT3, which is located in the mitochondria and regulates mitochondrial function through AMPK activation and AMPK-independent activation.244,253 In addition, a study showed that metformin reduced pain sensitivity in mice by upregulating AMPKα1 expression in the dorsal root ganglia (DRG).225 In a rat OA model induced by monosodium iodoacetate (MIA), metformin treatment decreased the expression of pain-related mediator CGRP in the DRG.243 In conclusion, metformin not only reduces blood glucose but also protects cartilage from degradation. Metformin is a promising therapeutic drug for OA.
mTOR pathway
The mTOR (mechanistic target of rapamycin kinase) is a serine/threonine-protein kinase that is evolutionarily conserved and regulated by various cellular signals. It plays a key role in growth, metabolism, autophagy, and other biological processes.254 mTOR, which forms subunits of two protein complexes referred to as mTOR Complex 1(mTORC1) and mTOR Complex 2 (mTORC2), belongs to the phosphatidylinositol 3-kinase (PI3K)-related kinase family due to its catalytic domain being similar to lipid kinases PI3K.255 mTORC1 contains three key components: mTOR, Raptor (regulatory protein associated with mTOR) that facilitates substrate recruitment and subcellular localization of mTORC1, and mLST8 (mammalian lethal with Sec13 protein 8), also known as GβL that contains catalytic domain of mTORC1. The two inhibitory subunits of mTORC1 are known as PRAS40 (proline-rich Akt substrate of 40 kDa) and DEPTOR (DEP domain containing mTOR interacting protein). Similar to mTORC1, mTORC2 also contains mTOR and mLST8, as well as the Rictor instead of DEPTOR, as its three main components, and mSin1 and Proctor1/2 as its regulatory subunits.256 mTORC1 boosts protein synthesis by phosphorylation of S6K1 (p70S6 kinase) and 4EBP (eIF4E binding protein) and regulates protein turnover through targeting proteasome-related protein ubiquitylation and degradation.257 It also promotes lipid and nucleotide synthesis in fast-growing and proliferating cells and facilitates cell growth by boosting the glucose metabolic shifts from oxidative phosphorylation to glycolysis. mTORC2 regulates cell proliferation and cell survival mainly by phosphorylating and activating Akt, a key regulator of insulin/PI3K pathway.258 In addition, mTORC2 controls cell growth and metabolism by phosphorylating AGC protein kinase family members. mTORC1 is regulated by growth factors and response to unfavorable growth conditions, such as DNA damage, hypoxia, low ATP levels, and depletion of amino acids.258 However, mTORC2 is primarily regulated by insulin levels.259 Dysregulation of mTOR can lead to many diseases, such as cancer, obesity, diabetes, and Alzheimer’s disease.255
Global ablation of mTOR causes embryonic lethal in mice.260 mTOR is crucial for the early stage of cartilage development and growth. The expression of mTOR is significantly upregulated in human OA cartilage and in articular chondrocytes of mouse experimental OA models.261 Conditional deletion of mTOR in articular cartilage upregulated chondrocyte autophagy and protected mice from DMM-induced OA.262 However, genetic evidence of the distinctive roles of mTORC1 and mTORC2 in articular cartilage and OA development is still missing. Activation of mTORC1 by cartilage-specific inactivation of Tsc1 (tuberous sclerosis complex 1), a mTORC1 upstream inhibitor, stimulated articular chondrocyte differentiation to initiate OA by downregulating parathyroid hormone (PTH)/PTH-related protein (PTHrP) receptor and FGFR3.263 The deletion of PPARγ (peroxisome proliferator-activated receptor-γ) in mouse chondrocytes increased mTOR expression, resulting in the overproduction of OA inflammatory and catabolic factors, which caused chondrocyte apoptosis, cartilage degradation, and accelerated OA progression.264 In contrast, PPARγ-mTOR double KO mice displayed delayed OA acceleration caused by the PPARγ loss [10].
Inhibition of mTOR expression in chondrocytes enhanced the expression of AMPK and the autophagy gene LC3 and ULK1.232 The interaction between ULK1/AMPK and the mTOR signaling pathways is a critical component in the regulation of autophagy (Fig. 5). Activation of mTORC1 decreased the phosphorylation of ULK-1, thus inhibiting the activation of ULK1, leading to the suppression of the autophagy.265 Pharmacological inhibition of mTOR by rapamycin enhanced cartilage matrix production and the content of collagen type II and decreased the expression of cartilage matrix-degrading proteins, including ADAMTS5 and IL-1β in articular cartilage.232
Abnormal mechanical loadings play an important role in the occurrence and progression of the OA.266 Abnormal mechanical stress caused degradation of articular cartilage by triggering the mTORC1 signaling, which modulated the apoptosis and autophagy of chondrocytes in TMJ (temporomandibular joint) both in vitro and in vivo.267 Subchondral bone has been reported to play a regulatory role in OA development. The mTORC1 was reported to be activated in the osterix-positive osteoblast in the subchondral bone of human and mouse OA. Eliminating TSC1 (tuberous sclerosis 1), a mTORC1 inhibitor, to constitutively activate mTORC1 in pre-osteoblasts stimulated osteosclerosis and CXCL12 secretion, thus exacerbating OA. Meanwhile, suppressing mTORC1 by deleting the mTORC1 component, Raptor, inhibited the formation of subchondral bone and degeneration of the cartilage of anterior cruciate ligament transection (ACLT) induced OA in mice.268
mTOR is also associated with the level of synovial inflammation.269 Rapamycin, a specific inhibitor of mTORC1, reduced ADAMTS5 and IL-1β levels in the synovitis of the knee joint in OA mice.232 microRNAs (miRNAs), as one of the epigenetic mechanisms that regulate gene expression,270 play a crucial role in OA. The miR-7, miR-27a, and miR-218-5p were reported to inhibit the activity of the PI3K/Akt/mTOR signaling pathway, thus enhancing autophagy and inhibiting the apoptosis of chondrocytes.271–273 In addition, exosomes derived from MSCs in the sub-patellar fat pad protected mice from DMM-induced OA partially by promoting autophagy through suppression of mTOR by miR-100-5p.274
Inhibition of the mTOR pathway by using rapamycin activated autophagy in the knee joint of mice and thus reduced the severity of experimental OA.232 Similarly, the hydroethanolic extract of Butea monosperma (BME) activated autophagy and inhibited IL-1β-induced MMP-3 and IL-6 expression by inhibiting the mTOR.275 Intra-articular injection of mTOR inhibitors, such as rapamycin, in the early stage of OA, is a promising therapeutic strategy for OA treatment.
HIFs pathway
Mammalian cells respond to low oxygen tension microenvironments by activating hypoxia-inducible factors (HIFs) to help the cell survival.276 Functional HIF proteins are heterodimers consisting of an α-subunit (HIF-α), which is encoded by HIF1α, HIF2α, or HIF3α three genes, and a β-subunit (HIF-β), also referred to as the ARNT (aryl hydrocarbon receptor nuclear translocator).277 When the oxygen tension is normal, HIF-1α is hydroxylated by three HIF-specific prolyl hydroxylases, PHD1, PHD2, and PHD3. The hydroxylated HIF-α protein is tagged by the pVHL (von Hippel-Lindau) factors with polyubiquitin, and subsequently targeted for degradation.277 When the cells are under hypoxic conditions, the activity of PHDs is inhibited, leading to impaired hydroxylation and accumulation of HIF-α proteins in the cytoplasm.277 The accumulated HIF-α protein is then translocated to the nucleus. It binds with HIF-β to function as a transcriptional factor to regulate hundreds of genes, including erythropoietin (EPO), glucose transporter-1 (GLUT1), and vascular endothelial growth factor A (VEGFA).277
HIF-1α is a crucial survival factor for hypoxic chondrocytes in vivo.278 It has been recently demonstrated that HIF-1α promotes hypoxic chondrocyte survival by suppressing mitochondrial respiration in the fetal growth plate in vivo.279–281 In contrast, the function of HIF-2α in endochondral bone development has shown controversial results. HIF-2α heterozygous global knockout mice showed impaired endochondral ossification by decreasing the Col10a1 expressing and suppressing hypertrophic differentiation and matrix degradation.282 On the other hand, it is reported that homozygous loss of HIF-2α in limb bud mesenchyme cells achieved by using conditional knockout strategy led to only a modest delay of endochondral bone development in the prenatal stage without affecting Col10a1 expression and accumulation. The delayed bone development phenotype was transient and not detectable in the postnatal stage in mice.283 The mechanism leading to this discrepancy requires further investigation.
Articular cartilage is an avascular tissue and has access to minimal oxygen.284,285 Therefore, articular chondrocytes are under hypoxia in physiological conditions, and accumulated HIF-1α and HIF-2α proteins were detected both in mice and human.286,287 The number of HIF-1α-positive articular chondrocytes was decreased at the early stages of post-traumatic OA mice models.285,288 HIF-1α protein inhibited Wnt signaling by blocking TCF4-β-catenin interaction and down-regulated MMP13 expression to protect articular cartilage loss that induces OA phenotype in mice.285,288 It suggests that HIF-1α acts as a protector of articular cartilage integrity in physiological conditions (Fig. 6). Several reports showed that HIF-1α and its target genes were expressed in normal human knee joint articular cartilage and were significantly increased in the cartilage and synovial fluid of joints of OA patients.286,289 In addition, HIF-1α-positive chondrocytes were increased with the severity of OA,286,289 and miRNA-411 were reported to target HIF-1α in regulating chondrocyte autophagy in OA.290,291 Then, HIF-1α was demonstrated to act as a protector to alleviate apoptosis and cell death in OA by promoting mitophagy in OA cartilage,292 which supports the notion that HIF-1α serves as a promising strategy for OA treatment. Activation of HIF signaling by conditional knocking out VHL in articular chondrocytes led to cartilage degradation in aged mice but did not affect the gross appearance of articular cartilage in young mice.293 The HIF-1α protein level was similar in VHL knockout articular cartilage compared to that in control mice, while the HIF2α protein was significantly increased.293 HIF-1α is downregulated in the articular chondrocytes at the early stage of OA but significantly upregulated at the late stage of OA in humans and mice. However, the effect of overexpression of HIF-1α in articular cartilage in transgenic mouse models remains to be determined to further identify the role of HIF-1α in OA development. The expression of HIF-2α showed a marked increase in OA cartilage compared to that on normal cartilage in mice and in humans, and HIF-2α-heterozygous deficient mice showed resistance to OA development.282,287 Overexpression of HIF-2α triggered the destruction of articular cartilage in mice and rabbits.287 In contrast to HIF-1α, HIF-2α was reported as a risk factor that directly induces the expression of OA risk factor Runx2 and catabolic genes, including MMP1, MMP3, MMP13, ADAMTS4, and NOS2, to promote cartilage destruction.287 There are few studies about the function of HIF-3α in cartilage and OA development. Besides hypoxia, the activation of the NF-κB pathway induced by cellular inflammation and the upregulation of mTORC1 could also induce the overexpression and excessive accumulation of HIFs protein in articular chondrocytes.294,295
Fig. 6.
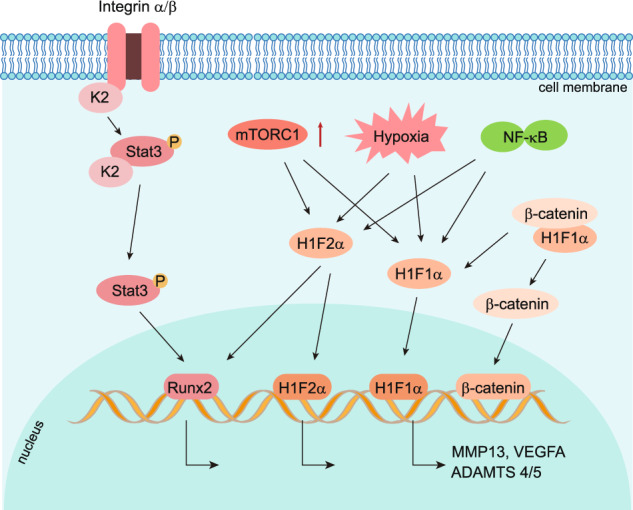
Role of focal adhesion and HIFs pathway in OA chondrocytes. Focal adhesion protein Kindlin-2(K2), which is co-localized with integrin α on the cell membrane, is highly expressed in adult articular chondrocytes. In OA chondrocytes, loss of Kindlin-2 promotes chondrocyte hypertrophy and catabolism by activating the phosphorylation of Stat3 and upregulating transcription factor Runx2. HIF-1α and HIF-2α are both activated by hypoxia. HIF-1α inhibits Wnt signaling by blocking β-catenin translocation to the nucleus and downregulates MMP13 expression in healthy articular cartilage in mice. HIF-1α λ is upregulated in OA chondrocytes and induces abnormal blood vessel invasion by upregulating VEGFA. HIF-2α directly induces the expression of OA risk factor, Runx2, and catabolic genes encoding MMP1, MMP3, MMP13, ADAMTS4, and NOS2, to promote cartilage destruction in OA chondrocytes. Besides hypoxia, the activation of the NF-κB pathway and the upregulation of mTORC1 induce the overexpression and excessive accumulation of HIFs protein in articular chondrocytes
Two new PHD2 inhibitors have been synthesized, known as TM6008 and TM 6089, to selectively inhibit PHD2, but not PHD1 and PHD3, to activate the HIF pathway.296 Other PHDs inhibitors, such as FK506-binding protein 38 (FKBP38) and desferrioxamine (DFO), have also been tested in animal models.296 However, the strategy of inhibiting PHD2 as a therapeutic target for OA is a double-edged sword since both HIF-1α and HIF-2α will be activated after the inhibition of PHD2. The benefit of accumulating HIF-1α could be neutralized by also activating the OA risk factor HIF-2α. Therefore, the specific HIF-2α inhibitor is an alternative promising therapeutic strategy. Resveratrol and YC-1 (3-(5’-hydroxymethyl-2’-furyl)-1-benzylindazole) were reported to prevent OA progression in vivo by suppression of HIF-2α indirectly in mice.297,298 Given that HIF-1α and HIF-2α have the opposite function in OA progression, a combination treatment of PHD2 inhibitor and specific HIF-2α inhibitor could be a potential strategy.
Focal adhesion pathway
Focal adhesions (FAs) are complex macromolecular assemblies that form dynamic mechanical links between intracellular actin bundles and the ECM to control a series of fundamental cellular processes, such as cell adhesion, migration, and signal transduction.299–303 Integrins are heterodimer transmembrane receptors consisting of one α subunit and one β subunit, which facilitate cell-cell and cell-ECM adhesion. After integrins anchoring with ECM proteins, many structural and signaling proteins, such as talin, kindlin-2, vinculin, paxillin, pinch1/2, zyxin, α-actinin, focal adhesion kinase (FAK), and phosphotyrosine proteins, are recruited to the cell-ECM contact sites to form FAs.299 During the last decade, cumulating evidence has pointed out that the FAs signaling plays a central role in controlling organogenesis, metabolism, and homeostasis.179,300–311 Genetic ablation of FAs-related genes in mesenchymal stem cells leads to severe chondrodysplasia, neonatal lethality, and skeletal abnormalities.303,311,312 More importantly, critical molecules of FAs, such as kindlin-2 and integrins, have been implicated in the pathogenesis of OA, considering their essential functions of mediating mechanotransduction, articular cartilage homeostasis, synovial activation, and subchondral bone remodeling.313–315 For instance, Lian and coworkers have reported that ECM protein type II collagen promoted the interaction between integrin β1 and Smad1, thereby suppressing articular chondrocyte hypertrophy during OA development.316 Moreover, changes in subchondral bone architecture altered the distribution of mechanical stress on articular cartilage and induced a talin-dependent cytoskeletal reorganization and the consequent increase of cell contractile forces and cell stiffness of articular chondrocytes, leading to αV integrin-mediated TGFβ activation.317 The latter further disrupted the cartilage homeostasis by enhancing the catabolic activity of articular chondrocytes, leading to OA progression.317,318 An increased expression of αVβ6 integrins was observed in OA synovial tissues, which also mediated TGF-β activation through their interaction with a protein fragment of vitronectin (amino acid 381–397).319 Furthermore, dysregulation of integrin αVβ3 and the integrin-associated receptor CD47 signaling promoted synovial inflammation, cartilage degradation, and OA progression.320 The roles of integrins in OA development and progression have been recently reviewed by Jin and colleagues.313
A recent study by our group has revealed a crucial role of key FA protein kindlin-2 in maintaining articular cartilage homeostasis in adult mice.308 We found that kindlin-2 is highly expressed in adult articular chondrocytes. Genetic deletion of kindlin-2 in these cells caused multiple OA-like phenotypes, including spontaneous cartilage degradation and synovial inflammation, subchondral bone sclerosis, and hyperalgesia. In in vitro and in vivo experiments, kindlin-2 deficiency promoted chondrocyte hypertrophy and catabolism by activating the Stat3-Runx2 pathway in articular chondrocytes (Fig. 6). Mechanistically, we found that kindlin-2 was robustly expressed in the mitochondria of articular chondrocytes, and loss of kindlin-2 resulted in mitochondrial dysfunction and excessive production of ROS. Moreover, we demonstrated that kindlin-2 is bound to the Stat3 protein to inhibit the ROS-mediated activation of the Stat3-Runx2 pathway in chondrocytes. Intraarticular injection of kindlin-2-expressing adeno-associated virus attenuated aging- and instability-induced OA damages in mice. Similarly, pharmacological inhibition of FAK decelerated articular cartilage degeneration and subchondral bone deterioration in a surgically induced rat OA model.321 Pharmacological inhibition of FAK relieved the degradation of articular cartilage of the mandibular condyle under mechanical loading by inhibiting the expression of pro-inflammatory cytokines and ECM-degrading enzymes.322 Miyauchi and coworkers showed that genetic deletion of the Hic-5 (hydrogen peroxide-inducible clone-5), a focal adhesion mechanosensitive adapter, alleviated the progression of OA through regulation of chondrocyte catabolism induced by mechanical stress.323
Taken together, current evidence suggests that the FAs signaling plays a critical role in the pathogenesis of OA. Further studies are needed to explore the molecular mechanisms underlying how alterations in the FAs signaling contribute to the initiation, development, and progression of OA.
TGFβ and BMP signaling pathways
Growth factor transforming growth factor β (TGFβ) superfamily consists of BMPs (bone morphogenetic proteins), TGFβs, growth/differentiation factors (GDFs), and other factors.324,325 TGFβ/BMP signaling is transduced by heteromeric transmembrane receptor complexes composed of type I and type II serine/threonine kinase receptor dimers. Seven Type I and five Type II receptors have so far been identified. Several type III receptors have been identified to stabilize the receptor complexes and facilitate the interaction between the ligand and receptor. TGFβ/BMP signaling activates both SMAD-dependent signaling (referred to as the canonical signaling pathway) and SMAD-independent signaling in cells. In the canonical pathway, transmembrane receptors are activated by specific ligands binding, followed by phosphorylation of R-SMAD (receptor-regulated Smads) proteins. Phosphorylated R-SMAD is then translocated to the nucleus and triggers transcriptions of downstream target genes. There are two sets of R-Smad complexes: one set consists of Smad1, Smad5, and Smad8 (also known as Smad9), and the other set is composed of Smad2 and Smad3.326 After receptor activation, these phosphorylated R-Smads are then combined with co-Mediator Smad (co-Smad) Smad4 to form a complex and transported to the nucleus to perform the function of transcriptional factor (Fig. 7). TGFβ/BMP can also activate SMAD-independent signaling pathways, including TAK1(TGFβ−activated kinase 1) and ERK1/2 (extracellular signal-regulated kinase 1 and 2).327,328
Fig. 7.
