

Abstract
Mutations in the gene Additional Sex-Combs Like 1 (ASXL1) are recurrent in myeloid malignancies as well as the pre-malignant condition clonal hematopoiesis, where they are universally associated with poor prognosis. However, the role of ASXL1 in myeloid lineage maturation is incompletely described. To define the role of ASXL1 in myelopoiesis, we employed single cell RNA sequencing and a murine model of hematopoietic-specific Asxl1 deletion. In granulocyte progenitors, Asxl1 deletion leads to hyperactivation of MYC and a quantitative decrease in neutrophil production. This loss of granulocyte production was not accompanied by significant changes in the landscape of covalent histone modifications. However, Asxl1 deletion results in a decrease in RNAPII promoter-proximal pausing in granulocyte progenitors, indicative of a global increase in productive transcription. These results suggest that ASXL1 inhibits productive transcription in granulocyte progenitors, identifying a new role for this epigenetic regulator in myeloid development.
Introduction
Recurrent mutations in the epigenetic regulator Additional Sex-Combs Like 1 (ASXL1) are associated with universally poor prognosis in myeloid malignancy1–4. Mutations in ASXL1 also occur in the premalignant condition clonal hematopoiesis, where they are associated with a high rate of progression to myeloid leukemia5–7. The association of ASXL1 mutations with myeloid lineage disease suggests a key role for this regulator in myeloid cells, yet little is known about the role of wildtype ASXL1 in normal myeloid lineage development.
One of the best studied roles of ASXL1 in the hematopoietic system is the regulation of the polycomb repressive complex 2 (PRC2). ASXL1 directs PRC2 to the HOXA locus, leading to deposition of repressive histone 3 lysine 27 trimethylation (H3K27me3)8,9. Knockdown of ASXL1 in leukemia cell lines results in a loss of H3K27me3 at the HOXA locus and upregulation of HOX genes, in particular HOXA9. Other studies in non-hematopoietic cells have shown that Asxl1 deletion fails to alter the landscape of H3K27me3 or other covalent histone modifications10. A challenge in interpreting these results is the increasingly appreciated context-specific roles that epigenetic regulators play in normal development11,12.
To gain insight into the normal biological role of ASXL1 in hematopoiesis, we utilized scRNA sequencing and CUT&Tag-based low-input epigenetic profiling to investigate the specific role of Asxl1 in myeloid cell development. These studies revealed that Asxl1 is required for terminal granulocyte production via regulation of global transcription in the committed neutrophil progenitor. Our studies demonstrate little variance in key covalent histone modifications and instead suggest a role for ASXL1 in modulating the activity of RNA Polymerase II. These findings provide an important basis for the investigation of the impact of mutant ASXL1 on hematopoiesis, identifying key cell types and biological processes that are uniquely dependent on ASXL1 function.
Results
Single Cell RNA Sequencing Defines Multiple Lineages in Murine Bone Marrow
To assess the impact of Asxl1 deletion on hematopoietic lineage outputs, we performed single cell RNA sequencing on lineage negative (Lin−) hematopoietic progenitor cells from Mx1-Cre Asxl1FL/FL or littermate control Cre negative mice (henceforth referred to as Asxl1Δ/Δ and Asxl1WT respectively). We examined two time points after induction of recombination via Poly I:C administration: 4 weeks (n=4/genotype) and greater than 6 months (n=3/genotype). We selected these timepoints as at a young age Asxl1Δ/Δ mice maintain normal peripheral blood counts, while they begin to decline in mice >6 months of age. In total, we profiled 53,579 cells, with data integration revealing 25 transcriptionally distinct subsets (Figure 1A, Figure S1A, Table S1–S2). To confirm the transcriptional identities assigned to clusters, we performed Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) for key surface markers associated with hematopoietic stem and progenitor cell identity (Figure 1B, Figure S1B)13. CITE-seq identified known stem and progenitor cell populations in a manner highly analogous to flow cytometry performed in parallel14,15. Collectively, these results establish a baseline map of the cellular constituents and lineages comprising murine bone marrow and represent a platform to investigate the role of ASXL1 in cell fate determination.
Figure 1. Single Cell RNA Reveals a Granulocyte Maturation Defect in Asxl1Δ/Δ mice at the point of cell cycle exit.
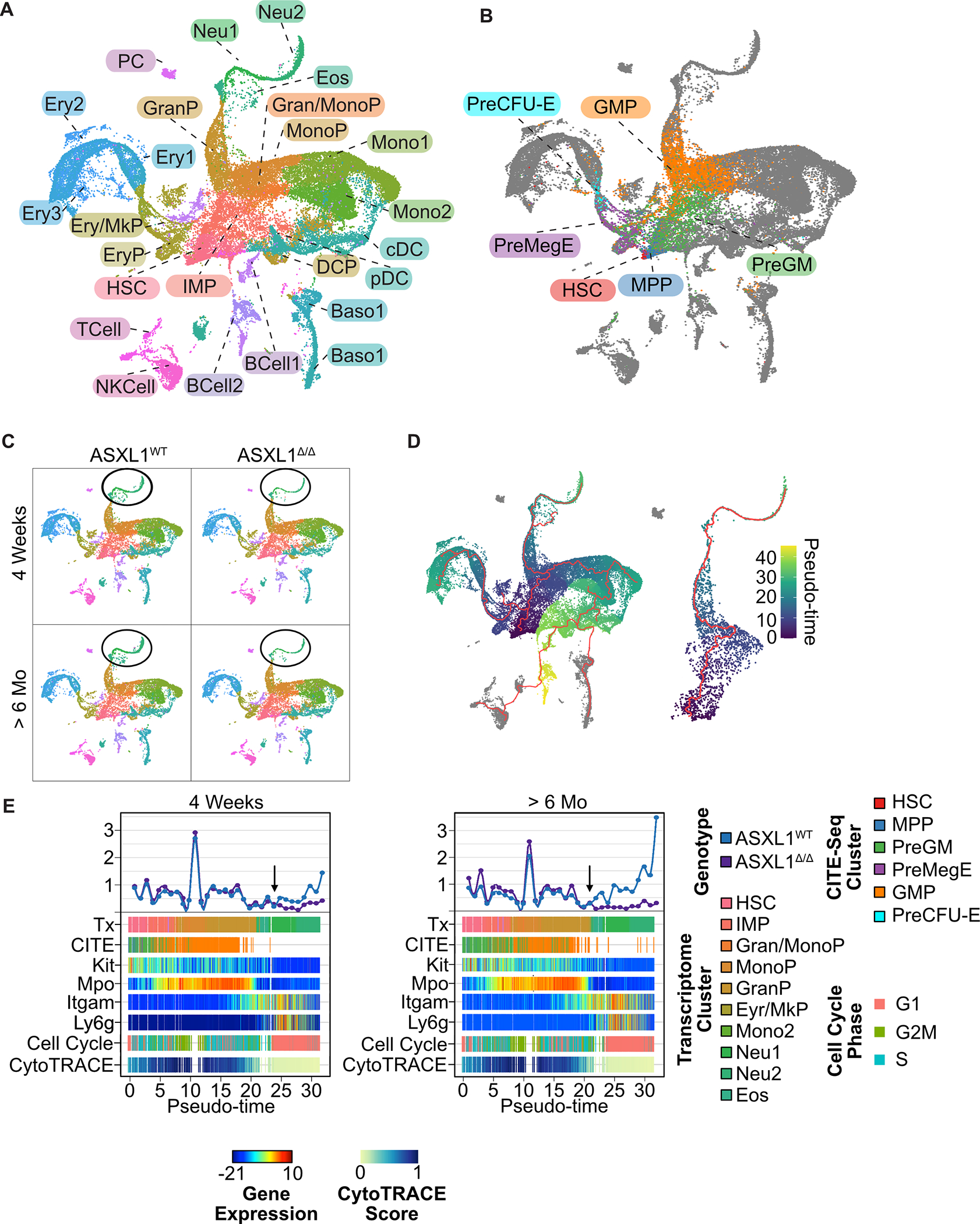
A. Bone marrow was isolated from Asxl1WT and Asxl1Δ/Δ mice 4 weeks and 6 months after induction of recombination with Poly I:C (n=3–4/group). Lineage positive cells were depleted using immunomagnetic purification and labeled with the indicated CITE-seq antibodies. Each individual mouse was labeled with a unique anti CD45 HTO antibody allowing for mouse-level bioinformatic deconvolution. Single cell transcriptional profiling was then performed using the Chromium platform (10X Genomics). UMAP projection demonstrating transcriptionally defined clusters identified using published datasets and data integration. Marker genes defining transcriptional cell clusters. B. UMAP projection demonstrating clusters defined by CITE-Seq. C. UMAP projections split by genotype (Asxl1WT and Asxl1Δ/Δ) and timepoint (4 weeks and 6 months) post induction of recombination. Circle surrounding terminal neutrophil populations D. Cell trajectories for all lineages and expanded view of granulocytic lineage. E. Linearized granulocyte trajectory showing transcriptional and CITE-seq clusters along with expression of key marker genes, cell cycle phase (phase), and CytoTRACE score. Arrows indicate point of divergence between Asxl1WT and Asxl1Δ/Δ.
