

Abstract
Multidrug resistance (MDR) proteins related to the ATP-binding cassette family are found in a very wide range of human tumors and result in therapeutic failure. The overexpression of efflux pumps such as ABCB1 is one of the mechanisms of MDR. This paper aims to develop a reliable quantitative structure-activity relationship (QSAR) model that best describes the correlation between the activity and the molecular structures in order to predict the inhibitory biological activity towards ABCB1. In this regard, a series of quinoline derivatives of 18 compounds were analyzed using different linear and non-linear machine learning (ML) regression methods including k-nearest neighbors (KNN), decision tree (DT), back propagation neural networks (BPNN) and gradient boosting-based (GB) methods. Their aim is to explain the origin of the activity of these investigated compounds and therefore, design new quinoline derivatives with higher effect on ABCB1. A total of 16 ML predictive models were developed on different number of 2D and 3D descriptors and were evaluated using the coefficient of determination (R2) and the root mean squared error (RMSE) statistical metrics. Among all developed models, A GB-based model in particular catboost achieved the highest predictive quality, with one descriptor, expressed by R2 and RMSE of 95% and 0.283 respectively. Molecular docking studies against the target crystal structure of the outward-facing p-glycoprotein (6C0V) revealed significant binding affinities via both hydrophobic and H-bond interactions with the relevant compounds. The 17 has shown the highest binding energy of −9.22 kcal/mol. Therefore, it can suggest that 17 may prove to be a valuable potential lead structure for the design and synthesis of more potent P-glycoprotein inhibitors for combination used with anti-cancer drugs for cancer multidrug resistance management.
Keywords: QSAR, Quinoline, PCA, Machine learning, Deep learning, Molecular docking
1. Introduction
All over the world, cancer ranks first among the causes of death [1]. It occurs because of disruption of the physiological functions of cells. A major challenge in developing effective therapy is the resistance of cells to multiple chemotherapy drugs [2]. The ABC (adenosine triphosphate) transporter super family, which transports cytotoxic agents and targeted anticancer drugs using ATP energy, is one of the major mechanisms of drug resistance [3,4,5].
Transporter B1 of the ATP-binding cassette family (ABCB1) is the commonest ABC protein able to mediate multidrug resistance. It is within a system of complex tissue and cellular features that help to bring about drug resistance in cancer cells [6,7,8,9]. In many cases, ABCB1 overexpression is the very first mechanism of resistance, which comes before the development of other mechanisms such as increased drug metabolism, drug target transformation, activation of DNA repair mechanisms, apoptosis checkpoint, and induction of EMT through cell proliferation and the ability to adapt to drug regimens [10]. Pgp (P-glycoprotein) has an impact on multidrug-resistant (MDR) cancer and the relationship between Pgp overexpression and MDR cancer has been proven in the literature [11,12,13,14,15,16]. The ability of Pgp to channel such diverse chemical classes is due, in part, to the numerous transport pathways through the protein that have been visualized using molecular dynamics simulations [17]. Work shows that overexpression of Pgp in cancers can be either intrinsic or acquired following drug treatment, depending on the tissue of origin [18].
The biological properties of some quinoline derivatives were found to be interesting and their pharmacological profile advantageous. It was remarked that those compounds with antitumor activity that contain a quinoline moiety perform as cytostatic agents or inhibitors of the topoisomerase-II enzyme, interfering with DNA replication [19,20,21,22,23].
In recent years, machine learning (ML) has drawn the attention of many researchers and has widely been used to develop QSAR models which allow a reliable prediction making of a targeted activity. Several powerful ML algorithms have recently been developed and proven to outperform the most commonly used regression algorithm in QSAR. Many ML models have been developed as each of them inherits a distinct regression algorithm and will therefore provide probable models with different performance. The idea here consists of making a comparative study of these models and choosing the one that performs best and that will guarantee quality predictions. El Hassan El Assiri has used multiple linear regression (MLR) and artificial neural networks to predict corrosion inhibitory activity of pyridazine-derivatives [24,25]. H. El Ghalia has also used MLR to predict the anticancer activity of 5.6.7-trimethoxy-N-aryl-2-styrylquinolin-4-amines [26]. Other methods were also used in QSAR modeling such as partial least squares, principal components regression methods and so on.
The molecular docking approach represents a new computational strategy to assess the binding affinity of docked molecules to receptors based on the scoring functions of mathematical algorithms, while the QSAR model produces new compounds with more precise pharmacological efficacy that can serve as effective future drug candidates. Both approaches are considered very effective in silico drug design and can be used separately or simultaneously [27].
The synthesis of novel compounds as potent modulators of ABCB1-induced drug resistance in mouse T-cell lymphoma has been previously reported by Baba, Y. F [28]. in our laboratory. These compounds were assessed for their cytotoxic effect and ABCB1 modulating properties against parental and ABCB1 overexpressing mouse T lymphoma cells. The findings of the rhodamine 123-accumulation assay in multidrug-resistant (MDR) mouse T lymphoma cells overexpressing the ATP-binding cassette B transporter protein will be used to construct QSAR models.
The main objective of this work is to develop mathematical QSAR models describing and predicting the inhibitory activity of quinoline derivatives against ABCB1 from different 2D and 3D molecular descriptors using different new ML regression methods. This is done using the molecular docking study to observe the binding mode of quinoline derivatives with anticancer effect in vitro to the active site of 6C0V, with the aim of synthesizing molecules with the desired biological response before carrying out the experimental synthesis protocol.
2. Materials and methods
2.1. Data set
In this study, 18 derivatives of 2-oxo-1, 2-dihydroquinoline-4-carboxylic acid were prepared by our Laboratory [28], each was synthetized by varying the halogen and the radical (Table 1) of the acid (Fig. 1), for their inhibitory activity against the ABCB1.
