

Abstract
The movement of ions in and out of neurons can exert significant effects on neighboring cells. Here we report several experimentally important consequences of activation of the optogenetic chloride pump, halorhodopsin. We recorded extracellular K+ concentration ([K+]extra) in neocortical brain slices prepared from young adult mice (both sexes) which express halorhodopsin in pyramidal cells. Strong halorhodopsin activation induced a pronounced drop in [K+]extra that persisted for the duration of illumination. Pharmacological blockade of K+ channels reduced the amplitude of this drop, indicating that it represents K+ redistribution into cells during the period of hyperpolarization. Halorhodopsin thus drives the inward movement of both Cl– directly, and K+ secondarily. When the illumination period ended, a rebound surge in extracellular [K+] developed over tens of seconds, partly reflecting the previous inward redistribution of K+, but additionally driven by clearance of Cl– coupled to K+ by the potassium-chloride cotransporter, KCC2. The drop in [K+]extra during light activation leads to a small (2-3 mV) hyperpolarization also of other cells that do not express halorhodopsin. Its activation therefore has both direct and indirect inhibitory effects. Finally, we show that persistent strong activation of halorhodopsin causes cortical spreading depolarizations (CSDs), both in vitro and in vivo. This novel means of triggering CSDs is unusual, in that the events can arise during the actual period of illumination, when neurons are being hyperpolarized and [K+]extra is low. We suggest that this fundamentally different experimental model of CSDs will open up new avenues of research to explain how they occur naturally.
SIGNIFICANCE STATEMENT Halorhodopsin is a light-activated electrogenic chloride pump, which has been widely used to inhibit neurons optogenetically. Here, we demonstrate three previously unrecognized consequences of its use: (1) intense activation leads to secondary movement of K+ ions into the cells; (2) the resultant drop in extracellular [K+] reduces excitability also in other, nonexpressing cells; and (3) intense persistent halorhodopsin activation can trigger cortical spreading depolarization (CSD). Halorhodopsin-induced CSDs can occur when neurons are hyperpolarized and extracellular [K+] is low. This contrasts with the most widely used experimental models that trigger CSDs with high [K+]. Both models, however, are consistent with the hypothesis that CSDs arise following net inward ionic movement into the principal neuron population.
Keywords: chloride, halorhodopsin, potassium, pyramidal cells, spreading depression
Introduction
The complexity of neuronal behavior derives in large measure from the fact that neurons do not act in isolation. This is most apparent in the form of synaptic interactions, but in certain circumstances, the neurons may also exert significant effects on neighboring cells through indirect means, arising from movement of ions in or out of the extracellular space (Jefferys, 1995). Neuronal firing and synaptic activity lead to large ionic movement across the neuronal membrane (Hodgkin and Huxley, 1952; Attwell and Laughlin, 2001), and if sufficiently intense, can result in significant redistribution of key ions, with depletion of extracellular Na+ and Ca2+ ions, and rises in extracellular K+ ions.
The development of optogenetics has provided a new set of research tools for investigating such matters, with novel ways to challenge neuronal networks (Deisseroth, 2011). Optogenetic studies have mostly involved network-level analyses, in which specific subpopulations of neurons are either activated or inactivated to dissect out their roles in large networks or whole animals. Cellular processes can also be examined using optogenetics; and in this regard, the light-activated ion pumps (Zhang et al., 2007; Chow et al., 2010) offer particularly interesting ways to challenge the cells experimentally, either in isolation or in tandem with ion channels. The light-activated chloride pump, halorhodopsin, can be used to drive chloride into neurons (Raimondo et al., 2012; Alfonsa et al., 2015), whereas chloride extrusion can be achieved by coupling the action of the light-activated proton pump and the opening of a chloride channel, using a co-operative opsin called Cl-out (Alfonsa et al., 2016). We reasoned that artificial chloride manipulation provides a further means of examining the relationship between chloride entry and delayed extracellular [K+] rises via the potassium chloride cotransporter (KCC2), that have to date only been examined by means of GABAergic activation (Viitanen et al., 2010; Shiri et al., 2015; Chang et al., 2018). Specifically, we hypothesized that halorhodopsin-mediated ion redistribution would lead to increased KCl efflux by KCC2, revealing itself as a surge in extracellular K+ concentration, [K+]extra, after illumination ends. To test this hypothesis, we used mice in which halorhodopsin was expressed in all pyramidal cells, under the Emx1 promoter (Gorski et al., 2002), and recorded changes in [K+]extra using ion-sensitive electrodes (Voipio et al., 1994).
These studies confirmed the relationship between intracellular [Cl–] and extracellular [K+] transients, but further revealed three other, previously unrecognized, consequences of activation of optogenetic pumps. The first is that intense activation of halorhodopsin leads to redistribution of K+ ions into neurons through K+ ion channels, on account of the hyperpolarization of a large population of neurons altering the balance between inward and outward fluxes of ions; this is particularly evident for K+ on account of it being highly permeant at resting Em, and its low basal extracellular concentration. An important consequence of the K+ redistribution is that the lowering of [K+]extra causes a hyperpolarization of all neurons, not just those that express the opsin; this represents a form of indirect inhibition by halorhodopsin. Finally, we report that halorhodopsin activation can also trigger cortical spreading depolarization (CSD) events. Our initial observations of halorhodopsin-triggered CSDs were of events arising immediately after the end of illumination, but of particular note were other instances, when CSDs arose even during the halorhodopsin activation, when most neurons in the network were strongly hyperpolarized and [K+]extra was low. As such, this novel way of triggering CSDs, by a hyperpolarizing photocurrent, is quite unlike most other known natural and experimental triggers (e.g., ischemia trauma, seizures) (Somjen, 2004; Ullah et al., 2015) where the common thread has been considered to be raised [K+]extra and strong depolarization (Tamim et al., 2021). The exception is the triggering of CSDs by application of hypotonic solution, leading to the suggestion that osmotic stress may be the critical factor (Chebabo et al., 1995; Somjen, 2004), a hypothesis that is consistent with the data we present here.
Materials and Methods
Ethical approval
All procedures performed were in accordance with the guidelines of the Home Office United Kingdom and Animals (Scientific Procedures) Act 1986 and approved by the Animal Welfare and Ethical Review Body at both Newcastle University and University College London. Male and female mice were used for experimentation.
