

Abstract
The combination of electrocyclizations and cycloadditions accounts for the formation of a range of fascinating natural products. Cascades consisting of 8π electrocyclizations, followed by 6π electrocyclization, and a cycloaddition are relatively common. We now report the synthesis of the tetramic acid PF-1018 through an 8π electrocyclization, the product of which is immediately intercepted by a Diels–Alder cycloaddition. The success of this pericyclic cascade was critically dependent on the substitution pattern of the starting polyene and could be rationalized through DFD calculations. The completion of the synthesis required the instalment of a trisubstituted double bond via radical deoxygenation. An unexpected byproduct formed through 4-exo-trig radical cyclization could be recycled through an unprecedented triflation/fragmentation.
Keywords: Diels–Alder Cycloaddition, Electrocyclization, Total Synthesis, Biomimetic Synthesis, Tetramic Acids
Graphical Abstract
The combination of an 8π−6π-electrocyclization cascade with a cycloaddition has been implicated in the biosynthesis of a range of natural products (Fig.1). In the well-known endriandric acid family, for instance, conrotatory 8π electrocyclization of 1 yields cyclooctatriene 2, which then undergoes disrotatory 6π electrocyclization to afford the bicyclo[4.2.0]octadiene endriandric acid G. This is followed by an intramolecular Diels–Alder reaction that leads to endriandric acid C (Fig. 1A).[1–3] In the case of kingianin A, the electrocyclization cascade yields cyclooctatriene 3 and then bicyclo[4.2.0]octadiene 5, which engages in intermolecular Diels–Alder dimerization (Fig. 1B).[4,5] Other examples that have been validated by biomimetic or bioinspired synthesis include ocellapyrone A,[6] where the electrocyclizations are followed by an intramolecular hetero Diels–Alder reaction, and orinocin,[7] where they are followed by a retro [2+2]-cycloaddition, resulting in “polyene splicing”.
Figure 1.

X-ray crystal structure of compounds 18 and 30.
The unusual natural product PF-1018 appears to stem from a fascinating variation of this pericyclic chemistry (Fig. 1C). The anionic lipid was isolated from a fungal strain identified as Humicola sp. 1018 in a screen against Plutella xylostella, the diamond back moth, which accounts for massive agricultural losses worldwide.[8,9] Its structure was established by NMR and X-ray analysis and was shown to feature a rare proline derived tetramic acid linked to a complex tricyclic hydrocarbon core. This core features six stereocenters, two of them quaternary, and three trisubstituted double bonds bearing methyl groups.
Biosynthetically, PF-1018 could originate, at least in principle, from the complex polyene 6 (Fig. 1C). A diastereoselective 8π electrocyclization would generate the cyclooctatriene 7. Unlike the cascades above, instead of a subsequent conrotatory 6π electrocyclization to afford a bicyclo[4.2.0]octadiene, the cyclooctatriene engages in an intramolecular cycloaddition with one of the disubstituted alkenes to generate the tricyclic hydrocarbon core of the molecule.
It should be noted that the endriandric acids and kingianin A were isolated as racemates and spontaneously form from their achiral polyene precursors 1 and 3.[1,4] By contrast, PF-1018 was isolated as a single enantiomer and diastereomer. It seems unlikely that the existing stereocenter in the hypothetical precursor 6 would govern the diastereoselectivity of the initial 8π electrocyclization. This indicates that the pericyclic cascades could already occur on a shorter intermediate, such as 8, within the chiral environment of a molecular assembly line (Fig. 1D).[10,11] The cyclization of an intermediate bound to a polyketide synthase–nonribosomal peptide synthetase (PKS-NRPS) via a thioester, could also explain why interception of the cyclooctadiene 9 via Diels–Alder reaction to afford 10 is favoured over a relatively fast 6π electrocyclization, which is essentially irreversible at ambient temperatures.[12–15]
Given these considerations, a biomimetic laboratory synthesis of PF-1018 poses considerable challenges. We have previously developed an 8π electrocyclization/Diels–Alder cascade that gave rise to the partially saturated tricyclic core of PF-1018.[16] However, our synthesis faltered because we were unable to install the remaining double bond in this highly hindered, caged intermediate. We now report the successful, asymmetric synthesis of PF-1018 that could overcome these issues through careful engineering of the cyclization substrate. It demonstrates that our strategy for the Diels–Alder interception of a cyclooctatriene is generalizable. It also features unusual and unexpected reactivity associated with the caged and congested tricyclic hydrocarbon core of our target.
