

Abstract
The Hippo signalling pathway is dysregulated across a wide range of cancer types and, although driver mutations that directly affect the core Hippo components are rare, a handful is found within pleural mesothelioma (PM). PM is a deadly disease of the lining of the lung caused by asbestos exposure. By pooling the largest‐scale clinical datasets publicly available, we here interrogate associations between the most prevalent driver mutations within PM and Hippo pathway disruption in patients, while assessing correlations with a variety of clinical markers. This analysis reveals a consistent worse outcome in patients exhibiting transcriptional markers of YAP/TAZ activation, pointing to the potential of leveraging Hippo pathway transcriptional activation status as a metric by which patients may be meaningfully stratified. Preclinical models recapitulating disease are transformative in order to develop new therapeutic strategies. We here establish an isogenic cell‐line model of PM, which represents the most frequently mutated genes and which faithfully recapitulates the molecular features of clinical PM. This preclinical model is developed to probe the molecular basis by which the Hippo pathway and key driver mutations affect cancer initiation and progression. Implementing this approach, we reveal the role of NF2 as a mechanosensory component of the Hippo pathway in mesothelial cells. Cellular NF2 loss upon physiological stiffnesses analogous to the tumour niche drive YAP/TAZ‐dependent anchorage‐independent growth. Consequently, the development and characterisation of this cellular model provide a unique resource to obtain molecular insights into the disease and progress new drug discovery programs together with future stratification of PM patients.
Keywords: YAP/TAZ, stratification, mesothelioma, BAP1, NF2
Stratifying pleural mesothelioma (PM) patients reveals active YAP/TAZ is associated with poor clinical outcomes.
An isogenic cell‐line model of PM driver mutations is developed that faithfully recapitulates clinical PM characteristics.
NF2 regulates YAP/TAZ mesothelial activity in response to stresses, including sensing mechanical cues, while BAP1 appears to regulate a stem‐cell‐like transcriptional program.
1. INTRODUCTION
Pleural mesothelioma (PM) is a rare cancer of the mesothelial pleural lining of the lung, most commonly associated with exposure to the carcinogen asbestos. PM is most common in males, likely due to more frequent exposure to asbestos fibres. 1 , 2 , 3 Despite the declining use of asbestos as an insulating and fire retardant agent in the developed world, 4 the prevalent presence of asbestos in buildings and the overall latency of upwards of 30 years from exposure to when mesothelioma presents clinically means that rates of PM have continued to increase. The peak incidence of PM is projected to occur within the next decade. 3 , 5 , 6 , 7 Consequently, the continued mining of asbestos and underdiagnosis of PM in the developing countries, 8 , 9 together with the realisation that some types of currently used nanofibers cause PM‐like cancers in rodent models, 10 strongly indicate that PM is a disease with a continued unmet clinical need. PM is characterised by a low mutational burden. 11 , 12 , 13 Patients who are diagnosed have a poor prognosis with survival rates one of the lowest of any cancer, while the current standard of care treatments extend survival by a matter of months. 7 Some progress has been made in recent years, with the successful combination of nivolumab and ipilimumab showing potential in extending median overall survival in a small subgroup of patients by up to 4 months. 14 However, current treatments are essentially palliative care. 15 As such, the search for a curative therapeutic regime remains elusive, highlighting the pressing need for the development of effective therapeutics to enable clinical management of the disease.
The difficulties in identifying effective therapeutics to manage PM are caused by late‐stage diagnosis and an infiltrative and therefore overall malignant phenotype. In addition, there is currently a lack of predictive preclinical models which inform disease positioning/patient stratification. An in‐depth understanding of the initial molecular drivers, as well as the complicit oncogenic pathways that facilitate the progression of this refractory cancer type, are consequently needed. 16 , 17 To this end, a number of studies have, through whole exome sequencing, examined in detail the molecular landscape of PM, 12 , 13 , 18 describing genomic and transcriptomic alterations most closely aligned with mesothelioma onset and initiation. These analyses have revealed that a number of loss‐of‐function mutations present at low frequency in other cancer types are frequently identified during the development of PM, with two tumour suppressors most notably lost regularly in PM patients: BAP1, which acts as a deubiquitinase 19 , 20 and NF2, a major upstream activator of the core kinase module of the Hippo signalling pathway. 21 , 22 Beyond NF2, a number of additional key upstream regulators of the Hippo pathway are inactivated at lower frequencies in PM. 12 , 13
The Hippo pathway consists of an upstream serine/threonine kinase module that when active, phosphorylates and thereby inhibits the transcriptional co‐activators YAP and TAZ, comprising the transcriptional module of the pathway. When YAP and TAZ are unphosphorylated, they localise to the nucleus and bind to TEAD family transcription factors, driving the expression of target genes. 23 , 24 , 25 The loss‐of‐function mutations in the upstream regulatory module of the Hippo pathway are consequently of relevance, as the loss of components within this kinase module is predicted to cause hyperactive YAP/TAZ. In other cancers and cell model systems, YAP and TAZ drive EMT, 26 , 27 , 28 migration/metastasis, 29 , 30 and chemoresistance, 31 , 32 while their expression and activation are associated with poor clinical outcomes across a range of cancer types. 33 , 34 , 35 , 36 This body of work reported here describes an integrated analysis of the largest scale studies of PM patients to date.
BAP1 and NF2 are the two most commonly mutated genes in PM and are loss‐of‐function mutated in up to 57% and 23% of cases respectively. 12 , 13 , 18 , 37 Mutation status within these genes, when considered in combination, represents a powerful prognostic indicator of PM 38 ; with the push towards realising precision oncology well underway, a sensible question is whether populations grouped according to the mutation status of these drivers might represent actionable subtypes within PM. Our work describes a pooled, detailed analysis of high‐throughput PM datasets 12 , 13 to explore potential stratification approaches. This approach allows us to investigate the link between the major driver mutations in PM and the Hippo signalling pathway. We additionally develop and characterise a pre‐clinical in vitro cellular model of PM that allows for follow‐up investigations into the dynamics and differential effects of the Hippo pathway and BAP1 dysregulation in PM.
2. RESULTS
2.1. Interrogating large‐scale databases to define patient subgroups in PM
This work describes the combination of two large PM databases, the first of which was generated by The Cancer Genome Atlas (TCGA), 12 consisting of 87 patients and the second of which includes 211 patients 13 (hereon referred to as TCGA cohort and Bueno cohort, respectively). As the occurrence of inactivating mutations in the upstream regulatory kinase module of the Hippo pathway is emerging as a common phenomenon associated with the development of PM, 39 , 40 patients were initially characterised according to Hippo pathway activation status. This was approached by initially assessing the mutation status of key Hippo pathway players, regularly identified in PM patients (Figure 1A). Secondly, the activity of YAP/TAZ, the transcriptional co‐activators within the downstream arm of the Hippo pathway, was approximated using a previously defined, curated gene set of bona fide YAP/TAZ transcriptional targets. 41
FIGURE 1.
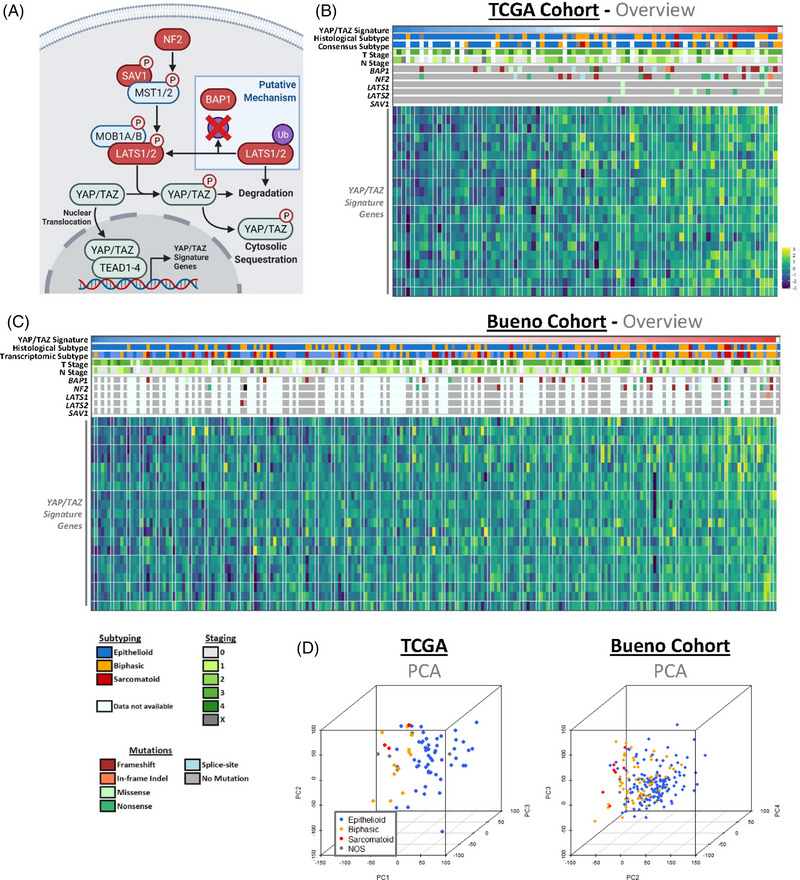
Defining distinct populations of pleural mesothelioma patients. (A) Schematic shows key components of the Hippo signalling pathway, alongside the suggested role BAP1 plays in its regulation (blue box). Elements reported as mutated in cases of pleural mesothelioma (PM) are highlighted in red. (B) Heatmap highlights the various clinical, genomic and transcriptomic features of the The Cancer Genome Atlas (TCGA) cohort of 87 PM patients. 12 Patients are ordered in ascending gene set variation analysis (GSVA) scoring of YAP/TAZ signature gene expression, 40 with the expression of individual genes also shown (bottom). Clinical outputs (top) include histological subtyping classifications, both original and as determined via consensus pathology review, and American Joint Committee on Cancer (AJCC) staging, while predicted Hippo kinase cascade inactivating mutation status (centre) are highlighted according to variant classification, with mutations in NF2, LATS1/2 and SAV1 included. (C) Heatmap, as in (B), details 212 patients constituting the Bueno et al. 13 cohort. Legend for both heatmaps (B, C), highlighting colour schema for subtyping, mutation status, and clinical staging of PM cases, shown below. (D) Principal component analysis (PCA) plots, depicted in 3D, show patients coloured according to histological subtype, with dimensionality reduction applied to full transcriptomes. This analysis highlights that broad transcriptional profiling is sufficient to distinguish between the different histological subtypes in both the consensus subtypes defined in the TCGA (left; n = 86) and Bueno et al. (right; n = 211) cohorts
