

Abstract
Lysosomal ion channels mediate ion flux from lysosomes and regulate membrane potential across the lysosomal membrane, which are essential for lysosome biogenesis, nutrient sensing, lysosome trafficking, lysosome enzyme activity, and cell membrane repair. As a cation channel, the transient receptor potential mucolipin 1 (TRPML1) channel is mainly expressed on lysosomes and late endosomes. Recently, the normal function of TRPML1 channels has been demonstrated to be important for the maintenance of cardiovascular and renal glomerular homeostasis and thereby involved in the pathogenesis of some cardiovascular and kidney diseases. In arterial myocytes, it has been found that Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP), an intracellular second messenger, can induce Ca2+ release through the lysosomal TRPML1 channel, leading to a global Ca2+ release response from the sarcoplasmic reticulum (SR). In podocytes, it has been demonstrated that lysosomal TRPML1 channels control lysosome trafficking and exosome release, which contribute to the maintenance of podocyte functional integrity. The defect or functional deficiency of lysosomal TRPML1 channels has been shown to critically contribute to the initiation and development of some chronic degeneration or diseases in the cardiovascular system or kidneys. Here we briefly summarize the current evidence demonstrating the regulation of lysosomal TRPML1 channel activity and related signaling mechanisms. We also provide some insights into the canonical and non-canonical roles of TRPML1 channel dysfunction as a potential pathogenic mechanism for certain cardiovascular and kidney diseases and associated therapeutic strategies.
Keywords: Lysosome, TRPML1 channel, NAADP, Autophagy, Exosomes, Atherosclerosis, Chronic kidney disease
13.1. Introduction
Transient receptor potential mucolipin 1 (TRPML1) channel is a cation channel expressed in late endosomes and lysosomes [1–4]. As a member of the TRPML family of ion channels, TRPML1 is coded by the gene MCOLN1. TRPML1 channel was identified in a search for genesis of mucolipidosis type IV (MLIV), one of lysosomal storage diseases [5, 6]. The major symptoms of MLIV include psychomotor retardation and visual impairment [7], which are associated with the defect of lysosome trafficking and lysosomal accumulation of lipofuscin and other macromolecules including phospholipids, gangliosides, and mucopolysaccharides [8, 9]. In this chapter, we will describe the common characteristics of lysosomal TRPML1 channel and summarize the regulatory mechanisms of TRPML1 channel and related cellular activities mediated by TRPML1 channels and their regulation. We will also present some recent findings regarding the pathogenic role of lysosomal TRPML1 channel deficiency or dysregulation in cardiovascular and glomerular diseases.
13.2. Characteristics of Lysosomal TRPML1 Channels
13.2.1. Subcellular Localization of Mammalian TRPML1 Channels
As a lysosomal transmembrane protein, TRPML1 channel has two di-leucine motifs for its trafficking to lysosome [2–4]. Deletion of these di-leucine motifs may induce the accumulation of TRPML1 channel in the plasma membrane [2–4]. Also, the expression of a dominant-negative variant of dynamin was found to induce the accumulation of TRPML1 channel in the plasma membrane through abrogation of clathrin-mediated endocytosis [3]. These findings indicate that TRPML1 channel may traffic from the plasma membrane to the lysosome. However, there are evidences showing the direct delivery of TRPML1 channel to the lysosome from the trans-Golgi without intermediate translocation to the plasma membrane [4, 10]. Interestingly, overactivation of TRPML1 channel may also result in its traffic from lysosomes to the plasma membrane. For example, elevated Ca2+ release through TRPML1 channel due to gene mutation enhanced lysosomal exocytosis and consequent translocation of TRPML1 channel to plasma membrane [11]. In Drosophila cells, activation of the mechanistic target of rapamycin complex-1 (mTORC1) also led to the elevated level of TRPML1 channel in the plasma membrane [12]. Moreover, three mammalian TRPMLs including TRPML1, 2, and 3 are capable of forming heteromultimers [3, 13–15], and the difference in biophysical properties and regulatory mechanisms between homomultimers and heteromultimers may enhance the functional diversity of these proteins [13].
13.2.2. Biophysical Properties of TRPML1 Channels
Various sensations and ligands participate in the regulation of TRP channel activity [16]. TRPMLs are relatively small proteins consisting of <600 amino acids, and the expectation of their molecular sizes is approximately 56–65 kDa. The different sizes of TRPML1 channel may range from 36 to 75 kDa due to cleavage and other modifications in various native cells [6, 17]. A similar topology to other TRP channels was found in TRPML1 channel. The ion channel is formed at the center of the canonical homotetrameric assembly which is formed by TRPML1 channels. Each subunit is composed of six transmembrane helices (S1–S6), two-pore helices (PH1 and PH2), and a luminal domain [18]. The characteristics of TRPML1 channel include a TRP channel-homologous region and an internal pore region for the passage of Ca2+ and Na+. Also, TRPML1 channel is characterized with the location of both NH2 and COOH terminal chains in cytosol. The activity of TRPML1 as a nonspecific cation channel is permeant to Ca2+ and H+ and thereby controlled by lysosomal luminal and cytosolic Ca2+ and H+ concentrations [19, 20].
The activity of TRPML1 reaches the maximum at pH 4.6, which is nearby the mean value of lysosomal lumen pH of mammalian cells [21, 22]. In Drosophila, the optimal pH for maximal activation of TRPML1 channel is pH 5.2, consistent with the mean luminal pH of insect lysosomes [23].
As channels insensitive to voltage but gated by ligands, TRPMLs have been extensively studied regarding their ion selectivity. For example, it has been demonstrated that human TRPML1 ion selectivity is Ba2+>Mn2+>Fe2+=Ca2+=Mg2+− > Ni2+ = Co2+ = Cd2+ > Zn2+ ⨠ Cu2+. In the presence of 30–105 mM Fe2+, its pS is 32–40 at pH 4.6 [21, 22]. Nevertheless, most of previous studies characterized lysosome TRPML1 channel in cell lines transfected with TRPML1 genes. Recently, by lipid bilayer reconstitution and single lysosome patch clamping, we have demonstrated the normal function of the TRPML1 channel in lysosomes isolated from rat hepatocytes, coronary arterial myocytes, human fibroblasts, and murine podocytes [24–27]. The native TRPML1 channels can be activated by Nicotinic acid adenine dinucleotide phosphate (NAADP). In addition, the opening of TRPML channel can be blocked by its specific TRPML1 channel blockers, TRPML1 siRNA, and neutralized antibody against TRPML1 channel.
Many studies have elucidated the physiological relevance of the Ca2+ conductivity of TRPML1 channel to the regulation of lysosome maturation, autophagic flux, lysosomal exocytosis, and Ca2+ signaling-related cellular activities [20, 28]. However, the functional significance of the conductance of TRPML1 channel to other ions beyond Ca2+ has not yet been well clarified. Based on the conductance of the TRMPL1 channel to Fe2+, it has been assumed that TRPML1 may contribute to the hematological and degenerative abnormality in MLIV via regulation of the cellular homeostasis of Fe2+ [11, 21]. Clearly, more studies are needed to explore the physiological relevance of the conductance of TRPML1 channel to other ions beyond Ca2+.
13.3. Agonists and Blockers of TRPML1 Channel
13.3.1. NAADP
As a metabolite of NADP+, NAADP has been reported to induce Ca2+ release from sea urchin egg microsomes [29–31]. Since then, it was demonstrated that NAADP is produced by the soluble protein Aplysia ADP-ribosylcyclase and its membrane-bound homologs, CD38 and CD157 [32–35]. These enzymes can exchange the terminal nicotinamide group of the NADP+ with nicotinic acid to produce NAADP through a base-exchange reaction, which has been found in various cells and tissues, such as smooth muscle cells, pancreatic acinar cells, human T lymphocytes, sea urchin eggs, and rat brain [36–39]. In our recent studies, we have demonstrated that CD38 and its cytosolic isoforms have been found to produce NAADP in mouse coronary artery in response to death receptor activation, endothelin, and oxidant stimulation [40, 41].
Our previous studies on redox signaling and lipid raft-associated transmembrane signaling mechanisms [40, 42, 43] indicate that some agonists or stimuli may generate an acid microenvironment in vascular cells. For example, both FasL and ET-1 have been found to preferably stimulate NAADP production in vascular smooth muscle cells (VSMCs) [41, 44], which may be associated with their ability to generate a local acidic microenvironment at the cell membrane, facilitating a base exchange reaction via CD38. The formation of this local acidic environment might be attributed to lipid raft clustering if agonist receptors are linked to lipid rafts. Previous studies have shown that the clustering of membrane lipid rafts may induce the activation of CD38 [42, 45–47]. The agonist-induced membrane raft clustering is usually initiated by lysosome fusion to the cell membrane, which translocates critical proteins to the cell membrane for raft clustering such as acid sphingomyelinase (ASM) and lysosomal vacuolar H+-ATPase. A local acidic environment is provided by lysosomal vacuolar H+-ATPase, which maintains the normal function of translocated ASM to produce ceramide for the construction of lipid raft platforms [43]. Such membrane microenvironment of acidic pH generated and maintained by vacuolar H+-ATPase is critical for the production of NAADP by CD38 within lipid raft platforms.
When we searched for the molecular target of NAADP action in lysosomes, reconstitution, and characterization of NAADP-sensitive Ca2+ channel from lysosome of rat liver was first done in our laboratory using lipid bilayer. In 2007, we have demonstrated a reconstituted Ca2+ channel with all the biophysical and pharmacological features of TRPML1 in liver lysosomes [26]. In bovine coronary arterial muscle cells, we further characterized this TRPML1 channel using lysosome preparations by the constitution in the lipid bilayer. In arterial muscle preparation, we have demonstrated that the reconstituted lysosomal channel is also a voltage-dependent Ca2+ channel [25]. It was found that NAADP activates TRPML1 channel reconstituted from the liver and coronary arterial muscle lysosome preparations in a concentration-dependent manner. In addition, high concentrations of NAADP may result in self-desensitization of TRPML1 channel. In the bilayer preparations containing coronary arterial muscle lysosomal channels, pretreatment with a subthreshold concentration of NAADP substantially attenuated the activity of these channels in response to higher concentrations of NAADP, which indicated the self-desensitization property of these lysosomal channels. This self-desensitization property of lysosomal ion channels has also been shown in the actions of NAADP as a Ca2+-releasing second messenger in other cells such as sea urchin egg fractions or intact egg cells [48–50].
Pharmacologically, these reconstituted lysosomal channels in both liver and coronary arterial lysosomes were blocked by commonly used antagonists of TRPML1 channels such as NAADP receptor antagonist PPADS, sodium channel antagonist amiloride, and dihydropyridine derivatives nifedipine and verapamil [26, 51]. Using gene silencing, deprivation of TRPML1 protein, and interference of its channel pore formation, NAADP was shown to induce Ca2+ release from the lysosomal store via TRPML1 channel-mediated Ca2+ release [25]. More recently, lysosomal TRPML1 channels have been reported to regulate local compartmental Ca2+ which importantly controls lysosome function such as trafficking or intracellular signaling [27].
13.3.2. Phosphoinositides
As a low-abundance lysosome-specific phosphoinositide [52–55], phosphatidylinositol-3,5-bisphosphate (PI(3,5)P2) can be generated from PI(3)P through PIKfyve/Fab1, a PI 5-kinase that locates in the lysosome of both yeast and mammalian cells [54, 56–58]. Several associated proteins, such as Fig4, Vac14, and Vac7, can enhance the activity of PIKfyve/Fab1 [53–55, 59]. Also, the myotubularin (MTM/MTMR)-family of PI-3 phosphatase can metabolize PI (3,5)P2 into PI(5)P [54, 58, 60]. A variety of neurodegenerative diseases can be induced by gene mutations of PI(3,5)P2-metabolizing enzymes and their regulators, such as Charcot–Marie–Tooth disease and amyotrophic lateral sclerosis [53, 54, 61]. Recently, it has been found that PI(3,5)P2 activates lysosome-localized TRPML1 channels specifically and potently [22, 62–64]. Enlarged lysosomes and vacuoles and deficient lysosome trafficking were found in both PI(3,5)P2-deficient cells and TRPML1-deficient cells [53–55, 59]. In PI(3,5)P2-deficient mouse fibroblasts, the enlargement of vacuole can be inhibited by overexpression of TRPML1 [62].
On the contrary, PI(4,5)P2 has been found to potently inhibit the opening of TRPML1 channel [64]. Overexpression and plasma membrane translocation of 5-phosphatase can potentiate whole-cell TRPML1 currents through degradation of PI(4,5)P2. Thus, inhibition by PI(4,5)P2 may serve as a mechanism to inactivate lysosome-operating TRPML1 channels upon plasma membrane insertion. It has been known that different PIP2 products may have different effects on TRPML1 channel activity, such as the inhibitory effects of the plasma membrane-specific phosphoinositides PI(4,5)P2, PI(3,4,5) P3, and PI(3,4)P2 and the stimulatory effect of lysosome-specific PI(3,5)P2. This suggests that the activity of TRPML1 channel is economically and efficiently regulated by these compartment-specific phosphoinositides.
In a recent study, the structure of mouse TRPML1 channel has been uncovered by single-particle cryo-electron microscopy after its embedding in nanodiscs [65]. Its structure indicates that the coupling of ligand binding to pore opening may depend on the binding of PIP2 to the N-terminus of the channel and the helix-turn-helix extension between S2 and S3. Under the closed condition, two equally distributed conformations of the S4–S5 linker have been observed, which implies that S4–S5 linker may mediate the gating of TRPML1 channel by PIP2.