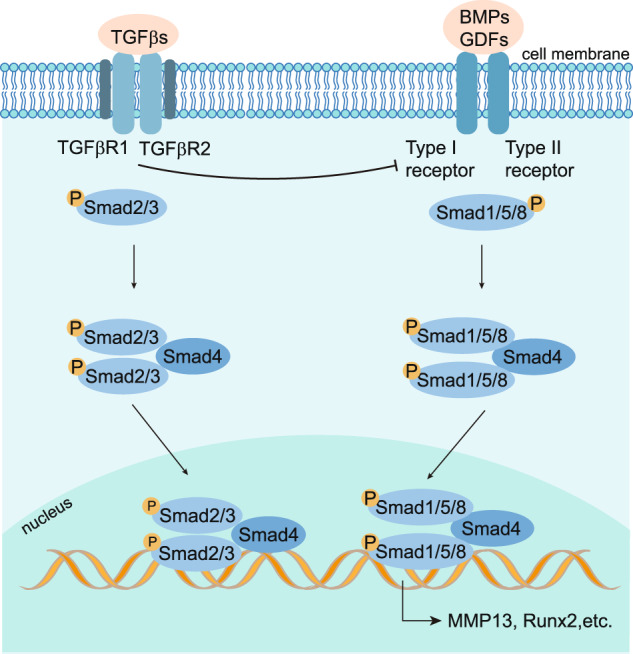
Schematic representation of TGFβ/BMP signaling pathway in OA chondrocytes. TGF-β/BMP family ligands bind to type I and type II receptors on the cell surface. TGF-βs bind TGFβRI and TGFβRII, and BMPs bind BMPRI and BMPRII. TGFβRIII is a coreceptor that facilitates interaction with TβRI and TβRII. TGF-βs induce the phosphorylation of Smad2/3, and BMPs mediate Smad1/5/8 phosphorylation. By forming complexes with Smad4, phosphorylated Smad2/3 and Smad1/5/8 translocate into the nucleus to regulate target gene expression
A series of studies have shown that TGFβ/BMPs are an indispensable signaling pathway for regulating cartilage homeostasis both through canonical and non-canonical pathways.329–331 TGFβ/BMPs signaling protects articular cartilage and participates in chondrocyte hypertrophy and ECM degradation.332 BMP2, BMP4, BMP6, and BMP7 are expressed in articular cartilage and induce the production and proliferation of ECM proteins, such as proteoglycan and collagen type II.333–338 BMP2 knockout mice formed un-mature knee joints and had dysfunctional meniscus and articular cartilage defects, leading to progressive OA with age.339 Although BMP7 had similar effects to BMP2, BMP7-Smad1/5 signaling was not associated with chondrocyte hypertrophy.340,341 BMP7 knockout mice exhibited increased proteoglycan loss and Mmp13 expression.342 Meanwhile, the signaling crosstalk between the BMP signaling pathway and the Wnt/β-catenin signaling pathway plays an important role in regulating cartilage homeostasis.343 Loss of TGFβR1 in mouse growth plate cartilage increased basal BMP activity, suggesting that TGFβR1inhibits BMP signaling in the development of the growth plate.344 However, the role of TGFβR1 and TGFβR2 in the homeostasis of articular cartilage is still unknown. How the non-canonical BMP pathway regulates the homeostasis of the articular cartilage and contributes to the OA onset and development remains to be determined.
The BMP signaling pathway is abnormal and unstable in OA patients.345 The expression of BMP-2 and BMP-4 in chondrocytes of articular cartilage of OA patients was significantly upregulated in the early stage and increased with OA degree. BMP-4 promoted the pathological remodeling of the osteochondral junction.346 OA significantly promoted BMP2 and MMP-13 expression in the subchondral bone of the experimental OA rat model. This increase was inhibited by intra-articular injection of noggin protein (a BMP2 inhibitor). Meanwhile, Noggin protein dramatically attenuated OA disease progression in early OA rat model.347 Moderate exercises increased the expression of BMP signaling pathway-associated proteins, such as BMP2, BMP6 and BMP receptor 2, and pSmad-5, thus inhibiting cartilage degeneration and attenuating the OA phenotype.348
More and more studies focus on the potential therapeutic targets of OA. miR-181a is critical for crosstalk between the BMP and Wnt/β-catenin signaling pathways and may become a target for OA therapy.349 BMP7-derived peptides ameliorated OA chondrocyte phenotype in vitro and attenuated cartilage degeneration in vivo.350 The clinical trial phase II of the intra-articular injection of BMP-7 for the treatment of knee OA patients was completed (NCT01111045) in 2011. BMP receptor type I (BMPRI) mimetic peptide CK2.1 promoted articular cartilage repair and inhibited chondrocyte hypertrophy through intra-articular injection.351 Several studies showed that BMP-2 and BMP-4 effectively decreased cartilage degeneration in combination with tissue engineering materials. Recombinant human BMP-2 promoted cartilage regeneration in injured cartilage in vitro and was evaluated in clinical trials (NCT00243295). Tissue engineering materials, such as porous Hydroxyapatite Collagen (HAp/Col), Conically GRADED scaffold of chitosan-hax Hydrogel/Poly (L-Lactide-co-Glycolide) (PLGA), and POLY caprolactone (PCL) scaffolds, combined with BMP-2 treatment greatly promoted cartilage regeneration and repaired the joint cartilage.352–354 BMP-3 inhibited cartilage regeneration in both partially and completely defective rabbit joint injury models by inducing ECM degradation, inhibited the expression of BMP-2 and BMP-4, and interfered with chondrocyte survival on the articular surface.355 The intra-articular injection of BMP-4 combined with muscle-derived stem cells (MDSCs) could efficiently repair articular cartilage damage in the experimental OA rat model.356 Regular intra-articular injections of BMP7 alleviated the damage to articular cartilage caused by strenuous running.357 The use of BMP6 combined with BMP2 or TGFB3 induced enhanced chondrogenesis of stem cells in vitro.358,359 The combined use of an osteogenic nanoparticulate mineralized glycosaminoglycan scaffold (MC-GAG) and BMP-9 promoted chondrogenic differentiation of primary human mesenchymal stem cells (hMSCs) by inducing increased expression of collagen II, aggrecan and cartilage oligomeric protein.360
FGF signaling pathways
The fibroblast growth factor (FGF) family is present in a wide range of animal species, including nematodes, zebrafish, mice, and humans.361 FGF signaling is involved in a variety of physiological processes, such as cell proliferation, migration, and differentiation.362 There are 18 FGFs in mammals, and they are FGFs1-10 and FGFs16-23. FGFs19, 21, and 23 are hormone-like FGFs that work in an endocrine fashion, and the other FGF members work in a paracrine fashion. FGFs ligands fulfill their functions through binding and activating FGF receptor (FGFR).362 The mammalian FGFR family consists of four members, FGFR1, FGFR2, FGFR3, and FGFR4. FGF signaling involves multiple downstream signaling pathways, including the RAS/MAPK, PI3K/AKT, and PLCγ pathways. In addition, FGF signaling can activate the STAT1/p21 pathway.363
FGF signaling plays a critical role in bone and cartilage development.363 FGFs/FGFRs function at various stages of bone and cartilage development from limb bud formation to long bone growth and maturation.363 FGFR1 and FGFR3 are the predominantly expressed FGF receptors in cartilage. FGFR2 expression is restricted to the pre-cartilage condensate zone, and FGFR2 can serve as an early marker of chondrocytes.364 During growth plate formation, FGFR1 is expressed in pre-hypertrophic and hypertrophic regions, and FGFR2 is observed in quiescent regions.365 FGFR3 was detected in the center of the mesenchymal condensation and all growth plate chondrocytes.364,365 At the same time, FGFs 1, 2, 5, 8, 9, 16–19, 21, and 23 are expressed in the growth plate chondrocytes, and FGFs 1, 2, 6, 7, 9, 18, 21, and 22 are expressed in the perichondrium.366
Previous studies have shown that FGFs 1, 2, 7, 8, 9,18, and 23 are the major ones associated with the pathogenesis of OA (Table 2), but they have diverse functions in the development of the joint disorder.367 The secretion and expression of FGF1 were significantly increased in the synovium in the late stages of the OA. FGF1 was reported to suppress ECM synthesis of the human articular chondrocytes and inhibit the expression of CCN2 (cellular communication network factor 2), which is an important factor that promotes the regeneration of damaged cartilage.368 FGF2 was reported to bind to perlecan, a heparan sulfate proteoglycan in the ECM, and functioned as a mechanosensor in articular cartilage.369 FGF2 binds to FGFR1 to upregulate the expression of MMP1 and MMP13 promoting matrix degradation through neuro-endocrine pathways in adult articular chondrocytes.370,371 FGF8 expression is upregulated in damaged synovium in a rabbit model of OA, and FGF8 was shown to enhance the production of proteases and prostaglandin E2 in inflamed synovial cells, thereby promoting cartilage degradation.372 The expression of FGF9 was downregulated in the human OA cartilage.373 Treatment of exogenous FGF9 attenuated cartilage degeneration but exacerbated the osteophyte formation in a mouse OA model.373 The function of FGF9 in articular cartilage remains to be defined. FGF18 was highly expressed in the superficial zone of articular cartilage and stimulated the expression and accumulation of type II collagen in articular chondrocytes to protect articular cartilage against degeneration.374–376 In addition, the FGF signaling pathway also plays an important role in synovitis. The synthesis and secretion of FGF1 were significantly increased in the synovial fibroblast in OA.377 FGFR2, which is one of the cognate receptors of FGF1, was upregulated in the synovial membrane in OA patients.377 FGF2 is a potent agent to promote the proliferation and chondrogenesis of synovial-derived stem cells.378 The expression of FGF8 was significantly upregulated in hyperplastic synovial cells and fibroblasts in the rabbit OA model.372 Deletion of FGFR1 in adult mouse articular chondrocytes inhibited the progression of articular cartilage degeneration, which was associated with MMP13 downregulation and FGFR3 upregulation.379 Zhou S et al. showed that deletion of FGFR3 in articular chondrocytes in mice resulted in OA-like defects in the temporomandibular joint, which were associated with upregulation of RUNX2 and Indian hedgehog (IHH).380 This suggests that targeting FGF may have a potential strategy for OA treatment. The clinical trials phase II of the only FGF-targeting drug for OA, recombinant human FGF18 (sprifermin), was finished in 2020 (NCT01919164). Intra-articular administration of 100 μg Sprifermin every 6 or 12 months significantly increased the thickness of the femorotibial joint cartilage after two years of treatment with no marked side effects.381
Table 2.
The expression changes of key proteins of each signaling pathway in the synovial joint
| Key protein | Expression location in human | Expression in human OA | Expression in animal OA | References |
|---|---|---|---|---|
| FGF1 | Synovial and articular chondrocytes | Upregulation | Upregulation in MIA-induced KOA in rats | PMID: 21835001 |
| FGF2 | Articular chondrocytes | Unknown | low-molecular-weight FGF2 isoform upregulates in spontaneous OA in mice; while loss of high-molecular-weight FGF2 isoforms downregulates in spontaneous OA in mice | PMID: 17960584 |
| FGF7 | OA meniscal cells | Upregulation | Upregulation in human OA meniscal cells | PMID: 20109188 |
| FGF8 | Unknown | Unknown | Produced by injured synovium in DMM-induced OA in rabbit | PMID: 18699993 |
| FGF9 | Articular cartilage | Downregulation | Downregulation in DMM-induced OA in mice; | PMID: 22841919 |
| FGF18 | Articular cartilage | Downregulation | Downregulation in DMM-induced OA in mice; | PMID: 24577103 |
| FGF23 | OA chondrocytes | Upregulation | Upregulation in posttraumatic OA in rats | PMID: 29718273 |
| FGFR1 | Articular cartilage | Upregulation | Upregulation in spontaneous and DMM-induced OA in mice; | PMID: 21835001 |
| FGFR2 | Articular cartilage | Upregulation | PMID: 26400350 | |
| FGFR3 | Articular cartilage; OA synovial tissue | Downregulation | Downregulation in spontaneous and DMM-induced OA in mice; | PMID: 27041063 |
OA osteoarthritis, KOA knee osteoarthritis, FRZB frizzled-related protein, DMM destabilization of the medial meniscus, MIA monosodium iodoacetate, ACLT anterior cruciate ligament transection, STR/ort mice a well-recognized mouse model which develops spontaneous osteoarthritis very similar to the human disease
Runx2
Runt-related transcription factor 2 (Runx2) is a runt domain-containing transcription factor that binds to DNA as a monomer or, with higher affinity, as a part of a heterodimeric complex.382 During skeletal development, Runx2 is predominantly expressed by pre-hypertrophic and hypertrophic chondrocytes and osteoblast-linage cells and functions as a potent inducer of hypertrophic chondrocyte differentiation as well as osteoblastic differentiation.383–389 Homozygous deficiency of Runx2 in mice caused neonatal lethality with a complete lack of skeletal ossification, whereas heterozygous Runx2 deficiency led to cleidocranial dysplasia, characterized by defective clavicle and skull formation.390,391 Although the expression of Runx2 is essential for skeletal development and bone remodeling, numerous studies have recently suggested that Runx2 also acts as a central pathological factor in OA initiation, development, and progression (Fig. 8). This notion is supported by: (1) elevated expression of Runx2 is a standard feature found in OA cartilage samples from humans and animals130,308; (2) chondrocyte-specific overexpression of Runx2 hastened the progression of posttraumatic OA in adult mice392; (3) genetic deletion of Runx2 in articular chondrocytes attenuated the severity of surgically-induced OA lesions in adult mice, and (4) Runx2 directly regulated the mRNA and protein expressions of chondrocyte hypertrophic marker Col10a1 and catabolic enzymes, such as Mmp13 and Adamts5.393,394 In addition, Runx2 can act as a downstream factor of multiple key signaling pathways, including Wnt/β-catenin, TGF-β/Smad, Indian hedgehog (Ihh), hypoxia-induced factor (HIF), and focal adhesion signaling pathways, to initiate the processes of hypertrophic chondrocyte differentiation and articular cartilage catabolism upon pathological stimulations.130 Despite a well-established role in articular cartilage, Runx2 has also been linked to pathological changes in various joint tissues, such as the meniscus, synovium, and subchondral bone.130 Notably, the involvement of Runx2 in OA pathogenesis has been extensively reviewed by Chen et al. and Zhao et al..130,395 Although recent studies have demonstrated a critical role of Runx2 in OA pathogenesis and highlighted it as a potential therapeutic target,130,308,393 currently, there are no Runx2-targeting drugs or therapies have been developed for the treatment of OA. Considering the essential roles of Runx2 in other organs and tissues,396–398 the recently developed chondrocytes-targeted delivery systems may serve as useful approaches for the specific delivery of Runx2 siRNAs or inhibitors, such as CADD552.399–402 Moreover, the CRISPR/Cas9 system has demonstrated high efficiency in gene editing in multiple joint tissues.403 Intraarticular injections of adeno-associated virus (AAV) expressing Runx2-targeting miRNA sequences significantly downregulated the expression of Runx2 and decelerated OA progression in mice.131 Nonetheless, new Runx2-targeting drugs still need to be designed, and further studies should be conducted to evaluate their therapeutic potential for OA treatment in animal models and humans.
Fig. 8.
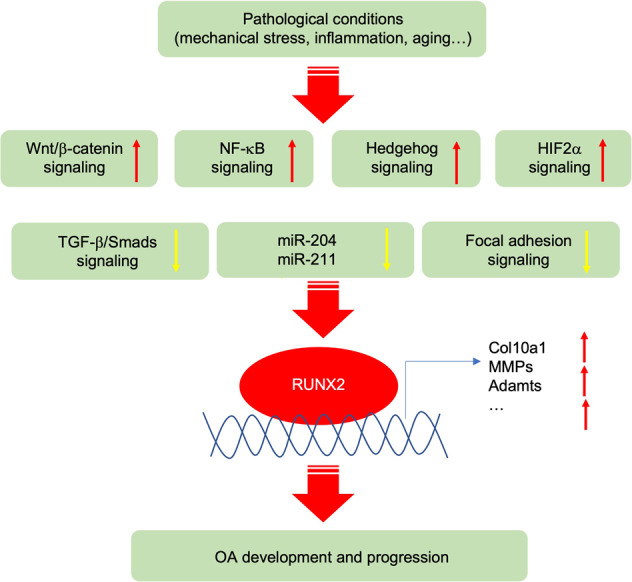
Runx2 in OA pathogenesis. A diagram showing the upstream and downstream of Runx2 in OA pathogenesis. Runt-related transcription factor 2 (Runx2) is upregulated by activation of Wnt/β-catenin, NF-κB, Hedgehog, HIF-2α signaling, and by suppression of TGF-β/Smads and focal adhesion signaling in OA chondrocytes. miR-204 and miR-211 inhibit the expression of Runx2 protein in articular chondrocytes in vivo. Red arrow: upregulated expression. Yellow arrow: downregulated expression
Alterations in the expression patterns of miRNAs were observed in serum and cartilage samples from OA patients, suggesting that miRNAs can serve as biomarkers for OA diagnosis and classification.404,405 In vitro and in vivo studies have demonstrated that miRNAs can regulate articular chondrocyte differentiation, matrix catabolism, inflammatory responses, autophagy, and chondrocyte apoptosis.406–411 miRNAs also control cellular signal transductions by mediating the expression levels of key transcriptional factors, such as Runx2. For instance, Huang and coworkers reported that two homologous microRNAs, i.e., miR-204 and miR-211, exerted multiple crucial functions in maintaining the homeostasis of articular cartilages via targeting the Runx2 expression.131 Genetic deletion of miR-204/211, either globally or selectively in mesenchymal progenitor cells, resulted in striking OA-like phenotypes, including deterioration of articular cartilage, osteophyte formation, subchondral bone sclerosis, and synovial hyperplasia.131 Notably, miRNAs participate in intracellular signal transduction and can also be utilized as mediators in miRNA-based intercellular communication. A recent study has revealed that miRNAs derived from subchondral osteoclasts can transfer to articular chondrocytes via exosomes, contributing to cartilage degeneration, osteochondral vessel invasion, and sensory innervation during OA development.412 These findings suggest that miRNAs play an important role in cartilage homeostasis and OA development.
Matrix metalloproteinases
Matrix metalloproteinases (MMPs) are a family of zinc-dependent proteolytic enzymes that cleave ECM proteins. One type of MMPs is called collagenase, including MMPs1, 8, and 13, which function as collagen-degrading enzymes and contribute to the catabolic turnover of articular cartilage. These MMPs, especially the MMP13, are closely associated with the ECM destruction of articular cartilage in OA. One substrate of MMP13 is type II collagen which is one of the most abundant structural components in articular cartilage. Compared to wild-type mice, MMP13 global knockout mice exhibited reduced articular cartilage degradation in DMM-induced mouse OA models, and constitutive overexpression of MMP13 in cartilage caused OA-like phenotypes in mice.413 Consistent with this, conditional knockout MMP13 in chondrocytes of adult mice decelerated progression at the early and middle stage of OA (8, 12, and 16 weeks post-meniscal-ligamentous injury, MLI, surgery).414 As the primary collagenase in OA pathogenesis, MMP13 was slightly upregulated in articular cartilage and significantly in the synovium at the early-stage OA, specifically at 1- and 3 weeks post-surgery of the rat medial meniscus transection (MMT) OA model.415 MMP2, MMP3, and MMP14 are significantly upregulated at one day, one week, and six weeks time points in articular cartilage of DMM-induced mouse OA models.416 Inhibiting MMP13 or other MMPs at the early- stage of OA could be a potential therapeutic strategy to prevent cartilage destruction and the OA initiation process. Several natural compounds, such as curcumin and resveratrol, were reported to suppress MMP13 expression. Some zinc-binding inhibitors specific to MMP-13, such as N-O-isopropyl sulfonamide-based hydroxamates, were shown to be effective in an in vitro cartilage degradation model.417 Additionally, several highly selective non-zinc binding MMP-13 inhibitors, including pyrimidines, biaryls, and diphenylethers, have been reported recently.418 The monoclonal antibody of MMP-13, which is highly specific over other MMPs, is a potential therapeutic approach,419 despite its difficulty penetrating the cartilage. PF152 has been demonstrated as an effective MMP-13 inhibitor that can decrease human cartilage degradation ex vivo in a dosage-dependent manner.420 MMPs inhibitor PG-530742 (PG-116800) completed its phase II clinical trials ten years ago. However, the results showed that its side effects make it unsuitable for clinical use in humans.421
ADAMTS and ADAMs
A Disintegrin and Metalloproteinase with Thrombospondin Motif (ADAMTS) are proteins that mediate the degradation of the ECM of chondrocytes leading to the distraction of cartilage integrity. ADAMTS family contains 19 secreted zinc metalloproteinases protein members. ADAMTS4/5 are the essential proteases expressed in human and animal OA cartilage tissues that play important roles in the degradation of aggrecan, the major component of cartilage ECM. Expression of ADAMTS4/5 was significantly elevated in articular chondrocytes even at the early-stage OA. ADAMTS5 deletion protected joints from cartilage destruction in surgically induced mouse OA model.422,423 The phase II clinical trials of the film-coated tablets GLPG1972/S201086, which is developed as an ADAMTS5 inhibitor for oral use for OA treatment finished in July 2021 (NCT03595618).In addition, anti-ADAMTS-5 Nanobody (M6495) for OA treatment finished the phase I clinical trials (NCT03224702) in May 2019. ADAMTS1, 2, 3, 7, 12, 14, and 16 were also upregulated at the early-stage OA. ADAMTS8 was expressed in articular cartilage, and its expression level was not changed in the articular cartilage of mouse OA models but increased in the human OA synovium.424,425 There are controversial reports about how ADAMTS15 expression level changes in OA cartilage. A Disintegrin and Metalloproteinases (ADAMS) are proteins responsible for removing membrane-located receptors. Out of 34 known ADAMS proteins, ADAM-8, -9, -10, -12, and -28 were reported to be overexpressed at the early stage of the OA articular cartilage.425 Expression of ADAM-17 was not markedly altered in human OA versus normal articular cartilages.426 ADAM-19 and -23 were upregulated in the late stage of the OA cartilage.427
Lubricin/proteoglycan-4 (PRG4)
Healthy articular cartilage is coated on the surface by lubricin molecules, which provide lubrication boundary and low friction of the cartilage surface. Lubricin encoded by the Prg4 (proteoglycan 4) gene is a secreted proteoglycan protein that is predominantly expressed by chondrocytes in the superficial zone of articular cartilage and synovial fibroblasts.428,429 Patients with a genetic mutation of the PRG4 gene have the CACP (camptodactyly-arthropathy-coxa vara-pericarditis) syndrome, characterized by childhood onset non-inflammatory joint disorder.430,431 Prg4 global knockout mice develop aging-related joint disorders with loss of chondrocytes in the superficial zone of articular cartilage and synovial cell hyperplasia.432 Running and fluid flow shear stress could promote the expression of Prg4 in the superficial zone chondrocytes in vivo and in vitro,433 suggesting the regulation of Prg4 expression can be partially controlled by mechanical forces. As a protective factor for joints, Prg4 is expressed by embryonic joint progenitors. Prg4 positive articular chondrocytes located on the surface of joint cartilage in adult mice have been demonstrated to be the progenitor for deeper layers of the mature articular cartilage.128,434 Exogenous recombinant human (rh) PRG4E was reported to promote ear wound closure and tissue regeneration by increasing VEGF expression and blood flow through a TLR (Toll-like receptor)-dependent mechanism in mice.435 Full-length recombinant human PRG4 (rhPRG4) produced by CHO-M cells and native human PRG4 (nhPRG4) purified from culture supernatants of human fibroblast-like synoviocytes from OA patients were also shown to bind to TLR2 and TLR4, mediating an anti-inflammatory factor.436 However, whether overexpressing Prg4 in the synovial joint can facilitate articular cartilage regeneration or prevent OA development remains to be determined. Lubricin was included as a potential biomarker in human synovial fluid in the diagnosis and progression of OA patients by a clinical observational study in 2022 (NCT02664870).