Table 1.
Substituents of 2-oxo-1, 2-dihydroquinoline- 4-carboxylic acid derivatives and their fluorescence activity ratio (FAR).
| Compounds | Structure | FAR |
|---|---|---|
| 1 |  |
1.29 |
| 2 |  |
1.43 |
| 3 |  |
1.53 |
| 4 |  |
1.23 |
| 5 | 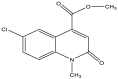 |
0.98 |
| 6 |  |
0.77 |
| 7 |  |
1.61 |
| 8 | 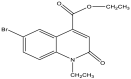 |
0.8 |
| 9 | 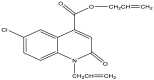 |
1.82 |
| 10 |  |
3.64 |
| 11 | 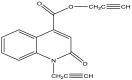 |
0.84 |
| 12 |  |
1.24 |
| 13 |  |
11.34 |
| 14 | 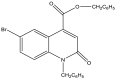 |
28.7 |
| 15 |  |
0.85 |
| 16 |  |
0.87 |
| 17 |  |
8.22 |
| 18 |  |
224.7 |
Fig. 1.

Structure of 2-oxo-1, 2-dihydroquinoline-4-carboxylic acid derivatives.
2.2. Calculation of descriptors
Molecular descriptors are a fundamental element of the QSAR theory, and are in a way the numerical description of a molecular structure. They are used to establish a relationship between the structure of a molecule and its biological activity. In this work, ChemDraw (v.18.2) and Molecular Operating Environment (MOE v.2009.10) were used to draw the molecules and generate 199 2D and 3D descriptors, some of which are shown in Table 2.
Table 2.
Values of Molecular descriptors.
| mol | FAR | b_rotR | dipoleX | lip_acc | logP (o/w) | PEOE_VSA-5 | PM3_HF | pmiZ | SlogP | AM1_HOMO | AM1_LUMO | apol | E | mr | vdw_area |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.29 | 0.07 | −0.49 | 4.00 | 0.96 | 27.13 | −89.97 | 74.19 | 1.11 | −9.16 | −1.18 | 25.77 | 13.32 | 4.98 | 175.36 |
| 2 | 1.43 | 0.06 | −0.65 | 4.00 | 1.59 | 27.13 | −96.03 | 173.70 | 1.76 | −9.22 | −1.29 | 27.29 | 13.36 | 5.46 | 192.94 |
| 3 | 1.53 | 0.06 | −0.20 | 4.00 | 1.79 | 27.13 | −81.68 | 375.34 | 1.87 | −9.24 | −1.35 | 28.16 | 12.57 | 5.71 | 204.71 |
| 4 | 1.23 | 0.12 | −1.17 | 4.00 | 1.42 | 27.13 | −80.80 | 73.62 | 1.22 | −9.02 | −0.97 | 31.96 | 35.74 | 5.96 | 224.84 |
| 5 | 0.98 | 0.11 | −0.52 | 4.00 | 2.05 | 27.13 | −86.95 | 422.78 | 1.87 | −9.06 | −1.15 | 33.47 | 34.41 | 6.45 | 242.42 |
| 6 | 0.77 | 0.21 | −0.83 | 4.00 | 2.10 | 27.13 | −91.81 | 400.35 | 2.00 | −8.97 | −0.92 | 38.15 | 32.81 | 6.91 | 259.31 |
| 7 | 1.61 | 0.20 | 0.00 | 4.00 | 2.73 | 27.13 | −97.36 | 498.44 | 2.65 | −8.97 | −1.14 | 39.66 | 30.41 | 7.40 | 276.89 |
| 8 | 0.80 | 0.20 | −0.14 | 4.00 | 2.93 | 27.13 | −81.63 | 634.13 | 2.76 | −9.03 | −1.15 | 40.53 | 31.04 | 7.65 | 288.66 |
| 9 | 1.82 | 0.27 | −0.09 | 4.00 | 3.37 | 27.13 | −45.24 | 1165.13 | 2.99 | −8.99 | −1.19 | 43.18 | 35.92 | 8.23 | 308.50 |
| 10 | 3.64 | 0.27 | −0.29 | 4.00 | 3.58 | 27.13 | −31.30 | 682.66 | 3.09 | −9.05 | −1.19 | 44.05 | 36.72 | 8.48 | 320.28 |
| 11 | 0.84 | 0.29 | −0.52 | 4.00 | 2.03 | 29.64 | 29.62 | 244.48 | 1.23 | −9.04 | −1.08 | 39.00 | 32.45 | 7.52 | 288.08 |
| 12 | 1.24 | 0.27 | −0.22 | 4.00 | 2.86 | 29.64 | 37.90 | 887.20 | 1.99 | −9.12 | −1.23 | 41.38 | 31.63 | 8.26 | 317.43 |
| 13 | 11.34 | 0.19 | −0.53 | 4.00 | 4.99 | 27.13 | −24.24 | 1658.82 | 4.89 | −8.97 | −1.04 | 58.42 | 71.03 | 10.97 | 365.84 |
| 14 | 28.70 | 0.19 | −0.86 | 4.00 | 5.83 | 27.13 | −15.42 | 3349.24 | 5.66 | −9.09 | −1.13 | 60.80 | 70.02 | 11.72 | 395.20 |
| 15 | 0.85 | 0.19 | −0.10 | 6.00 | 2.45 | 38.50 | −9.46 | 1434.52 | 3.68 | −8.87 | −0.77 | 55.76 | 73.39 | 10.65 | 362.13 |
| 16 | 0.87 | 0.19 | 0.77 | 6.00 | 3.08 | 38.50 | −12.54 | 283.35 | 4.34 | −9.02 | −1.23 | 57.27 | 73.28 | 11.15 | 379.71 |
| 17 | 8.22 | 0.19 | 0.62 | 6.00 | 3.29 | 38.50 | −0.46 | 622.57 | 4.45 | −8.99 | −1.08 | 58.14 | 73.45 | 11.40 | 391.48 |
| 18 | 224.70 | 0.22 | 0.94 | 6.00 | 4.31 | 24.93 | 10.66 | 1109.62 | 5.26 | −9.21 | −1.15 | 58.14 | 75.45 | 11.51 | 387.86 |
Note: The full name of descriptors can found in annex immediately after the conclusion.