Assessing these various metrics, the broad make‐up of both datasets is consistent and both patient cohorts across datasets are directly comparable (Figure 1B,C), with no over‐representation of subtype/clinical stage or mutations in either. Principal component analysis (PCA) of patient transcriptomes grouped by histological subtype reveals a general clustering of subtype groups (Figure 1D and Figure S1A), which points to the potential of broad transcriptional profiles to predict simple clinical outputs. In order to explore patient stratification, various clinical outputs were assessed upon the grouping of patients according to the mutation status of two key driver mutations within PM: NF2 and BAP1. Interestingly, although these genes are frequently loss‐of‐function mutated in cases of PM, no single distinct mutation is common amongst patients. Considering specific alterations in amino acids of protein products, two patients exhibited aberrant stop codons at both arginine 57 and tyrosine 153 in NF2, while two patients each harboured serine 10 to arginine, cysteine 91 to glycine, and asparagine 645 to lysine, while all other residue shifts in NF2 and BAP1 were unique. Interestingly, two, 13 and 13 out of 36 impactful NF2 mutations include missense mutations, nonsense mutations and frameshift indels respectively, while out of 54 impactful BAP1 mutations, 9, 10, and 25 were missense mutations, nonsense mutations, and frameshift indels respectively. The remainder of the mutations include in‐frame indels and splice‐site mutations, indicating a high frequency of highly disruptive genomic perturbations within these driver genes. When grouping patients according to the mutation status of these drivers, initial PCA highlights that patient groups do not readily cluster according to broad transcriptional profiles (Figure S1B). Further analyses reveal that there are no significant associations between mutation status and transcriptomic subtypes (Figure S1C), a molecular subgrouping approach which mirrors histological subtypes, 13 clinical stages (Figure S1D), or overall patient survival (Figure S1E). The absence of any transcriptomic clustering or clear association with prognostic indicators suggests that profiling patients according to mutation status may not be meaningful clinically; however, in spite of the lack of significance, trends suggest that BAP1 mutation may loosely associate with earlier‐stage PM and the epithelioid subtype, an observation which is corroborated by previous studies. 42 , 43 , 44 , 45
2.2. YAP/TAZ activation status predicts prognosis in PM patients
Notably, NF2 is the most frequently mutated Hippo pathway component in PM, however, multiple key members of the upstream core kinase cascade (LATS1, LATS2, SAV1), are also loss‐of‐function mutated 13 , 18 , 39 (Figure 1A–C). These observations suggest that Hippo kinase cascade inactivation (and therefore YAP/TAZ activation) likely plays a major role in PM onset, while both NF2 (also known as Merlin) and BAP1 have been tied to the Hippo pathway as a bona fide key regulator 46 , 47 and a putative effector of core kinase members, 48 respectively. If the loss of either NF2 or BAP1 mediates oncogenesis via the perturbation of Hippo signalling, the failure to take into account the additional Hippo kinase module inactivating events present in PM may have confounded previous efforts to stratify patients according to mutation status. Collectively, these observations suggest that subgrouping patients according to YAP/TAZ activation status may be a more biologically meaningful stratification strategy.
Upregulation of YAP1 and WWTR1, the genes that encode YAP and TAZ respectively, have previously been used as a marker of Hippo pathway inactivation, 49 , 50 however this is an indirect measure, as YAP/TAZ are predominantly regulated via protein levels and subcellular location. 51 , 52 LATS1/2 mediated phosphorylation of YAP and TAZ leads to the cytoplasmic sequestering of these two transcriptional co‐activators, whereas unphosphorylated YAP and TAZ translocate to the nucleus and bind to TEAD family transcription factors (Figure 1A). 51 , 52 Within the patient cohorts, it is possible to approximate a more direct metric of activity via quantification of levels of total YAP relative to phosphorylated, inactive YAP at the protein level. 53 Utilising this approach, protein quantification within tumour samples in the TCGA cohort 12 via reverse phase protein array (RPPA) reveals that presumed active YAP levels are higher in patients affected with any common, canonical Hippo kinase module inactivating mutation (NF2, LATS1/2, SAV1) (Figure 2A). As further validation of this inferred approach for the transcriptional Hippo pathway activity within this subgrouping, the collective expression of YAP/TAZ downstream targets, as defined by TCGA, 41 were assessed. This 22‐gene signature is comprised of target genes associated with a variety of cellular functions, including two primary markers of YAP/TAZ activity CCN1 (or CYR61) and CCN2 (or CTGF), with gene‐set expression quantified via gene set variation analysis (GSVA), allowing a single signature metric for each patient. This analysis shows a robust upregulation of the YAP/TAZ signature within patients harbouring a canonical Hippo kinase module inactivating mutation, both in each individual patient cohort (Figure S2A) and upon pooling all patients into a single dataset (Figure 2B). Although BAP1 is not part of the Hippo pathway, a recent report highlights that BAP1 functions as a stabiliser of LATS1 and LATS2 via its deubiquitinase activity in a pancreatic cancer cell‐line model, hence acting as an activator of the Hippo kinase module (Figure 1A). 48 Loss of BAP1 in PM is associated with a slight increase in expression of YAP/TAZ targets in patients, though this is not conserved in both patient cohorts (Figure S2B).
FIGURE 2.
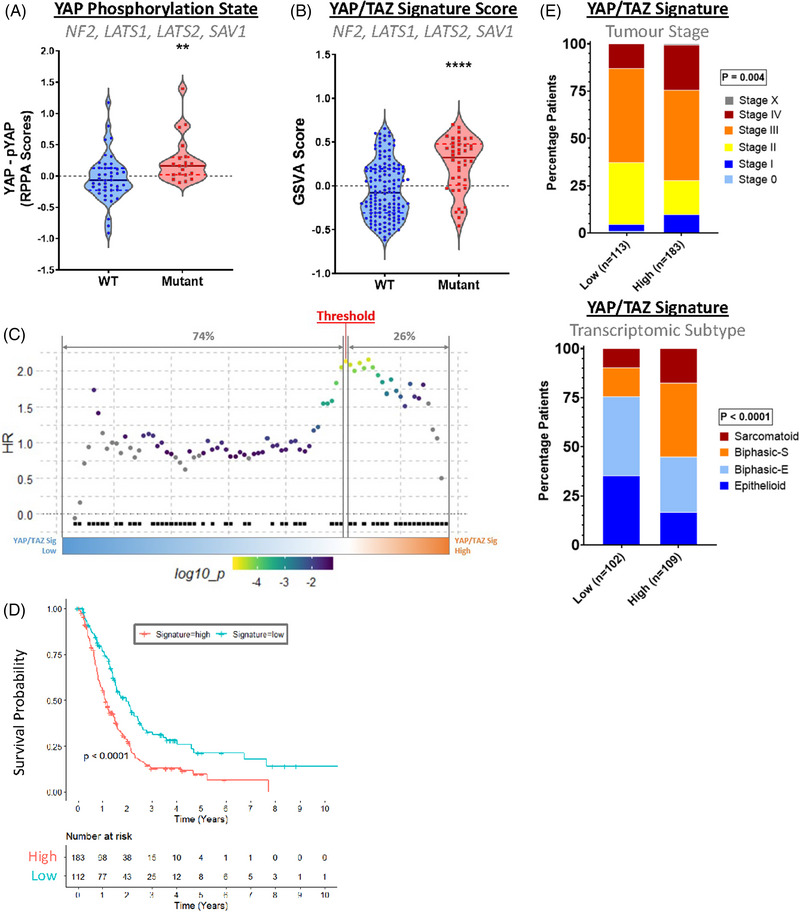
Expression of YAP/TAZ signature genes correlates with poor clinical outcomes in pleural mesothelioma (PM) patients. (A) Violin plot shows relative levels of unphosphorylated YAP in patient material, thereby approximating activity. This was calculated by subtracting levels of YAP phosphorylated at serine 127, an inhibitory post‐translational modification, from total YAP protein levels, recorded in reverse phase protein array (RPPA) data in The Cancer Genome Atlas (TCGA) dataset (n = 63). The mutant grouping includes Hippo pathway components NF2, LATS1, LATS2 and SAV1. (B) Violin plot shows gene set variation analysis (GSVA) scores of YAP/TAZ signature gene expression in patients split according to Hippo kinase cascade inactivating mutation status. A significant collective overexpression of these genes is observed in patients with mutations in genes associated with Hippo signalling when both cohorts are combined (n = 184). (C) SurvivALL plot shows the chosen methodology of setting thresholds based on the YAP/TAZ signature. Patients from the TCGA cohort are ordered according to the increasing signature, as determined by GSVA scores, with hazard ratios (HRs) for all possible thresholds shown as a scatterplot. Deaths are denoted by black squares, while the scale shows the colouring of points according to HRs. The thresholding that yields the highest HR is highlighted (red) and the percentage of patients classed as signature low and signature high are shown above (grey). (D) Kaplan‐Meier curve shows the overall survival of patients split according to YAP/TAZ signature thresholds. Patients classed as YAP/TAZ signature high experience a reduction in overall survival when both datasets are grouped. HR = 1.75, n = 296, the threshold set at 38%. (E) Barplots show T staging (top) in grouped datasets and consensus subtype (bottom) in the Bueno et al. cohort (n = 211, threshold set at 52%), with patients, split according to YAP/TAZ signature. Patients categorised as signature high generally exhibit more advanced clinical stages and more aggressive subtypes. p‐Values in (A) and (B) were determined by the Mann‐Whitney U test, p‐value and HR for (D) were calculated via log‐rank test and Cox proportional hazard model respectively, while p‐values for (E) were calculated via Fisher's exact test. **p < .01 and ****p < .0001 relative to wild‐type (WT)
With a reliable metric for determining Hippo kinase module inactivation established, it is possible to consider an effective strategy for stratifying patients meaningfully according to YAP/TAZ activity. Conventionally, subgrouping patients according to a continuous variable involves setting arbitrary thresholds, typically splitting patients around a median score. While a commonly employed approach, this is not necessarily relevant, as it fails to take sufficiently into account the biology surrounding a phenomenon. As a means to address this shortcoming, we employed SurvivAll, 54 a package that maximises the hazard ratio of high/low subgroups via an iterative grouping process (Figure 2C). Applying the resulting subgroups to the pooled dataset reveals a significant and dramatic decrease in median survival in YAP/TAZ high vs low patients (median survival 13.32 months vs. 23.52 months) (Figure 2D). Additionally, YAP/TAZ activity is found to be increased in both late‐stage PM and the aggressive sarcomatoid subtype (Figure 2E). These results are furthermore consistent between both cohorts when assessed separately (Figure S2C,D), validating the observation that active YAP/TAZ is a predictor of poor clinical outcome in PM.