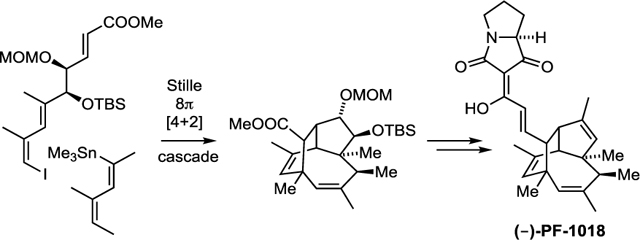
Our synthesis started with the asymmetric addition of Brown’s chiral allyl borane 12[17] to the known aldehyde 11. This gave the mono methoxymethyl (MOM)-protected diol 13 in decent yield and with excellent enantiomeric purity. Subsequent olefin cross-metathesis with methyl acrylate was achieved through sequential addition of the second-generation Hoveyda−Grubbs catalyst over several hours to overcome low conversion. This was followed by protection of the secondary alcohol as a silyl ether and afforded the unsaturated ester 14. The stage was now set for the key step of the synthesis, which commenced with cross-coupling of 14 with the known vinyl stannane 15 under Stille−Liebeskind conditions. The initial cross-coupling product 16 was not isolated but was heated gradually in solution from 65° C to 125 °C and then maintained for 20 hours at this temperature to promote the 8π electrocyclization (16 → 17) and Diels–Alder interception (17 → 18). Under these conditions we could isolate the tricycle 18 in 42% yield. The X-ray structure of 18 is shown in Fig. 1 confirming the configuration and highly congested nature of this caged compound. With 18 in hand, our next task was to convert the doubly protected diol into the trisubstituted C2,C3 double bond of PF-1018 (original nomenclature). To this end, we first cleaved the silyl ether at C2 to afford the secondary alcohol 19 and then activated its hydroxy group as the benzyl xanthate 20. Barton−McCombie deoxygenation under highly optimized conditions (see below) then gave MOM ether 21. Cleavage of this ether using a modification of Fujioka’s-conditions,[18] followed by oxidation with Dess−Martin periodinane, yielded ketone 22. Conversion to the enol triflate, followed by Negishi coupling, installed the required olefinic double bond. Subsequent reduction of the ester with DIBAL then gave the primary alcohol 23.
In the final stage of our synthesis, 23 was oxidized to an unstable aldehyde, which was condensed with tetramic acid phosphonate 25.[19] This gave (−)-PF-1018 as an inseparable 3.5:1 mixture of (E) and (Z) isomers with respect to the C19-C2’ double bond.The analytical data of our synthetic samples were identical with those reported for the natural product, with the exception of the optical rotation, which is significantly lower in our case. However, we have confirmed the high optical purity of alcohol 19 by Mosher ester analysis (see Supporting Information) and it seems unlikely that the final steps of the synthesis result in partial racemization. The difference in optical rotation probably stems from variations of the (E)/(Z) ratio of the C19-C2’ double bond (3.2:1 reported for the natural sample).[9]
The preparation of tetramic acid phosphonate 25 from proline through a modified route is shown in the lower portion of Fig. 2. Acetylation and conversion to the Weinreb amide gave 24. A subsequent Dieckmann condensation then afforded an unstable pyrrolizidinedione,[20] which was coupled with diethyl phosphonoacetic acid to afford the tetramic acid phosphonate 25.[19,21] This building block was also condensed with octanal to afford the recently isolated anionic lipid pyreudione C.[20,22]
Figure 2.

Density functional theory calculated transition state structures of pericyclic cascade. Hydrogens on protecting groups are omitted for clarity, silicon shown in wheat colour.
The key step of our synthesis warrants further analysis. Given the many possible isomers that can be formed, we consider this a remarkable result both in terms of selectivity and yield (see SI Fig. S1). In preliminary studies, we had established that the relative stereochemistry at C2 and C3, as well as the size of the silyl group at C2 are crucial to achieve high yields in the cascade.[16] Bicyclo[4.2.0]octadienes resulting from competing 6π electrocyclization cascades were never observed but are likely destroyed via formal retro [2+2] cycloaddition under our optimized conditions.[7] Other isomers could not be isolated either.