2.3. The inhibitory activity response
The rhodamine 123 accumulation assay is a fluorescence detection system that uses verapamil as a reference inhibitor of the ABCB1 efflux pump [29]. The fluorescence intensity of the selected cell population was measured with a Partec CyFlow cytometer (Partec, Munster, Germany). The average fluorescence intensity was calculated for MDR and T-lymphoma cell lines from treated parental mice compared to untreated cells 30,31. The fluorescence activity ratio (FAR) was calculated based on the following equation relating the measured fluorescence values:
2.4. Data split
The data consisted of 18 molecules descriptors was randomly split into training and testing sets. The former consisted of 15 molecules that span the entire chemical space for all the data. While the consisted of three molecules within the Applicability Domain (i.e., the range of the training set).
2.4.1. Data exploration by means of PCA
Principal component analysis (PCA) permits us to verify redundancy and collinearity between the studied descriptors and to carry out a comparative statistical study between the proposed mathematical models such as partial least squares regression (PLS) and stepwise multiple linear regression (SMLR) with the aim of correlating activity with molecular structure [32].
2.4.2. Partial least squares regression
PLS leads to a robust statistical solution when the independent variables are strongly related to each other, or if the independent variables outnumber the observations. PLS is an alternating regression method, which generates its solutions based on the linear transformation of a large number of original descriptors to a small number of new orthogonal terms, called latent variables [33,34]. Therefore, this methodology is considered a standard statistical.
2.4.3. Stepwise multiple linear regression
This approach uses the MLR variant commonly, which generates a multi-term linear equation, although not all of the independent variables are used. This method is well suited to be used when the number of descriptors is large and the main descriptors are unknown [35]. MLR is based on the assumption that the dependent variable is linearly related to some independent variables according to the following relationship.
Whereas Y represents the dependent variable (biological activity to be predicted), Xi representing the independent variables (molecular descriptors), n indicating the number of molecular descriptors, a0 showing the constant in the previous equation, ai being the coefficient of descriptors.
2.5. Data modeling
In general, QSAR data consists of a large number of features that reflect physico-chemical properties of molecules and which not all correlate to the target. Moreover, it is often characterized by high redundancy, which frequently leads to instability and increases the variance of ML models. The primary goal of data modeling is to develop prediction models that can accurately describe the FAR target variation based on descriptors that have a high, moderate, or low correlation to it. This helps identify the key property of the molecule that affect its activity, suggest a structure or molecule with a specific activity as well as understand the interaction between functional groups in a molecule. Data modeling was performed, on centered and scaled data, with python core (v3.10) where several machine learning (ML) algorithms were used to develop predictive regression models including, decision tree (DT), k nearest neighbor (KNN), gradient boosting-based (GB) models, back propagation artificial neural network (BPNN) [36,37,38].
2.5.1. Back propagation neural network
Back propagation neural network is a commonly known method for developing predictive models from large datasets. It is a neural network (NN) that is primarily trained through using back-propagation (BP) algorithm. It consists of three sorts of layers (input, hidden, and output layers), each of which is composed of one or more neurons characterized by an activation function and bias (Fig. 2). Neurons in one layer are interconnected to those in the next layer by connections known as synaptic weights, resulting in a network that attempts to mimic the human brain. The BP learning algorithm typically comprises of two successive phases which work to minimize the difference between measured values and the Network output by tuning weights and biases in iterative manner: forward-propagation of information and backpropagation of error. Proper tuning of the weights and biases allows reduce error and make the model reliable. The number of neurons in each layer, appropriate activation functions, the number of layers, the maximum number of iterations. Are all hyperparameters to be tuned in BPNN. In this study, the tuning was performed using a meta-heuristic optimization algorithm called Simulated annealing provided by hyper opt module [39].
Fig. 2.

Back propagation algorithm.
2.5.2. K nearest neighbor and decision tree regressors
KNN and DT are two regression methods used in this work because of their simplicity and performance in providing efficient predictive models. They are non-parametric regression methods able to quickly identify the relationship between descriptors and the target. The implementation of DT can also be done without scaling the descriptors and is not largely influenced by the presence of outliers in the data [40].
2.5.3. Gradient boosting algorithms
Gradient boosting is based on the assumption that combining the best next model with the prior models lowers overall prediction errors. Gb algorithms are a family of open-source ensemble methods. They have been widely used for several purposes including, high-precision and adoptable recommender systems building, weather prediction, features selection in regression problems and so on. They allow development of performant and stable predictive models by training a sequence of weak models based on decision trees, each of which compensates for the errors of its predecessors, in contrast to many ML models that concentrate on high quality prediction achieved by a single model. In this work, three GB regression algorithms were used: eXtreme gradient boosting (XGBoost), light gradient boosting machine (LGBM) and categorical boosting (CatBoost) [41].