13.3.3. Synthetic Agonists and Blockers
Xu and his associates developed an elegant approach using patch-clamp techniques and recorded Fe2+ and Ca2+ channel activity directly from the lysosome membrane in different cell types with TRPML1 transgene. They have discovered that TRPML1 is a proton-impermeable and inwardly rectifying cation channel, which is dually permeable to Ca2+ and Fe2+/Mn2+ [11, 21, 62]. More recently, the same group used a genetically encoded Ca2+ indicator (GCaMP3) attached directly to TRPML1 to directly measure Ca2+ release from lysosomes via TRP-ML1 [66], which further confirms the nature of TRPML1 as a lysosomal Ca2+ release channel. These studies have demonstrated that all three members of the mammalian TRPML subfamily, TRPML1–3, are activated by mucolipin synthetic agonist 1 (ML-SA1). More recently, it has been revealed that ML-SA1 may have therapeutic potential against some lysosome storage diseases, in which TRPML1 expression and function may be diminished. For example, the defect in lysosomal function was found in cells from patients with Niemann–Pick (NP) disease, one of the lysosome storage diseases that resulted in alterations in Ca2+ and Fe2+ homoeostasis, defects in lipid trafficking, and impairments in autophagosome–lysosome fusion and/or lysosome reformation [67, 68]. Similar pathological changes can also be observed in MLIV, which led to a broad examination of TRPML1 channel function in cells with NP disease type A and type C (NPA and NPC) mutations and cells lack of NPC1 gene (NPC1−/− cells). It was found that the activity of TRPML1 channel was remarkably inhibited by NPA and NPC mutations or NPC1 gene deletion, suggesting that reduction of TRPML1 channel activity is a consequence of NP disease [67]. Mechanically, the activity of the TRPML1 channel was directly inhibited by sphingomyelin, which is abnormally accumulated in lysosomes in all NP disease cells. Moreover, the lysosomal function in NPC1−/− cells was recovered by enhancement of TRPML1 expression or activation of TRPML1 by ML-SA1 [67]. In a recent study, it has been reported that Duchenne muscular dystrophy in mouse models can be ameliorated by ML-SA5 through activation of TRPML1 channel [69]. Therefore, the pharmacological intervention of the TRPML1 channel by synthetic agonist may be an effective strategy to rescue lysosomal function in various lysosomal storage disorders. However, the mechanism mediating the action of ML-SA1 on TRPML1 remains unclear. Before the discovery of ML-SAs, previous studies showed that SF-22 and SF-51 have stimulatory effects on TRPML1 channel [67, 70]. It has been found that ML-SA1 induced more potent activation of TRPML1 channel compared to SF-51 [67]. As an mTOR inhibitor, rapamycin has been reported to have direct activating action on TRPML1 channels [71]. All these activators can be used to study TRPML1 channel activity and its physiological and pathological relevance in different cells or tissues.
It was found that amiloride, ruthenium red, nifedipine, 2APB, and SKF 96365 as commonly used cation channel and/or Ca2+ channel blockers failed to inhibit TRPML1 or TRPML3. Verapamil and La3+, however, have been demonstrated to block both TRPML1 channel and TRPML3 channel [25, 72, 73]. ML-SI1, a novel selective TRPML1 inhibitor, has also been found to block the opening of TRPML1 channel, leading to inhibition of particle uptake and lysosomal exocytosis in bone marrow-derived macrophages [74]. More recently, ML-SI3 and ML-SI6 have been reported to inhibit TRPML1 channel activity [69] in a more specific manner, which can be used as tool drugs for studies on these lysosomal channels.
13.4. Associated Proteins of TRPML1 Channel
13.4.1. ALG-2
A recent study has shown that TRPML1 channel interacts with apoptosis-linked gene 2 (ALG-2) in a Ca2+-dependent manner, which impacts endolysosomal vesicle trafficking [75]. As a Ca2+-binding protein, ALG-2 belongs to the penta-EF-hand protein family [76]. The conformation of ALG-2 is changed in response to Ca2+, which enhances its affinities to its binding partners [76–78]. It has been found that ALG-2 binds to the amino-terminal tail of TRPML1 channel in a Ca2+-dependent manner. Furthermore, mutation of the ALG-2-binding domain in TRPML1 channel may result in the disarrangement of its distribution and function, suggesting that TRPML1 channel activity is regulated by ALG-2. More recently, there is evidence showing that lysosomal distribution is controlled by ALG-2 as a Ca2+ effector of the TRPML1 channel [79]. The overexpression of ALG-2 in fibroblasts resulted in a dramatic perinuclear distribution of lysosomes, which was abolished by inhibition of TRPML1 channel. On the other hand, activation of TRPML1 channel with ML-SA1 increased the amount of ALG-2 on the lysosomal membrane. Importantly, ALG-2 pulled down dynamitin, a dynactin complex component, in coimmunoprecipitation assays in a Ca2+-independent manner [79]. We have also found that NAADP induces Ca2+ release through TRPML1 channels via ALG-2 (data not published). In mouse coronary arterial myocytes, ALG-2 was found abundantly expressed in the cytosol. Using confocal microscopy, FasL and ET-1, two well-known NAADP stimulators were found to increase the colocalization coefficient of ALG-2-GFP with TRPML1-RFP in these arterial myocytes. Immunoprecipitation with anti-ALG-2 antibody, the interaction of ALG-2 with TRPML1 channel in the lysosome, was confirmed, which was enhanced upon NAADP stimulation. In addition, recombinant ALG-2 protein binding with NAADP as receptors was significantly reduced by NAADP, and NAADP-induced lysosomal Ca2+ release was blocked by ALG-2 siRNA. These results demonstrate that ALG-2 indeed directly binds to NAADP and thereby may mediate the action of NAADP-induced lysosomal Ca2+ release via lysosomal TRPML1 channels.
13.4.2. Hsc70
The chaperone-mediated autophagy (CMA) selectively determines the recycle of soluble cytosolic proteins [80]. The selectivity of CMA depends on the recognition of a targeting motif in soluble cytosolic proteins by a chaperone, which is essential for the transportation of these proteins to the lysosomal surface [80]. As a constitutively expressed member of the 70-kDa family of chaperones, the heat shock cognate protein of 70 kDa (Hsc70) recognizes the substrate of CMA in the cytoplasm [81]. Recently, it has been demonstrated that Hsc70 interacts with TRPML1 channel in yeast two-hybrid and co-immunoprecipitation experiments [82]. In starvation studies, a working model has been suggested that lysosomal TRPML1 activity upon starvation may release Ca2+ to promote lysosomal Hsc70 recruitment. This TRMPL1–Hsc70 interaction enhances CMA and shifts metabolism from an anabolic toward a catabolic, thereby exerting nutrient-liberating action [79]. In macrophages of MLIV patients, the defect of CMA has been found in response to serum withdrawal, indicating that the interaction between TRPML1 channel and Hsc70 is essential for CMA. Furthermore, the reduction of lysosome-associated membrane protein type 2A is associated with the elevation of oxidized proteins in MLIV fibroblasts. All these results tell us that the delivery of substrates to the lysosomal surface depends on TRPML1 channels as a docking site for Hsc70 during CMA [82].
13.4.3. LAPTM
More recently, a novel interaction between the TRPML1 channel and the members of the lysosome-associated protein transmembrane (LAPTM) family has been identified [83]. In ARPE-19 cells, LAPTM4a, LAPTM4b, and LAPTM5 showed very high degrees of colocalization with the TRPML1 channel in late endosomes and lysosomes [83]. Functionally, overexpression of LAPTM4b caused enlargement of late endosomes and lysosomes, which can be prevented by overexpression of TRPML1 channels [83]. In HeLa cells, dysfunction of LAPTM4 resulted in a phenotype similar to lysosomal storage disease found in MLIV patients [83]. Correspondingly, a previous study has shown that expressions of LAPTM4a and LAPTM5 in MLIV cells increased threefold and sevenfold, respectively [84]. This changes the compensation attempted by MLIV cells due to the lack of LAPTM activity. In summary, LAPTM–TRPML1 interaction is important for the normal function of lysosomes and that the defect of this interaction is implicated in the pathogenesis of MLIV.
13.5. Regulatory Mechanisms of TRPML1 Channel Activity
13.5.1. Cathepsin B
The lysosomal proteases contribute to antimicrobial host defense against infection. As lysosomal proteases, cathepsins provide antimicrobial host defense against infection through degradation of intracellular bacteria within lysosomes. The reduction of capacity to kill bacteria has been reported in macrophages lacking cathepsin D during pneumococcal infection [85]. Also, the susceptibility to infections by Porphyromonas gingivalis and Staphylococcus aureus was amplified by the knockout of the cathepsin E gene [86]. Moreover, cathepsin L has been reported to inhibit the infection of Mycoplasma pulmonis [87]. On the contrary, there is evidence indicating that the activity of cathepsin B may contribute to bacterial infection.
TRPML1 channel may be inactivated after cleavage by lysosomal proteases. Recently, it has been reported that cathepsin B mediates the critical or final cleavage of TRPML1 channel [88]. The cleavage of the TRPML1 channel constitutes a regulatory mechanism to limit the duration of TRPML1 channel activity, which is important to the maintenance of lysosomal ionic homeostasis. In a recent study, the elevation of resistance to Francisella novicida, a cytosolic bacterial pathogen, has been found in mice and macrophages lacking cathepsin B activity [89]. Mechanically, defect in cathepsin B function inhibited mTOR activity and prevented cleavage of TRPML1 channel. In response to these changes, transcription of lysosomal and autophagy genes increased lysosomal biogenesis and enhanced the activity of autophagy initiation kinase ULK1 for bacteria clearance, which depended on transcription factor EB (TFEB). Interestingly, it has been found that inhibition of TRPML1 channel expression leads to the leak of lysosomal protease cathepsin B into the cytoplasm [90]. The leak of cathepsin B is associated with apoptosis, which can be prevented by pharmacological inhibition of cathepsin B activity. These findings indicate that modulation of cathepsin B activity may be a potential therapeutic strategy to enhance host immunity against certain bacterial infections. Also, cathepsin B might be a therapeutic target for the treatment of debilitating lysosome storage diseases.
13.5.2. TOR–TFEB Signaling Pathway
In autophagy, unwanted cellular components are digested for the generation of catabolites that are used for housekeeping biosynthesis processes in response to nutrient starvation. A recent study has reported that the activity of TRPML1 is potently and rapidly increased upon nutrient starvation [91]. In this study, starvation-induced boost of lysosomal degradation capability was completely abolished by pharmacological inhibition or genetic deletion of TRPML1, indicating the essential role of TRPML1 channel in the lysosomal adaptation to nutrient starvation [91]. Similar to nutrient starvation, the complete suppression of mTORC1 function remarkably increased TRPML1 channel-mediated Ca2+ release [91]. Mechanically, TFEB nuclear translocation is triggered after nutrient starvation or complete inhibition of mTORC1 [91, 92]. The expression of TRPML1 channel is upregulated by nuclear translocation or overexpression of TFEB [91, 93]. Furthermore, TOR as a nutrient-sensitive protein kinase has been found to directly target and inactivate the TRPML1 channel through phosphorylation [94]. The mutations of phosphorylation sites of the TRPML1 channel blocked the inhibition of TRPML1 channel activity by TOR [94].
13.5.3. Phosphorylation
The function of protein is importantly regulated by post-translational modifications. Phosphorylation and dephosphorylation events play vital roles in the regulation of the activity of many TRP channels. Serine, threonine, and tyrosine residues are targets of TRP phosphorylation, which is catalyzed by various kinases, including tyrosine kinase, PKA (protein kinase A), PKC (protein kinase C), PKG (protein kinase G), and CaMKII (Ca2+/calmodulin-dependent protein kinase II) [95]. In a recent study, it has been demonstrated that the activity of TRPML1 channel is modulated by phosphorylation [96]. FSK-induced activation of PKA enhanced phosphorylation of TRPML1 channel, leading to inhibition of TRPML1 channel activity. On the contrary, the activity of the TRPML1 channel was remarkably amplified after the inhibition of PKA by H89. These results indicate that PKA may be involved in the regulation of lysosomal function through modulation of TRPML1 channel activity.
13.5.4. Regulation of TRPML1 Channel Activity by Sphingolipids
Ceramide is the central core in sphingolipid metabolism and plays an essential role in normal cell and tissue homeostasis and in the development of numerous diseases [97, 98]. The hydrolysis of sphingomyelin by acid sphingomyelinase (ASM) leads to the production of ceramide, which preferentially occurs in lysosomes or other acidic vesicles. Lysosomal acid ceramidase (AC) metabolizes ceramide into sphingosine which can be phosphorylated by sphingosine kinase for the generation of sphingosine-1-phosphate (S1P) [99, 100]. Recent studies have shown that sphingomyelin accumulation inhibits lysosomal TRPML1 channel-mediated Ca2+ release and thereby leads to impaired lysosome trafficking and lysosomal storage disease as shown in Niemann–Pick disease [67, 101]. On the contrary, lysosomal Ca2+ release through the TRPML1 channel and associated lysosome trafficking are enhanced by sphingosine. More recently, we have demonstrated that sphingolipid metabolism by AC regulates TRPML1 channel activity [24]. The defect in AC function may block TRPML1 channel, leading to lysosome dysfunction and increased exosome release from podocytes [24]. These findings suggest that sphingolipid signaling plays a pivotal role in the regulation of lysosomal TRPML1 channel activity and thereby determines Ca2+-dependent lysosome trafficking and fusion to multivesicular bodies (MVBs) that govern exosome release. However, it remains unknown how sphingolipids alter the activity of TRPML1 channel. Further study on this regulatory mechanism would be beneficial to our understanding of the regulation of TRPML1 channel under both physiological and pathological conditions.
13.5.5. Redox Regulation of TRPML1 Channel Activity
A variety of active oxygen-containing compounds are generally called reactive oxygen species (ROS), including hydrogen peroxide (H2O2), superoxide anion (O2−), and free radical (superoxide and hydroxyl radicals) [102]. Biomacromolecules involved in different cellular activities can be damaged by ROS [103]. On the contrary, ROS is essential for the redox signaling cascade in many crucial cellular processes [104]. A simultaneous balance of ROS is maintained by cellular oxidation and antioxidation. Cellular damage can be induced by oxidative stress when oxidation exceeds antioxidation [105]. There is increasing evidence showing that the boost of autophagy may be attributed to the production of ROS, which is a vital defensive mechanism against cellular stress [106, 107]. Under conditions such as ischemia, hypoxia, and nutrient starvation, autophagy may be induced by mitochondrial ROS [108–110]. Nevertheless, autophagosome accumulation was observed after induction of autophagy by ROS [111–113], which indicated that lysosome-dependent autophagosome degradation may be interfered. Therefore, the actions of ROS on the molecules controlling lysosomal functions, such as TRPML1 channel, need to be studied.
A recent study has focused on the regulation of TRPML1 channel by ROS or oxidants in some cell lines [113]. The actions of a variety of commonly used oxidants on the TRPML1 channel have been tested in this study. It was found that chloramine-T, a nonselective strong oxidant, activated TRPML1 channel with a potency comparable to those of PI(3,5)P2 and ML-SA1. Furthermore, several other commonly used oxidants, including NaOCl, H2O2, N-chlorosuccinimide, t-butyl hydroperoxide (TBHP), and thimerosal, have been found to activate TRPML1 channel less potently. Cysteine-modifying oxidants such as DTNP and DTNB24, however, failed to induce the opening of TRPML1 channel. Similarly, SNAP, a NO-donor, and 4-HNE25, a reactive lipid peroxidation intermediate, did not affect the activity of TRPML1 channel. However, among these different oxidants, only H2O2 mimics the mitochondrial ROS while the dose of H2O2 in this study was too high (10 mM). Furthermore, various forms of ROS or oxidants that produced effects on TRPML1 channel activity vary a lot in this study, which made the conclusion of this study uncertain. At least, ROS from different resources and ROS at different levels may play diverse roles in the regulation of TRPML1 channel activity. Functionally, carbonyl cyanide m-chlorophenylhydrazone (CCCP), a mitochondrial respiration inhibitor commonly used to induce ROS production, was found to induce autophagic clearance of damaged mitochondria through activation of TRPML1 channel [113]. Conversely, another previous study has shown that CCCP induced autophagosome accumulation and apoptosis through activation of TRPML1 channel [114]. Recently, we have tested whether ROS affects TRPML1 channel activity in murine podocytes (data not published). It was found that pretreatment of H2O2 (100 μM) totally blocked the opening of TRPML1 channel induced by ML-SA1. Moreover, pretreatment with homocysteine (Hcy), an inducer of endogenous ROS production [115], remarkably attenuated Ca2+ release through the TRPML1 channel induced by ML-SA1. These results indicate that ROS may act as a danger factor by inhibiting TRPML1 channel activity under pathological conditions, such as hyperhomocysteinemia (hHcy). In this regard, it has been found that many compounds, including N-acetylcysteine, Ferulic acid, and Trehalose, reduce ROS and enhance autophagy simultaneously [116–118]. These findings may support the possibility that ROS inhibits lysosome function through blockade of TRPML1 channels.
