

Summary
RNA methylation at adenosine N6 (m6A) is one of the most common RNA modifications, impacting RNA stability, transport and translation. Previous studies uncovered RNA destabilization in amyotrophic lateral sclerosis (ALS) models, in association with accumulation of the RNA-binding protein TDP43. Here, we show that TDP43 recognizes m6A RNA, and that RNA methylation is critical for both TDP43 binding and autoregulation. We also observed extensive RNA hypermethylation in ALS spinal cord, corresponding to methylated TDP43 substrates. Emphasizing the importance of m6A for TDP43 binding and function, we identified several m6A factors that enhance or suppress TDP43-mediated toxicity via single-cell CRISPR/Cas9 in primary neurons. The most promising modifier—the canonical m6A reader YTHDF2—accumulated within ALS spinal neurons, and its knockdown prolonged the survival of human neurons carrying ALS-associated mutations. Collectively, these data show that m6A modifications modulate RNA binding by TDP43, and that m6A is pivotal for TDP43-related neurodegeneration in ALS.
eTOC blurb
Mislocalization of the essential RNA binding protein TDP43 is characteristic of ALS. McMillan, Gomez et al. show that RNA binding by TDP43 is strongly influenced by RNA methylation at adenosine N6 (m6A). They also uncover marked RNA hypermethylation in ALS, and connect TDP43-mediated toxicity with the m6A reader YTHDF2.
Graphical Abstract
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease resulting in the death of upper and lower motor neurons1. Limited therapeutic options exist for ALS, and the underlying pathological mechanisms remain unclear. Considerable variability in clinical, biochemical, and genetic features also complicate identification of therapeutic targets. Despite this, over 95% of ALS cases exhibit cytoplasmic inclusions of the RNA binding protein TDP43 (TAR binding protein of 43 kDa), and mutations in TARDBP, the gene encoding TDP43, result in familial disease in 2–5% of individuals2–6. These observations imply that strategies targeting TDP43 may be relevant to the large majority of those with ALS.
TDP43 is a predominantly nuclear RNA binding protein critical for RNA splicing, transport, translation, and stability. Consistent with this, cytoplasmic mislocalization and nuclear clearing of TDP43 in ALS are closely associated with RNA missplicing, impaired translation, and widespread RNA instability5,7–9. We previously found that TDP43 deposition primarily destabilizes mRNAs encoding ribosomal proteins and oxidative phosphorylation enzymes5. Notably, these same mRNAs are upregulated in neurons lacking the RNA methyltransferase-like protein 14 (METTL14)10. This enzyme is part of a “writer” complex that methylates RNA at adenosine N6 (N6-methyladenosine methylation, or m6A)11,12. These marks can be removed by demethylases (“erasers”) and recognized by a class of RNA binding proteins (“readers”) that, like TDP43, function in RNA splicing, transport, translation and stability13,14. TDP43 also physically interacts with both writers and readers15–18. Together, these findings suggest that TDP43 may recognize m6A-modified RNA, raising the possibility that TDP43 mislocalization and aggregation in ALS may preferentially affect RNA substrates carrying m6A marks.
Here, we answer the question of whether TDP43 binds m6A-modified RNA through several approaches. We show not only that the majority of TDP43 substrates carry m6A modifications, but also that the m6A reader protein YTHDF2 facilitates TDP43-related toxicity in rodent and human neuron models of ALS. Supporting the connection between TDP43 pathology and m6A RNA, we detected extensive RNA hypermethylation in postmortem spinal cord tissue from sporadic ALS patients. These data highlight a fundamental link between m6A RNA modifications and ALS pathogenesis, potentially mediated by TDP43-dependent misprocessing of m6A-modified RNA.
Results
TDP43 binds m6A-modified RNA
YTH domains, common to m6A reader proteins, exhibit selective recognition of m6A-modified RNA, but previous evidence indicates that other functional domains such as RNA recognition motifs (RRMs) present in TDP43 and other members of the hnRNP family can also bind m6A-modified RNA11,14. Specifically, hnRNP-C19 and hnRNP-A2/B120 recognize m6A-modified RNA, and the presence of m6A modifications enhances their affinity for RNA substrates. Given this, we asked whether TDP43, which shows high affinity for UG rich sequences21–24, also recognizes m6A-modified RNA. First, we measured the UG density surrounding experimentally verified m6A sites, determined by m6A antibody cross-linking induced methylation (CIMS; Fig. 1A) and truncation (CITS; Fig. 1B)25. UG density is significantly greater immediately upstream of m6A sites, whereas random segments of non-methylated genes showed no apparent change in UG density surrounding the m6A site (Fig. 1A, B). These data indicate a possible connection between UG-rich TDP43 recognition motifs and m6A sites. To pursue this concept further and empirically determine if TDP43 recognizes m6A-modifed RNA in mammalian cells, we overexpressed TDP43 fused with HaloTag, a multifunctional adapter protein that facilitates TDP43 isolation by immunoaffinity purification26, in HEK293T cells. As a positive control, we also overexpressed a fusion of HaloTag and YTHDF2, a verified m6A reader protein14. We then isolated each protein by immunoaffinity purification, collected bound RNA and assessed total and m6A RNA by dot blot (Fig. 1C). TDP43-HaloTag and YTHDF2-HaloTag, but not HaloTag itself, pulled down m6A RNA (Fig. 1D), showing that TDP43 and YTHDF2 are both capable of binding m6A RNA.
Figure 1: TDP43 recognizes m6A-modified RNA.
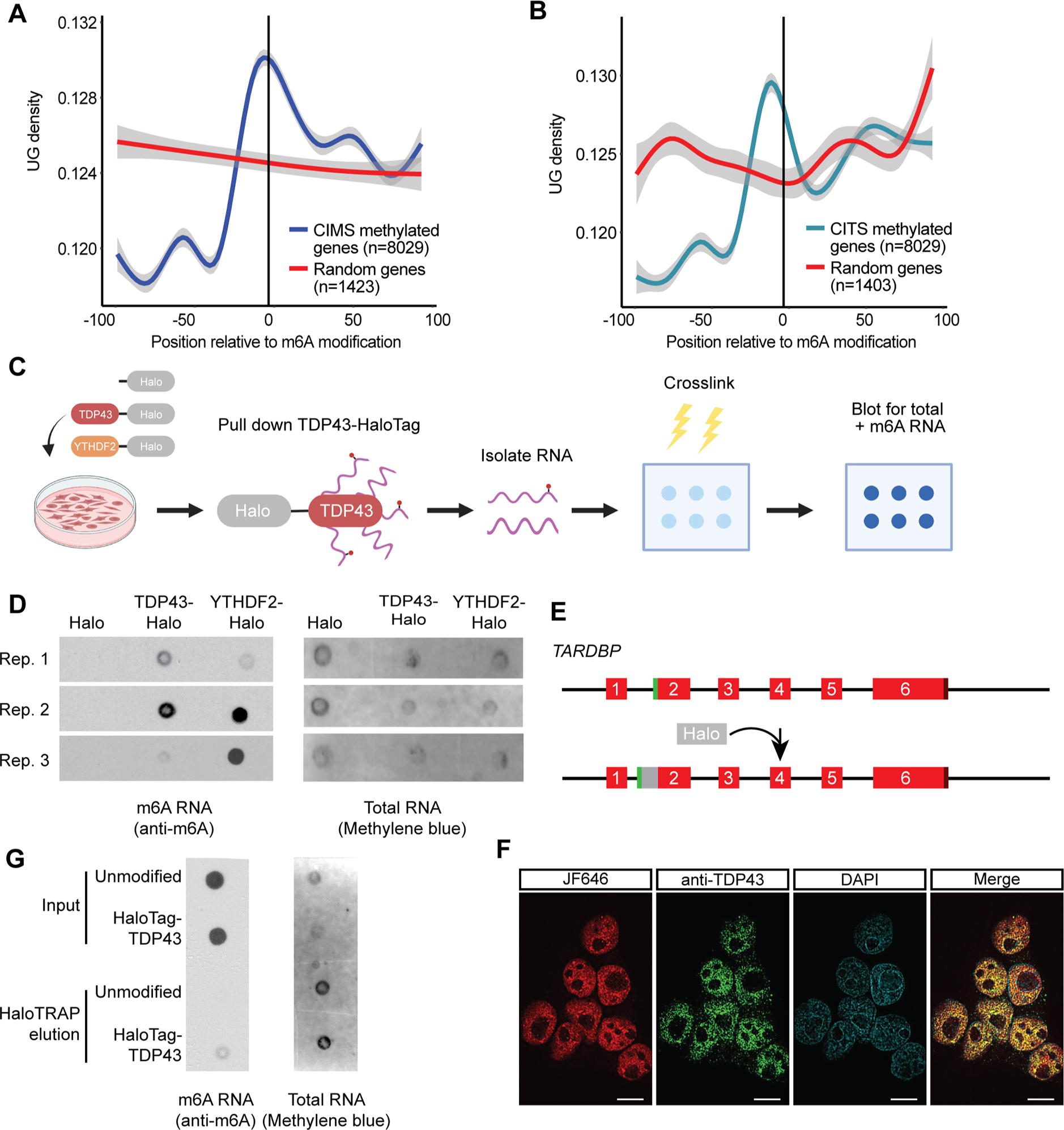
UG density 100bp up- and downstream of m6A modifications identified by cross-linking induced mutation sites (CIMS; A) or cross-linking induced truncation sites (CITS; B) in relation to random sequences (red line). Grey shading, 95% CI. (C) Schematic of HaloTag immunoprecipitation and dot blot procedure. (D) Dot blot for total RNA (detected by methylene blue) or m6A-modified RNA (detected by anti-m6A antibody) following HaloTag immunoaffinity purification in HEK293T cells overexpressing HaloTag, TDP43-HaloTag or YTHDF2-HaloTag from 3 biological replicates. (E) Diagram illustrating insertion of HaloTag downstream of the TARDBP start codon, encoding HaloTag-TDP43. (F) Halo-TDP43 HEK293T cells labeled live with JF646 dye (red), then fixed, permeabilized, and immunostained for TDP43 (green) prior to imaging. DAPI (blue) marks the nucleus. Scale bar, 10μm. (G) Dot blot for total and m6A RNA isolated by immunoaffinity purification of endogenous HaloTag-TDP43 or overexpressed HaloTag. Empty space between lanes was clipped in (D) and (E). Additional replicates shown in Fig. S1.
Because protein overexpression can introduce non-specific interactions, we engineered a line of HEK293T cells in which endogenous TDP43 was labeled at the N-terminus with HaloTag (Fig. 1E). Correct insertion of HaloTag was verified by Sanger sequencing, and analysis of HaloTag-TDP43 function using the CFTR splicing reporter uncovered no loss-of-function effects (Fig. S1A). In addition, live-cell labeling of HaloTag-TDP43 HEK293T cells with JF646, a far-red dye that covalently binds HaloTag, showed strong overlap with TDP43 immunostaining (Fig. 1F). As before, we detected m6A modified RNA via dot blot among transcripts pulled down by HaloTag-TDP43 (Fig. 1G and Fig. S1B–C), confirming that endogenously expressed TDP43 can bind m6A modified RNA.
TDP43 substrates are enriched in m6A modifications
To explicitly define the identity, number, and location of m6A modifications in TDP43 substrates, we took advantage of DART-seq (deamination adjacent to RNA modification targets, followed by next generation RNA sequencing)27 (Fig. 2A). This method highlights specific m6A modifications without the need for m6A antibodies, which may have limited sensitivity and specificity in discriminating m6A from other RNA modifications (i.e., m6Am). Briefly, DART-seq involves the overexpression of a chimeric fusion protein consisting of the m6A-binding YTH domain, and the deaminating enzyme APOBEC1. As illustrated previously11,12,25,27, m6A modifications occur in the context of a highly conserved motif (DRACH, where D can be A, G or T; R is A or G; and H is A, C or T). Within this motif, the methylated adenosine (A) residue is followed by an obligate cytosine (C). Recognition of m6A by the APOBEC1-YTH fusion results in deamination of the C immediately 3’ to the modified A residue, generating uracil (U) bases that are read as thymine (T) during sequencing. Thus, C-T transitions occurring within a DRACH motif immediately adjacent to an A signify m6A modifications. In combination with immunoaffinity purification of endogenous HaloTag-TDP43 from the HEK293T cells we created, DART-seq enables us to define not only the RNA substrates of native HaloTag-TDP43, but also the location and m6A modification status of each substrate.
Figure 2: Site-specific identification of m6A-modified TDP43 substrates.
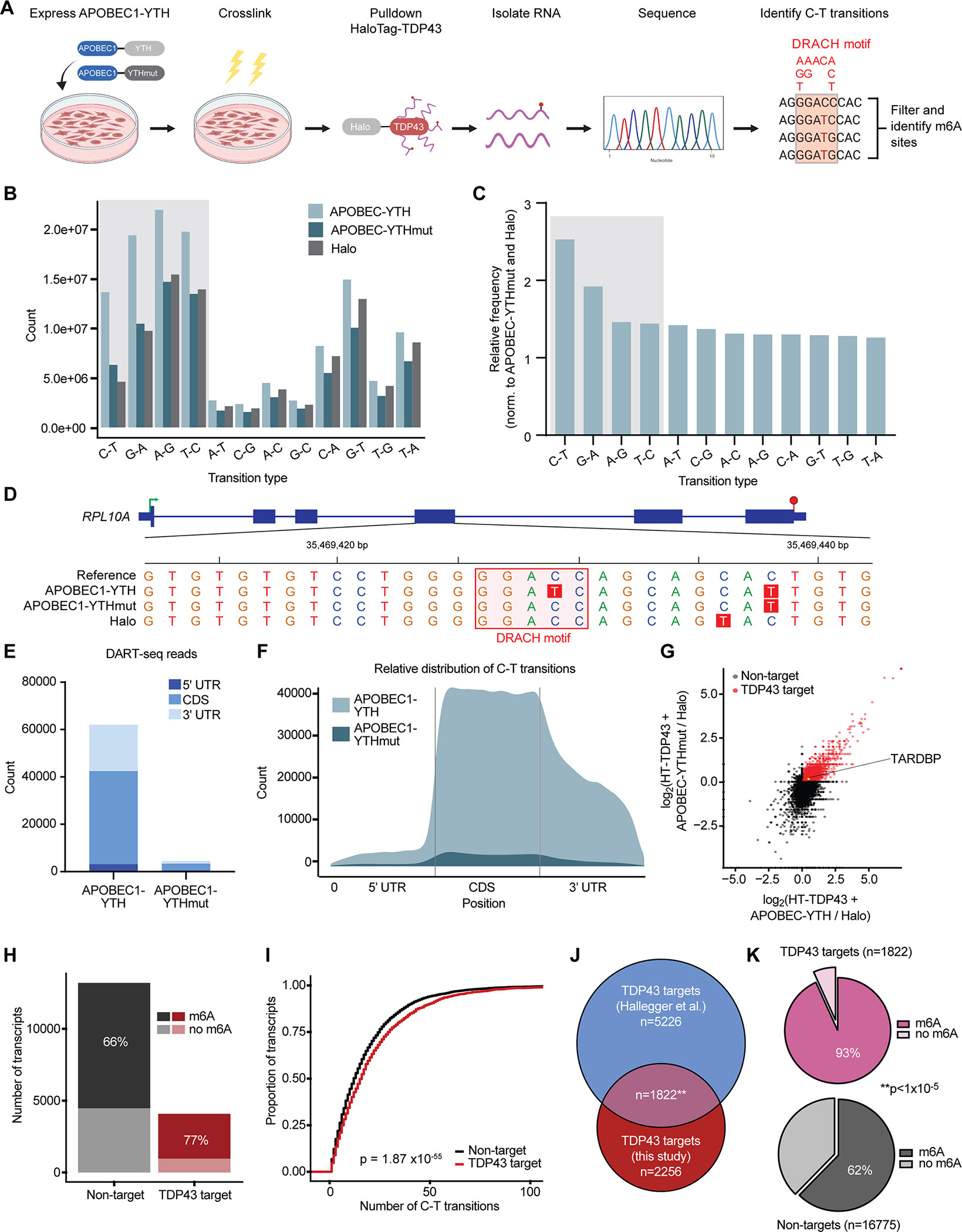
(A) Schematic of DART-seq in HaloTag-TDP43 HEK293T cells. D=A/G/T, R=A/G, H=A/C/T. Absolute counts (B) and relative frequency (C) of base pair transitions observed by RNA-seq in each condition. Shaded boxes represent transition types expected from APOBEC1 activity. (D) Example m6A sites identified by DART-seq in RPL10A. C-T transitions are highlighted in red, and DRACH motifs in pink. Green arrow, transcription start site; red hexagon, transcription stop site; thick blue bars, coding exons; thin blue bars, UTR. Absolute count (E) and relative distribution (F) of DART-seq reads in cells expressing APOBEC1-YTH and APOBEC1-YTHmut. (G) Scatter plot of TDP43 targets, determined by fold enrichment in precipitated RNA from HaloTag-TDP43 cells (expressing APOBEC1-YTH and APOBEC1-YTHmut) compared to cells transfected with HaloTag. Red dots signify transcripts showing ≥2-fold enrichment in both APOBEC1-YTH and APOBEC1-YTHmut expressing cells. (H) Stacked bar graph showing percentage of m6A modified RNA in TDP43 targets (red) and non-targets (black). (I) Cumulative distribution of RNA methylation in TDP43 targets (red) and non-targets (black). p=1.87×10−55 Kolmogorov Smirnov test. (J) Euler diagram depicting overlap between TDP43 targets identified in this study, and those identified previously by Hallegger et al.28. **p=1.5×10−117, hypergeometric test. (K) Pie charts demonstrating the percentage of methylated RNA among TDP43 targets (pink) and non-targets (grey). **p<1×10−5 chi-square test.