Other signaling factors
There are several other signaling cascades that have essential roles in the OA onset and development, such as notch signaling. Notch signaling was upregulated in mouse and human OA cartilage.437 Notch2 gain of function mutation in articular cartilage increased the severity of post-trauma OA by crosstalk with NF-κB, Wnt, and TGFβ signaling.438,439 Transient overexpression of NICD (NOTCH1 intracellular domain) led to enhanced synthesis of ECM and promoted the maintenance of articular cartilage, while constitutional overexpression of NICD resulted in early and progressive OA lesions in mice.437 Activation of Hes1, an essential mediator of Notch signaling, suppressed articular cartilage degradation and OA development by decreasing the Adamts5 and Mmp13 expression.440 The endoplasmic reticulum (ER) stress-triggered unfolded protein response (UPR) signaling has been identified as a contributing factor to OA pathology. Older OA patients developed ER stress in the early-stage OA when there was a higher synthesis of ECM proteins.441 How the UPR signaling mediated cell survival and the chronic ER stress-initiated apoptosis in cartilage and synovium contribute to the onset and development of OA needs to be investigated.
Clinical therapy and clinical trials
So far there is no effective cure for OA. The OA treatment approaches are divided into physical modalities, pharmacologic treatments, and surgical treatments.442 Several new therapies have also recently been developed. In the early stages of OA, treatment focuses on reducing pain and joint stiffness. Subsequently, treatment mainly focuses on maintaining joint physical function.443,444 In summary, OA treatment aims to reduce the disease symptoms and slow its progression.
Non-pharmacological treatment
Weight loss
Excessive body weight or obesity is a major risk factor for OA.445 Greater body weight adversely affects joint structure by adding additional load to the joints during daily activities and causing increases in the expression and production of enzymes that degrade the joints or increase joint inflammation.446 Weight loss is recommended for overweight or obese patients with low-extremity osteoarthritis.447
Exercise
According to the recommendations from the International Association for the Study of OA (OARSI), exercise is considered a core approach to the treatment of OA and is recommended for all patients.448 Exercise has been extensively studied as a treatment for OA.448 Uthman and colleagues found that exercise reduced painful movements and improved physical function in OA patients.449 The most common exercises used to treat OA include aquatic exercise,450 aerobic exercise,451 resistance exercise,451 multimodal exercise,452 and combination exercise.452,453
Assistive devices
OA patients often need assistive devices to compensate for decreased strength and impaired pain during exercise.454 Common devices include splints, braces, canes, functional shoes, and other training equipment.455 While there are some positive results from clinical studies, the need for assistive devices and their long-term safety remains in doubt.456
Physical therapy
Physiotherapy has significant therapeutic effects on OA, including therapeutic ultrasound, electrical stimulation, phototherapy, hydrotherapy, magnetotherapy, cryotherapy, and thermotherapy.457–461 Physical therapy provides significant relief of symptoms of OA, including pain, edema, and joint motion disturbances, and is suitable for emergency management in the acute phase.460 Instructing patients to use thermal agents has been recommended as a self-management strategy by the recent American College of Rheumatology Clinical Guidelines.462
Acupuncture
Acupuncture is a non-pharmacological treatment method in Chinese medicine.463,464 Acupuncture has analgesic and functional restorative effects in treating OA.463 The therapeutic effect of acupuncture may come from modulating inflammatory factors.463,465 However, there is evidence of uncertainty in the treatment of OA with acupuncture, particularly a significant difference between electro-acupuncture and hand acupuncture.466 In addition, non-pharmacological strategies, including health education, and lifestyle changes, such as diet, postural correction, and self-management, are important measures to prevent OA.467,468
Pharmacologic treatment
Currently, no drugs can alter the progression of OA and prevent long-term disability.469 Current international guidelines recommend medications for the treatment of OA that revolve solely around reducing the burden of the disease (symptomatic effects) and altering the natural course of the disease by slowing or stopping the biological process of tissue damage.469,470 The following classes of drugs are currently used to treat OA of the knee: non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, opioids, symptomatic, chondroprotective agents, and anti-cytokines.470–472
Paracetamol (acetaminophen) is the first-line analgesic for clinical pain control of arthritis.470 The safety profile of paracetamol relative to other analgesics, such as non-steroidal anti-inflammatory drugs (NSAIDs), has led to its increased use, resulting in paracetamol becoming one of the most common drugs used in OA treatment.470 However, there is evidence that paracetamol is associated with an increased risk of gastrointestinal, cardiovascular, and renal disease, as well as mortality.473 NSAIDs are commonly used anti-inflammatory and analgesic drugs in the treatment of OA.474,475 NSAIDs, including ketorolac, and COX-2 inhibitors, such as diclofenac, ibuprofen, celecoxib, and rofecoxib,474,476 have finished phase 4 clinical trial (NCT01444365) in April 2012. Acetaminophen has been found to be effective in the treatment of OA, and acetaminophen is usually safe and may be better in situations where NSAIDs are contraindicated, has finished phase 4 clinical trial (NCT00635349) in April 2009.477–479 Glucocorticoids are another important class of drugs used in the OA treatment480,481 and finished the phase 4 clinical trial (NCT05291650) in Dec. 2022. When signs of inflammation appear, intra-articular injections of glucocorticoids can very quickly eliminate joint effusion and provide analgesia.481,482 Glucocorticoids include methylprednisolone acetate, trenbolone acetate (TA), betamethasone acetate (BA) and betamethasone sodium phosphate (BP), hexamethonium trenbolone (TH) and dexamethasone (DEX).482–484 They exhibit a complex biological activity, including anti-inflammatory and immunosuppressive effects that block the production of pro-inflammatory cytokines, leukocyte recruitment, and activation.485,486 Steroid injections should be used with caution in diabetic patients who are already hyperglycemic.487,488 Chondroprotective agents include montelukast, placebo, glucosamine, chondroitin sulfate, and hyaluronic acid.489–491 Montelukast and placebo entered the early phase I clinical trial (NCT04572256) in Feb. 2021. The mechanisms of action of individual factors have not been fully elucidated, and they range from inhibition of inflammation and injury receptor blockade to potential alterations in the viscoelastic properties of cartilage tissue.490–492 These agents slow the radiographic progression of knee osteoarthritis.492–494 Anti-cytokine treatments have also been considered.495 Many of the drugs used to treat OA are designed to counteract the pro-inflammatory, matrix-destroying effects of cytokines.495 Infliximab, a neutralizing antibody to TNF-α, finished the phase 4 clinical trial (NCT01144143) in June 2010. Further therapeutic approaches include the use of anti-TNF-α antibodies or the use of anti-inflammatory enzymes, such as IL-4, IL-10, IL-13, and TNF-β.496–499 Strontium ranelate inhibits subchondral bone resorption by modulating the activity of osteoprotegerin (OPG), RANK ligands, and MMPs produced by osteoblasts.500,501 Strontium may have a direct effect on cartilage and promotes the synthesis of proteoglycans and thus stimulates cartilage matrix formation in vitro.500,502 Intermittent injections of PTH (iPTH) reduce OA pain and slow the progression of OA by reducing subchondral bone degeneration and cartilage degeneration.503 In addition, iPTH reduces sensory innervation and PGE2 levels in subchondral bone and improves subchondral bone remodeling via PTH1R on Nestin+ MSC.503 Teriparatide entered the Phase 2 clinical trial (NCT03072147) in Mar. 2017. Nerve growth factor (NGF) is thought to modulate signals that control the expression of peripheral and central pain substances and enable neighboring nociceptive neurons to respond to stimuli.478,504–506 Tanezumab is a highly selective immunoglobulin G2 osteoarthritis13 antibody against NGF507,508 and finished the phase 3 clinical trial (NCT02709486) in Nov. 2018. Several studies have shown that tanezumab is more effective in improving knee pain, stiffness, and function in patients with moderate to severe knee osteoarthritis compared to placebo.508–510
Biological treatment
Regenerative therapy has emerged as one of the most recent and rapidly evolving strategies for treating OA.511,512 Platelet-rich plasma extracted from blood samples provides important growth factors which may facilitate OA recovery.511–513 Platelet-rich plasma led to clinical improvements 12 months after injection and completed the phase 4 clinical trial (NCT02984228) in Aug. 2022.511,513 Mesenchymal stem cells (MSCs) related strategies for OA treatment have been extensively studied due to their ability to self-renew, immunomodulatory properties, and potential multilineage differentiation ability (especially toward chondrocytes).514–517 Intra-articular injections of autologous MSCs, usually derived from bone marrow or adipose tissue, finished the phase 4 clinical trial (NCT04675359) in Jul. 2021.514,517,518 Several clinical studies using MSCs-based therapy for regeneration of cartilage in knee OA have reported positive results.519,520 Kim and co-workers injected adipose-derived MSCs into 49 patients and measured treatment outcomes by the IKDC (International Knee Documentation Committee) scores, Tegner activity scores, and overall patient satisfaction with the procedure.521 Results showed that the mean scores of IKDC and Tegner activity were significantly improved both preoperatively and postoperatively, while they noted that patient age and lesion size were important factors affecting clinical outcomes.521 In 2019, Chahal et al. published a non-randomized, open-label, dose-escalating phase I/II clinical study,522 which included twelve 40–65-year-old patients who received different doses of bone marrow-derived MSCs. The follow-up trials from baseline to 12 months indicate that patients showed significant improvements in KOOS and WOMAC hardness scores, quality of life, and symptoms 12 months after BM-MSC treatment.522 In addition, patients treated with higher doses showed better outcomes than those treated with lower doses.522 Furthermore, gene therapy is a new therapeutic approach tested in clinical for OA treatment in recent years. TGF-β1 gene therapy using retroviruses was approved for allogeneic chondrocytes in vitro.523 Kim and coworkers reported the clinical safety and efficacy of the intra-articular injection of tissue gene-c (TG-C) in OA patients. TG-C is a cell-based gene therapy that uses un-transduced and transduced chondrocytes that are retroviral transduction of overexpressing TGF-β1.524 They concluded that TG-C was associated with statistically significant improvements in function and pain in patients with knee OA and started the phase III clinical trial (NCT03291470) in Oct. 2021.
Surgical treatment
As OA continues to worsen, surgery becomes necessary when basic treatment and medications are ineffective.525 The goal of surgery for OA patients is to reduce pain, decrease disability and improve quality of life.526 With the help of modern technology, patients can usually bear weight on the affected limb immediately after surgery.527
Knee arthroscopy
Knee arthroscopy is the most used clinical procedure with minimal invasion to treat OA.528 It can be used to detect injuries of joints, repair injured soft tissues, such as ligaments and tendons, and bones, and remove inflamed and damaged tissue. For articular cartilage, surgical strategies include scraping (debris removal and chondroplasty), repair (microfracture and perforation), or restoration (autologous chondrocyte graft, autologous osteochondral graft, and allogeneic osteochondral graft).529
Artificial joint replacement
Arthroplasty is currently an effective clinical treatment for advanced knee OA. It effectively eliminates pain, corrects joint deformity, and improves knee function.530 However, clinical studies have found that some patients still do not recover satisfactorily after surgery and cannot fully straighten the knee.531 Cartilage injury, ligament injury, postoperative infection, and postoperative deep vein thrombosis in the lower extremity are all factors that influence the outcome of surgical treatment of knee OA, and early postoperative prevention of complications is crucial.531,532
Conclusions and perspectives
OA is a tremendously complex synovial whole-joint disorder, and how OA is initiated and developed remains poorly understood. Research on the cellular and molecular mechanisms of OA is still in the beginning phase. We have summarized from the current knowledge changes of key molecules in the essential signaling pathways in the articular cartilage, synovial membrane, subchondral bone, and synovial fluids of OA patients and animal models (Table 3), as well as the potential therapeutic reagents that have been reported (Table 4). Wnt, TNF, TGFβ/BMP, FGF pathway receptors, FA proteins, and other factors that are located on the chondrocyte membrane sense and transduce biochemical and mechanical signals. Activated signaling pathways and regulators, such as AMPK, mTOR, FGF, BMP, HIFs, and NF-κB, via crosstalk and feedback mechanisms in a complicated network, regulate the expression of key downstream factors, such as Runx2, MMP13, ADAMTS4/5, Prg4, and other factors, in articular chondrocytes and synovium. The initiation factors of OA are various, including excessive mechanical loading, inflammatory factors, aging and etc., which lead to the different primary effects of early-stage OA with unique molecular signaling signatures. The destruction and erosion of the articular cartilage, the synovial hyperplasia and inflammation, the abnormal angiogenesis of the synovial joint, the subchondral bone disturbance, and the instability of the ligaments and tendons could all contribute together or respectfully to the onset and progression of the disease. Scientists and clinic doctors are still debating, and there is still no conclusion on which one happens earlier than the others during the initiation of OA. No matter which pathological factor is the first dominant over others, or they contribute equally to the progression of OA; it has been well accepted that interference at the early stage of OA to prevent its progression will be a more efficient and effective strategy for better outcomes than focusing on the medical treatments and joint replacement surgery at the late and end-stage of the disease. Therefore, targeting critical signaling pathways and key molecules that are significantly changed at the early stage of the disease to control crosstalk between molecular pathways and the interaction of different compartments of synovial joint is critical for future research.
Table 3.
Therapeutic reagents and their strategies and target genes have been reported for each OA pathogenic signaling pathway
| Signaling pathway | Key protein | Articular cartilage | Synovial membrane | Subchondral bone | Joint cavity (synovial fluid) | References |
|---|---|---|---|---|---|---|
| Wnt | Wnt | Increated in DMM-induced OA in mice; Increased in human OA cartilage | Increased in human OA cartilage | unknown | Increated in DMM-induced OA in mice; Increased in human OA cartilage | PMID: 22682243;27147711 |
| WISP-1 | Increased in human OA cartilage | Increased in human OA cartilage | Increased in human OA cartilage | Increased in human OA cartilage | PMID: 19180479 | |
| β-catenin | Increased in human OA cartilage | Increased in human OA cartilage | Increased in human OA cartilage | Increased in human OA cartilage | PMID: 19180479;15866164 | |
| Dickkopf-1 | unknown | unknown | unknown | Decreased in human OA cartilage | PMID: 28678406 | |
| AMPK pathway | AMPKα | Decreased in DMM-induced OA mouse model and Human knee OA chondrocytes. | Decreased in acute gouty arthritis (AGA) rats. | unknown | unknown |
PMID: 25940958;30585453 |
| AMPKβ | unknown | unknown | unknown | unknown | n.a | |
| AMPKγ | unknown | unknown | unknown | unknown | n.a | |
| NF-κB | RelA/p65 | P65 protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p65 translocates to the nucleus in human OA cartilage | P65 protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p66 translocates to the nucleus in human OA cartilage | P65 protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p67 translocates to the nucleus in human OA cartilage | P65 protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p68 translocates to the nucleus in human OA cartilage | PMID: 31842396 |
| RelB | RelB protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; RelB translocates to the nucleus in human OA cartilage | RelB protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; RelB translocates to the nucleus in human OA cartilage | RelB protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; RelB translocates to the nucleus in human OA cartilage | RelB protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; RelB translocates to the nucleus in human OA cartilage | PMID: 31842396 | |
| c-Rel | c-Rel protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; c-Rel translocate to the nucleus in human OA cartilage | c-Rel protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; c-Rel translocate to the nucleus in human OA cartilage | c-Rel protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; c-Rel translocate to the nucleus in human OA cartilage | c-Rel protein is released, phosphorylated, and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; c-Rel translocate to the nucleus in human OA cartilage | PMID: 31842396 | |
| NF-κB1/p105p50 | The precursor protein p105 opposed to degradation and release of active p50; p50 protein is released and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p50 translocate to the nucleus in human OA cartilage | The precursor protein p105 opposed to degradation and release of active p50; p50 protein is released and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p51 translocates to the nucleus in human OA cartilage | The precursor protein p105 opposed to degradation and release of active p50; p50 protein is released and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p52 translocates to the nucleus in human OA cartilage | The precursor protein p105 opposed to degradation and release of active p50; p50 protein is released and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p53 translocates to the nucleus in human OA cartilage | PMID: 31842396 | |
| NF-κB2/p100p52 | The precursor protein p100 opposed to degradation and release of active p52; p50 protein is released and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p52 translocates to the nucleus in human OA cartilage | The precursor protein p100 opposed to degradation and release of active p52; p50 protein is released and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice; p53 translocates to the nucleus in human OA cartilage | The precursor protein p100 opposed to degradation and release of active p52; p50 protein is released and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice p54 translocate to the nucleus in human OA cartilage | The precursor protein p100 opposed to degradation and release of active p52; p50 protein is released and translocated from the cytoplasm to the nucleus in DMM-induced OA in mice p55 translocate to the nucleus in human OA cartilage | PMID: 31842396 | |
| HIF pathway | HIF1a | Decreased in DMM-induced OA in mice; Increased in human OA cartilage. | Upregulated in MIA-induced KOA in rats. | Upregulated in osteoblasts in human OA subchondral bone. | Increased in human OA cartilage. | PMID: 27122313; 28123469 |
| HIF2a | Increased in STR/ort and DMM mouse OA model. Increased in human OA cartilage. | Unknown | Unknown | Unknown | PMID: 28123469; 24999110 | |
| HIF3a | Unknown | Unknown | Unknown | Unknown | ||
| mTOR | mTOR | Increased in DMM-induced OA in mice; Increased in human OA cartilage. | Unknown | No change | Unknown | PMID:24651621 |
| Focal adhesion pathway | Kindlin-2 | Highly expressed in healthy articular cartilage; Decreased in aged and OA cartilage | Unknown | Unknown | Unknown | https://www.nature.com/articles/s43587-021-00165-w |
| Talin | Decreased in human OA cartilage | Unknown | Unknown | Unknown | ||
| Vinculin | Decreased in human OA cartilage | Unknown | Unknown | Unknown | ||
| Integrin αVβ3 | Unknown | Elevated in synovial membrane in DMM mouse model. | Unknown | Unknown | PMID:31534047 | |
| Focal adhesion kinase | Unknown | Unknown | Elevated in subchondral bone in ACLT rat model. | Unknown | PMID:32324278 | |
| BMP pathway | BMP-2 | Increased in human OA cartilage; Down-regulated in DMM-induced OA in rats | Unknown | Increased in human OA cartilage; Increased in ACLT-induced OA in rats | Decreased in patients with OA | PMID: 35216203; 32290085; 29429984 |
| BMP-4 | Increased in human OA cartilage; downregulated in DMM-induced OA in rats | Unknown | Increased in human OA cartilage | Unknown | PMID: 35216203 | |
| BMP-6 | Upregulated in DMM-induced OA in rats | Unknown | Unknown | Unknown |
PMID: 34680516 |
|
| BMP-7 | Decreased in human OA cartilage; upregulated in DMM-induced OA in rats | Unknown | Unknown | Unknown | PMID: 29980690; 26010756 | |
| GDF-5 | Down-regulated in DMM-induced OA in rats | Unknown | Unknown | Unknown |
PMID: 26010756 |
FRZB frizzled-related protein, IκB inhibitors of NF-κB, IKK IκB kinases, HIF hypoxia-induced factor, PHD HIF prolyl hydroxylases, AAV adeno-associated virus
Table 4.
