

Abstract
In acute myeloid leukemia (AML), p53 tumor suppressor activity can be reduced due to enhanced expression of MDM2 which promotes the degradation of p53. In TP53 wild-type malignancies, therapy with small molecule antagonists of MDM2 results in antileukemic activity. Current treatment strategies, however, have been limited by poor tolerability and incomplete clinical activity. We have developed a proteolysis-targeting chimera (PROTAC) MS3227 that targets MDM2 by recruiting the E3 ligase Von Hippel-Lindau, resulting in proteasome-dependent degradation of MDM2. In WT TP53 leukemia cell lines, MS3227 led to activation of p53 targets p21, PUMA, and MDM2 and resulted in cell-cycle arrest, apoptosis, and decreased viability. The catalytic PROTAC MS3227 led to more potent activation when compared to a stoichiometric inhibitor, in part by dampening the negative feedback mechanism in the p53 – MDM2 circuit. The effectiveness of MS3227 was also observed in primary patient specimens with selectivity towards leukemic blasts. The addition of MS3227 enhanced the activity of other anti-leukemic agents including azacytidine, cytarabine, and venetoclax. In particular, MS3227 treatment was shown to downregulate MCL-1, a known mediator of resistance to venetoclax. A PROTAC-based approach may provide a means of improving MDM2 inhibition to gain greater therapeutic potential in AML.
Introduction
P53 pathway dysregulation occurs in acute myeloid leukemia (AML) and other myeloid diseases through several different mechanisms [1, 2]. Alterations in the TP53 gene itself, including mutations and deletions are present in less than a quarter of AML cases with the majority occurring in therapy-related AML or AML associated with myelodysplastic syndrome changes [3]. In TP53 wild-type (WT) leukemia the p53 pathway can be downregulated through alternate mechanisms such as increased activity of Mouse Double Mutant 2 homolog (MDM2). MDM2 downregulates p53 activity by inhibiting p53 dependent transcription, facilitating its nuclear export, and promoting its degradation [4]. As a target of p53 transcription itself, MDM2 modulates activity through an autoregulatory feedback mechanism. In up to half of leukemias MDM2 is overexpressed and is associated with unfavorable karyotype [5]. MDM2 is therefore a therapeutic target in TP53 WT AML. Small molecule antagonists against MDM2, termed nutlins, have been investigated in clinical trials of AML and myeloproliferative neoplasms (MPNs) [6, 7]. Nutlins and other similar inhibitors disrupt the interaction of MDM2 and p53 by binding to the MDM2 p53-binding pocket [8]. These compounds have significant clinical activity in AML and MPN, and higher expression levels of MDM2 in AML blasts have been shown to correlate with clinical response [9]. These agents, however, are associated with gastrointestinal and hematologic toxicities [10]. Alternative agents that can provide better specificity or change pharmacodynamic properties are needed to better target this pathway.
A novel approach to the targeting of oncogenic proteins is the use of PROteolysis TArgeting Chimeras (PROTACs) which facilitate chemical knockdown [11]. PROTACs connect a protein of interest to an E3 ubiquitin ligase via a linker — inducing formation of a ternary complex that initiates ubiquitination and subsequent degradation. This PROTAC-induced degradation is catalytic in nature, acting in a sub-stoichiometric fashion which allows for greater potency than corresponding inhibitors and has been shown to prevent the development of resistance [12]. Unlike stoichiometric inhibitors, this activity may also counterbalance feedback mechanisms as in the p53-MDM2 axis - specifically, the accumulation of MDM2 itself as p53 is activated. MDM2 PROTACs have been previously developed using Cereblon (CRBN) as the E3 ligase [13–15]. However, immunomodulatory imide drugs (IMiD)-based targeting agents are possibly compromised by neosubstrates that are also degraded [16]. The consequences of degrading these substrates are still unclear. For example, prolonged IMiD exposure has been associated with the risk of venous thrombosis and secondary malignancies [17, 18]. To more efficiently target MDM2 in AML we have developed an MDM2 VHL-based degrader, MS3227, which we show to be a selective degrader of MDM2 with enhanced anti-leukemic activity.
Materials / Methods
Cell culture
The HEL, MOLM-13, MOLT-4, THP-1, U-937, OCI-AML3, SIGM5, K-562, NOMO-1 cell lines were maintained in RPMI-1640 + 10% FBS; MV4–11 and UKE-1 in IMDM + 10% FBS; solid tumor cell lines as follows: HCT 116 in RPMI + 10% FBS, MCF7 in IMDM + 10% FBS, and Caco-2, A549, HepA2 in DMEM + 10–20% FBS. Cells were obtained from ATCC and DSMZ. OP9-DL1 was maintained in αMEM without nucleosides + 20%FBS + 1% L-glutamine on 0.1% gelatin coated flasks. AMG 232 was supplied by MedChemExpress. Puromycin was used at 1–2μg/mL. MLN4924 (Chemietek) and MG132 (Selleckchem) were used at 1 μM.
Cell proliferation assay
Cell viability and proliferation was performed using the Promega CellTiter-Glo Luminescent Cell Viability Assay (#G924A).
Western blot analysis
Protein lysates were prepared with cell lysis buffer with protease and phosphatase inhibitors and protein concentrations were determined using Bradford assays. Proteins were run on NuPAGE Bis-Tris gels and transferred to nitrocellulose membranes. Membranes were blocked in 5% non-fat milk in 1X TBST and incubated with respective primary antibodies overnight at 4°C (1:1000 dilution), followed by an anti-rabbit or anti-mouse secondary IgG antibody. Protein bands were then detected using chemiluminescence-detection and the signal was visualized using a digital imager (BioRad ChemiDoc). Densitometry was performed using ImageJ analysis.