We transfected HaloTag-TDP43 HEK293T cells with APOBEC1-YTH or a mutated variant of APOBEC1-YTH that is unable to bind m6A modified RNA (APOBEC1-YTHmut). To control for non-specific RNA binding by HaloTag, we also transfected separate cultures of unmodified HEK293T cells with HaloTag alone. We then cross-linked RNA and protein using UV light, isolated HaloTag and HaloTag-TDP43 by immunoaffinity purification, sequenced the co-precipitated RNA and compared the resulting data to a reference database to identify base-pair transitions (Fig. 2A). As expected based on the deaminase activity of APOBEC1, C-T and G-A transitions—and to a lesser extent antisense T-C and A-G transitions—were strongly enriched in APOBEC1-YTH expressing cells in comparison to those expressing APOBEC1-YTHmut or HaloTag alone (Fig. 2B, C). These transitions occurred both within and outside of canonical DRACH motifs (Fig. 2D), but to increase specificity we limited further analyses to transitions that arose in the context of a DRACH motif. In doing so, we noted profound enrichment for high-confidence m6A sites in cells expressing APOBEC1-YTH, compared to those expressing APOBEC1-YTHmut and HaloTag (Fig. 2E, F). The majority of detected m6A sites fall within the coding sequence (CDS) of HaloTag-TDP43 target RNAs, rather than the untranslated regions (5’ or 3’UTRs) or intronic segments (Fig. S2A, B). This pattern is consistent with the previously observed exclusion of m6A sites from introns, but differs from the expected concentration of m6A modifications in the 3’UTR immediately downstream of the stop codon12,25,27. We suspect that the unusual distribution of m6A sites likely reflects the differences between HaloTag-TDP43 substrates investigated here, and total mRNA used in prior studies.
Emphasizing the sensitivity of this approach for capturing TDP43 target RNAs, we identified 2,256 transcripts that were significantly enriched by HaloTag-TDP43 pulldown in APOBEC1-YTH and APOBEC1-YTHmut expressing cells, compared to cells expressing HaloTag alone (Fig. 2G). In keeping with our previous data suggesting that TDP43 recognizes m6A-modified RNA, the majority of HaloTag-TDP43 target RNAs were methylated (Fig. 2H), and also displayed a higher degree of methylation in comparison to non-targets. (Fig. 2I). To ensure that our results are not confounded by potential RNA-HaloTag interactions or the immunoaffinity purification itself, we compared our HaloTag-TDP43 substrates to a published dataset of TDP43 targets in HEK293 cells identified by CLIP-seq28. In the process, we identified 1822 high-confidence TDP43 substrates (p=1.5×10−117 for the overlap, hypergeometric test), over 90% (1699) of which were m6A-modified (Fig. 2J, K). By gene ontology (GO), methylated TDP43 target RNAs were highly enriched for componets of the nuclear pore complex (Fig. S2C), a structure with intricate ties to ALS pathogenesis. Furthermore, protein-protein interaction (PPI) network prediction using STRING29 indicated strong enrichment for several additional disease-associated pathways, including apoptosis and p53 signaling30, the ribosome5, long-term potentiation31, VEGF signaling32 and RNA transport33,34 (Fig. S2D). In comparison, only 62% of non-targets were methylated (Fig. 2K, p<1×10−5 for the comparison with TDP43 targets; chi-square test). These results not only confirm that TDP43 recognizes m6A modified RNA, but also indicate a preference of TDP43 for methylated transcripts.
RNA methylation modulates TDP43 binding and autoregulation
To explore the functional impact of RNA methylation on TDP43 binding and function, we re-examined the DART-seq results, concentrating on the relationship between identified m6A sites and UG-rich TDP43 recognition motifs. In doing so, we observed a striking overlap between m6A modifications and UG-rich sequences within the 3’UTR of HaloTag-TDP43 substrates (Fig. S2E). Indeed, 74% of m6A sites located within 20 nt of a TDP43 motif are found within the 3’UTR of HaloTag-TDP43 targets (Fig. S2F). This subset of 237 transcripts was highly enriched for genes whose expression is regulated by TDP43 (Fig. S2G). Using qRT-PCR, we confirmed the sensitivity of 3 candidate transcripts (TARDBP and MAPK14, both with 3’UTR m6A sites; and XIST, which is methylated and a TDP43 target but lacks a UTR) to TDP43 overexpression (Fig. S2H–J). In each case, YTHDF2 knockdown partially mitigated the effect of TDP43 overexpression, consistent with the incomplete YTHDF2 reduction in these cells (Fig. S2K). Together, these results emphasize the functional relevance of m6A and YTHDF2 for TDP43-dependent gene regulation.
We next focused on TARDBP, a HaloTag-TDP43 substrate RNA that showed prominent methylation based on our DART-seq experiments. The TARDBP transcript, which encodes TDP43 itself, exhibited a clear C-T transition within the context of a 3’UTR DRACH motif (Fig. 3A). We focused on this m6A site for three reasons: First, it is located within the TDP43 binding region (TBR)35, a section of the 3’UTR that is both necessary and sufficient for TDP43 recognition. Second, the m6A site is adjacent to a UG-rich stretch resembling the consensus GUGUGU motif commonly recognized by TDP4336,37. Third, the m6A site lies within a 34 nucleotide region (denoted CLIP34nt) that binds TDP43 in vitro and in cellulo38 (Fig. 3A).
Figure 3: m6A modifications influence TDP43 binding and autoregulation.
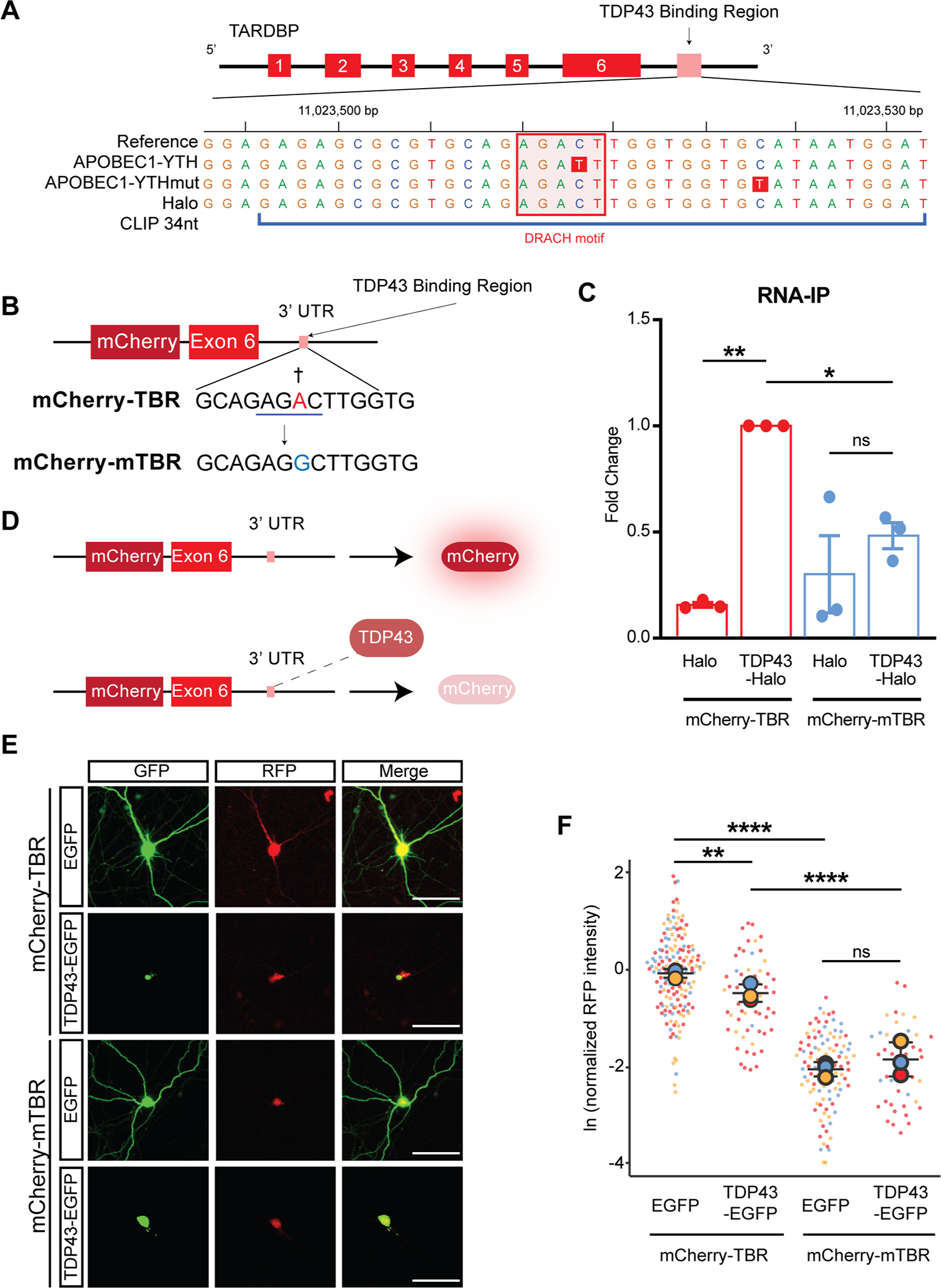
(A) TARDBP gene map, illustrating TDP43 binding region (TBR), the location of the DRACH motif (pink square), and the C-T transition (red box) identified by DART-seq within this domain, representing an m6A site. (B) Schematic of the TARDBP minigene reporter, consisting of the mCherry ORF upstream of TARDBP exon 6 and 3.4 Kb of the TARDBP 3’ UTR. The A residue adjacent to the putative m6A site in the WT reporter (mCherry-TBR) was mutated to a G, precluding methylation in the mutant reporter (mCherry-mTBR). Red, methylated residue; blue line, DRACH motif; †, putative m6A site. (C) Immunoaffinity purification of HaloTag-TDP43 from HEK293T cells expressing mCherry-TBR or mCherry-mTBR, followed by qRT-PCR for reporter RNA. (D) Outline of TDP43 autoregulation assay. Excess TDP43 binds to the TBR, triggering splicing, destabilization, and reduced mCherry fluorescence. (E) Primary rodent neurons were transfected with WT or mutant reporters, together with EGFP or TDP43-EGFP. After 7d, mCherry fluorescence was assessed by fluorescence microscopy. Scale bar, 20 μm. (F) Normalized RFP (mCherry) intensity at day 7. mCherry-TBR+GFP n=160, mCherry-TBR+TDP43(WT)-GFP n=58, mCherry-mTBR+GFP n=105, mCherry-mTBR+TDP43(WT)-GFP n=44. Data in D plotted as mean ± SD, color coded by biological replicate. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; one-way ANOVA with Tukey’s test.
To probe the importance of this m6A modification for TDP43 binding and function, we utilized a minigene reporter in which TARDBP exon 6 and a portion of the 3’UTR containing the TBR is fused to mCherry (Fig. 3B; mCherry-TBR)7,39,40. Using site-directed mutagenesis, we changed the methylated adenosine to a guanosine, thereby blocking methylation of the mutated reporter (mCherry-mTBR). Both reporters were then expressed in HaloTag-TDP43 HEK293T cells or HEK293T cells overexpressing HaloTag. Following HaloTag immunoaffinity purification, bound RNA was separated and assessed by qRT-PCR (Fig. 3C). HaloTag-TDP43 efficiently pulled down the mCherry-TBR reporter but not the m6A-deficient mCherry-mTBR reporter despite no change in reporter input levels (Fig. S3A), demonstrating that this m6A site is required for TDP43 recognition.
Two possibilities may account for the apparent effect of m6A on TDP43 binding: a direct influence on binding, resulting from an increase in affinity of TDP43 for m6A vs. unmodified RNA; or an indirect effect arising from enhanced accessibility of the TDP43 binding motif in m6A-modified RNA. Several observations argue for the latter (indirect) possibility: First, TDP43 typically recognizes UG-rich motifs rather than the DRACH sequence associated with m6A36,37. Second, TDP43 exhibits canonical RRM motifs rather than the more m6A-specific YTH domains found in most direct m6A reader proteins41. Third, hnRNP-C19, a heterogeneous ribonucleoprotein that is structurally and functionally related to TDP43, is an indirect m6A binding protein. To examine these possibilities in more detail, we created short (14 nt) synthetic RNA probes corresponding to the TBR with or without methylation of the predicted m6A site and performed electromobility shift assays (EMSAs) with recombinant TDP43 (Fig. S3B, C). In contrast to results from RNA-immunoprecipitation studies (Fig. 3B), methylation of the TBR probe had little effect on TDP43 binding. In fact, we observed a slight (~2-fold) reduction in binding affinity for the m6A-modified probe in comparison to the unmodified probe. Using RNAstructure prediction software42, we noted no clear deviation in CLIP34nt secondary structure associated with methylation (Fig. S3D, E), arguing against a clear conformational change facilitating TDP43 binding. These studies suggest that m6A modifications indirectly affect TDP43 binding through an unknown mechanism.