Interestingly, the TRPML1 channel has been found to mediate the removal of ROS [113, 119]. In RPE1 (retinal pigmented epithelial 1) cells, inhibition of TRPML1 channel expression remarkably increased lipid peroxidation, which indicated the elevation of ROS production [119]. Also, depolarization and morphological changes in mitochondria were induced by siRNA of TRPML1 channel in these cells [119]. In another study, repolarization of mitochondrial membrane potential after CCCP treatment was much slower in MLIV cells compared with wild-type cells [113]. The basal level of ROS in MLIV fibroblasts was much more than the basal level of ROS in wild-type cells. Constitutive elevation of ROS and chronic inhibition of TRPML1 were observed in ML-IV cells and NPC cells [67]. Furthermore, blockade of TRPML1 channel by ML-SI4 markedly enhanced CCCP-induced ROS production in HeLa cells [113]. Collectively, these findings unveil that inhibition of TRPML1 channels by ROS and clearance of ROS by TRPML1 channel may form a balance under normal conditions, and this balance may be broken by pathological stimuli.
In addition to the controversy of the role of ROS in TRPML1 channel regulation, the mechanisms by which ROS control TRPML1 channel activity are under exploration. For example, previous studies have demonstrated that ROS enhanced the activity of Cathepsin B while having no effects on the expression of Cathepsin B [120, 121]. The enhancement of Cathepsin B activity by ROS may lead to inhibition of TRPML1 channel activity through increased cleavage of TRPML1 channels. Furthermore, it has been found that sanguinarine (SNG), a naturally occurring benzophenanthridine alkaloid, can induce ROS production and apoptotic cell death in human leukemic cells [122]. In addition, ceramide accumulation due to activation of ASM and inhibition of AC may be the mechanism mediating SNG-induced ROS production and apoptosis in human leukemic cells [122]. In MDA-MB-231 cells, the combination of C6-ceramide and DM-102, an AC inhibitor, was reported to enhance ROS production remarkably [123]. These findings indicate the complex interactions in the regulatory system of TRPML1 channel activity. For our better understanding of the impact of ROS on TRPML1 channels, further studies are imperative on this regulatory pathway. Regulatory mechanisms of TRPML1 channel activity are summarized in Fig. 13.1.
Fig. 13.1.
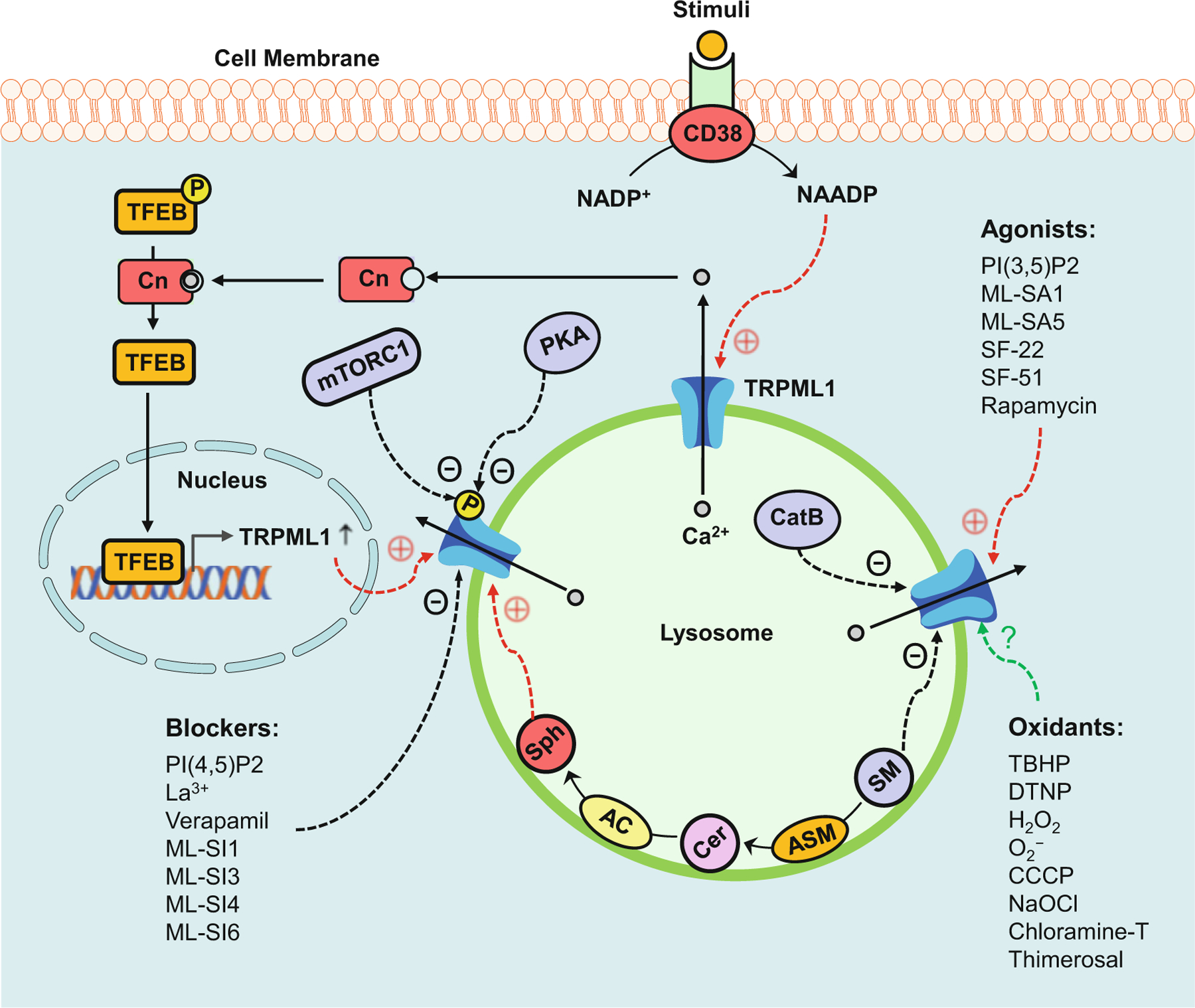
Regulatory mechanisms of TRPML1 channel activity. TRPML1 channel agonists such as PI(3,5)P2 and ML-SA1 may induce the opening of this channel. TRPML1 channel blockers such as PI(4,5)P2 or ML-SI1 may block this channel. In response to stimuli such as FasL and ET-1, CD38 converts NADP+ to NAADP that may activate the TRPML1 channel. PKA and mTORC1 are responsible for the phosphorylation of the TRPML1 channel which may result in dysfunction of this channel. Ca2+ released through the TRPML1 channel activates calcineurin (Cn) which is responsible for the dephosphorylation of TFEB. Dephosphorylated TFEB translocates to the nucleus to initiate the transcription of lysosomal and autophagic genes. Sphingolipids have different effects on TRPML1 channel activity, with inhibition by sphingomyelin (SM), no effect from ceramide (Cer), and enhancement by sphingosine (Sph). Cathepsin B mediates the critical or final cleavage of TRPML1 channel which results in dysfunction of this channel. The effects of oxidants on the TRPML1 channel remain controversial
13.6. Functions of TRPML1 Channels in Health and Diseases
13.6.1. Lysosomal pH Control
The regulatory effect of the TRPML1 channel on lysosomal pH remains debatable. It has been reported that TRPML1 channel may mediate the release of H+ from the lysosomal lumen [88, 124–126]. The dysfunction of TRPML1 channel may lower the lysosomal pH. Also, it was found that elevation of the lysosomal pH by Nigericin or Chloroquine attenuated the lysosomal storage in MLIV cells [125]. However, another study found that treatment with Nigericin or Chloroquine had no significant effect on the lysosomal storage in MLIV cells [127]. Moreover, there is evidence showing that TRPML1 channel is not permeable to H+ [21, 73]. Taken together, the exact role of TRPML1 channel in the regulation of lysosomal pH remains unclear and further studies are necessary.
13.6.2. Fusion and Fission of Cell Membrane
Previous studies have reported that TRPML1 channel activity is essential to cellular activities associated with vesicular trafficking, such as autophagy, lysosomal exocytosis, and the formation of early endosomes [20]. All these cellular functions require the fusion and fission of the membrane. In this regard, defect in TRPML1 channel or PI(3,5)P2 has been found to block lysosome-to-Golgi retrograde trafficking, a process requiring membrane fission [2, 8, 54–58, 128–131]. Also, it has been reported that membrane-fusion processes including exocytosis and fusion of lysosome and autophagosome are dependent on the TRPML1 channel and PI(3,5) P2-metabolizing enzymes [11, 54, 58, 132, 133]. Protein complexes are recruited by the local elevation of PI(3,5)P2, leading to the generation of membrane curvature and consequent membrane fusion and fission [58]. Also, the activity of TRPML1 can be enhanced by the local elevation of PI(3,5)P2, leading to the increase in juxtaorganellar Ca2+ that affects SNARE proteins and lipid bilayer fusion [58, 134] via binding to putative Ca2+ sensor proteins such as ALG-2 [75] and Synaptotagmin/CaM [135]. These findings have shown that the normal function of the TRPML1 channel is essential for membrane fission and fusion.
13.6.3. Autophagy
Lysosome as a primary digestive organelle is responsible for the degradation of membrane proteins, membrane polysaccharides, complex lipids, endocytosed membranes, and autophagocytosed organelles. In recent studies, it has been found that a number of neurodegenerative disorders may be attributed to impaired lysosomal function and consequent autophagic deficiency [136–139]. As a lysosomal storage disorder, MLIV is featured by severe ophthalmological and neurological abnormalities caused by defective lysosomal transport of membrane components. The accumulation of enlarged vacuoles containing phospholipids, sphingolipids, and acid mucopolysaccharides was observed in fibroblasts obtained from MLIV patients [140–143], suggesting that the transport of protein and lipids is dependent on TRPML1 channel. It has been reported that the fusion of lysosome and autophagosome is inhibited in fibroblasts of MLIV patients [144]. TRPML1 channel was found to mediate the fusion of lysosome and autophagosome [20, 144, 145]. Also, Ca2+ released through the TRPML1 channel was found to activate calcineurin (Cn) which is responsible for the dephosphorylation of TFEB. Then, dephosphorylated TFEB translocated to the nucleus to initiate the transcription of lysosomal and autophagic genes [146, 147]. Thus, TRPML1 and TFEB form a positive feedback loop in which activation of TRPML1 or TFEB enhances the action of the other factor [147, 148]. Recently, it has been found that TRPML1 contributes to autophagosome biogenesis, which is attributed to the induction of the Beclin1/VPS34 autophagic complex, the activation of calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ), the boost of AMP-activated protein kinase (AMPK), and the generation of phosphatidylinositol 3-phosphate (PI3P) [149]. These findings suggest that the TRPML1 channel plays an important role in the regulation of autophagy and the control of cellular metabolism.
13.6.4. Lysosomal Exocytosis
Previous studies have shown that the TRPML1 channel mediates the exocytosis of lysosomes [150–152]. Also, the elevation of TRPML1 channel activity due to gene mutations may induce constitutive lysosomal exocytosis, leading to remarkable translocation of TRPML1 channels to the plasma membrane [11]. Furthermore, it has been found that the TRPML1 channel contributes to the construction of tubular extensions on the cell surface as a consequence of lysosomal exocytosis [153]. In mouse macrophages, the transport of major histocompatibility complex II to the plasma membrane and the escape of endocytosed macromolecules from the lysosomes requires the normal function of the TRPML1 channel [130]. Moreover, lysosomal exocytosis is promoted by upregulation of TRPML1 channel expression due to overexpression of TFEB [148, 154], confirming the critical role of the TRPML1 channel in lysosomal exocytosis. A recent study has shown that the TRPML1 channel is required for focal exocytosis of lysosomes to the site of phagosome formation at the plasma membrane, indicating the contribution of the TRPML1 channel to phagocytosis [74]. Loss or inhibition of TRPML1 channel function decreased the phagocytosis of senescent and apoptotic cell corpses [74]. In the Drosophila MLIV model, lack of TRPML1 channel resulted in deficient phagocytic uptake of apoptotic neurons, leading to the precipitous onset of neurodegeneration [126].
13.6.5. Mitochondrial Function
There is increasing evidence suggesting the functional crosstalk between lysosome and mitochondrion [155, 156]. In unbiased protein interaction studies, the physical interaction between the TRPML1 channel and several mitochondrial proteins was observed [157]. Also, aberrations in mitochondrial function, morphology, and Ca2+ buffering capacity were found in cells lacking the TRPML1 channel [158]. Furthermore, genetic deletion of TRPML1 channel induced neuronal cell loss in the brains of Drosophila adults, which was attributed to dysfunction of the mitochondrion, accumulation of mitochondria with dissipated electrochemical membrane potentials, diminished autophagic clearance of damaged mitochondria, and elevation of ROS [126]. In a recent study, loss of mitochondrial membrane potential and the enhancement of ROS production were observed in mammalian cells lacking the TRPML1 channel [119].
13.6.6. Triggering of Large Ca2+ Release from Sarcoplasmic Reticulum
As a ubiquitous Ca2+ messenger, NAADP acts as one of the most potent intracellular Ca2+ mobilizing molecules [35, 159–161]. The Ca2+ mobilizing action of NAADP is even stronger than that induced by commonly known Ca2+ mobilizing second messengers IP3 and cADPR [30, 31]. It has been reported that the ryanodine receptor (RyR) is a possible target for the action of NAADP in mobilizing Ca2+ from intracellular stores [162–167]. However, other studies agreed more with the findings that in sea urchin eggs, there is an acidic compartment related to lysosomes [168]. In recent studies, NAADP has been demonstrated to first activate Ca2+ bursts as a triggering mechanism and then lead to global Ca2+ mobilization through IP3Rs and RyRs in the sarcoplasmic reticulum (SR), a so-called two-pool mechanism. It is assumed that the NAADP-sensitive Ca2+ store or acidic Ca2+ store is responsible for a localized signal, where the latter triggers Ca2+-induced Ca2+ release (CICR) to cause global Ca2+ increases through RyRs or IP3Rs on the SR [41, 169, 170]. Recent studies in our laboratory have shown that NAADP may not directly activate RyRs on the SR in VSMCs [25, 26, 44]. Instead, we have demonstrated that NAADP induces Ca2+ release through the lysosomal TRPML1 channel, which leads to large Ca2+ release through RyRs on the SR [25].