2.3. Driver mutations differentially regulate Hippo signalling in an isogenic, preclinical model of PM
With YAP/TAZ activity established as a likely driver of PM progression, we sought to establish an isogenic model to probe Hippo pathway dysregulation in non‐malignant mesothelial cells in vitro. Two primary targets were selected for CRISPR‐Cas9 mediated knockout (KO) due to their high frequency of deletion in PM; NF2, to explore its role as a principal regulator of the Hippo pathway and BAP1, in order to assess its impact on YAP/TAZ activity, as well as more broadly, the effect of its loss on cancer progression, in the context of mesothelioma. Successful targeting of NF2 and BAP1 in the mesothelial cell line MeT‐5A is highlighted by the complete loss of protein in two independently established clones (Figure 3A). In order to characterise these isogenic KO clones, including that loss of either BAP1 or NF2 functionally recapitulates the expected biological impact within MeT‐5A cells, a range of downstream effects was assessed (Figure 3B).
FIGURE 3.
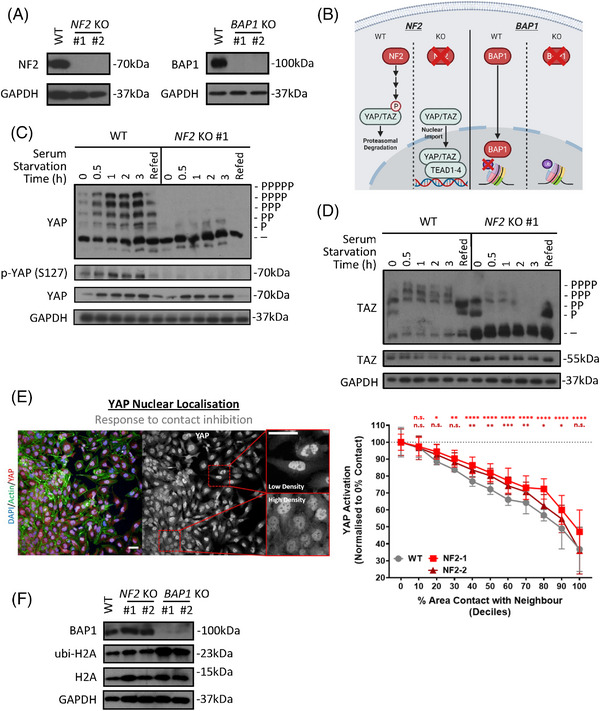
Development of an isogenic in vitro mesothelioma cell model of NF2 and BAP1 loss. (A) Western blots show successful CRISPR‐mediated knockout of NF2 (left) and BAP1 (right) in the mesothelial derived cell‐line, MeT‐5A. #1 and #2 label two independently generated knockout (KO) clones for each gene. (B) Schematic depicts well‐established functions of NF2 (left) and BAP1 (right) and therefore strategies of functional CRISPR KO validation, downstream of NF2 and BAP1. (C) Phos‐tag‐based western blots (top) show the phosphorylation status of YAP in response to serum starvation across a range of time points. Responses in wild‐type (WT) MeT‐5A cells (left) are compared to NF2 KO #1 (right), with a clear loss of response evident in NF2 deficient cells. The same lysates as analysed by Phos‐tag are also developed on a regular Western blot (bottom), where the lysates are probed for levels of phosphor (S127)‐YAP, total YAP and GAPDH (loading control). Refed condition refers to cells starved of serum for 3 hours, before adding serum‐replete medium for 1 h. (D) Phos‐tag‐based western blots as in (C) but probed for TAZ, show phosphorylation status of TAZ in response to serum starvation, alongside total TAZ levels, in WT and NF2 KO MeT‐5A cells. (E) Representative maximum projection images (left) obtained on the Operetta high‐content imaging platform show differential YAP nuclear compartmentalisation according to relative density in WT MeT‐5A cells. Scale bar = 50 μm. Scatter‐plot (right) shows the relative decrease in YAP nuclear localisation, as determined by the intensity of nuclear relative to cytoplasmic YAP, as percentage cell‐cell contact increases, with cells binned into deciles of percentage cell‐cell contact. NF2 KO MeT‐5A cells (red) are less sensitive to fall‐off of YAP nuclear localisation as cells approach confluency than WT cells (grey), as determined by two‐way analysis of variance (ANOVA), adjusted for multiple comparisons. n = 8, with three wells acting as technical triplicates for each biological replicate. 300–9000 cells counted/well and a total of 76,901, 76,970, and 65,653 cells were quantified for WT, NF2 KO clones #1 and #2 respectively. Error bars show SD from mean and significance levels illustrated above. n.s. = Not significant, *p < .05, **p < .01, ***p < .001 and ****p < .0001 relative to 0% contact with neighbour. (F) Western blot shows an increase in levels of (ubi)quityl‐histone H2A, an established BAP1 substrate, in BAP1 KO cells, compared to WT and NF2 KO clones
NF2, a protein associated with the plasma membrane/cytoskeleton and which is known to temporally relocate to cell junctions, 55 , 56 , 57 activates the Hippo kinase cascade in response to a range of stresses. 52 , 58 Firstly, serum starvation in multiple cell types, including HEK293A and U2OS cells, leads to LATS1/2 activation, and therefore YAP and TAZ phosphorylation and inactivation, 59 , 60 a phenomenon mediated by NF2. 61 In the mesothelial context, a similar effect is observed, using Phos‐tag based western blots, a technique that allows for visualising YAP and TAZ phosphorylation status. 59 , 62 These Phos‐tag based gels reveal that NF2 KO in MeT‐5A cells abrogates the serum starvation mediated YAP and TAZ phosphorylation readily observed in wild‐type (WT) cells (Figure 3C,D and Figure S3A). Of note, WT MeT‐5A cells exhibit a slight increase in levels of phosphorylated YAP/TAZ relative to NF2 KO cells at steady state conditions; however, this effect is minor when compared to the difference seen upon serum starvation, with both WT MeT‐5A and independently generated NF2 KO clones containing very little detectable phosphorylated YAP/TAZ under serum‐replete conditions (Figure 3C and Figure S3A). Re‐expression of NF2 in KO cells (Figure S3B) is sufficient to restore this serum starvation‐induced phosphorylation of YAP observed in WT cells (Figure S3C,D). We next used antibodies against YAP that are specific and verified for immunofluorescence‐based assays. 63 , 64 We used these on a confocal based high‐content imaging platform to quantify the nuclear localisation of YAP in MeT‐5A cells, to utilise as an activity marker for YAP. Surprisingly, this revealed that at steady state conditions, WT MeT‐5A display similar levels of nuclear YAP as compared to NF2 KO cells (Figure S3E). Although unexpected, this is consistent with Phos‐tag‐based results (Figure 3C) showing high levels of active (unphosphorylated) YAP in WT cells at steady state, suggesting that NF2 may be inactive in these cells when cultured sparsely, with high levels of nutrients, and on plastic. Beyond mediating the activation of the Hippo pathway on serum deprivation, NF2 additionally orchestrates the response to contact inhibition, 65 , 66 which is the cellular response to cell‐cell contact and subsequent inhibition of growth. The use of a high‐content imaging system allows us to obtain large‐scale cellular datasets including percentage cell‐cell contact metrics for each cell, facilitating the analysis of nuclear, and thereby active YAP levels in response to contact inhibition. As cells approach confluency, WT MeT‐5A cells exhibit a robust decrease in nuclear:cytoplasmic ratios of YAP, indicating a reduction in activity, while NF2 KO cells are markedly less sensitive to this effect (Figure 3E and Figure S3F). A decrease in nuclear YAP is, however, still observed in NF2 KO MeT‐5A cells, consistent with previous findings showing additional NF2‐independent activation of the Hippo kinase cascade in response to contact inhibition. 61
While BAP1 is reported to deubiquitinate a range of targets, 67 , 68 , 69 it is most commonly associated with monoubiquitinated histone H2A (H2AK119Ub). 19 , 70 In order to validate the functional loss of BAP1 in MeT‐5A KO cells, we assessed levels of histone H2A ubiquitination at lysine residue 119. A robust increase in levels of ubiquitination of H2A is observed in BAP1 KO cells, but not in NF2 KO cells (Figure 3F). Recently, BAP1 was reported to deubiquitinate and thereby stabilise the LATS kinases in certain contexts. 48 We, therefore, sought to establish if this effect is conserved in mesothelial cells. However, within mesothelial cells, BAP1 loss does not associate with a decrease in levels of LATS1/LATS2, as we conversely find that BAP1 loss drives an increase in both LATS1/LATS2 protein (Figure S3G). Despite being an inhibitory component of YAP/TAZ transcriptional activity, LATS2, which encodes the LATS2 kinase, is a direct target of YAP/TAZ and its expression is upregulated when the Hippo transcriptional module is active, 41 driving a negative feedback loop within the Hippo pathway. 71 , 72 In order to assess whether LATS2 protein levels may be increased as a result of increased LATS2 expression due to YAP/TAZ activation, we quantified LATS2 expression in BAP1 KO relative to WT MeT‐5A cells, revealing a consistent, minor upregulation (Figure S3H). This suggests that cells may respond by activating the tumour‐suppressive Hippo kinase cascade as a negative feedback mechanism 72 , 73 in order to respond to a loss of the tumour suppressor BAP1.