2.6. Predictive models validation and evaluation
After predictive model development, a validation process is required to ensure that the models are performing the way it was intended and that it accurately predict the target. In this work, the validation process was performed in two ways: internal and external. The former was performed using Leave-One-Out Cross Validation (LOOCV) to prevent the model under and over-fitting and perform the hyperparameters tuning. While the later was performed using unseen data and it aimed to test the model capability in making the right predictions in the future. Models evaluation was used to estimate the developed models performance in training, internal and external validation. It was performed using two common statistical metrics: the coefficient of determination (R2) and the root mean squared error (RMSE). The best model is characterized with high R2, low RMSE and low variance between the train, internal and external validation. Computational formulas are provided in (1(1) and (2)(2):
| (1) |
| (2) |
where yi, ȳ, ŷ, and N are respectively a measured value, the average value of all measured values, the predicted value, the total number of samples.
2.7. Molecular docking methodology
The docking process was further investigated between the studied molecules with the best FAR values and the crystal structure of human P-glycoprotein in the outward-facing ATP-bound conformation. All scores achieved during the process of molecular docking have been computed and presented using the Molecular Operating Environment (MOE) software. The structures of these compounds were constructed using ChemDraw 18.2 software.
The protein data bank (https://www.rcsb.org/pdb) was used to recover and generate the target crystal structure of outward-facing p-glycoprotein (PDB code = 6C0V). This multi-drug transporter permeability (P)-glycoprotein is a transporter of adenosine triphosphate (ATP) binding cassettes that accounts for clinical resistance to chemotherapy. As such, P-glycoprotein extrudes toxic molecules and drugs from cells through ATP-powered conformational changes. To accomplish the optimization, all water-bound cofactors and ligands were detached from the protein structure and the hydrogen atoms were finally attached. The active sites have been sequestered and taken as dummy atoms. The MMFF94x force field was adopted to assign all parameters and charges. Following the generation of the alpha site spheres using the MOE site search module, the structural model of the molecules was docked to the surface of the cancer protein interior through the MOE DOCK module. The London dG notation function was used to execute the dock notation in the MOE software and two unrelated refinement methods were then used for the upgrade. Auto-rotating links were then authorized for the top ten link poses that were targeted for analysis to obtain the highest possible score. The database browser was then utilized to match the docking poses to the ligand in the co-crystallized structure along with acquiring the RMSD of the docking pose. Then, the binding free energy and hydrogen bonds between the synthesized molecules and the amino acid residues of the receptors were computed to rank the binding affinity of the molecules to the protein molecules under study. The interaction types together with the RMSD of the (native) ligand in the receptor structure were assumed as the default-docking model.
3. Results and discussion
3.1. Descriptors exploration
A heatmap of correlation shown in (Fig. 3) was used as a tool to provide an overview of the relationship between descriptors. It shows the presence of positive and negative high correlations (red and blue zones which correlation coefficient in absolute error is close to 1); this correlation means that there is a curse of redundancy of information in our data which often leads to the instability of ML models. To overcome this curse, two approaches were used: features extraction by means of partial least squares regression, and feature selection using embedded methods based on random selection. However, the figure also illustrates descriptors which have no significant correlation with any descriptor presented by the sky blue, green and yellow zones (correlation coefficient ranging between −0.5 and 0.5).
Fig. 3.

Heatmap of correlation.
3.2. Principal component analysis
The PCA was conducted on centered and scaled data in order to identify and select descriptors that correlate to FAR. For this, it was made on 199 descriptors and the sixteen principal components obtained are displayed in Fig. 4.
Fig. 4.

The principal components and their variances.
Ten descriptors which have high correlation (Fig. 5) with the component explaining much our target variation were selected to be used for development of SMLR and PLS models.
Fig. 5.

Bar plot of the correlation coefficients between FAR and the most ten correlated descriptors and the principal component that best explain the FAR.
3.3. Partial least squares regression analysis
Partial least squares regression widely used in the case of a high redundancy in the data. It has the ability to distinguish highly informative features from redundant and uninformative ones and therefore helps construct reduced models that retains key features which have a relation with the response.
The resulting PLS model expression, together with the statistical parameter values, is represented by the following equation:
FAR = −3.74 + 2.03 × b_rotR - 1.01 × dipoleX - 0.14 × lip_acc +1.64 × logP (o/w) - 0.04 × PEOE_VSA-5 + 0.01 × PM3_HF + 0.01 × pmiZ + 1.33 × SlogP - 0.01 × SlogP_VSA4 - 0.02 × SMR_VSA6.
Based on the descriptors given in the PLS model equation, the importance of each individual descriptor in relation to the standardized regression coefficients is displayed in Fig. 6.
Fig. 6.

Standardized coefficients versus variables in the proposed PLS model.
We observe in Fig. 6, that FAR importance based on molecular structure varies from descriptor to another. Nevertheless, since the descriptors in the PLS model have different units, these standardized coefficients are estimates without real scale, suggesting that these standardized coefficients are not useful for determining the true relative importance and significance of each descriptor in the regression analysis. Furthermore, their value is restricted to determining the positive or negative contribution of molecular indices to the property under study.
Returning to the statistical parameters and the result obtained in Fig. 7, we see that the model achieved a high performance in the training phase: a high R2 value of 99% and a low RMSE value of 0.37. However, the model attained a performance much lower than that obtained in the training (R2 = 40%, RMSE = 5.86) which means that it has overfitted the data. The weak statistical results of the PLS model led us to evaluate other statistical models, such as stepwise multiple linear regression and machine learning models.
Fig. 7.

Relationship between the observed Far values and those predicted by the PLS model.
3.4. Stepwise multiple linear regression (SMLR) analysis
Stepwise multiple linear regression is widely regarded as being one of the most fundamental modeling methods recognized in the QSAR field. The ten descriptors resulting from the PCA are an input file for a stepwise selection based on MLR analysis. SMLR consists of treating the links between the dependent quantitative variable to be explained (FAR) and the independent explanatory quantitative variables (descriptors), the expression of the established SMLR model, accompanied with the values of the statistical parameters of five selected descriptors is represented by the following equation:
FAR = 4.51–7.31 × b_rotR - 2.92 × dipoleX + 3.57 × lip_acc +10.44 × logP (o/w) + 4.99 × SMR_VSA6.