Slice preparation
We used both WT C57BL/6 mice, and also mice expressing eNpHR3.0 within the pyramidal cell population, generated by cross-breeding homozygous Emx1-cre mice (The Jackson Laboratory, stock #005628) with mice containing a floxed STOP cassette in front of an eNpHR3.0/EYFP domain (The Jackson Laboratory, stock #014539; both maintained on the C57BL/6 background). Emx1-promoter yields gene expression only in pyramidal cells in neocortex, in adult mice, although it can drive gene expression in glia in other brain areas, and early in development (Gorski et al., 2002). Experiments were performed on mice 1-8 months of age, of both sexes. Mice were sacrificed by cervical dislocation, brains were removed and placed in cold cutting solution containing the following (in mm): 3 MgCl2; 126 NaCl; 2.6 NaHCO3; 3.5 KCl; 1.26 NaH2PO4; 10 glucose; 400 µm horizontal sections were made on a Leica VT1200 vibratome (Leica Microsystem). Slices were stored at room temperature, in an interface holding chamber for 1-4 h before experimentation. Solutions were bubbled with carbogen (95% O2 and 5% CO2) in aCSF containing the following (in mm): 2 CaCl2; 1 MgCl2; 126 NaCl; 26 NaHCO3; 3.5 KCl; 1.26 NaH2PO4; 10 glucose.
In vitro extracellular recordings
Extracellular recordings were performed using an interface recording chamber. Slices were placed in the recording chamber and perfused with aCSF supplemented in different experiments with various combinations of the following drugs: 1 μm TTX (Abcam), 10 μm VU 0463271 (Tocris Bioscience), 10 mm tetraethylammonium (TEA) (Sigma), 2 mm Ba2+ (Fisons), as indicated in Results. Recordings were obtained using aCSF-filled ∼1-3 mΩ borosilicate glass microelectrodes (GC120TF-10; Harvard apparatus) placed in deep layers of neocortex. Extracellular potassium, [K+]extra, was measured using single-barreled K+-selective microelectrodes. The pipettes were pulled from nonfilamented borosilicate glass (Harvard Apparatus), and the glass was exposed to vapor of dimethyl-trimethyl-silylamine (Sigma-Aldrich), baking at 200°C for 40 min. The pipettes were then backfilled with aCSF. A short column of the K+ sensor (Potassium ionophore I, cocktail B; Sigma-Aldrich, #99373) was taken into the tip of the salinized pipette by using slight suction. The recordings through the K+-sensor electrode were referenced to a second electrode filled with aCSF. From the differential signal from a custom build amplifier, we calculated the [K+]extra from calibration recordings made in an open bath, using sudden increments in [K+]extra. This provided a scaling factor S, of 55-59 mV, where the K+ concentration at a given moment in time, t, was calculated from the differential voltage, V(t), as follows:
[K]baseline for our experiments was 3.5 mm. The temperature of the chamber and perfusate was maintained at 33°C–36°C using a closed circulating heater (FH16D, Grant Instruments). The solutions were perfused at the rate of 3 ml/min by a Watson Marlow 501U peristaltic pump (Watson-Marlow Pumps). The direct current local field potential (LFP) signal was unfiltered and amplified to a 10× output with a custom build amplifier. These waveform signals were digitized with a Micro 1401-3 ADC board (Cambridge Electronic DesignK) and Spike2 version 7.10 software (Cambridge Electronic Design). Signals were sampled at 10 kHz. Recordings were analyzed using a custom-written code in MATLAB (The MathWorks).
In vitro patch-clamp recordings
Brain slices were prepared, from the same mice strain (HaloFl × Emx1-Cre), as follows. Mice were anesthetized by intraperitoneal injection of ketamine (75 mg kg−1, Ketalar Injection, Pfizer) combined with medetomidine (1 mg kg−1, Domitor, Janssen Animal Health), and perfused through the heart using a chilled and aerated “sucrose aCSF” (in mm as follows: 228 sucrose; 26 NaHCO3; 4 MgCl2; 3 KCl; 1.25 NaH2PO4; 10 glucose) for 1 min, before removing the brain, and making 350 µm coronal brain slices in the same sucrose solution. Slices were then incubated in a submerged chamber in conventional 1 mm Mg2+/2 mm Ca2+ aCSF, at room temperature. For the recordings, slices were transferred to a recording chamber held on a movable top plate (Scientifica) with mounted electrode micromanipulators (Scientifica). Slices were visualized, and illuminated, through a UMPlanFL N 40×, 0.5 NA objective (Olympus), using a Scientifica upright microscope fitted with a spinning disk confocal attachment (Visitech) and a Hamamatsu C9100 EM camera (Hamamatsu Photonics) and coordinated using Visitech software. The temperature in the recording chamber was maintained at 32°C-34°C by warming the perfusate (delivered at 3-5mls/min) using a heated sleeve immediately before the recording chamber, and with the additional aid of a plate heater (Warner Instruments). Whole-cell recordings of neurons from the temporal association area were made using 4-7 mΩ pipettes. Pipettes were filled with a KMeSO4-based internal solution containing the following (mm): 125 KMeSO4, 6 NaCl, 10 HEPES, 2.5 Mg-ATP, 0.3 Na2-GTP. Osmolarity and pH of all the internal solutions used were adjusted to 284 mOsm and 7.4. The offset of the patch-clamp amplifier was zeroed before patching the cells, and we adjusted the subsequent measurement of membrane potential, therefore, by the liquid junction potential, which was calculated using LJPcalc software (https://swharden.com/LJPcalc) to be 9.6 mV, according to the stationary Nernst–Planck equation (Marino et al., 2014). The eNPHR3.0 was conjugated to an eYFP fluorescent marker, allowing us to discriminate between cells that expressed the opsin (pyramidal cells), and those that did not (presumptive interneurons and some glia). After going whole cell, we recorded in current-clamp mode, first testing the optogenetic response to a short (200 ms) illumination with 561 nm light, and then the firing response to a current injected through the patch electrode. We then assessed the response to a more intense, and sustained illumination with both 488 and 568 nm wavelength light together. We used this combination because previous work indicated that concurrent illumination with both yellow and blue light yields a more sustained eNPHR3.0 photocurrent because the blue light refreshes the opsin (Han and Boyden, 2007). In nonexpressing, fast-spiking interneurons, we finally tested the firing response to 200 pA current either in darkness or under steady-state illumination with 488 and 568 nm light. Patch-clamp data were collected using a Multiclamp 700B amplifier (Molecular Devices, Invitrogen), digitized using a 1401 AD converter and recorded onto a Dell Precision Desktop computer. Recordings were analyzed offline, using custom-written codes in Matlab2018b. The input–output functions were fitted with sigmoidal equation, as follows:
Where F is the firing rate, I is the current, and the fitting procedure used a nonlinear least squares process to derive the other three parameters: maximal firing rate (Fmax), the current giving half-maximal firing (σ), and a parameter that dictates the slope (N; note, high values give shallower slopes).