Antibodies (Cell Signaling Technology catalog #): Vinculin (13901), p21 (2947), p53 (48818), GAPDH (97166), Actin (3700), VHL (68547), MDM2 (86934), MDMX (3703264), PUMA (98672), MCL-1 (94296), Tubulin (2146)
Lentivirus CRISPR and Transgene
The VHL was edited using LentiCRISPRv2 puro (Addgene). VHL guide RNA were inserted using the target sequence: TCTGAAAGAGCGATGCCTCC. Lentivirus was produced in 293T cells co-transfected with lentivirus plasmid, MD2G, and psPAX plasmids using JetPrime reagent. Using polybrene, target cells were transduced with lentivirus. MDM2 was cloned using pDONR223_MDM2_WT and pLenti PGK Puro DEST (w529–2) from Addgene.
Flow-cytometric Analyses
Cell apoptosis was assessed using FITC Annexin V (640945, Biolegend) and Propidium Iodide. Immunophenotyping was performed using the following antibodies: CD34 (clone 561), CD45 (HI30), and CD33 (P67.6). Data was collected on an Attune Flow Cytometer (ThermoFisher Scientific) and analyzed using FCS Express.
Primary AML samples and culture
Patients at the Mount Sinai Hospital provided informed consent for biological samples research, in accordance with the Declaration of Helsinki. Primary AML cells were obtained through the Hematological Malignancies Tissue Bank of Tisch Cancer Institute through an IRB-approved protocol. AML cells were grown on OP9-DL1 (OP9) cells [19], in a gelatin-coated 48 well plates, irradiated at 3000 rad. Primary AML cells were then seeded at 5×104 cells/mL density in IMDM and cytokines: SCF and FLT3 at 50 ng/mL, IL3, IL6, GM-CSF, G-CSF at 20 ng/mL, penicillin-streptomycin, FBS 2% and 2-mercaptoethanol at 50 μM.
Statistical Analysis
Analysis was performed using GraphPad Prism software and Microsoft Excel. Data are presented as mean +/− standard deviation. Statistical significance was calculated using the Student t-test. IC50 determination was performed using a log-transformed, non-linear regression curve fit analysis. Differences with p < 0.05 were considered statistically significant. TCGA gene expression data was analyzed using Firebrowse software from the Broad Institute. Synergy calculations were made using SynergyFinder [20] using the Bliss and Zero interaction potency (ZIP) models, with stochastic, independent effects.
Chemical Synthesis and Characterization of Compounds
The synthetic procedures and the characterization of novel compounds 2–12 (Schemes S1 & S2); as well as 1H-NMR and LCMS spectra of compounds 5 (MS3227) and 12 (MS3227N) (Fig. S4–S7A) are described and included in the supplementary information section.
Results
Design, synthesis, and evaluation of VHL-based MDM2 PROTAC degraders
To design a PROTAC degrader for MDM2, we selected AMG 232 as the small molecule ligand scaffold. AMG 232 binds MDM2 in the interaction pocket with p53 and inhibits ubiquitination and degradation of p53 [21]. It binds specifically to MDM2 and not MDMX to an appreciable extent. To select the E3 binding partner for MDM2 PROTAC design, we assessed the expression of VHL in the TCGA RNA-seq data set. AMLs have relatively higher expression of VHL compared to other cancer subtypes and normal tissues (Fig. 1A). Thus, the selection of VHL could potentially target MDM2 inhibition in AML cells over other cell types.
Figure 1. Design and Structure Activity Relationship of MDM2 degraders.

A. RNA-seq data from TCGA database of various tumor types analyzed by FireBrowse (Broad Institute) for VHL expression. RPKM – Reads Per Kilobase of transcript, per Million mapped reads. LAML, Acute myeloid leukemia, Other tumor types as per TCGA study abbreviations.
B. Western blot for MDM2 and actin in U-937 cells treated with MDM2 PROTAC candidate compounds at 250 nM and 1 μM for 24hr.
C. Structure of compound 5, MS3227. Blue, MDM2 binder; black, linker; red, VHL binder.
The co-crystal structure of MDM2 in complex with a close analog of AMG 232 (PDB code: 4OAS) revealed that the isopropyl sulfonyl or carboxylic acid groups of AMG 232 (Fig. S1A) are solvent exposed and suitable positions for the linker attachment. However, since the carboxylic acid moiety of AMG 232 forms a key hydrogen bonding interaction with MDM2, we elected to use the sulfonyl group as a handle for attachment. We used compound 1, a known analog of AMG 232 with a piperazine sulfonyl group [22] for the design of putative MDM2 PROTAC degraders. Linkers play a crucial role in the development of PROTACs, and we designed compounds featuring different linkers as VHL-recruiting MDM2 degraders (Fig. S1B). Compounds with linkers composed of alkylene (compounds 2–6, Fig. S1B) or polyethylene glycol (PEG) (compounds 7–11, Fig. S1B) with varying lengths were synthesized. The synthesis (Scheme S1) and spectral characterization of these and related compounds were obtained (Fig. S1–S6). With these putative degraders, we next assessed the MDM2 protein levels by western blot (WB) in the human leukemia cell line, U-937 after treatment with 250 nM and 1 μM of the candidate compounds for 24 hrs (Fig. 1B). Compound 2 with the shortest 2-carbon linker showed no obvious degradation, while the compounds 3 and 4 bearing 3- and 6-carbon linkers, respectively, showed degradation at 1 μM only or limited degradation. The most effective MDM2 degradation was seen with compounds 5 and 6, featuring longer carbon chain linkers (8- and 9-carbons, respectively) at both concentrations. The same trend was seen in U-937 cells over-expressing MDM2 (Fig. S7A). On the other hand, compounds featuring PEG linkers (compounds 7-11) were not effective at all in degrading MDM2. Therefore, based on this SAR study, we have selected compound 5 (MS3227) for further characterization (Fig. 1C).