The expression and changes of FGF signaling in human and animal synovial joint and OA
| Signaling pathway | Therapeutic strategy | Target gene | Therapeutic reagent | References |
|---|---|---|---|---|
| Wnt | Inhibition of Wnt protein | Wnt | SM04690 | PMID: 28978991 |
|
Inhibit Wnt signaling in an indirect way by upregulating FRZB expression |
FRZB | Verapamil | PMID: 24658359 | |
| AMPK pathway | Upregulated phosphorylated AMPK (AMPK activator) | AMPKα1 | Metformin; | PMID:32156705; 25940958 |
| AMPK-mTOR signaling pathway | Vitamin D | PMID:31815524 | ||
| AMPK/PGC-1α pathway. | Puerarin | PMID:29961054 | ||
| AMPK-Nrf2 Signaling | Luteolin | PMID:35154568 | ||
| Promoted SIRT1 expression. | SIRT1 | Safflower yellow (SY) | PMID:32871523 | |
| 17β-E2 | PMID:33613450 | |||
| NF-κB | Induce expression of IκB, causing increased cytosolic retention of NF-κB | I-κB | Glucocorticoids, NF-κB ODN, NF-κB morpholinos. | PMID:10760263 |
| Inhibit IKK activity significantly, preventing IκB phosphorylation | IKK | Non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin, salycilate, ibuprofen, and sulindac | PMID:11641233 | |
| Preventing IκBα degradation | 20 S proteasome complex | Bortezomib, Cyclosporin A, sc-514, lactacystin. | PMID:15135296 | |
| Diminution of levels | p65, p50 | siRNA, AAV | PMID:11160335 | |
| Inhibit Nuclear translocation of NF-κB subunits p65, p50 | p65, p50 | FK-506, BMS-205820, I-κB super repressor, Tat-srIκBα. | PMID:8112299 | |
| Inhibit the NF-κB DNA-binding activity | p65, p50 | Glucocorticoids, NF-κB ODN, NF-κB morpholinos. | PMID:10760263 | |
| HIFs pathway | Inhibition of PHD protein (PHD inhibitor) | PHD2 | TM6008, TM6089 | PMID: 28682438 |
| FK506-binding Protein | FKBP38 | PMID: 28682438 | ||
| PHD1/2/3 | DFO, DMOG | PMID: 28682438 | ||
| Inhibition of HIF2a protein (HIF2 inhibitor) | The silent information regulator 2 type 1 (SIRT1) | Resveratrol | PMID: 25737402 | |
| HIF2a | YC-1 | PMID: 25737402 | ||
| mTOR | Inhibition of mTOR | mTOR | Rapamycin | PMID:22084394 |
| The hydromethanolic extract of Butea monosperma (BME) | PMID: 28987943 | |||
| Focal adhesion pathway | Intra-articular injection of Kindlin-2-expressing AAV | Fermt2 | AAV | https://www.nature.com/articles/s43587-021-00165-w |
| Inhibition of FAK (FAK inhibitor) | PTK2 | PF-0455487; | PMID:32558123 | |
| Y15 | PMID:32324278 | |||
| BMP pathway | Inhibition of BMP2 protein (BMP2 inhibitor) | BMP2 | Noggin | PMID: 32290085 |
| Inhibition of BMP4 protein | Rspondin2 | Mianserin | PMID: 30808932 | |
| MiR-181a | PMID: 34778258 | |||
| Activation of BMP7 pathway | Smad1/5 | BMP7-derived peptide | PMID: 33850953 | |
| Activation of BMPRIa pathway | BMPRIa | CK2.1 | PMID: 28420447 |
KOA knee osteoarthritis, DMM destabilization of the medial meniscus, MIA monosodium iodoacetate
The multiple layers of complexity of the pathogenesis of OA and limited knowledge of the pathogenetic molecular signaling pathways and specific mechanisms have made the therapeutic pharmacological targeting of OA extremely difficult. At this point, in the future, a comprehensive understanding of alterations in distinct signaling pathways and expression of key factors in the superficial, middle and deep zones of articular cartilage and synovial membrane at different stages of OA triggered by different factors should be investigated in great detail. The whole picture of functions and regulations of each pathological signaling and key factors in various early-stage OA conditions could help us to develop more specific resolutions to halt or reverse the OA disease.
Acknowledgements
We apologize to the authors whose relevant studies have not been included in this review article. This work was supported, in part, by the National Key Research and Development Program of China Grants (2019YFA0906004), the National Natural Science Foundation of China Grants (82230081, 82250710175, 82172375, 81991513, and 82261160395), the Shenzhen Stable Support Plan Program for Higher Education Institutions (20200925150409001), the Shenzhen Fundamental Research Program (JCYJ20220818100617036), the Shenzhen Key Laboratory of Cell Microenvironment Grant (ZDSYS20140509142721429) and the Guangdong Provincial Science and Technology Innovation Council Grant (2017B030301018).
Author contributions
Q.Y. and G.X. conceptualized and wrote the outline of the manuscript. Q.Y., X.W., C.T., W.G., M.C., M.Q., Y.Z., T.H., and S.C. did the literature search and wrote the manuscript draft. G.X. reviewed and edited the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Qing Yao, Xiaohao Wu, Chu Tao, Weiyuan Gong
Contributor Information
Qing Yao, Email: yaoq@sustech.edu.cn.
Guozhi Xiao, Email: xiaogz@sustech.edu.cn.
References
- 1.Hunter, D. J. & Bierma-Zeinstra, S. Osteoarthritis. Lancet393, 1745–1759 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Quicke, J. G., Conaghan, P. G., Corp, N. & Peat, G. Osteoarthritis year in review 2021: epidemiology & therapy. Osteoarthr. Cartil.30, 196–206 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Little, C. B. & Hunter, D. J. Post-traumatic osteoarthritis: from mouse models to clinical trials. Nat. Rev. Rheumatol.9, 485–497 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Leung, Y. Y. & Thumboo, J. An overview of OA research in two urban APLAR populations. Int. J. Rheum. Dis.14, 130–135 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Lu, K. et al. Molecular signaling in temporomandibular joint osteoarthritis. J. Orthop. Transl.32, 21–27 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prieto-Alhambra, D. et al. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: influences of age, gender and osteoarthritis affecting other joints. Ann. Rheum. Dis.73, 1659–1664 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Luna, A., Otahal, A. & Nehrer, S. Mesenchymal stromal cell-derived extracellular vesicles - silver linings for cartilage regeneration? Front. Cell Dev. Biol.8, 593386 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nedunchezhiyan, U. et al. Obesity, inflammation, and immune system in osteoarthritis. Front. Immunol.13, 907750 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallace, I. J. et al. Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc. Natl Acad. Sci. USA114, 9332–9336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turkiewicz, A. et al. Current and future impact of osteoarthritis on health care: a population-based study with projections to year 2032. Osteoarthr. Cartil.22, 1826–1832 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Pereira, D. et al. The effect of osteoarthritis definition on prevalence and incidence estimates: a systematic review. Osteoarthr. Cartil.19, 1270–1285 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Li, Y., Xie, W., Xiao, W. & Dou, D. Progress in osteoarthritis research by the national natural science foundation of China. Bone Res10, 41 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Safiri, S. et al. Global, regional and national burden of osteoarthritis 1990–2017: a systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis.79, 819–828 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Long, H. et al. Prevalence trends of site-specific osteoarthritis from 1990 to 2019: findings from the global burden of disease study 2019. Arthritis Rheumatol.74, 1172–1183 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arslan, I. G. et al. Estimating incidence and prevalence of hip osteoarthritis using electronic health records: a population-based cohort study. Osteoarthr. Cartil.30, 843–851 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Jin, Z. Y. et al. Incidence trend of five common musculoskeletal disorders from 1990 to 2017 at the global, regional and national level: results from the global burden of disease study 2017. Ann. Rheum. Dis.79, 1014–1022 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Disease, G. B. D., Injury, I. & Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet392, 1789–1858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abbafati, C. et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet396, 1204–1222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swain, S. et al. Trends in incidence and prevalence of osteoarthritis in the United Kingdom: findings from the Clinical Practice Research Datalink (CPRD). Osteoarthr. Cartil.28, 792–801 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Tie, X.-J. et al. Prevalence of knee osteoarthritis in the middle-aged and elderly in China: </br> a Meta-analysis. Chin. J. Tissue Eng. Res.22, 650–656 (2018). [Google Scholar]
- 21.Tang, X. et al. The prevalence of symptomatic knee osteoarthritis in china: results from the China health and retirement longitudinal study. Arthritis Rheumatol.68, 648–653 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Kang, X. et al. The high prevalence of knee osteoarthritis in a rural Chinese population: the Wuchuan osteoarthritis study. Arthritis Rheum.61, 641–647 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun, X. et al. Osteoarthritis in the middle-aged and elderly in china: prevalence and influencing factors. Int. J. Environ. Res. Public Health16, 4701 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang, Y. et al. Comparison of the prevalence of knee osteoarthritis between the elderly Chinese population in Beijing and whites in the United States: The Beijing Osteoarthritis Study. Arthritis Rheum.44, 2065–2071 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Nevitt, M. C. et al. Very low prevalence of hip osteoarthritis among Chinese elderly in Beijing, China, compared with whites in the United States: the Beijing osteoarthritis study. Arthritis Rheum.46, 1773–1779 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Zhang, Y. et al. Lower prevalence of hand osteoarthritis among Chinese subjects in Beijing compared with white subjects in the United States: the Beijing Osteoarthritis Study. Arthritis Rheum.48, 1034–1040 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Chen, H. et al. Trends and patterns of knee osteoarthritis in China: a longitudinal study of 17.7 million adults from 2008 to 2017. Int. J. Environ. Res. Public Health18, 8864 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu, D., Peat, G., Bedson, J. & Jordan, K. P. Annual consultation incidence of osteoarthritis estimated from population-based health care data in England. Rheumatology54, 2051–2060 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliveria, S. A., Felson, D. T., Reed, J. I., Cirillo, P. A. & Walker, A. M. Incidence of symptomatic hand, hip, and knee osteoarthritis among patients in a health maintenance organization. Arthritis Rheum.38, 1134–1141 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Johnson, V. L. & Hunter, D. J. The epidemiology of osteoarthritis. Best. Pract. Res Clin. Rheumatol.28, 5–15 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Liu, Q. et al. Knee symptomatic osteoarthritis, walking disability, nsaids use and all-cause mortality: population-based wuchuan osteoarthritis study. Sci. Rep.7, 3309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cross, M. et al. The global burden of hip and knee osteoarthritis: estimates from the Global Burden of Disease 2010 study. Ann. Rheum. Dis.73, 1323–1330 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Lo, J., Chan, L. & Flynn, S. A systematic review of the incidence, prevalence, costs, and activity and work limitations of amputation, osteoarthritis, rheumatoid arthritis, back pain, multiple sclerosis, spinal cord injury, stroke, and traumatic brain injury in the united states: a 2019 update. Arch. Phys. Med. Rehab.102, 115–131 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, S. X. et al. Healthcare resource utilization and costs by age and joint location among osteoarthritis patients in a privately insured population. J. Med Econ.20, 1299–1306 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Kiadaliri, A. A., Lohmander, L. S., Moradi-Lakeh, M., Petersson, I. F. & Englund, M. High and rising burden of hip and knee osteoarthritis in the Nordic region, 1990–2015: findings from the Global Burden of Disease Study 2015. Acta Orthop.89, 177–183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Long, H. et al. Burden of osteoarthritis in China, 1990–2017: findings from the Global Burden of Disease Study 2017. Lancet Rheumatol.2, e164–e172 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Kassebaum, N. J. et al. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet388, 1603–1658 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu, Q., Wang, S., Lin, J. & Zhang, Y. The burden for knee osteoarthritis among Chinese elderly: estimates from a nationally representative study. Osteoarthr. Cartil.26, 1636–1642 (2018). [DOI] [PubMed] [Google Scholar]
- 39.O’Brien, M. S. & McDougall, J. J. Age and frailty as risk factors for the development of osteoarthritis. Mech. Ageing Dev.180, 21–28 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Sacitharan, P. K. Ageing and Osteoarthritis. Subcell. Biochem91, 123–159 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Loeser, R. F., Collins, J. A. & Diekman, B. O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol.12, 412–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coryell, P. R., Diekman, B. O. & Loeser, R. F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol.17, 47–57 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coppe, J. P. et al. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem.286, 36396–36403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freund, A., Patil, C. K. & Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J.30, 1536–1548 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuchida, A. I. et al. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res. Ther.16, 441 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hui, W. et al. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis.75, 449–458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blanco, F. J., Rego, I. & Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol.7, 161–169 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Scott, J. L. et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis.69, 1502–1510 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greene, M. A. & Loeser, R. F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil.23, 1966–1971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raud, B. et al. Level of obesity is directly associated with the clinical and functional consequences of knee osteoarthritis. Sci. Rep.10, 3601 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reyes, C. et al. Association between overweight and obesity and risk of clinically diagnosed knee, hip, and hand osteoarthritis: a population-based cohort study. Arthritis Rheumatol.68, 1869–1875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conde, J. et al. Identification of novel adipokines in the joint. Differential expression in healthy and osteoarthritis tissues. PLoS ONE10, e0123601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang, H. et al. Adiponectin, may be a potential protective factor for obesity-related osteoarthritis. Diabetes Metab. Syndr. Obes.15, 1305–1319 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang, M. J. et al. Enhancement of the synthesis of n-3 PUFAs in fat-1 transgenic mice inhibits mTORC1 signalling and delays surgically induced osteoarthritis in comparison with wild-type mice. Ann. Rheum. Dis.73, 1719–1727 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Sekar, S. et al. Saturated fatty acids promote chondrocyte matrix remodeling through reprogramming of autophagy pathways. Nutrition54, 144–152 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Dai, Y. et al. A low proportion n-6/n-3 PUFA diet supplemented with Antarctic krill (Euphausia superba) oil protects against osteoarthritis by attenuating inflammation in ovariectomized mice. Food Funct.12, 6766–6779 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Peshkova, M. et al. Gender-related aspects in osteoarthritis development and progression: a review. Int. J. Mol. Sci.23, 2767 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Price, M. D. & Herndon, J. H. Gender differences in osteoarthritis. Menopause16, 624–625 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Yan, Y. S. et al. Sex steroids and osteoarthritis: a mendelian randomization study. Front. Endocrinol.12, 683226 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tschon, M., Contartese, D., Pagani, S., Borsari, V. & Fini, M. Gender and sex are key determinants in osteoarthritis not only confounding variables. a systematic review of clinical data. J. Clin. Med.10, 3178 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laitner, M. H., Erickson, L. C. & Ortman, E. Understanding the impact of sex and gender in osteoarthritis: assessing research gaps and unmet needs. J. Women’s Health (Larchmt.)30, 634–641 (2021). [DOI] [PubMed] [Google Scholar]
- 62.Blaker, C. L., Clarke, E. C. & Little, C. B. Using mouse models to investigate the pathophysiology, treatment, and prevention of post-traumatic osteoarthritis. J. Orthop. Res.35, 424–439 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Papalia, R. et al. Sport activity as risk factor for early knee osteoarthritis. J. Biol. Regulators Homeost. Agents33, 29–37 (2019). XIX Congresso Nazionale S I C O O P Societa’ Italiana Chirurghi Ortopedici Dell’ospedalita’ Privata Accreditata. [PubMed] [Google Scholar]
- 64.Astephen Wilson, J. L. & Kobsar, D. Osteoarthritis year in review 2020: mechanics. Osteoarthr. Cartil.29, 161–169 (2021). [DOI] [PubMed] [Google Scholar]
- 65.Andersson, J. K., Hagert, E. & Brittberg, M. Cartilage injuries and posttraumatic osteoarthritis in the wrist: a review. Cartilage13, 156S–168S (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harlaar, J., Macri, E. M. & Wesseling, M. Osteoarthritis year in review 2021: mechanics. Osteoarthr. Cartil.30, 663–670 (2022). [DOI] [PubMed] [Google Scholar]
- 67.Boer, C. G. et al. Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell184, 4784–4818 e4717 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ratneswaran, A. & Kapoor, M. Osteoarthritis year in review: genetics, genomics, epigenetics. Osteoarthr. Cartil.29, 151–160 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Aubourg, G., Rice, S. J., Bruce-Wootton, P. & Loughlin, J. Genetics of osteoarthritis. Osteoarthr. Cartil.30, 636–649 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Loughlin, J. Translating osteoarthritis genetics research: challenging times ahead. Trends Mol. Med.28, 176–182 (2022). [DOI] [PubMed] [Google Scholar]
- 71.Young, D. A., Barter, M. J. & Soul, J. Osteoarthritis year in review: genetics, genomics, epigenetics. Osteoarthr. Cartil.30, 216–225 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fayet, M. & Hagen, M. Pain characteristics and biomarkers in treatment approaches for osteoarthritis pain. Pain. Manag.11, 59–73 (2021). [DOI] [PubMed] [Google Scholar]
- 73.Katz, J. N., Arant, K. R. & Loeser, R. F. Diagnosis and treatment of hip and knee osteoarthritis: a review. JAMA325, 568–578 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seifert, O. & Baerwald, C. Interaction of pain and chronic inflammation. Z. Rheumatol.80, 205–213 (2021). [DOI] [PubMed] [Google Scholar]
- 75.Baral, P., Udit, S. & Chiu, I. M. Pain and immunity: implications for host defence. Nat. Rev. Immunol.19, 433–447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wood, M. J., Miller, R. E. & Malfait, A. M. The genesis of pain in osteoarthritis: inflammation as a mediator of osteoarthritis pain. Clin. Geriatr. Med.38, 221–238 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Conaghan, P. G., Cook, A. D., Hamilton, J. A. & Tak, P. P. Therapeutic options for targeting inflammatory osteoarthritis pain. Nat. Rev. Rheumatol.15, 355–363 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Vincent, T. L. Peripheral pain mechanisms in osteoarthritis. Pain161, S138–S146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu, H., Huang, T., Lu, W. W., Tong, L. & Chen, D. Osteoarthritis pain. Int. J. Mol. Sci.23, 4642 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Malfait, A. M., Miller, R. E. & Miller, R. J. Basic mechanisms of pain in osteoarthritis: experimental observations and new perspectives. Rheum. Dis. Clin. North Am.47, 165–180 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tong, L. et al. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res10, 60 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burr, D. B. & Gallant, M. A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol.8, 665–673 (2012). [DOI] [PubMed] [Google Scholar]
- 83.Funck-Brentano, T. & Cohen-Solal, M. Subchondral bone and osteoarthritis. Curr. Opin. Rheumatol.27, 420–426 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Maruotti, N., Corrado, A. & Cantatore, F. P. Osteoblast role in osteoarthritis pathogenesis. J. Cell. Physiol.232, 2957–2963 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hu, W., Chen, Y., Dou, C. & Dong, S. Microenvironment in subchondral bone: predominant regulator for the treatment of osteoarthritis. Ann. Rheum. Dis.80, 413–422 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han, X. et al. Abnormal subchondral trabecular bone remodeling in knee osteoarthritis under the influence of knee alignment. Osteoarthr. Cartil.30, 100–109 (2022). [DOI] [PubMed] [Google Scholar]
- 87.Zhang, L. et al. Expansion of myeloid-derived suppressor cells contributes to metabolic osteoarthritis through subchondral bone remodeling. Arthritis Res. Ther.23, 287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Amir, G., Pirie, C. J., Rashad, S. & Revell, P. A. Remodelling of subchondral bone in osteoarthritis: a histomorphometric study. J. Clin. Pathol.45, 990–992 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Benske, J., Schunke, M. & Tillmann, B. Subchondral bone formation in arthrosis. Polychrome labeling studies in mice. Acta Orthop. Scand.59, 536–541 (1988). [DOI] [PubMed] [Google Scholar]