The developed model achieved high quality performance during the training phase, expressed by high R2 and low RMSE of 90% and 2.377 respectively. However, during the CV the model performance has decreased significantly (R2 = 48%, RMSE = 5.325) while during the test the model has not explained any amount of the response variability (R2 = 0%, RMSE = 32.546) (Table 3 and Fig. 8). This means it is statistically not acceptable.
Table 3.
Summary of result model analysis.
| R2 | RMSE | |
|---|---|---|
| Train | 90% | 2.377 |
| CV | 48% | 5.325 |
| Test | 0% | 32.546 |
Fig. 8.

Correlation of FAR and predicted FAR.
3.5. Machine learning models with feature selection
A summary of developed ML models on different numbers of descriptors (NVAR) is presented in Table 4. It shows that these models have achieved higher performance than SMLR and PLS. in general, most of the models have a good performance in all development and validation phases: training, CV and testing. An objective comparison between them reveals that the KNN (M2), the LGBM (M4), CATBOOST (M8) and BPNN (M15) models outperformed all the other developed models showing simultaneously high predictive performance and low variance through the training and testing phases.
Table 4.
Summary of developed ML regression models.
| Regressor | Model | Train |
CV |
Test |
NVAR | |||
|---|---|---|---|---|---|---|---|---|
| R2 | RMSE | R2 | RMSE | R2 | ||||
| KNN | M1 | 94 | 0.316 | 91 | 0.394 | 97 | 8 | |
| M2 | 93 | 0.345 | 93 | 0.352 | 96 | 2 | ||
| LGBM | M3 | 92 | 0.353 | 90 | 0.403 | 97 | 8 | |
| M4 | 94 | 0.302 | 93 | 0.352 | 97 | 3 | ||
| XGBOOST | M5 | 93 | 0.332 | 90 | 0.397 | 97 | 11 | |
| M6 | 92 | 0.353 | 90 | 0.403 | 97 | 159 | ||
| CATBOOST | M7 | 95 | 0.277 | 91 | 0.376 | 96 | 4 | |
| M8 | 95 | 0.283 | 93 | 0.346 | 96 | 1 | ||
| DT | M9 | 92 | 0.353 | 90 | 0.403 | 97 | 20 | |
| M10 | 100 | 0.076 | 91 | 0.387 | 92 | 9 | ||
| BPNN | 1 hidden layer | M11 | 94 | 0.305 | 91 | 0.384 | 95 | 62 |
| M12 | 93 | 0.342 | 88 | 0.437 | 91 | 20 | ||
| 2 hidden layers | M13 | 92 | 0.354 | 91 | 0.392 | 96 | 159 | |
| M14 | 95 | 0.28 | 92 | 0.374 | 97 | 20 | ||
| 3 hidden layers | M15 | 98 | 0.168 | 94 | 0.324 | 97 | 24 | |
| M16 | 100 | 0.006 | 93 | 0.337 | 70 | 186 | ||
However, a comparison between models with respect to the computational cost and the number of descriptors shows that M15 is not recommended because of its complexity, larger number of descriptors and a higher computational cost. Therefore, the M2, M4 and M8 are the most performant predictive models which can be used for the prediction of biological activity against ABCB1 from a low number of descriptors.
3.6. Docking study
With the intention of targeting more potent molecules, various molecular docking studies were undertaken using MOE software to virtual screen molecular binding modes of four prepared compounds, such as 10, 12, 13 and 17, in the P-glycoprotein pocket. Here, the ligands were docked to the encoded protein 6C0V loaded from the PDB. Ten different interaction positions were authorized for each molecule with the protein and the ranking poses were generated by the scoring functions which are given in Table 5. The 17 obtained the highest score, and the result was −9.22 kcal/mol. The list of hydrogen bonds between the compounds and the selected protein coenzymes is given in Table 6. The best-fitting pose that was adopted by the enzyme-calmed compound 6C0V is displayed in Fig. 9.
Table 5.
Docking score and energy of the compounds and 6C0V protein.
| Compound | S | Rmsd_refine | E_conf | E_place | E_score1 | E_refine | E_score2 |
|---|---|---|---|---|---|---|---|
| 10 | −7.905 | 1.025 | 1.394 | −75.559 | −9.623 | −18.183 | −7.905 |
| −7.775 | 0.800 | −2.839 | −86.373 | −9.463 | −23.079 | −7.775 | |
| −7.645 | 1.608 | −1.793 | −49.818 | −9.905 | −22.728 | −7.645 | |
| −7.543 | 2.688 | −0.888 | −49.161 | −10.039 | −22.203 | −7.543 | |
| −7.414 | 2.085 | −2.084 | −58.509 | −9.664 | −17.422 | −7.414 | |
| 12 | −7.373 | 1.741 | −9.593 | −47.854 | −9.930 | −12.378 | −7.373 |
| −7.297 | 1.012 | −5.187 | −79.545 | −9.838 | −12.151 | −7.297 | |
| −7.243 | 0.885 | −12.536 | −67.845 | −9.334 | −6.729 | −7.243 | |
| −6.997 | 1.031 | −17.548 | −68.131 | −9.505 | −18.468 | −6.997 | |
| −6.652 | 1.332 | −14.182 | −84.596 | −9.484 | −10.754 | −6.652 | |
| 13 | −7.947 | 1.486 | 36.845 | −68.307 | −11.132 | 5.557 | −7.947 |
| −7.789 | 1.970 | 43.578 | −54.778 | −10.129 | −8.370 | −7.789 | |
| −7.724 | 1.956 | 37.138 | −72.143 | −11.943 | −6.981 | −7.724 | |
| −7.555 | 2.616 | 49.700 | −61.442 | −10.441 | −3.158 | −7.555 | |
| −7.129 | 1.281 | 37.123 | −80.267 | −10.301 | −1.612 | −7.129 | |
| 17 | −9.222 | 2.289 | 48.015 | −56.777 | −11.020 | −17.708 | −9.222 |
| −8.637 | 2.336 | 37.203 | −68.444 | −11.376 | −8.698 | −8.637 | |
| −8.545 | 2.083 | 50.419 | −62.657 | −11.611 | −7.534 | −8.545 | |
| −8.371 | 2.000 | 48.119 | −58.199 | −11.726 | −4.530 | −8.371 | |
| −8.057 | 1.413 | 48.004 | −47.495 | −10.832 | −4.235 | −8.057 |
Table 6.