MS3227 is a PROTAC Degrader of MDM2
To further characterize MS3227’s PROTAC activity, we selected the U-937 cell line that has a TP53 inactivating mutation. Since MDM2 is itself a p53 transcription target, use of a TP53 mutant cell line would allow for more direct assessment of MS3227’s effect on MDM2 degradation without implicating feedback mechanisms that would secondarily impact MDM2 levels. MS3227 treatment resulted in the concentration dependent depletion of MDM2 and was effective at 100 nM and 250 nM (Fig. 2A and quantified in Fig. S7B). This depletion was specific to MDM2 and not the related protein MDMX (Fig. 2A and quantified Fig. S7B). Since MDM2 is itself an E3 ligase and could theoretically target VHL for ubiquitination, we also demonstrated that VHL did not significantly change (Fig. 2A). It has been reported that higher concentrations of PROTAC degraders can cause a “hook effect” that results in binary complexes rather than the ternary complex of E3 ligase, degrader molecule, and target [23]. This would lead to inefficient target depletion at higher concentrations due to separate ligase and target sequestration. We observed this phenomenon at 10 μM of MS3227 (Fig. S7C). Rapid degradation of MDM2 with MS3227 treatment (500 nM) occurred within 1 hr (Fig. 2B).
Figure 2. MS3227 acts as an MDM2 PROTAC.

A. Western blot of U-937 cells treated with various concentrations (nM) of MS3227 for 24 hr and probed for expression of MDM2, VHL, MDMX, and vinculin.
B. Western blot of U-937 cells treated with MS3227 at 500 nM treated at various time points (hrs) and probed for MDM2 and vinculin.
C. (left) Western blot of U-937 cells treated with MS3227 (500 nM) and / or the proteasome inhibitor MG132 (1 μM) for 1 hr and probed for MDM2 and vinculin. (right) Western blot of U-937 cells treated with MS3227 (100 nM) for 1 hr and / or the neddylation inhibitor MLN4924 (1 μM) for 1 hr pre-treatment and probed for MDM2 and actin.
D. Western blot of U-937 cells treated with MS3227 (500 nM) and a negative control MS3227N (500 nM) for 1 hr and probed for MDM2 and actin.
E. Western blot of U-937 cells treated with MS3227 (250 nM) and / or a VHL binding ligand (VHL-lig (Ac-VHL1) 2 μM) for 1 hr and probed for MDM2 and vinculin.
F. Western blot of U-937 cells modified with control lentivirus or with CRISPR lentivirus sgRNA for VHL and probed for VHL and actin.
G. U-937 VHL CRISPR KO cells treated with MS3227 (500 nM) at various time points (hrs) and probed for MDM2 and vinculin.
Binding of p53 increases the stability of MDM2 [24], and treatment with small molecule MDM2 inhibitors that bind in a similar fashion likewise increases MDM2 [25]. We thus compared AMG 232 and MS3227 in terms of MDM2 protein accumulation in other cell lines. In the colon cancer cell line HCT 116 and the leukemia cell line THP-1, AMG 232 treatment resulted in increased MDM2 while treatment with MS3227 led to relatively lower MDM2 levels at the same concentration (Fig. S7D).
To further assess the mechanism of MS3227 action, co-treatment with the proteasomal inhibitor MG132 at 1 μM blocked the ability of MS3227 to deplete MDM2, indicating that proteasomal degradation is necessary (Fig. 2C). Similarly, pre-treatment with the neddylation inhibitor MLN4924 at 1 μM for 1 hr also blocked MS3227 degradation of MDM2, indicating that E3 ubiquitin ligase activity (Cullin activation) is necessary (Fig. 2C). The dependence of PROTAC activity on VHL binding was tested using a negative control compound (MS3237N). MS3227N (compound 12, Fig. S2) was designed to be incapable of binding VHL by changing the VHL binding portion of MS3227 to its diastereomer [26] and keeping the linker and MDM2 binding portion of MS3227N identical to that of MS3227 (Scheme S2). This negative control compound did not degrade MDM2 at a concentration of 500 nM (Fig. 2D). Next, we employed a competitive binder of VHL, using a VHL ligand (Ac-VHL1), to assess the VHL-dependence of MS3227 activity. Co-treatment of MS3227 with an excess of VHL ligand [27] blocked MS3227 depletion of MDM2 (Fig. 2E). We also assessed genetic knock-out of VHL on the activity of MS3227. Using CRISPR methods we targeted VHL in the U-937 cell line (Fig. 2F). When testing MS3227 at 500 nM in this cell line, MS3227 was no longer able to deplete MDM2 (Fig. 2G), and an increase in MDM2 was observed. As MS3227 contains the MDM2 binding AMG 232 analog, its binding would stabilize MDM2 in the absence of VHL, leading to its accumulation. These data demonstrate that MS3227 works as an MDM2 degrader in leukemia cells in a concentration-, time- and proteasome-dependent manner.