- 90.Hu, Y., Chen, X., Wang, S., Jing, Y. & Su, J. Subchondral bone microenvironment in osteoarthritis and pain. Bone Res.9, 20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hugle, T. & Geurts, J. What drives osteoarthritis?-synovial versus subchondral bone pathology. Rheumatology56, 1461–1471 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Dieppe, P., Cushnaghan, J., Young, P. & Kirwan, J. Prediction of the progression of joint space narrowing in osteoarthritis of the knee by bone scintigraphy. Ann. Rheum. Dis.52, 557–563 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mansell, J. P., Tarlton, J. F. & Bailey, A. J. Biochemical evidence for altered subchondral bone collagen metabolism in osteoarthritis of the hip. Br. J. Rheumatol.36, 16–19 (1997). [DOI] [PubMed] [Google Scholar]
- 94.Bolbos, R. I. et al. Relationship between trabecular bone structure and articular cartilage morphology and relaxation times in early OA of the knee joint using parallel MRI at 3 T. Osteoarthr. Cartil.16, 1150–1159 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ding, R., Zhang, N., Wang, Q. & Wang, W. Alterations of the subchondral bone in osteoarthritis: complying with Wolff’s law. Curr. Rheumatol. Rev.18, 178–185 (2022). [DOI] [PubMed] [Google Scholar]
- 96.Fazzalari, N. L. & Parkinson, I. H. Fractal properties of subchondral cancellous bone in severe osteoarthritis of the hip. J. Bone Miner. Res.12, 632–640 (1997). [DOI] [PubMed] [Google Scholar]
- 97.Arden, N. K., Griffiths, G. O., Hart, D. J., Doyle, D. V. & Spector, T. D. The association between osteoarthritis and osteoporotic fracture: the Chingford Study. Br. J. Rheumatol.35, 1299–1304 (1996). [DOI] [PubMed] [Google Scholar]
- 98.Hannan, M. T., Anderson, J. J., Zhang, Y., Levy, D. & Felson, D. T. Bone mineral density and knee osteoarthritis in elderly men and women. The Framingham Study. Arthritis Rheum.36, 1671–1680 (1993). [DOI] [PubMed] [Google Scholar]
- 99.Mathiessen, A. & Conaghan, P. G. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res. Ther.19, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smith, M. D., Triantafillou, S., Parker, A., Youssef, P. P. & Coleman, M. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J. Rheumatol.24, 365–371 (1997). [PubMed] [Google Scholar]
- 101.Goldring, M. B. & Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol.23, 471–478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pelletier, J. P., Martel-Pelletier, J. & Abramson, S. B. Osteoarthritis, an inflammatory disease: potential implication for the selection of new therapeutic targets. Arthritis Rheum.44, 1237–1247 (2001). [DOI] [PubMed] [Google Scholar]
- 103.Maglaviceanu, A., Wu, B. & Kapoor, M. Fibroblast-like synoviocytes: Role in synovial fibrosis associated with osteoarthritis. Wound Repair Regen.29, 642–649 (2021). [DOI] [PubMed] [Google Scholar]
- 104.Thomson, A. & Hilkens, C. M. U. Synovial macrophages in osteoarthritis: the key to understanding pathogenesis? Front. Immunol.12, 678757 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Krenn, V. et al. Grading of chronic synovitis-a histopathological grading system for molecular and diagnostic pathology. Pathol. Res Pract.198, 317–325 (2002). [DOI] [PubMed] [Google Scholar]
- 106.Krenn, V. et al. 15 years of the histopathological synovitis score, further development and review: a diagnostic score for rheumatology and orthopaedics. Pathol. Res. Pract.213, 874–881 (2017). [DOI] [PubMed] [Google Scholar]
- 107.Bonnet, C. S. & Walsh, D. A. Osteoarthritis, angiogenesis and inflammation. Rheumatology44, 7–16 (2005). [DOI] [PubMed] [Google Scholar]
- 108.Benito, M. J., Veale, D. J., FitzGerald, O., van den Berg, W. B. & Bresnihan, B. Synovial tissue inflammation in early and late osteoarthritis. Ann. Rheum. Dis.64, 1263–1267 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol.115, 911–919 (2005). [DOI] [PubMed] [Google Scholar]
- 110.Zapata-Linares, N., Eymard, F., Berenbaum, F. & Houard, X. Role of adipose tissues in osteoarthritis. Curr. Opin. Rheumatol.33, 84–93 (2021). [DOI] [PubMed] [Google Scholar]
- 111.Belluzzi, E. et al. Systemic and local adipose tissue in knee osteoarthritis. J. Cell. Physiol.232, 1971–1978 (2017). [DOI] [PubMed] [Google Scholar]
- 112.Xie, C. & Chen, Q. Adipokines: new therapeutic target for osteoarthritis? Curr. Rheumatol. Rep.21, 71 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chang, J. et al. Systemic and local adipose tissue in knee osteoarthritis. Osteoarthr. Cartil.26, 864–871 (2018). [DOI] [PubMed] [Google Scholar]
- 114.Collins, K. H. et al. Adipose tissue is a critical regulator of osteoarthritis. Proc. Natl Acad. Sci. USA118, e2021096118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Frazier, T. et al. Non-homologous use of adipose-derived cell and tissue therapies: osteoarthritis as a case study. Bone Rep.17, 101601 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sandell, L. J. & Aigner, T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res.3, 107–113 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hwang, H. S. & Kim, H. A. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int. J. Mol. Sci.16, 26035–26054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Janczi, T. et al. ADAM15 in apoptosis resistance of synovial fibroblasts: converting Fas/CD95 death signals into the activation of prosurvival pathways by calmodulin recruitment. Arthritis Rheumatol.71, 63–72 (2019). [DOI] [PubMed] [Google Scholar]
- 119.Li, Q. et al. Effects of TNFR1 gene silencing on early apoptosis of marbofloxacin-treated chondrocytes from juvenile dogs. Toxicology422, 53–59 (2019). [DOI] [PubMed] [Google Scholar]
- 120.Del Carlo, M. Jr. & Loeser, R. F. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum.46, 394–403 (2002). [DOI] [PubMed] [Google Scholar]
- 121.Wang, G. et al. Cathelicidin antimicrobial peptide (CAMP) gene promoter methylation induces chondrocyte apoptosis. Hum. Genomics15, 24 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang, J., Hao, X., Chi, R., Qi, J. & Xu, T. Moderate mechanical stress suppresses the IL-1beta-induced chondrocyte apoptosis by regulating mitochondrial dynamics. J. Cell Physiol.236, 7504–7515 (2021). [DOI] [PubMed] [Google Scholar]
- 123.Sun, Y. et al. G protein coupled estrogen receptor attenuates mechanical stress-mediated apoptosis of chondrocyte in osteoarthritis via suppression of Piezo1. Mol. Med.27, 96 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Castrogiovanni, P., Ravalli, S. & Musumeci, G. Apoptosis and autophagy in the pathogenesis of osteoarthritis. J. Invest. Surg.33, 874–875 (2020). [DOI] [PubMed] [Google Scholar]
- 125.Yang, J. et al. Targeting cell death: pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev. Biol.9, 789948 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hu, J. et al. Loganin ameliorates cartilage degeneration and osteoarthritis development in an osteoarthritis mouse model through inhibition of NF-kappaB activity and pyroptosis in chondrocytes. J. Ethnopharmacol.247, 112261 (2020). [DOI] [PubMed] [Google Scholar]
- 127.Ulrich-Vinther, M., Maloney, M. D., Schwarz, E. M., Rosier, R. & O’Keefe, R. J. Articular cartilage biology. J. Am. Acad. Orthop. Surg.11, 421–430 (2003). [DOI] [PubMed] [Google Scholar]
- 128.Kozhemyakina, E. et al. Identification of a Prg4-expressing articular cartilage progenitor cell population in mice. Arthritis Rheumatol.67, 1261–1273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sun, M. M. & Beier, F. Chondrocyte hypertrophy in skeletal development, growth, and disease. Birth Defects Res. C. Embryo Today102, 74–82 (2014). [DOI] [PubMed] [Google Scholar]
- 130.Chen, D., Kim, D. J., Shen, J., Zou, Z. & O’Keefe, R. J. Runx2 plays a central role in osteoarthritis development. J. Orthop. Transl.23, 132–139 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huang, J. et al. The microRNAs miR-204 and miR-211 maintain joint homeostasis and protect against osteoarthritis progression. Nat. Commun.10, 2876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mobasheri, A. et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol.13, 302–311 (2017). [DOI] [PubMed] [Google Scholar]
- 133.Zheng, L., Zhang, Z., Sheng, P. & Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev.66, 101249 (2021). [DOI] [PubMed] [Google Scholar]
- 134.Nusse, R. & Clevers, H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell169, 985–999 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Kadowaki, T., Wilder, E., Klingensmith, J., Zachary, K. & Perrimon, N. The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev.10, 3116–3128 (1996). [DOI] [PubMed] [Google Scholar]
- 136.Reya, T. & Clevers, H. Wnt signalling in stem cells and cancer. Nature434, 843–850 (2005). [DOI] [PubMed] [Google Scholar]
- 137.Valenta, T., Hausmann, G. & Basler, K. The many faces and functions of β-catenin. EMBO J.31, 2714–2736 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schunk, S. J., Floege, J., Fliser, D. & Speer, T. WNT-β-catenin signalling - a versatile player in kidney injury and repair. Nat. Rev. Nephrol.17, 172–184 (2021). [DOI] [PubMed] [Google Scholar]
- 139.Alok, A. et al. Wnt proteins synergize to activate β-catenin signaling. J. Cell Sci.130, 1532–1544 (2017). [DOI] [PubMed] [Google Scholar]
- 140.MacDonald, B. T., Tamai, K. & He, X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell17, 9–26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nusse, R. & Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell169, 985–999 (2017). [DOI] [PubMed] [Google Scholar]
- 142.Zhu, M. et al. Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum.58, 2053–2064 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nalesso, G. et al. WNT16 antagonises excessive canonical WNT activation and protects cartilage in osteoarthritis. Ann. Rheum. Dis.76, 218–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tong, W. et al. Wnt16 attenuates osteoarthritis progression through a PCP/JNK-mTORC1-PTHrP cascade. Ann. Rheum. Dis.78, 551–561 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Guo, X. et al. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev.18, 2404–2417 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Loughlin, J. et al. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc. Natl Acad. Sci. USA101, 9757–9762 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lories, R. J. et al. Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum.56, 4095–4103 (2007). [DOI] [PubMed] [Google Scholar]
- 148.Akiyama, H. et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev.18, 1072–1087 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Harada, N. et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J.18, 5931–5942 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wu, Q. et al. Beta-catenin, cartilage, and osteoarthritis. Ann. N. Y. Acad. Sci.1192, 344–350 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhu, M. et al. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res.24, 12–21 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang, M. et al. Activation of beta-catenin signalling leads to temporomandibular joint defects. Eur. Cells Mater.28, 223–235 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhou, Y., Wang, T., Hamilton, J. L. & Chen, D. Wnt/beta-catenin signaling in osteoarthritis and in other forms of arthritis. Curr. Rheumatol. Rep.19, 53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Xia, C. et al. Activation of beta-catenin in Col2-expressing chondrocytes leads to osteoarthritis-like defects in hip joint. J. Cell. Physiol.234, 18535–18543 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li, W., Xiong, Y., Chen, W. & Wu, L. Wnt/beta-catenin signaling may induce senescence of chondrocytes in osteoarthritis. Exp. Ther. Med20, 2631–2638 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yuasa, T. et al. Transient activation of Wnt/{beta}-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am. J. Pathol.175, 1993–2003 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yasuhara, R. et al. Roles of beta-catenin signaling in phenotypic expression and proliferation of articular cartilage superficial zone cells. Lab. Investig.91, 1739–1752 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Blom, A. B. et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum.60, 501–512 (2009). [DOI] [PubMed] [Google Scholar]
- 159.Honsawek, S. et al. Dickkopf-1 (Dkk-1) in plasma and synovial fluid is inversely correlated with radiographic severity of knee osteoarthritis patients. BMC Musculoskelet. Disord.11, 257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.van den Bosch, M. H. et al. Brief report: induction of matrix metalloproteinase expression by synovial Wnt signaling and association with disease progression in early symptomatic osteoarthritis. Arthritis Rheumatol.69, 1978–1983 (2017). [DOI] [PubMed] [Google Scholar]
- 161.Oh, H., Chun, C. H. & Chun, J. S. Dkk-1 expression in chondrocytes inhibits experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum.64, 2568–2578 (2012). [DOI] [PubMed] [Google Scholar]
- 162.Funck-Brentano, T. et al. Dkk-1-mediated inhibition of Wnt signaling in bone ameliorates osteoarthritis in mice. Arthritis Rheumatol.66, 3028–3039 (2014). [DOI] [PubMed] [Google Scholar]
- 163.Snelling, S. J. et al. Dickkopf-3 is upregulated in osteoarthritis and has a chondroprotective role. Osteoarthr. Cartil.24, 883–891 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Conde, J. et al. Dickkopf-3 (DKK3) signaling in IL-1alpha-challenged chondrocytes: involvement of the NF-kappaB pathway. Cartilage13, 925S–934S (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.van den Bosch, M. H. et al. Induction of canonical Wnt signaling by synovial overexpression of selected wnts leads to protease activity and early osteoarthritis-like cartilage damage. Am. J. Pathol.185, 1970–1980 (2015). [DOI] [PubMed] [Google Scholar]
- 166.Clevers, H. & Nusse, R. Wnt/β-catenin signaling and disease. Cell149, 1192–1205 (2012). [DOI] [PubMed] [Google Scholar]
- 167.Li, Y. et al. The expression of osteopontin and Wnt5a in articular cartilage of patients with knee osteoarthritis and its correlation with disease severity. Biomed. Res. Int2016, 9561058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Onuora, S. Osteoarthritis: Wnt inhibitor shows potential as a DMOAD. Nat. Rev. Rheumatol.13, 634 (2017). [DOI] [PubMed] [Google Scholar]
- 169.Deshmukh, V. et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil.26, 18–27 (2018). [DOI] [PubMed] [Google Scholar]
- 170.De Palma, A. & Nalesso, G. WNT signalling in osteoarthritis and its pharmacological targeting. Handb. Exp. Pharmacol.269, 337–356 (2021). [DOI] [PubMed] [Google Scholar]
- 171.Deshmukh, V. et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr. Cartil.27, 1347–1360 (2019). [DOI] [PubMed] [Google Scholar]
- 172.Lietman, C. et al. Inhibition of Wnt/beta-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight3, e96308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Held, A. et al. Targeting beta-catenin dependent Wnt signaling via peptidomimetic inhibitors in murine chondrocytes and OA cartilage. Osteoarthr. Cartil.26, 818–823 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Takamatsu, A. et al. Verapamil protects against cartilage degradation in osteoarthritis by inhibiting Wnt/beta-catenin signaling. PLoS One9, e92699 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xia, H. et al. Jiawei Yanghe decoction ameliorates cartilage degradation in vitro and vivo via Wnt/beta-catenin signaling pathway. Biomed. Pharmacother.122, 109708 (2020). [DOI] [PubMed] [Google Scholar]
- 176.Li, K. et al. Tyrosine kinase Fyn promotes osteoarthritis by activating the beta-catenin pathway. Ann. Rheum. Dis.77, 935–943 (2018). [DOI] [PubMed] [Google Scholar]
- 177.Chen, M. et al. Inhibition of beta-catenin signaling causes defects in postnatal cartilage development. J. Cell Sci.121, 1455–1465 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jimi, E. & Ghosh, S. Role of nuclear factor-kappaB in the immune system and bone. Immunol. Rev.208, 80–87 (2005). [DOI] [PubMed] [Google Scholar]
- 179.Chen, S. et al. Kindlin-2 inhibits Nlrp3 inflammasome activation in nucleus pulposus to maintain homeostasis of the intervertebral disc. Bone Res.10, 5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Marcu, K. B., Otero, M., Olivotto, E., Borzi, R. M. & Goldring, M. B. NF-kappaB signaling: multiple angles to target OA. Curr. Drug Targets11, 599–613 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hoffmann, A. & Baltimore, D. Circuitry of nuclear factor κB signaling. Immunol. Rev.210, 171–186 (2006). [DOI] [PubMed] [Google Scholar]
- 182.Oeckinghaus, A. & Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol.1, a000034 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yasuda, T. Activation of Akt leading to NF-κB up-regulation in chondrocytes stimulated with fibronectin fragment. Biomed. Res.32, 209–215 (2011). [DOI] [PubMed] [Google Scholar]
- 184.Arra, M., Swarnkar, G., Alippe, Y., Mbalaviele, G. & Abu-Amer, Y. IκB-ζ signaling promotes chondrocyte inflammatory phenotype, senescence, and erosive joint pathology. Bone Res.10, 12 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Huang, B., Yang, X. D., Lamb, A. & Chen, L. F. Posttranslational modifications of NF-kappaB: another layer of regulation for NF-kappaB signaling pathway. Cell Signal.22, 1282–1290 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Iotsova, V. et al. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat. Med.3, 1285–1289 (1997). [DOI] [PubMed] [Google Scholar]
- 187.Caron, M. M. et al. Activation of NF-κB/p65 facilitates early chondrogenic differentiation during endochondral ossification. PLoS One7, e33467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kanegae, Y., Tavares, A. T., Izpisúa Belmonte, J. C. & Verma, I. M. Role of Rel/NF-kappaB transcription factors during the outgrowth of the vertebrate limb. Nature392, 611–614 (1998). [DOI] [PubMed] [Google Scholar]
- 189.Wu, S., Fadoju, D., Rezvani, G. & De Luca, F. Stimulatory effects of insulin-like growth factor-I on growth plate chondrogenesis are mediated by nuclear factor-kappaB p65. J. Biol. Chem.283, 34037–34044 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nakatomi, C. et al. Constitutive activation of the alternative NF-κB pathway disturbs endochondral ossification. Bone121, 29–41 (2019). [DOI] [PubMed] [Google Scholar]
- 191.Kobayashi, H. et al. Biphasic regulation of chondrocytes by Rela through induction of anti-apoptotic and catabolic target genes. Nat. Commun.7, 13336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ulivi, V., Giannoni, P., Gentili, C., Cancedda, R. & Descalzi, F. p38/NF-kB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes. J. Cell Biochem.104, 1393–1406 (2008). [DOI] [PubMed] [Google Scholar]
- 193.Yoon, D. S. et al. TLR4 downregulation by the RNA-binding protein PUM1 alleviates cellular aging and osteoarthritis. Cell Death Differ.29, 1364–1378 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kapoor, M., Martel-Pelletier, J., Lajeunesse, D., Pelletier, J. P. & Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol.7, 33–42 (2011). [DOI] [PubMed] [Google Scholar]
- 195.Goldring, M. B. et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur. Cell Mater.21, 202–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li, W. et al. Role of HIF-2α/NF-κB pathway in mechanical stress-induced temporomandibular joint osteoarthritis. Oral Dis.28, 2239–2247 (2021). [DOI] [PubMed]
- 197.Zheng, T., Li, Y., Zhang, X., Xu, J. & Luo, M. Exosomes derived from miR-212-5p overexpressed human synovial mesenchymal stem cells suppress chondrocyte degeneration and inflammation by targeting ELF3. Front. Bioeng. Biotechnol.10, 816209 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lai, Y. et al. Kindlin-2 loss in condylar chondrocytes causes spontaneous osteoarthritic lesions in the temporomandibular joint in mice. Int. J. Oral. Sci.14, 33 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Goldring, M. B. & Marcu, K. B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther.11, 224 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Deng, Y. et al. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat. Commun.9, 4564 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yin, M. J., Yamamoto, Y. & Gaynor, R. B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature396, 77–80 (1998). [DOI] [PubMed] [Google Scholar]
- 202.Chen, S. et al. Moderate fluid shear stress regulates heme oxygenase-1 expression to promote autophagy and ECM homeostasis in the nucleus pulposus cells. Front Cell Dev. Biol.8, 127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Scheinman, R. I., Gualberto, A., Jewell, C. M., Cidlowski, J. A. & Baldwin, A. S. Jr. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol. Cell Biol.15, 943–953 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Aitken, D. et al. A randomised double-blind placebo-controlled crossover trial of HUMira (adalimumab) for erosive hand OsteoaRthritis - the HUMOR trial. Osteoarthr. Cartil.26, 880–887 (2018). [DOI] [PubMed] [Google Scholar]
- 205.Kloppenburg, M. et al. Etanercept in patients with inflammatory hand osteoarthritis (EHOA): a multicentre, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis.77, 1757–1764 (2018). [DOI] [PubMed] [Google Scholar]
- 206.Wang, S. X. et al. Safety, tolerability, and pharmacodynamics of an anti-interleukin-1alpha/beta dual variable domain immunoglobulin in patients with osteoarthritis of the knee: a randomized phase 1 study. Osteoarthr. Cartil.25, 1952–1961 (2017). [DOI] [PubMed] [Google Scholar]