Asxl1 Deletion Results in an Early Loss of Bone Marrow Granulocytic Progenitors
Asxl1Δ/Δ mice are known to develop peripheral neutropenia when they reach >6 months of age, however at earlier timepoints they maintain peripheral blood neutrophil counts within the normal range9. In contrast, scRNA-seq of bone marrow progenitors reveals a marked defect in the output of the granulocyte lineage 4 weeks after the induction of recombination that worsens with aging (Figure 1C, Figure S1C, D). To investigate potential mechanisms underlying reduced neutrophil maturation in the Asxl1Δ/Δ mice, we utilized Monocle 3 to perform a lineage tracing analysis, which placed all cells along a maturation trajectory and assigned pseudo-time values (Figure 1D, left panel). We then identified cells assigned to the neutrophil trajectory consisting of a subset of cells from the HSC, IMP, Gran/MonoP, GranP, Neu1, and Neu2 clusters (Figure 1D, right panel). We confirmed the transcriptional identity of cells assigned to the neutrophil lineage via comparison to published transcriptional datasets16 (Figure S2A). Considering the cells in pseudotime order reveals uniform cell counts in all clusters prior to the committed neutrophils, confirming that the defect in maturation occurs at the post-GMP stage (pseudotime 20, Figure 1E). Analysis of other hematopoietic lineages revealed that only terminal neutrophils showed a significant change in cell number in Asxl1Δ/Δ mice as compared to littermate controls (Figure S1C).
To understand why Asxl1 deletion specifically impacts the production of neutrophils, we used gene expression module scores to assign cells to specific phases of the cell cycle (Figure S2B). This revealed that decreased neutrophil production occurs precisely at the point of cell cycle exit in the Asxl1 deficient mice (Figure 1E). We next performed a CytoTRACE analysis which uses transcriptional diversity to predict developmental potential17. This algorithm uses the number of expressed genes (transcripts per cell) to assign a differentiation state score. Global transcriptional diversity increased as cells transitioned from the HSC compartment into the progenitor compartment and then fell rapidly as cells undergo terminal differentiation (Figure S2B). In the neutrophil lineage, transcript diversity fell rapidly in the GranP population, immediately before the cell cycle exit and the decrease in neutrophil output in the Asxl1Δ/Δ mice (Figure 1E). Collectively, these data show that Asxl1 deletion impacts neutrophil lineage output at a point of marked remodeling of the transcriptome as cells prepare for terminal cell cycle exit.
To validate the granulocyte lineage defect associated with Asxl1 deletion, we examined the capacity of Asxl1-deficient bone marrow to produce mature neutrophils in ex-vivo culture (Figure S2E, F). Neutrophil progenitors were produced in normal numbers from Asxl1Δ/Δ bone marrow (Figure S2E), but these progenitors failed to differentiate in normal numbers upon withdrawal of stemness-promoting cytokines (Figure S2F). While Asxl1Δ/Δ mice demonstrate largely normal peripheral blood neutrophil counts, they demonstrate reduced peripheral granulocytosis upon in vivo G-CSF challenge suggesting a functional bone marrow defect (Figure S2G).
Asxl1 Deletion Results in Perturbation of the Myc Network in Maturing Granulocytes
To elucidate the dynamics of transcriptional networks that occur in response to Asxl1 deletion, we performed a network analysis using CausalPath adapted to scRNA-seq (Figure 2A, Table S3)18. CausalPath evaluates transcriptional differences in the context of curated protein-protein relations and biological pathways, highlighting coordinated changes in signaling networks. The number of predicted relations between source and target proteins is an approximation of network complexity (Figure 2B). Differential network complexity between Asxl1Δ/Δ and Asxl1WT mice increased with maturation up to the point of the GranP cluster, decreasing substantially in the later stages of neutrophil development (Figure 2B). To identify key regulators in the network, we quantified the number of relations by source protein in each cluster. In the stem and progenitor clusters (HSC, IMP, Gran/MonoP, and GranP), this unbiased analysis revealed a complex regulatory network nucleated by the master proliferative regulator Myc, with maximal activity in the GranP cluster (Figure 2C). Asxl1Δ/Δ cells within the neutrophil trajectory demonstrated marked upregulation of Myc and MYC-targets consistent with the inability of these cells to exit the proliferative GranP cluster (Figure 2D). Asxl1Δ/Δ mice showed upregulation of Myc expression throughout the neutrophil trajectory (Figure 2E) and fail to appropriately downregulate Myc in the terminal GranP population as cells approach the point of cell cycle exit at pseudotime 20 (Figure 1E, 2F–G). These data collectively suggest that Asxl1 acts to restrict Myc expression and activity in late GranP cells, with Asxl1 deletion preventing terminal granulocyte maturation. To evaluate a potential role for MYC in restraining granulocytic differentiation, we overexpressed Myc in murine Hoxb8-ER immortalized progenitors, a granulocytic progenitor cell line that undergoes differentiation upon estrogen withdrawal. Myc-expressing cells showed reduced upregulation of mature surface markers after estrogen withdrawal, as compared to empty vector control cells, showing that inappropriate MYC expression is sufficient to arrest granulocyte maturation (Figure S2H).
Figure 2. Asxl1 Deletion Leads to Hyperactivation of a Myc Transcriptional Signature in Granulocyte Progenitors.