In vivo extracellular recordings
Animals were anesthetized with urethane (20% w/v in saline for injection at 0.1 ml/g of mouse) and placed on a stereotaxic frame with heating pad, monitored with a rectal probe to maintain body temperature. A cranial window was created on the left hemisphere and an LFP electrode was placed into the somatosensory cortex. DC LFP recordings were performed as in the in vitro experiments, with the electrode ∼1 mm deep into the tissue. No mice were used past 4 h from urethane injection.
Optogenetic illumination for extracellular recordings
Optogenetic illumination was performed using a fiber-coupled LED light at 565 nm (Thorlabs, M565F3) and driven by an LED driver (Thorlabs, LEDD1) placed in trigger mode. Light output was measured at an average of 9 mW. The LED light was positioned just above the superficial layers of the neocortex for the slice experiments and pointed toward the deep layers, where the extracellular recordings were performed. For the in vivo experiments, the LED was placed in the somatosensory cortex, just above the tissue and ∼2 mm from the recording electrode. Optogenetic activation was from 90 s to 5 min depending on the experiment.
In vivo recordings in awake head-fixed mice using graphene micro-transistor arrays
C57BL/6 mice (∼3-4 months of age), were injected with 500 nl of rAAV5-hSyn-eNpHR3.0-eYFP (UNC Vector Core) unilaterally into the primary motor cortex (M1) and headbars attached. Three weeks later, we initiated rounds of habituation to the Neurotar fixation frame (Neurotar). Once the animal was fully habituated, a second surgery was performed to allow a craniotomy over the ipsilateral somatosensory and visual cortex. Upon recovery from this surgery (3-4 h), the animal was placed in the Neurotar frame, a 16-channel graphene micro-transistor array positioned over the craniotomy (dura intact), and a fiber optic cannula, connected to a Thorlabs green (594 nm) LED positioned over a small bur hole made over the M1 area. Optogenetic or pinprick induction of CSD was induced in the motor cortex and electrographically recorded using the graphene transistor arrays over ipsilateral somatosensory and visual cortex (for full surgical, experimental and graphene transistor data analysis details, see Masvidal-Codina et al., 2021).
Statistics
Statistical analysis of electrophysiology was performed using GraphPad Prism (GraphPad Software). Data were analyzed with a Mann–Whitney test, a Kruskal-Wallis with a Dunn's multiple comparisons test, or a paired t test where appropriate. Figures of electrophysiology traces were created in Matlab2015b or 2018b (The MathWorks). Statistics are expressed as the mean ± SEM.
Results
Activation of halorhodopsin causes secondary redistribution of K+ ions
Previous work showed that intense GABAergic activation is followed by a surge in [K+]extra (Chang et al., 2018; Viitanen et al., 2010; Shiri et al., 2015). The explanation for this is that Cl– ions which moved into neurons through the GABA receptors are then extruded coupled to K+ via KCC2. This hypothesized mechanism predicts that a similar surge in [K+]extra should occur if neurons are artificially loaded with Cl– using the optogenetic chloride pump, halorhodopsin (Raimondo et al., 2012; Alfonsa et al., 2015). We prepared brain slices from adult mice which expressed halorhodopsin in all pyramidal cells, under the Emx1 promoter, and measured [K+]extra during and after a 90 s illumination epoch (Fig. 1). In all slices, we recorded large increases in [K+]extra, peaking at a mean 8.0 ± 0.44 mm (mean ± SEM; n = 14 slices). The surge was relatively slow, peaking only 59.9 ± 10.4 s after the end of the period of illumination. We also observed a drop in [K+]extra that persisted throughout the entire period of illumination. Brain slices from WT mice showed no change in [K+]extra during periods of equivalent illumination (n = 3 slices; Fig. 1A).
Figure 1.

Activation of halorhodopsin in principal cell class induces strong modulation of extracellular [K+] during and after illumination. A, Recordings of extracellular [K+] in brain slices in which either halorhodopsin (blue, top) was expressed in all pyramidal cells, or in a slice with no opsin expression (black, bottom). Yellow bar represents the time of illumination (90 s period). Light activation of halorhodopsin induced a very marked reduction in [K+]extra, followed by an even larger, rebound increase. Brain slices without opsins showed no light-induced fluctuations in [K+]extra. B, The peak extracellular [K+], after the end of the period of halorhodopsin activation (mean peak = 7.98 mm). Lines represent mean +/– std.
We first investigated the nature of the drop in [K+]extra during the period of illumination. We performed these experiments in the presence of TTX, to rule out any possible contribution arising from altered levels of neuronal firing during activation of halorhodopsin. In the presence of TTX alone, the [K+]extra dropped to 1.71 ± 0.08 mm (ΔKextra = ∼−1.8 mm from the baseline level of 3.5 mm), compared with 2.8 ± 0.09 mm in TEA (ΔKextra = ∼−0.7 mm), and 2.85 ± 0.11 mm when Ba2+ was added (ΔKextra = ∼−0.65 mm; Fig. 2). Glial K+ channels are relatively insensitive to TEA, so the fact that the addition of Ba2+ ions was not significantly different from the effect of TEA alone suggests that most of the drop in [K+]extra occurs via a hyperpolarization-driven redistribution of K+ into neurons. This is consistent with the pattern of opsin expression in these animals, driven under the Emx1 promoter, which in adults, appears limited to the pyramidal population (Gorski et al., 2002). Whole-cell patch-clamp recordings of halorhodopsin-expressing pyramidal neurons recorded light-induced hyperpolarization by 24.5 ± 1.5 mV (mean ± SEM; range 10-34 mV, 23 cells, from 6 mice) from resting membrane potential (−62.1 ± 1.5 mV; during intense illumination Em = −84.9 ± 2.5 mV; range = −64 to −104 mV; Fig. 3A).
Figure 2.

The main movement of K+ ions from the extracellular space, induced by opsin activation is through K+ channels. A, Recordings of extracellular [K+] during a period of halorhodopsin activation in the presence of TTX and additionally TEA and Ba2+, to block different K+ channels. Note the far smaller change in extracellular [K+] when K channels were blocked. B, Measurements of the minimum level of extracellular [K+] in the different pharmacological conditions (p = 0.0021 for TTX vs TTX + TEA and p = 0.0015 for TTX vs TTX/TEA vs TTX/TEA/Ba2+, Kruskal–Wallis test with a Dunn's multiple comparisons test). Lines represent mean +/– std.
Figure 3.