One consequence of TDP43 binding to the TBR is downregulation of TDP43 expression—this autoregulatory feedback loop is crucial for maintaining TDP43 homeostasis36,43–46. The mCherry-TBR reporter includes the majority of exon 6 and the TARDBP 3’UTR, facilitating investigations of TDP43 autoregulation in response to engineered mutations and genetic modifiers7,39,40. Binding of TDP43 to mCherry-TBR reporter results in 3’UTR splicing and a reduction of mCherry fluorescence (Fig. 3D), likely due to a combination of nuclear retention of the spliced transcript and nonsense mediated mRNA decay (NMD)45. Consistent with this, TDP43-EGFP overexpression reduces mCherry fluorescence in primary neurons transfected with the mCherry-TBR reporter (Fig. 3E, F). In contrast, mCherry fluorescence intensity of the m6A-deficient mCherry-mTBR reporter was unaffected by TDP43-EGFP expression (Fig. 3F). Thus, not only is the TBR m6A site required for recognition of the mCherry-TBR reporter, but it also affects the efficiency of TDP43 autoregulation.
RNA hypermethylation in ALS spinal cord
Based on data in Figs. 1 and 2 suggesting that TDP43 binds methylated RNA, and the prominent effect of RNA methylation on TDP43 autoregulation (Fig. 3), we questioned whether RNA methylation may be disrupted in sporadic ALS (sALS), a disorder characterized by nuclear clearance and cytoplasmic accumulation of TDP432–4. To answer this question, we obtained fresh frozen spinal cord from 4 sALS patients and 3 age-matched controls (Table S1), isolated RNA from these samples, and quantitatively assessed methylation using an m6A array (Fig. 4A).
Figure 4: RNA hypermethylation in ALS patient spinal cord.
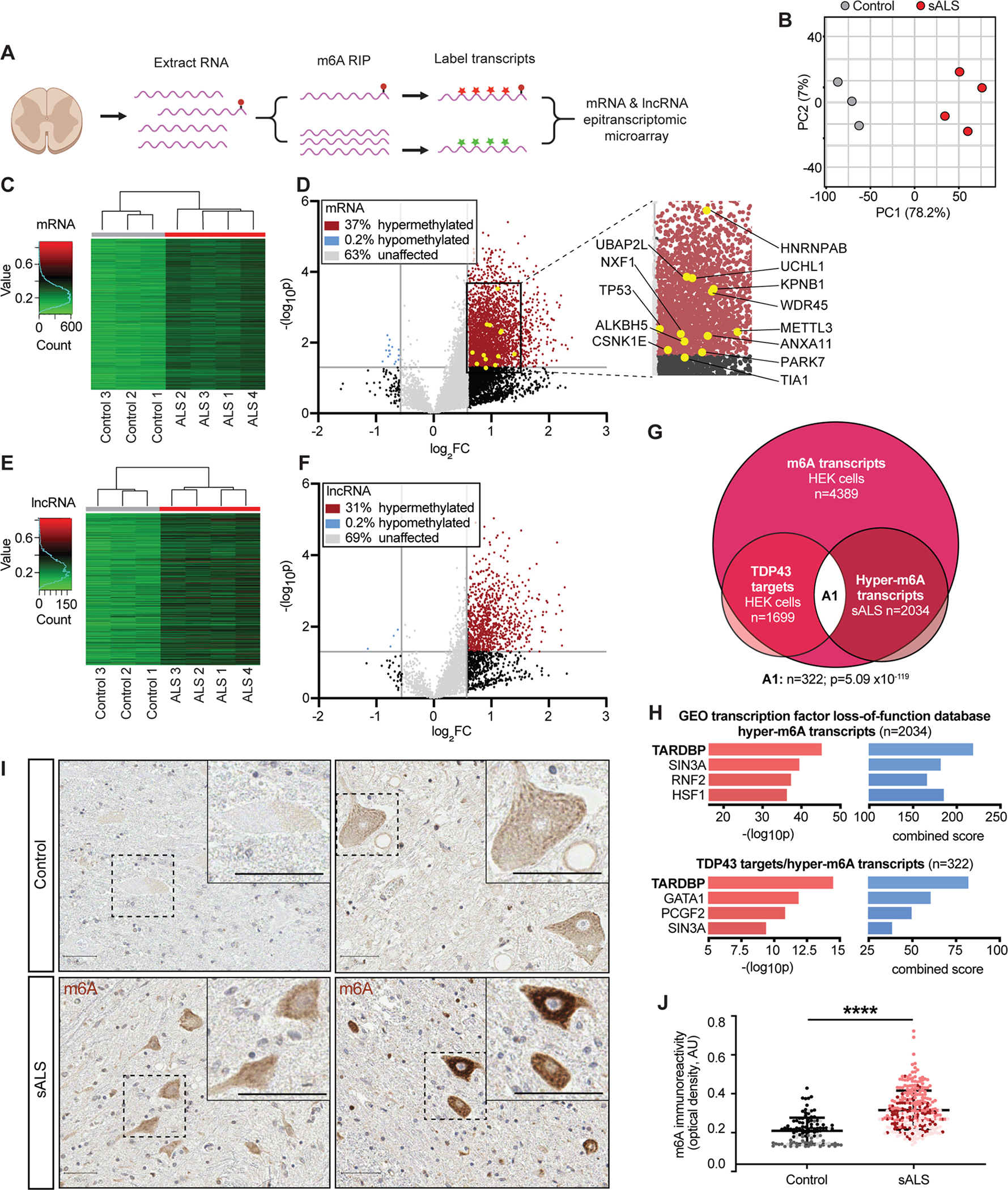
(A) Diagram of epitranscriptomic array used to assess RNA methylation in control and ALS spinal cord. (B) Principal component analysis (PCA) plot comparing methylation levels in control (grey) and ALS (red) samples. (C) Hierarchical clustering of methylation profiles from control and ALS mRNA samples. (D) Volcano plot depicting fold change in mRNA methylation in ALS compared to control spinal cord. (E) Hierarchical clustering of lncRNA methylation in control and ALS samples. (F) Volcano plot showing fold change in lncRNA methylation in ALS compared to control spinal cord. In D and F, grey horizontal vertical lines represent p=0.05 and fold change (FC)=2. (G) Euler diagram demonstrating overlap (n=322, p=5.09×10−119, hypergeometric test) among TDP43 substrates and methylated mRNA identified in HEK293T cells, in additional to hypermethylated mRNA determined via m6A array in sALS spinal cord (comparisons were limited to the subset of transcripts expressed in both HEK293T cells and human spinal cord, nTPM>2). (H) Bar plots showing enrichment for TDP43-regulated genes among the 2034 transcripts hypermethylated in sALS spinal cord, and the 322 TDP43 targets hypermethylated in sALS (A1 in G). Combined score=(log10p*Z-score). (I) Immunohistochemistry staining for m6A in control and sALS spinal cord. Scale bars, 50 μm. (J) m6A antibody reactivity in spinal neurons from control (n=110 neurons) and sALS (n=277 neurons) sections. Plot shows mean ± SD, color coded by patient. ****p<0.0001 via Mann-Whitney test.
Principal component analysis (PCA; Fig. 4B) and hierarchical clustering (Fig. 4C) demonstrated distinct patterns of mRNA methylation in sALS vs. control sections. These studies also suggested widespread mRNA hypermethylation in sALS samples, with 37% of assayed m6A sites displaying increased methylation in sALS (Fig. 4D), whereas <1% of transcripts were hypomethylated. We detected a similar pattern of hypermethylation for lncRNA (Fig. 4E, F), indicating a broad phenomenon not limited to protein-coding mRNAs.
To determine which of the hypermethylated transcripts in ALS are m6A-modified TDP43 substrates, we examined the overlap between the DART-seq results from HEK293T cells (Fig. 2), and hypermethylated transcripts from sALS spinal cord (Fig. 4G). Within the 2184 hypermethylated transcripts detected in both HEK293T cells and spinal cord, 2034 (93%) were also identified as m6A-modified RNAs by DART-seq, indicating excellent agreement between the two approaches (p~0, hypergeometric test). These transcripts were enriched for protein kinases and RNA binding proteins, several of which are also associated with ALS (CSNK1E, TIA1, hnRNPAB, ANXA11), other neurodegenerative diseases (PARK7, WDR45), basic processes such as stress granule formation (UBAP2L), and nucleocytoplasmic transport (NXF1, KPNB1). By cross-referencing these transcripts with m6A-modified RNAs pulled-down by HaloTag-TDP43 (Fig. 2), we identified 322 hypermethylated TDP43-target RNAs (Fig. 4G)—these transcripts were likewise enriched in kinases and RNA binding proteins, and included several factors linked with ALS pathogenesis (TP53, UCHL1) or RNA methylation itself (METTL3, HNRNPC). Similar to observations from HEK293T cells (Fig. S2), gene ontology and PPI analysis of hypermethylated TDP43 substrates indicated an enrichment for proteins involved apoptosis, RNA transport and splicing, and VEGF signaling (Fig. S4A). Remarkably, the 322 hypermethylated TDP43 targets, as well as the larger set of 2034 hypermethylated transcripts in sALS spinal cord, displayed strong enrichment for RNAs whose expression is regulated by TDP43 (Fig. 4H). Thus, a broad range of transcripts are hypermethylated in sALS spinal cord, many of which are functionally regulated by TDP43 and overlap with m6A-modified TDP43 target RNAs highlighted by DART-seq.
To validate these results, and to examine the distribution of RNA hypermethylation in sALS, we immunostained control and sALS spinal cord using antibodies against m6A (Fig. 4I). Moderate cytoplasmic staining for m6A was detected in large neurons located within the anterior horn of control spinal cord. In comparison, we observed pronounced and often punctate m6A staining in anterior horn neurons from sALS sections, with an approximate 1.5-fold increase in m6A staining intensity for sALS spinal neurons compared to controls (Fig. 4J). Supporting the ability of TDP43 to recognize m6A-modified RNA, we also noted distinct overlap between m6A and cytoplasmic TDP43 inclusions using multiplexed immunohistochemistry (Fig. S4B–D). These data provide independent verification of results from DART-seq (Fig. 2) and the epitranscriptomic array (Fig. 4C–F), emphasizing the association between TDP43 and hypermethylated RNA in sALS spinal cord, while further suggesting that m6A-modified RNA accumulates predominantly within spinal neurons harboring TDP43 pathology in sALS.
YTHDF2 knockout mitigates TDP43-related neurotoxicity
Considering TDP43’s ability to recognize m6A-modified RNA, and the substantial RNA hypermethylation noted in sALS spinal cord, we questioned whether RNA methylation is relevant for TDP43-mediated neurotoxicity. We therefore designed a platform that would enable us to readily screen m6A-related factors using CRISPR/Cas9 in a TDP43-based neuron model of ALS/FTD7,39,47,48. As proof of principle, single-guide (sg)RNAs targeting the neuronal transcription factor NeuN or a control (lacZ) were cloned into a vector that also encodes enhanced green fluorescent protein (EGFP) and Cas9 nuclease. Rodent primary mixed cortical neurons were transfected with this vector, and cells were fixed and immunostained for NeuN after 5d. Confirming the utility of this platform for single-cell gene knockouts, we observed a marked reduction in NeuN immunoreactivity only in transfected cells marked by EGFP fluorescence, and only in neurons that received sgRNAs against NeuN (Fig. 5A, B).
Figure 5: A single-cell CRISPR-based platform emphasizes the contribution of m6A factors to TDP43-dependent neurotoxicity.
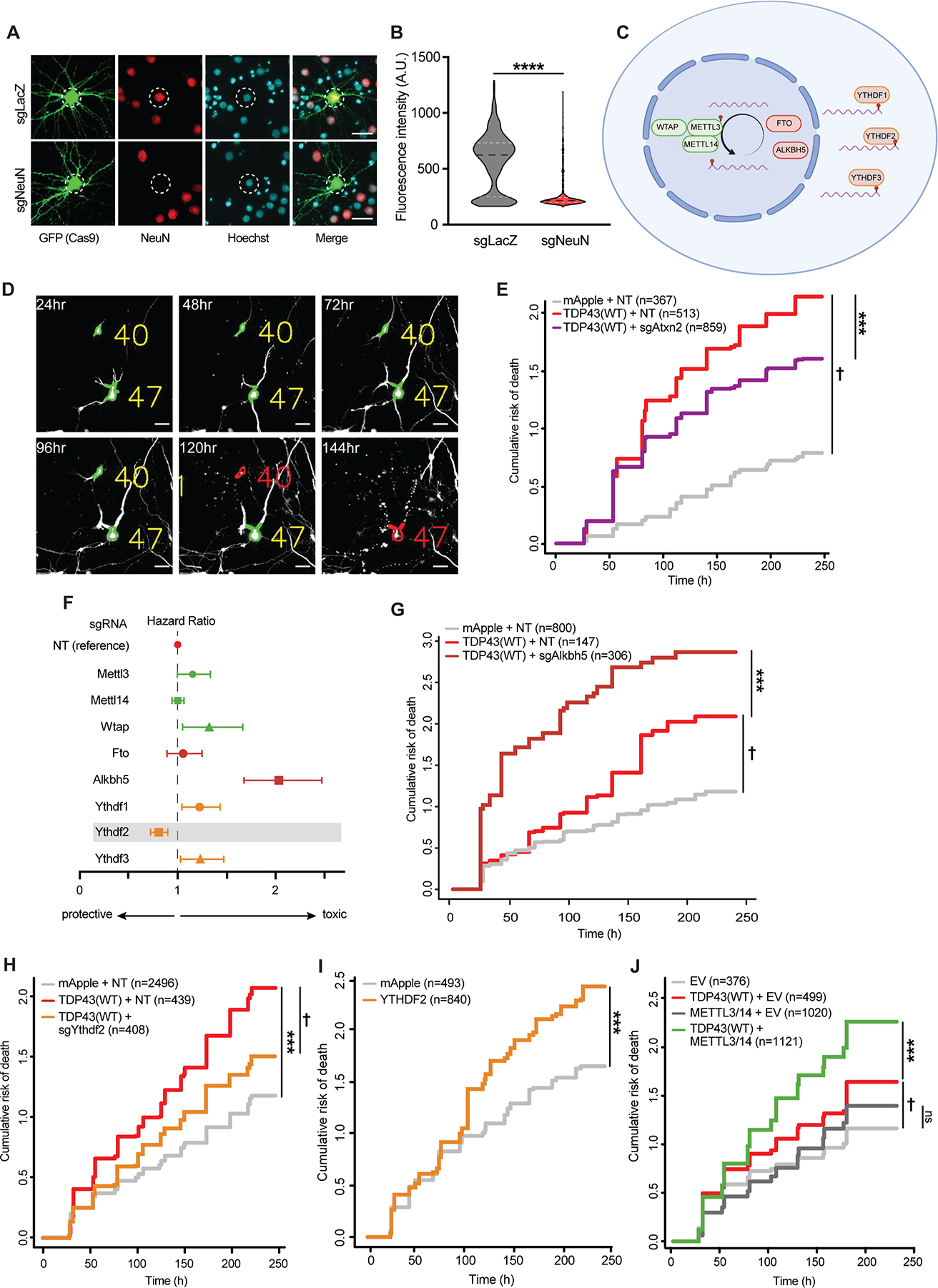
(A) Rodent primary neurons were transfected with plasmids encoding Cas9-2A-EGFP and sgRNA targeting NeuN or negative control (LacZ). 5d after transfection, neurons were fixed and immunostained for NeuN (red). White dashed circles indicate nucleus, determined via Hoechst staining (blue). (B) NeuN antibody reactivity in EGFP(+) neurons expressing sgLacZ (n=565) or sgNeuN (n=654), ****p<0.0001 by Mann-Whitney. (C) Schematic depicting m6A writers (green), erasers (red), and readers (orange) targeted by CRISPR/Cas9. (D) Primary neurons expressing EGFP and TDP43-mApple were imaged at 24h intervals by fluorescence microscopy, and their time of death determined by automated survival analysis. Individual neurons (yellow) are tracked until their time of death (red). Scale bar, 20μm. (E) Expression of sgRNA targeting Atxn2 (sgAtxn2) mitigates TDP43-related toxicity in primary neurons. NT, non-targeting. †p<2.0×10−16, hazard ratio (HR)=3.45; ***p=5.81×10−4, HR=0.80. (F) Forest plot depicting HR for TDP43-overexpressing neurons upon knockdown of m6A writers (green), erasers (dark red), and readers (orange), in comparison to NT control. Dashed line indicates the survival of neurons expressing TDP43-mApple+NT sgRNA. Values >1 indicate increased toxicity, whereas values <1 denote relative protection. Error bars, 95% CI. (G) Alkbh5 knockout significantly increases TDP43 associated toxicity. †p=3.11×10−5, HR=1.59; ***p=2.65×10−11, HR=2.03. (H) Ythdf2 knockout extends survival in TDP43-expressing neurons. ***p<2.0×10−16, HR=1.69; †p=6.2×10−6, HR=0.71. (I) YTHDF2 overexpression is toxic to neurons. ***p=3.07×10−5, HR=1.30. (J) METTL3/14 overexpression enhances TDP43-dependent toxicity in neurons. †p=5.53×10−4, HR=1.32; ***p =4.16×10−6, HR=1.31. p values in E, G-J determined via Cox proportional hazards analysis, with a minimum 3 of biological replicates.