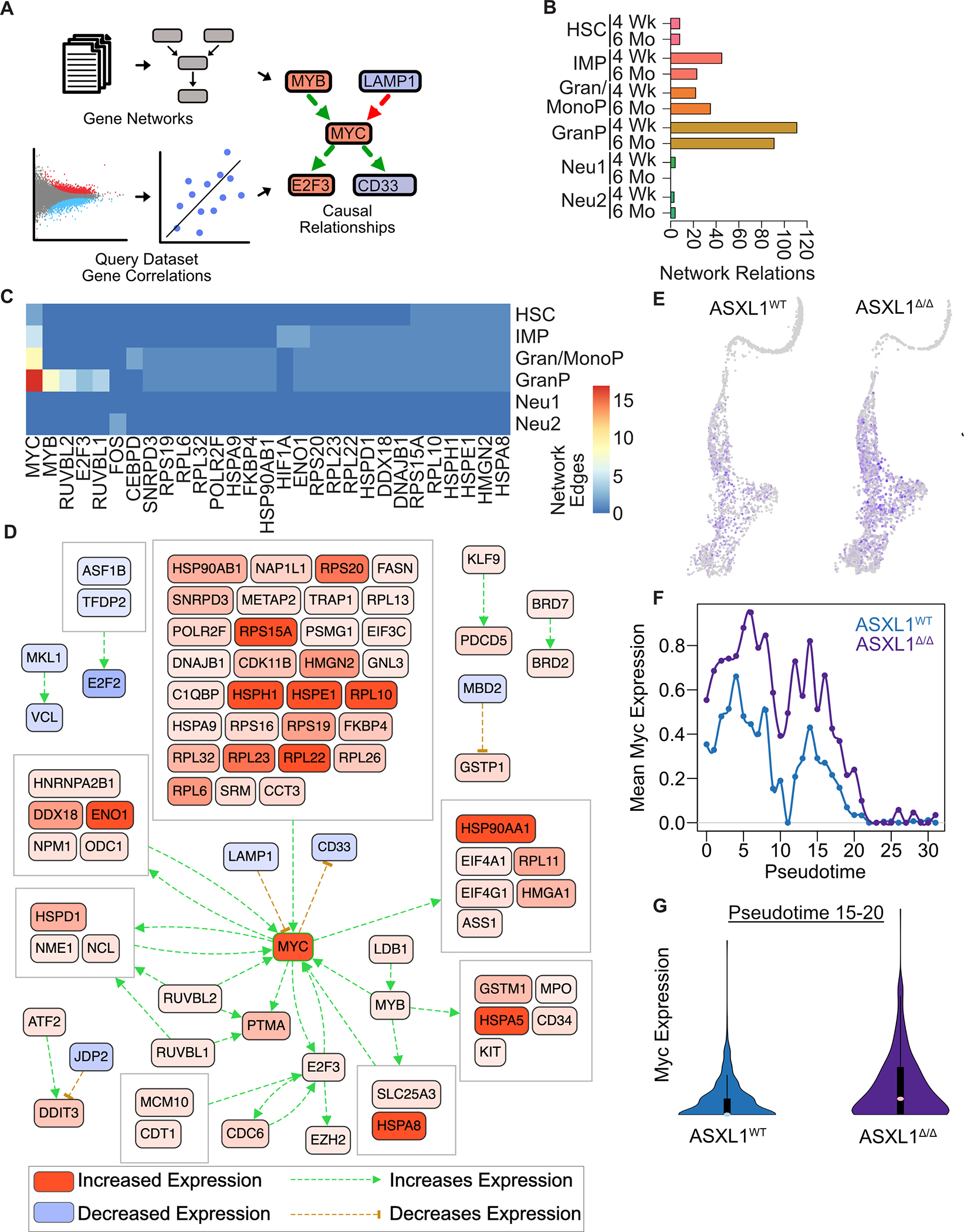
A. Schematic of CausalPath analysis which utilizes literature derived gene regulatory networks and gene correlations from the query dataset to predict causal relationships. B. Number of network edges as a measure transcriptional network complexity between Asxl1WT and Asxl1Δ/Δ mice. C. Differential MYC network activity in Asxl1Δ/Δ mice relative to wild type controls along the granulocytic lineage 6 months post induction of recombination. Network with a reduced FDR threshold (<0.025) shown to reduce complexity for the purposes of visualization. D. Differential network activation in granulocyte progenitor population 6 months post induction of recombination. E. Feature plots of Myc expression in Asxl1WT and Asxl1Δ/Δ mice. Note the increased density and intensity of blue MYC expressing cells in Asxl1Δ/Δ mice. F. Myc expression along granulocyte trajectory. G. Myc expression in terminal granulocyte progenitors (Pseudotime 15–20).
As Asxl1 mutations are common in myelodysplastic syndrome (MDS) and aplastic anemia, both of which are associated with decreased RBC production, we evaluated transcriptional changes in erythroid progenitors from Asxl1Δ/Δ mice 19,20. This analysis revealed a similar activation of the MYC transcriptional network with a peak in the Ery2 population and a subsequent decrease in the more mature Ery3 population (Figure S3). This suggests that Asxl1 deletion produces similar transcriptional changes in multiple lineages and that these changes may underpin the bone marrow failure observed in ASXL1-mutant aplastic anemia and MDS.
Asxl1 Deletion is not Accompanied by Marked Changes in Covalent Histone Modifications in Granulocytic Progenitors.
Prior work in cell lines and partially fractionated bone marrow has extensively characterized the role of Asxl1 in regulating PRC2 mediated deposition of repressive H3K27me3 histone modifications8,9. To characterize the dynamics of this chromatin modification during granulocyte maturation, we developed a flow cytometric sorting strategy to purify myeloid progenitors, granulocytic progenitors and mature bone marrow neutrophils (Figure S4A). We then utilized Cleavage Under Targets and Tagmentation (CUT&Tag) based genome wide profiling of H3K27me3 across the trajectory of granulocyte maturation21.
Comparison of genome wide binding profiles revealed clear clustering by cell type with a high degree of similarity between Asxl1Δ/Δ and Asxl1WT populations. Uncommitted myeloid progenitors showed the highest degree of variability between genotypes, but these differences were lost with further maturation (Figure 3A). Differential binding analysis between genotypes revealed the greatest differences in myeloid progenitors, again with normalization in more mature cell populations (Figure S4B, Table S4–7). Gene ontology analysis of these differentially marked regions in myeloid progenitors revealed enrichment of terms associated with RNA polymerase activity and multiple neuronal specific ion channels, consistent with a role for Asxl1 in mediating PRC2-dependent repression of neuronal phenotypes in immature hematopoietic cells (Figure S4C, Table S8). Crucially, few differences in H3K27me3 signal were observed in GranP cells, where our single cell analysis demonstrated the most robust transcriptional changes. In both Asxl1WT and Asxl1Δ/Δ cells, we found marked global increases in H3K27me3 signal with granulocytic maturation (Figure 3B–C, Figure S4D–E). This was accompanied by developmentally appropriate deposition of H3K27me3 at loci associated with non-granulocyte cell fates including Ebf1 (lymphoid), Gata2 (erythroid) and Irf8 (monocyte). Conversely, H3K27me3 levels at genes associated with cell proliferation, such as Ccnd1 and Id2, show marked increases in H3K27me3 with maturation. Finally, the Hoxa locus showed clear heterochromatic spread across the Hoxa9 gene associated with the transition between myeloid progenitors and granulocyte precursors. Collectively these data argue that the transcriptional dysregulation observed in Asxl1 deficient GranP cells is not due to altered deposition of H3K27me3.