- 207.Yan, Y., Zhou, X. E., Xu, H. E. & Melcher, K. Structure and physiological regulation of AMPK. Int J. Mol. Sci.19, 3534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Petursson, F. et al. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res. Ther.15, R77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Terkeltaub, R., Yang, B., Lotz, M. & Liu-Bryan, R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum.63, 1928–1937 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ge, Y. et al. Estrogen prevents articular cartilage destruction in a mouse model of AMPK deficiency via ERK-mTOR pathway. Ann. Transl. Med.7, 336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Qiu, L. et al. Effect of Electroacupuncture on Synovial M 1/M 2 Macrophage Polarization in Rats with Acute Gouty Arthritis. Zhen Ci Yan Jiu43, 767–772 (2018). [DOI] [PubMed] [Google Scholar]
- 212.Zhou, S. et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci. Rep.7, 43245 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mevel, E. et al. Systemic inhibition or global deletion of CaMKK2 protects against post-traumatic osteoarthritis. Osteoarthr. Cartil.30, 124–136 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Momcilovic, M., Hong, S. P. & Carlson, M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J. Biol. Chem.281, 25336–25343 (2006). [DOI] [PubMed] [Google Scholar]
- 215.Neumann, D. Is TAK1 a direct upstream kinase of AMPK? Int. J. Mol. Sci.19, 2412 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cheng, J. et al. Inhibition of transforming growth factor beta-activated kinase 1 prevents inflammation-related cartilage degradation in osteoarthritis. Sci. Rep.6, 34497 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wang, Y., Zhao, X., Lotz, M., Terkeltaub, R. & Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol.67, 2141–2153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang, J. et al. AMPK: implications in osteoarthritis and therapeutic targets. Am. J. Transl. Res.12, 7670–7681 (2020). [PMC free article] [PubMed] [Google Scholar]
- 219.Zhu, H. et al. CCAL1 enhances osteoarthritis through the NF-kappaB/AMPK signaling pathway. FEBS Open Bio10, 2553–2563 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang, Y. et al. Inhibition of glycolysis ameliorate arthritis in adjuvant arthritis rats by inhibiting synoviocyte activation through AMPK/NF-small ka, CyrillicB pathway. Inflamm. Res.69, 569–578 (2020). [DOI] [PubMed] [Google Scholar]
- 221.Shang, L. & Wang, X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy7, 924–926 (2011). [DOI] [PubMed] [Google Scholar]
- 222.Husa, M., Petursson, F., Lotz, M., Terkeltaub, R. & Liu-Bryan, R. C/EBP homologous protein drives pro-catabolic responses in chondrocytes. Arthritis Res. Ther.15, R218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Feng, K., Chen, Z., Pengcheng, L., Zhang, S. & Wang, X. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J. Cell Physiol.234, 18192–18205 (2019). [DOI] [PubMed] [Google Scholar]
- 224.Zhao, X. et al. Peroxisome proliferator-activated receptor gamma coactivator 1alpha and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheumatol.66, 3073–3082 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Li, J. et al. Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann. Rheum. Dis.79, 635–645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Feng, X. et al. Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging (Albany NY)12, 1087–1103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wang, C. et al. Metformin mitigates cartilage degradation by activating AMPK/SIRT1-mediated autophagy in a mouse osteoarthritis model. Front. Pharm.11, 1114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Story, G. M. et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell112, 819–829 (2003). [DOI] [PubMed] [Google Scholar]
- 229.Wang, S. et al. Negative regulation of TRPA1 by AMPK in primary sensory neurons as a potential mechanism of painful diabetic neuropathy. Diabetes67, 98–109 (2018). [DOI] [PubMed] [Google Scholar]
- 230.Kong, C. et al. Active vitamin D activates chondrocyte autophagy to reduce osteoarthritis via mediating the AMPK-mTOR signaling pathway. Biochem. Cell Biol.98, 434–442 (2020). [DOI] [PubMed] [Google Scholar]
- 231.Kunanusornchai, W. et al. Chitosan oligosaccharide suppresses synovial inflammation via AMPK activation: an in vitro and in vivo study. Pharm. Res.113, 458–467 (2016). [DOI] [PubMed] [Google Scholar]
- 232.Carames, B. et al. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis.71, 575–581 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wang, C. et al. Safflower yellow alleviates osteoarthritis and prevents inflammation by inhibiting PGE2 release and regulating NF-kappaB/SIRT1/AMPK signaling pathways. Phytomedicine78, 153305 (2020). [DOI] [PubMed] [Google Scholar]
- 234.Wang, L. et al. Puerarin attenuates osteoarthritis via upregulating AMP-activated protein kinase/proliferator-activated receptor-gamma coactivator-1 signaling pathway in osteoarthritis rats. Pharmacology102, 117–125 (2018). [DOI] [PubMed] [Google Scholar]
- 235.Ma, T. et al. Bilobalide exerts anti-inflammatory effects on chondrocytes through the AMPK/SIRT1/mTOR pathway to attenuate aclt-induced post-traumatic osteoarthritis in rats. Front. Pharm.13, 783506 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.He, M. et al. Metformin prevents or delays the development and progression of osteoarthritis: new insight and mechanism of action. Cells11, 3012 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Nageeb, M. M., Saadawy, S. F. & Attia, S. H. Breast milk mesenchymal stem cells abate cisplatin-induced cardiotoxicity in adult male albino rats via modulating the AMPK pathway. Sci. Rep.12, 17554 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Yerevanian, A. & Soukas, A. A. Metformin: mechanisms in human obesity and weight loss. Curr. Obes. Rep.8, 156–164 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wang, Y. et al. Association between metformin use and disease progression in obese people with knee osteoarthritis: data from the Osteoarthritis Initiative-a prospective cohort study. Arthritis Res. Ther.21, 127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ameen, O., Samaka, R. M. & Abo-Elsoud, R. A. A. Metformin alleviates neurocognitive impairment in aging via activation of AMPK/BDNF/PI3K pathway. Sci. Rep.12, 17084 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zhang, Y. et al. Neuromedin B receptor stimulation of Cav3.2 T-type Ca(2+) channels in primary sensory neurons mediates peripheral pain hypersensitivity. Theranostics11, 9342–9357 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Xing, H. et al. Metformin mitigates cholesterol accumulation via the AMPK/SIRT1 pathway to protect osteoarthritis chondrocytes. Biochem. Biophys. Res. Commun.632, 113–121 (2022). [DOI] [PubMed] [Google Scholar]
- 243.Na, H. S. et al. Metformin attenuates monosodium-iodoacetate-induced osteoarthritis via regulation of pain mediators and the autophagy-lysosomal pathway. Cells10, 681 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Song, Y., Wu, Z. & Zhao, P. The effects of metformin in the treatment of osteoarthritis: current perspectives. Front. Pharm.13, 952560 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Nagata, K. et al. Runx2 and Runx3 differentially regulate articular chondrocytes during surgically induced osteoarthritis development. Nat. Commun.13, 6187 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lin, C. C. et al. Metformin enhances cisplatin cytotoxicity by suppressing signal transducer and activator of transcription-3 activity independently of the liver kinase B1-AMP-activated protein kinase pathway. Am. J. Respir. Cell Mol. Biol.49, 241–250 (2013). [DOI] [PubMed] [Google Scholar]
- 247.Zheng, Z., Bian, Y., Zhang, Y., Ren, G. & Li, G. Metformin activates AMPK/SIRT1/NF-kappaB pathway and induces mitochondrial dysfunction to drive caspase3/GSDME-mediated cancer cell pyroptosis. Cell Cycle19, 1089–1104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Luo, X., Hu, R., Zheng, Y., Liu, S. & Zhou, Z. Metformin shows anti-inflammatory effects in murine macrophages through Dicer/microribonucleic acid-34a-5p and microribonucleic acid-125b-5p. J. Diabetes Investig.11, 101–109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Tian, R. et al. Metformin ameliorates endotoxemia-induced endothelial pro-inflammatory responses via AMPK-dependent mediation of HDAC5 and KLF2. Biochim. Biophys. Acta Mol. Basis Dis.1865, 1701–1712 (2019). [DOI] [PubMed] [Google Scholar]
- 250.Lin, Z. et al. JUNB-FBXO21-ERK axis promotes cartilage degeneration in osteoarthritis by inhibiting autophagy. Aging Cell20, e13306 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Hwang, H. Y., Shim, J. S., Kim, D. & Kwon, H. J. Antidepressant drug sertraline modulates AMPK-MTOR signaling-mediated autophagy via targeting mitochondrial VDAC1 protein. Autophagy17, 2783–2799 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Guo, H. et al. Induction of autophagy via the ROS-dependent AMPK-mTOR pathway protects copper-induced spermatogenesis disorder. Redox Biol.49, 102227 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wang, C. et al. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci. Trends12, 605–612 (2019). [DOI] [PubMed] [Google Scholar]
- 254.Klionsky, D. J. The autophagy connection. Dev. Cell19, 11–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Mossmann, D., Park, S. & Hall, M. N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer18, 744–757 (2018). [DOI] [PubMed] [Google Scholar]
- 257.Holz, M. K., Ballif, B. A., Gygi, S. P. & Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell123, 569–580 (2005). [DOI] [PubMed] [Google Scholar]
- 258.Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science307, 1098–1101 (2005). [DOI] [PubMed] [Google Scholar]
- 259.Liu, P. D. et al. PtdIns(3,4,5)P-3-dependent activation of the mTORC2 kinase complex. Cancer Discov.5, 1194–1209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Gangloff, Y. G. et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell. Biol.24, 9508–9516 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pal, B., Endisha, H., Zhang, Y. & Kapoor, M. mTOR: a potential therapeutic target in osteoarthritis? Drugs R. D.15, 27–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Zhang, Y. et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis.74, 1432–1440 (2015). [DOI] [PubMed] [Google Scholar]
- 263.Zhang, H. et al. mTORC1 activation downregulates FGFR3 and PTH/PTHrP receptor in articular chondrocytes to initiate osteoarthritis. Osteoarthr. Cartil.25, 952–963 (2017). [DOI] [PubMed] [Google Scholar]
- 264.Vasheghani, F. et al. PPAR gamma deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann. Rheum. Dis.74, 569–578 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol.13, 132–U171 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bijlsma, J. W. J., Berenbaum, F. & Lafeber, F. P. J. G. Osteoarthritis: an update with relevance for clinical practice. Lancet377, 2115–2126 (2011). [DOI] [PubMed] [Google Scholar]
- 267.Yang, H. X. et al. MTORC1 coordinates the autophagy and apoptosis signaling in articular chondrocytes in osteoarthritic temporomandibular joint. Autophagy16, 271–288 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Lin, C. X. et al. Activation of mTORC1 in subchondral bone preosteoblasts promotes osteoarthritis by stimulating bone sclerosis and secretion of CXCL12. Bone Res.7, 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Karonitsch, T. et al. mTOR senses environmental cues to shape the fibroblast-like synoviocyte response to inflammation. Cell Rep.23, 2157–2167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Moutinho, C. & Esteller, M. MicroRNAs and epigenetics. Adv. Cancer Res.135, 189–220 (2017). [DOI] [PubMed] [Google Scholar]
- 271.Jiang, H. T., Dai, J., Zhang, C., Sun, H. L. & Tang, X. M. Circ_0045714 alleviates TNF-alpha-induced chondrocyte injury and extracellular matrix degradation through miR-218-5p/HRAS axis. J. Bioenerg. Biomembr.53, 97–107 (2021). [DOI] [PubMed] [Google Scholar]
- 272.Zhou, X. D. et al. Down-regulated ciRS-7/up-regulated miR-7 axis aggravated cartilage degradation and autophagy defection by PI3K/AKT/mTOR activation mediated by IL-17A in osteoarthritis. Aging Us12, 20163–20183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Cai, C. et al. MiR-27a promotes the autophagy and apoptosis of IL-1 beta treated-articular chonarocytes in osteoarthritis through PI3K/AKT/mTOR signaling. Aging Us11, 6371–6384 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 274.Wu, J. Y. et al. miR-100-5p-abundant exosomes derived from infrapatellar fat pad MSCs protect articular cartilage and ameliorate gait abnormalities via inhibition of mTOR in osteoarthritis. Biomaterials206, 87–100 (2019). [DOI] [PubMed] [Google Scholar]
- 275.Ansari, M. Y., Khan, N. M. & Haqqi, T. M. A standardized extract of Butea monosperma (Lam.) flowers suppresses the IL-1 beta-induced expression of IL-6 and matrix-metalloproteases by activating autophagy in human osteoarthritis chondrocytes. Biomed. Pharmacother.96, 198–207 (2017). [DOI] [PubMed] [Google Scholar]
- 276.Dunwoodie, S. L. The role of hypoxia in development of the Mammalian embryo. Dev. Cell17, 755–773 (2009). [DOI] [PubMed] [Google Scholar]
- 277.Semenza, G. L. Hypoxia-inducible factors in physiology and medicine. Cell148, 399–408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Schipani, E. et al. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev.15, 2865–2876 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Yao, Q. et al. Suppressing mitochondrial respiration is critical for hypoxia tolerance in the fetal growth plate. Dev. Cell49, 748–763 e747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Long, F. Less is more: ditching mitochondria saves hypoxic cartilage. Dev. Cell49, 656–658 (2019). [DOI] [PubMed] [Google Scholar]
- 281.Yao, Q., Parvez-Khan, M. & Schipani, E. In vivo survival strategies for cellular adaptation to hypoxia: HIF1alpha-dependent suppression of mitochondrial oxygen consumption and decrease of intracellular hypoxia are critical for survival of hypoxic chondrocytes. Bone140, 115572 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Saito, T. et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med.16, 678–686 (2010). [DOI] [PubMed] [Google Scholar]
- 283.Araldi, E., Khatri, R., Giaccia, A. J., Simon, M. C. & Schipani, E. Lack of HIF-2alpha in limb bud mesenchyme causes a modest and transient delay of endochondral bone development. Nat. Med.17, 25–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Pfander, D. & Gelse, K. Hypoxia and osteoarthritis: how chondrocytes survive hypoxic environments. Curr. Opin. Rheumatol.19, 457–462 (2007). [DOI] [PubMed] [Google Scholar]
- 285.Ummarino, D. Osteoarthritis: hypoxia protects against cartilage loss by regulating Wnt signalling. Nat. Rev. Rheumatol.12, 315 (2016). [DOI] [PubMed] [Google Scholar]
- 286.Pfander, D., Cramer, T. & Swoboda, B. Hypoxia and HIF-1alpha in osteoarthritis. Int. Orthop.29, 6–9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Yang, S. et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med.16, 687–693 (2010). [DOI] [PubMed] [Google Scholar]
- 288.Bouaziz, W. et al. Interaction of HIF1alpha and beta-catenin inhibits matrix metalloproteinase 13 expression and prevents cartilage damage in mice. Proc. Natl Acad. Sci. USA113, 5453–5458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Qing, L. et al. Expression of hypoxia-inducible factor-1alpha in synovial fluid and articular cartilage is associated with disease severity in knee osteoarthritis. Exp. Ther. Med.13, 63–68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Lu, J. et al. Hypoxia inducible factor-1alpha is a regulator of autophagy in osteoarthritic chondrocytes. Cartilage13, 1030S–1040S (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Yang, F., Huang, R., Ma, H., Zhao, X. & Wang, G. miRNA-411 regulates chondrocyte autophagy in osteoarthritis by targeting hypoxia-inducible factor 1 alpha (HIF-1alpha). Med. Sci. Monit.26, e921155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Hu, S. et al. Stabilization of HIF-1alpha alleviates osteoarthritis via enhancing mitophagy. Cell Death Dis.11, 481 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Weng, T. et al. Loss of Vhl in cartilage accelerated the progression of age-associated and surgically induced murine osteoarthritis. Osteoarthr. Cartil.22, 1197–1205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Rius, J. et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature453, 807–811 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Chen, J., Crawford, R. & Xiao, Y. Vertical inhibition of the PI3K/Akt/mTOR pathway for the treatment of osteoarthritis. J. Cell. Biochem.114, 245–249 (2013). [DOI] [PubMed] [Google Scholar]
- 296.Fernandez-Torres, J., Zamudio-Cuevas, Y., Martinez-Nava, G. A. & Lopez-Reyes, A. G. Hypoxia-Inducible Factors (HIFs) in the articular cartilage: a systematic review. Eur. Rev. Med. Pharmacol. Sci.21, 2800–2810 (2017). [PubMed] [Google Scholar]
- 297.Li, W., Cai, L., Zhang, Y., Cui, L. & Shen, G. Intra-articular resveratrol injection prevents osteoarthritis progression in a mouse model by activating SIRT1 and thereby silencing HIF-2alpha. J. Orthop. Res.33, 1061–1070 (2015). [DOI] [PubMed] [Google Scholar]
- 298.Xia, Y., Choi, H. K. & Lee, K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur. J. Med. Chem.49, 24–40 (2012). [DOI] [PubMed] [Google Scholar]
- 299.Burridge, K. Focal adhesions: a personal perspective on a half century of progress. FEBS J.284, 3355–3361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Cao, H. et al. Focal adhesion protein Kindlin-2 regulates bone homeostasis in mice. Bone Res.8, 2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Wang, Y. et al. Focal adhesion proteins Pinch1 and Pinch2 regulate bone homeostasis in mice. JCI Insight4, e131692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Fu, X. et al. Kindlin-2 regulates skeletal homeostasis by modulating PTH1R in mice. Signal Transduct. Target Ther.5, 297 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Wu, C. et al. Kindlin-2 controls TGF-beta signalling and Sox9 expression to regulate chondrogenesis. Nat. Commun.6, 7531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Gao, H. et al. Lipoatrophy and metabolic disturbance in mice with adipose-specific deletion of kindlin-2. JCI Insight.4, e128405 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Gao, H. et al. Pinch loss ameliorates obesity, glucose intolerance, and fatty liver by modulating adipocyte apoptosis in mice. Diabetes70, 2492–2505 (2021). [DOI] [PubMed] [Google Scholar]
- 306.Gao, H. et al. Kindlin-2 haploinsufficiency protects against fatty liver by targeting Foxo1 in mice. Nat. Commun.13, 1025 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Guo, L. et al. Kindlin-2 regulates mesenchymal stem cell differentiation through control of YAP1/TAZ. J. Cell Biol.217, 1431–1451 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wu, X. et al. Kindlin-2 preserves integrity of the articular cartilage to protect against osteoarthritis. Nat. Aging2, 332–347 (2022). [DOI] [PubMed]
- 309.Wu, X. et al. Kindlin-2 deletion in osteoprogenitors causes severe chondrodysplasia and low-turnover osteopenia in mice. J. Orthop. Transl.32, 41–48 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Zhu, K. et al. Kindlin-2 modulates MafA and beta-catenin expression to regulate beta-cell function and mass in mice. Nat. Commun.11, 484 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Lei, Y. et al. LIM domain proteins Pinch1/2 regulate chondrogenesis and bone mass in mice. Bone Res.8, 37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Raducanu, A., Hunziker, E. B., Drosse, I. & Aszodi, A. Beta1 integrin deficiency results in multiple abnormalities of the knee joint. J. Biol. Chem.284, 23780–23792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Jin, H. et al. Mechanistic insight into the roles of integrins in osteoarthritis. Front. Cell Dev. Biol.9, 693484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Qin, L., Liu, W., Cao, H. & Xiao, G. Molecular mechanosensors in osteocytes. Bone Res.8, 23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Qin, L. et al. Roles of mechanosensitive channel Piezo1/2 proteins in skeleton and other tissues. Bone Res.9, 44 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Lian, C. et al. Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin beta1-SMAD1 interaction. Bone Res.7, 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Zhen, G. et al. Mechanical stress determines the configuration of TGFbeta activation in articular cartilage. Nat. Commun.12, 1706 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Zhen, G. et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med.19, 704–712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Ciregia, F. et al. Modulation of alphaVbeta6 integrin in osteoarthritis-related synovitis and the interaction with VTN(381-397 a.a.) competing for TGF-beta1 activation. Exp. Mol. Med.53, 210–222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Wang, Q. et al. Dysregulated integrin alphaVbeta3 and CD47 signaling promotes joint inflammation, cartilage breakdown, and progression of osteoarthritis. JCI Insight.4, e128616 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Wu, H. et al. Specific inhibition of FAK signaling attenuates subchondral bone deterioration and articular cartilage degeneration during osteoarthritis pathogenesis. J. Cell Physiol.235, 8653–8666 (2020). [DOI] [PubMed] [Google Scholar]
- 322.Yanoshita, M. et al. FAK inhibition protects condylar cartilage under excessive mechanical stress. Oral. Dis.26, 1736–1746 (2020). [DOI] [PubMed] [Google Scholar]