We then designed a small-scale screen targeting the major m6A writers (METTL3, METTL14, WTAP), erasers (FTO, ALKBH5), and readers (YTHDF1, YTHDF2, YTHDF3) (Fig. 5C) in a neuronal model of TDP43-related disease7,39,47,48. sgRNAs targeting each factor were expressed in rodent primary mixed cortical neurons along with the red fluorescent protein mApple or TDP43 fused with mApple (TDP43-mApple; Fig. 5D). Overexpression of TDP43 recapitulates key aspects of ALS/FTD pathophysiology in neurons, including progressive neurodegeneration accompanied by TDP43 mislocalization and aggregation49. To track TDP43-dependent neuron loss, we employed automated longitudinal microscopy50,51. Here, hundreds of transfected neurons are simultaneously imaged at regular 24h intervals. Image segmentation algorithms detect neurons based on their unique morphology and determine their time of death through characteristic changes including soma rounding, neurite retraction and fragmentation, and cellular blebbing (Fig. 5D).
Transient transfection in this manner results in a wide distribution of expression levels, ranging from 1–7-fold endogenous TDP43 (Fig. S5A). As in previous studies, we observed dose-dependent TDP43-mediated toxicity in primary neurons, with the greatest risk of death observed in neurons with the highest TDP43 expression (Fig. S5B, C). Therefore, to maximize our ability to identify genetic modifiers and avoid avoiding supraphysiological effects, we focused on neurons within the lowest TDP43 expression (<2-fold endogenous levels).
In cells expressing non-targeting (NT) sgRNA, TDP43-mApple overexpression resulted in a >3-fold increase in the risk of death compared to neurons transfected with mApple alone (Fig. 5E). As a positive control, we cotransfected neurons with sgRNAs targeting Atxn2, one of the strongest genetic modifiers of TDP43-dependent toxicity52–54. Co-expression of Atxn2 sgRNA significantly suppressed TDP43-mediated toxicity (Fig. 5E and Fig. S5D), validating the use of this system for identifying disease modifiers. We then assessed whether knockout of the m6A writers, erasers or readers listed above are capable of modulating neuron loss due to TDP43 overexpression (Fig. 5F). In control neurons expressing mApple alone, we observed baseline increases in the risk of death upon knockdown of several factors (Fig. S5E), suggesting that many m6A-related components are essential. Among these factors, knockout of Wtap, Alkbh5, Ythdf1, and Ythdf3 significantly enhanced TDP43-related toxicity (Fig. 5G and Fig. S5F). Only Ythdf2 knockout reduced TDP43-mediated toxicity (Fig. 5H), suggesting that TDP43 may act in concert with YTHDF2 to elicit neurodegeneration.
In a complementary set of experiments, we overexpressed a subset of factors in neurons and followed their survival by automated microscopy. Analogous to TDP43, overexpression of YTHDF2 resulted in significant toxicity compared to the negative control (Fig. 5I). Conversely, combined overexpression of METTL3 and METTL14 had little effect on their own (Fig. 5J). To determine if METTL3/METTL14 might act synergistically with TDP43, we expressed TDP43-mApple at low concentrations, resulting in more subtle toxicity than in previous experiments. METTL3/METTL14 overexpression significantly enhanced TDP43-related toxicity in this paradigm (Fig. 5J). Considering the ability of TDP43 to bind m6A-modified RNA, and the increase in RNA methylation stimulated by METTL3/METTL14 overexpression55, these data imply that RNA hypermethylation facilitates TDP43-related neurodegeneration in ALS/FTD models.
YTHDF2 in sALS spinal cord and human iPSC-derived neurons
We detected RNA hypermethylation in sALS spinal cord samples by m6A array (Fig. 4) and found that genetic ablation of the m6A reader YTHDF2 reduced TDP43-dependent toxicity in a neuronal model of disease (Fig. 5H). Furthermore, YTHDF2 overexpression was sufficient to elicit toxicity in rodent primary neurons (Fig. 5I). To determine if YTHDF2, like TDP43, is mislocalized and/or accumulates in ALS, we immunostained for YTHDF2 in control and ALS post-mortem spinal cord (Fig. 6A, B) and frontal cortex (Fig. S6A, B). In controls, YTHDF2 displayed the expected uniform, cytoplasmic distribution within neurons and other cell types. Not only was YTHDF2 staining significantly more intense within sALS spinal neurons, but we also noted punctate accumulations of YTHDF2 within many of these cells (Fig. 6A, B) that overlapped with TDP43 pathology (Fig. S6C). Strong YTHDF2 staining was also noted within layer IV-V neurons from ALS frontal cortex (Fig. S6A), but puncta were not as evident in these cells as in spinal motor neurons (Fig. S6C). These results show that YTHDF2 accumulates in association with TDP43 pathology and RNA hypermethylation in sALS spinal neurons, potentially contributing to TDP43-mediated toxicity and disease pathogenesis.
Figure 6: YTHDF2 reduction extends neuronal survival in human neuron disease models.
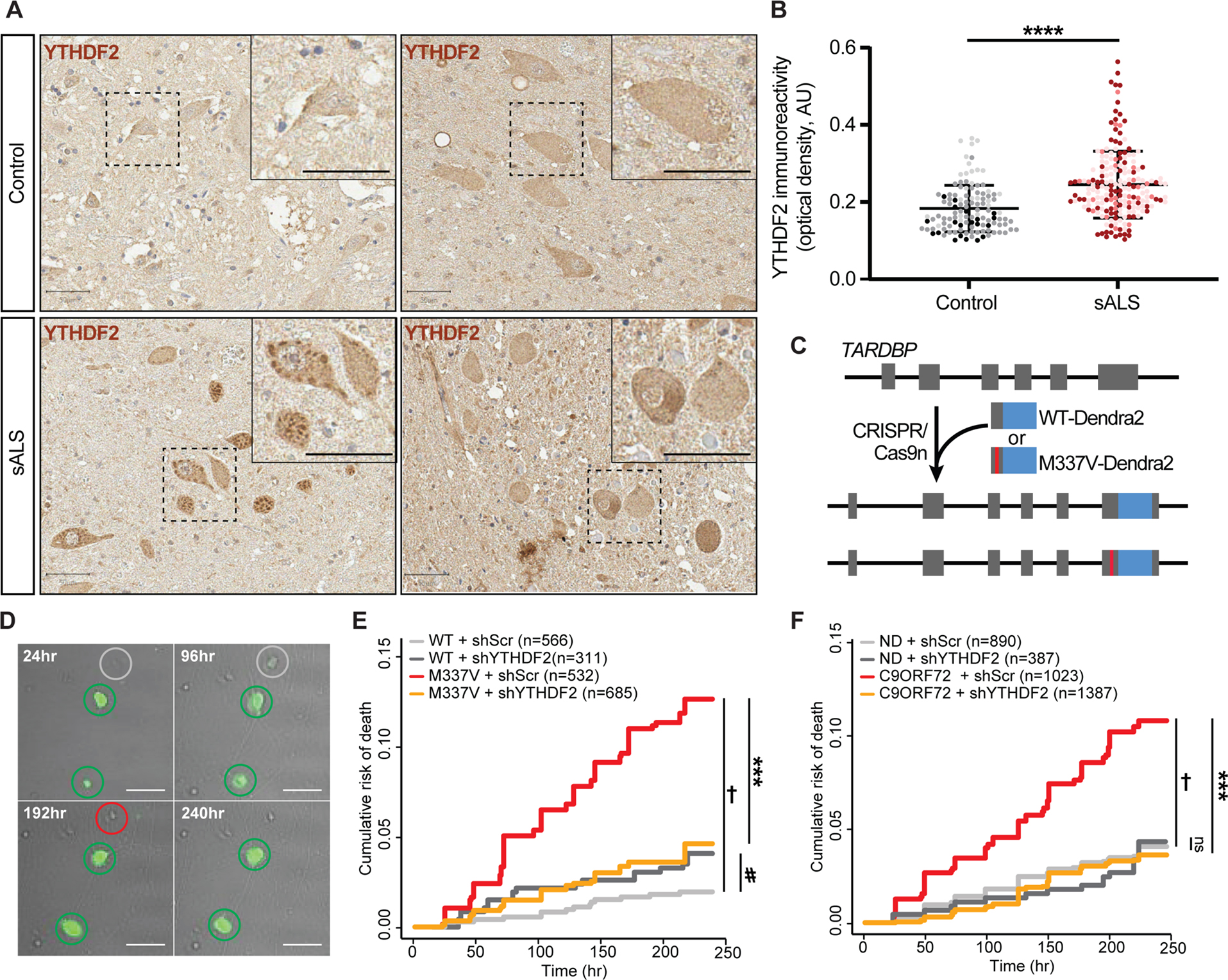
(A) YTHDF2 immunostaining in control and sALS spinal cord. Scale bar, 50 μm. (B) YTHDF2 immunoreactivity in spinal neurons from control (n=117 neurons) and sALS (n=193 neurons) samples. Plot shows mean ± SD, color coded by sample. ****p<0.0001 via Mann-Whitney test. (C) Strategy used to create isogenic iPSCs expressing native TDP43(WT)-Dendra2 or TDP43(M337V)-Dendra2. (D) Representative images of untransduced (grey) and transduced (green) iNeurons expressing shRNA against YTHDF2 (shYTHDF2). Time of death (red circles) for each cell is used to determine cumulative risk of death, plotted in (E) and (F). Scale bar, 20μm. YTHDF2 knockdown significantly extended the survival of TDP43(M337V)-Dendra2 iNeurons (E; †p=8.42×10−12, HR=6.25; ***p=4.82×10−9, HR=0.32; #p=0.08, HR=1.84) as well as mutant C9ORF72 iNeurons (F, †p=1.42×10−11, HR=2.85; ***p=1.42×10−16, HR=0.32). ns, not significant. Values in (E, F) calculated by Cox proportional hazards analysis, with a minimum 3 biological replicates.
The overabundance of YTHDF2 in sALS spinal cord, together with data from Fig. 5 suggesting neuroprotection upon Ythdf2 knockout in primary neurons, imply that YTHDF2 could be a therapeutic target for sALS. To pursue this further, we examined the effect of YTHDF2 knockdown in human neuron disease models. The ALS-associated TDP43(M337V) mutation was introduced into the native TARDBP locus of control human induced pluripotent stem cells (iPSCs) by CRISPR/Cas956. Simultaneously, endogenous TDP43 was labeled at the C-terminus with Dendra2, facilitating identification and selection of clonal cell populations harboring the M337V mutation. We also generated isogenic WT iPSCs in which native TDP43 was fused to Dendra2 without introduction of a pathogenic mutation (Fig. 6C). iPSCs were then differentiated into forebrain-like neurons (iNeurons) through the induced expression of master transcription factors Ngn1-2, as described previously40,56,57. Neurons were tracked by longitudinal microscopy over a 10d period, and their survival assessed by semi-automated tracking software developed specifically for these purposes (Fig. 6D). Upon neurotrophic factor withdrawal, we detected a significant increase in the risk of death for TDP43(M337V) iNeurons in comparison to controls (Fig. 6E). As in our primary neuron disease model, YTHDF2 knockdown via lentiviral shRNA delivery substantially extended the survival of TDP43(M337V) iNeurons without adversely affecting WT iNeurons (Fig. 6E).
We also evaluated the effect of YTHDF2 knockdown in 3 separate lines of human iNeurons carrying the C9ORF72 hexanucleotide expansion, the most prevalent cause of familial ALS and FTD in Northern Europe and North America58–60. C9ORF72 mutant iNeurons displayed a ~3-fold elevation in the risk of death upon neurotrophic factor withdrawal compared to control neurons (Fig. 6F and Fig. S6D). As in TDP43(M337V) iNeurons, shRNA-mediated knockdown of YTHDF2 significantly prolonged the survival of C9ORF72 mutant iNeurons but had no effect on non-disease (ND) iNeurons (Fig. 6F and Fig. S6D). Together with our data from rodent primary neurons (Fig. 5), these findings emphasize the neuroprotective potential of YTHDF2 knockdown in ALS/FTD disease models featuring TDP43 pathology.
Discussion
Here, we show not only that TDP43 recognizes m6A-modified RNA, but also that the majority of TDP43 substrates exhibit m6A marks. We also demonstrated a spatial correlation between 3’UTR m6A modifications and TDP43 recognition motifs, and found that methylation strongly influences the binding of TDP43 to its RNA targets as well as TDP43 autoregulation. In ALS, a disease characterized by cytoplasmic TDP43 mislocalization and aggregation, we also observed striking RNA hypermethylation compared to disease controls. Consistent with a primary role for RNA hypermethylation in ALS pathogenesis, knockout or knockdown of the m6A reader YTHDF2 mitigated toxicity in primary rodent and human iPSC-derived neuron models of ALS and FTD, while overexpression of the m6A writers METTL3 and METTL14 exacerbated TDP43-mediated neuron loss. Together, these findings underscore the importance of m6A modifications for RNA binding by TDP43 and emphasize the potential contribution of TDP43’s actions on m6A-modified RNA to the development of ALS and FTD.