2.4. BAP1 loss in vitro recapitulates general transcriptomic dysregulation observed in PM patients
With an in vitro model established and functionally validated at the molecular level, an important question remains: do these cell lines retain features intrinsic to patient tumours? In order to explore this, NanoString nCounter gene expression assays were conducted across MeT‐5A KO lines in order to quantify the expression of a panel of 1540 genes involved in cancer progression and immune signalling. Viewing expression broadly across the full panel reveals that separate clones from individual genotypes exhibit similar patterns of expression and cluster separately (Figure S4A). To further validate this observation, PCA demonstrates that variance in general transcription is driven by the various KOs, with clones of each KO exhibiting convergent transcriptional profiles (Figure 4A).
FIGURE 4.
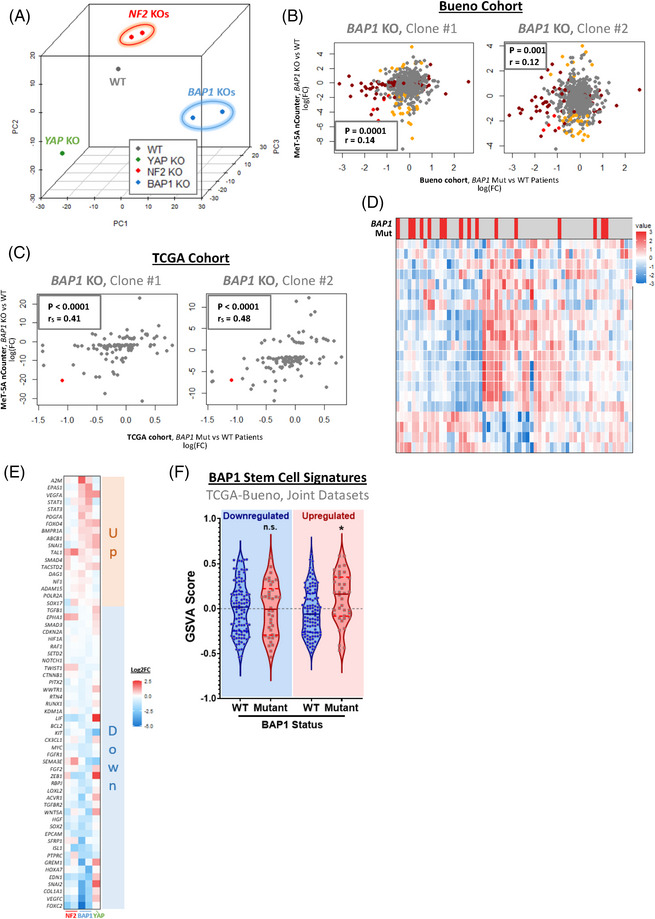
Gene dysregulation associated with pleural mesothelioma (PM) mutation status is preserved in in vitro models of BAP1 loss. (A) 3D principal component analysis (PCA) plot shows MeT‐5A genotypes coloured according to knockout (KO) target and condition, with dimensionality reduction applied to the expression of 770 genes included in the NanoString PanCancer Progression nCounter panel. This highlights that transcriptional profiles diverge according to genotype and are consistent between paired KO clones. (B) Scatter plots show a correlation between fold changes in genes when comparing BAP1 mutant versus wild‐type (WT) patients (x‐axis) and when comparing BAP1 KO MeT‐5A cells to WT (y‐axis). Genes are coloured according to significance, with genes significantly differentially expressed in both clones of cell‐line models (orange), patients (dark red), and both (red) highlighted. There is a weak, though statistically significant positive correlation in fold‐changes calculated in patients compared to the in vitro model. (C) Scatter plots, as in (B), show correlation statistics of fold‐changes, comparing all genes quantified in the NanoString panel applied to MeT‐5A cell lines. In order to reduce noise, only genes identified to be reliably dysregulated in both KO clones in vitro are included, with genes significantly dysregulated in patient cohorts highlighted in red. A stronger positive correlation is observed when this additional complexity is removed. (D) Heat‐map shows the quantification of the expression of genes identified as significantly differentially expressed (|log(FC)| > 2.5 in both KO clones) in BAP1 KO relative to WT MeT‐5A cells, as determined by NanoString, in patients from both cohorts grouped. The majority of those genes included are downregulated on BAP1 KO in the in vitro model (with the exception of IL18 and NR4A3), which is mirrored in BAP1 mutant patients. Expression is shown in arbitrary units, with log(CPM) centred and scaled across each gene. As BAP1 mutations are associated with a slight increase in YAP/TAZ activity, all patients with additional Hippo kinase module inactivating mutations (NF2, LATS1/2, SAV1) were excluded from the analysis. (E) Heatmap shows dysregulation of genes comprising stem cell maintenance, differentiation, and proliferation GO gene sets across MeT‐5A KO genotypes. Expression is shown as log2FC relative to WT MeT‐5A, with genes ordered according to the degree of dysregulation in BAP1 KO cells. Up‐regulated (red) and down‐regulated (blue) gene sets were inferred according to the relative expression of BAP1 loss. (F) Violin plot shows collective dysregulation of stem‐cell associated gene sets downregulated (left) and upregulated (right) in BAP1 KO MeT‐5A cells within PM patients, as quantified via gene set variation analysis (GSVA). Patients combined from The Cancer Genome Atlas (TCGA) and Bueno datasets were grouped according to BAP1 mutation status, with tumours exhibiting BAP1 loss‐of‐function mutations (red) displaying a marked upregulation of the upregulated gene set as compared to WT BAP1 samples (blue). As in (d) patients with Hippo kinase module inactivating mutations were excluded from the analysis. Correlation coefficients and p‐values in (B) and (C) were determined by Pearson and Spearman methods respectively, while p‐values in (F) were calculated via Dunnet's multiple comparison tests
To compare the impact of the loss of tumour suppressors in vitro relative to in‐patient samples, we performed differential expression analysis to calculate fold‐changes in gene expression in BAP1 WT vs BAP1 mutant patients. These were then compared to fold‐changes calculated in WT vs BAP1 KO MeT‐5A cells, with a significant, though relatively weak (mean r = 0.13), positive correlation in fold‐changes observed in both BAP1 KO clones (Figure 4B). This highlights that the broad dysregulation of transcription affected by BAP1 deletion in patients is generally preserved in BAP1 KO MeT‐5A cells. In order to limit the noise inherent to transcriptomic analysis, with the majority of non‐differentially expressed genes potentially masking the correlation of genes truly disrupted upon BAP1 loss, correlations were recalculated with genes limited to just those identified to be dysregulated within the cell‐line model. This reveals a relatively strong (r = 0.41 and 0.48 in KO clones #1 and #2, respectively) significant correlation between patient and cell‐line dysregulation (Figure 4C). Similarly, the clustering of patients according to an expression of genes identified to be highly dysregulated within BAP1 KO cell lines shows a clear grouping of BAP1 mutant patients (Figure S4B), which is even more pronounced when patients with Hippo kinase cascade inactivating mutations are excluded from analysis (Figure 4D). Additionally, splitting genes dysregulated in vitro into BAP1 KO up‐ or down‐regulated gene sets and quantifying collective expression in patients categorised according to NF2 and BAP1 mutation status reveals that gene sets are selectively up‐or down‐regulated in BAP1 mutant patients (Figure S4C), mirroring regulation in MeT‐5A cells. Taken together, these findings further validate that the impact of BAP1 loss on widespread gene regulation in patients is retained within monocultured cells. This is evident despite the lack of an immune component or stroma, including tumour‐associated fibroblasts, suggesting that perturbation of expression is tumour cell‐intrinsic.
Animal models have shown that BAP1 is required for the switch from pluripotent to differentiated cells across a range of developmental lineages, including the mesoderm. 74 In the context of cancers, such as uveal melanoma, 75 , 76 , 77 BAP1 loss has been proposed as a driver of characteristics associated with cancer stem cells. 78 To assess the role BAP1 depletion plays in mesothelial cells, we assessed the expression of genes associated with stem cell maintenance, differentiation, and proliferation, as determined via a search of gene ontology terms 79 , 80 across our NanoString data, within the generated BAP1 KO MeT‐5A cells (Figure 3A). This revealed a selection of genes both weakly and strongly dysregulated in BAP1 KO relative to WT cells (Figure 4E). Interestingly, this dysregulation appears to differentially affect homologues and functionally related genes: for example, SNAI1, which encodes the SNAIL transcription factor, a major effector of the stem‐like phenotype in multiple cancer types, 81 , 82 is found to be upregulated, while its homologue with shared functionality within cancer, SNAI2 83 (encoding SLUG) is downregulated on BAP1 loss (Figure 4E). While both act in combination to promote EMT and metastasis, 84 , 85 there is evidence that in certain contexts, each has a distinct role in driving cancer progression. 86 , 87 In patients with PM, expression of SNAI1 and SNAI2 is not correlated, as SNAI1 is more frequently highly expressed within PM effusions. 88 Similarly, EPAS1, which encodes the HIF‐2α transcription factor, a major regulator of the cellular hypoxic response, is upregulated on BAP1 loss, while HIF1A, encoding HIF‐1α is downregulated. While both HIF transcription factors are known to contribute to tumorigenicity and cancer progression by orchestrating the tumour hypoxic response, 89 HIF‐1α is generally considered a driver of metabolic reprogramming, 90 with HIF‐2α coordinating a broader set of genes, including a variety of stem cell factors. 89 This regulatory role of HIF‐2α is evidenced by the decrease in stem cell markers NANOG and SOX2 and stem cell proliferation on EPAS1 silencing in human embryonic stem cells. 91 Interestingly, expression of YAP/TAZ signature genes, which are also known to potentiate EMT and cancer stem cell maintenance, 26 , 53 , 92 are similarly dysregulated. Within MeT‐5A cells, eight of 22 genes comprising the signature are upregulated and seven downregulated in BAP1 KO relative to WT cells (Figure S4D), with the remainder of target genes inconsistently dysregulated between BAP1 KO clones.