MS3227 activates the p53 pathway activity with greater potency than a small molecule MDM2 antagonist
MDM2 acts as an inhibitor of the p53 pathway, and depletion of MDM2 is expected to activate p53 and its downstream targets. We compared the activity of MS3227 to the MDM2 antagonist AMG 232. Although both agents are able to activate p53 in the WT TP53 MOLM-13 leukemia cell line, p53 protein levels were induced to a higher level with MS3227 compared to AMG 232 at similar concentrations (Fig. 3A). Levels of p53-transciptional target CDKN1A (p21) were increased and more robustly induced in MS3227 than AMG232 treated samples (Fig. 3A). This was associated with cell cycle arrest and apoptosis (Fig. S7E, S7F). Apoptosis and cell killing occurred in a concentration dependent manner (Fig. S7G). MDM2 itself is a transcriptional target of p53 and therefore treatment with either MS3227 or AMG 232 leads to elevation of expression of p53 and MDM2. MDM2 levels, however, reached similar levels in the face of even higher p53 and p21 accumulation following MS3227 treatment because MS3227 but not AMG 232 shortens the half-life of MDM2 (Fig. 3A). Similar results were observed in the MOLT-4 leukemia cell line with more potent induction of p21 by MS3227 despite lower levels of MDM2 at longer time points (Fig. S8A). We confirmed p53 transcriptional activity by measuring MDM2 mRNA levels and observed increased transcripts 4 hrs after treatment with MS3227 (Fig. 3B). In contrast TP53 transcript levels were largely unchanged despite increased protein (Fig. 3B), consistent with the proposed mechanism of MS3227 on protein stability.
Figure 3. MS3227 activates the p53 pathway and with higher potency than standard inhibitor.
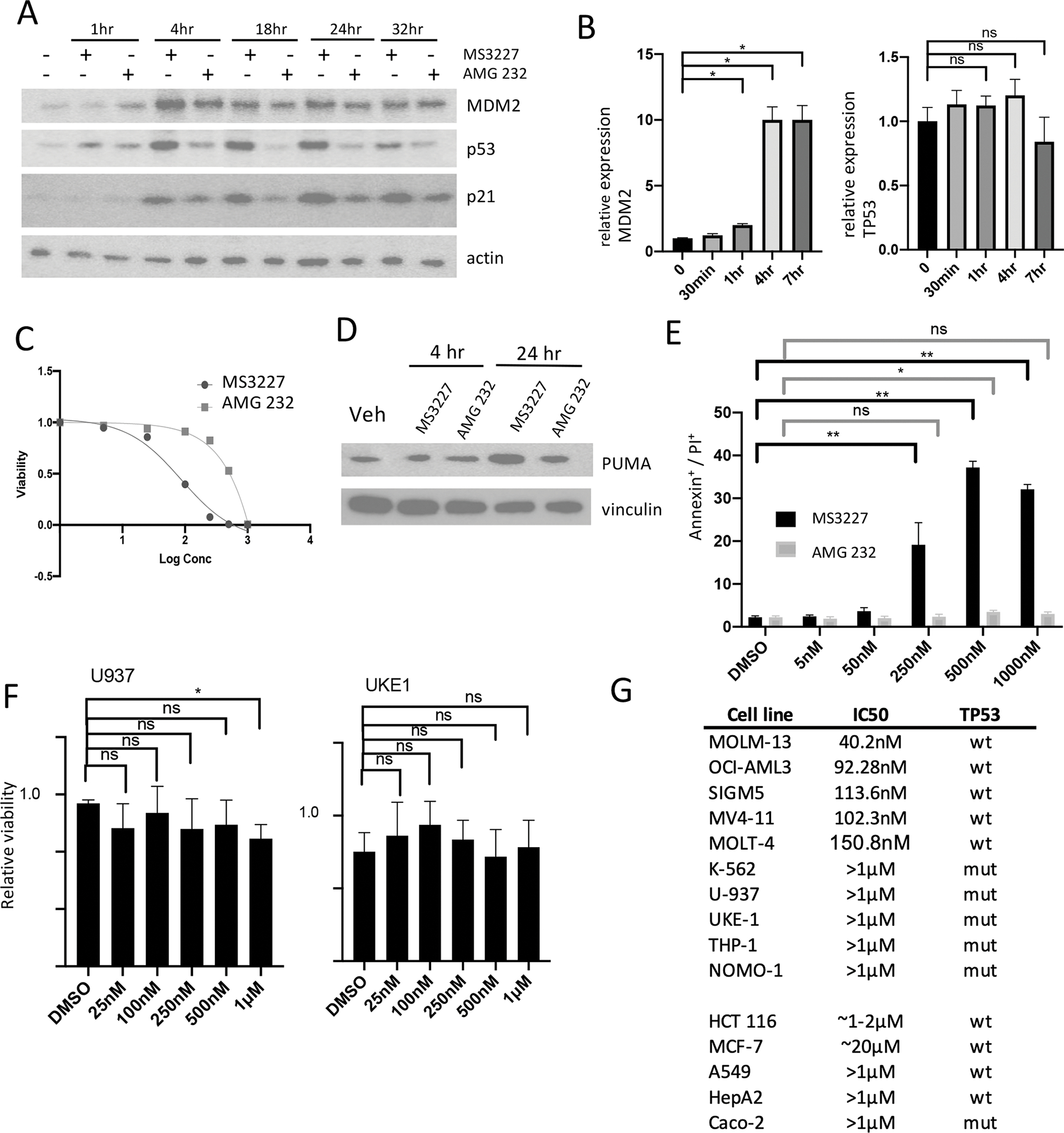
A. Western blot of MOLM-13 cells treated with MS3227 (250 nM) or AMG 232 (250 nM) at various time points and probed for MDM2, p53, p21, and vinculin.
B. qRT-PCR analysis of MOLM-13 cells treated with MS3227 (250 nM) at various time points and assessed for relative RNA-expression of MDM2 and TP53.
C. Dose-response curve cell viability for MOLM-13 cells treated with MS3227 or AMG 232 for 3 days.
D. Western blot of MOLM-13 cells treated with MS3227 or AMG 232 at 4 hr and 24 hr and probed for PUMA and vinculin.
E. Bar graph of MOLM-13 cells with flow cytometry for Annexin+ / PI+ cells following treatment with MS3227 or AMG 232 at various concentrations after 24 hr wash-out.