Our study builds on several lines of evidence hinting at a connection between TDP43 and RNA methylation. m6A writers (METTL3, METTL14, WTAP) and erasers (FTO, ALKBH5) primarily localize to nuclear speckles61,62, membraneless organelles that facilitate peri-transcriptional RNA processing and splicing. TDP43 is also concentrated within speckles63, and interacts with several m6A writer and reader proteins, including METTL3, YTHDF1, YTHDF2 and hnRNPC15,64,65. Furthermore, TDP43 was among a series of proteins that exhibited preferential binding to m6A-modified bait RNAs66. Supporting this, we found that both overexpressed and endogenously labeled TDP43 bind m6A-modified RNA. Combining affinity purification of native HaloTag-TDP43 with antibody-free detection of m6A through DART-seq, we clarified the location and number of m6A sites within TDP43 substrate RNAs. Importantly, DART-seq is not limited by the specificity or sensitivity of m6A antibodies, instead relying on APOBEC1-YTH induced C-T transitions to indicate m6A sites. In contrast to the previously noted enrichment of m6A marks within the proximal 3’UTR11,12,25,27, TDP43 targets displayed a broad distribution of m6A modifications across the CDS and 3’UTR. It is unclear whether this pattern is unique to TDP43 substrate RNAs, or whether a similar distribution would be observed in targets of related RNA binding proteins. We observed few m6A sites within the introns of TDP43 target RNAs, consistent with the relative lack of intronic m6A modifications noted in previous studies67–69. Given the relative concentration of TDP43 binding sites within introns and 3’UTRs36,37, and our finding that m6A modifications cluster within the CDS and 3’UTR of TDP43 target RNAs, it stands to reason that the overlap between TDP43 binding sites and m6A modifications is greatest within the 3’UTR of these transcripts. Our studies indicate that the expression and/or stability of this subset of TDP43 target RNAs is strongly influenced by conditions that enhance or reduce m6A modifications.
Previous studies as well as our DART-seq experiments identified key m6A modifications located within the TBR, a region of the TARBDP transcript that is essential for recognition and autoregulation by TDP43 itself38,43. In keeping with the association between m6A modifications and UG-rich domains recognized by TDP43 (Fig. 1), these m6A marks are adjacent to TDP43 binding motifs within the TBR. Mutagenesis of the methylated A within a reporter containing the TBR led to a reduction in TDP43 binding and ineffective autoregulation, suggesting that m6A modifications are crucial for TDP43 recognition and events downstream of binding. Even so, in vitro electromobility shift assays involving recombinant TDP43 and a short (14 nt) m6A-modified probe failed to show methylation-dependent increases in TDP43 binding, implying that m6A modifications indirectly enhance RNA recognition by TDP43. Similar relationships have been observed for hnRNP-C19 and IGF2BP370, suggesting that indirect changes in motif accessibility upon methylation can promote recognition by TDP43 and other RNA binding proteins.
In prior work, we observed extensive RNA destabilization in human iPSCs overexpressing TDP435. Transcripts encoding components of the ribosome and oxidative phosphorylation pathways were most heavily represented among TDP43-destabilized RNAs, a pattern that was mirrored in iPSCs carrying ALS/FTD-associated C9ORF72 mutations. Notably, these same families of transcripts accumulate upon targeted deletion of the m6A writer METTL1410, indicating that they are regulated by m6A modification. Given these observations, together with the degree of RNA hypermethylation we observed in sALS spinal cord, the ability of TDP43 to recognize m6A-modified RNA, and the overlap between TDP43 pathology, methylated RNA and YTHDF2 in patient-derived material, we suspect that TDP43 mislocalization and accumulation may lead to RNA destabilization in ALS through an m6A-dependent mechanism. Although this hypothesis requires further investigation, as an example we explored the potential for m6A modifications to influence TDP43 autoregulation, a phenomenon that involves the nuclear retention and destabilization of TARDBP transcripts upon their recognition by TDP435,36,44–46. m6A marks located within the TBR influence TDP43 binding and are crucial for proper autoregulation of a reporter. In light of recent data highlighting the possible contribution of TDP43 autoregulation to ALS/FTD pathogenesis46,71, these observations draw attention to the potential importance of m6A modifications for physiological TDP43 function as well as its dysfunction in disease.
Among different tissue types, the central nervous system displays some of the highest baseline levels of m6A RNA, and these modifications are essential for proper neuronal development and maturation12,72–74. Total m6A levels within the nervous system rise with age, and previous evidence suggests that neurodegenerative diseases independent of ALS and FTD are likewise characterized by RNA hypermethylation75,76. We observed significant hypermethylation in end-stage tissue from ALS patients compared to age-matched controls; although we do not believe age is responsible for the observed RNA hypermethylation, other factors may be playing a part. For example, astrocytosis and microgliosis are common features of ALS as well as other neurodegenerative diseases, and it is possible that much of the observed RNA hypermethylation is a consequence of such neuroinflammation77.
Importantly, YTHDF2 overlapped with TDP43 pathology in sALS spinal cord, and was also the only m6A-related factor that emerged from our limited CRISPR-based screen for modulators of TDP43-mediated toxicity. This platform—which combines longitudinal fluorescence microscopy, automated survival analysis, and single-cell genetic knockouts—allowed us to rapidly interrogate most m6A readers, writers, and erasers for their effects on neuronal survival. In doing so, we found that knockout of the m6A reader Ythdf2 mitigated TDP43-related toxicity in rodent primary neurons, while YTHDF2 overexpression itself was lethal. Furthermore, YTHDF2 knockdown in human iNeurons carrying ALS/FTD-associated mutations in TARDBP and C9ORF72 prolonged cellular survival. YTHDF2 is one of the primary m6A reader proteins associated with RNA degradation. Through direct interactions with effectors such as CNOT178 (a component of the CCR4-NOT deadenylation complex), HRSP1279 (associated with ribonuclease P-dependent RNA cleavage), and UPF180 (a key factor involved in nonsense mediated RNA decay), YTHDF2 effectively facilitates the clearance of m6A-modified RNA substrates. Notably, UPF1 overexpression prevents neurodegeneration in several models ALS/FTD models, including those involving TDP4339, the related RNA binding protein FUS81,82, and C9ORF72 mutations83–86. These observations, together with the apparent accumulation of hypermethylated RNA and YTHDF2 in ALS spinal cord, indicate that YTHDF2 may be a novel therapeutic target in ALS and FTD.
Limitations of the study.
Data on m6A-modified TDP43 targets were obtained from cultured HEK293T cells, but it is possible that TDP43 substrates and their methylation status may be distinct in motor neurons and other vulnerable cell types in ALS and FTD. Furthermore, RNA methylation patterns in ALS post-mortem tissue reflect end-stage disease; the extent and distribution of RNA methylation may be very different in individuals with mild or moderate disease. Lastly, epitranscriptomic arrays allow for quantitative comparison of m6A-modified loci, but are limited to m6A sites included within the arrays. Future studies may therefore focus on cell type-specific TDP43 targets and methylation patterns in staged ALS/FTD tissue and model systems, identified via unbiased approaches.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sami Barmada (sbarmada@umich.edu).
Materials availability
Plasmids and other unique reagents generated in this study are available by contacting the lead author, Sami Barmada (sbarmada@umich.edu).
Data and code availability
DART-seq data have been deposited at GEO (SUB12370770) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Epitranscriptomic array data are included in supplemental data files 1 and 2 of the publication.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-TDP43 | Proteintech | Cat# 10782-2-AP; RRID: AB_2892214 |
| Rabbit monoclonal anti-NeuN | Cell Signaling Technologies | Cat# 24307T RRID: AB_2651140 |
| Rabbit polyclonal anti-YTHDF2 | Proteintech | Cat# 24744-1-AP; RRID: AB_2687435 |
| Rabbit polyclonal anti-m6A | Millipore Sigma | Cat# ABE572; RRID: AB_2892213 |
| Rabbit monoclonal anti-m6A | Cell Signaling Technologies | Cat# 56593S; RRID: AB_2799515 |
| Rabbit polyclonal anti-m6A | Millipore Sigma | Cat# ABE572-I-100UG; RRID: AB_2892214 |
| Goat polyclonal anti-rabbit Alexa Fluor 488 | ThermoFisher | Cat# A-11034; RRID: AB_2576217 |
| Goat polyclonal anti-rabbit HRP | Jackson ImmunoResearch Labs Inc. | Cat# 111-035-003; RRID: AB_2313567 |
| Bacterial and virus strains | ||
| GIPZ Non-silencing Lentiviral shRNA Control | Horizon Discovery | #RHS4346 |
| GIPZ Lentiviral Human YTHDF2 shRNA | Horizon Discovery | #RHS4430-200182983 |
| Biological samples | ||
| Human post-mortem sample; Control 1 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 66 Sex: M |
| Human post-mortem sample; Control 2 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 85 Sex: M |
| Human post-mortem sample; Control 3 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 68 Sex: M |
| Human post-mortem sample; sALS 1 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 65 Sex: M |
| Human post-mortem sample; sALS 2 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 64 Sex: M |
| Human post-mortem sample; sALS 3 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 65 Sex: M |
| Human post-mortem sample; sALS 4 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 81 Sex: M |
| Human post-mortem sample; Control 4 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 76 Sex: F |
| Human post-mortem sample; Control 5 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 88 Sex: M |
| Human post-mortem sample; Control 6 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 56 Sex: M |
| Human post-mortem sample; ALS 1 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 66 Sex: F |
| Human post-mortem sample; ALS 2 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 64 Sex: M |
| Human post-mortem sample; ALS 3 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 68 Sex: M |
| Human post-mortem sample; Control 7 | University of Maryland Brain and Tissue Bank https://www.medschool.umaryland.edu/btbank/ | Experiment: m6A array Age: 48 Sex: M |
| Human post-mortem sample; ALS 4 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 76 Sex: M |
| Human post-mortem sample; ALS 5 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 60 Sex: F |
| Human post-mortem sample; ALS 6 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 99 Sex: M |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant TDP43(WT) | Flores et al., 20197 | N/A |
| Deposited data | ||
| DART-seq data from HEK293T cells | This manuscript | https://www.ncbi.nlm.nih.gov/geo/ (SUB12370770) |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 |
| Primary rat neurons | Charles River Laboratories | Long-Evans rat (Crl:LE) |
| Human iPSCs | University of Michigan ALS Repository | 1021, 0883, 0312 |
| Human iPSCs | Cedars Sinai iPSC Repository | Cs29i, Cs29 |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene TARDBP WT; Tag TDP43-Dendra2-Carboxyl term | Weskamp et al., 202041 | Line 1021 Identifier: WT |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene TARDBP Mutation Isogenic M337V; Tag TDP43-Dendra2 Carboxyl term | Weskamp et al., 202041 Sidibé et al., 202157 |
Line 1021 Identifier: M337V |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene C9ORF72 Mutation | Weskamp et al., 202041 | Line 312 Identifier: C9ORF72 (#1) |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene C9ORF72 Mutation | Weskamp et al., 202041 | Line 883 Identifier: C9ORF72 (#2) |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene C9ORF72 Mutation | Weskamp et al., 202041 | Line Cs29i Identifier: C9ORF72 (#3) |
| Human iPSCs (CLYBL:TO-hNgn1/21); Gene C9ORF72 Mutation Isogenic corrected | Weskamp et al., 202041 Ho et al., 2021104 |
Line Cs29i Identifier: C9ORF72 ISO (#3) |
| Oligonucleotides | ||
| See Table S4 for complete list | ||
| Recombinant DNA | ||
| See Table S2 for complete list | ||
| Software and algorithms | ||
| RNAstructure software | Kierzek et al., 202243 | https://rna.urmc.rochester.edu/RNAstructure.html |
| Automated imaging and survival analysis | Malik et al., 2018101 | DOI: 10.5281/zenodo.7438867 DOI: 10.5281/zenodo.7438874 |
| Biorender | N/A | https://Biorender.com |
| Other | ||
| HaloTag ligand (JF646 dye) | Janelia Farms | https://www.janelia.org |
All original code has been deposited in Github and Zenodo, and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
All schematics created with Biorender.com.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293T cell culture
Human embryonic kidney (HEK) 293T cells were cultured in DMEM (GIBCO), 10% FBS, 100 units/mL Penicillin/Streptomycin at 37°C in 5% CO2. HEK293T cells are originally female in origin, are easily transfected, and have been transformed with SV40 T-antigen.
CRISPR/Cas9 integration of HaloTag into TARDBP locus in HEK293T cells
Oligos complementary to the target region (Table S4) were annealed, digested, and ligated into the BbsI site in the pX335 vector (Addgene, #42335) according to the protocol available from Addgene. 1.25 μg of each vector was transfected into HEK293T cells using Lipofectamine 2000 (ThermoFisher, #11668019) together with 2.5 μg of a plasmid encoding the HaloTag open reading frame flanked by 400 bp of sequence homologous to regions immediately upstream and downstream of the TARDBP start codon, according to the manufacturer’s instructions. Following transfection, cells were split at a low density, allowing transfected cells to establish individual colonies. Cells were screened for nuclear fluorescence after incubation with JF635 dye, as described previously40,87. Positive cells were carefully scraped/aspirated using a P200 pipet tip and transferred to a new dish. This process was repeated until 100% of cells displayed nuclear fluorescence after JF635 application, and correct integration of the HaloTag cassette into the TARDBP locus was confirmed by PCR and Sanger sequencing. HaloTag-TDP43 HEK293T cells were cultured on plates coated with 0.1% gelatin (Sigma, #G2500) in DMEM (GIBCO), 10% FBS, 100 units/mL Penicillin/Streptomycin at 37°C in 5% CO2.
iNeuron differentiation
Day 0: Induced pluripotent stem cells were washed in PBS and incubated in prewarmed accutase (Sigma, #A6964) at 37°C for 8 min. Four volumes of E8 media (ThermoFisher, #A1517001) were added to the plate, and the cells were collected and pelleted at 200×g for 5 min. The media was aspirated, and the pellet was resuspended in 1mL of fresh E8 media. Cells were counted using a hemocytometer, diluted, plated at a density of 20,000 cells/mL in E8 media with ROCK inhibitor and incubated at 37°C overnight. Day 1: Media was changed to N2 media (1x N2 Supplement (Gibco, #17502- 048), 1x NEAA Supplement (Gibco, #11140-050), 10 ng/mL BDNF (Peprotech, #450-02), 10 ng/mL NT3 (Peprotech, #450-03), 0.2 μg/mL laminin (Sigma, #L2020), 2 mg/mL doxycycline (Sigma, #D3447) in E8 media). Day 2: Media was changed to transition media ((1x N2 Supplement, 1x NEAA Supplement, 10 ng/mL BDNF, 10 ng/ml NT3, 0.2 μg/mL laminin, 2 mg/mL doxycycline in half E8 media, half DMEM F12 (Gibco, #11320-033)). Day 3: Media was changed into B27 media (1x B27 Supplement (Gibco, #17504-044), 1x Glutamax Supplement (Gibco, #35050-061), 10 ng/mL BDNF, 10 ng/mL NT3, 0.2 μg/mL laminin, and 1x Culture One (Gibco, #A33202-01) in Neurobasal-A (Gibco, #12349-015)). On day 6 cells were transduced with the appropriate virus (prepared by University of Michigan Vector Core, Table S2) and cells were sustained in the same culture medium for the remainder of the experiment. Day 14: Imaging began for survival experiments and iNeurons were monitored over the course of 10 days.