To explain such reactivities and selectivities of the pericyclic cascade, density functional theory calculations were employed at the SMD(DMF)-ωB97X-D/def2-QZVPP//SMD(DMF)-ωB97X-D/6–311+G(d,p) level of theory (see SI for computational details).[23–33]
These calculations indicate that the polyene 16 can engage in two conrotatory 8π electrocyclizations to form 17 and its diastereomer 17’ via TS-1 and TS-2, respectively (Fig. 2, S2). The cyclooctatrienes 17 and 17’ are nearly isoenergetic with their precursor 16 (Figure S3), resulting in an initial equilibrium between the species 16, 17, and 17’. The subsequent intramolecular cycloaddition of 17, forming 18 via TS-3, is 4.7 kcal·mol−1 lower in Gibbs-free energy than the diastereomeric cycloaddition via TS-4. Consequently, the equilibrium mixture of 16, 17, and 17’ will funneled through TS-3 to selectively form the isolated product 18. The Diels–Alder transition states TS-3 and TS-4 are both stabilized by favourable secondary orbital interactions between the diene and ester oxygen of the dienophile. The difference in Gibbs-free energy between TS-3 and TS-4 (ΔΔG‡ = 4.7 kcal·mol−1) is due to the MOM-ether being forced into a pseudo-axial position in TS-4. In TS-3, both the MOM-ether and silyl-ether groups on the emerging cyclopentane are in pseudo-equatorial positions. The silyl ether greatly affects the energy difference between the Diels–Alder cycloaddition via TS-3 and a competing 6π electrocyclization via TS-5. Decreasing the size of this substituent would favour the 6π electrocyclization according to our calculations (Fig. 2, S4, S5a). A substrate with the double bond in place at C2,3 will also predominately form the 6π-product, suggesting that an enzyme is necessary for the Diels–Alder reaction to occur in Nature (Fig. S5b).
A step in our synthesis that delivered unexpected but interesting results was the Barton−McCombie deoxygenation. (Scheme 3 and Table 1). Standard conditions involving the methyl xanthate 26 only gave a low yield of the desired product 21. It was accompanied by substantial amounts of methyl ether 28 (byproduct previously reported in these reactions)[34–36] and, to our surprise, cyclobutane 29. The latter results from a rare 4-exo-trig radical cyclization, which generates a secondary radical.[37] This unusual selectivity presumably results from the constrained nature of the caged precursor. Increasing the concentration of tributyl tin hydride suppressed this cyclization but also led to higher yields of the unwanted methyl ether 28. This problem could be finally overcome by variation of the xanthate: use of the larger benzyl xanthate 20 to promote fragmentation gave a good yield of the desired intermediate 21.[38]
Scheme 3 and Table 1:

Products of the Barton−McCombie deoxygenation.
Cyclobutane 29 was not a waste product but could be converted into a late-stage intermediate of our synthesis through an unusual fragmentation (Scheme 4). We reasoned that ketone 30 could fragment after electrophilic activation of the carbonyl compound with triflic anhydride to yield unsaturated vinyl triflate 31.[39,40] Deprotection and oxidation of 29 with Dess−Martin periodinane gave cyclobutyl ketone 30, the X-ray structure of which is shown in Fig 1 (right). It confirmed that the carbonyl group, the C,C-bond, and the requisite C-H bond are indeed stereoelectronically well aligned to allow for smooth formation of the two π bonds. Attempts to convert 30 into vinyl triflate 31 however, were unsuccessful and led to decomposition. We assigned this result to the interreference of the methyl ester, which was accordingly reduced to afford primary alcohol 32. Cleavage of the MOM-ether, DIBAL-reduction, selective protection of the primary alcohol with a bulky silyl group, and oxidation of the secondary alcohol then yielded cyclobutyl ketone 32. Treatment of this ketone with triflic anhydride in the presence of a sterically hindered pyridine base elicited the desired fragmentation and gave vinyl triflate 33. Although the methyl group could also be deprotonated and is less hindered, only a small amount the corresponding exo-methylene isomer was observed. To the best of our knowledge, a fragmentation of this type with concomitant enol triflate formation has only been reported once in the literature.[40] Negishi cross-coupling of 33 with dimethyl zinc and deprotection then afforded primary alcohol 23, which could be used in the final steps of our total synthesis.