Although the two-pool mechanism is attractive in its interpretation of a two-phase Ca2+ release induced by NAADP, some issues remain to be addressed. For example, a long delay of the second phase Ca2+ release (seconds to minutes) has been confirmed in NAADP-induced CICR [41, 171, 172], which is very different from the classical CICR reported previously [173–177]. Between lysosomal clusters and a subpopulation of SR, the lysosome–SR junction has been found to form a trigger zone, where NAADP-induced lysosomal Ca2+ release through the TRPML1 channel activates a global Ca2+ response via CICR in pulmonary VSMCs and some other cells [41, 44, 169]. In collaboration with Dr. van Breemen, who has extensive experience in characterizing plasma membrane–SR and mitochondria–SR junctions in smooth muscle cells under resting and contracting condition [178, 179], we performed electron microscopy and found that there are lysosome–SR junctions around 30–80 nm in VSMCs, which are relatively large compared to plasma membrane–SR and mitochondria–SR junctions. However, these Lysosome–SR junctions are rather heterogeneous within arterial myocytes (Fig. 13.2a). Based on the dimensional characteristics of the lysosome–SR junction obtained by electron microscopy, we built a three-dimensional reconstruction of a typical lysosome–SR junction, including lysosome (green), SR (yellow), TRPML1 channel (blue), SERCA2 pumps (red), and Ca2+ (white) (Fig. 13.2b). Given the great mobility of lysosomes in cytosol, lysosome–SR junction possibly depends on lysosomal movement toward SR [25]. It has been proposed that lysosomal aggregation around SR contributes to the global Ca2+ release following small Ca2+ bursts from lysosomes. Although lysosomal Ca2+ release through TRPML1 channel may not be enough to induce global Ca2+ release from the SR, it may be enough to drive lysosomal movement toward SR. When these clustered lysosomes work together, global Ca2+ release from the SR is activated [25]. This interaction of lysosome and SR was also proposed later by Zhu et al. in pulmonary VSMCs [172]. Furthermore, we have demonstrated that NAADP stimulates lysosome trafficking through its Ca2+ mobilizing action [27].
Fig. 13.2.
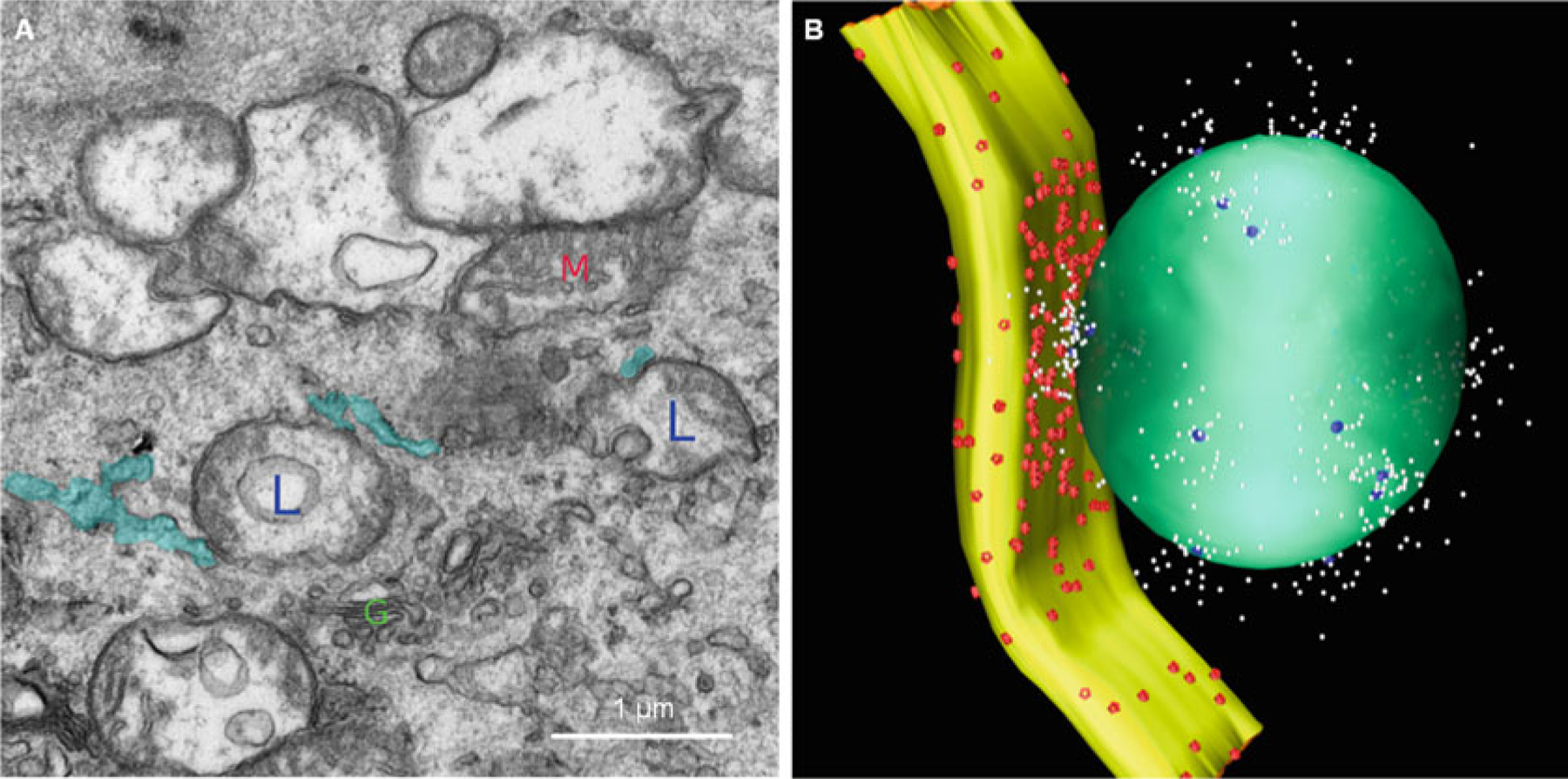
Lysosome–SR junction. The image obtained by electron microscopy shows the structure of the lysosome–SR junction. Lysosomes are labeled with L; SRs are labeled with blue color; the mitochondrion is labeled with M; the Golgi is labeled with G. Based on the dimensional characteristics of the lysosome–SR junction obtained by electron microscopy, we built a three-dimensional reconstruction of a typical lysosome–SR junction, including lysosome (green), SR (yellow), TRPML1 channel (blue), SERCA2 pumps (red), and Ca2+ (white)
Recently, the two-pool mechanism has been found to regulate vasoconstriction. In this regard, Evans and associates first reported that intracellular dialysis of NAADP induced spatially restricted “bursts” of Ca2+ release, leading to global Ca2+ wave and contraction in pulmonary artery smooth muscle cells [180]. Depletion of SR Ca2+ stores with thapsigargin and inhibition of RyRs with ryanodine both blocked the global Ca2+ waves by NAADP [180]. In VSMCs, this two-pool mechanism has been demonstrated to function in response to different agonists such as FasL and ET-1 or by delivery of NAADP into the cells [41, 44]. In coronary arteries, we also found that ET-1-induced NAADP production mobilized intracellular Ca2+, which depends on the normal function of the lysosome [44]. The lysosome function inhibitor bafilomycin A1 and NAADP antagonist PPADS substantially blocked ET-1-induced maximal coronary arterial constriction. These results indicate that NAADP-induced Ca2+ release through lysosomal TRPML1 channel may contribute to ET-1-induced Ca2+ mobilization in CASMCs and consequent vasoconstriction of coronary arteries [44]. More recently, we further demonstrated that FasL increased the production of NAADP but failed to induce vasoconstriction in coronary arterial preparation. However, IP3-producing agonist U46619-induced coronary arterial contraction was significantly enhanced by FasL, suggesting that arterial contraction may be sensitized by elevation of NAADP production [41]. CICR in VSMCs is shown in Fig. 13.3.
Fig. 13.3.
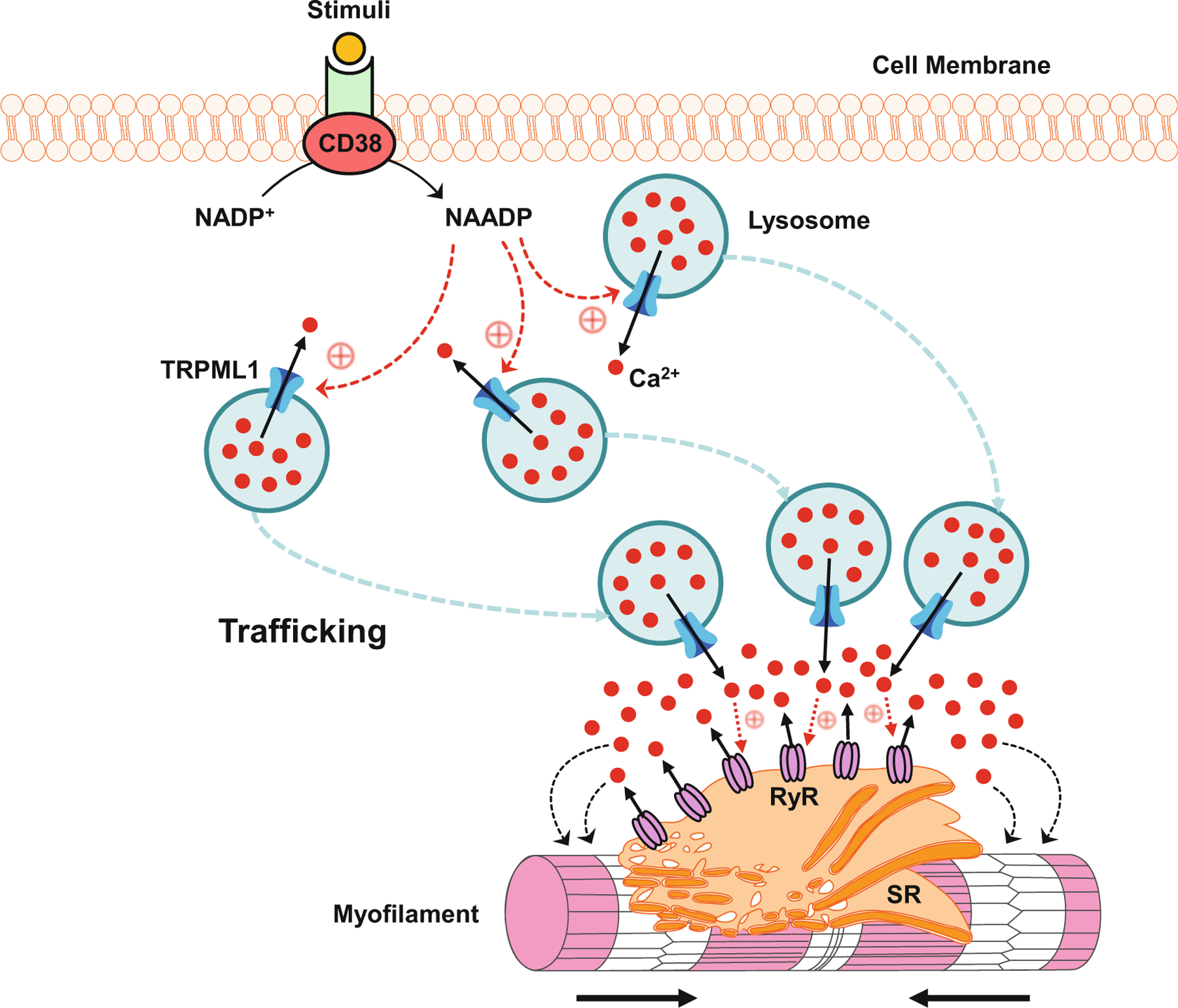
CICR in VSMCs. In response to stimuli such as FasL and ET-1, CD38 converts NADP+ to NAADP which may activate TRPML1 channel. Ca2+ released through the TRPML1 channel drives lysosome trafficking toward the SR, where aggregated lysosomes further release Ca2+ to activate RyRs to produce large Ca2+ release from the SR, leading to the contraction of myofilament
13.6.7. Podocyte Differentiation and Podocytopathy
According to previous studies, the differentiation and maturation of podocytes are highly dependent on normal autophagy [181–183]. Our recent studies have demonstrated that autophagic flux or autophagy maturation is importantly attributed to lysosomal function in mouse podocytes and that lysosome dysfunction may result in autophagic deficiency and consequent podocyte dedifferentiation [183, 184]. Recently, we have demonstrated that CD38 controls lysosome function and thereby regulates autophagic flux in podocytes [183]. It was found that inhibition of CD38 attenuated the fusion of lysosome and autophagosome, leading to autophagosome accumulation in podocytes. Moreover, NAADP, the product of CD38, has been shown to play an essential role in the autophagic flux in podocytes. PPADS, an antagonist of the NAADP receptor, decreased GPN-induced lysosomal Ca2+ release and inhibited autophagic flux in podocytes. These results indicate that NAADP-induced lysosomal Ca2+ release through the TRPML1 channel may contribute to autophagic flux in podocytes. Furthermore, we have demonstrated that dysfunction of CD38 enhances podocyte dedifferentiation, leading to glomerular injury and sclerosis [184]. These findings indicate that CD38–NAADP–TRPML1 signaling pathway may be a therapeutic target for podocyte dedifferentiation and glomerular diseases due to autophagic deficiency.
Previous studies have disclosed that obesity is a risk factor for chronic kidney disease (CKD) and end-stage renal disease (ESRD) [185, 186]. In adolescents with severe obesity, an early glomerular injury may be detected using urinary sphingolipid excretion as a parameter, which is proposed according to a recent clinical study reporting that urinary sphingolipid excretion occurs in adolescents with severe obesity despite the absence of microalbuminuria [187]. The elevated urinary sphingolipids include ceramides, sphingomyelin, and glycosphingolipids. In the development of obesity-induced glomerular diseases, exosomes may play an important role, given the recent discovery that ceramide-enriched exosome stimulates further damage in response to danger signals [188–190]. As one of the extracellular vesicles (EVs), the exosome is released after the fusion of MVB to the plasma membrane [191]. The involvement of exosomes in cell communication and the pathogenesis of different diseases has been extensively studied [192–194]. There is evidence that lysosomal function determines the fate of various intracellular vesicles, including phagosomes, autophagosomes, and MVBs [27, 183, 195, 196]. Recently, we have reported that lysosome trafficking and lysosome–MVB interaction in podocytes are controlled by TRPML1 channel-mediated Ca2+ release [72]. Dysfunction of lysosomal AC may block the TRPML1 channel. The lack of Ca2+ release through the TRPML1 channel may decrease lysosome–MVB interaction, leading to enhancement of exosome secretion from podocytes. Under pathological conditions, enhancement of podocyte-derived exosome release due to blockade of TRPML1 channel may contribute to podocyte injury and glomerular damage. Regulations of autophagic flux and exosome release by TRPML1 channels in podocytes are summarized in Fig. 13.4.