- 323.Miyauchi, A. et al. Alleviation of murine osteoarthritis by deletion of the focal adhesion mechanosensitive adapter, Hic-5. Sci. Rep.9, 15770 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Salazar, V. S., Gamer, L. W. & Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol.12, 203–221 (2016). [DOI] [PubMed] [Google Scholar]
- 325.Zhang, Y. & Que, J. BMP signaling in development, stem cells, and diseases of the gastrointestinal tract. Annu Rev. Physiol.82, 251–273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Heldin, C. H., Miyazono, K. & ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature390, 465–471 (1997). [DOI] [PubMed] [Google Scholar]
- 327.Wang, R. N. et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis.1, 87–105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Lowery, J. W. & Rosen, V. The BMP pathway and its inhibitors in the skeleton. Physiol. Rev.98, 2431–2452 (2018). [DOI] [PubMed] [Google Scholar]
- 329.Ashraf, S. et al. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthr. Cartil.24, 196–205 (2016). [DOI] [PubMed] [Google Scholar]
- 330.Deng, Z. H., Li, Y. S., Gao, X., Lei, G. H. & Huard, J. Bone morphogenetic proteins for articular cartilage regeneration. Osteoarthr. Cartil.26, 1153–1161 (2018). [DOI] [PubMed] [Google Scholar]
- 331.Thielen, N. G. M., van der Kraan, P. M. & van Caam, A. P. M. TGFbeta/BMP signaling pathway in cartilage homeostasis. Cells8, 969 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Zhong, L., Huang, X., Karperien, M. & Post, J. N. The regulatory role of signaling crosstalk in hypertrophy of MSCs and human articular chondrocytes. Int. J. Mol. Sci.16, 19225–19247 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Blaney Davidson, E. N. et al. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res. Ther.9, R102 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.van Beuningen, H. M., Glansbeek, H. L., van der Kraan, P. M. & van den Berg, W. B. Differential effects of local application of BMP-2 or TGF-beta 1 on both articular cartilage composition and osteophyte formation. Osteoarthr. Cartil.6, 306–317 (1998). [DOI] [PubMed] [Google Scholar]
- 335.Zhou, N. et al. BMP2 induces chondrogenic differentiation, osteogenic differentiation and endochondral ossification in stem cells. Cell Tissue Res.366, 101–111 (2016). [DOI] [PubMed] [Google Scholar]
- 336.Flechtenmacher, J. et al. Recombinant human osteogenic protein 1 is a potent stimulator of the synthesis of cartilage proteoglycans and collagens by human articular chondrocytes. Arthritis Rheum.39, 1896–1904 (1996). [DOI] [PubMed] [Google Scholar]
- 337.Luyten, F. P., Chen, P., Paralkar, V. & Reddi, A. H. Recombinant bone morphogenetic protein-4, transforming growth factor-beta 1, and activin A enhance the cartilage phenotype of articular chondrocytes in vitro. Exp. Cell Res210, 224–229 (1994). [DOI] [PubMed] [Google Scholar]
- 338.Bobacz, K. et al. Expression of bone morphogenetic protein 6 in healthy and osteoarthritic human articular chondrocytes and stimulation of matrix synthesis in vitro. Arthritis Rheum.48, 2501–2508 (2003). [DOI] [PubMed] [Google Scholar]
- 339.Gamer, L. W. et al. The Role of Bmp2 in the maturation and maintenance of the murine knee joint. J. Bone Min. Res.33, 1708–1717 (2018). [DOI] [PubMed] [Google Scholar]
- 340.Caron, M. M. et al. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthr. Cartil.21, 604–613 (2013). [DOI] [PubMed] [Google Scholar]
- 341.Caron, M. M. et al. BAPX-1/NKX-3.2 acts as a chondrocyte hypertrophy molecular switch in osteoarthritis. Arthritis Rheumatol.67, 2944–2956 (2015). [DOI] [PubMed] [Google Scholar]
- 342.Abula, K. et al. Elimination of BMP7 from the developing limb mesenchyme leads to articular cartilage degeneration and synovial inflammation with increased age. FEBS Lett.589, 1240–1248 (2015). [DOI] [PubMed] [Google Scholar]
- 343.Papathanasiou, I., Malizos, K. N. & Tsezou, A. Bone morphogenetic protein-2-induced Wnt/beta-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res. Ther.14, R82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Wang, W. et al. The TGFbeta type I receptor TGFbetaRI functions as an inhibitor of BMP signaling in cartilage. Proc. Natl. Acad. Sci. USA116, 15570–15579 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Zhai, G. Clinical relevance of biochemical and metabolic changes in osteoarthritis. Adv. Clin. Chem.101, 95–120 (2021). [DOI] [PubMed] [Google Scholar]
- 346.Perez-Lozano, M. L. et al. Gremlin-1 and BMP-4 overexpressed in osteoarthritis drive an osteochondral-remodeling program in osteoblasts and hypertrophic chondrocytes. Int. J. Mol. Sci.23, 2084 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Chien, S. Y. et al. Noggin Inhibits IL-1beta and BMP-2 expression, and attenuates cartilage degeneration and subchondral bone destruction in experimental osteoarthritis. Cells9, 927 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Iijima, H. et al. Physiological exercise loading suppresses post-traumatic osteoarthritis progression via an increase in bone morphogenetic proteins expression in an experimental rat knee model. Osteoarthr. Cartil.25, 964–975 (2017). [DOI] [PubMed] [Google Scholar]
- 349.Melnik, S., Hofmann, N., Gabler, J., Hecht, N. & Richter, W. MiR-181a targets RSPO2 and regulates bone morphogenetic protein - WNT signaling crosstalk during chondrogenic differentiation of mesenchymal stromal cells. Front. Cell Dev. Biol.9, 747057 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Caron, M. M. J. et al. Discovery of bone morphogenetic protein 7-derived peptide sequences that attenuate the human osteoarthritic chondrocyte phenotype. Mol. Ther. Methods Clin. Dev.21, 247–261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Akkiraju, H. et al. CK2.1, a bone morphogenetic protein receptor type Ia mimetic peptide, repairs cartilage in mice with destabilized medial meniscus. Stem Cell Res. Ther.8, 82 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Taniyama, T. et al. Repair of osteochondral defects in a rabbit model using a porous hydroxyapatite collagen composite impregnated with bone morphogenetic protein-2. Artif. Organs39, 529–535 (2015). [DOI] [PubMed] [Google Scholar]
- 353.Han, F. X. et al. A pilot study of conically graded chitosan-gelatin hydrogel/PLGA scaffold with dual-delivery of TGF-beta 1 and BMP-2 for regeneration of cartilage-bone interface. J. Biomed. Mater. Res. Part B-Appl. Biomater.103, 1344–1353 (2015). [DOI] [PubMed] [Google Scholar]
- 354.Jeong, C. G., Zhang, H. N. & Hollister, S. J. Three-dimensional polycaprolactone scaffold-conjugated bone morphogenetic protein-2 promotes cartilage regeneration from primary chondrocytes in vitro and in vivo without accelerated endochondral ossification. J. Biomed. Mater. Res. Part A100a, 2088–2096 (2012). [DOI] [PubMed] [Google Scholar]
- 355.Zhang, Z. et al. The functions of BMP3 in rabbit articular cartilage repair. Int. J. Mol. Sci.16, 25934–25946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Li, H. S. et al. The superior regenerative potential of muscle-derived stem cells for articular cartilage repair is attributed to high cell survival and chondrogenic potential. Mol. Ther.-Methods Clin. Dev.3, 16065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Sekiya, I. et al. Periodic knee injections of BMP-7 delay cartilage degeneration induced by excessive running in rats. J. Orthop. Res.27, 1088–1092 (2009). [DOI] [PubMed] [Google Scholar]
- 358.Mendes, L. F. et al. Combinatorial analysis of growth factors reveals the contribution of bone morphogenetic proteins to chondrogenic differentiation of human periosteal cells. Tissue Eng. Part C.22, 473–486 (2016). [DOI] [PubMed] [Google Scholar]
- 359.Choi, S., Cho, T. J., Kwon, S. K., Lee, G. & Cho, J. Chondrogenesis of periodontal ligament stem cells by transforming growth factor-beta 3 and bone morphogenetic protein-6 in a normal healthy impacted third molar. Int. J. Oral. Sci.5, 7–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Ren, X. Y. et al. Nanoparticulate mineralized collagen scaffolds and BMP-9 induce a long-term bone cartilage construct in human mesenchymal stem cells. Adv. Healthc. Mater.5, 1821–1830 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Thisse, B. & Thisse, C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev. Biol.287, 390–402 (2005). [DOI] [PubMed] [Google Scholar]
- 362.Beenken, A. & Mohammadi, M. The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov.8, 235–253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Ornitz, D. M. & Itoh, N. New developments in the biology of fibroblast growth factors. WIREs Mech. Dis.14, e1549 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Im, H. J. et al. Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase Cdelta pathways in human adult articular chondrocytes. J. Biol. Chem.282, 11110–11121 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Orr-Urtreger, A., Givol, D., Yayon, A., Yarden, Y. & Lonai, P. Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development113, 1419–1434 (1991). [DOI] [PubMed] [Google Scholar]
- 366.Mohammadi, M., Olsen, S. K. & Ibrahimi, O. A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev.16, 107–137 (2005). [DOI] [PubMed] [Google Scholar]
- 367.Kisand, K., Tamm, A. E., Lintrop, M. & Tamm, A. O. New insights into the natural course of knee osteoarthritis: early regulation of cytokines and growth factors, with emphasis on sex-dependent angiogenesis and tissue remodeling. A pilot study. Osteoarthr. Cartil.26, 1045–1054 (2018). [DOI] [PubMed] [Google Scholar]
- 368.El-Seoudi, A. et al. Catabolic effects of FGF-1 on chondrocytes and its possible role in osteoarthritis. J. Cell Commun. Signal.11, 255–263 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Vincent, T. L., McLean, C. J., Full, L. E., Peston, D. & Saklatvala, J. FGF-2 is bound to perlecan in the pericellular matrix of articular cartilage, where it acts as a chondrocyte mechanotransducer. Osteoarthr. Cartil.15, 752–763 (2007). [DOI] [PubMed] [Google Scholar]
- 370.Im, H.-J. et al. Basic fibroblast growth factor accelerates matrix degradation via a neuro-endocrine pathway in human adult articular chondrocytes. J. Cell. Physiol.215, 452–463 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 371.Nummenmaa, E., Hämäläinen, M., Moilanen, T., Vuolteenaho, K. & Moilanen, E. Effects of FGF-2 and FGF receptor antagonists on MMP enzymes, aggrecan, and type II collagen in primary human OA chondrocytes. Scand. J. Rheumatol.44, 321–330 (2015). [DOI] [PubMed] [Google Scholar]
- 372.Uchii, M. et al. Role of fibroblast growth factor 8 (FGF8) in animal models of osteoarthritis. Arthritis Res. Ther.10, R90 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Zhou, S. et al. Exogenous fibroblast growth factor 9 attenuates cartilage degradation and aggravates osteophyte formation in post-traumatic osteoarthritis. Osteoarthr. Cartil.24, 2181–2192 (2016). [DOI] [PubMed] [Google Scholar]
- 374.Rockel, J. S. et al. Hedgehog inhibits β-catenin activity in synovial joint development and osteoarthritis. J. Clin. Investig.126, 1649–1663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Mori, Y. et al. Identification of fibroblast growth factor-18 as a molecule to protect adult articular cartilage by gene expression profiling. J. Biol. Chem.289, 10192–10200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Meloni, G. R. et al. Recombinant human FGF18 preserves depth-dependent mechanical inhomogeneity in articular cartilage. Eur. Cells Mater.38, 23–34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Li, R. et al. Upregulation of fibroblast growth factor 1 in the synovial membranes of patients with late stage osteoarthritis. Genet. Mol. Res.14, 11191–11199 (2015). [DOI] [PubMed] [Google Scholar]
- 378.Kim, J. H., Lee, M. C., Seong, S. C., Park, K. H. & Lee, S. Enhanced proliferation and chondrogenic differentiation of human synovium-derived stem cells expanded with basic fibroblast growth factor. Tissue Eng. Part A17, 991–1002 (2011). [DOI] [PubMed] [Google Scholar]
- 379.Weng, T. et al. Genetic inhibition of fibroblast growth factor receptor 1 in knee cartilage attenuates the degeneration of articular cartilage in adult mice. Arthritis Rheum.64, 3982–3992 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Deng, C., Wynshaw-Boris, A., Zhou, F., Kuo, A. & Leder, P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell84, 911–921 (1996). [DOI] [PubMed] [Google Scholar]
- 381.Hochberg, M. C. et al. Effect of intra-articular sprifermin vs placebo on femorotibial joint cartilage thickness in patients with osteoarthritis: the FORWARD randomized clinical trial. Jama322, 1360–1370 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Xiao, G., Jiang, D., Gopalakrishnan, R. & Franceschi, R. T. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J. Biol. Chem.277, 36181–36187 (2002). [DOI] [PubMed] [Google Scholar]
- 383.Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol.149, 313–323 (2018). [DOI] [PubMed] [Google Scholar]
- 384.Renny, T. F. et al. Multiple signaling pathways converge on the Cbfa1/Runx2 transcription factor to regulate osteoblast differentiation. Connect Tissue Res.44, 109–116 (2003). [PMC free article] [PubMed] [Google Scholar]
- 385.Xiao, G. et al. Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast-specific osteocalcin gene expression. J. Biol. Chem.280, 30689–30696 (2005). [DOI] [PubMed] [Google Scholar]
- 386.Zhao, Z., Zhao, M., Xiao, G. & Franceschi, R. T. Gene transfer of the Runx2 transcription factor enhances osteogenic activity of bone marrow stromal cells in vitro and in vivo. Mol. Ther.12, 247–253 (2005). [DOI] [PubMed] [Google Scholar]
- 387.Yu, S. et al. General transcription factor IIA-gamma increases osteoblast-specific osteocalcin gene expression via activating transcription factor 4 and runt-related transcription factor 2. J. Biol. Chem.283, 5542–5553 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Zheng, Q. et al. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Biol.162, 833–842 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Enomoto, H. et al. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J. Biol. Chem.275, 8695–8702 (2000). [DOI] [PubMed] [Google Scholar]
- 390.Otto, F. et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell89, 765–771 (1997). [DOI] [PubMed] [Google Scholar]
- 391.Tsuji, K., Komori, T. & Noda, M. Aged mice require full transcription factor, Runx2/Cbfa1, gene dosage for cancellous bone regeneration after bone marrow ablation. J. Bone Min. Res.19, 1481–1489 (2004). [DOI] [PubMed] [Google Scholar]
- 392.Catheline, S. E. et al. Chondrocyte-specific RUNX2 overexpression accelerates post-traumatic osteoarthritis progression in adult mice. J. Bone Min. Res.34, 1676–1689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Liao, L. et al. Deletion of Runx2 in articular chondrocytes decelerates the progression of DMM-induced osteoarthritis in adult mice. Sci. Rep.7, 2371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Li, F. et al. Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. J. Bone Min. Res.26, 2899–2910 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Zhao, W. et al. Runx2 and microRNA regulation in bone and cartilage diseases. Ann. N. Y. Acad. Sci.1383, 80–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Tosa, I. et al. Postnatal Runx2 deletion leads to low bone mass and adipocyte accumulation in mice bone tissues. Biochem. Biophys. Res. Commun.516, 1229–1233 (2019). [DOI] [PubMed] [Google Scholar]
- 397.Adhami, M., Ghori-Javed, F. Y., Chen, H., Gutierrez, S. E. & Javed, A. Runx2 regulates the gene network associated with insulin signaling and energy homeostasis. Cells Tissues Organs194, 232–237 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Chopin, M. et al. RUNX2 mediates plasmacytoid dendritic cell egress from the bone marrow and controls viral immunity. Cell Rep.15, 866–878 (2016). [DOI] [PubMed] [Google Scholar]
- 399.Kim, M. S., Gernapudi, R., Choi, E. Y., Lapidus, R. G. & Passaniti, A. Characterization of CADD522, a small molecule that inhibits RUNX2-DNA binding and exhibits antitumor activity. Oncotarget8, 70916–70940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Ji, M. L. et al. Precise targeting of miR-141/200c cluster in chondrocytes attenuates osteoarthritis development. Ann. Rheum. Dis.80, 356–366 (2021). [DOI] [PubMed] [Google Scholar]
- 401.Wei, Y. et al. Targeting cartilage EGFR pathway for osteoarthritis treatment. Sci. Transl. Med.13, eabb3946 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Wei, Y. et al. Phospholipase A(2) inhibitor-loaded micellar nanoparticles attenuate inflammation and mitigate osteoarthritis progression. Sci. Adv.7, eabe6374 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Zhao, L. et al. Exploration of CRISPR/Cas9-based gene editing as therapy for osteoarthritis. Ann. Rheum. Dis.78, 676–682 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Coutinho de Almeida, R. et al. RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Ann. Rheum. Dis.78, 270–277 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Ntoumou, E. et al. Serum microRNA array analysis identifies miR-140-3p, miR-33b-3p and miR-671-3p as potential osteoarthritis biomarkers involved in metabolic processes. Clin. Epigenet.9, 127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Zhang, H., Song, B. & Pan, Z. Downregulation of microRNA-9 increases matrix metalloproteinase-13 expression levels and facilitates osteoarthritis onset. Mol. Med. Rep.17, 3708–3714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Mao, G. et al. Exosomal miR-95-5p regulates chondrogenesis and cartilage degradation via histone deacetylase 2/8. J. Cell Mol. Med.22, 5354–5366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Zhao, X., Li, H. & Wang, L. MicroRNA-107 regulates autophagy and apoptosis of osteoarthritis chondrocytes by targeting TRAF3. Int. Immunopharmacol.71, 181–187 (2019). [DOI] [PubMed] [Google Scholar]
- 409.Song, J. et al. MicroRNA-181b regulates articular chondrocytes differentiation and cartilage integrity. Biochem. Biophys. Res. Commun.431, 210–214 (2013). [DOI] [PubMed] [Google Scholar]
- 410.Lian, W. S. et al. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis.9, 919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Meng, F. et al. MicroRNA-320 regulates matrix metalloproteinase-13 expression in chondrogenesis and interleukin-1beta-induced chondrocyte responses. Osteoarthr. Cartil.24, 932–941 (2016). [DOI] [PubMed] [Google Scholar]
- 412.Liu, J. et al. Exosomal transfer of osteoclast-derived miRNAs to chondrocytes contributes to osteoarthritis progression. Nat. Aging1, 368–384 (2021). [DOI] [PubMed] [Google Scholar]
- 413.Little, C. B. et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum.60, 3723–3733 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Wang, M. et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther.15, R5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Salazar-Noratto, G. E., De Nijs, N., Stevens, H. Y., Gibson, G. & Guldberg, R. E. Regional gene expression analysis of multiple tissues in an experimental animal model of post-traumatic osteoarthritis. Osteoarthr. Cartil.27, 294–303 (2019). [DOI] [PubMed] [Google Scholar]
- 416.Chang, J. C. et al. Global molecular changes in a tibial compression induced ACL rupture model of post-traumatic osteoarthritis. J. Orthop. Res.35, 474–485 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Nuti, E. et al. N-O-isopropyl sulfonamido-based hydroxamates: design, synthesis and biological evaluation of selective matrix metalloproteinase-13 inhibitors as potential therapeutic agents for osteoarthritis. J. Med; Chem.52, 4757–4773 (2009). [DOI] [PubMed] [Google Scholar]
- 418.De Savi, C. et al. Selective non zinc binding inhibitors of MMP13. Bioorg. Med. Chem. Lett.21, 4215–4219 (2011). [DOI] [PubMed] [Google Scholar]
- 419.Gege, C. et al. Discovery and evaluation of a non-Zn chelating, selective matrix metalloproteinase 13 (MMP-13) inhibitor for potential intra-articular treatment of osteoarthritis. J. Med. Chem.55, 709–716 (2012). [DOI] [PubMed] [Google Scholar]
- 420.Settle, S. et al. Cartilage degradation biomarkers predict efficacy of a novel, highly selective matrix metalloproteinase 13 inhibitor in a dog model of osteoarthritis: confirmation by multivariate analysis that modulation of type II collagen and aggrecan degradation peptides parallels pathologic changes. Arthritis Rheum.62, 3006–3015 (2010). [DOI] [PubMed] [Google Scholar]
- 421.Krzeski, P. et al. Development of musculoskeletal toxicity without clear benefit after administration of PG-116800, a matrix metalloproteinase inhibitor, to patients with knee osteoarthritis: a randomized, 12-month, double-blind, placebo-controlled study. Arthritis Res. Ther.9, R109 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Glasson, S. S. et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature434, 644–648 (2005). [DOI] [PubMed] [Google Scholar]
- 423.Stanton, H. et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature434, 648–652 (2005). [DOI] [PubMed] [Google Scholar]
- 424.Gardiner, M. D. et al. Transcriptional analysis of micro-dissected articular cartilage in post-traumatic murine osteoarthritis. Osteoarthr. Cartil.23, 616–628 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Yang, C. Y., Chanalaris, A. & Troeberg, L. ADAMTS and ADAM metalloproteinases in osteoarthritis - looking beyond the ‘usual suspects’. Osteoarthr. Cartil.25, 1000–1009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Dunn, S. L. et al. Gene expression changes in damaged osteoarthritic cartilage identify a signature of non-chondrogenic and mechanical responses. Osteoarthr. Cartil.24, 1431–1440 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Djouad, F. et al. Microenvironmental changes during differentiation of mesenchymal stem cells towards chondrocytes. Arthritis Res. Ther.9, R33 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Jay, G. D. & Waller, K. A. The biology of lubricin: near frictionless joint motion. Matrix Biol.39, 17–24 (2014). [DOI] [PubMed] [Google Scholar]