F. Bar graph of U-937 and UKE-1 cells with flow cytometry for Annexin+/PI+ cells following treatment with MS3227 at various concentrations (25 nM to 1 μM) after 2 days.
G. Table of cancer cell lines and IC50 as determined by cell-titer glo viability assay after treatment for 2 to 3 days with MS3227, and cell line TP53 mutation status. > 1 μM, IC50 not reached.
ns, not significant; * p<.05; ** p<.01
To assess the effect on leukemic cell growth we tested MOLM-13 cells treated with MS3227 and AMG 232. Both agents inhibited leukemia cell proliferation but MS3227 (IC50= ~50 nM) was more potent compared to AMG 232 (IC50= ~250 nM) (Fig. 3C). The dose response curve also suggests feedback mechanisms that may change the kinetics of MDM2 inhibition and p53 activation with AMG 232 compared to MS3227, with non-linear responses to PROTAC activity compared to stoichiometric inhibition (Fig. S8B). This growth inhibition was associated with induction of PUMA, a pro-apoptotic Bcl-2 family protein 24 hrs post treatment (Fig. 3D). We assessed p53 transcriptional targets CDKN1A (p21) and PUMA and similar relative potency results were observed (Fig. S8C, Fig. S8D). An increase in potency of MS3227 to induce apoptosis as compared to AMG 232 was also observed in MOLT-4 leukemia cells (Fig. S8E). We also assessed pharmacodynamic properties of MS3227 by treating MOLM-13 cells with MS3227 or AMG 232, changing to drug free media for 24 hrs, and then assessing cell death. After this washout period, more MS3227 treated cells continued to undergo apoptosis as compared to treatment with AMG 232, suggesting a greater sustained effect by the degrader (Fig. 3E).
MS3227 growth inhibition and potency are dependent on TP53 mutational status and VHL expression
We assessed the growth inhibitory activity of MS3227 in the TP53 mutant leukemia cell lines U-937 and UKE-1. Both cell lines were relatively resistant to the effects of MS3227 up to concentrations of 1 μM (Fig. 3F). Similarly, MS3227 did not increase apoptosis in the TP53 mutant MPN-derived HEL cell line (Fig. S8F). TP53 mutant lines were relatively resistant to the growth inhibitory effects of MS3227 up to 1 μM compared to TP53 WT cell lines that had IC50 concentrations ranged between 40nM-150nM (Fig. 3G, upper). We also tested a series of TP53 wild-type and mutant solid tumor cell lines (Fig. 3G, lower). HCT 116 demonstrated an IC50 ~1–2 μM, consistent with induction of p53 and downstream targets (Fig. S8G). Other cell lines were relatively resistant. For MCF7 cells we also tested higher concentrations with IC50 ~20 μM (Fig. S8H). Thus, leukemia cell lines in general were more sensitive to MS3227 treatment than the solid tumor lines studied.
Since VHL is required for MS3227’s PROTAC activity, we assessed whether loss or decreased expression of VHL would affect drug sensitivity. We generated a MOLM-13 cell culture with CRISPR VHL knockdown which resulted in two- to three-fold decreased potency of MS3227 (Fig. S8I).
MS3227 is effective against primary AML samples
In order to evaluate the activity of MS3227 against primary AML cells we utilized a long-term primary cell culture system whereby mononuclear cells are cultured on a murine stromal cell line (OP9-DL1). After 9 days of treatment samples were assayed for viability. Primary patient AML cells underwent significant apoptosis and cell death when treated with MS3227 at 250–500 nM (Fig. 4A). As expected, MS3227 was not active in an AML sample with deletion of TP53 (Fig. 4B). Sensitivity to MS3227 was observed in samples with disparate mutational profiles and cytogenetic abnormalities (Fig. 4C). In comparison to AMG 232, MS3227 induced an equal or greater degree of apoptosis and cell death in primary AML specimens (Fig. 4D). Mononuclear cells from the bone marrow of normal healthy donors were not more sensitive to MS3227 (Fig. 4E). In this co-culture system we also observed selectivity of MS3227 towards CD34+ CD33+ leukemic blasts as compared to the stem-progenitor CD34+ CD33− population (Fig. 4F Leuk E and Fig. S9A Leuk G). Although WT TP53 predicted for MS3227 sensitivity in most cases there may be alternative resistance mechanisms as we observed resistant AMLs without known TP53 mutations or deletions (Fig. 4C, 4F Leuk F).
Figure 4. MS3227 is active in primary leukemia specimens.

A & B. Bar graph of primary (A) TP53 wild-type (B) TP53 mutant AML cells in OP9 co-culture for Annexin+/PI+ cells following treatment with MS3227 at 250 nM and 500 nM for 9 days.
C. Table of primary AML cells used with mutation profile, karyotype, and sensitivity or resistance to MS3227. cnLOH (Array), copy neutral loss of heterozygosity by Array testing.
D. Bar graph of primary TP53 wild-type AML cells in OP9 co-culture with for Annexin+/PI+ cells following treatment with MS3227 or AMG 232 at 250 nM for 9 days.
E. Bar graph of normal donor bone marrow (BM) CD34+ cells in OP9 co-culture for Annexin+/PI+ cells following treatment with MS3227 at 250 nM for 9 days.
F. Flow cytometry plot of primary AML samples treated with vehicle or MS3227 at 100 nM for 6 days (Leuk E) and 250 nM for 4 days (Leuk F). Samples were analyzed for fraction of AML cells (CD33+CD34+ in Leuk E, CD33+CD34− in Leuk F) and stem-progenitor cells (CD33−CD34+). Comparisons made between Vehicle and MS3227 treated samples.
ns, not significant; * p<.05; ** p<.01.