Figure 3. ASXL1 Deletion Does Not Substantially Impact the Landscape of Covalent Histone Modifications in Developing Myeloid Cells.
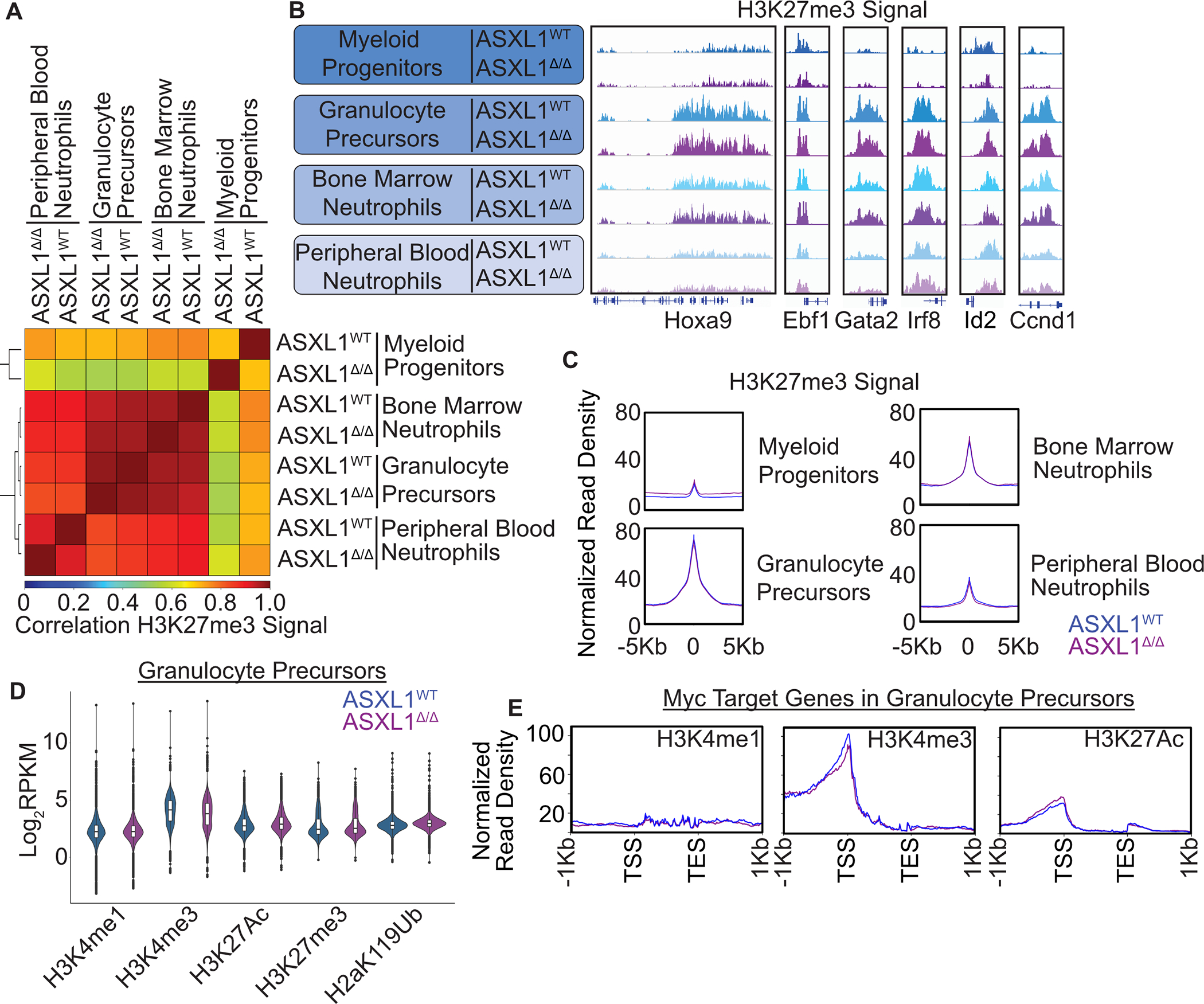
A. Bone marrow from Asxl1WT and Asxl1Δ/Δ mice was harvested 6 months after induction of recombination via poly I:C administration, maturing granulocyte lineage cells isolated by FACS, and subjected to CUT&Tag for H3K27me3. Spearman Correlation of Genome-wide H3K27me3 signal in indicated bone marrow sub-populations. B. Representative tracks of H3K27me3 signal in indicated bone marrow sub-populations. C. Mean normalized read density at H3K27me3 peaks in indicated bone marrow sub populations. D. RPKM values for reads in peaks for indicated histone marks in CD133 purified granulocytic progenitors. E. Mean normalized read density for indicated histone marks at Myc target genes as measured by CUT&Tag in CD133-granulocytic progenitors from CausalPath analysis in Figure 2.
To better characterize the epigenetic landscape of Asxl1 deficient granulocytic progenitors, we searched our single cell dataset for GranP specific surface markers that were not differentially expressed between genotypes. We identified Prom1/CD133 which is known to mark immature granulocytic cells, as a marker of this sub population (Figure S5A)22. Magnetic bead-based purification of CD133 positive cells yielded a KIT+/Ly6g positive cell population consistent with an immature granulocyte phenotype (Figure S5B). Bulk RNA sequencing revealed increased expression of MYC target genes in Asxl1Δ/Δ cells (Figure S5C–D, Table S9). To comprehensively profile the chromatin landscape of Asxl1Δ/Δ GranPs, we performed CUT&Tag for multiple covalent histone modifications enabling the global segmentation of chromatin into distinct functional domains. Specifically, we examined H3K4me1 which is broadly associated with enhancers, H3K4me3 which is present at active promoters, H3K27Ac which is present at active enhancers and promoters. In addition, we assessed H3K27me3 to correlate our results with our prior findings in sorted cells. Finally, we profiled H2aK119Ub which is associated with PRC1-mediated gene repression and can specifically be altered by ASXL1 via activation of the BAP1 deubiquitinase23. Globally, the total abundance of all marks was not substantially altered by Asxl1 deletion, nor were substantial numbers of differential peaks detected (Figure 3D, Figure S5 G–H, Table S10–12). To establish whether Asxl1 regulates MYC network activity via the deposition of covalent histone modifications, we assessed read pileup at MYC target genes identified as differentially expressed in our scRNAseq dataset. This revealed no significant differences in histone mark pileup at MYC target genes between Asxl1WT and Asxl1Δ/Δ cells (Figure 3E, Figure S5F). Collectively, these data argue against differential accumulation of histone modifications as a major mechanism of Asxl1-dependent regulation of MYC target genes.