Ethics statement
All vertebrate animal work was approved by the Committee on the Use and Care of Animals (UCUCA) at the University of Michigan. All experiments were performed in accordance with UCUCA guidelines and designed to minimize animal use. Rats (Rattus norvegicus) were housed single in chambers equipped with environmental enrichment and cared for by veterinarians from the Unit for Laboratory Animal Medicine at the University of Michigan. All individuals were trained and approved in the care of long-term maintenance of rodents, in accordance with the NIH-supported Guide for the Care and Use of Laboratory Animals. All personnel handling the rats and administering euthanasia were properly trained in accordance with the University of Michigan Policy for Education and Training of Animal Care and Use Personnel. Euthanasia followed the recommendations of the Guidelines on Euthanasia of the American Veterinary Medical Association. Brains from individual pups in each litter were pooled to maximize cell counts prior to plating; as a result, primary cortical neurons used for all studies include an even mix of cells from both male and female pups.
METHOD DETAILS
Plasmids
pGW1-GFP, pGW1-TDP43(WT)-GFP, pGW1-mApple47,49, pGW1-Halo, pGW1-TDP43(WT)-Halo7,40, and pCAGGS-TDPBR-mCherry7,39,40 were created as previously described.
To generate pGW1-YTHDF2-Halo, the YTHDF2 ORF was PCR amplified from pcDNA-flag-YTHDF2 (Addgene, #52300). The resulting amplicon was digested with AgeI and XbaI and cloned into cut sites upstream of HaloTag insert in pGW1-Halo.
To create pgW1-YTHDF2-2A-GFP, the YTHDF2 ORF was PCR amplified from pcDNA-flag-YTHDF2 (Addgene, #52300). The resulting amplicon was digested with KpnI and SalI and cloned into cut sites upstream of the 2A in the pGW1-2A-GFP vector.
pCAGGS-TDPBR (mTBR)-mCherry, was created via site directed mutagenesis from pCAGGS-TDPBR-mCherry using the Pfu Ultra high-fidelity polymerase (Agilent Technologies, #600380) according to manufacturer’s protocols to change the A into a G in clip34nt sequence in the TDPBR.
HaloTag immunoprecipitation
HEK293T cells were transfected with pGW1-Halo, pGW1-TDP43-Halo, or pGW1-YTHDF2-Halo using Lipofectamine 2000 (ThermoFisher, #11668019) following the manufacturer’s protocol. Approximately forty-eight hours post-transfection, cell pellets were harvested and lysed using a syringe in 100 μL lysis buffer (50 mM Tris-HCl, 150 mm NaCl, 1% Triton-X100, 0.1% NaDeoxycholate) and incubated on ice for 30 min. Following lysis, tubes were centrifuged at 17000×g for 5 min and pellet discarded. 200 g for lysate was added to 25 μL of HaloTrap beads (Chromotek, #ota-10), prewashed with 500 μL of 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA centrifuged at 2500×g for 5 minutes. Lysate and bead mix was incubated rotating overnight at 4°C. Following incubation, the mixture was centrifuged at 2500×g for 5 min and supernatant discarded. HaloTrap beads were washed 3 times for 5 min at 2500×g in 500 μL of wash buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton-X100, 0.1% NaDeoxycholate, 0.05% IGEPAL). To elute RNA, Trizol (ThermoFisher, #15596026) was added directly to the beads and RNA was isolated using phenol-chloroform extraction.
m6A dot blot
For m6A dot blot, isolated RNA was boiled at 95°C for 3 minutes to denature RNA then immediately chilled on ice. RNA was added in 1 μL drops to a BrightStar nylon membrane (ThermoFisher, #AM10102) and crosslinked at 1200 μJ [x100] for 2 min twice. After crosslinking, membranes were stained with methylene blue (0.04% methylene blue in 0.5M sodium acetate pH 5.2) until circles were visible (approximately 5 min). Membranes were washed 3x with water quickly to remove background stain and imaged. Afterwards, membranes were washed several times with water to remove methylene blue staining, then blocked in 5% milk in 1x PBS + 0.02% Tween-20 at room temperature for 1 hour. Primary antibody (rabbit anti-m6A antibody 1:500, Cell Signaling #56593) was added to blocking solution, and membranes rocked overnight at 4°C. Blots were then washed in wash buffer (1x PBS + 0.02% Tween-20) 3x for 5 min and incubated at room temperature with blocking buffer containing goat anti-rabbit HRP secondary antibody (Jackson Immunoresearch labs, #111-035-003, 1:5000). Blots were rinsed in wash buffer 3x for 5 min, then incubated in Pierce ECL Western blotting substrate (ThermoFisher, #32106) for 1 min before exposure to CL-XPosure™ Film (Fisher, #PI34090). For endogenous Halo-TDP43 pulldowns, the procedure was followed as before starting from cell pellets of Halo-TDP43 HEK293T or unmodified HEK293T cells.
DART-seq
Halo-TDP43 HEK293T cells were transfected with pCMV-APOBEC1-YTH, pCMV-APOBEC1-YTHmut, or pGW1-Halo using Lipofectamine 2000 (ThermoFisher, #11668027) following the manufacturer’s protocol. Approximately 48 hours post-transfection, cells were washed with cold PBS (Invitrogen) then crosslinked with UV light (254 nm, 150 mJ/cm2). Following crosslinking, cells were harvested and lysed using a syringe in 100 μL lysis buffer supplemented RNAse inhibitor (Invitrogen, #N8080119) (50 mM Tris-HCl, 150 mM NaCl, 1% Triton-X100, 0.1% NaDeoxycholate) and incubated on ice for 30 min. 200 μg for lysate was added to 25 μL of HaloTrap beads, prewashed with 500 μL of equilibration buffer supplemented with RNAse inhibitor (10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA) centrifuged at 2500×g for 5 minutes. Lysate and bead mix was incubated rotating overnight at 4°C with Baseline Zero DNAse (Fisher Scientific, #NC1424104). Following incubation, the mixture was centrifuged at 2500×g for 5 min and supernatant discarded. HaloTrap beads were washed 3 times for 5 min at 2500×g in 500 μL of wash buffer supplemented with RNAse inhibitor (50 mM Tris-HCl, 150 mM NaCl, 1% Triton-X100, 0.1% NaDeoxycholate, 0.05% IGEPAL). To elute RNA, Trizol was added directly to the beads and RNA was isolated using phenol-chloroform extraction. Once extracted, RNA was submitted to Advanced Genomics Core at University of Michigan for RNA sequencing.
Next-generation sequencing
cDNA libraries were prepared from Trizol extracted, DNA-digested samples using the Illumina Stranded Total RNA Prep with Ribo-Zero Plus kit (Illumina, #20040525). Paired end sequencing was carried out on an Illumina NovaSeq (S4) 300 cycle sequencer at the University of Michigan Advanced Genomics Core. Samples were sequenced at an average depth of 15.6 million unique reads per sample.
m6A array
Human spinal cord samples were homogenized in Trizol and RNA was extracted using phenol-chloroform extraction for the m6A mRNA & lncRNA Epitranscriptomic microarray (8×60K, Arraystar, Rockville, MD, USA). 1–3 μg total RNA and m6A spike-in control mixture were added to 300 μL 1xIP buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% NP40, 40U/μL RNase Inhibitor) containing 2 μg anti-m6A rabbit polyclonal antibody (Synaptic Systems, #202003). The reaction was incubated with head-over-tail rotation at 4°C for 2 hours. 20 uL DynabeadsTM M-280 Sheep Anti-Rabbit IgG (Invitrogen, #11203D) suspension per sample was blocked with freshly prepared 0.5% BSA at 4°C for 2 hours, washed three times with 300 μL 1xIP buffer, and resuspended in the total RNA-antibody mixture prepared above. The RNA binding to the m6A-antibody beads was carried out with head-over-tail rotation at 4°C for 2 hours. The beads were then washed three times with 500 μL 1xIP buffer and twice with 500 μL Wash buffer (50 mM Tris-HCl, pH 7.4, 50 mM NaCl, 0.1% NP40, 40 U/μL RNase Inhibitor (Enzymatics, #Y9240L). The enriched RNA was eluted with 200 μL Elution buffer (10 mM Tris-HCl, pH7.4, 1 mM EDTA, 0.05% SDS, 40U Proteinase K) at 50°C for 1 hour. The RNA was extracted by acid phenol-chloroform and ethanol precipitated. The “IP” RNA and “Sup” RNAs were added with equal amount of calibration spike-in control RNA, separately amplified and labeled with Cy3 (for “Sup”) and Cy5 (for “IP”) using Arraystar Super RNA Labeling Kit (Arraystar, #AL-SE-005). The synthesized cRNAs were purified by RNeasy Mini Kit (QIAGEN, #74105). The concentration and specific activity (pmol dye/μg cRNA) were measured with NanoDrop ND-1000. 2.5 μg of Cy3 and Cy5 labeled cRNAs were mixed. The cRNA mixture was fragmented by adding 5 μL 10x Blocking Agent and 1 μL of 25x Fragmentation Buffer, heated at 60°C for 30 min, and combined with 25 μL 2x Hybridization buffer. 50 μL hybridization solution was dispensed into the gasket slide and assembled to the m6A-mRNA & lncRNA Epitranscriptomic Microarray slide. The slides were incubated at 65°C for 17 hours in an Agilent Hybridization Oven. The hybridized arrays were washed, fixed and scanned using an Agilent Scanner G2505C. Comparisons with DART-seq results (Fig. 4G) included a limited set of transcripts (2184 out of 5646) that were expressed in both human spinal cord and HEK293T cells [normalized transcripts per million (TPM)>2], based on datasets made available through the Human Protein Atlas.
Longitudinal microscopy and automated survival analysis
Cortices from embryonic day (E)19–20 Long-Evans rat embryos were dissected and disassociated, and primary neurons were plated at a density of 6×105 cells/ml in 96-well plates, as described previously7,39,46,84. For CRISPR candidate screen, on in vitro day (DIV) 4, neurons were transfected with 25 ng of plasmids containing sgRNAs for respective genes, 50 ng of pGW1-mApple or pGW1-TDP43-mApple, and 25 ng of pGW1-EGFP to mark cell bodies using Lipofectamine 2000 (Invitrogen #52887. Following transfection, cells were placed in Neurobasal Complete Media (Neurobasal (Gibco, #21103-049), 1x B27, 1x Glutamax, 100 units/mL Pen Strep (Gibco, #15140-122)) and incubated at 37°C in 5% CO2.
m6A CRISPR candidate screen
Oligos complementary to m6A pathway component genes were created using ChopChop88–90 (https://chopchop.cbu.uib.no/) then annealed, digested, and ligated into the BbsI site of pSpCas9(BB)-2A-GFP plasmid (Addgene, #48138) according to manufacturer’s protocol. 25 ng of plasmids containing sgRNAs for respective genes were transfected into rat primary neurons on DIV 4 using Lipofectamine 2000 (ThermoFisher, #11668027) along with 50 ng of TDP43(WT)-EGFP or mApple then survival was measured over the course of 10 days. Outcomes were calculated based on Cox proportional hazard values and related to expression of TDP43(WT)-EGFP to determine if knockout of target was beneficial or toxic.
Neurons were imaged as described previously7,39,47,48 using a Nikon Eclipse Ti inverted microscope with PerfectFocus3a 20X objective lens and either an Andor iXon3 897 EMCCD camera or Andor Zyla4.2 (+) sCMOS camera. A Lambda 421 multi-LED light source (Sutter) with 5 mm liquid light guide (Sutter) was used to illuminate samples, and custom scripts written in Beanshell for use in μManager controlled all stage movements, shutters, and filters. Custom ImageJ/Fiji macros and Python scripts were used to identify neurons and draw both cellular and nuclear regions of interest (ROIs) based upon size, morphology, and fluorescence intensity. Fluorescence intensity of labeled proteins was used to determine protein localization or abundance. Custom Python scripts were used to track ROIs over time, and cell death marked a set of criteria that include rounding of the soma, loss of fluorescence and degeneration of neuronal processes.
Immunocytochemistry
HEK293T cells were plated on glass coverslips (Fisher Scientific, #1254580) and grown overnight at 37°C with 5% CO2. The following day, JF646 (Promega, #GA1120) was added to HEK293T media at 1:10,000 dilution and added to cells for 30 minutes. After 30 minutes, JF646 containing media was removed and washed twice with regular HEK293T media for 15 minutes each. Following wash out, coverslips were washed once with 1x PBS (Gibco, #14200-075) and fixed in 4% PFA for 10 minutes. Cells were then permeabilized with PBS + 0.1% Triton-X-100 for 20 minutes at room temperature, treated with 10 mM glycine for 20 minutes at room temperature, and then blocked in blocking buffer (PBS + 0.1% Triton-X-100, 2% fetal calf serum, and 3% BSA) for 1 hour at room temperature. Coverslips were then incubated overnight with blocking buffer + rabbit anti-TDP43 antibody (Proteintech, #10782-2-AP) at 1:500 to stain for TDP43. Following primary incubation, coverslips were washed three times with PBS for 5 minutes then stained with secondary antibody goat anti-rabbit AF488 (ThermoFisher, #A-11008) at 1:250 for 1 hour at room temperature in PBS + 0.1% Triton-X-100, 2% fetal calf serum, and 3% BSA. After secondary incubation, coverslips were washed three times for 5 minutes in PBS then stained with Hoechst 1:20,000 to mark nucleus. The coverslips were mounted on coverslips and allowed to dry overnight before imaging. Coverslips were imaged using confocal mode on ONI Nanoimager.
Immunohistochemistry
Immunostaining was accomplished using the Dako Autostainer Link 48 (Agilent, USA). Anti-YTHDF2 antibody (Proteintech, #24744-1-AP, 1:300) or anti-m6A antibody (Synaptic Systems #202003, 1:100) were used with the Dako High pH Target Retrieval Solution (Tris/EDTA, pH 9; Agilent, USA) (20 minutes, 97°C) and the Dako Envision Flex Plus Mouse Link Kit (Agilent, USA) to detect the antibody along with the Dako DAB (Agilent, USA). Whole-slide images were generated by the University of Michigan Digital Pathology group within the Department of Pathology using a Leica Biosystems Aperio AT2 scanner equipped with a 0.75 NA Plan Apo 20x objective; 40x scanning is achieved using a 2x optical magnification changer. Resolution is 0.25 microns per pixel for 40x scans. Focus during the scan is maintained using a triangulated focus map built from individual focus points determined in a separate step before scanning is started. Proprietary software is used for image processing during acquisition. Image analysis performed using QuPath software91 and mean intensity calculated for stained cells.
RT-PCR and quantitative RT-PCR
Total RNA was extracted using Trizol following the manufacturer’s protocol. To synthesize cDNA, 500 ng of total RNA was used in a 20 μL reaction volume with the Bio-Rad iScript cDNA synthesis kit (Bio-Rad, #1708890) according to the manufacturer’s protocol. The reactions were incubated at 25°C for 5 min, 46°C for 20 min, and 95°C for 1 min. For quantitative RT-PCR (qRT-PCR), reactions were carried out using Step One Plus Realtime PCR system (Applied Biosystems). Reactions were carried out using PowerUp™ SYBR™ Green Master Mix (ThermoFisher #A25742), with 1 μM primers, and 1 μL cDNA, according to manufacturer’s protocol. Relative gene expression was calculated using the ΔΔCt method. Values obtained from qRT-PCR were plotted in GraphPad Prism.