MS3227 has cooperative effects with other therapeutic agents used to treat AML
To assess the potential for additive effects with other treatments, we tested MS3227 in combination with other agents presently used to treat AML, azacytidine and cytarabine, in cell lines sensitive to MS3227. In MOLM-13 cells, MS3227 demonstrated cooperative effects with cytarabine but not azacytidine (Fig. 5A and Fig. S9B). Using drug synergy calculations [28], MS3227 at 100nM and cytarabine at 25nM demonstrated positive cooperative activity. In OCI-AML3 cells, MS3227 had positive cooperative effects at 25 nM with cytarabine at 250 nM, and with azacytidine at 2.5 μM (Fig. 5B and Fig. S9B). Thus, even at lower doses, MS3227 could potentially enhance the activity of common AML agents.
Figure 5. MS3227 cooperates with other leukemia therapies for greater efficacy.
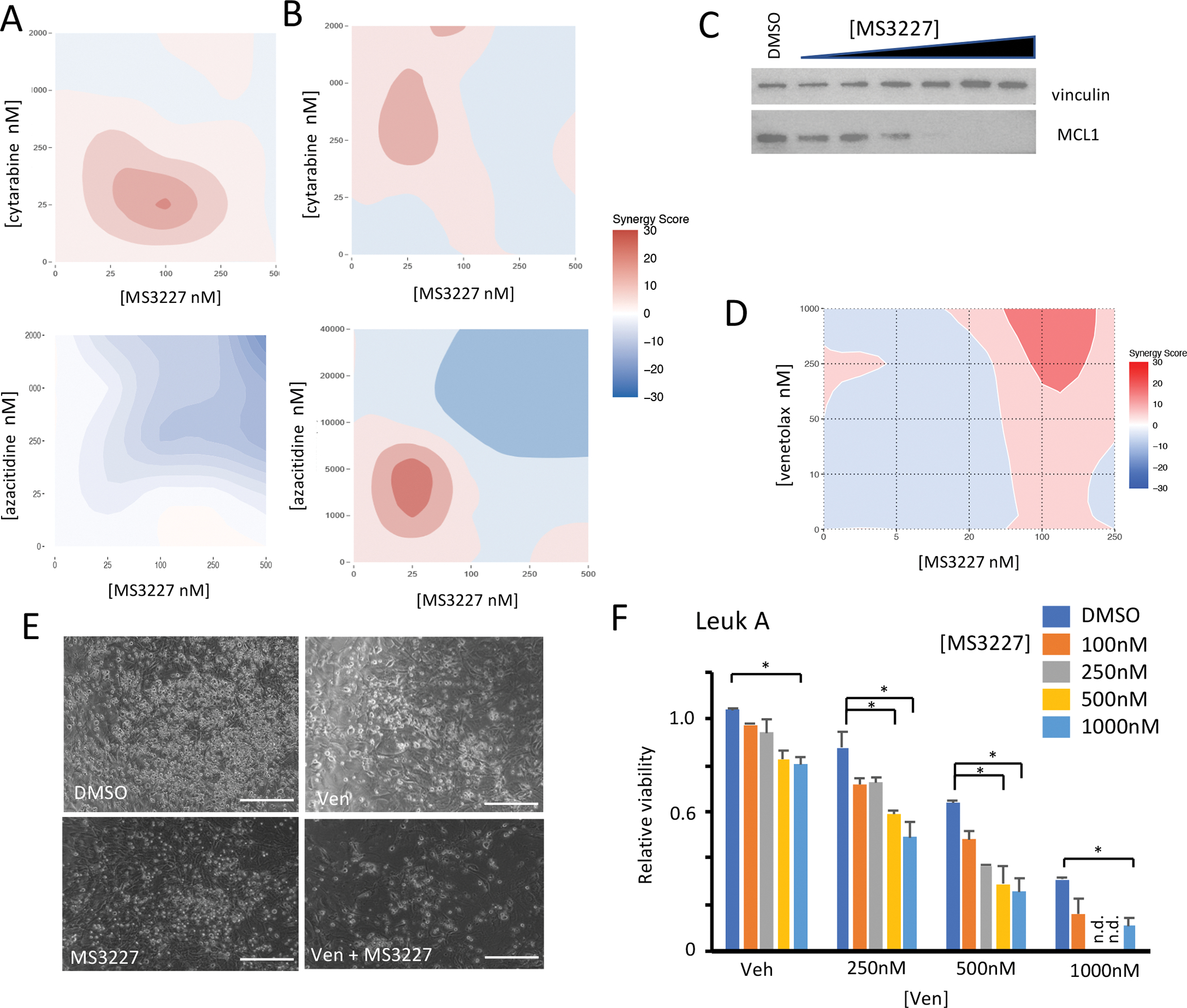
A. Synergy analysis of MOLM-13 cells treated with combination MS3227 and cytarabine (top) and azacytidine (bottom) for 3 days.
B. Synergy analysis of OCI-AML3 cells treated with combination MS3227 and cytarabine (top) and azacytidine (bottom) for 3 days.
C. Western blot of OCI-AML3 cells treated with MS3227 at various concentrations for 1 day and probed for MCL1 and vinculin.
D. Synergy analysis of OCI-AML3 cells treated with combination MS3227 and venetoclax for 3 days.
E. Images of primary TP53 wild-type AML cells treated with Venetoclax 500 nM (Ven), MS3227 100 nM, or combination for 2 days. Scale bar 150 μM.
F. Bar graph of primary TP53 wild-type AML cells treated with various combinations of Venetoclax (Ven) and MS3227 for 2 days for relative viability.
* p<.05; ** p<.01, n.d. not done.