Beyond this dysregulation, a variety of functionally similar genes associated with cancer progression were identified as upregulated in BAP1 KO MeT‐5A cells. This includes STAT1 and STAT3, which encode two STAT family transcription factors, both of which are involved in the maintenance of healthy and cancer stem cells, 93 , 94 while STAT3 additionally has been proposed as a target for treatment in PM. 95 PDGFA and VEGFA, which both code for mitogenic growth factors known to stimulate mesenchymal proliferation associated with tumorigenesis are additionally upregulated in BAP1 KO cells. 96 To assess whether the dysregulation of stem cell‐associated genes observed in MeT‐5A cells is reflected in PM patients, we quantified and analysed the collective expression of both up‐ and down‐regulated gene sets in the joint TCGA and Bueno datasets. This revealed that the upregulated gene set is preserved within PM patients, with a collective expression of these genes also upregulated in patients with BAP1 mutations (Figure 4F). These observations suggest that this upregulated module of genes associated with stem cell maintenance may facilitate PM initiation by driving a stem‐like phenotype, as observed on BAP1 loss in uveal melanoma cancer cells and patients. 78 , 97
2.5. Exploring the transcriptional impact of NF2 and BAP1 loss in mesothelial cells
In order to infer the putative mechanisms of action by which NF2 and BAP1 loss regulates the observed transcriptional effect (Figure 4A), we assessed the top dysregulated genes within MeT‐5A KO cells, as determined by NanoString nCounter analysis. Within NF2 KOs, this includes a variety of genes that, to our knowledge have not previously been directly associated with NF2, the Hippo pathway, or PM, including KISS1. KISS1 is highly upregulated in MeT‐5A cells upon loss of NF2 and encodes the Kisspeptin family of G‐protein coupled receptor ligands. KISS1 has been reported to act as a context‐dependent inhibitor of metastasis or tumour promoter in a range of cancers, 98 , 99 , 100 as well as a disruptor of proliferation and invasion in PM cell lines. Beyond upregulated targets, NF2 KO was identified to decrease the expression of CXCL8, which encodes interleukin‐8.
Expression of potential transcriptional regulated targets upon NF2 loss was validated via qPCR, which showed a close corroboration of dysregulation as those observed via NanoString (Figure S5A). Interestingly, there is a clear inverse correlation between directionality and the extent of dysregulation when comparing NF2 KO MeT‐5A cells to YAP KO (Figure S5B), with genes upregulated on NF2 loss downregulated when YAP is lost and vice versa. This antagonistic relationship between NF2 and YAP, alongside NF2's established role as an upstream regulator of the Hippo pathway, 21 suggests that YAP/TAZ transcriptional co‐activators and the TEAD family of transcription factors 30 , 53 , 101 are likely effectors of NF2 KO transcriptional dysregulation in our in vitro based MeT‐5A cellular PM model. To investigate this, we utilised the Cistrome Data Browser 102 to visualise the binding of TEAD members to genes dysregulated on NF2 loss in cells across a wide range of widely‐used cancer cell lines, including MSTO‐211H, a cell line derived from PM tissue. This revealed peaks for TEAD1 and TEAD4 on both KISS1 and CXCL8 promotors (Figure S5C), suggesting that within cancer cells, TEAD transcription factors facilitate the expression of genes both up‐ and down‐regulated on NF2 loss. To facilitate the assessment of a broader selection of genes, we utilised Lisa (epigenetic Landscape In Silico deletion Analysis). 103 Lisa is a tool developed to infer transcriptional regulators in silico likely to coordinate the regulation of gene sets, these analyses in Lisa combine publicly available ChIP‐seq data for specific regulators with markers of chromatin accessibility. 103 Gene sets comprising potential transcriptional targets downstream of NF2 were generated by grouping together all genes identified as dysregulated on NF2 KO (|log2FC| > 1). Both upregulated and downregulated genes were included, as it appears that transcription factors mediating the transcriptional dysregulation of NF2 loss do not coordinate unidirectional changes (Figure S5C). This analysis reveals the likely involvement of a number of transcription factors within the downstream transcriptional components of the Hippo pathway, as well as those known to associate with this component (Table 1). Most notably, this includes YAP and TEAD1, with YAP's regulatory role most significantly determined in the mesothelioma cell line, NCI‐H2052 (top two p‐values, each < .0001) and TEAD1 also yielding a highly significant association (p < .0001) in the MSTO mesothelioma line. Additional transcriptional regulators found significantly associated with gene‐set regulation include STAT3, which is known to bind to and regulate YAP/TAZ, 104 , 105 and FOSL2 and JUN, two components of the AP‐1 transcriptional complex, which co‐localises with YAP, TAZ, and TEAD at transcriptional enhancer sites. 53 , 106 Importantly, all three of these regulators recruit YAP/TAZ, orchestrating the transcription of YAP/TAZ target genes in transformed cells. 107 Collectively, these findings further reinforce NF2 loss in the mesothelial context as a driver of YAP/TAZ‐TEAD mediated transcriptional perturbation.
TABLE 1.
Lisa's analysis of NF2 knockout (KO) differentially expressed genes
| Transcription factor | Rank | 1st Sample p‐value | 2nd Sample p‐value | 3rd Sample p‐value | 4th Sample p‐value | 5th Sample p‐value |
|---|---|---|---|---|---|---|
| STAT3 | 1 | 5.22E‐16 | 1.48E‐05 | 2.11E‐05 | 1.05E‐03 | 5.80E‐03 |
| SRPK2 | 2 | 5.15E‐08 | 5.59E‐01 | |||
| RELA | 3 | 5.67E‐08 | 3.65E‐07 | 1.54E‐06 | 5.87E‐06 | 7.36E‐06 |
| NR3C1 | 4 | 8.59E‐07 | 1.74E‐05 | 6.06E‐05 | 9.94E‐05 | 1.39E‐04 |
| YAP1 | 5 | 9.23E‐07 | 2.46E‐06 | 5.44E‐04 | 2.34E‐03 | 8.45E‐03 |
| FOSL2 | 6 | 1.84E‐06 | 5.92E‐05 | 1.36E‐04 | 1.87E‐04 | 2.47E‐04 |
| SMAD3 | 7 | 3.02E‐06 | 1.10E‐04 | 1.57E‐04 | 2.04E‐04 | 2.81E‐04 |
| JUN | 8 | 3.94E‐06 | 2.42E‐04 | 2.46E‐04 | 1.16E‐03 | 2.47E‐03 |
| TEAD1 | 9 | 5.21E‐06 | 2.19E‐05 | 3.08E‐05 | 8.05E‐05 | 1.62E‐01 |
| TAL1 | 10 | 1.64E‐05 | 1.37E‐04 | 1.25E‐01 | 1.85E‐01 | 2.23E‐01 |
Results from Lisa's analysis, 103 show the top 10 most significant p‐values across corresponding transcriptional regulators (including both transcription factors and chromatin regulators). The top five most significant samples, which consist of cell lines in which ChIP‐seq data corresponding to that regulator have been deposited in the Cistrome database, are included for each regulator.
2.6. NF2 loss in mesothelial cells cause YAP/TAZ hyperactivation in response to stress
While in vitro BAP1 loss recapitulates the transcriptional disruption evident in BAP1 mutant patient populations, the same phenomenon is not readily observed when compared with NF2 KO cells (Figure S4E). This recapitulates that under steady‐state growth, with cells cultured in serum at relatively low density on plastic, NF2 does not appear to be highly active in WT MeT‐5A cells, with NF2 loss having minimal impact on YAP/TAZ phosphorylation and nuclear localisation (Figure 3C and Figure S3E), while NF2 KO cells display a transcriptional profile more similar to WT MeT‐5A cells relative to other genotypes (Figure 4A and Figure S4A). In order to assess a relevant metric of gene dysregulation on NF2 loss, expression of YAP/TAZ signature genes, which are consistently upregulated in patients with Hippo kinase module inactivating mutations (Figure 2B), was quantified in NF2 KO cells. To further contextualise this given NF2's confirmed role as a regulator of Hippo signalling in response to stresses associated with the tumour microenvironment, including serum starvation (Figure 3C) and contact inhibition (Figure 3E), expression was assessed at increasing cell densities. This analysis reveals that, as NF2 KO cells become more confluent, they exhibit a consistent general upregulation of signature genes 41 relative to WT MeT‐5A cells, with a mean increase in expression of the entire signature of 85% and 115% across two NF2 KO clones compared to WT cells (Figure 5A). These data closely resemble those from NF2 mutant patients (Figure 2B), indicating that Hippo kinase cascade activation in response to contact inhibition is impaired on NF2 loss in vitro.
FIGURE 5.