- 429.Marcelino, J. et al. CACP, encoding a secreted proteoglycan, is mutated in camptodactyly-arthropathy-coxa vara-pericarditis syndrome. Nat. Genet.23, 319–322 (1999). [DOI] [PubMed] [Google Scholar]
- 430.Bahabri, S. A. et al. The camptodactyly-arthropathy-coxa vara-pericarditis syndrome: clinical features and genetic mapping to human chromosome 1. Arthritis Rheum.41, 730–735 (1998). [DOI] [PubMed] [Google Scholar]
- 431.Ciullini Mannurita, S. et al. CACP syndrome: identification of five novel mutations and of the first case of UPD in the largest European cohort. Eur. J. Hum. Genet.22, 197–201 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Rhee, D. K. et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J. Clin. Investig.115, 622–631 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Ogawa, H., Kozhemyakina, E., Hung, H. H., Grodzinsky, A. J. & Lassar, A. B. Mechanical motion promotes expression of Prg4 in articular cartilage via multiple CREB-dependent, fluid flow shear stress-induced signaling pathways. Genes Dev.28, 127–139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Lefebvre, V. & Bhattaram, P. Prg4-expressing cells: articular stem cells or differentiated progeny in the articular chondrocyte lineage? Arthritis Rheumatol.67, 1151–1154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Krawetz, R. J. et al. Proteoglycan 4 (PRG4) treatment enhances wound closure and tissue regeneration. NPJ Regen. Med.7, 32 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Alquraini, A. et al. The interaction of lubricin/proteoglycan 4 (PRG4) with toll-like receptors 2 and 4: an anti-inflammatory role of PRG4 in synovial fluid. Arthritis Res. Ther.17, 353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Liu, Z. et al. A dual role for NOTCH signaling in joint cartilage maintenance and osteoarthritis. Sci. Signal.8, ra71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Zanotti, S., Yu, J., Bridgewater, D., Wolf, J. M. & Canalis, E. Mice harboring a Hajdu Cheney Syndrome mutation are sensitized to osteoarthritis. Bone114, 198–205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Monteagudo, S. & Lories, R. J. A Notch in the joint that exacerbates osteoarthritis. Nat. Rev. Rheumatol.14, 563–564 (2018). [DOI] [PubMed] [Google Scholar]
- 440.Sugita, S. et al. Transcription factor Hes1 modulates osteoarthritis development in cooperation with calcium/calmodulin-dependent protein kinase 2. Proc. Natl Acad. Sci. USA112, 3080–3085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Venn, G., Billingham, M. E. & Hardingham, T. E. Increased proteoglycan synthesis in cartilage in experimental canine osteoarthritis does not reflect a permanent change in chondrocyte phenotype. Arthritis Rheum.38, 525–532 (1995). [DOI] [PubMed] [Google Scholar]
- 442.Abramoff, B. & Caldera, F. E. Osteoarthritis: pathology, diagnosis, and treatment options. Med Clin. North Am.104, 293–311 (2020). [DOI] [PubMed] [Google Scholar]
- 443.Krishnan, Y. & Grodzinsky, A. J. Cartilage diseases. Matrix Biol.71–72, 51–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Kon, E. et al. Bone marrow lesions and subchondral bone pathology of the knee. Knee Surg. Sports Traumatol. Arthrosc.24, 1797–1814 (2016). [DOI] [PubMed] [Google Scholar]
- 445.Arden, N. K. et al. Non-surgical management of knee osteoarthritis: comparison of ESCEO and OARSI 2019 guidelines. Nat. Rev. Rheumatol.17, 59–66 (2021). [DOI] [PubMed] [Google Scholar]
- 446.Messier, S. P. et al. Intentional weight loss in overweight and obese patients with knee osteoarthritis: is more better? Arthritis Care Res.70, 1569–1575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Skou, S. T. & Roos, E. M. Physical therapy for patients with knee and hip osteoarthritis: supervised, active treatment is current best practice. Clin. Exp. Rheumatol.37, 112–117 (2019). [PubMed] [Google Scholar]
- 448.Gay, C., Chabaud, A., Guilley, E. & Coudeyre, E. Educating patients about the benefits of physical activity and exercise for their hip and knee osteoarthritis. Systematic literature review. Ann. Phys. Rehabil. Med.59, 174–183 (2016). [DOI] [PubMed] [Google Scholar]
- 449.Uthman, O. A. et al. Exercise for lower limb osteoarthritis: systematic review incorporating trial sequential analysis and network meta-analysis. BMJ347, f5555 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Bartels, E. M. et al. Aquatic exercise for the treatment of knee and hip osteoarthritis. Cochrane Database Syst. Rev.3, Cd005523 (2016). [DOI] [PubMed] [Google Scholar]
- 451.Vincent, K. R., Vasilopoulos, T., Montero, C. & Vincent, H. K. Eccentric and concentric resistance exercise comparison for knee osteoarthritis. Med Sci. Sports Exerc.51, 1977–1986 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Nguyen, C., Lefèvre-Colau, M. M., Poiraudeau, S. & Rannou, F. Rehabilitation (exercise and strength training) and osteoarthritis: a critical narrative review. Ann. Phys. Rehabil. Med.59, 190–195 (2016). [DOI] [PubMed] [Google Scholar]
- 453.Goh, S. L. et al. Efficacy and potential determinants of exercise therapy in knee and hip osteoarthritis: a systematic review and meta-analysis. Ann. Phys. Rehabil. Med.62, 356–365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Smith, T. O. et al. Assistive devices, hip precautions, environmental modifications and training to prevent dislocation and improve function after hip arthroplasty. Cochrane Database Syst. Rev.7, Cd010815 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Fernandes, L. et al. EULAR recommendations for the non-pharmacological core management of hip and knee osteoarthritis. Ann. Rheum. Dis.72, 1125–1135 (2013). [DOI] [PubMed] [Google Scholar]
- 456.Healy, A., Farmer, S., Pandyan, A. & Chockalingam, N. A systematic review of randomised controlled trials assessing effectiveness of prosthetic and orthotic interventions. PLoS One13, e0192094 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Dantas, L. O., Osani, M. C. & Bannuru, R. R. Therapeutic ultrasound for knee osteoarthritis: a systematic review and meta-analysis with grade quality assessment. Braz. J. Phys. Ther.25, 688–697 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Huang, Z. et al. Effectiveness of low-level laser therapy in patients with knee osteoarthritis: a systematic review and meta-analysis. Osteoarthr. Cartil.23, 1437–1444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Dias, J. M. et al. Hydrotherapy improves pain and function in older women with knee osteoarthritis: a randomized controlled trial. Braz. J. Phys. Ther.21, 449–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Flynn, D. M. Chronic musculoskeletal pain: nonpharmacologic, noninvasive treatments. Am. Fam. Physician102, 465–477 (2020). [PubMed] [Google Scholar]
- 461.Brosseau, L. et al. Thermotherapy for treatment of osteoarthritis. Cochrane Database Syst. Rev.2003, Cd004522 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Hochberg, M. C. Osteoarthritis year 2012 in review: clinical. Osteoarthr. Cartil.20, 1465–1469 (2012). [DOI] [PubMed] [Google Scholar]
- 463.Corbett, M. S. et al. Acupuncture and other physical treatments for the relief of pain due to osteoarthritis of the knee: network meta-analysis. Osteoarthr. Cartil.21, 1290–1298 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Kelly, R. B. & Willis, J. Acupuncture for pain. Am. Fam. Physician100, 89–96 (2019). [PubMed] [Google Scholar]
- 465.Tu, J. F. et al. Efficacy of intensive acupuncture versus sham acupuncture in knee osteoarthritis: a randomized controlled trial. Arthritis Rheumatol.73, 448–458 (2021). [DOI] [PubMed] [Google Scholar]
- 466.Shi, G. X. et al. Effect of electro-acupuncture (EA) and manual acupuncture (MA) on markers of inflammation in knee osteoarthritis. J. Pain. Res.13, 2171–2179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Kolasinski, S. L. et al. 2019 American College of Rheumatology/arthritis foundation guideline for the management of osteoarthritis of the hand, hip, and knee. Arthritis Rheumatol.72, 220–233 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Zhang, W. et al. OARSI recommendations for the management of hip and knee osteoarthritis, part II: OARSI evidence-based, expert consensus guidelines. Osteoarthr. Cartil.16, 137–162 (2008). [DOI] [PubMed] [Google Scholar]
- 469.Mao, L. et al. Targeted treatment for osteoarthritis: drugs and delivery system. Drug Deliv.28, 1861–1876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Hochberg, M. C. et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res.64, 465–474 (2012). [DOI] [PubMed] [Google Scholar]
- 471.Rannou, F., Pelletier, J. P. & Martel-Pelletier, J. Efficacy and safety of topical NSAIDs in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Semin. Arthritis Rheum.45, S18–S21 (2016). [DOI] [PubMed] [Google Scholar]
- 472.Scanzello, C. R., Moskowitz, N. K. & Gibofsky, A. The post-NSAID era: what to use now for the pharmacologic treatment of pain and inflammation in osteoarthritis. Curr. Rheumatol. Rep.10, 49–56 (2008). [DOI] [PubMed] [Google Scholar]
- 473.Roberts, E. et al. Paracetamol: not as safe as we thought? A systematic literature review of observational studies. Ann. Rheum. Dis.75, 552–559 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Xie, P. et al. Safety, tolerability, and pharmacokinetics of ibuprofenamine hydrochloride spray (NSAIDs), a new drug for rheumatoid arthritis and osteoarthritis, in healthy chinese subjects. Drug Des. Dev. Ther.15, 629–638 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Kloppenburg, M. Hand osteoarthritis-nonpharmacological and pharmacological treatments. Nat. Rev. Rheumatol.10, 242–251 (2014). [DOI] [PubMed] [Google Scholar]
- 476.Dougados, M. Why and how to use NSAIDs in osteoarthritis. J. Cardiovasc. Pharm.47, S49–S54 (2006). [DOI] [PubMed] [Google Scholar]
- 477.Bannwarth, B. Acetaminophen or NSAIDs for the treatment of osteoarthritis. Best. Pract. Res. Clin. Rheumatol.20, 117–129 (2006). [DOI] [PubMed] [Google Scholar]
- 478.D’Arcy, Y. et al. Treating osteoarthritis pain: mechanisms of action of acetaminophen, nonsteroidal anti-inflammatory drugs, opioids, and nerve growth factor antibodies. Postgrad. Med.133, 879–894 (2021). [DOI] [PubMed] [Google Scholar]
- 479.Zhu, X. et al. Comparative effectiveness of glucosamine, chondroitin, acetaminophen or celecoxib for the treatment of knee and/or hip osteoarthritis: a network meta-analysis. Clin. Exp. Rheumatol.36, 595–602 (2018). [PubMed] [Google Scholar]
- 480.Deyle, G. D. et al. Physical therapy versus glucocorticoid injection for osteoarthritis of the knee. N. Engl. J. Med.382, 1420–1429 (2020). [DOI] [PubMed] [Google Scholar]
- 481.Block, J. A. Are intraarticular glucocorticoids safe in osteoarthritis? Arthritis Rheumatol.74, 181–183 (2022). [DOI] [PubMed] [Google Scholar]
- 482.Savvidou, O. et al. Glucocorticoid signaling and osteoarthritis. Mol. Cell Endocrinol.480, 153–166 (2019). [DOI] [PubMed] [Google Scholar]
- 483.Black, R. & Grodzinsky, A. J. Dexamethasone: chondroprotective corticosteroid or catabolic killer? Eur. Cell Mater.38, 246–263 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Chen, L. et al. Long term usage of dexamethasone accelerating accelerates the initiation of osteoarthritis via enhancing chondrocyte apoptosis and the extracellular matrix calcification and apoptosis of chondrocytes. Int. J. Biol. Sci.17, 4140–4153 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Zhou, J. et al. Treatment of hip osteoarthritis with glucocorticoids. Ann. Rheum. Dis.78, e100 (2019). [DOI] [PubMed] [Google Scholar]
- 486.Riis, R. G. C. et al. The effects of intra-articular glucocorticoids and exercise on pain and synovitis assessed on static and dynamic magnetic resonance imaging in knee osteoarthritis: exploratory outcomes from a randomized controlled trial. Osteoarthr. Cartil.25, 481–491 (2017). [DOI] [PubMed] [Google Scholar]
- 487.McAlindon, T. E. et al. Effect of intra-articular triamcinolone vs saline on knee cartilage volume and pain in patients with knee osteoarthritis: a randomized clinical trial. JAMA317, 1967–1975 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Li, B. et al. Baicalein alleviates osteoarthritis by protecting subchondral bone, inhibiting angiogenesis and synovial proliferation. J. Cell Mol. Med.25, 5283–5294 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Fransen, M. et al. Glucosamine and chondroitin for knee osteoarthritis: a double-blind randomised placebo-controlled clinical trial evaluating single and combination regimens. Ann. Rheum. Dis.74, 851–858 (2015). [DOI] [PubMed] [Google Scholar]
- 490.Clegg, D. O. et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N. Engl. J. Med.354, 795–808 (2006). [DOI] [PubMed] [Google Scholar]
- 491.Xiong, F. et al. pH-responsive and hyaluronic acid-functionalized metal-organic frameworks for therapy of osteoarthritis. J. Nanobiotechnol.18, 139 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Bruyère, O., Altman, R. D. & Reginster, J. Y. Efficacy and safety of glucosamine sulfate in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Semin. Arthritis Rheum.45, S12–S17 (2016). [DOI] [PubMed] [Google Scholar]
- 493.Su, K. et al. Comparison of hyaluronic acid and PRP intra-articular injection with combined intra-articular and intraosseous PRP injections to treat patients with knee osteoarthritis. Clin. Rheumatol.37, 1341–1350 (2018). [DOI] [PubMed] [Google Scholar]
- 494.Tang, J. Z. et al. Platelet-rich plasma versus hyaluronic acid in the treatment of knee osteoarthritis: a meta-analysis. J. Orthop. Surg. Res.15, 403 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Malemud, C. J. Cytokines as therapeutic targets for osteoarthritis. BioDrugs18, 23–35 (2004). [DOI] [PubMed] [Google Scholar]
- 496.Urech, D. M. et al. Anti-inflammatory and cartilage-protecting effects of an intra-articularly injected anti-TNF{alpha} single-chain Fv antibody (ESBA105) designed for local therapeutic use. Ann. Rheum. Dis.69, 443–449 (2010). [DOI] [PubMed] [Google Scholar]
- 497.Millward-Sadler, S. J. et al. Integrin-regulated secretion of interleukin 4: a novel pathway of mechanotransduction in human articular chondrocytes. J. Cell Biol.145, 183–189 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Georgescu, L., Vakkalanka, R. K., Elkon, K. B. & Crow, M. K. Interleukin-10 promotes activation-induced cell death of SLE lymphocytes mediated by Fas ligand. J. Clin. Invest.100, 2622–2633 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Nees, T. A. et al. Synovial cytokines significantly correlate with osteoarthritis-related knee pain and disability: inflammatory mediators of potential clinical relevance. J. Clin. Med.8, 1343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Han, W., Fan, S., Bai, X. & Ding, C. Strontium ranelate, a promising disease modifying osteoarthritis drug. Expert Opin. Investig. Drugs26, 375–380 (2017). [DOI] [PubMed] [Google Scholar]
- 501.Yu, H. et al. Strontium ranelate promotes chondrogenesis through inhibition of the Wnt/β-catenin pathway. Stem Cell Res. Ther.12, 296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Apostu, D. et al. Systemic drugs with impact on osteoarthritis. Drug Metab. Rev.51, 498–523 (2019). [DOI] [PubMed] [Google Scholar]
- 503.Sun, Q. et al. Parathyroid hormone attenuates osteoarthritis pain by remodeling subchondral bone in mice. Elife10, e66532 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Neogi, T. et al. Observed efficacy and clinically important improvements in participants with osteoarthritis treated with subcutaneous tanezumab: results from a 56-week randomized NSAID-controlled study. Arthritis Res. Ther.24, 78 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Wise, B. L., Seidel, M. F. & Lane, N. E. The evolution of nerve growth factor inhibition in clinical medicine. Nat. Rev. Rheumatol.17, 34–46 (2021). [DOI] [PubMed] [Google Scholar]
- 506.Schnitzer, T. J. & Marks, J. A. A systematic review of the efficacy and general safety of antibodies to NGF in the treatment of OA of the hip or knee. Osteoarthr. Cartil.23, S8–S17 (2015). [DOI] [PubMed] [Google Scholar]
- 507.Karsdal, M. A. et al. Serological biomarker profiles of rapidly progressive osteoarthritis in tanezumab-treated patients. Osteoarthr. Cartil.27, 484–492 (2019). [DOI] [PubMed] [Google Scholar]
- 508.Jayabalan, P. & Schnitzer, T. J. Tanezumab in the treatment of chronic musculoskeletal conditions. Expert Opin. Biol. Ther.17, 245–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Oo, W. M. & Hunter, D. J. Nerve growth factor (NGF) inhibitors and related agents for chronic musculoskeletal pain: a comprehensive review. BioDrugs35, 611–641 (2021). [DOI] [PubMed] [Google Scholar]
- 510.Gondal, F. R., Bilal, J. & Kent Kwoh, C. Tanezumab for the treatment of osteoarthritis pain. Drugs Today58, 187–200 (2022). [DOI] [PubMed] [Google Scholar]
- 511.Paget, L. D. A. et al. Effect of platelet-rich plasma injections vs placebo on ankle symptoms and function in patients with ankle osteoarthritis: a randomized clinical trial. JAMA326, 1595–1605 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Katz, J. N. Platelet-rich plasma for osteoarthritis and achilles tendinitis. JAMA326, 2012–2014 (2021). [DOI] [PubMed] [Google Scholar]
- 513.Belk, J. W. et al. Platelet-rich plasma versus hyaluronic acid for knee osteoarthritis: a systematic review and meta-analysis of randomized controlled trials. Am. J. Sports Med.49, 249–260 (2021). [DOI] [PubMed] [Google Scholar]
- 514.Pas, H. I. et al. Stem cell injections in knee osteoarthritis: a systematic review of the literature. Br. J. Sports Med.51, 1125–1133 (2017). [DOI] [PubMed] [Google Scholar]
- 515.Harrell, C. R., Markovic, B. S., Fellabaum, C., Arsenijevic, A. & Volarevic, V. Mesenchymal stem cell-based therapy of osteoarthritis: current knowledge and future perspectives. Biomed. Pharmacother.109, 2318–2326 (2019). [DOI] [PubMed] [Google Scholar]
- 516.Kim, Y. G., Choi, J. & Kim, K. Mesenchymal stem cell-derived exosomes for effective cartilage tissue repair and treatment of osteoarthritis. Biotechnol. J.15, e2000082 (2020). [DOI] [PubMed] [Google Scholar]
- 517.De Bari, C. & Roelofs, A. J. Stem cell-based therapeutic strategies for cartilage defects and osteoarthritis. Curr. Opin. Pharm.40, 74–80 (2018). [DOI] [PubMed] [Google Scholar]
- 518.Ng, J. et al. Stem cell-directed therapies for osteoarthritis: the promise and the practice. Stem Cells38, 477–486 (2020). [DOI] [PubMed] [Google Scholar]
- 519.Shapiro, S. A., Kazmerchak, S. E., Heckman, M. G., Zubair, A. C. & O’Connor, M. I. A prospective, single-blind, placebo-controlled trial of bone marrow aspirate concentrate for knee osteoarthritis. Am. J. Sports Med.45, 82–90 (2017). [DOI] [PubMed] [Google Scholar]
- 520.Pers, Y. M. et al. Adipose mesenchymal stromal cell-based therapy for severe osteoarthritis of the knee: a phase I dose-escalation trial. Stem Cells Transl. Med.5, 847–856 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Kim, Y. S., Choi, Y. J. & Koh, Y. G. Mesenchymal stem cell implantation in knee osteoarthritis: an assessment of the factors influencing clinical outcomes. Am. J. Sports Med.43, 2293–2301 (2015). [DOI] [PubMed] [Google Scholar]
- 522.Chahal, J. et al. Bone marrow mesenchymal stromal cell treatment in patients with osteoarthritis results in overall improvement in pain and symptoms and reduces synovial inflammation. Stem Cells Transl. Med.8, 746–757 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Zhu, Y. et al. Crosstalk between Smad2/3 and specific isoforms of ERK in TGF-β1-induced TIMP-3 expression in rat chondrocytes. J. Cell Mol. Med.21, 1781–1790 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Kim, M. K. et al. A multicenter, double-blind, phase iii clinical trial to evaluate the efficacy and safety of a cell and gene therapy in knee osteoarthritis patients. Hum. Gene Ther. Clin. Dev.29, 48–59 (2018). [DOI] [PubMed] [Google Scholar]
- 525.Liddle, A. D., Pegg, E. C. & Pandit, H. Knee replacement for osteoarthritis. Maturitas75, 131–136 (2013). [DOI] [PubMed] [Google Scholar]
- 526.Dieppe, P. Management of osteoarthritis of the hip and knee joints. Curr. Opin. Rheumatol.5, 487–493 (1993). [DOI] [PubMed] [Google Scholar]
- 527.Sheikh, A. & Schweitzer, M. Pre- and postoperative assessment in joint preserving and replacing surgery. Rheum. Dis. Clin. North Am.35, 651–673 (2009). [DOI] [PubMed] [Google Scholar]
- 528.Katz, J. N., Brownlee, S. A. & Jones, M. H. The role of arthroscopy in the management of knee osteoarthritis. Best. Pract. Res. Clin. Rheumatol.28, 143–156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Li, M. H., Xiao, R., Li, J. B. & Zhu, Q. Regenerative approaches for cartilage repair in the treatment of osteoarthritis. Osteoarthr. Cartil.25, 1577–1587 (2017). [DOI] [PubMed] [Google Scholar]
- 530.Lo, C. W. T. et al. Risk factors for falls in patients with total hip arthroplasty and total knee arthroplasty: a systematic review and meta-analysis. Osteoarthr. Cartil.27, 979–993 (2019). [DOI] [PubMed] [Google Scholar]
- 531.Jayakumar, P. et al. Comparison of an artificial intelligence-enabled patient decision aid vs educational material on decision quality, shared decision-making, patient experience, and functional outcomes in adults with knee osteoarthritis: a randomized clinical trial. JAMA Netw. Open4, e2037107 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Nishiwaki, T. et al. Pelvic tilt displacement before and after artificial hip joint replacement surgery. J. Arthroplast.33, 925–930 (2018). [DOI] [PubMed] [Google Scholar]