Fig. 13.4.
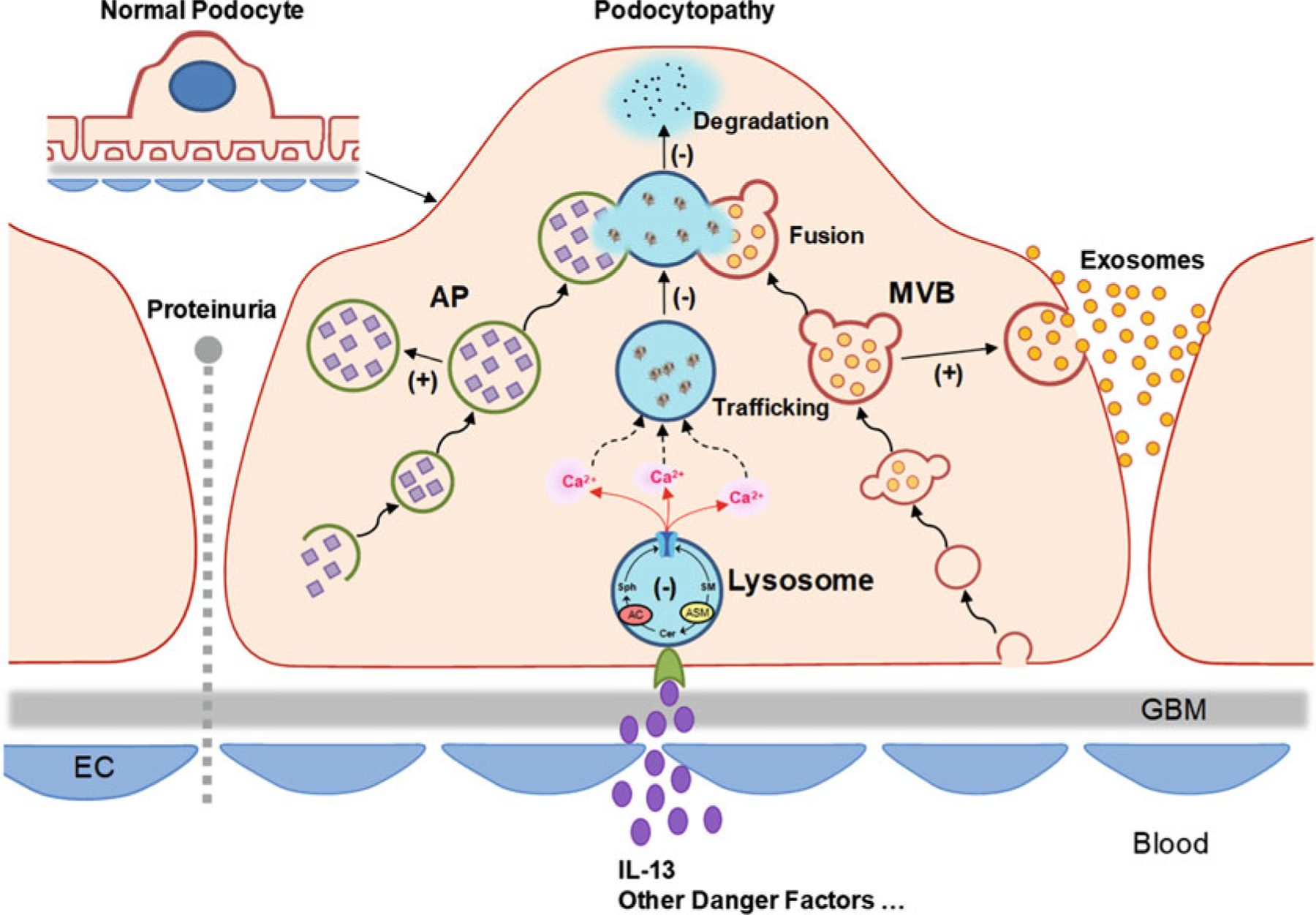
Regulations of autophagic flux and exosome release by TRPML1 channels in podocytes. Autophagosomes (APs) and multivesicular bodies (MVBs) can fuse with and deliver content to lysosomes for degradation, which is regulated by lysosome trafficking. This lysosome-mediated regulatory mechanism constitutively controls the fate of APs and MVBs. Sphingolipids have different effects on TRPML1 channel activity, with inhibition by sphingomyelin (SM), no effect from ceramide (Cer), and enhancement by sphingosine (Sph). Ca2+ released through TRPML1 channel drives lysosome trafficking to and fusion with APs or MVBs. AC deficiency or inhibition may undermine lysosome–AP and lysosome–MVB interactions, leading to accumulation of APs and increased release of exosomes, which in turn triggers or promotes podocyte phenotypic transition and podocytopathy
13.6.8. Exosome Release and Arterial Medial Calcification
The process of apatite calcium salts accumulate and deposit within the vascular wall is known as arterial calcification which has been reported to be associated with atherosclerosis, diabetes mellitus, aging, and CKD [197]. Arterial calcification is anatomically classified into intimal and medial calcification [198]. In arterial intimal calcification, artery occlusion often occurs due to lipid accumulation, inflammation, and fibrosis, which is featured by irregularly scattered deposits in the atherosclerotic plaques [199]. In arterial medial calcification (AMC), the elevation of arterial stiffness is attributed to continuous deposition of hydroxyapatite in the absence of inflammatory cells along the internal elastic lamina [200, 201].
In recent studies, arterial SMC-derived exosomes in the vascular interstitial space have been reported to contribute to the development of arterial medial calcification [202–204]. Given the important role of the lysosomal TRPML1 channel in the regulation of exosome release [72], we hypothesized that dysfunction of the lysosomal TRPML1 channel may block lysosome trafficking and fusion to MVBs in arterial SMCs, leading to enhanced exosome release from arterial SMCs. A recent study in our laboratory has demonstrated that the normal function of lysosomal AC may be essential for the maintenance of contractile phenotype and lysosome-dependent degradation of MVBs in arterial SMCs [205]. The SMC-specific deletion of the Asah1 gene was found to result in enhanced exosome secretion, SMC phenotypic transition, and AMC. The regulatory role of AC is attributed to the action of AC-associated sphingolipids on the lysosomal TRPML1-mediated Ca2+ release. In arterial SMCs lack of Asah1 gene, the dysfunction of the TRPML1 channel led to reduced lysosome–MVB interaction and enhanced exosome release, which contributed to the development of AMC [205]. Furthermore, a recent study has demonstrated that phenotypic transition of arterial medial SMCs can be enhanced by deletion of Mcoln1 gene in a mouse model of AMC [206]. The deletion of the Mcoln1 gene induced an abnormality of lysosome positioning and elevation of exosome release, which contributed to the development of AMC [206]. These findings have further confirmed that the normal function of the lysosomal TRPML1 channel is essential for the maintenance of contractile phenotype and lysosome-dependent degradation of MVBs in arterial SMCs.
13.6.9. Lysosome-Mediated Autophagic Flux and Atherogenesis
Acute experiments in cells and isolated vessels have indicated that NAADP is involved in the development of hypertension and pulmonary hypertension [159]. However, so far there is no direct evidence showing that this CD38-derived second messenger is implicated in any vascular diseases. Given the diversity of cell types in the artery wall and the involvement of autophagy in different vascular cell functions, the role of autophagy in the development of atherosclerosis is complex [207–209]. It has been demonstrated that autophagy can generate either protective or detrimental effect on atherosclerosis, which is determined by the status of autophagy and the stage of atherosclerosis [210, 211]. Damaged components in the arterial wall can be cleaned up by autophagy, leading to the recovery of cells from the damage in response to atherosclerotic stimuli. Moreover, autophagosomes can engulf defective or damaged mitochondria for autophagic degradation, which inhibits proapoptotic protein release and cell apoptosis [212–214]. This autophagic flux may protect arterial cells from atherogenic injury. However, damaged lysosomes may release hydrolases, engage as part of oxidative stress, and enhance cellular damages in response to acute or persistent oxidative stress during atherosclerosis [211, 215–217]. Enhanced or reduced autophagy plays different roles in the development of atherosclerosis depending on the different cells involved. For example, cholesterol transport out of macrophages is increased by autophagy, which may reduce foam cell formation by prevention of lipid droplet formation. Enhanced autophagic death of macrophages also attenuates foam cell formation, which may inhibit atherosclerotic injury. Autophagy contributes to the maintenance of differentiated, quiescent, and contractile phenotype of arterial SMCs, leading to the inhibition of cell proliferation and prevention of fibrosis. Nevertheless, the death of arterial SMCs may be induced by excessive autophagy, which increases the instability of atherosclerotic plaques. In endothelial cells (ECs); however, excessive activation of autophagy may induce damage of the endothelium and initiate atherogenic injury [215, 217–220]. Although these previous studies have demonstrated the complex role of augmented autophagy in atherosclerosis, the role of autophagic deficiency in the pathogenesis of atherosclerosis remains unknown.
In our recent studies, we have demonstrated that autophagic deficiency also importantly contributes to atherogenesis. In vivo, it was found that CD38 gene knockout amplified Western diet-induced autophagosome accumulation in coronary arterial media [221]. In vitro, both deletion of CD38 gene and inhibition of TRPML1 channel expression remarkably attenuated fusion of lysosome and autophagosome, leading to autophagosome accumulation in CAMs. Defects in the CD38–NAADP–TRPML1 signaling pathway promoted CAM dedifferentiation and stimulated the production of the extracellular matrix, which contributed to the development of atherosclerosis [221]. Another study has shown that CD38 is essential for the normal function of nuclear factor E2-related factor 2 (Nrf2) in CAMs [222]. The dedifferentiation of CAMs is attributed to the downregulation of Nrf2 in CD38 gene knockout CAMs [222]. As an event observed in the early stage of atherosclerosis, increased collagen I deposition in the extracellular matrix may be caused by excess formation or decreased degradation or both of collagen I in CAMs [223]. In this regard, we have demonstrated that CD38–NAADP–TRPML1 signaling pathway plays an important role in the regulation of autophagy and thereby controls collagen metabolism [195]. The collagen I deposition and arterial wall thickening in mice fed with Western diet were enhanced by CD38 gene knockout [195]. As a hallmark in the development of atherosclerosis, cholesterol accumulation in macrophages was worsened by blockade of CD38–NAADP–TRPML1 signaling pathway [224]. Compared to wild-type mice, Western diet-induced atherosclerosis and lysosomal cholesterol sequestration in macrophages were more severe in CD38 gene knockout mice [224]. Taken together, these findings have demonstrated the importance of CD38–NAADP–TRPML1 signaling pathway in the maintenance of normal function of the coronary artery. The lysosome trafficking and autophagic flux are dependent on TRPML1 channelmediated Ca2+ release, which is essential for the homeostasis of CAMs.
13.7. Concluding Remarks
There is increasing evidence that the lysosomal TRPML1 channel is implicated in the regulation of cardiovascular and glomerular functions and may be involved in the development of cardiovascular and glomerular diseases. TRPML1 channel-mediated lysosome Ca2+ bursts activate CICR that is an important regulatory mechanism of vasoconstriction. Furthermore, TRPML1 channel regulates various cellular activities, including autophagy, lysosomal exocytosis, apoptosis, lipid transportation, and exosome release. All these studies have provided innovative insights into the physiology and Pathobiology of TRPML1 channels, which may help develop therapeutic strategies preventing the development of vascular diseases such as arterial calcification and atherosclerosis. In addition, dysfunction of TRPML1 channels has also been implicated in the development of a variety of glomerular diseases due to their induction of podocyte dysfunction and injury, resulting in glomerular sclerosis and ultimate ESRD. A deeper mechanistic investigation is of the utmost importance to understand how pathological stimuli regulate lysosomal TRPML1 channel activity, which may further promote the development of more effective therapies for the prevention or treatment of chronic degenerative cardiovascular and glomerular diseases.