Asxl1 Localizes to Regions of Active Transcription and Regulates RNA-Polymerase II Activity
Given the strong MYC-dependent gene expression signature we observed in scRNA seq and bulk RNA seq datasets as well as the role of MYC in regulating RNAPII pause release, we hypothesized that ASXL1 might be modifying gene expression through direct regulation of RNAPII24,25. We therefore compared ASXL1 ChIP-seq signal from cKIT+ murine bone marrow progenitors with covalent histone modifications profiled in CD133 positive cells26. In addition, we profiled RNAPII binding genome wide in CD133+ neutrophil progenitors. Examining the global signal, we saw the greatest degree of correlation between ASXL1 and RNAPII signal and the least correlation with H3K27me3 (Figure 4A). Indeed, ASXL1 was largely distributed at the transcriptional start sites of actively transcribed genes. To assess the activity of RNAPII in Asxl1Δ/Δ granulocytic progenitors, we examined the RNAPII pause index in which a ratio of proximal promoter read density is divided by the read density across the remainder of the gene body (Figure 4B)25. We observed that Asxl1Δ/Δ granulocyte progenitors had consistently lower pause indices than Asxl1WT cells (Figure 4C–F, Table S13). These changes in RNAPII pausing behavior occur primarily at RNAPII-MYC co-bound genes rather than genes with only an RNAPII peak (Figure 4C, D, E). These results are consistent with ASXL1 playing a role in modulating the activity of RNAPII10.
Figure 4. ASXL1 Deletion Leads to an Increase in RNAPII Pause-Release.
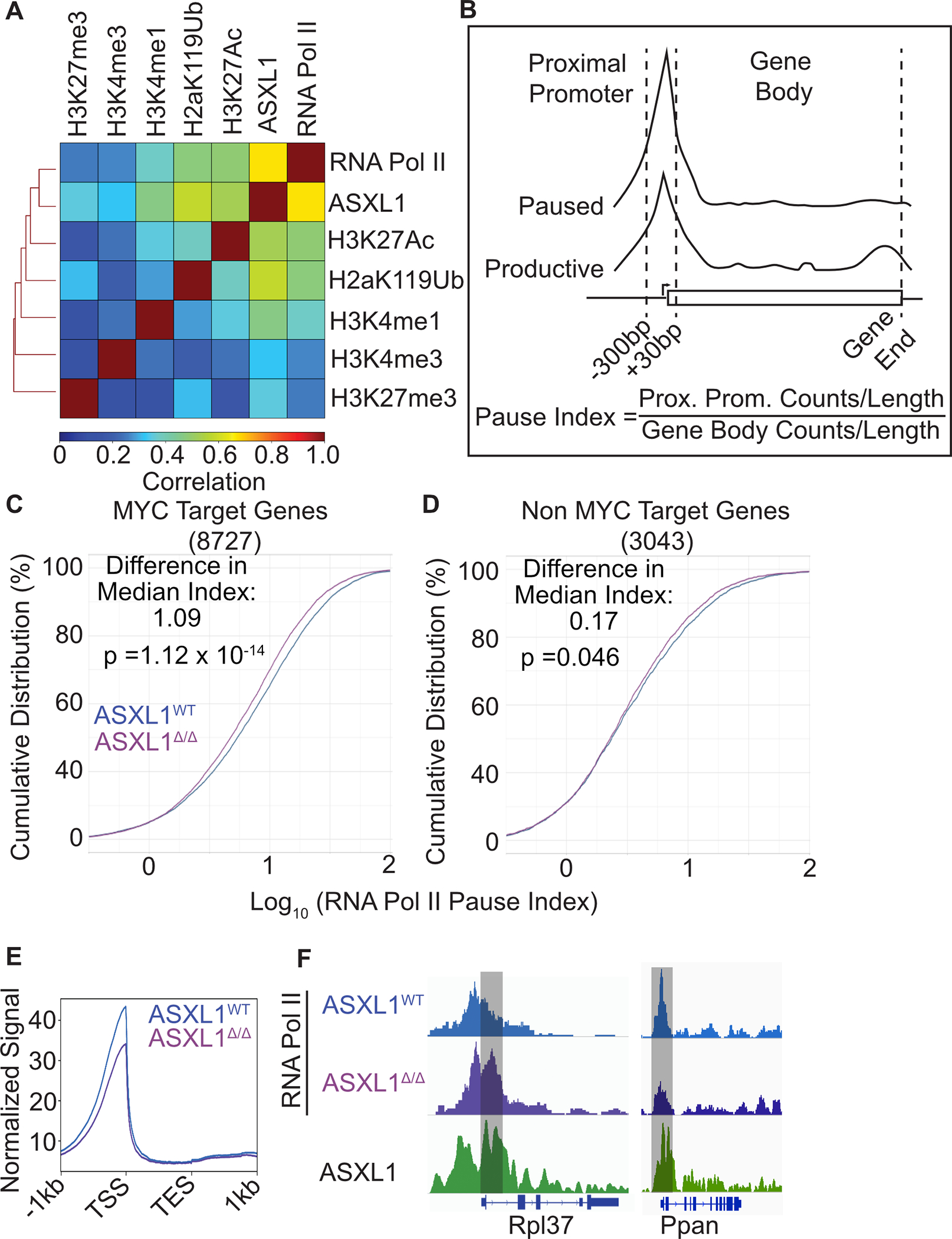
A. Spearman correlation of histone mark or RNAPII CUT&Tag in CD133 positive granulocyte progenitors from Asxl1WT mice (n=2–4/group) and published ASXL1 ChIP-seq. B. Schematic depicting the calculation of RNAPII pause-index. C. Cumulative distribution of RNAPII pause-indices at genes bound by Myc and RNAPII in Asxl1WT and Asxl1Δ/Δ mice (n=4/group). D. Cumulative distribution of RNAPII pause-indices at genes bound by RNAPII and lacking a Myc peak in Asxl1WT and Asxl1Δ/Δ mice (n=4/group). Median pause index difference between Asxl1WT and Asxl1Δ/Δ conditions. Statistical significance between RNAPII pause-index distributions in Asxl1WT and Asxl1Δ/Δ conditions evaluated using a KS-test. E. Mean signal at all genes bound by RNAPII. Representative RNAPII tracks from genes with differential RNAPII pausing in from Asxl1WT and Asxl1Δ/Δ mice.
Discussion
The processes of granulopoiesis involves tightly coordinated epigenetic and transcriptional changes associated with lineage commitment and terminal differentiation16,27. Given that neutrophils have very specific biological functions and peripheral blood half-life of hours to a few days, they require the expression of a relatively narrow subset of specific genes 28. Coordinated cell cycle exit and cessation of proliferation is likely necessary to prevent the malignant transformation of this high output lineage, which produces an estimated one-hundred billion mature neutrophils daily28. The proliferative balance of this lineage is disrupted in myeloid malignancy, where both over and under-proliferation are observed. In both myeloproliferative neoplasms and myelodysplastic syndromes, recurrent mutations in ASXL1 are observed, suggesting a specific connection between this gene and myeloid lineage cells1,29. Despite extensive work on the biology of ASXL1 mutations, little is known about the role of this global regulator in normal hematopoiesis. A clearer definition of the role of ASXL1 in normal granulocyte development is critical to understanding the behavior of ASXL1 mutations in myeloid leukemias and in the development of effective therapy options. Using single cell RNA sequencing and chromatin profiling with CUT&Tag we establish that ASXL1 plays a critical role in the regulation of active transcription, with deletion resulting in the activation of a Myc-dependent proliferative program and loss of terminal differentiation potential. These results ascribe a previously uncharacterized role to ASXL1 as a regulator of RNA polymerase activity in granulocytic lineage cells. Further, they identify a new mechanism for ASXL1 in hematopoietic development, and crucially suggest key mechanisms for oncogenic ASXL1 mutations, particularly in granulocytic leukemias where they are common.