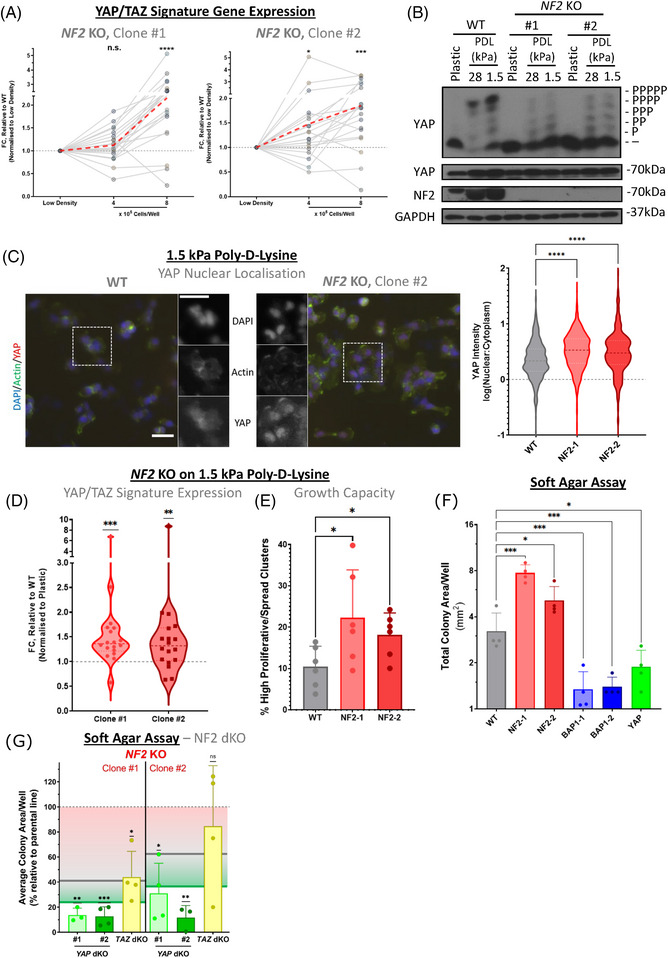
Culturing in conditions representative of the tumour microenvironment results in selective upregulation of YAP activity in cells deficient in NF2. (A) Line‐plot shows trends in expression of YAP/TAZ signature genes, as determined via quantitative polymerase chain reaction (qPCR), at increasing cell confluences. Each point represents the logFC of a single gene, relative to wild‐type (WT) expression at the same confluency and normalised to low density. The red‐dashed line shows the mean logFC calculated across the 22‐gene signature, with a clear increase in expression observed in both NF2 knockout (KO) clones #1 (left) and #2 (right). logFCs were calculated by taking mean values across biological replicates (n = 5). (B) Phos‐tag‐based western blot shows the phosphorylation status of YAP in response to culturing MeT‐5A cells on a soft substrate. WT and NF2 KO cells were seeded on poly‐D‐lysine coated plastic, 28 kPa, and 1.5 kPa ESS plates and left overnight before harvesting. WT MeT‐5A cells exhibit a clear induction of YAP phosphorylation (upshift) on both 28 and 1.5 kPa substrates, while the same response in NF2 deficient cells is markedly reduced. (C) Representative images (left), obtained via EVOS FL Auto 2 Imaging System, show dysregulation of YAP/TAZ activity, as determined by nuclear localisation, when culturing MeT‐5A cells on 1.5 kPa poly‐D‐lysine. Scale bar = 50 μm. A violin plot (right) shows quantification of YAP nuclear localisation, which is decreased on soft substrate relative to plastic (Figure S3E), with activity less diminished in NF2 KO cells (>500 cells shown per genotype across four biological replicates). (D) Violin plot (left) shows the relative expression of YAP/TAZ signature genes, as determined via qPCR, in NF2 KO cells, as compared to WT MeT‐5A (points represent mean expression per gene across three biological replicates). An increase in expression is observed, similar to the upregulation of YAP activity in the same condition (B). (E) Bar‐plot shows the percentage of highly proliferative/spread cell clusters on 1.5 kPa poly‐D‐lysine ESS plates. Clusters were determined as highly proliferative/spread if they exhibited areas > 10 times the median at time‐point 0 (n = 6). (F) Bar plots show quantification of the total area of colonies of MeT‐5A genotypes cultured in soft agar, with statistical significance relative to WT computed (n = 4). (G) Bar plots, as in (E), show the total area of colonies of dKO cells on an NF2 KO background, cultured in soft agar. The area is normalised to corresponding parental NF2 KO cells, with significance computed in comparison to parental genotype (n = 3–4). Annotations are included to highlight the mean relative total area of parental NF2 KO cells (red line), WT MeT‐5A cells (grey line), and YAP KO cells (green line). p‐Values in (A), (C) and (D) were determined via a one‐sample Wilcoxon signed rank test, in (E) via Kruskal‐Wallis test, while p‐values in (F) were calculated via Dunnet's multiple comparison tests, with data log‐transformed to adjust for the skewness of soft agar assay results. Finally, p‐values for (G) were obtained using one sample t‐test, against a hypothetical mean of 100%. n.s. = Not significant, * p < .05, *** p < .001 and **** p < .0001 relative to low density (A, E, F), WT (C, D), or parental genotype (G)
The stiffness of cell culture plastics is well in excess of 100 kPa, approximating 106 kPa, 108 , 109 which is much stiffer than tissue. To more faithfully in vitro recapitulate the environment at the onset of PM, cells were next cultured on poly‐D‐lysine with an elasticity index of 1.5 kPa, which matches the mechanical properties of pleural tissue. 110 , 111 Phos‐tag based western‐blots revealed that YAP phosphorylation is increased when cells are cultured on soft poly‐D‐lysine, with a clear shift particularly observed when comparing WT MeT‐5A cells cultured on 1.5 and 28 kPa substrate, relative to cells cultured on plastic (Figure 5B). However, this effect was considerably less pronounced in cells lacking NF2, with only a minor increase in YAP phosphorylation (Figure 5B). Similarly, while YAP nuclear localisation is also generally decreased on soft substrate, NF2 KO is less affected than WT MeT‐5A cells (Figure 5C and Figure S3E). Our quantification highlights a significant increase in nuclear YAP signal in NF2 KO relative to WT cells (Figure 5C), similar to when culturing at high cell density (Figure 3E). A concomitant relative upregulation of the YAP/TAZ signature is also observed in NF2 KO cells (Figure 5D), further validating the activation of YAP/TAZ reflected in the dysregulation of the signature in NF2 mutant patients (Figure 2B). Beyond this, after 48 h growth on a soft 1.5 kPa substrate, ∼20% of NF2 KO cell clusters reached >10 times the median size at time point 0, as compared to 10% of WT cells (Figure 5E). This higher percentage of large colonies upon NF2 loss indicates either a greater proliferative capacity on a soft substrate or a resistance to the expected decrease in the cytoplasmic area observed when cells are cultured on surfaces with low elastic moduli. 112 , 113 Moreover, NF2 KO cells exhibit greater anchorage‐independent growth, as determined by soft agar assays (Figure 5F), which combined indicate a greater potential for tumorigenicity on NF2 loss. For these functional assays, cells are cultured in 0.35% soft agar, which is reported as having Young's modulus around 30–50 kPa, 114 , 115 a stiffness where we observe a clear increase in active YAP in NF2 KO compared to WT cells (Figure 5B). In order to establish if the phenotypes observed in NF2 KO cells are mediated by, and dependent on, YAP or TAZ, we established NF2‐YAP double knockout (dKO) and NF2‐TAZ dKO MeT‐5A cells (Figure S3J). The capacity to readily grow in soft agar is markedly decreased in YAP KO, as well as in NF2‐YAP dKO) cells (Figure 5F,G). While NF2‐YAP dKO is sufficient to inhibit colony growth in soft agar, NF2‐TAZ dKO has a modest impact, with total area coverage roughly equivalent to WT MeT‐5A cells (Figure 5G). These observations suggest that NF2 KO MeT‐5A cells’ capacity for anchorage‐independent growth is primarily driven by hyperactive YAP, with cells lacking NF2 exhibiting dependence on YAP for this trait. This indicates transcriptional addiction, a common feature of tumour cells. 116 , 117 Despite YAP's primary role in facilitating anchorage‐independent growth, we anticipate that targeting both YAP and TAZ might be more effective and may be necessary in a clinical setting. 53 Interestingly, BAP1 loss decreases anchorage‐independent growth (Figure 5F), with decreased total colony coverage on soft agar, even as compared to YAP KO MeT‐5A cells. This might reveal that cellular BAP1 loss is only advantageous in settings where the cancer cells are under additional stresses, 118 such as upon nutrient limitations in the cancer niche and the onset of the Warburg effect. 118
Although transcriptional dysregulation via YAP/TAZ is observed on NF2 KO in cells cultured at low cell density on plastic (Figure S5A–C), the addition of stresses, some of which were previously associated with NF2 activation, drives a greater degree of relative YAP/TAZ activation and subsequent transcriptional effects. These findings, therefore, highlight the role NF2 plays as a facilitator of the Hippo pathway's mechanosensory component, 53 , 119 acting to inhibit YAP/TAZ activity in response to serum starvation (Figure 3C,D), cell density (Figure 3E), and decreasing substrate stiffness (Figure 5B–D), with NF2 loss rendering cells less sensitive to growth on soft substrate. Within the scope of PM, the increase in context‐dependent YAP/TAZ activation on NF2 loss likely translates to a decreased sensitivity to and enhanced proliferative potential within, a tumour niche, with a concurrently increased capacity for anchorage‐independent growth.