CFTR minigene assay
HEK293 cells were co-transfected with EGFP or TDP43(WT) and the CFTR minigene using Lipofectamine 2000. 48h post-transfection, total RNA was extracted using Trizol according to manufacturer’s protocol. cDNA was synthesized using the Bio-Rad iScript cDNA synthesis kit according the manufacturer’s protocol, and RT-PCR was accomplished as described previously23.
Purification of recombinant TDP43
TDP43(WT) was expressed in BL21 DE3 E. coli cells from the plasmid pE-6xHis-SUMO-TDP43(WT) (a gift from Dr. James Shorter). Induction was carried out with 1 mM isopropyl-b-D-1-thiogalactopyranoside and cells were grown at 15°C for 16h. Cell pellets were resuspended in lysis buffer (50 mM HEPES, 2% Triton X-100, 300 mM NaCl, 5% glycerol, 50 mM imidazole, 2 mM BME, EDTA-free protease inhibitor cocktail, 5 mM pepstatin, and 20 mg/mL lysozyme) and incubated on ice for 30 minutes. Following sonication on ice, cell lysates were centrifuged for 20 min at 11,000 × g at 4°C. Recombinant protein was purified by binding to Ni-NTA resin (Qiagen #30210), rinsed with 25 mL of wash buffer 4 times (50 mM HEPES, 2% Triton X-100, 300 mM NaCl, 5% glycerol, 50 mM imidazole, and 2 mM BME), and released with 3 mL of elution buffer (50 mM HEPES, 500 mM NaCl, 300 mM imidazole, 5% glycerol, and 5 mM DTT) at room temperature (RT), collecting five 2 mL fractions. Protein was dialyzed twice for 1h in 1 L of final buffer (50 mM HEPES and 500 mM NaCl), and dialyzed once in 1 L of final buffer overnight at 4°C.
Electromobility shift assays (EMSAs)
Binding assays were performed with purified full-length recombinant TDP43 protein and either unmodified or m6A modified ssRNA labeled probes tagged with a 5’ 800nm infrared (IR) moiety (IDT; Table S4). Binding reactions were performed in binding buffer (12.5 mM HEPES, pH 7.8, 50 mM KCl, 2.5 mM MgCl2, 0.5 mM TCEP, 25 mg/mL BSA, 0.01% NP-40) with 50% glycerol, 1 mg/ml poly-dIdC, 500 pM of labeled probe, and recombinant protein (concentrations indicated in figure legends). Reactions were incubated on ice for 5 min followed by 25 min at RT. Electrophoresis of 6% acrylamide gels were performed at 60V. Images were acquired using the LI-COR Odyssey platform.
QUANTIFICATION AND STATISTICAL ANALYSIS
DART-seq data processing and analysis
Raw reads were quality and adapter trimmed with Cutadapt (v2.3)92 with default parameters, and aligned to the GRCh38 human genome assembly with bwa-mem (v0.7.17)93 with default parameters. Single nucleotide transition analysis was performed as previously described27. Briefly, aligned reads were deduplicated, sorted by genome coordinate, parsed to BED format, and collapsed by PCR replicate using the CLIP Tool Kit (CTK) suite94. Mutations were extracted and replicates were merged, preserving replicate information. The CTK suite script “CIMS.pl” was then used to generate coverage, mutation rate, and a false discovery statistic. Resulting BED files were filtered in R for entries with a minimum mutation rate of 2, a minimum read count of 10 per replicate, and a mutation/read threshold of 0.1–0.6. Finally, sense C to T (and antisense G to A) transitions were filtered for presence of the surrounding DRACH motif using custom scripts and the GenomicRanges, BSgenome, Biostrings, and gUtils R packages.
The distribution of mutational transitions was calculated using custom scripts adapted from the MetaPlotR Perl/R suite95 and visualized with ggplot2. Motif logos were generated before and after DRACH filtration using ggseqlogo. Linear U-G dinucleotide density was calculated using custom R scripts. Briefly, sequences were collapsed to binary representations of UG/GU (1) or non-UG/GU (0) dyads. For UG15 density, a continuous sliding average was calculated in 15-nucleotide windows along the target sequence such that a tract of uniform UG alternation corresponds to a UG15 density of 1. Sliding averages were rescaled to the basepair length of the target sequence and used to generate site-of-interest-centered density plots using ggplot2. Gene ontology analyses were accomplished via STRING or Enrichr29,96. Euler diagrams were created using eulerr in R97.
Statistical Analysis
Statistical analysis performed using GraphPad Prism 9 or Superplots98,99. For primary neuron survival analysis, the open-source R survival package was used to determine hazard ratios and statistical significance between conditions through Cox proportional hazards analysis7,40,51,100. All survival studies include biological replicates that were treated identically, but the experiments were performed at separate times. To combine the data from biological replicates, we stratified the results based on experimental date; similar approaches are utilized in meta-analyses that incorporate date from multiple clinical trials101.
Supplementary Material
Supplemental data file 1: Hyper- and hypomethylated mRNAs in ALS vs. control spinal cord (related to Fig. 4)
Supplemental data file 2: Hyper- and hypomethylated lncRNAs in ALS vs. control spinal cord (related to Fig. 4)
Highlights.
The RNA binding protein TDP43 preferentially binds N6-methyladenosine (m6A) modified RNA
Post-mortem samples from ALS patients display widespread RNA hypermethylation
Methylated RNA overlaps with TDP43 pathology in ALS patient spinal cord
Reduction of the m6A reader YTHDF2 mitigates TDP43-mediated toxicity in ALS models
Acknowledgements
We thank all the patients that made this work possible. Skin samples from participants were collected and de-identified through the Michigan Institute for Clinical and Health Research (MICHR, UL1TR000433) via an IRB-approved protocol (HUM00028826). We thank Stephen Goutman, Director of the University of Michigan (UM) ALS Clinic and Biorepository, Eva Feldman and Crystal Pacut from the Program for Neurology Research and Discovery. We also acknowledge Matthew Perkins, Kathy Toy, John Moran, Ahmed Malik, and Kaitlin Weskamp for their assistance and suggestions.
This work was supported by National Institutes of Health (R01NS097542 and R01NS113943 to SJB; and P30AG072931 to the UM Brain Bank and Alzheimer’s Disease Research Center), the family of Angela Dobson and Lyndon Welch, the A. Alfred Taubman Medical Research Institute, the Danto Family, Ann Arbor Active Against ALS, and the Robert Packard Center for ALS Research. Immunohistochemistry was performed at the Rogel Cancer Center Tissue and Molecular Pathology Shared Resource Laboratory at UM (NIH P30 CA04659229).
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Charcot J., and Joffory A (1869). Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arcives Physiol. Neurol. Pathol. 2, 744–754. [Google Scholar]
- 2.Neumann M (2009). Molecular neuropathology of TDP-43 proteinopathies. Int. J. Mol. Sci. 10, 232–246. 10.3390/ijms10010232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ou SH, Wu F, Harrich D, García-Martínez LF, and Gaynor RB (1995). Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 69, 3584–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buratti E, and Baralle FE (2008). Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 13, 867–878. [DOI] [PubMed] [Google Scholar]
- 5.Tank EM, Figueroa-Romero C, Hinder LM, Bedi K, Archbold HC, Li X, Weskamp K, Safren N, Paez-Colasante X, Pacut C, et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 9, 2845. 10.1038/s41467-018-05049-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weskamp K, and Barmada SJ (2018). TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res. 1693, 67–74. 10.1016/j.brainres.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flores BN, Li X, Malik AM, Martinez J, Beg AA, and Barmada SJ (2019). An Intramolecular Salt Bridge Linking TDP43 RNA Binding, Protein Stability, and TDP43-Dependent Neurodegeneration. Cell Rep. 27, 1133–1150.e8. 10.1016/j.celrep.2019.03.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci, 1. 10.1038/s41593-018-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22, 180–190. 10.1038/s41593-018-0293-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koranda JL, Dore L, Shi H, Patel MJ, Vaasjo LO, Rao MN, Chen K, Lu Z, Yi Y, Chi W, et al. (2018). Mettl14 Is Essential for Epitranscriptomic Regulation of Striatal Function and Learning. Neuron 99, 283–292.e5. 10.1016/J.NEURON.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 12.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646. 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sommer S, Lavi U, and Darnell JE (1978). The absolute frequency of labeled N-6-methyladenosine in HeLa cell messenger RNA decreases with label time. J. Mol. Biol. 124, 487–499. 10.1016/0022-2836(78)90183-3. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freibaum BD, Chitta RK, High AA, and Taylor JP (2010). Global Analysis of TDP-43 Interacting Proteins Reveals Strong Association with RNA Splicing and Translation Machinery. J. Proteome Res. 9, 1104–1120. 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo F, Jiao F, Song Z, Li S, Liu B, Yang H, Zhou Q, and Li Z (2015). Regulation of MALAT1 expression by TDP43 controls the migration and invasion of non-small cell lung cancer cells in vitro. Biochem. Biophys. Res. Commun. 465, 293–298. 10.1016/J.BBRC.2015.08.027. [DOI] [PubMed] [Google Scholar]
- 17.Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, and Qian S-B (2015). Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 526, 591–594. 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coker H, Wei G, and Brockdorff N (2018). m6A modification of non-coding RNA and the control of mammalian gene expression. Biochim. Biophys. Acta - Gene Regul. Mech 10.1016/J.BBAGRM.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T (2015). N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560. 10.1038/NATURE14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, and Tavazoie SF (2015). HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell 162, 1299–1308. 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buratti E, Dörk T, Zuccato E, Pagani F, Romano M, and Baralle FE (2001). Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784. 10.1093/EMBOJ/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buratti E, and Baralle FE (2001). Characterization and Functional Implications of the RNA Binding Properties of Nuclear Factor TDP-43, a Novel Splicing Regulator ofCFTR Exon 9 *. J. Biol. Chem. 276, 36337–36343. 10.1074/JBC.M104236200. [DOI] [PubMed] [Google Scholar]
- 23.Ayala YM, Pagani F, and Baralle FE (2006). TDP43 depletion rescues aberrant CFTR exon 9 skipping. FEBS Lett. 580, 1339–1344. 10.1016/J.FEBSLET.2006.01.052. [DOI] [PubMed] [Google Scholar]
- 24.Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF, and Allain FHT (2013). Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449. 10.1038/NSMB.2698. [DOI] [PubMed] [Google Scholar]
- 25.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, and Jaffrey SR (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.England CG, Luo H, and Cai W (2015). HaloTag Technology: A Versatile Platform for Biomedical Applications. Bioconjug. Chem. 26, 975–986. 10.1021/acs.bioconjchem.5b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer KD (2019). DART-seq: an antibody-free method for global m6A detection. Nat. Methods, 1–6. 10.1038/s41592-019-0570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hallegger M, Chakrabarti AM, Lee FCY, Lee BL, Amalietti AG, Odeh HM, Copley KE, Rubien JD, Portz B, Kuret K, et al. (2021). TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell 184, 4680–4696.e22. 10.1016/J.CELL.2021.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al. (2021). The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605. 10.1093/NAR/GKAA1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maor-Nof M, Shipony Z, Lopez-Gonzalez R, Nakayama L, Zhang YJ, Couthouis J, Blum JA, Castruita PA, Linares GR, Ruan K, et al. (2021). p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 184, 689–708.e20. 10.1016/J.CELL.2020.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho WY, Navakkode S, Liu F, Soong TW, and Ling SC (2020). Deregulated expression of a longevity gene, Klotho, in the C9orf72 deletion mice with impaired synaptic plasticity and adult hippocampal neurogenesis. Acta Neuropathol. Commun. 8. 10.1186/S40478-020-01030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, Van Marion I, et al. (2003). VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat. Genet. 2003 344 34, 383–394. 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- 33.Wang S, Latallo MJ, Zhang Z, Huang B, Bobrovnikov DG, Dong D, Livingston NM, Tjoeng W, Hayes LR, Rothstein JD, et al. (2021). Nuclear export and translation of circular repeat-containing intronic RNA in C9ORF72-ALS/FTD. Nat. Commun. 12. 10.1038/S41467-021-25082-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castelli LM, Cutillo L, Souza CDS, Sanchez-Martinez A, Granata I, Lin YH, Myszczynska MA, Heath PR, Livesey MR, Ning K, et al. (2021). SRSF1-dependent inhibition of C9ORF72-repeat RNA nuclear export: genome-wide mechanisms for neuroprotection in amyotrophic lateral sclerosis. Mol. Neurodegener. 16. 10.1186/S13024-021-00475-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koh CWQ, Goh YT, and Goh WSS (2019). Atlas of quantitative single-base-resolution N6-methyl-adenine methylomes. Nat. Commun. 10, 5636. 10.1038/s41467-019-13561-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling S-C, Sun E, Wancewicz E, Mazur C, et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Župunski V, et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458. 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhardwaj A, Myers MP, Buratti E, and Baralle FE (2013). Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res. 41, 5062–5074. 10.1093/nar/gkt189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barmada SJ, Ju S, Arjun A, Batarse A, Archbold HC, Peisach D, Li X, Zhang Y, Tank EMH, Qiu H, et al. (2015). Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc. Natl. Acad. Sci. U. S. A. 112, 7821–7826. 10.1073/PNAS.1509744112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weskamp K, Tank EM, Miguez R, McBride JP, Gómez NB, White M, Lin Z, Gonzalez CM, Serio A, Sreedharan J, et al. (2020). Shortened TDP43 isoforms upregulated by neuronal hyperactivity drive TDP43 pathology in ALS. J. Clin. Invest. 130, 1139–1155. 10.1172/JCI130988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao S, Sun H, and Xu C (2018). YTH Domain: A Family of N6-methyladenosine (m6A) Readers. Genomics. Proteomics Bioinformatics 16, 99–107. 10.1016/J.GPB.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kierzek E, Zhang X, Watson RM, Kennedy SD, Szabat M, Kierzek R, and Mathews DH (2022). Secondary structure prediction for RNA sequences including N6-methyladenosine. Nat. Commun. 2022 131 13, 1–10. 10.1038/s41467-022-28817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ayala YM, De Conti L, Avendaño-Vázquez SE, Dhir A, Romano M, D’Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, et al. (2011). TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288. 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Budini M, and Buratti E (2011). TDP-43 autoregulation: Implications for disease. J. Mol. Neurosci. 45, 473–479. 10.1007/S12031-011-9573-8. [DOI] [PubMed] [Google Scholar]