Interaction table between the compounds and 6C0V protein.
| Compounds | Ligand | Receptor | Interaction | Distance | E (kcal/mol) | |||
|---|---|---|---|---|---|---|---|---|
| 10 | O | 15 | NH1 | ARG | 148 | H-acceptor | 3.33 | −1 |
| NH2 | ARG | 148 | 2.88 | −4.8 | ||||
| O | 25 | NE2 | GLN | 824 | 2.74 | −0.7 | ||
| NZ | LYS | 1000 | 2.89 | −8.4 | ||||
| 12 | O | 15 | ND2 | ASN | 820 | H-acceptor | 3.08 | −2.8 |
| 13 | C | 13 | OD1 | ASP | 886 | H-donor | 3.41 | −0.8 |
| 17 | O | 21 | ND2 | ASN | 183 | H-acceptor | 3.17 | −1.7 |
| 6-ring | NE2 | GLN | 882 | pi-H | 4.18 | −1.1 | ||
Fig. 9.

3D docking, site view & 2D of compound 17 and 6C0V protein.
MOE is one of the most important molecular docking mechanisms used to recognize a precise docking study between compounds and target proteins. The compound 17 exhibited a high docking score by hydrogen π-stacking with the 6-membered ring of Gln 882. The interaction distance and stabilization energy were respectively equal to 4.18 A° and −1.1 kcal/mol and by hydrogen bonding of the oxygen atom contained in the ester function to the amino acid residue Asn 183. This hydrogen bond is at about 3.17 A° and the energy stabilization amounts to −1.7 K cal/mol. Through these bonds obtained with the key amino acid residues of the binding pockets highlighted based on the above results, the target receptor structure could be stabilized. The docking pose showing the highest affinity is regarded as the best docking conformation. Similarly, in view of these crucial interactions at the molecular level, MOE was able to match the experimentally observed binding modes, thus identifying the particular conformation of the target and ligand. By comparing the results obtained with the others of biological activity carried out by our laboratory [42], we note that they are the same since the compound 17 that has the best biological activity obtained the best-docked conformation.
4. Conclusion
QSAR modeling was performed to develop models for the prediction of the inhibitory effect of quinoline-derivatives on one of the most studied human adenosine-triphosphate (ATP)-dependent efflux transporters that encodes a multidrug resistance protein, called ABCB1 gene. Through this study, several machine learning methods were tested to identify relevant descriptors and develop reliable models including partial least squares regression, stepwise multiple linear regression, back propagation neural networks. The results obtained showed that a catboost model statistically outperformed the other used methods, achieving a R2 and RMSE of 95% and 0.28 respectively. Based on the computational study, compound 17 was found to possess the maximum binding affinity to the target protein of outward-facing p-glycoprotein with a binding energy equal to −9.22 kcal/mol. According to the above fact of the laboratory results, this compound can be proposed as a lead structure for the design and synthesis of more potent P-glycoprotein inhibitors for combination used with anti-cancer drugs for cancer multidrug resistance management.
Indeed, in this work, descriptors were identified by QSAR models to effectively predict the P-glycoprotein inhibitory effect as well as to guide the design of new of 2-oxo 1, 2-dihydroquinoline-4- carboxylic acid derivatives for potential applications in cancer multidrug resistance area. This will help to facilitate the drug development process and minimize the cost of synthesis in pharmaceutical chemistry laboratories.
Annex: 1
| Abbreviation of molecular descriptors | description |
|---|---|
| b_rotR | Fraction of rotatable bonds |
| dipoleX | Dipole moment |
| lip_acc | Lipinski acceptor count |
| logP (o/w) | Log octanol/water partition coefficient |
| PEOE_VSA-5 | Total positive surface area |
| PM3_HF | Heat of formation |
| pmiZ | Principal moment of inertia |
| SlogP | partition coefficient |
| AM1_HOMO | Homo energy |
| AM1_LUMO | Lumo energy |
| apol | Sum of atomic polarizabilities |
| E | Potential energy |
| mr | Molar refractivity |
| Vdw_area | Van der Waals surface area |
Annex: 2
| Abbreviation | description |
|---|---|
| S | The finale score of GBVI/WSA binding free energy |
| Rmsd_refine | The mean square deviation after refinement |
| E_place | Score of the placement phase |
| E_conf | Energy conformer |
| E_refine | Score refinement |
| E_scor1 | Score the first step of notation |
Author contribution statement
Mouad Lahyaoui: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Wrote the paper.
Abderrahim Diane: Performed the experiments; Wrote the paper.
Hafsa El-Idrissi, Taoufiq Saffaj, Ihssane Bouchaib: Performed the experiments.