The light-induced drop in extracellular [K+] indirectly affects the membrane potential and excitability of cells that do not express halorhodopsin. A, Whole-cell current-clamp recordings of a pyramidal cell (black), a fast-spiking interneuron (red), and a glial cell, made from neocortical brain slices prepared from mice that express halorhodopsin in all pyramidal cells, but not in other cell classes. In each case, intense light activation was delivered diffusely to an area of several hundred microns around the cell location, through the microscope objective. Ai, The very large-amplitude hyperpolarization in the pyramidal cells (the baseline Em for the three recordings is superimposed, for ease of comparison of the light-induced effect; pyramidal Em = −63 mV; interneuronal Em = −70.5 mV; glial Em = −68.5 mV), and the far smaller effect on the nonexpressing cell classes. Aii, The same plots normalized to the maximal drop, to illustrate the difference in time course of the effects on Em. B, Bar charts represent the difference in the amplitude (Bi, p = 7.2 × 10−14; Student's t test) and the time to peak (Bii, p = 3.2 × 10−10; Student's t test) of the hyperpolarization in the Halo-expressing pyramidal cells (n = 23), and the nonexpressing interneurons (n = 15). Error bars represent s.e.m.
Nonspecific inhibitory effects of halorhodopsin activation
We further reasoned that the drop in the extracellular [K+] may have indirect effects also on nonexpressing cells because the reduced [K+]extra would be expected to lower their Em. To examine this possibility, we prepared brain slices in which halorhodopsin was expressed in the pyramidal population, but made targeted patch-clamp recordings of nonexpressing cells (Fig. 3): nonpyramidal neurons interneurons (n = 15), indicated by firing patterns and the lack of a prominent halorhodopsin response, and also glia (n = 2), indicated by nonfiring response to current injection. We found that, during these periods of illumination, there was also a small, but significant hyperpolarization of nonexpressing cells of 2.0 ± 0.3 mV (n = 17 cells, comprising 2 glia [hyperpolarized by 2.0 and 3.6 mV, respectively] and 15 interneurons [Em = −62.4 ± 1.0 mV], from 6 mice; range of hyperpolarizing effect = 0-3.6 mV), which was both significantly different from the null response (p = 6.9 × 10−6, Student's t test) and also significantly smaller than the pyramidal response (p = 7.2 × 10−14; Student's t test). No change was seen during illumination in WT brain slices, which lack halorhodopsin. A possible explanation for the small hyperpolarizing effect in the nonpyramidal population is that there was a low level of “breakthrough” expression of halorhodopsin in these cells. If that were the explanation, though, the kinetics of the change in Em would be comparable to that seen in pyramidal cells, since this is dictated primarily at the molecular level, rather than the level of expression. Notably, the time course of the hyperpolarization of “nonexpressing” cells was very slow, reaching its peak level at 1061 ± 150 ms (n = 13 interneurons, discounting the two cells that showed no discernible shift in Em; the two glia reached peak effect at 800 and 870 ms, respectively) after the start of illumination, in marked contrast to a rapid peak hyperpolarization achieved in pyramidal cells, which occurred at just 47.6 ± 5.6 ms (n = 23), which was highly significantly faster than in nonexpressing cells (p = 3.2 × 10−10, Student's t test). The slow evolving hyperpolarizing effect in the nonexpressing cells had a similar time course to the drop in [K+]extra, and so we concluded that this was indeed because of indirect effects, and not because of breakthrough expression in cells without the Emx1 promoter.
We reasoned that the drop in [K+]extra may also influence neuronal firing in nonexpressing neurons. To test this, we measured the input–output function, in fast-spiking interneurons, in response to step changes in somatically injected current, in baseline conditions and during a sustained period of pyramidal-halorhodopsin activation (n = 9 cells, 6 mice; Fig. 4). Consistently, we found that firing rates were reduced, in these nonexpressing cells, when the tissue was illuminated, consistent with our results regarding the drop in [K+]extra (Fig. 2) and also of Em in nonexpressing cells (Fig. 3). This was the case if the effect was quantified either by the total number of spikes in the 1 s current injection (Fig. 4Bi) or as the maximal instantaneous firing rate (reciprocal of the shortest interspike interval) (Fig. 4Bii). For each cell, we fitted a sigmoidal function (Fig. 4Ci) to the input–output functions for both conditions (light vs no light; for fitting details, see Materials and Methods). In every case, the half-maximal current was increased during periods of illumination to activate halorhodopsin in the pyramidal population (Fig. 4Cii,Ciii; 9 of 9 cells; mean increase = 12.7 ± 3.3%; significantly different from null response, p = 0.0049). Across the 9 cells, there was no significant change in either the maximal firing rate (p = 0.20) or the slope of the fit (p = 0.37), in the two conditions (light vs no light).
Figure 4.

Indirect suppression of firing in nonexpressing, fast-spiking interneurons. A, Firing response to current injection (200 pA, 1 s) in a fast-spiking interneuron that did not express halorhodopsin in control conditions (black) or while there was concurrent activation of halorhodopsin in the local pyramidal cells. B, Input–output functions of a different fast-spiking interneuron, plotted as the total number of action potentials during the 1s period of current injection (Bi) or as the maximal instantaneous firing rate (reciprocal of the shortest interspike interval) (Bii). In both cases, the input–output function was derived without illumination (black) and then with concurrent activation of halorhodopsin in the pyramidal population (red, “light”). Ci, The input–output function (see Materials and Methods). Cii, Paired (light/no light) values of the half-maximal current (I50) for each cell, and Ciii, the same data, normalized to the no-light I50 (p < 10−6, paired t test).
A large rebound surge in [K+]extra follows halorhodopsin activation
We next examined the nature of the large rebound surge in peak [K+]extra, following halorhodopsin activation (range 2.0-7.1 mm above baseline, 3.5 mm). One component of the rebound surge will be extrusion of K+ ions that moved into the neurons during the period of illumination. Notably, however, in a set of experiments with 90 s illumination, when the illumination-induced lowering of [K+]extra reaches a steady state, there was no correlation between the amplitude of the illumination-induced drop in [K+]extra, and the rebound surge in [K+]extra (R2 = 0.006, p = 0.79).
A second explanation is that the rebound [K+]extra surge was caused by rebound spiking following a period of hyperpolarization. However, the large post-halorhodopsin surge persisted in the presence of the Na+ channel blocker, TTX, indicating that rebound spiking was not the source of the rebound peak in [K+]extra. In contrast, the peak was substantially reduced, although not abolished, by blocking the K+- Cl– cotransporter, KCC2, using VU0463271 (p = 0.0037, Kruskal–Wallis test with a Dunn's multiple comparisons test). The postillumination peak in [K+]extra was also reduced by the K+ channel blocker, TEA (Fig. 5C,D; p = 0.0064, Kruskal–Wallis test with a Dunn's multiple comparisons test). The time to the peak of the rebound in [K+]extra was delayed by blocking KCC2 (Fig. 5C, p = 0.0014, two-tailed paired t test), but not by blocking K+ channels (Fig. 5D, p = 0.61, two-tailed paired t test). Together, our data showing a large surge in [K+]extra, following a period of halorhodopsin chloride-loading, are entirely consistent with the previous report of a secondary rise in [K+]extra, following chloride-loading occurring naturally caused by intense GABAergic activation (Viitanen et al., 2010).