Scheme 4:

Fragmentation of a cyclobutyl ketone and recycling of cyclobutane side product 29.
In summary, we have achieved the first total synthesis of the unusual polyketide tetramic acid PF-1018. Our 15-step asymmetric synthesis is marked by a bioinspired cascade that combines an 8π electrocyclization with a Diels–Alder cycloaddition. Computational studies indicate that the size of substituent at C2 determines whether a 6π electrocyclization or Diels–Alder will be preferred, while the stereochemistry at C3 determines facial selectivity of the cycloaddition. This synthesis also rests on an unusual strategy to install a trisubstituted double bond. In the course this sequence, we observed a 4-exo-trig radical cyclization that generated an unexpected tetracycle. The product of this radical cyclization could be recycled by way on a stereoelectronically favourable fragmentation that simultaneously generated a trisubstituted double bond and a vinyl triflate. Future work will be directed towards exploring the biosynthesis, biological activity and structure activity-relationships of PF-1018 and other anionic lipids that feature proline-derived tetramic acids, such as talarotoxin[41] and the pyreudiones.[10,20,42]
Supplementary Material
Scheme 1.

Electrocyclization-Cycloaddition cascades in natural product chemistry.
Scheme 2.

Total synthesis of (−)-PF-1018 and Pyreudione C.
Acknowledgements
We thank Petr Tatarskiy for his synthesis work, Dr. Peter Mayer (LMU) and Prof. Chunhua Hu (NYU) for X-ray structure analysis and Dr. Chin Lin for assistance with NMR measurements. The TCI cryoprobe used for NMR acquisition was supported by the National Institutes of Health (OD016343). We would also like to thank the National Science Foundation (NSF CHE-1806581 to K.N.H.) for funding. All computational resources were provided by the Institute for Digital Research and Education. C.S.J. is supported by generous funding through the Saul Winstein Fellowship.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Bandaranayake WM, Banfield JE, Black DSC, J. Chem. Soc., Chem. Commun 1980, 902–903. [Google Scholar]
- [2].Banfield JE, Black DSC, Johns SR, Willing RI, Aust. J. Chem 1982, 35, 2247–2256. [Google Scholar]
- [3].Nicolaou KC, Petasis NA, Uenishi J, Zipkin RE, J. Am. Chem. Soc 1982, 104, 5557–5558. [Google Scholar]
- [4].Leverrier A, Dau METH, Retailleau P, Awang K, Guéritte F, Litaudon M, Org. Lett 2010, 12, 3638–3641. [DOI] [PubMed] [Google Scholar]
- [5].Lim HN, Parker KA, Org. Lett 2013, 15, 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Miller AK, Trauner D, Angewandte Chemie International Edition 2005, 44, 4602–4606. [DOI] [PubMed] [Google Scholar]
- [7].Müller M, Kusebauch B, Liang G, Beaudry CM, Trauner D, Hertweck C, Angewandte Chemie International Edition 2006, 45, 7835–7838. [DOI] [PubMed] [Google Scholar]
- [8].Zalucki MP, Shabbir A, Silva R, Adamson D, Shu-Sheng L, Furlong MJ, J Econ Entomol 2012, 105, 1115–1129. [DOI] [PubMed] [Google Scholar]
- [9].Gomi S, Imamura K-I, Yaguchi T, Kodama Y, Minowa N, Koyama M, J. Antibiot 1994, 47, 571–580. [DOI] [PubMed] [Google Scholar]
- [10].Klapper M, Braga D, Lackner G, Herbst R, Stallforth P, Cell Chem Biol 2018, 25, 659–665.e9. [DOI] [PubMed] [Google Scholar]
- [11].Kasahara K, Miyamoto T, Fujimoto T, Oguri H, Tokiwano T, Oikawa H, Ebizuka Y, Fujii I, ChemBioChem 2010, 11, 1245–1252. [DOI] [PubMed] [Google Scholar]