References
- 1.Manzoni M, Monti E, Bresciani R, Bozzato A, Barlati S, Bassi MT, Borsani G (2004) Overexpression of wild-type and mutant mucolipin proteins in mammalian cells: effects on the late endocytic compartment organization. FEBS Lett 567(2–3):219–224 [DOI] [PubMed] [Google Scholar]
- 2.Pryor PR, Reimann F, Gribble FM, Luzio JP (2006) Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic 7(10):1388–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venkatachalam K, Hofmann T, Montell C (2006) Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J Biol Chem 281(25):17517–17527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vergarajauregui S, Puertollano R (2006) Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 7(3):337–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bassi MT, Manzoni M, Monti E, Pizzo MT, Ballabio A, Borsani G (2000) Cloning of the gene encoding a novel integral membrane protein, mucolipidin- and identification of the two major founder mutations causing mucolipidosis type IV. Am J Hum Genet 67(5):1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Acierno JS Jr, Bove C, Kaneski CR, Nagle J, Bromley MC, Colman M, Schiffmann R, Slaugenhaupt SA (2000) Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet 9(17):2471–2478 [DOI] [PubMed] [Google Scholar]
- 7.Amir N, Zlotogora J, Bach G (1987) Mucolipidosis type IV: clinical spectrum and natural history. Pediatrics 79(6):953–959 [PubMed] [Google Scholar]
- 8.Chen CS, Bach G, Pagano RE (1998) Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc Natl Acad Sci U S A 95(11):6373–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldin E, Blanchette-Mackie EJ, Dwyer NK, Pentchev PG, Brady RO (1995) Cultured skin fibroblasts derived from patients with mucolipidosis 4 are auto-fluorescent. Pediatr Res 37(6):687–692 [DOI] [PubMed] [Google Scholar]
- 10.Miedel MT, Weixel KM, Bruns JR, Traub LM, Weisz OA (2006) Posttranslational cleavage and adaptor protein complex-dependent trafficking of mucolipin-1. J Biol Chem 281(18):12751–12759 [DOI] [PubMed] [Google Scholar]
- 11.Dong XP, Wang X, Shen D, Chen S, Liu M, Wang Y, Mills E, Cheng X, Delling M, Xu H (2009) Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem 284(46):32040–32052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong CO, Li R, Montell C, Venkatachalam K (2012) Drosophila TRPML is required for TORC1 activation. Curr Biol 22(17):1616–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curcio-Morelli C, Zhang P, Venugopal B, Charles FA, Browning MF, Cantiello HF, Slaugenhaupt SA (2010) Functional multimerization of mucolipin channel proteins. J Cell Physiol 222(2):328–335 [DOI] [PubMed] [Google Scholar]
- 14.Zeevi DA, Frumkin A, Offen-Glasner V, Kogot-Levin A, Bach G (2009) A potentially dynamic lysosomal role for the endogenous TRPML proteins. J Pathol 219(2):153–162 [DOI] [PubMed] [Google Scholar]
- 15.Zeevi DA, Lev S, Frumkin A, Minke B, Bach G (2010) Heteromultimeric TRPML channel assemblies play a crucial role in the regulation of cell viability models and starvation-induced autophagy. J Cell Sci 123(Pt 18):3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venkatachalam K, Montell C (2007) TRP channels. Annu Rev Biochem 76:387–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamaguchi S, Jha A, Li Q, Soyombo AA, Dickinson GD, Churamani D, Brailoiu E, Patel S, Muallem S (2011) Transient receptor potential mucolipin 1 (TRPML1) and two-pore channels are functionally independent organellar ion channels. J Biol Chem 286(26):22934–22942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmiege P, Fine M, Blobel G, Li X (2017) Human TRPML1 channel structures in open and closed conformations. Nature 550(7676):366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong XP, Wang X, Xu H (2010) TRP channels of intracellular membranes. J Neurochem 113 (2):313–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venkatachalam K, Wong CO, Zhu MX (2015) The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58(1):48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H (2008) The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455(7215):992–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng X, Huang Y, Lu Y, Xiong J, Wong CO, Yang P, Xia J, Chen D, Du G, Venkatachalam K, Xia X, Zhu MX (2014) Drosophila TRPML forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane. J Biol Chem 289 (7):4262–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swetha MG, Sriram V, Krishnan KS, Oorschot VM, ten Brink C, Klumperman J, Mayor S (2011) Lysosomal membrane protein composition, acidic pH and sterol content are regulated via a light-dependent pathway in metazoan cells. Traffic 12(8):1037–1055 [DOI] [PubMed] [Google Scholar]
- 24.Yuan X, Bhat OM, Lohner H, Zhang Y, Li PL (2019) Endothelial acid ceramidase in exosome-mediated release of NLRP3 inflammasome products during hyperglycemia: evidence from endothelium-specific deletion of Asah1 gene. Biochim Biophys Acta Mol Cell Biol Lipids 1864(12):158532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang F, Jin S, Yi F, Li PL (2009) TRP-ML1 functions as a lysosomal NAADP-sensitive Ca2+ release channel in coronary arterial myocytes. J Cell Mol Med 13(9B):3174–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang F, Li PL (2007) Reconstitution and characterization of a nicotinic acid adenine dinucleotide phosphate (NAADP)-sensitive Ca2+ release channel from liver lysosomes of rats. J Biol Chem 282 (35):25259–25269 [DOI] [PubMed] [Google Scholar]
- 27.Zhang F, Xu M, Han WQ, Li PL (2011) Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(−/−) cells. Am J Physiol Cell Physiol 301(2):C421–C430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li PL, Zhang Y, Abais JM, Ritter JK, Zhang F (2013) Cyclic ADP-ribose and NAADP in vascular regulation and diseases. Messenger (Los Angel) 2(2):63–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chini EN, Beers KW, Dousa TP (1995) Nicotinate adenine dinucleotide phosphate (NAADP) triggers a specific calcium release system in sea urchin eggs. J Biol Chem 270(7):3216–3223 [DOI] [PubMed] [Google Scholar]
- 30.Clapper DL, Walseth TF, Dargie PJ, Lee HC (1987) Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J Biol Chem 262 (20):9561–9568 [PubMed] [Google Scholar]
- 31.Lee HC, Aarhus R (1995) A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem 270(5):2152–2157 [DOI] [PubMed] [Google Scholar]
- 32.Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC (1995) ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J Biol Chem 270(51):30327–30333 [DOI] [PubMed] [Google Scholar]
- 33.Galione A (1993) Cyclic ADP-ribose: a new way to control calcium. Science 259(5093):325–326 [DOI] [PubMed] [Google Scholar]
- 34.Lee HC (1997) Mechanisms of calcium signaling by cyclic ADP-ribose and NAADP. Physiol Rev 77 (4):1133–1164 [DOI] [PubMed] [Google Scholar]
- 35.Lee HC (2005) Nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated calcium signaling. J Biol Chem 280(40):33693–33696 [DOI] [PubMed] [Google Scholar]
- 36.Ge ZD, Li PL, Chen YF, Gross GJ, Zou AP (2002) Myocardial ischemia and reperfusion reduce the levels of cyclic ADP-ribose in rat myocardium. Basic Res Cardiol 97(4):312–319 [DOI] [PubMed] [Google Scholar]
- 37.Ge ZD, Zhang DX, Chen YF, Yi FX, Zou AP, Campbell WB, Li PL (2003) Cyclic ADP-ribose contributes to contraction and Ca2+ release by M1 muscarinic receptor activation in coronary arterial smooth muscle. J Vasc Res 40(1):28–36 [DOI] [PubMed] [Google Scholar]
- 38.Lee HC, Aarhus R (2000) Functional visualization of the separate but interacting calcium stores sensitive to NAADP and cyclic ADP-ribose. J Cell Sci 113 (Pt 24):4413–4420 [DOI] [PubMed] [Google Scholar]
- 39.Li PL, Tang WX, Valdivia HH, Zou AP, Campbell WB (2001) cADP-ribose activates reconstituted ryanodine receptors from coronary arterial smooth muscle. Am J Physiol Heart Circ Physiol 280(1): H208–H215 [DOI] [PubMed] [Google Scholar]
- 40.Xu M, Zhang Y, Xia M, Li XX, Ritter JK, Zhang F, Li PL (2012) NAD(P)H oxidase-dependent intracellular and extracellular O2*-production in coronary arterial myocytes from CD38 knockout mice. Free Radic Biol Med 52(2):357–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang F, Xia M, Li PL (2010) Lysosome-dependent Ca(2+) release response to Fas activation in coronary arterial myocytes through NAADP: evidence from CD38 gene knockouts. Am J Physiol Cell Physiol 298(5):C1209–C1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia SJ, Jin S, Zhang F, Yi F, Dewey WL, Li PL (2008) Formation and function of ceramide-enriched membrane platforms with CD38 during M1-receptor stimulation in bovine coronary arterial myocytes. Am J Physiol Heart Circ Physiol 295(4):H1743–H1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu M, Xia M, Li XX, Han WQ, Boini KM, Zhang F, Zhang Y, Ritter JK, Li PL (2012) Requirement of translocated lysosomal V1 H(+)-ATPase for activation of membrane acid sphingomyelinase and raft clustering in coronary endothelial cells. Mol Biol Cell 23(8):1546–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang F, Zhang G, Zhang AY, Koeberl MJ, Wallander E, Li PL (2006) Production of NAADP and its role in Ca2+ mobilization associated with lysosomes in coronary arterial myocytes. Am J Physiol Heart Circ Physiol 291(1):H274–H282 [DOI] [PubMed] [Google Scholar]
- 45.Deaglio S, Vaisitti T, Billington R, Bergui L, Omede P, Genazzani AA, Malavasi F (2007) CD38/CD19: a lipid raft-dependent signaling complex in human B cells. Blood 109(12):5390–5398 [DOI] [PubMed] [Google Scholar]
- 46.Munoz P, Navarro MD, Pavon EJ, Salmeron J, Malavasi F, Sancho J, Zubiaur M (2003) CD38 signaling in T cells is initiated within a subset of membrane rafts containing Lck and the CD3-zeta subunit of the T cell antigen receptor. J Biol Chem 278 (50):50791–50802 [DOI] [PubMed] [Google Scholar]
- 47.Zilber MT, Setterblad N, Vasselon T, Doliger C, Charron D, Mooney N, Gelin C (2005) MHC class II/CD38/CD9: a lipid-raft-dependent signaling complex in human monocytes. Blood 106(9):3074–3081 [DOI] [PubMed] [Google Scholar]
- 48.Aarhus R, Dickey DM, Graeff RM, Gee KR, Walseth TF, Lee HC (1996) Activation and inactivation of Ca2+ release by NAADP+. J Biol Chem 271 (15):8513–8516 [DOI] [PubMed] [Google Scholar]
- 49.Bach G (2005) Mucolipin 1: endocytosis and cation channel—a review. Pflugers Arch 451(1):313–317 [DOI] [PubMed] [Google Scholar]
- 50.Genazzani AA, Empson RM, Galione A (1996) Unique inactivation properties of NAADP-sensitive Ca2+ release. J Biol Chem 271(20):11599–11602 [DOI] [PubMed] [Google Scholar]
- 51.Yusufi AN, Cheng J, Thompson MA, Burnett JC, Grande JP (2002) Differential mechanisms of Ca (2+) release from vascular smooth muscle cell microsomes. Exp Biol Med (Maywood) 227 (1):36–44 [DOI] [PubMed] [Google Scholar]
- 52.Bonangelino CJ, Nau JJ, Duex JE, Brinkman M, Wurmser AE, Gary JD, Emr SD, Weisman LS (2002) Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol 156 (6):1015–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS, Meisler MH (2007) Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448 (7149):68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dove SK, Dong K, Kobayashi T, Williams FK, Michell RH (2009) Phosphatidylinositol 3,5-bisphosphate and Fab1p/PIKfyve underPPIn endo-lysosome function. Biochem J 419(1):1–13 [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Zolov SN, Chow CY, Slutsky SG, Richardson SC, Piper RC, Yang B, Nau JJ, Westrick RJ, Morrison SJ, Meisler MH, Weisman LS (2007) Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci U S A 104(44):17518–17523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Botelho RJ, Efe JA, Teis D, Emr SD (2008) Assembly of a Fab1 phosphoinositide kinase signaling complex requires the Fig4 phosphoinositide phosphatase. Mol Biol Cell 19(10):4273–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duex JE, Nau JJ, Kauffman EJ, Weisman LS (2006) Phosphoinositide 5-phosphatase Fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell 5(4):723–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poccia D, Larijani B (2009) Phosphatidylinositol metabolism and membrane fusion. Biochem J 418 (2):233–246 [DOI] [PubMed] [Google Scholar]
- 59.Jin N, Chow CY, Liu L, Zolov SN, Bronson R, Davisson M, Petersen JL, Zhang Y, Park S, Duex JE, Goldowitz D, Meisler MH, Weisman LS (2008) VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J 27(24):3221–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen J, Yu WM, Brotto M, Scherman JA, Guo C, Stoddard C, Nosek TM, Valdivia HH, Qu CK (2009) Deficiency of MIP/MTMR14 phosphatase induces a muscle disorder by disrupting Ca(2+) homeostasis. Nat Cell Biol 11(6):769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, Everett L, Lenk GM, McKenna-Yasek DM, Weisman LS, Figlewicz D, Brown RH, Meisler MH (2009) Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet 84(1):85–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H (2010) PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca (2+) release channels in the endolysosome. Nat Commun 1:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng X, Xiong J, Lu Y, Xia X, Zhu MX (2014) Differential mechanisms of action of the mucolipin synthetic agonist, ML-SA1, on insect TRPML and mammalian TRPML1. Cell Calcium 56(6):446–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang X, Li X, Xu H (2012) Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc Natl Acad Sci U S A 109 (28):11384–11389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Q, She J, Zeng W, Guo J, Xu H, Bai XC, Jiang Y (2017) Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 550 (7676):415–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, Fini M, McCubrey JA (2012) Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget 3(4):371–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, Xu H (2012) Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3:731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weiss N (2012) Cross-talk between TRPML1 channel, lipids and lysosomal storage diseases. Commun Integr Biol 5(2):111–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu L, Zhang X, Yang Y, Li D, Tang K, Zhao Z, He W, Wang C, Sahoo N, Converso-Baran K, Davis CS, Brooks SV, Bigot A, Calvo R, Martinez NJ, Southall N, Hu X, Marugan J, Ferrer M, Xu H (2020) Small-molecule activation of lysosomal TRP channels ameliorates Duchenne muscular dystrophy in mouse models. Sci Adv 6(6):eaaz2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen CC, Keller M, Hess M, Schiffmann R, Urban N, Wolfgardt A, Schaefer M, Bracher F, Biel M, Wahl-Schott C, Grimm C (2014) A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat Commun 5:4681. [DOI] [PubMed] [Google Scholar]