3. DISCUSSION
The Hippo signalling pathway is closely tied to a range of oncogenic pathways across a wide variety of cancer types 51 and is found frequently, mutationally perturbed in PM relative to other cancers. 58 Although incidence rates of PM are relatively low and there is, therefore, a limited selection of well‐annotated, high‐throughput clinical datasets available to analyse, we here show with confidence, via pooling the largest PM datasets available, 12 , 13 that Hippo signalling disruption is closely associated with worse prognosis in PM patients. The inclusion of the larger Bueno cohort 13 as part of these analyses facilitates enhancing the statistical power of included tests, increasing confidence while expanding on recent work which assessed the clinical impact of Hippo dysregulation within the numerically smaller TCGA cohort. 40 , 41 , 120 These findings are also consistent with a recent study showing that NF2 deletion tends to be positively selected for during tumour evolution and might be a late‐stage event, 121 all suggesting that activation of the Hippo transcriptional module may be a limiting factor for PM progression and metastasis. Our preclinical modelling of NF2 loss supports this observation, with in vitro NF2 knockout in mesothelial cells facilitating resistance to YAP/TAZ inactivation in response to serum starvation, contact inhibition, and culturing on soft substrate (Figures 3C–E and 5A–C). This is of relevance in a clinical setting as nutrient deprivation is a recurring feature of solid tumours as a result of poor vascularisation and high energy demand, 122 while tumour tissues in PM are typified by hypoxia, suggesting limited vascularisation and hence nutrients. 123 , 124 Loss of contact inhibition is a critical and primary hallmark of cancer cells, 125 while tumour growth, infiltration and metastasis are all limited by contact inhibition of proliferation. 126 , 127 Additionally, the establishment of the tumour microenvironment and metastatic niche over time is associated with extracellular matrix remodelling and a concurrent change in stiffness. 128 , 129 Our data point to NF2 being a cardinal sensor of mechanotransductive processes in mesothelial cells, and that the loss of NF2 therefore might play an integral part in mesothelioma during disease progress. This possibility is reinforced by the YAP addiction observed in NF2 KO cells when assessing anchorage‐independent growth in vitro (Figure 5G). These findings collectively point to a function of Hippo pathway kinase inactivation to promote tumour growth under conditions of stress commonly experienced within tumour tissue, with YAP and TAZ known to generally enhance cell growth and survival under nutrient‐limited conditions, 63 , 130 resistance to therapeutics 53 and motility in response to cytoskeletal tension. 131
Considering the importance of perturbed Hippo signalling in driving PM, therapeutic interventions that inhibit the downstream effectors of the pathway are likely to be instrumental in future treatment regimens. 53 Previous studies have revealed potential in inhibiting the individual protein products of various YAP/TAZ signature genes in diverse cancer types, 132 , 133 an example of such is bemcentinib, an inhibitor of AXL whose expression is directly mediated by the Hippo transcriptional module, which has been combined with pembrolizumab in recent clinical trials in PM. 134 However, as the Hippo pathway regulates the expression of a wide variety of genes associated with oncogenic pathways, a likely more productive approach may be to directly target the transcriptional effectors (YAP, TAZ and the TEAD transcription factors). 53 Efforts to target this transcriptional module identified the benzoporphyrin derivative verteporfin as a disruptor of the YAP‐TEAD complex with anti‐cancer potential. 135 , 136 However the drug is effective at high concentrations and appear to lack specificity 137 limiting its clinical utility in cancers. In recent years, there has been a major push to identify viable drugs that directly target the transcriptional module of the Hippo pathway and can be used in the clinic, either by disrupting the interaction between YAP/TAZ and TEAD, 138 , 139 or by targeting TEAD stability. 140 While the various novel therapeutics identified in this fashion have yet to be fully validated in a clinical setting, early findings are promising, 53 and with some already in early‐stage clinical trials. The wide interest in targeting YAP/TAZ‐TEAD in cancers, 53 and the range of currently developed compounds targeting this transcriptional modality highlight the need for relevant preclinical models. The ability to evaluate these compounds effectively in order to achieve their full therapeutic potential in the near future includes the further development and assessment of their mechanism of action. These translational efforts rely on the development of scalable and clinically relevant model systems, such as the one developed here.
Although we present evidence that BAP1 loss in certain instances may be involved in partially activating the Hippo transcriptional module (Figures S2B and S4D), the mechanism by which this is effected in vitro in mesothelial cells is unclear, though not mediated via degradation of LATS proteins as previously described within the pancreatic cancer context 48 (Figure S3G,H). A core role for BAP1 is to act as a deubiquitinase as part of the PR‐DUB complex 20 and BAP1 has been described as a regulator of a variety of epigenetic markers. 118 This, therefore, suggests that BAP1's involvement in regulating the Hippo pathway is likely indirect, possibly as a result of the transcriptional changes effected by epigenomic alterations on BAP1 loss; this is all the more relevant as the primary metric with which YAP/TAZ activity is quantified clinically in this study is transcriptional in nature (Figures 1, 2 and 5A–D). Additionally, BAP1's ability to modulate expression broadly via Polycomb repressive complexes 20 may explain the faithful recapitulation of patient dysregulation in vitro, in the absence of non‐tumour cell interactions. This observation is particularly relevant as these epigenetic writers tend to exhibit context sensitivity. 141
Interestingly, we find that BAP1 loss in vitro leads to a reduction in invasive potential. This finding is consistent with previous studies, which show that, while ectopic BAP1 expression may lead to enhanced anchorage‐independent growth in renal cancer cells, 142 BAP1 depletion via KO or KD inhibits proliferation, colony formation, and anchorage‐independence in cells from multiple cancer types. 67 , 78 , 143 Mechanistically, BAP1 loss has been proposed to act by driving dedifferentiation and stem‐like characteristics associated with early cancer rather than tumorigenicity. 78 Within the mesothelial context, we find that BAP1 loss leads to the dysregulation of a range of genes involved in stem cell specification, with a module of these genes consistently upregulated within both the generated isogenic cell‐line model and tumour tissue (Figure 4E,F). Additional work is required to determine how this dysregulation drives dedifferentiation within mesothelial cells and how this may affect PM progression. However, the decrease in invasiveness observed on BAP1 KO in vitro may in part explain the observation in other studies that BAP1 loss predicts improved relative prognosis in PM cases. 45 , 144 Although the same effect is not apparent in the larger, combined cohorts (Figure S1E), this may be limited by inferring BAP1 loss by mutation status alone. Taken together, this highlights BAP1's complex role as a tumour suppressor, 118 whereby its high frequency of loss strongly implicates its involvement in driving PM onset, while in the mesothelial context inhibits anchorage‐independent growth (Figure 5F) and sensitises tumour cells to therapeutic intervention. 45
In conclusion, this work serves to reinforce the role that Hippo pathway dysregulation plays in driving PM progression, which is increasingly relevant as inhibitors of the transcriptional module of the pathway are likely to become clinically available. 53 We also highlight the potential in stratifying PM patients according to biomarkers of YAP/TAZ activity rather than driver mutation status, while developing, establishing, and validating a robust isogenic preclinical model. This isogenic cellular PM model is a unique system and provides a platform to allow for further discoveries and in‐depth insights into harnessing the therapeutic potential of loss of BAP1 and NF2 in mesothelioma, as well as provide a detailed understanding of BAP1 and the Hippo pathway's fundamental biological functions. Currently, the cellular model system has not yet been analysed in vivo. Noteworthy, a range of gene‐sets, including cytokines as well as ECM components, are widely dysregulated (Figure 4A). Xenografts using the developed model system may shed further light on the interplay between PM driver mutations and the onset/progression of PM, including revealing critical insights into the interplay between the cancer cells and components of the stroma including the immune system. Consequently, we envision that the isogenic preclinical cellular model developed and characterised here is likely to be impactful in the context of developing future stratified approaches in PM.
4. MATERIALS AND METHODS
4.1. Culture maintenance
MeT‐5A mesothelial cells were cultured in RPMI‐1640 (31870‐025; Gibco) supplemented with 10 mM HEPES Buffer solution (15630‐049; Gibco), 2 mM L‐Glutamine (25030‐024; Gibco), 10% foetal bovine serum (FBS) (10500‐064; Gibco), with 100 units/ml of penicillin and 100 μg/ml of streptomycin (15140‐122; Gibco). HEK293T cells were kindly gifted by Professor Kun‐Liang Guan (UCSD) and cultured in high glucose Dulbecco's Modified Eagle Medium (21969‐035; Gibco) supplemented with 2 mM L‐Glutamine (25030‐024; Gibco), 10% FBS (10500‐064; Gibco). All cells were cultured in the presence of 100 units/ml of penicillin and 100 μg/ml of streptomycin (15140‐122; Gibco) and incubated at 37°C with 20% O2. For modelling soft tissue substrate, cells were cultured on 100 μg/ml poly‐D‐lysine (P7886; Sigma‐Aldrich), chosen due to its inertness with regards to the Hippo pathway. 64 Poly‐D‐lysine was coated on 1.5 and 28 kPa Ibidi ESS plates (81291; Ibidi) and cells were cultured overnight before imaging and protein‐lysate/RNA harvesting. Cells were subject to routine checks for mycoplasma using a MycoAlert Mycoplasma Detection Kit (LT07‐318; Lonza).
4.2. Gene knockout and re‐expression
CRISPR‐Cas9 mediated KO was carried out using the following guide RNA (gRNA): 5′‐GTCCATGGTGACGATCCTCA‐3′ for NF2 #1; 5′‐GAGTTCAATTGCGAGGTAAC‐3′ for NF2 #2; 5′‐AAACGGACCGGCGCTCTTCGATCC‐3′ for BAP1 #1; 5′‐CACCGGATCGAAGAGCGCCGGTCC‐3′ for BAP1 #2; 5′‐CACCGCATCAGATCGTGCACGTCCG‐3′ for YAP1 (YAP); and 5′‐ CACCGTGTCTAGGTCCTGCGTGACG‐3′ for WWTR1 (TAZ). gRNA sequences were ligated to the pSpCas9(BB)‐2A‐Puro CRISPR construct (PX459; Addgene, #48139) as described in Rausch et al. 62 MeT‐5A cells were electroporated for 20 ms at 1400 V, pulsed once using the Neon electroporation transfection system (MPK10096; Invitrogen). After overnight recovery, the cells were selected for 48 h with puromycin to enrich for cells that had taken up the plasmid. Cells were then taken out of selection for 24 h before fluorescence‐activated cell sorting (FACS) into single wells of a 96‐well plate. Clonal expansion and replicate plating were carried out after FACS‐based single‐cell sorting. KO validation was performed by western blotting. NF2 re‐expression in NF2 KO MeT‐5A cells was achieved via lentiviral transduction using a pBABE puro system. Lentivirus was produced in HEK293T cells and harvested at 48 and 72 h post‐transfection, where the supernatant is filtered with low binding 0.45 μm SFCA filters (Corning, 431220), and added to polybrene supplemented MeT‐5A cells directly or stored at ‐80°C for later use. Selection after transduction/transfection was achieved with puromycin treatment.