Figure 5.
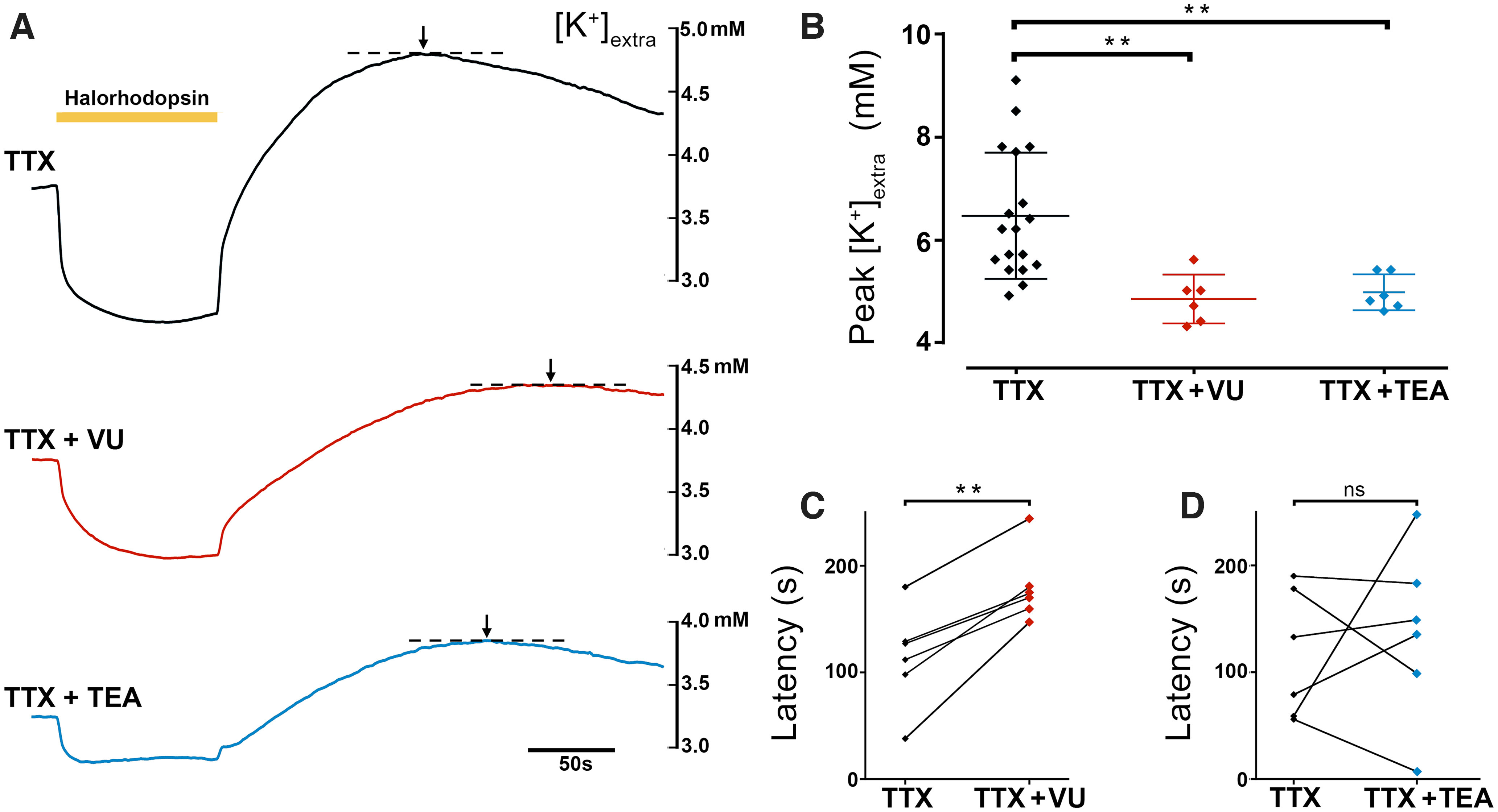
Halorhodopsin-induced K+ redistribution involves KCC2 and voltage-activated K+ channels. A, Recordings of [K+]extra during and after a 90 s period of halorhodopsin activation (yellow bar) in the presence of the Na+ channel blocker, TTX (blue, top), or the KCC2 blocker, VU0463271 (red, middle), or the blocker of voltage-gated K+ channels, TEA (black, bottom). B, The peak [K+]extra, after the end of illumination, for the three different drugs (p = 0.0037 for TTX vs TTX + VU and p = 0.0064 for TTX vs TTX + TEA, Kruskal–Wallis test with a Dunn's multiple comparisons test). Lines represent mean +/– std. C, The time to the peak [K+]extra, following a 90 s period of halorhodopsin activation, with and without the presence of the KCC2 blocker, VU0463271. Blockade of KCC2 causes a significant delay in the time to the peak (p = 0.0014, two-tailed paired t test). All experiments were performed in the presence of the Na+ channel blocker, TTX, to exclude any contribution to the [K+] fluxes made by rebound neuronal spiking. D, Similar measurements made with and without the voltage-gated K+ channel blocker, TEA, showing no significant effect.
Induction of spreading depolarization events by halorhodopsin activation
While performing these investigations of halorhodopsin-induced K+ redistribution, we observed that CSD events often occurred shortly after periods of illumination (Fig. 6A). This happened rarely with shorter illumination epochs (10-30 s); but when the period of illumination were extended to 90 s, CSDs occurred in ∼33% of trials. Notably, we were able to induce CSDs also in the presence of TTX alone (n = 5 brain slices, from 4 mice), with TTX, TEA and Ba2+ together (to additionally block K+ channels, n = 3 brain slices, from 2 mice), with TTX and KCC2 blocked using VU 0463271 (1 brain slice), and with blockade of both glutamatergic and GABAergic neurotransmission in the presence of TTX (1 brain slice).
Figure 6.