Numerous possible mechanisms have been proposed by which ASXL1 might mediate gene expression. Given the homeotic phenotypes associated with germline ASXL1 mutations, it was logically assumed that the primary role of ASXL1 in the hematopoietic system was the regulation of HOX genes with changes in repressive H3K27me3 serving as an intermediate 8,30. This was particularly compelling given the known role of HOX gene dysregulation in the pathogenesis of acute leukemia31–33. Indeed, in leukemia cell lines, ASXL1 deletion is associated with a loss of repressive H3K27me3 and marked upregulation of HOXA9. However, results from in vivo models are more complex with several plausible interpretations. LSKs from Asxl1 deficient mice show marked transcriptional changes but only modest changes in Hoxa9 expression9. Western blotting in bulk splenocytes showed that Asxl1 deletion was accompanied by a significant reduction in total H3K27me3, a finding with several possible interpretations. Asxl1 deletion results in a multi-lineage maturation defect, increasing the relative abundance of immature cells in bulk populations. As H3K27me3 levels increase with maturation (Figure 3), it is possible that this decrease in signal results from population changes (i.e. more immature cells with less global H3K27me3) rather than cell specific differences. Consistent with this, we found no changes in abundance of H3K27me3 in sorted cell populations, arguing for key PRC2-independent functions of Asxl1 in hematopoietic development. Indeed, in other non-hematopoietic cell types such as bone marrow stromal cells, Asxl1 has been shown to have minimal impact on the deposition of covalent histone modifications and also similarly regulates RNA polymerase II activity10.
A major challenge in the study of preleukemic driver mutations such as those in Asxl1 is that in isolation they evoke relatively mild changes in cell phenotype. This is perhaps unsurprising as these mutations can exist for years to decades without the development of overt malignancy. Understanding this biology requires high resolution methods capable of dissecting transcriptional dynamics at the single cell level. Our study demonstrates that ASXL1 inhibits a MYC transcriptional network during myeloid development with Asxl1 deletion resulting in maturation arrest. It is unclear whether ASXL1 regulates MYC activity directly or the observed expression changes are the result of ASXL1 modulation of RNAPII. Furthermore, these transcriptional changes occur directly at the point of cell cycle exit during granulocytic maturation, but a causal relationship cannot easily be established between these transcriptional changes and the failure of granulocytic maturation in Asxl1 Δ/Δ mice.
The precise mechanism of ASXL1 mutations and whether they are loss or gain of function remains an area of debate, with evidence suggesting both poor expression of ASXL1 truncations as well as neomorphic interactions with BRD48,34. Irrespective, work has largely focused on the role of ASXL1 as a regulator of covalent histone modifications. Our work suggests that the regulation of RNAPII is an important contributor to the normal function of ASXL1 in the hematopoietic system. The development of therapeutic strategies to target ASXL1 mutant clones is dependent on a comprehensive understanding of the underlying pathogenic mechanisms. Therefore, in future studies it will be critical to address the impact of mutant ASXL1 on the regulation of MYC target gene expression profiles and the activity of RNAPII.
A limitation of our study is that it does not provide a direct mechanistic link between Asxl1 deletion, dysregulation of RNAPII at MYC target genes and a failure of granulocyte maturation. To our knowledge, none of the available cell line models of murine neutrophil differentiation ever upregulate surface expression of Ly6G, instead arresting at a developmental stage before the Ly6G positive CD133 positive granulocyte progenitor. Presenting challenges in the study of protein complex formation in this transient developmentally intermediate cell population. Ultimately, a mechanistic dissection of this biology will require the development of new model systems in which large numbers of this cell type can be grown, enabling the study of RNAPII complex assembly in the setting of Asxl1 deletion or mutation.
Methods
Mice
Mx-1 Cre (003556) and Asxl1Flox (025665) mice were obtained from The Jackson Laboratories. Poly I:C (Sigma) was dissolved in PBS and injected intraperitoneally at 12 mg/kg on day 1, 3 and 5 starting at 6 weeks of age. Male mice were utilized for all experiments. For in vivo GCSF treatment studies, 6-month-old Asxl1WT and Asxl1Δ/Δ mice were treated with recombinant human G-CSF (TBO-Filgrastim, Teva) at 250 μg/kg every 12 hours via intraperitoneal injection. CBCs were collected via automated blood counter (Scil Vet). All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committee of Oregon Health & Science University (Protocol #TR01_IP00000482).
Flow Cytometry
The following antibodies were utilized for FACS according to the manufacturer instructions: CD150 BV421 (TC15-12F12.2, BioLegend), CD105 PE-CF594 (MJ7/18, BD), CD41a APC-Cy7 (MWReg30, Biolegend), CD16/32 PerCP e710 (93, eBioscience), Sca1 PE (D7, Biolegend), cKIT PE-Cy7 (2B8, Biolegend), Lineage APC (BD), Ly6G e450 (1A8-Ly6G, ebioscience). Stained cells were analyzed on a FACSAria III flow cytometer, LSRFortessa flow cytometer and Influx cell sorter (BD).
Magnetic Cell Separation
Bone marrow cells were incubated with anti-mouse direct lineage depletion cocktail (Miltenyi). Cells were separated using an autoMACS cell separator (Miltenyi). For purification of CD133 positive progenitors, bone marrow was incubated with a biotinylated anti CD133 antibody (Clone 315-2C11, Biolegend), followed by Streptavidin MicroBeads (Miltenyi). Peripheral blood neutrophils were isolated from 200 μL of peripheral blood using Ly6G MicroBeads (Miltenyi). In both cases, cells were separated using MACS LS columns (Miltenyi).
Ex-vivo Neutrophil Generation
Lineage negative bone marrow (Lin−) cells were isolated by magnetic cell separation as above. Cells were maintained at 3–5 ×105/mL throughout the culture process. Cells were initially cultured in IMDM +20% FBS (AtlantaBiologics) in recombinant murine IL-3 and SCF (Peprotech), both at 50 ng/mL. After 3 days, recombinant murine G-CSF (Peprotech) at 50 ng/mL was added to the culture. On day 6, cells were washed 4X in PBS and resuspended in IMDM +20% FBS and 50 ng/mL G-CSF. Viable cell counts were determined by trypan blue exclusion.
Single Cell RNA Sequencing and CITE-seq
Lineage negative bone marrow cells (5×105/mouse) were suspended in FACS staining buffer and incubated for 30 minutes with the following CITE-Seq primary antibodies and cell hash tag antibodies: CD45 (M1/24), CD150 (TC15-12F12.2), CD105 (MJ7/18), CD41a, CD16/32 (93), Sca1 (D7), cKIT (2B8) (BioLegend). The CD45 antibody barcode was specific to each mouse, all other antibody barcodes were specific to the primary antibody. After washing, cells from each mouse were mixed in equal proportions. At the 4-week timepoint, cells from n=4/genotype were mixed while at the >6 mo timepoint, n=3/genotype were used. Mixed cell populations were loaded onto the Chromium Controller (10X Genomics) according to the manufacturer’s instructions. Libraries were sequenced on a HiSeq2500 or HiSeq-X (Illumina) using 100 BP PE sequencing.