- 71.Zhang X, Chen W, Gao Q, Yang J, Yan X, Zhao H, Su L, Yang M, Gao C, Yao Y, Inoki K, Li D, Shao R, Wang S, Sahoo N, Kudo F, Eguchi T, Ruan B, Xu H (2019) Rapamycin directly activates lysosomal mucolipin TRP channels independent of mTOR. PLoS Biol 17(5):e3000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li G, Huang D, Hong J, Bhat OM, Yuan X, Li PL (2019) Control of lysosomal TRPML1 channel activity and exosome release by acid ceramidase in mouse podocytes. Am J Physiol Cell Physiol 317(3):C481–C491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu H, Delling M, Li L, Dong X, Clapham DE (2007) Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc Natl Acad Sci U S A 104 (46):18321–18326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, Gregg E, Azar M, Zhuo Y, Garrity AG, Gao Q, Slaugenhaupt S, Pickel J, Zolov SN, Weisman LS, Lenk GM, Titus S, Bryant-Genevier M, Southall N, Juan M, Ferrer M, Xu H (2013) A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell 26 (5):511–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vergarajauregui S, Martina JA, Puertollano R (2009) Identification of the penta-EF-hand protein ALG-2 as a Ca2+-dependent interactor of mucolipin-1. J Biol Chem 284(52):36357–36366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lo KW, Zhang Q, Li M, Zhang M (1999) Apoptosis-linked gene product ALG-2 is a new member of the calpain small subunit subfamily of Ca2+-binding proteins. Biochemistry 38(23):7498–7508 [DOI] [PubMed] [Google Scholar]
- 77.Maki M, Kitaura Y, Satoh H, Ohkouchi S, Shibata H (2002) Structures, functions and molecular evolution of the penta-EF-hand Ca2+-binding proteins. Biochim Biophys Acta 1600(1–2):51–60 [DOI] [PubMed] [Google Scholar]
- 78.Tarabykina S, Mollerup J, Winding P, Berchtold MW (2004) ALG-2, a multifunctional calcium binding protein? Front Biosci 9:1817–1832 [DOI] [PubMed] [Google Scholar]
- 79.Li X, Rydzewski N, Hider A, Zhang X, Yang J, Wang W, Gao Q, Cheng X, Xu H (2016) A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol 18 (4):404–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bejarano E, Cuervo AM (2010) Chaperone-mediated autophagy. Proc Am Thorac Soc 7(1):29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chiang HL, Terlecky SR, Plant CP, Dice JF (1989) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246(4928):382–385 [DOI] [PubMed] [Google Scholar]
- 82.Venugopal B, Mesires NT, Kennedy JC, Curcio-Morelli C, Laplante JM, Dice JF, Slaugenhaupt SA (2009) Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol 219 (2):344–353 [DOI] [PubMed] [Google Scholar]
- 83.Vergarajauregui S, Martina JA, Puertollano R (2011) LAPTMs regulate lysosomal function and interact with mucolipin 1: new clues for understanding mucolipidosis type IV. J Cell Sci 124(Pt 3):459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bozzato A, Barlati S, Borsani G (2008) Gene expression profiling of mucolipidosis type IV fibroblasts reveals deregulation of genes with relevant functions in lysosome physiology. Biochim Biophys Acta 1782 (4):250–258 [DOI] [PubMed] [Google Scholar]
- 85.Bewley MA, Marriott HM, Tulone C, Francis SE, Mitchell TJ, Read RC, Chain B, Kroemer G, Whyte MK, Dockrell DH (2011) A cardinal role for cathepsin d in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog 7(1):e1001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsukuba T, Yamamoto S, Yanagawa M, Okamoto K, Okamoto Y, Nakayama KI, Kadowaki T, Yamamoto K (2006) Cathepsin E-deficient mice show increased susceptibility to bacterial infection associated with the decreased expression of multiple cell surface Toll-like receptors. J Biochem 140(1):57–66 [DOI] [PubMed] [Google Scholar]
- 87.Xu X, Greenland J, Baluk P, Adams A, Bose O, McDonald DM, Caughey GH (2013) Cathepsin L protects mice from mycoplasmal infection and is essential for airway lymphangiogenesis. Am J Respir Cell Mol Biol 49(3):437–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, Muallem S, Soyombo A (2005) TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem 280(52):43218–43223 [DOI] [PubMed] [Google Scholar]
- 89.Qi X, Man SM, Malireddi RK, Karki R, Lupfer C, Gurung P, Neale G, Guy CS, Lamkanfi M, Kanneganti TD (2016) Cathepsin B modulates lysosomal biogenesis and host defense against Francisella novicida infection. J Exp Med 213(10):2081–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Colletti GA, Miedel MT, Quinn J, Andharia N, Weisz OA, Kiselyov K (2012) Loss of lysosomal ion channel transient receptor potential channel mucolipin-1 (TRPML1) leads to cathepsin B-dependent apoptosis. J Biol Chem 287 (11):8082–8091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant-Genevier M, Southall NT, Marugan J, Ferrer M, Xu H (2015) Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci U S A 112(11):E1373–E1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31(5):1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325(5939):473–477 [DOI] [PubMed] [Google Scholar]
- 94.Onyenwoke RU, Sexton JZ, Yan F, Diaz MC, Forsberg LJ, Major MB, Brenman JE (2015) The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem J 470(3):331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yao X, Kwan HY, Huang Y (2005) Regulation of TRP channels by phosphorylation. Neurosignals 14 (6):273–280 [DOI] [PubMed] [Google Scholar]
- 96.Vergarajauregui S, Oberdick R, Kiselyov K, Puertollano R (2008) Mucolipin 1 channel activity is regulated by protein kinase A-mediated phosphorylation. Biochem J 410(2):417–425 [DOI] [PubMed] [Google Scholar]
- 97.Gault CR, Obeid LM, Hannun YA (2010) An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 688:1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gulbins E, Li PL (2006) Physiological and pathophysiological aspects of ceramide. Am J Physiol Regul Integr Comp Physiol 290(1):R11–R26 [DOI] [PubMed] [Google Scholar]
- 99.Bhat OM, Yuan X, Li G, Lee R, Li PL (2018) Sphingolipids and redox signaling in renal regulation and chronic kidney diseases. Antioxid Redox Signal 28(10):1008–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Futerman AH, Hannun YA (2004) The complex life of simple sphingolipids. EMBO Rep 5(8):777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Piccoli E, Nadai M, Caretta CM, Bergonzini V, Del Vecchio C, Ha HR, Bigler L, Dal Zoppo D, Faggin E, Pettenazzo A, Orlando R, Salata C, Calistri A, Palu G, Baritussio A (2011) Amiodarone impairs trafficking through late endosomes inducing a Niemann-Pick C-like phenotype. Biochem Pharmacol 82(9):1234–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Siti HN, Kamisah Y, Kamsiah J (2015) The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc Pharmacol 71:40–56 [DOI] [PubMed] [Google Scholar]
- 103.Huang J, Lam GY, Brumell JH (2011) Autophagy signaling through reactive oxygen species. Antioxid Redox Signal 14(11):2215–2231 [DOI] [PubMed] [Google Scholar]
- 104.Droge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82(1):47–95 [DOI] [PubMed] [Google Scholar]
- 105.Skowronska M, Albrecht J (2013) Oxidative and nitrosative stress in ammonia neurotoxicity. Neurochem Int 62(5):731–737 [DOI] [PubMed] [Google Scholar]
- 106.Scherz-Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36(1):30–38 [DOI] [PubMed] [Google Scholar]
- 107.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26(7):1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Filomeni G, De Zio D, Cecconi F (2015) Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22(3):377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mi Y, Xiao C, Du Q, Wu W, Qi G, Liu X (2016) Momordin Ic couples apoptosis with autophagy in human hepatoblastoma cancer cells by reactive oxygen species (ROS)-mediated PI3K/Akt and MAPK signaling pathways. Free Radic Biol Med 90:230–242 [DOI] [PubMed] [Google Scholar]
- 110.Scherz-Shouval R, Shvets E, Elazar Z (2007) Oxidation as a post-translational modification that regulates autophagy. Autophagy 3(4):371–373 [DOI] [PubMed] [Google Scholar]
- 111.Kim RJ, Hah YS, Sung CM, Kang JR, Park HB (2014) Do antioxidants inhibit oxidative-stress-induced autophagy of tenofibroblasts? J Orthop Res 32(7):937–943 [DOI] [PubMed] [Google Scholar]
- 112.Liu GY, Jiang XX, Zhu X, He WY, Kuang YL, Ren K, Lin Y, Gou X (2015) ROS activates JNK-mediated autophagy to counteract apoptosis in mouse mesenchymal stem cells in vitro. Acta Pharmacol Sin 36(12):1473–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, Delling M, Marugan J, Ferrer M, Xu H (2016) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7:12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morelli MB, Amantini C, Tomassoni D, Nabissi M, Arcella A, Santoni G (2019) Transient receptor potential mucolipin-1 channels in glioblastoma: role in patient’s survival. Cancers (Basel) 11(4):525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Abais JM, Xia M, Li G, Gehr TW, Boini KM, Li PL (2014) Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free Radic Biol Med 67:211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chowdhury S, Ghosh S, Das AK, Sil PC (2019) Ferulic acid protects hyperglycemia-induced kidney damage by regulating oxidative insult, inflammation and autophagy. Front Pharmacol 10:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cui J, Tang L, Hong Q, Lin S, Sun X, Cai G, Bai XY, Chen X (2019) N-Acetylcysteine ameliorates gentamicin-induced nephrotoxicity by enhancing autophagy and reducing oxidative damage in miniature pigs. Shock 52(6):622–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang XY, Yang H, Wang MG, Yang DB, Wang ZY, Wang L (2017) Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis 8(10):e3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Coblentz J, St Croix C, Kiselyov K (2014) Loss of TRPML1 promotes production of reactive oxygen species: is oxidative damage a factor in mucolipidosis type IV? Biochem J 457(2):361–368 [DOI] [PubMed] [Google Scholar]
- 120.Rybicka JM, Balce DR, Chaudhuri S, Allan ER, Yates RM (2012) Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J 31(4):932–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rybicka JM, Balce DR, Khan MF, Krohn RM, Yates RM (2010) NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci U S A 107 (23):10496–10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rahman A, Thayyullathil F, Pallichankandy S, Galadari S (2016) Hydrogen peroxide/ceramide/Akt signaling axis play a critical role in the antileukemic potential of sanguinarine. Free Radic Biol Med 96:273–289 [DOI] [PubMed] [Google Scholar]
- 123.Flowers M, Fabrias G, Delgado A, Casas J, Abad JL, Cabot MC (2012) C6-ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res Treat 133(2):447–458 [DOI] [PubMed] [Google Scholar]
- 124.Miedel MT, Rbaibi Y, Guerriero CJ, Colletti G, Weixel KM, Weisz OA, Kiselyov K (2008) Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J Exp Med 205(6):1477–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K (2006) TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem 281 (11):7294–7301 [DOI] [PubMed] [Google Scholar]
- 126.Venkatachalam K, Long AA, Elsaesser R, Nikolaeva D, Broadie K, Montell C (2008) Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell 135(5):838–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kogot-Levin A, Zeigler M, Ornoy A, Bach G (2009) Mucolipidosis type IV: the effect of increased lysosomal pH on the abnormal lysosomal storage. Pediatr Res 65(6):686–690 [DOI] [PubMed] [Google Scholar]
- 128.Cheng X, Shen D, Samie M, Xu H (2010) Mucolipins: intracellular TRPML1–3 channels. FEBS Lett 584(10):2013–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Duex JE, Tang F, Weisman LS (2006) The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol 172(5):693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thompson EG, Schaheen L, Dang H, Fares H (2007) Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol 8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Treusch S, Knuth S, Slaugenhaupt SA, Goldin E, Grant BD, Fares H (2004) Caenorhabditis elegans functional orthologue of human protein h-mucolipin-1 is required for lysosome biogenesis. Proc Natl Acad Sci U S A 101(13):4483–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ferguson CJ, Lenk GM, Meisler MH (2009) Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet 18 (24):4868–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Puertollano R, Kiselyov K (2009) TRPMLs: in sickness and in health. Am J Physiol Renal Physiol 296 (6):F1245–F1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roth MG (2004) Phosphoinositides in constitutive membrane traffic. Physiol Rev 84(3):699–730 [DOI] [PubMed] [Google Scholar]
- 135.Luzio JP, Pryor PR, Bright NA (2007) Lysosomes: fusion and function. Nat Rev Mol Cell Biol 8 (8):622–632 [DOI] [PubMed] [Google Scholar]
- 136.Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179(3):485–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, Morrison KE, Pall HS, Hardiman O, Collinge J, Shaw PJ, Fisher EM, MRC Proteomics in ALS Study; FReJA Consortium (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67(6):1074–1077 [DOI] [PubMed] [Google Scholar]
- 138.Reid E, Connell J, Edwards TL, Duley S, Brown SE, Sanderson CM (2005) The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Hum Mol Genet 14(1):19–38 [DOI] [PubMed] [Google Scholar]
- 139.Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37(8):806–808 [DOI] [PubMed] [Google Scholar]
- 140.Berman ER, Livni N, Shapira E, Merin S, Levij IS (1974) Congenital corneal clouding with abnormal systemic storage bodies: a new variant of mucolipidosis. J Pediatr 84(4):519–526 [DOI] [PubMed] [Google Scholar]
- 141.Goldin E, Cooney A, Kaneski CR, Brady RO, Schiffmann R (1999) Mucolipidosis IV consists of one complementation group. Proc Natl Acad Sci U S A 96(15):8562–8566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Riedel KG, Zwaan J, Kenyon KR, Kolodny EH, Hanninen L, Albert DM (1985) Ocular abnormalities in mucolipidosis IV. Am J Ophthalmol 99 (2):125–136 [DOI] [PubMed] [Google Scholar]
- 143.Slaugenhaupt SA, Acierno JS Jr, Helbling LA, Bove C, Goldin E, Bach G, Schiffmann R, Gusella JF (1999) Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. Am J Hum Genet 65(3):773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R (2008) Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet 17 (17):2723–2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ahuja M, Park S, Shin DM, Muallem S (2016) TRPML1 as lysosomal fusion guard. Channels (Austin) 10(4):261–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Medina DL, Ballabio A (2015) Lysosomal calcium regulates autophagy. Autophagy 11(6):970–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17(3):288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332 (6036):1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Scotto Rosato A, Montefusco S, Soldati C, Di Paola S, Capuozzo A, Monfregola J, Polishchuk E, Amabile A, Grimm C, Lombardo A, De Matteis MA, Ballabio A, Medina DL (2019) TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat Commun 10(1):5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.LaPlante JM, Falardeau J, Sun M, Kanazirska M, Brown EM, Slaugenhaupt SA, Vassilev PM (2002) Identification and characterization of the single channel function of human mucolipin-1 implicated in mucolipidosis type IV, a disorder affecting the lysosomal pathway. FEBS Lett 532(1–2):183–187 [DOI] [PubMed] [Google Scholar]
- 151.LaPlante JM, Sun M, Falardeau J, Dai D, Brown EM, Slaugenhaupt SA, Vassilev PM (2006) Lysosomal exocytosis is impaired in mucolipidosis type. IV. Mol Genet Metab 89(4):339–348 [DOI] [PubMed] [Google Scholar]
- 152.LaPlante JM, Ye CP, Quinn SJ, Goldin E, Brown EM, Slaugenhaupt SA, Vassilev PM (2004) Functional links between mucolipin-1 and Ca2+-dependent membrane trafficking in mucolipidosis IV. Biochem Biophys Res Commun 322 (4):1384–1391 [DOI] [PubMed] [Google Scholar]