Venetoclax, a BCL2 inhibitor, is widely used in AML. Expression of MCL1, an anti-apoptotic BCL-2 family member, has been reported to contribute to venetoclax resistance [29]. The MDM2-p53 pathway can regulate MCL1 expression and targeting this pathway may affect venetoclax resistance [30]. We tested this possibility by treating leukemia cells with MS3227 and observed that MCL1 expression did decrease in a dose-dependent manner (Fig. 5C). Leukemia cell lines have different levels of sensitivity to venetoclax, and the OCI-AML3 line is relatively resistant, with lack of significant response up to 1 μM [31]. To assess whether MS3227 can cooperate with venetoclax and overcome resistance, we tested combination treatments. With 100 nM of MS3227 we observed cooperative activity with 1 μM of venetoclax (Fig. 5D and Fig. S9B). While single agent venetoclax lacked appreciable ability to inhibit leukemic cell growth, with the addition of MS3227, inhibition of proliferation was greater than with either agent alone.
We next tested this combination in primary patient cells, specifically from a patient who had previously been exposed to a hypomethylating agent and venetoclax combination and progressed. MS3227 had limited single agent effect; while venetoclax retained efficacy in this in vitro assay (Fig. 5E, 5F). However, the combination with MS3227 proved to be more effective with enhanced leukemic cell death, achieving greatest inhibition with the combination (Fig. 5E, 5F). By contrast, treatment of a patient sample that was TP53 mutant failed to demonstrate any combinatorial benefit (Fig. S9C). We tested additional primary AML samples and demonstrated that expression of CDKN1A (p21) and PUMA are enhanced with the combination (Fig. S9D).
Discussion
Dysregulation of the p53 pathway is common across many cancer types, and as a tumor suppressor, its function can be disrupted by somatic mutations and deletions in AML and MPNs. In the majority of AML and MPN cases, TP53 remains wild-type. Other mechanisms act to suppress its activity, and thus, therapeutic approaches that reactivate this pathway may be active in myeloid malignancies. MDM2 is an inhibitor of p53 function. Small molecule inhibition of MDM2 occurs through binding of the p53 interaction pocket. These agents have been used in clinical trials for hematologic malignancies as single agents and in combinations and have demonstrated activity. However, side-effects and incomplete responses have plagued further development.
PROTACs are an emerging class of heterobifunctional small molecules that induce target protein degradation, utilizing the natural cell ubiquitin-proteasome machinery. This mechanism of targeted protein degradation is catalytic in nature and can lead to greater potency than standard small molecule inhibitors. It also leads to distinct pharmacodynamic properties that can support alternative dosing schedules. Another potential advantage to PROTAC design is the added selectivity due to the required E3 ligase in addition to the target protein.
We examined TCGA data and noted increased expression of VHL in AML cells as compared to other cancer types and normal tissues. Utilizing VHL as the E3 ligase for PROTAC design may enable greater specificity toward leukemic cells as compared to normal hematopoietic cells as well as non-hematopoietic tissues. Through SAR studies we identified MS3227 as a potent PROTAC MDM2 degrader. Functionally, we demonstrated that MS3227 degrades MDM2 in a concentration dependent manner that requires proteasomal and ubiquitin ligase activities and VHL binding. In TP53 WT leukemia cell lines, MS3227 is able to activate the p53 pathway, induce transcription of down-stream targets, and promote apoptosis. We tested MS3227 in primary AML samples and confirmed activity in samples that harbored various cytogenetic and mutational profiles. Furthermore, MS3227 had greater potency than AMG 232. This may be due, at least in part, to activity against the negative feedback mechanism that upregulates MDM2. For MDM2 stoichiometric inhibitors, as the p53 pathway is being activated additional MDM2 is produced that may act as a sink for small molecule binders and thus limits the extent to which the pathway can be activated. Furthermore, this build-up of MDM2 can be active against p53 once the inhibitor is washed out or metabolized. In contrast, MS3227 can act catalytically and the degree to which MDM2 is upregulated by p53 activation is blunted. We showed that MS3227 can achieve greater potency by limiting the accumulation of MDM2 with a given degree of p53 activation.
In our studies, TP53 mutation status was the main determinant of response, although additional mutations or properties also affect drug efficacy. We also confirmed that reduced VHL expression diminished the potency of MS3227. A potential resistance mechanism to MS3227 could therefore be loss of the VHL which functions as a tumor suppressor. We observed greater sensitivity to MS3227 in leukemia cell lines compared to solid tumor lines which suggests that AML cell function and viability may be particularly dependent on this pathway. Since combination therapies are likely required to treat AML, we tested MS3227 in combination with cytarabine and azacytidine. We demonstrated that MS3227 could combine with these standard AML therapeutics to inhibit leukemia cell growth. The p53 pathway is also known to regulate multiple pathways involved in apoptosis. Specifically, activation of p53 by MS3227 could deplete MCL-1, a pro-survival factor and resistance mechanism to the Bcl-2 inhibitor venetoclax. We demonstrated that combination treatment with MS3227 and venetoclax in leukemia cell lines and primary AML cells led to improved efficacy.
Our study has implications for drug development for AML and MPN patients. VHL-based PROTACs may be a means to achieve greater specificity against AML compared to off-target tissues. In instances such as MDM2, where there is negative feedback regulation, PROTAC degradation may be more effective than a standard binding inhibitor. Other non-enzymatic, potential oncogenic functions of MDM2 on metabolism and transcription are also inhibited by PROTACs that are not affected by existing inhibitors [32]. It has the potential to target p53-independent effects of MDM2 that may contribute to its oncogenic potential [33, 34]. The PROTAC MS3227 can cooperate with other anti-leukemic therapies to target the MDM2-p53 pathway, and these combination therapies may provide greater therapeutic potential and overcome resistance mechanisms when treating AML and myeloid malignancy patients.