- 45.Eréndira Avendaño-Vázquez S, Dhir A, Bembich S, Buratti E, Proudfoot N, and Baralle FE (2012). Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 26, 1679–1684. 10.1101/GAD.194829.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koyama A, Sugai A, Kato T, Ishihara T, Shiga A, Toyoshima Y, Koyama M, Konno T, Hirokawa S, Yokoseki A, et al. (2016). Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 44, 5820–5836. 10.1093/NAR/GKW499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D, et al. (2014). Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 10, 677. 10.1038/NCHEMBIO.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EM-H, Li X, Miguez R, Dayton RD, Tamir S, Klein RL, et al. (2018). TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep. 8, 4606. 10.1038/s41598-018-22858-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, and Finkbeiner S (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 30, 639–649. 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arrasate M, and Finkbeiner S (2005). Automated microscope system for determining factors that predict neuronal fate. Proc. Natl. Acad. Sci. U. S. A. 102, 3840–3845. 10.1073/PNAS.0409777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weskamp K, Safren N, Miguez R, and Barmada S (2019). Monitoring Neuronal Survival via Longitudinal Fluorescence Microscopy. J. Vis. Exp. 2019. 10.3791/59036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, et al. (2017). Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. 10.1038/nature22038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, Hung G, Rigo F, Bennett CF, Otis TS, et al. (2017). Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 544, 362. 10.1038/NATURE22044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scoles DR, and Pulst SM (2019). Antisense therapies for movement disorders. Mov. Disord. 34, 1112–1119. 10.1002/MDS.27782. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95. 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sidibé H, Khalfallah Y, Xiao S, Gómez NB, Fakim H, Tank EMH, Tomasso G. Di, Bareke E, Aulas A, McKeever PM, et al. (2021). TDP-43 stabilizes G3BP1 mRNA: relevance to amyotrophic lateral sclerosis/frontotemporal dementia. Brain 144, 3461. 10.1093/BRAIN/AWAB217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernandopulle MS, Prestil R, Grunseich C, Wang C, Gan L, and Ward ME (2018). Transcription-factor mediated differentiation of human iPSCs into neurons. Curr. Protoc. cell Biol. 79, e51. 10.1002/CPCB.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257. 10.1016/J.NEURON.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NCA, Flynn H, Adamson J, et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. 10.1016/J.NEURON.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghasemi M, and Brown RH (2018). Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med 8. 10.1101/CSHPERSPECT.A024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bokar JA, Shambaugh ME, Polayes D, Matera AG, and Rottman FM (1997). Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3, 1233. [PMC free article] [PubMed] [Google Scholar]
- 62.Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, Adhikari S, Shi Y, Lv Y, Chen Y-S, et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Casafont I, Bengoechea R, Tapia O, Berciano MT, and Lafarga M (2009). TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J. Struct. Biol. 167, 235–241. 10.1016/J.JSB.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 64.Liu N, Parisien M, Dai Q, Zheng G, He C, and Pan T (2013). Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856. 10.1261/rna.041178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 8, 284–296. 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Edupuganti RR, Geiger S, Lindeboom RGH, Shi H, Hsu PJ, Lu Z, Wang S-Y, Baltissen MPA, Jansen PWTC, Rossa M, et al. (2017). N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 24, 870–878. 10.1038/nsmb.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE, and Darnell RB (2017). m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006. 10.1101/GAD.301036.117/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Louloupi A, Ntini E, Conrad T, and Ørom UAV (2018). Transient N-6-Methyladenosine Transcriptome Sequencing Reveals a Regulatory Role of m6A in Splicing Efficiency. Cell Rep. 23, 3429–3437. 10.1016/J.CELREP.2018.05.077. [DOI] [PubMed] [Google Scholar]
- 69.Wei G, Almeida M, Pintacuda G, Coker H, Bowness JS, Ule J, and Brockdorff N (2021) Acute depletion of METTL3 implicates N 6-methyladenosine in alternative intron/exon inclusion in the nascent transcriptome. Genome Res. 31, 1395–1408. 10.1101/gr.271635.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun L, Fazal FM, Li P, Broughton JP, Lee B, Tang L, Huang W, Kool ET, Chang HY, and Zhang QC (2019). RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 26, 322. 10.1038/S41594-019-0200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.White MA, Kim E, Duffy A, Adalbert R, Phillips BU, Peters OM, Stephenson J, Yang S, Massenzio F, Lin Z, et al. (2018). TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 21, 552–563. 10.1038/S41593-018-0113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang CG, et al. (2017). m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 18, 2622. 10.1016/J.CELREP.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li F, Yi Y, Miao Y, Long W, Long T, Chen S, Cheng W, Zou C, Zheng Y, Wu X, et al. (2019). N6-methyladenosine Modulates Nonsense-mediated mRNA Decay in Human Glioblastoma. Cancer Res, canres.2868.2018. 10.1158/0008-5472.CAN-18-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Widagdo J, Zhao QY, Kempen MJ, Tan MC, Ratnu VS, Wei W, Leighton L, Spadaro PA, Edson J, Anggono V, et al. (2016). Experience-Dependent Accumulation of N6-Methyladenosine in the Prefrontal Cortex Is Associated with Memory Processes in Mice. J. Neurosci. 36, 6771. 10.1523/JNEUROSCI.4053-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shafik AM, Zhang F, Guo Z, Dai Q, Pajdzik K, Li Y, Kang Y, Yao B, Wu H, He C, et al. (2021). N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease. Genome Biol. 22, 17. 10.1186/s13059-020-02249-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang L, Lin W, Zhang C, Ash PEA, Verma M, Kwan J, van Vliet E, Yang Z, Cruz AL, Boudeau S, et al. (2021). Interaction of tau with HNRNPA2B1 and N 6-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 81, 4209–4227.e12. 10.1016/J.MOLCEL.2021.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luo J, Xu T, and Sun K (2021). N6-Methyladenosine RNA Modification in Inflammation: Roles, Mechanisms, and Applications. Front. cell Dev. Biol. 9. 10.3389/FCELL.2021.670711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng D, Ezzeddine N, Chen CYA, Zhu W, He X, and Shyu A. Bin (2008). Deadenylation is prerequisite for P-body formation and mRNA decay in mammalian cells. J. Cell Biol. 182, 89. 10.1083/JCB.200801196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park OH, Ha H, Lee Y, Boo SH, Kwon DH, Song HK, and Kim YK (2019). Endoribonucleolytic Cleavage of m6A-Containing RNAs by RNase P/MRP Complex. Mol. Cell. 10.1016/J.MOLCEL.2019.02.034. [DOI] [PubMed] [Google Scholar]
- 80.Boo SH, Ha H, Lee Y, Shin M-K, Lee S, and Kim YK (2022). UPF1 promotes rapid degradation of m6A-containing RNAs. Cell Rep. 39, 110861. 10.1016/J.CELREP.2022.110861. [DOI] [PubMed] [Google Scholar]
- 81.Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, Bosco DA, Hayward LJ, Brown RH, Lindquist S, et al. (2011). A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 9. 10.1371/JOURNAL.PBIO.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kamelgarn M, Chen J, Kuang L, Jin H, Kasarskis EJ, and Zhu H (2018). ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. U. S. A. 115, E11904–E11913. 10.1073/PNAS.1810413115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu W, Bao P, Jiang X, Wang H, Qin M, Wang R, Wang T, Yang Y, Lorenzini I, Liao L, et al. Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. 10.1093/brain/awz070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun Y, Eshov A, Zhou J, Isiktas AU, and Guo JU (2020). C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat. Commun. 11. 10.1038/S41467-020-17129-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ortega JA, Daley EL, Kour S, Samani M, Tellez L, Smith HS, Hall EA, Esengul YT, Tsai YH, Gendron TF, et al. (2020). Nucleocytoplasmic Proteomic Analysis Uncovers eRF1 and Nonsense-Mediated Decay as Modifiers of ALS/FTD C9orf72 Toxicity. Neuron 106, 90–107.e13. 10.1016/J.NEURON.2020.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zaepfel BL, Zhang Z, Maulding K, Coyne AN, Cheng W, Hayes LR, Lloyd TE, Sun S, and Rothstein JD (2021). UPF1 reduces C9orf72 HRE-induced neurotoxicity in the absence of nonsense-mediated decay dysfunction. Cell Rep. 34. 10.1016/J.CELREP.2021.108925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, et al. (2017). A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987. 10.1038/NMETH.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Montague TG, Cruz JM, Gagnon JA, Church GM, and Valen E (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401–W407. 10.1093/NAR/GKU410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Labun K, Montague TG, Gagnon JA, Thyme SB, and Valen E (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 44, W272–W276. 10.1093/NAR/GKW398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, and Valen E (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174. 10.1093/NAR/GKZ365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Reports 2017 71 7, 1–7. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. 10.14806/EJ.17.1.200. [DOI] [Google Scholar]
- 93.Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. 10.1093/BIOINFORMATICS/BTP698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shah A, Qian Y, Weyn-Vanhentenryck SM, and Zhang C (2017). CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567. 10.1093/BIOINFORMATICS/BTW653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Olarerin-George AO, and Jaffrey SR (2017). MetaPlotR: a Perl/R pipeline for plotting metagenes of nucleotide modifications and other transcriptomic sites. Bioinformatics 33, 1563–1564. 10.1093/BIOINFORMATICS/BTX002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 1, e90. 10.1002/CPZ1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.J, L. (2020). eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses.
- 98.Lord SJ, Velle KB, Dyche Mullins R, and Fritz-Laylin LK (2020). Reproducibility: SuperPlots: Communicating reproducibility and variability in cell biology. J. Cell Biol. 219. 10.1083/JCB.202001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goedhart J (2021). SuperPlotsOfData - A web app for the transparent display and quantitative comparison of continuous data from different conditions. Mol. Biol. Cell 32, 470–474. 10.1091/MBC.E20-09-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Malik AM, Miguez RA, Li X, Ho YS, Feldman EL, and Barmada SJ (2018). Matrin 3-dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. Elife 7. 10.7554/ELIFE.35977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Giganti MJ, Luz PM, Caro-Vega Y, Cesar C, Padgett D, Koenig S, Echevarria J, McGowan CC, and Shepherd BE (2015). A Comparison of Seven Cox Regression-Based Models to Account for Heterogeneity Across Multiple HIV Treatment Cohorts in Latin America and the Caribbean. AIDS Res. Hum. Retroviruses 31, 496. 10.1089/AID.2014.0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 811. 2013;8(11):2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ho R, Workman MJ, Mathkar P, et al. (2021). Cross-Comparison of Human iPSC Motor Neuron Models of Familial and Sporadic ALS Reveals Early and Convergent Transcriptomic Disease Signatures. Cell Syst. 12, 159–175.e9. doi: 10.1016/J.CELS.2020.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data file 1: Hyper- and hypomethylated mRNAs in ALS vs. control spinal cord (related to Fig. 4)
Supplemental data file 2: Hyper- and hypomethylated lncRNAs in ALS vs. control spinal cord (related to Fig. 4)
Data Availability Statement
DART-seq data have been deposited at GEO (SUB12370770) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Epitranscriptomic array data are included in supplemental data files 1 and 2 of the publication.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-TDP43 | Proteintech | Cat# 10782-2-AP; RRID: AB_2892214 |
| Rabbit monoclonal anti-NeuN | Cell Signaling Technologies | Cat# 24307T RRID: AB_2651140 |
| Rabbit polyclonal anti-YTHDF2 | Proteintech | Cat# 24744-1-AP; RRID: AB_2687435 |
| Rabbit polyclonal anti-m6A | Millipore Sigma | Cat# ABE572; RRID: AB_2892213 |
| Rabbit monoclonal anti-m6A | Cell Signaling Technologies | Cat# 56593S; RRID: AB_2799515 |
| Rabbit polyclonal anti-m6A | Millipore Sigma | Cat# ABE572-I-100UG; RRID: AB_2892214 |
| Goat polyclonal anti-rabbit Alexa Fluor 488 | ThermoFisher | Cat# A-11034; RRID: AB_2576217 |
| Goat polyclonal anti-rabbit HRP | Jackson ImmunoResearch Labs Inc. | Cat# 111-035-003; RRID: AB_2313567 |
| Bacterial and virus strains | ||
| GIPZ Non-silencing Lentiviral shRNA Control | Horizon Discovery | #RHS4346 |
| GIPZ Lentiviral Human YTHDF2 shRNA | Horizon Discovery | #RHS4430-200182983 |
| Biological samples | ||
| Human post-mortem sample; Control 1 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 66 Sex: M |
| Human post-mortem sample; Control 2 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 85 Sex: M |
| Human post-mortem sample; Control 3 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 68 Sex: M |
| Human post-mortem sample; sALS 1 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 65 Sex: M |
| Human post-mortem sample; sALS 2 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 64 Sex: M |
| Human post-mortem sample; sALS 3 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 65 Sex: M |
| Human post-mortem sample; sALS 4 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 81 Sex: M |
| Human post-mortem sample; Control 4 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 76 Sex: F |
| Human post-mortem sample; Control 5 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 88 Sex: M |
| Human post-mortem sample; Control 6 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 56 Sex: M |
| Human post-mortem sample; ALS 1 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 66 Sex: F |
| Human post-mortem sample; ALS 2 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 64 Sex: M |
| Human post-mortem sample; ALS 3 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 68 Sex: M |
| Human post-mortem sample; Control 7 | University of Maryland Brain and Tissue Bank https://www.medschool.umaryland.edu/btbank/ | Experiment: m6A array Age: 48 Sex: M |
| Human post-mortem sample; ALS 4 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 76 Sex: M |
| Human post-mortem sample; ALS 5 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 60 Sex: F |
| Human post-mortem sample; ALS 6 | University of Michigan Brain Bank https://www.brainbank.umich.edu/ | Experiment: m6A array Age: 99 Sex: M |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant TDP43(WT) | Flores et al., 20197 | N/A |
| Deposited data | ||
| DART-seq data from HEK293T cells | This manuscript | https://www.ncbi.nlm.nih.gov/geo/ (SUB12370770) |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 |
| Primary rat neurons | Charles River Laboratories | Long-Evans rat (Crl:LE) |
| Human iPSCs | University of Michigan ALS Repository | 1021, 0883, 0312 |
| Human iPSCs | Cedars Sinai iPSC Repository | Cs29i, Cs29 |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene TARDBP WT; Tag TDP43-Dendra2-Carboxyl term | Weskamp et al., 202041 | Line 1021 Identifier: WT |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene TARDBP Mutation Isogenic M337V; Tag TDP43-Dendra2 Carboxyl term | Weskamp et al., 202041 Sidibé et al., 202157 |
Line 1021 Identifier: M337V |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene C9ORF72 Mutation | Weskamp et al., 202041 | Line 312 Identifier: C9ORF72 (#1) |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene C9ORF72 Mutation | Weskamp et al., 202041 | Line 883 Identifier: C9ORF72 (#2) |
| Human iPSCs (CLYBL:TO-hNgn1/2); Gene C9ORF72 Mutation | Weskamp et al., 202041 | Line Cs29i Identifier: C9ORF72 (#3) |
| Human iPSCs (CLYBL:TO-hNgn1/21); Gene C9ORF72 Mutation Isogenic corrected | Weskamp et al., 202041 Ho et al., 2021104 |
Line Cs29i Identifier: C9ORF72 ISO (#3) |
| Oligonucleotides | ||
| See Table S4 for complete list | ||
| Recombinant DNA | ||
| See Table S2 for complete list | ||
| Software and algorithms | ||
| RNAstructure software | Kierzek et al., 202243 | https://rna.urmc.rochester.edu/RNAstructure.html |
| Automated imaging and survival analysis | Malik et al., 2018101 | DOI: 10.5281/zenodo.7438867 DOI: 10.5281/zenodo.7438874 |
| Biorender | N/A | https://Biorender.com |
| Other | ||
| HaloTag ligand (JF646 dye) | Janelia Farms | https://www.janelia.org |
All original code has been deposited in Github and Zenodo, and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
All schematics created with Biorender.com.