Kandri Rodi Youssef: Performed the experiments; Contributed reagents, materials, analysis tools or data.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Data availability statement
Data included in article/supplementary material/referenced in article.
The authors declare no competing interests.
Footnotes
Declaration of interest's statement.
References
- 1.Plummer M., De Martel C., Vignat J., Ferlay J., Bray F., Franceschi S. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Global Health. 2016;4:e609. doi: 10.1016/S2214-109X(16)30143-7. –e616. [DOI] [PubMed] [Google Scholar]
- 2.Chen Z., Shi T., Zhang L., Deng M., Huang C., Hu T., Jiang L., Li J. Cancer Lett.; 2015. Mammalian Drug Efflux Transporters of the ATP Binding Cassette (ABC) Family in Multidrug Resistance: a Review of the Past Decade. [DOI] [PubMed] [Google Scholar]
- 3.Konig J., Muller F., Fromm M.F. Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol. Rev. 2013;65:944–966. doi: 10.1124/pr.113.007518. [DOI] [PubMed] [Google Scholar]
- 4.Levatic J., Curak J., Kralj M., Smuc T., Osmak M., Supek F. Accurate models for P-gp drug recognition induced from a cancer cell line cytotoxicity screen. J. Med. Chem. 2013;56:5691–5708. doi: 10.1021/jm400328s. [DOI] [PubMed] [Google Scholar]
- 5.Li S., Zhang W., Yin X., Xing S., Xie H.Q., Cao Z., et al. Binding cassette (ABC) transporters conferring multi-drug resistance. Anti Cancer Agents Med. Chem. 2015;15:423–432. [PubMed] [Google Scholar]
- 6.Atalay C., Demirkazik A., Gunduz U. Role of ABCB1 and ABCC1 gene induction on survival in locally advanced breast cancer. J. Chemother. 2008;20:734–739. doi: 10.1179/joc.2008.20.6.734. [DOI] [PubMed] [Google Scholar]
- 7.Vasan N., Baselga J., Hyman D.M. A view on drug resistance in cancer. Nature. 2019;575:299–309. doi: 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohammad I.S., He W., Yin L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed. Pharmacother. 2018;100:335–348. doi: 10.1016/j.biopha.2018.02.038. [DOI] [PubMed] [Google Scholar]
- 9.P-glycoprotein transporter in drug development. EXCLI Journal. 2016;15:113–118. doi: 10.17179/excli2015-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gromicho M., Magalhaes M., Torres F., Dinis J., Fernandes A.R., Rendeiro P., et al. Instability of mRNA expression signatures of drug transporters in chronic myeloid leukemia patients resistant to imatinib. Oncol. Rep. 2013;29:741–750. doi: 10.3892/or.2012.2153. [DOI] [PubMed] [Google Scholar]
- 11.Hardy J., Selkoe D.J. The Amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 12.Hartz A.M.S., Miller D.S., Bauer B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-β in a mouse model of Alzheimer's disease. Mol. Pharmacol. 2010;77:715–723. doi: 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hennessy M., Spiers J.P. A primer on the mechanics of P-glycoprotein the multidrug transporter. Pharmacol. Res. 2007;55:1–15. doi: 10.1016/j.phrs.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Klepsch F., Vasanthanathan P., Ecker G.F. Ligand and structure-based classification models for prediction of P-glycoprotein inhibitors. J. Chem. Inf. Model. 2014;54:218–229. doi: 10.1021/ci400289j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishna R., Mayer L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal-using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharmaceut. Sci. 2000;11:265–283. doi: 10.1016/s0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 16.Kuhnke D., Jedlitschky G., Grube M., Krohn M., Jucker M., Mosyagin I., et al. MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer's amyloid-β peptides - implications for the mechanisms of Aβ clearance at the blood-brain barrier. Brain Pathol. 2007;17:347–353. doi: 10.1111/j.1750-3639.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCormick J.W., Vogel P.D., Wise J.G. Multiple drug transport pathways through human P-glycoprotein. Biochemistry. 2015;54:4374–4390. doi: 10.1021/acs.biochem.5b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eckford P.D., Sharom F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- 19.Scott D.A., Balliet C.L., Cook D.J., Davies A.M., Gero T.W., Omer C.A., Poondru S., Theoclitou M.E., Tyurin B., Zinda M.J. Identification of 3-amido-4-anilinoquinolines as potent and selective inhibitors of CSF-1R kinase. Bioorg. Med. Chem. Lett. 2009;19:697–700. doi: 10.1016/j.bmcl.2008.12.046. [DOI] [PubMed] [Google Scholar]
- 20.Marganakop S.B., Kamble R.R., Taj T., Kariduraganvar M.Y. An efficient one-pot cyclization of quinoline thiosemicarbazones to quinolines derivatives with 1,3,4-thiadiazole as anticancer and anti-tubercular agents. Med. Chem. Res. 2010;21:185–191. [Google Scholar]
- 21.Ma X., Wu Y., Yang X., Li Y., Huang Y., Lee R.J., Bai T., Luo Y. A novel 1,2-dihydroquinoline anticancer agent and its delivery to tumor cells using cationic liposomes. Anticancer Res. 2016;36:2105–2111. [PubMed] [Google Scholar]