- 153.LaPlante JM, Falardeau JL, Brown EM, Slaugenhaupt SA, Vassilev PM (2011) The cation channel mucolipin-1 is a bifunctional protein that facilitates membrane remodeling via its serine lipase domain. Exp Cell Res 317(6):691–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, Palmieri M, Polishchuk R, Puertollano R, Ballabio A (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 21(3):421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Terman A, Gustafsson B, Brunk UT (2006) The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact 163 (1–2):29–37 [DOI] [PubMed] [Google Scholar]
- 156.Terman A, Gustafsson B, Brunk UT (2006) Mitochondrial damage and intralysosomal degradation in cellular aging. Mol Asp Med 27(5–6):471–482 [DOI] [PubMed] [Google Scholar]
- 157.Spooner E, McLaughlin BM, Lepow T, Durns TA, Randall J, Upchurch C, Miller K, Campbell EM, Fares H (2013) Systematic screens for proteins that interact with the mucolipidosis type IV protein TRPML1. PLoS One 8(2):e56780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jennings JJ Jr, Zhu JH, Rbaibi Y, Luo X, Chu CT, Kiselyov K (2006) Mitochondrial aberrations in mucolipidosis Type IV. J Biol Chem 281 (51):39041–39050 [DOI] [PubMed] [Google Scholar]
- 159.Evans AM, Wyatt CN, Kinnear NP, Clark JH, Blanco EA (2005) Pyridine nucleotides and calcium signalling in arterial smooth muscle: from cell physiology to pharmacology. Pharmacol Ther 107(3):286–313 [DOI] [PubMed] [Google Scholar]
- 160.Galione A (2006) NAADP, a new intracellular messenger that mobilizes Ca2+ from acidic stores. Biochem Soc Trans 34(Pt 5):922–926 [DOI] [PubMed] [Google Scholar]
- 161.Yamasaki M, Churchill GC, Galione A (2005) Calcium signalling by nicotinic acid adenine dinucleotide phosphate (NAADP). FEBS J 272 (18):4598–4606 [DOI] [PubMed] [Google Scholar]
- 162.Dammermann W, Guse AH (2005) Functional ryanodine receptor expression is required for NAADP-mediated local Ca2+ signaling in T-lymphocytes. J Biol Chem 280(22):21394–21399 [DOI] [PubMed] [Google Scholar]
- 163.Gerasimenko J, Maruyama Y, Tepikin A, Petersen OH, Gerasimenko O (2003) Calcium signalling in and around the nuclear envelope. Biochem Soc Trans 31(Pt 1):76–78 [DOI] [PubMed] [Google Scholar]
- 164.Gerasimenko JV, Maruyama Y, Yano K, Dolman NJ, Tepikin AV, Petersen OH, Gerasimenko OV (2003) NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J Cell Biol 163(2):271–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hohenegger M, Suko J, Gscheidlinger R, Drobny H, Zidar A (2002) Nicotinic acid-adenine dinucleotide phosphate activates the skeletal muscle ryanodine receptor. Biochem J 367(Pt 2):423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Langhorst MF, Schwarzmann N, Guse AH (2004) Ca2+ release via ryanodine receptors and Ca2+ entry: major mechanisms in NAADP-mediated Ca2+ signaling in T-lymphocytes. Cell Signal 16(11):1283–1289 [DOI] [PubMed] [Google Scholar]
- 167.Mojzisova A, Krizanova O, Zacikova L, Kominkova V, Ondrias K (2001) Effect of nicotinic acid adenine dinucleotide phosphate on ryanodine calcium release channel in heart. Pflugers Arch 441 (5):674–677 [DOI] [PubMed] [Google Scholar]
- 168.Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A (2002) NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 111(5):703–708 [DOI] [PubMed] [Google Scholar]
- 169.Kinnear NP, Boittin FX, Thomas JM, Galione A, Evans AM (2004) Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J Biol Chem 279(52):54319–54326 [DOI] [PubMed] [Google Scholar]
- 170.Kinnear NP, Wyatt CN, Clark JH, Calcraft PJ, Fleischer S, Jeyakumar LH, Nixon GF, Evans AM (2008) Lysosomes co-localize with ryanodine receptor subtype 3 to form a trigger zone for calcium signalling by NAADP in rat pulmonary arterial smooth muscle. Cell Calcium 44(2):190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Evans AM, Cannell MB (1997) The role of L-type Ca2+ current and Na+ current-stimulated Na/Ca exchange in triggering SR calcium release in guinea-pig cardiac ventricular myocytes. Cardiovasc Res 35(2):294–302 [DOI] [PubMed] [Google Scholar]
- 172.Zhu MX, Ma J, Parrington J, Calcraft PJ, Galione A, Evans AM (2010) Calcium signaling via two-pore channels: local or global, that is the question. Am J Physiol Cell Physiol 298(3):C430–C441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fleischer S, Inui M (1989) Biochemistry and biophysics of excitation-contraction coupling. Annu Rev Biophys Biophys Chem 18:333–364 [DOI] [PubMed] [Google Scholar]
- 174.Franco L, Zocchi E, Calder L, Guida L, Benatti U, De Flora A (1994) Self-aggregation of the transmembrane glycoprotein CD38 purified from human erythrocytes. Biochem Biophys Res Commun 202 (3):1710–1715 [DOI] [PubMed] [Google Scholar]
- 175.Galione A, Lee HC, Busa WB (1991) Ca(2+)-induced Ca2+ release in sea urchin egg homogenates: modulation by cyclic ADP-ribose. Science 253 (5024):1143–1146 [DOI] [PubMed] [Google Scholar]
- 176.Galione A, White A, Willmott N, Turner M, Potter BV, Watson SP (1993) cGMP mobilizes intracellular Ca2+ in sea urchin eggs by stimulating cyclic ADP-ribose synthesis. Nature 365(6445):456–459 [DOI] [PubMed] [Google Scholar]
- 177.Hirst DG, Kennovin GD, Flitney FW (1994) The radiosensitizer nicotinamide inhibits arterial vasoconstriction. Br J Radiol 67(800):795–799 [DOI] [PubMed] [Google Scholar]
- 178.Dai J, Kuo KH, Leo JM, van Breemen C, Lee CH (2005) Rearrangement of the close contact between the mitochondria and the sarcoplasmic reticulum in airway smooth muscle. Cell Calcium 37(4):333–340 [DOI] [PubMed] [Google Scholar]
- 179.Poburko D, Kuo KH, Dai J, Lee CH, van Breemen C (2004) Organellar junctions promote targeted Ca2+ signaling in smooth muscle: why two membranes are better than one. Trends Pharmacol Sci 25(1):8–15 [DOI] [PubMed] [Google Scholar]
- 180.Boittin FX, Galione A, Evans AM (2002) Nicotinic acid adenine dinucleotide phosphate mediates Ca2+ signals and contraction in arterial smooth muscle via a two-pool mechanism. Circ Res 91(12):1168–1175 [DOI] [PubMed] [Google Scholar]
- 181.Asanuma K, Mundel P (2003) The role of podocytes in glomerular pathobiology. Clin Exp Nephrol 7 (4):255–259 [DOI] [PubMed] [Google Scholar]
- 182.Li G, Li CX, Xia M, Ritter JK, Gehr TW, Boini K, Li PL (2015) Enhanced epithelial-to-mesenchymal transition associated with lysosome dysfunction in podocytes: role of p62/Sequestosome 1 as a signaling hub. Cell Physiol Biochem 35(5):1773–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Xiong J, Xia M, Xu M, Zhang Y, Abais JM, Li G, Riebling CR, Ritter JK, Boini KM, Li PL (2013) Autophagy maturation associated with CD38-mediated regulation of lysosome function in mouse glomerular podocytes. J Cell Mol Med 17 (12):1598–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Boini KM, Xia M, Xiong J, Li C, Payne LP, Li PL (2012) Implication of CD38 gene in podocyte epithelial-to-mesenchymal transition and glomerular sclerosis. J Cell Mol Med 16(8):1674–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Boini KM, Zhang C, Xia M, Han WQ, Brimson C, Poklis JL, Li PL (2010) Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim Biophys Acta 1801 (12):1294–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hall JE, Henegar JR, Dwyer TM, Liu J, Da Silva AA, Kuo JJ, Tallam L (2004) Is obesity a major cause of chronic kidney disease? Adv Ren Replace Ther 11 (1):41–54 [DOI] [PubMed] [Google Scholar]
- 187.Davis S, Nehus E, Inge T, Zhang W, Setchell K, Mitsnefes M (2018) Effect of bariatric surgery on urinary sphingolipids in adolescents with severe obesity. Surg Obes Relat Dis 14(4):446–451 [DOI] [PubMed] [Google Scholar]
- 188.Kakazu E, Mauer AS, Yin M, Malhi H (2016) Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J Lipid Res 57 (2):233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Podbielska M, Szulc ZM, Kurowska E, Hogan EL, Bielawski J, Bielawska A, Bhat NR (2016) Cytokine-induced release of ceramide-enriched exosomes as a mediator of cell death signaling in an oligodendroglioma cell line. J Lipid Res 57(11):2028–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang G, Dinkins M, He Q, Zhu G, Poirier C, Campbell A, Mayer-Proschel M, Bieberich E (2012) Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): potential mechanism of apoptosis induction in Alzheimer disease (AD). J Biol Chem 287(25):21384–21395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hessvik NP, Llorente A (2018) Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 75(2):193–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bruno S, Porta S, Bussolati B (2016) Extracellular vesicles in renal tissue damage and regeneration. Eur J Pharmacol 790:83–91 [DOI] [PubMed] [Google Scholar]
- 193.Erdbrugger U, Le TH (2016) Extracellular vesicles in renal diseases: more than novel biomarkers? J Am Soc Nephrol 27(1):12–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pomatto MAC, Gai C, Bussolati B, Camussi G (2017) Extracellular vesicles in renal pathophysiology. Front Mol Biosci 4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bao JX, Zhang QF, Wang M, Xia M, Boini KM, Gulbins E, Zhang Y, Li PL (2017) Implication of CD38 gene in autophagic degradation of collagen I in mouse coronary arterial myocytes. Front Biosci (Landmark Ed) 22:558–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Xu M, Li X, Walsh SW, Zhang Y, Abais JM, Boini KM, Li PL (2013) Intracellular two-phase Ca2+ release and apoptosis controlled by TRP-ML1 channel activity in coronary arterial myocytes. Am J Physiol Cell Physiol 304(5):C458–C466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109(6):697–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Demer LL, Tintut Y (2008) Vascular calcification: pathobiology of a multifaceted disease. Circulation 117(22):2938–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ehara S, Kobayashi Y, Yoshiyama M, Shimada K, Shimada Y, Fukuda D, Nakamura Y, Yamashita H, Yamagishi H, Takeuchi K, Naruko T, Haze K, Becker AE, Yoshikawa J, Ueda M (2004) Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation 110(22):3424–3429 [DOI] [PubMed] [Google Scholar]
- 200.Mackey RH, Venkitachalam L, Sutton-Tyrrell K (2007) Calcifications, arterial stiffness and atherosclerosis. Adv Cardiol 44:234–244 [DOI] [PubMed] [Google Scholar]
- 201.Shroff RC, Shanahan CM (2007) The vascular biology of calcification. Semin Dial 20(2):103–109 [DOI] [PubMed] [Google Scholar]
- 202.Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez-Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ, Shanahan CM (2015) Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 116(8):1312–1323 [DOI] [PubMed] [Google Scholar]
- 203.Kapustin AN, Schoppet M, Schurgers LJ, Reynolds JL, McNair R, Heiss A, Jahnen-Dechent W, Hackeng TM, Schlieper G, Harrison P, Shanahan CM (2017) Prothrombin loading of vascular smooth muscle cell-derived exosomes regulates coagulation and calcification. Arterioscler Thromb Vasc Biol 37(3):e22–e32 [DOI] [PubMed] [Google Scholar]
- 204.Kapustin AN, Shanahan CM (2016) Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J Physiol 594(11):2905–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bhat OM, Li G, Yuan X, Huang D, Gulbins E, Kukreja RC, Li PL (2020) Arterial medial calcification through enhanced small extracellular vesicle release in smooth muscle-specific Asah1 gene knockout mice. Sci Rep 10(1):1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bhat OM, Yuan X, Camus S, Salloum FN, Li PL (2020) Abnormal lysosomal positioning and small extracellular vesicle secretion in arterial stiffening and calcification of mice lacking mucolipin 1 gene. Int J Mol Sci 21(5):1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Fasano T, Pisciotta L, Bocchi L, Guardamagna O, Assandro P, Rabacchi C, Zanoni P, Filocamo M, Bertolini S, Calandra S (2012) Lysosomal lipase deficiency: molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Mol Genet Metab 105(3):450–456 [DOI] [PubMed] [Google Scholar]
- 208.Ryter SW, Lee SJ, Smith A, Choi AM (2010) Autophagy in vascular disease. Proc Am Thorac Soc 7(1):40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Seedorf U, Wiebusch H, Muntoni S, Christensen NC, Skovby F, Nickel V, Roskos M, Funke H, Ose L, Assmann G (1995) A novel variant of lysosomal acid lipase (Leu336-->Pro) associated with acid lipase deficiency and cholesterol ester storage disease. Arterioscler Thromb Vasc Biol 15(6):773–778 [DOI] [PubMed] [Google Scholar]
- 210.Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM (2005) The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy 1(1):23–36 [DOI] [PubMed] [Google Scholar]
- 211.Martinet W, De Meyer GR (2009) Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 104 (3):304–317 [DOI] [PubMed] [Google Scholar]
- 212.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119(6):753–766 [DOI] [PubMed] [Google Scholar]
- 213.Kim JS, Nitta T, Mohuczy D, O’Malley KA, Moldawer LL, Dunn WA Jr, Behrns KE (2008) Impaired autophagy: a mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology 47 (5):1725–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA (2007) Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117 (7):1782–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jia G, Cheng G, Gangahar DM, Agrawal DK (2006) Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol 84(5):448–454 [DOI] [PubMed] [Google Scholar]
- 216.Martinet W, De Meyer GR (2008) Autophagy in atherosclerosis. Curr Atheroscler Rep 10(3):216–223 [DOI] [PubMed] [Google Scholar]
- 217.Xu K, Yang Y, Yan M, Zhan J, Fu X, Zheng X (2010) Autophagy plays a protective role in free cholesterol overload-induced death of smooth muscle cells. J Lipid Res 51(9):2581–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jia G, Cheng G, Agrawal DK (2007) Autophagy of vascular smooth muscle cells in atherosclerotic lesions. Autophagy 3(1):63–64 [DOI] [PubMed] [Google Scholar]
- 219.Schrijvers DM, De Meyer GR, Herman AG, Martinet W (2007) Phagocytosis in atherosclerosis: molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res 73(3):470–480 [DOI] [PubMed] [Google Scholar]
- 220.Verheye S, Martinet W, Kockx MM, Knaapen MW, Salu K, Timmermans JP, Ellis JT, Kilpatrick DL, De Meyer GR (2007) Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol 49(6):706–715 [DOI] [PubMed] [Google Scholar]
- 221.Zhang Y, Xu M, Xia M, Li X, Boini KM, Wang M, Gulbins E, Ratz PH, Li PL (2014) Defective autophagosome trafficking contributes to impaired autophagic flux in coronary arterial myocytes lacking CD38 gene. Cardiovasc Res 102(1):68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Xu M, Li XX, Wang L, Wang M, Zhang Y, Li PL (2015) Contribution of Nrf2 to atherogenic phenotype switching of coronary arterial smooth muscle cells lacking CD38 gene. Cell Physiol Biochem 37 (2):432–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Adiguzel E, Ahmad PJ, Franco C, Bendeck MP (2009) Collagens in the progression and complications of atherosclerosis. Vasc Med 14 (1):73–89 [DOI] [PubMed] [Google Scholar]
- 224.Xu X, Yuan X, Li N, Dewey WL, Li PL, Zhang F (2016) Lysosomal cholesterol accumulation in macrophages leading to coronary atherosclerosis in CD38(−/−) mice. J Cell Mol Med 20(6):1001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]