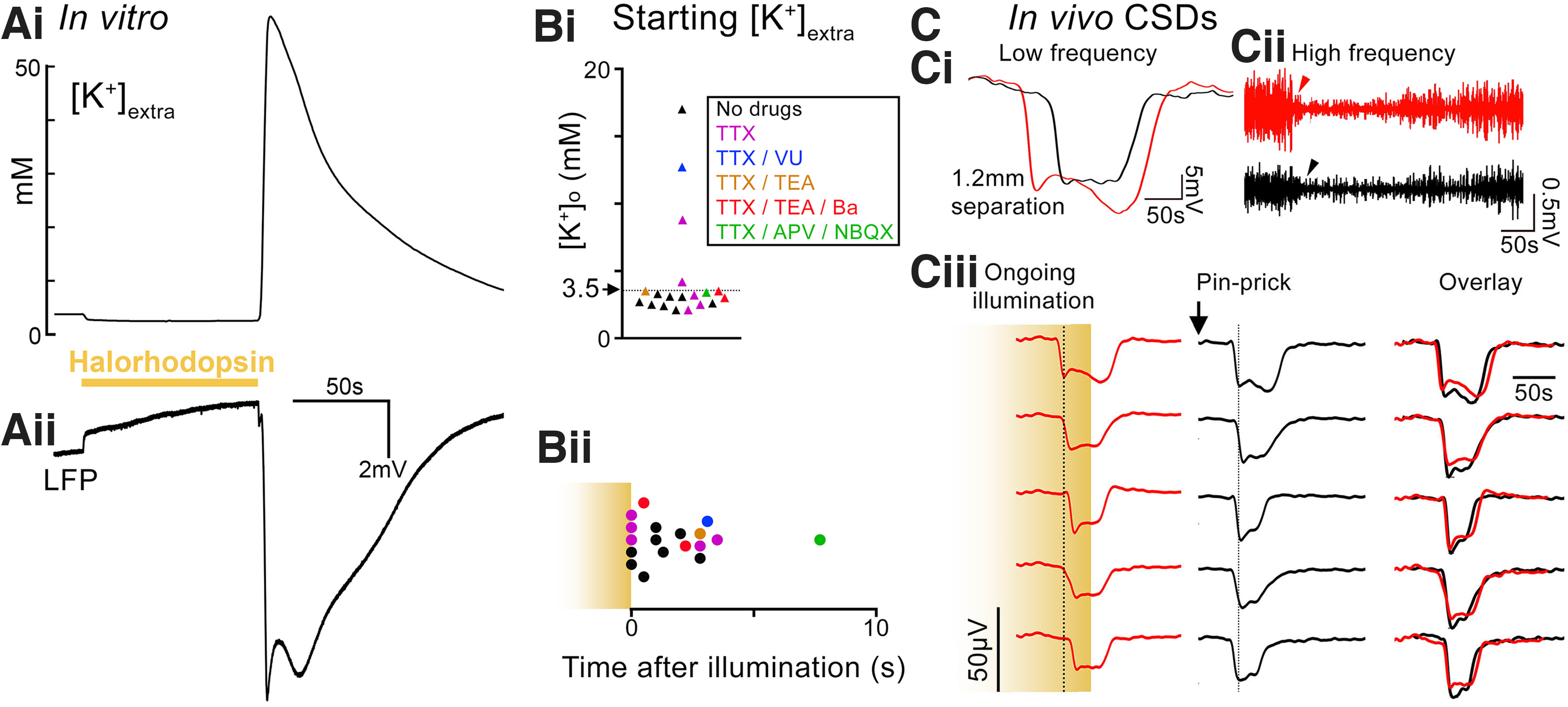
Prolonged activation of halorhodopsin in pyramidal populations can trigger CSDs. A, Concurrent recordings of extracellular K+ concentration (Ai), and the LFP (Aii), during a 90 s activation of halorhodopsin in the pyramidal population, in which a CSD event developed soon after the illumination period ended. B, The [K+]extra at the start of the CSD for the different drug groupings (Bi), and its time of initiation (Bii) relative to the end of a 90-s-long period of halorhodopsin activation (each data point is a single brain slice). C, Example traces of a CSD triggered in vivo, recorded using a graphene multitransistor array, which started while illumination was still ongoing (illumination was started ∼250 s before the start of the event). Top panel represents both low-frequency (Ci) and high-frequency (Cii) components, to illustrate how these different bandwidths can help distinguish between depolarization (the voltage deflection in Ci) and depression (the reduced high-frequency component in Cii). Ciii, Illustration of the propagation across multiple electrodes. Dotted line is aligned to the maximal deflection of the leading electrode, to help visualize the relative delays of onset in the other electrodes. A second CSD was triggered locally by a pinprick injury, and recorded through the same array. Right, The overlaid electrophysiological signatures of the two events, recorded through the different transistors, to illustrate the similar time course for the Halo- and pinprick-induced events, at each individual site (for the right panels, we aligned the recordings to their start). Calibration: 50 s, 50 µV.
Using synchronous measurements of the LFP and also the [K+]extra, we estimated that at the moment the CSD started, the median [K+]extra locally was 3.1 mm (interquartile range = 2.5-3.5 mm; full range = 2.1-17 mm; 19 brain slices, from 8 mice; Fig. 6B), and occurred shortly after (1.73 ± 1.91 s latency) the end of a period of illumination (typically <100 s illumination). Remarkably, if the illumination was maintained, then CSDs occurred in every slice (n = 6 slices, from 2 mice) with a latency of 526 ± 79.6 s. In contrast, CSDs were not induced by 600 s illumination with 565 nm light either in brain slices from WT mice (n = 6 slices, from 1 mouse) or from mice expressing channelrhodopsin-eYFP (n = 12 slices, from 2 mice). In each case, light was delivered at a spot, and notably, post hoc staining for DAPI, NeuN, and GFAP revealed no inhomogeneity of stain at that site, either in halorhodopsin slices that experienced CSDs (n = 12 slices, from 2 animals) or from the WT and channelrhodopsin-eYFP mice. The significance of CSDs occurring during periods of halorhodopsin activation is that, at this time, the expressing population of pyramidal neurons is hyperpolarized by the direct action of the opsin, and the [K+]extra is below baseline.
Notably, spreading depolarizations could be induced using this same method, in vivo, in anesthetized adult mice expressing halorhodopsin in pyramidal neurons. Illumination was directed onto the cortical surface, having performed a craniotomy, and with the dura removed to allow penetration of the electrode. In these experiments, the light fiber was out of the brain, fractionally above its surface. The period of illumination required to induce CSDs varied from animal to animal, but once we had established the requisite duration in an individual animal, this same stimulation consistently triggered CSDs thereafter, having allowed at least 15 min recovery between events (n = 4 mice).