- [12].Rolf Huisgen, Alexander Dahmen, Helmut Huber, J. Am. Chem. Soc 1967, 89, 7130–7131. [Google Scholar]
- [13].Huisgen R, Dahmen A, Huber H, Tetrahedron Lett. 1969, 10, 1461–1464. [Google Scholar]
- [14].Thomas BE, Evanseck JD, Houk KN, Isr. J. Chem 1993, 33, 287–293. [Google Scholar]
- [15].Patel A, Houk KN, J. Org. Chem 2014, 79, 11370–11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Webster R, Gaspar B, Mayer P, Trauner D, Org. Lett 2013, 15, 1866–1869. [DOI] [PubMed] [Google Scholar]
- [17].Brown HC, Jadhav PK, Bhat KS, J. Am. Chem. Soc 1988, 110, 1535–1538. [Google Scholar]
- [18].Fujioka H, Okitsu T, Sawama Y, Murata N, Li R, Kita Y, J. Am. Chem. Soc 2006, 128, 5930–5938. [DOI] [PubMed] [Google Scholar]
- [19].Tan SWB, Chai CLL, Moloney MG, Org. Biomol. Chem 2014, 12, 1711–1716. [DOI] [PubMed] [Google Scholar]
- [20].Klapper M, Götze S, Barnett R, Willing K, Stallforth P, Angewandte Chemie International Edition 2016, 55, 8944–8947. [DOI] [PubMed] [Google Scholar]
- [21].Rosen T, Fernandes PB, Marovich MA, Shen L, Mao J, Pernet AG, J. Med. Chem 1989, 32, 1062–1069. [DOI] [PubMed] [Google Scholar]
- [22].Klapper M, Paschold A, Zhang S, Weigel C, Dahse H-M, Götze S, Pace S, König S, Rao Z, Reimer L, et al. , ACS Chem. Biol 2019, 14, 1693–1697. [DOI] [PubMed] [Google Scholar]
- [23].Marenich AV, Cramer CJ, Truhlar DG, J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- [24].Krishnan R, Binkley JS, Seeger R, Pople JA, J. Chem. Phys 1980, 72, 650–654. [Google Scholar]
- [25].Rassolov VA, Pople JA, Ratner MA, Windus TL, J. Chem. Phys 1998, 109, 1223–1229. [Google Scholar]
- [26].Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA, J. Chem. Phys 1982, 77, 3654–3665. [Google Scholar]
- [27].Dill JD, Pople JA, J. Chem. Phys 1975, 62, 2921–2923. [Google Scholar]
- [28].Hehre WJ, Ditchfield R, Pople JA, J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- [29].Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PVR, Journal of Computational Chemistry 1983, 4, 294–301. [Google Scholar]
- [30].Blaudeau J-P, McGrath MP, Curtiss LA, Radom L, J. Chem. Phys 1997, 107, 5016–5021. [Google Scholar]
- [31].Schäfer A, Huber C, Ahlrichs R, J. Chem. Phys 1994, 100, 5829–5835. [Google Scholar]
- [32].Weigend F, Ahlrichs R, Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- [33].Chai J-D, Head-Gordon M, Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- [34].Bensasson CS, Cornforth SJ, Du M-H, Hanson JR, Chem. Commun 1997, 1509–1510. [Google Scholar]
- [35].Cristol SJ, Klein MW, Hendewerk MH, Daussin RD, J. Org. Chem 1981, 46, 4992–4998. [Google Scholar]
- [36].Srikrishna A, Gharpure SJ, J. Org. Chem 2001, 66, 4379–4385. [DOI] [PubMed] [Google Scholar]
- [37].Majetich G, Grove JL, Org. Lett 2009, 11, 2904–2907. [DOI] [PubMed] [Google Scholar]
- [38].Barton DHR, Crich D, Löbberding A, Zard SZ, Tetrahedron 1986, 42, 2329–2338. [Google Scholar]
- [39].Kato M, Watanabe M, Awen BZ, J. Org. Chem 1993, 58, 5145–5152. [Google Scholar]
- [40].Martínez García, Fernández Herrera, Sánchez García A, M J, An. Quim 1979, 75, 723. [Google Scholar]
- [41].Ishii K, Itoh T, Kobayashi K, Horie Y, Ueno Y, Appl. Environ. Microbiol 1995, 61, 941–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Petersen LM, Frisvad JC, Knudsen PB, Rohlfs M, Gotfredsen CH, Larsen TO, The Journal of Antibiotics 2015, 68, 603–608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.