- 22.Godlewska J., Luniewski W., Zagrodski B., Kaczmarek L., Bielawska-Pohl A., Dus D., Wietrzyk J., Opolski A., Siwko M., Jaromin A., Jakubiak A., Kozubek A., Peczyska-Czoch W. Biological evaluation of ω-(dialkylamino)alkyl derivatives of 6H-indolo[2,3-b]quinoline–Novel cytotoxic DNA topoisomerase II inhibitors. Anticancer Res. 2005;25:2857–2868. [PubMed] [Google Scholar]
- 23.Sharma V., Mehta D.K., Das R. Synthetic methods of quinoline derivatives as potent anticancer agents. Mini Rev. Med. Chem. 2017;17:1557–1572. doi: 10.2174/1389557517666170510104954. [DOI] [PubMed] [Google Scholar]
- 24.El Assiri E.H., Driouch M., Lazrak J., Bensouda Z., Elhaloui A., Sfaira M.…Taleb M. Development and validation of QSPR models for corrosion inhibition of carbon steel by some pyridazine derivatives in acidic medium. Heliyon. 2020;6(10) doi: 10.1016/j.heliyon.2020.e05067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Assiri, El Hassan, Driouch Majid, Zakaria Bensouda, Jhilal F., Saffaj Taoufiq, Sfaira M., Younes Abboud. Quantum chemical and QSPR studies of bis-benzimidazole derivatives as corrosion inhibitors by using electronic and lipophilic descriptors. Desalination Water Treat. 2018;111:208–225. [Google Scholar]
- 26.El Ghalia H., Amina G., El Aissouq A., Oussama C., Abdelkrim O., Mohammed B. A quantitative study of the structure-activity relationship and molecular docking of 5.6.7- trimethoxy-N-aryl-2-styrylquinolin-4-amines as potential anticancer agents using quantum chemical descriptors and statistical methods. J. Mol. Struct. 2022:133794. [Google Scholar]
- 27.Surabhi S., Singh B.K. Computer aided drug design: an overview. J.D.D.T. 2018;8(5):504–509. [Google Scholar]
- 28.Baba Y.F., Misbahi H., Rodi Y.K., Ouzidan Y., Essassi E.M., Vincze K.…Mazzah A. Chemical Data Collections; 2020. 2-oxo-1, 2-Dihydroquinoline-4-Carboxylic Acid Derivatives as Potent Modulators of ABCB1-Related Drug Resistance of Mouse T-Lymphoma Cells. [Google Scholar]
- 29.Forster S., Thumser A.E., Hood S.R., Plant N. Characterization of Rhodamine-123 as a tracer dye for use in in vitro drug transport assays. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dominguez-Alvarez E., Gajdacs M. Identification of Selene-compounds with promising properties to reverse cancer multidrug resistance. Bioorg. Med. Chem. Lett. 2016;26:2821–2824. doi: 10.1016/j.bmcl.2016.04.064. [DOI] [PubMed] [Google Scholar]
- 31.Gajdacs M., Spengler G., Sanmartin C., Marc M.A., Handzlik J., Dominguez-Alvarez E. Selenoesters and selenoanhydrides as novel multidrug resistance reversing agents: a confirmation study in a colon cancer MDR cell line. Bioorg. Med. Chem. Lett. 2017;27:797–802. doi: 10.1016/j.bmcl.2017.01.033. [DOI] [PubMed] [Google Scholar]
- 32.Bro R., Smilde A.K. Principal component analysis. Anal. Methods. 2014;6(9):2812–2831. [Google Scholar]
- 33.Wold S., Johansson E., Cocchi M. In: 3D QSAR in Drug Design: Theory, Methods and Applications. Kubinyi H., editor. ESCOM Science Publishers; Leiden: 1993. PLS: partial least squares projections to latent structures; pp. 523–550. [Google Scholar]
- 34.Wold S., Sjostrom M., Eriksson L. PLS-regression: a basic tool of chemometrics. Chemometr. Intell. Lab. Syst. 2001;58(2):109–130. [Google Scholar]
- 35.Berk R.A. SAGE Publications Ltd; London: 2003. Some Popular Extensions of Multiple Regression Analyses: a Constructive Critique; pp. 125–150. [Google Scholar]
- 36.Reda R., Saffaj T., Derrouz H., Itqiq S.E., Bouzida I., Saidi O.…El Hadrami E.M. Comparing CalReg performance with other multivariate methods for estimating selected soil properties from Moroccan agricultural regions using NIR spectroscopy. Chemometr. Intell. Lab. Syst. 2021;211 [Google Scholar]
- 37.Reda R., Saffaj T., Itqiq S.E., Bouzida I., Saidi O., Yaakoubi, El Hadrami E.M. Predicting soil phosphorus and studying the effect of texture on the prediction accuracy using machine learning combined with near-infrared spectroscopy. Spectrochim. Acta Mol. Biomol. Spectrosc. 2020 doi: 10.1016/j.saa.2020.118736. [DOI] [PubMed] [Google Scholar]
- 38.Reda R., Saffaj T., Ilham B., Saidi O., Issam K., Brahim L., El Hadrami E.M. Chemometrics and Intelligent Laboratory Systems; 2019. A Comparative Study between a New Method and Other Machine Learning Algorithms for Soil Organic Carbon and Total Nitrogen Prediction Using Near Infrared Spectroscopy. [Google Scholar]
- 39.Goh A.T.C. Back-propagation neural networks for modeling complex systems. Artif. Intell. Eng. 1995;9(3):143–151. [Google Scholar]
- 40.Am. Statistician. 1992;46(3):175–185. [Google Scholar]
- 41.Natekin A., Knoll A. Gradient boosting machines, a tutorial. Front. Neurorob. 2013;7 doi: 10.3389/fnbot.2013.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baba Y.F., Misbahi H., Rodi Y.K., Ouzidan Y., Essassi E.M., Vincze K.…Mazzah A. Chemical Data Collections; 2020. 2-oxo-1, 2-Dihydroquinoline-4-Carboxylic Acid Derivatives as Potent Modulators of ABCB1-Related Drug Resistance of Mouse T-Lymphoma Cells. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data included in article/supplementary material/referenced in article.
The authors declare no competing interests.