In a final set of in vivo experiments, we recorded CSD propagation in awake head-fixed mice (n = 3) using multichannel graphene micro-transistor arrays (Masvidal-Codina et al., 2021), thereby avoiding any confounders that may arise from anesthesia. We recorded events using a 4 × 4 graphene microarray placed on the cortical surface. Data were collected at 16 bits, allowing a wide dynamic range in DC recordings (without saturating the signal), while retaining adequate voltage resolution. As such, these devices allow wide-bandwidth detection of electrographic signals and are suitable for recording spreading depolarizations and higher-frequency activity concurrently (Fig. 6Ci and Fig. 6Cii, respectively). We examined how Halo-induced CSDs compared with those triggered by physical trauma, in this case, by a brief pinprick to the cortical surface. In these recordings, CSDs induced by the two different methods appeared virtually identical at the individual transistors (Fig. 6Ciii). We concluded therefore that, although the method of induction differed, once established, the actual events were equivalent.
Discussion
The direct effect of illumination of halorhodopsin is well established: that it drives an inward movement of chloride ions, causing the cell to become hyperpolarized. This has been used in many previous studies to inhibit neuronal activity. Here we show that there are also indirect effects, and that these may have useful experimental value for probing neuronal pathophysiological mechanisms. The first of these is that sustained halorhodopsin activation causes a lowering of extracellular K+ levels. In all such cases, a steady state occurs when inward ionic movement equals the outward movement. In normal baseline conditions, there is a continuous small drift of K+ ions out of the cell through leak channels, driven by the difference between the membrane potential, Em, and EK. Since this is a steady state, it must be balanced by continual inward movement of K+ ions by other means, the most important of which is the Na+-K+ ATPase (Larsen et al., 2014). During the illumination, hyperpolarization alters the leak current driving force (EK – Em), reducing the outward movement of K+ initially, and even reversing its direction (to an inward movement) once Em drops below EK. The time course of the K+ redistribution is likely to have a complex relationship to the action of the halorhodopsin pump, since it is affected both by its hyperpolarizing, and also its chloride-loading, actions; the latter effect will increase K+ extrusion via KCC2, even during the period of illumination, and so counter the K+ entry previously mentioned. There may further be redistribution of K+ through the glial network that could affect how quickly the system reaches a steady state.
A consequence of this drop in [K+]extra is a measurable reduction in excitability of nonexpressing neurons in the local vicinity. This consequence is important to recognize when interpreting the inhibitory consequences of halorhodopsin, because not all the effects may be attributable to the specific subpopulation of neurons expressing the opsin. Having said that, it is also pertinent that our experiments used tissue with halorhodopsin expressed in the pyramidal population, which constitutes ∼80% of the neuronal population, in murine neocortex. The effect is likely to be far weaker with sparse expression patterns, and also will critically depend on the intensity of illumination.
We further show that, following cessation of halorhodopsin stimulation, there was a very marked rebound rise in [K+]extra. This likely constitutes, in part, a return to the extracellular space of some of the K+ that had entered the cell. The size of the overshoot beyond resting [K+]extra indicates that this is further boosted by cells extruding K+ coupled to Cl– extrusion, mediated by KCC2, as has been shown in several previous publications following intense GABAergic activation (Viitanen et al., 2010). The rebound [K+]extra may also contribute to the excess network excitability following halorhodopsin, which has previously been noted (Raimondo et al., 2012; Alfonsa et al., 2015).
An interesting and unanticipated finding was the final effect, which is that protracted, intense activation of halorhodopsin proves to be a very reliable means of triggering CSDs, both in vitro and in vivo. The significance of this new way of triggering CSDs is that it differs fundamentally from what has been a prevailing hypotheses of CSD initiation: the critical point is that instances of CSDs arising during the actual period of illumination demonstrate that they can develop from a point where all the pyramidal cells are hyperpolarized and the [K+]extra is actually below baseline levels (i.e., <3.5 mm). CSDs are thought to underlie spreading migraine auras (Lauritzen et al., 2011), and have also been described following both ischemic and hemorrhagic strokes (Dreier, 2011; Lemale et al., 2022), and seizures (Mody et al., 1987; Hablitz and Heinemann, 1989; Dreier and Reiffurth, 2015; Tamim et al., 2021). They were first described by Leao (1944, 1947), with the early realization that they could be triggered by increasing [K+]extra, leading to the idea that a strong depolarization was the means of induction and propagation. Latterly, a more nuanced view is evolving (Somjen, 2004; Dreier and Reiffurth, 2015; Lemale et al., 2022), taking into account other ways in which CSDs may be triggered, such as raised osmotic tension within neurons. Our main findings are consistent with this hypothesis, by describing a process whereby a continuous electrogenic pump, driving first Cl–, and secondarily K+ ions into neurons, triggers the CSD, but fall short of a conclusive proof of the theory. New experimental models, however, often provide the impetus for new insights, and we suggest that halorhodopsin induction could fuel research into how CSDs arise naturally. One important instance is where CSDs arise from seizure activity. Persistently intense neuronal activity, as occurs during seizures, results in a large net movement of ions into cells, the opening of pannexin channels in neuronal membranes (Thompson et al., 2008), allowing movement of large molecules, such as fluorescent dyes, and likely also allowing water to follow. This, in turn, leads to cell swelling and a substantial drop in the extracellular space (Iwasa et al., 1980; Andrew and MacVicar, 1994; Somjen, 2004). Indeed, the change in the tissue impedance because of this redistribution can be used to characterize the spatial-temporal extent of pathologic discharges (Witkowska-Wrobel et al., 2021). An important research question remains regarding the relationship of CSDs with seizure activity (Dreier and Reiffurth, 2015). Superficially, seizure activity and halorhodopsin activation appear rather different in terms of the effects on membrane potential but share an important feature that, in both, there is a net influx of ions into neurons.
Footnotes
The work was supported by Biotechnology and Biological Sciences Research Council Grant BB/P019854/1, Medical Research Council Grant MR/R005427/1, and European Union's Horizon 2020 Research and Innovation Program Grant Agreement 881603 (GrapheneCore3). A.C. was supported by National Institute for Health and Care Research, Academic Clinical Fellowship in Neurology. We thank the support staff in the animal facilities at both Newcastle University and University College London; Dr. Anton Guimera-Brunet and Dr. Eduard Masvidal-Codina (Institut de Mircroelectronica de Barcelona, CNM-CSIC) for providing the graphene micro-transistor arrays used in this study; Dr. Martin Smith (University College London) for technical assistance in preparing the mice for the graphene micro-transistor array in vivo experiments; and Joe Raimondo for commenting on drafts of this manuscript and discussion.
The authors declare no competing financial interests.