Bulk RNA Sequencing
Total RNA was extracted from approximately 5×105 CD133 purified progenitors using an RNeasy micro kit (Qiagen) according to the manufacturer’s instructions. Libraries were prepared using a TruSeq library prep kit (Illumina) according to the manufacturer’s instructions. Libraries were sequenced on a HiSeq2500 (Illumina) using SE 100BP sequencing.
CUT&Tag
Sorted or purified cells (1×104 to 1×105) were washed in CUT&Tag wash buffer 2X (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 1× Protease inhibitor cocktail). Concanavalin A magnetic coated beads (Bangs Laboratories) were activated by washing in binding buffer (20 mM HEPES, pH 7.5, 10 mM KCL, 1 mM CaCl2, 1 mM MnCl2) 2X. To bind the cells to the beads, 10 uL of beads were added to each sample and rotated end over end for 7 minutes. Using a magnetic stand, supernatant was removed and primary antibody was added to each sample at a 1:50 dilution in antibody buffer (20 mM HEPES pH 7.5, 150mM NaCl, 0.5 mM Spermidine, 1× Protease inhibitor cocktail, 0.05% digitonin, 2 mM EDTA, 0.1% BSA). The following primary antibodies were utilized: H3K27Ac (ab4729, Abcam), H3K4me1 (#5326, Cell Signaling Technologies), H3K4me3 (#9751, Cell Signaling Technologies), H3K27me3 (#9733, Cell Signaling Technologies), H2aK119Ub (#8240, Cell Signaling Technologies). Cells were incubated on a nutator at 4°C overnight, then antibody was removed and a guinea-pig anti rabbit secondary antibody was added at 1:100 (Antibodies Online). Samples were incubated on a nutator at room temperature for 1 hour. Secondary antibody was removed and the samples were washed twice in digitonin-wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 1× Protease inhibitor cocktail, 0.05% digitonin). pA-Tn5 transposase was diluted 1:250 in digitonin-300 buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 0.5 mM Spermidine, 1× Protease inhibitor cocktail, 0.01% digitonin) and samples were incubated on the nutator for 45 minutes at room temperature. Samples were then washed twice with digitonin-300 buffer and then resuspended in tagmentation buffer (digitonin 300 buffer supplemented with 1 mM MgCl2) and incubated at 37°C for 1 hour. DNA was then extracted by phenol:chloroform extraction. Samples were then amplified by PCR using custom nextera primers at 400 nM and NEBNext HiFi 2x PCR Master Mix (New England Biolabs) 35. PCR conditions were as follows: 72°C for 5 minutes; 98°C for 30 seconds; 14 cycles of 98°C for 10 sec, 63°C for 10 sec; and 72°C for 1 minute. Libraries were purified with AMPure Beads (Beckman) and sequenced on a NextSeq 550 sequencer (Illumina) using 37 PE sequencing.
Data Analysis
Single Cell RNA Sequencing Analysis
Sequencing output from 10X mRNA, ADT and HTO sequencing libraries were aligned to the murine genome (mm10) using CellRanger. Filtered feature matrices were analyzed using Seurat 36. Cells were demultiplexed and assigned to individual mice using Seurat function HTODemux. The gating strategy for the CITE-seq is presented in Figure S1. Cells expressing greater than 20% mitochondrial RNA were excluded as non-viable. Individual experiments were integrated using Seurat SCT integration37. Initial cluster identification was performed by comparison to reference bulk mouse transcriptomes in haemosphere.org; annotations are listed in Table S138. The trajectory graph was identified, cells ordered in pseudotime, and lineage trajectories assigned using Monocle339,40. CytoTRACE analysis was performed as previously reported using expression matrices and the CytoTRACE R package17.
Causal Path Analysis
Causal path statistically evaluates and mechanistically grounds relationships observed among transcriptomic, proteomic and phosphoproteomic measurements to yield inferences of activity flow across data points18. We utilized a prototype version of Causal Path adapted to scRNA-seq as input data. Normalized counts were formatted per the causal path input specifications and the algorithm was run with the following parameters: fdr-threshold-for-data-significance = 0.1 protein; value-transformation = significant-change-of-mean; minimum-sample-size = 3; calculate-network-significance = true; permutations-for-significance = 1000; use-network-significance-for-causal-reasoning = true. Causal networks were visualized using the Newt Editor41. For network legibility, the neutrophil lineage was limited to interactions with both Source and Target p < 0.025, and the erythrocyte network was limited to interactions with both the Source and Target p < 2 × 10−10; the complete data set is included in Table S3.
Bulk RNA Sequencing Analysis
Raw reads were trimmed with Trimmomatic42 and aligned with STAR 43. Differential expression analysis was performed using DESeq244. Raw p values were adjusted for multiple comparisons using the Benjamini-Hochberg method. GO analysis was performed using Enrichr45.
CUT&Tag Analysis
CUT&Tag libraries were aligned to the mouse genome (mm10) using Bowtie246. BAM files were downsampled to the lowest common denominator of read depth across conditions using Samtools47. To avoid issues with false positives inherent to CUT&Tag data, peaks were called on a merged BAM file containing equal representation from all conditions using MACS248. The following q-value cutoffs were used for each specific histone mark to ensure capture of high confidence peaks only: H3K4me1 q= 0.00001; H3K4me3 q= 0.0001 for peaks, q= 1×10−26 to exclude low abundance peaks at enhancers; H3K27Ac q=0.001, H3K27me3 q=0.001, H2aK119Ub q= 0.000001, Rbp1 q=0.01. Differential peaks were identified utilizing the Bioconductor package Diffbind using the default parameters49. Global signal correlation and heatmaps were generated using DeepTools50. RNAPolII pause indices were calculating by generating counts tables for each gene in a window from −30 to +100 around each TSS and then from the remainder of the gene bodies. Counts were divided by the region length and the pause index was calculated by dividing the TSS values by those for the corresponding gene body. Published ChIP-seq data was downloaded from Cistrome DB with the following accession numbers: ASXL1 76682, H3K4me1 88060, H3K4me3 76226, H3K27Ac 66607, Rbp1 9153951–54.
Supplementary Material
Acknowledgements
We would like to thank Dr. Marilynn Chow-Castro for her thoughtful discussions and Hannah Manning for her contributions related to the Causal path analysis; the OHSU Massively Parallel Sequencing Shared Resource for scRNA-seq library prep using their 10X Genomics Chromium Controller and performing short read sequencing assays. This project was supported by funding from the Cancer Early Detection and Research center (CEDAR) at Oregon Health and Science University’s Knight Cancer Institute, funding from the Edward P. Evans Foundation to TPB and R01HL157147 from the NCI to JEM and a LLS Scholar Award to JEM".