Supplementary Material
Acknowledgements
AHS is supported by the Gilead Sciences Research Scholar Program. BKM is supported by a grant from the MPN Research Foundation. JJ acknowledges the support by an endowed professorship by the Icahn School of Medicine at Mount Sinai. Biospecimens and data were provided through the HMTB. The HMTB is as an independent tissue bank repository for hematological malignancies and related disorders. The HMTB is administered and functions under the auspices of the NCI-designated Tisch Cancer Institute (TCI) at the Icahn School of Medicine at Mount Sinai.
Funding
This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG grants 1S10OD025132 and 1S10OD028504.
Footnotes
Author Disclosures / Competing Interests
J.J. is a cofounder and equity shareholder in Cullgen, Inc. and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc. Y.X. is a cofounder, equity shareholder and a current employee of Cullgen, Inc. J.L. is a current employee of Cullgen, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc. and Cullinan Oncology, Inc.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
- 1.Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012; 119: 2114–2121. [DOI] [PubMed] [Google Scholar]
- 2.Marcellino BK, Hoffman R, Tripodi J, Lu M, Kosiorek H, Mascarenhas J, et al. Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv 2018; 2: 3581–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica 2013; 98: 908–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci 1999; 55: 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bueso-Ramos CE, Yang Y, deLeon E, McCown P, Stass SA, Albitar M. The human MDM-2 oncogene is overexpressed in leukemias. Blood 1993; 82: 2617–2623. [PubMed] [Google Scholar]
- 6.Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res 2008; 14: 5318–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stein EM, DeAngelo DJ, Chromik J, Chatterjee M, Bauer S, Lin CC, et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin Cancer Res 2022; 28: 870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848. [DOI] [PubMed] [Google Scholar]
- 9.Reis B, Jukofsky L, Chen G, Martinelli G, Zhong H, So WV, et al. Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica 2016; 101: e185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khurana A, Shafer DA. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: perspectives on the therapeutic potential of idasanutlin (RG7388). Onco Targets Ther 2019; 12: 2903–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dale B, Cheng M, Park KS, Kaniskan HU, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer 2021; 21: 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambrosini M, Fuca G, Duca M, Damian S, De Santis F, Corti F, et al. Targeted protein degraders from an oncologist point of view: The Holy Grail of cancer therapy? Crit Rev Oncol Hematol 2022; 169: 103532. [DOI] [PubMed] [Google Scholar]
- 13.Fang Y, Liao G, Yu B. Small-molecule MDM2/X inhibitors and PROTAC degraders for cancer therapy: advances and perspectives. Acta Pharm Sin B 2020; 10: 1253–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang B, Wu S, Liu J, Yang K, Xie H, Tang W. Development of selective small molecule MDM2 degraders based on nutlin. Eur J Med Chem 2019; 176: 476–491. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L, et al. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J Med Chem 2019; 62: 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 2016; 535: 252–257. [DOI] [PubMed] [Google Scholar]
- 17.Palumbo A, Rajkumar SV, Dimopoulos MA, Richardson PG, San Miguel J, Barlogie B, et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia 2008; 22: 414–423. [DOI] [PubMed] [Google Scholar]
- 18.Kotchetkov R, Masih-Khan E, Chu CM, Atenafu EG, Chen C, Kukreti V, et al. Secondary primary malignancies during the lenalidomide-dexamethasone regimen in relapsed/refractory multiple myeloma patients. Cancer Med 2017; 6: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duy C, Teater M, Garrett-Bakelman FE, Lee TC, Meydan C, Glass JL, et al. Rational Targeting of Cooperating Layers of the Epigenome Yields Enhanced Therapeutic Efficacy against AML. Cancer Discov 2019; 9: 872–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017; 33: 2413–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem 2014; 57: 1454–1472. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Zhu J, Liu JJ, Chen X, Mihalic J, Deignan J, et al. Optimization beyond AMG 232: discovery and SAR of sulfonamides on a piperidinone scaffold as potent inhibitors of the MDM2-p53 protein-protein interaction. Bioorg Med Chem Lett 2014; 24: 3782–3785. [DOI] [PubMed] [Google Scholar]
- 23.Casement R, Bond A, Craigon C, Ciulli A. Mechanistic and Structural Features of PROTAC Ternary Complexes. Methods Mol Biol 2021; 2365: 79–113. [DOI] [PubMed] [Google Scholar]
- 24.Peng Y, Chen L, Li C, Lu W, Agrawal S, Chen J. Stabilization of the MDM2 oncoprotein by mutant p53. J Biol Chem 2001; 276: 6874–6878. [DOI] [PubMed] [Google Scholar]
- 25.Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem 2013; 56: 5979–5983. [DOI] [PubMed] [Google Scholar]
- 26.Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2016; 113: 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galdeano C, Gadd MS, Soares P, Scaffidi S, Van Molle I, Birced I, et al. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J Med Chem 2014; 57: 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng S, Wang W, Aldahdooh J, Malyutina A, Shadbahr T, Tanoli Z, et al. SynergyFinder Plus: Toward Better Interpretation and Annotation of Drug Combination Screening Datasets. Genomics Proteomics Bioinformatics 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, et al. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017; 32: 748–760 e746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 2014; 4: 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein AM, de Queiroz RM, Venkatesh D, Prives C. The roles and regulation of MDM2 and MDMX: it is not just about p53. Genes Dev 2021; 35: 575–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wienken M, Dickmanns A, Nemajerova A, Kramer D, Najafova Z, Weiss M, et al. MDM2 Associates with Polycomb Repressor Complex 2 and Enhances Stemness-Promoting Chromatin Modifications Independent of p53. Mol Cell 2016; 61: 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cisse MY, Pyrdziak S, Firmin N, Gayte L, Heuillet M, Bellvert F, et al. Targeting MDM2-dependent serine metabolism as a therapeutic strategy for liposarcoma. Sci Transl Med 2020; 12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.