References
- Alfonsa H, Merricks EM, Codadu NK, Cunningham MO, Deisseroth K, Racca C, Trevelyan AJ (2015) The contribution of raised intraneuronal chloride to epileptic network activity. J Neurosci 35:7715–7726. 10.1523/JNEUROSCI.4105-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonsa H, Lakey JH, Lightowlers RN, Trevelyan AJ (2016) Cl– out is a novel cooperative optogenetic tool for extruding chloride from neurons. Nat Commun 7:13495. 10.1038/ncomms13495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew RD, MacVicar BA (1994) Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience 62:371–383. 10.1016/0306-4522(94)90372-7 [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145. 10.1097/00004647-200110000-00001 [DOI] [PubMed] [Google Scholar]
- Chang M, Dian JA, Dufour S, Wang L, Moradi Chameh H, Ramani M, Zhang L, Carlen PL, Womelsdorf T, Valiante TA (2018) Brief activation of GABAergic interneurons initiates the transition to ictal events through post-inhibitory rebound excitation. Neurobiol Dis 109:102–116. 10.1016/j.nbd.2017.10.007 [DOI] [PubMed] [Google Scholar]
- Chebabo SR, Hester MA, Aitken PG, Somjen GG (1995) Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res 695:203–216. 10.1016/0006-8993(95)00778-o [DOI] [PubMed] [Google Scholar]
- Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE, Boyden ES (2010) High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463:98–102. 10.1038/nature08652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K (2011) Optogenetics. Nat Methods 8:26–29. 10.1038/nmeth.f.324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP (2011) The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17:439–447. 10.1038/nm.2333 [DOI] [PubMed] [Google Scholar]
- Dreier JP, Reiffurth C (2015) The stroke-migraine depolarization continuum. Neuron 86:902–922. 10.1016/j.neuron.2015.04.004 [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22:6309–6314. 10.1523/JNEUROSCI.22-15-06309.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hablitz JJ, Heinemann U (1989) Alterations in the microenvironment during spreading depression associated with epileptiform activity in the immature neocortex. Brain Res Dev Brain Res 46:243–252. 10.1016/0165-3806(89)90288-5 [DOI] [PubMed] [Google Scholar]
- Han X, Boyden ES (2007) Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS One 2:e299. 10.1371/journal.pone.0000299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF (1952) Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol 116:449–472. 10.1113/jphysiol.1952.sp004717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa K, Tasaki I, Gibbons RC (1980) Swelling of nerve fibers associated with action potentials. Science 210:338–339. 10.1126/science.7423196 [DOI] [PubMed] [Google Scholar]
- Jefferys JG (1995) Nonsynaptic modulation of neuronal activity in the brain: electric currents and extracellular ions. Physiol Rev 75:689–723. 10.1152/physrev.1995.75.4.689 [DOI] [PubMed] [Google Scholar]
- Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N (2014) Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4.1 to hippocampal K(+) clearance and volume responses. Glia 62:608–622. 10.1002/glia.22629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ (2011) Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 31:17–35. 10.1038/jcbfm.2010.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao AA (1944) Spreading depression of activity in the cerebral cortex. J Neurophysiol 7:359–390. 10.1152/jn.1944.7.6.359 [DOI] [PubMed] [Google Scholar]
- Leao AA (1947) Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol 10:409–414. 10.1152/jn.1947.10.6.409 [DOI] [PubMed] [Google Scholar]
- Lemale CL, Luckl J, Horst V, Reiffurth C, Major S, Hecht N, Woitzik J, Dreier JP (2022) Migraine aura, transient ischemic attacks, stroke, and dying of the brain share the same key pathophysiological process in neurons driven by Gibbs-Donnan forces, namely spreading depolarization. Front Cell Neurosci 16:837650. 10.3389/fncel.2022.837650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M, Misuri M, Brogioli D (2014) A new open source software for the calculation of the liquid junction potential between two solutions according to the stationary Nernst-Planck equation. arXiv 14033640v2. [Google Scholar]
- Masvidal-Codina E, Smith TM, Rathore D, Gao Y, Illa X, Prats-Alfonso E, Corro ED, Calia AB, Rius G, Martin-Fernandez I, Guger C, Reitner P, Villa R, Garrido JA, Guimerà-Brunet A, Wykes RC (2021) Characterization of optogenetically-induced cortical spreading depression in awake mice using graphene micro-transistor arrays. J Neural Eng 18:055002. 10.1088/1741-2552/abecf3 [DOI] [PubMed] [Google Scholar]
- Mody I, Lambert JD, Heinemann U (1987) Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol 57:869–888. 10.1152/jn.1987.57.3.869 [DOI] [PubMed] [Google Scholar]
- Raimondo JV, Kay L, Ellender TJ, Akerman CJ (2012) Optogenetic silencing strategies differ in their effects on inhibitory synaptic transmission. Nat Neurosci 15:1102–1104. 10.1038/nn.3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiri Z, Manseau F, Levesque M, Williams S, Avoli M (2015) Interneuron activity leads to initiation of low-voltage fast-onset seizures. Ann Neurol 77:541–546. 10.1002/ana.24342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG (2004) Ions in the brain: normal function, seizures and stroke. Oxford: Oxford UP. [Google Scholar]
- Tamim I, Chung DY, de Morais AL, Loonen IC, Qin T, Misra A, Schlunk F, Endres M, Schiff SJ, Ayata C (2021) Spreading depression as an innate antiseizure mechanism. Nat Commun 12:2206. 10.1038/s41467-021-22464-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, MacDonald JF, MacVicar BA (2008) Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 322:1555–1559. 10.1126/science.1165209 [DOI] [PubMed] [Google Scholar]
- Ullah G, Wei Y, Dahlem MA, Wechselberger M, Schiff SJ (2015) The role of cell volume in the dynamics of seizure, spreading depression, and anoxic depolarization. PLoS Comput Biol 11:e1004414. 10.1371/journal.pcbi.1004414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viitanen T, Ruusuvuori E, Kaila K, Voipio J (2010) The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol 588:1527–1540. 10.1113/jphysiol.2009.181826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voipio J, Pasternack M, MacLeod K (1994) Ion-sensitive microelectrodes. In: Microelectrode techniques: the Plymouth workshop handbook (Ogden D, ed). Cambridge, UK: Company of Biologists. [Google Scholar]
- Witkowska-Wrobel A, Aristovich K, Crawford A, Perkins JD, Holder D (2021) Imaging of focal seizures with Electrical Impedance Tomography and depth electrodes in real time. Neuroimage 234:117972. 10.1016/j.neuroimage.2021.117972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, Deisseroth K (2007) Multimodal fast optical interrogation of neural circuitry. Nature 446:633–639. 10.1038/nature05744 [DOI] [PubMed] [Google Scholar]