Footnotes
Competing Interests
B.J.D. potential competing interests-- SAB: Aileron Therapeutics, Therapy Architects (ALLCRON), Cepheid, Vivid Biosciences, Celgene, RUNX1 Research Program, Novartis, Gilead Sciences (inactive), Monojul (inactive); SAB & Stock: Aptose Biosciences, Blueprint Medicines, EnLiven Therapeutics, Iterion Therapeutics, Third Coast Therapeutics, GRAIL (SAB inactive); Scientific Founder: MolecularMD (inactive, acquired by ICON); Board of Directors & Stock: Amgen; Board of Directors: Burroughs Wellcome Fund, CureOne; Joint Steering Committee: Beat AML LLS; Founder: VB Therapeutics; Research Funding: EnLiven Therapeutics; Clinical Trial Funding: Novartis, Bristol-Myers Squibb, Pfizer; Royalties from Patent 6958335 (Novartis exclusive license) and OHSU and Dana-Farber Cancer Institute (one Merck exclusive license and one CytoImage, Inc. exclusive license). The remaining authors have no conflicts to report.
Data Availability:
The datasets generated during this study are available at GEO under the accession number GSE158184.
References:
- 1.Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci M-J, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol 2012; 5: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim T, Tyndel MS, Zhang Z, Ahn J, Choi S, Szardenings M et al. Exome sequencing reveals DNMT3A and ASXL1 variants associate with progression of chronic myeloid leukemia after tyrosine kinase inhibitor therapy. Leukemia Research 2017; 59: 142–148. [DOI] [PubMed] [Google Scholar]
- 3.Pratcorona M, Abbas S, Sanders MA, Koenders JE, Kavelaars FG, Erpelinck-Verschueren CAJ et al. Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. 1 2012; 97: 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tefferi A, Guglielmelli P, Lasho TL, Rotunno G, Finke C, Mannarelli C et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia 2014; 28: 1494–1500. [DOI] [PubMed] [Google Scholar]
- 5.Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018; 559: 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371: 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371: 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012; 22: 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med 2013; 210: 2641–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang P, Chen Z, Li R, Guo Y, Shi H, Bai J et al. Loss of ASXL1 in the bone marrow niche dysregulates hematopoietic stem and progenitor cell fates. Cell Discovery 2018; 4: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izzo F, Lee SC, Poran A, Chaligne R, Gaiti F, Gross B et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat Genet 2020; 52: 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Viny AD, Bowman RL, Liu Y, Lavallée V-P, Eisman SE, Xiao W et al. Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation. Cell Stem Cell 2019; 25: 682–696.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H et al. Simultaneous epitope and transcriptome measurement in single cells. Nature Methods 2017; 14: 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 2013; 13: 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pronk CJH, Rossi DJ, M\a ansson R, Attema JL, Norddahl GL, Chan CKF et al. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 2007; 1: 428–442. [DOI] [PubMed] [Google Scholar]
- 16.Muench DE, Olsson A, Ferchen K, Pham G, Serafin RA, Chutipongtanate S et al. Mouse models of neutropenia reveal progenitor-stage-specific defects. Nature 2020; 582: 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 2020; 367: 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babur Ö, Luna A, Korkut A, Durupinar F, Siper MC, Dogrusoz U et al. Causal interactions from proteomic profiles: molecular data meets pathway knowledge. bioRxiv 2018; : 258855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang J, Ge M, Lu S, Shi J, Li X, Zhang J et al. Mutations of ASXL1 and TET2 in aplastic anemia. Haematologica 2015; 100: e172–e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangaonkar AA, Gangat N, Al-Kali A, Elliott MA, Begna KH, Hanson CA et al. Prognostic impact of ASXL1 mutations in patients with myelodysplastic syndromes and multilineage dysplasia with or without ring sideroblasts. Leukemia Research 2018; 71: 60–62. [DOI] [PubMed] [Google Scholar]
- 21.Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 2019; 10: 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim M-H, Yang D, Kim M, Kim S-Y, Kim D, Kang S-J. A late-lineage murine neutrophil precursor population exhibits dynamic changes during demand-adapted granulopoiesis. Sci Rep 2017; 7: 39804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asada S, Goyama S, Inoue D, Shikata S, Takeda R, Fukushima T et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat Commun 2018; 9: 2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eberhardy SR, Farnham PJ. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem 2002; 277: 40156–40162. [DOI] [PubMed] [Google Scholar]
- 25.Price DH. Regulation of RNA Polymerase II Elongation by c-Myc. Cell 2010; 141: 399–400. [DOI] [PubMed] [Google Scholar]
- 26.Nagase R, Inoue D, Pastore A, Fujino T, Hou H-A, Yamasaki N et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J Exp Med 2018; 215: 1729–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H et al. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 2016; 537: 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol 2010; 31: 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Wilmot B, Bottomly D, Dao K-HT, Stevens E, Eide CA et al. Genomic landscape of neutrophilic leukemias of ambiguous diagnosis. Blood 2019; 134: 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milne TA, Sinclair DA, Brock HW. The Additional sex combs gene of Drosophila is required for activation and repression of homeotic loci, and interacts specifically with Polycomb and super sex combs. Mol Gen Genet 1999; 261: 753–761. [DOI] [PubMed] [Google Scholar]
- 31.Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 2013; 27: 1000–1008. [DOI] [PubMed] [Google Scholar]
- 32.Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes & Development 2003; 17: 2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pineault N, Helgason CD, Lawrence HJ, Humphries RK. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp Hematol 2002; 30: 49–57. [DOI] [PubMed] [Google Scholar]
- 34.Yang H, Kurtenbach S, Guo Y, Lohse I, Durante MA, Li J et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018; 131: 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buenrostro JD, Corces R, Wu B, Schep AN, Lareau C, Majeti R et al. Single-cell epigenomics maps the continuous regulatory landscape of human hematopoietic differentiation. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 2018; 36: 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM et al. Comprehensive Integration of Single-Cell Data. Cell 2019; 177: 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi J, Baldwin TM, Wong M, Bolden JE, Fairfax KA, Lucas EC et al. Haemopedia RNA-seq: a database of gene expression during haematopoiesis in mice and humans. Nucleic Acids Res 2019; 47: D780–D785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA et al. Reversed graph embedding resolves complex single-cell trajectories. Nature Methods 2017; 14: 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology 2014; 32: 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balci H, Siper MC, Saleh N, Safarli I, Roy L, Kilicarslan M et al. Newt: a comprehensive web-based tool for viewing, constructing and analyzing biological maps. Bioinformatics 2021; 37: 1475–1477. [DOI] [PubMed] [Google Scholar]
- 42.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016; 44: W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012; 481: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 2016; 44: W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fong N, Saldi T, Sheridan RM, Cortazar MA, Bentley DL. RNA Pol II Dynamics Modulate Co-transcriptional Chromatin Modification, CTD Phosphorylation, and Transcriptional Direction. Mol Cell 2017; 66: 546–557.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hauri S, Comoglio F, Seimiya M, Gerstung M, Glatter T, Hansen K et al. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep 2016; 17: 583–595. [DOI] [PubMed] [Google Scholar]
- 53.Morgan MAJ, Rickels RA, Collings CK, He X, Cao K, Herz H-M et al. A cryptic Tudor domain links BRWD2/PHIP to COMPASS-mediated histone H3K4 methylation. Genes Dev 2017; 31: 2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Savitsky P, Krojer T, Fujisawa T, Lambert J-P, Picaud S, Wang C-Y et al. Multivalent Histone and DNA Engagement by a PHD/BRD/PWWP Triple Reader Cassette Recruits ZMYND8 to K14ac-Rich Chromatin. Cell Rep 2016; 17: 2724–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at GEO under the accession number GSE158184.