4.3. Western blotting
Cell lysates were harvested and analysed using home‐cast sodium dodecyl sulfate (SDS) polyacrylamide gels to perform western blots as in Hansen et al. 63 PageRuler prestained Protein Ladder (26616; Thermo Scientific) was used to provide a scale for protein size and separate proteins transferred from gels to Immobilon‐P PVDF membranes (IPVH00010; Millipore). After transfer, the membrane was blocked in 5% milk in tris‐buffered saline‐T (TBS‐T) (150 mM NaCl, 20 mM Tris, 0.1% TWEEN 20), washed with TBS‐T, and incubated with primary antibodies diluted in TBS‐T supplemented with 2% bovine serum albumin (Fisher Bioreagents). Membranes were then washed and incubated for 1 h with secondary antibodies specific to the primary antibody isotype, conjugated to Horseradish Peroxidase (HRP; P044801‐2 and P044701‐2, Agilent). Finally, Immobilon Western Chemiluminescent HRP Substrate (WBKLS0500; Millipore) was used to produce a visible signal from HRP, which was subsequently developed using X‐ray film (MOL7016; SLS). Phos‐tag gels were generated by adding Phos‐tag reagent (304‐93521; Alpha Laboratories) and 10 mM MnCl2 added to each SDS polyacrylamide gel. Primary antibodies used were as follows: NF2 (D1D8; CST), BAP1 (13271S; CST), YAP (ab52771; Abcam), phospho‐YAP (Ser127; 13008, CST), histone H2A (12349; CST), ubiquityl‐histone H2A (Lys119; 8240, CST), LATS1 (3477; CST), LATS2 (3477; CST), and GAPDH (SC‐47724; Santa Cruz) used as a loading control for samples.
4.4. Quantitative polymerase chain reaction and NanoString nCounter
Cells were seeded in a 6‐well plate and allowed to adhere overnight. RNA was harvested from plates and purified using the RNeasy Mini Kit (Qiagen) according to the manufacturer's specifications. Purified RNA was quantified via NanoDrop spectrophotometer and kept frozen at ‐80°C until needed. RNA was submitted to the University of Edinburgh's Host and Tumour Profiling Unit (HTPU) for nCounter analysis with PanCancer Progression and Immune Profiling panels used, conducted according to the manufacturer's specifications. Each panel consisted of 770 genes, with an overlap of 157 gene targets between panels; 719 and 486 genes were detected in any sample in the Progression and Immune Profiling panels, respectively. For quantitative polymerase chain reaction (qPCR), cDNA was generated using 100 ng of RNA as input with a High‐Capacity cDNA Reverse Transcription Kit (4368814; Applied Biosystems), with the reaction carried out according to all manufacturer's instructions. cDNA was frozen at ‐20°C until required for qPCR, with assays carried out using Brilliant III Ultra‐Fast SYBR Green QPCR Master Mix (600883; Agilent) and custom IDT primers, designed using templates deposited on PrimerBank 145 all according to manufacturer's directions. Assays were performed on a QuantStudio 5 Real‐Time PCR System, with data processed and analysed using R statistical software. Primer sequences were as follows: 5′‐GCTCGTTGAGTGAACGGCT‐3′ and 5′‐CATGAGCTAGTACAACATGAGGG‐3′ for AMOTL2; 5′‐AGTAGAGGAACTGGTCACTGG‐3′ and 5′‐TGTTTCTCGCTTTTCCACTGTT‐3′ for ANKRD1; 5′‐CCAAGGTGAGCTTTCCCTCG‐3′ and 5′‐CCTACTAGACCATAGGTCGTCGT‐3′ for ARHGEF17; 5′‐TAGAACAGCCCTTCAGAAAGTGA‐3′ and 5′‐CGGGGTTGTCTCGACTTAAAAA‐3′ for ASAP1; 5′‐GTGGGCAACCCAGGGAATATC‐3′ and 5′‐GTACTGTCCCGTGTCGGAAAG‐3′ for AXL; 5′‐CCCTGTGACGAGTCCAAGTG‐3′ and 5′‐GGTTCCGTAAATCCCGAAGGT‐3′ for CRIM1; 5′‐GAGGCAGAAGTACGGGGTTG‐3′ and 5′‐CAGGAATCACGGTTTCATGCT 3′ for DOCK5; 5′‐GGCGCTTCAGGCACTACAA‐3′ and 5′‐TTGATTGACGGGTTTGGGTTC‐3′ for F3; 5′‐GCTGGTGGACCTAGTACAATGG‐3′ and 5′‐CTTACGAGCCGGTCGAAGTTG‐3′ for FJX1; 5′‐AATGCCACTCGCCCTACAC‐3′ and 5′‐CGTTCTGGTGCAAGTAGCTCT‐3′ for FOXF2; 5′‐GAGAGCAGAAGACCGAAAGGA‐3′ and 5′‐CACAACACCACGTTATCGGG‐3′ for GADD45A; 5′‐AGAGCACAGATACCCAGAACT‐3′ and 5′‐GGTGATTCAGTGTGTCTTCCATT‐3′ for IGFBP3; 5′‐ACTTTTCCTGCCACGACTTATTC‐3′ and 5′‐GATGGCTGTTTTAACCCCTCA‐3′ for LATS2; 5′‐TAATTGGCACGGCGACTGTAG‐3′ and 5′‐GGAGATCAGCTTGTACGGCAG‐3′ for MYOF; 5′‐GCCTGGGAGCTTACGATTTTG‐3′ and 5′‐TAGTGCCCTGGTACTGGTCG‐3′ for NT5E; 5′‐CGCCCAAGCCCCTAATGAAG‐3′ and 5′‐TCCCTCCGTATGTGCATCAGA‐3′ for NUAK2; 5′‐ATGCCTTTTGGTCTGAAGCTC‐3′ and 5′‐CCCTGTGCTTTCCACCGAC‐3′ for PTPN14; 5′‐GGGGAACAGTTGAGTAAAACCA‐3′ and 5′‐ACAATTTTTCCATACGGTTGGCA‐3′ for RBMS3; and 5′‐CAGCACACTCGATATGGACCA‐3′ and 5′‐CCTCGGGCTCAGGATAGTCT‐3′ for TGFB2. All gene expression values were normalized to Hypoxanthine Phosphoribosyltransferase 1 (HPRT1) expression, with primer sequence 5′‐AGAATGTCTTGATTGTGGAAGA‐3′ and 5′‐ACCTTGACCATCTTTGGATTA‐3′.
4.5. Immunofluorescence
For steady‐state imaging, cells were seeded on 96‐well μClear plates (655090; Grenier). For mixed models, NF2 KO cells were stained with CellTracker Red CMTPX Dye (C34552; Invitrogen) for 30 min before seeding. Cells were fixed using 4% formaldehyde (28906; Thermo Scientific), rinsed with PBS and then permeabilized in IF buffer (2.5% FBS and 0.3% Triton X‐100 in PBS). Fixed, permeabilised cells were next incubated overnight with a specific YAP antibody 64 (ab52771; Abcam), before multiple PBS washes and further incubation with DAPI, phalloidin conjugated to Alexa Fluor 488 (A12379; Invitrogen) and Alexa Fluor 555/647‐conjugated goat anti‐rabbit secondary antibody, (A‐21428 and A27040; Invitrogen). Fluorescence was then imaged using the Operetta or Opera Phenix Plus high‐content imaging systems (PerkinElmer) using a 20x objective for 96‐well plates. Eight biological replicates were conducted with cells seeded at 7500 cells per well and imaged at a range of time‐points. 25 fields were imaged per well, with three wells per sample/condition to act as technical controls. At the time of imaging, wells contained between 300 and 9000 cells. Cell features were computed using the Columbus image data storage and analysis system (PerkinElmer) and statistical analysis was then carried out using R and plotted with GraphPad Prism. Variance across samples was adjusted by removing outlying biological replicates, with outliers specified as values greater than 2 median absolute deviations from the median of biological replicates. Ibidi ESS plates were imaged using the EVOS FL Auto 2 Imaging System (Invitrogen) at 10x magnification, with individual cells sampled from the total population to allow for identical numbers across four biological replicates for statistical testing.
4.6. Data acquisition and processing
TCGA‐MESO data were accessed and downloaded using the GDCquery function, while TCGA RPPA data were obtained using the GDAC Firehose repository (https://gdac.broadinstitute.org/). Raw data from the Bueno cohort were obtained using the pyEGA3 download client and aligned using the STAR aligner 146 with default settings. HTSeq 147 was used to generate count tables post alignment, with normalisation via trimmed mean of M‐values implemented via the edgeR package. 148 All data were then imported into the R environment for additional analyses, with pooled data obtained by combining both TCGA 12 and Bueno et al. 13 cohorts and normalising with the ComBat function within the SVA package. 149 Differential expression analysis was conducted on un‐normalised count data using DESeq2. 150
4.7. Stiffness and soft agar assays
The growth of cells on poly‐D‐lysine coated ESS plates was assessed by seeding 500 cells into cloning rings (8 mm × 8 mm; C1059‐1EA, SLS). Cells were fixed 24 or 72 hours post‐seeding (representing 0‐ and 48‐h time‐points, respectively) and stained with phalloidin conjugated to Alexa Fluor 488, in order to infer cell coverage. Plates were imaged at 10x magnification with the EVOS FL Auto 2 Imaging System, with cell cluster boundaries and corresponding cluster areas then computed using CellProfiler. For soft agar assays, MeT‐5A cells were split and resuspended in 0.35% agar dissolved in 2X strength MeT‐5A culture medium, prepared using powdered RPMI‐1640 (51800‐019; Gibco). Cell suspensions in 2 ml agar were then seeded onto 6‐well plates coated with 1% agar, with 5,000 cells per well. After one week of growth, cells were fixed and stained using a mixture of crystal violet (0.5%, 11435027; Thermo Scientific) and methanol. Bright‐field images of plates were then captured using the EVOS FL Auto 2 Imaging System at 10x magnification, with ImageJ used to quantify the total colony area/well. For both ESS plate and soft agar assays, image capture was automated, with 35%–50% of wells (300–500 fields/well total) imaged and stitched together.
CONFLICT OF INTEREST
The authors declare they have no conflict of interest.
Supporting information
Supplementary Figure 1| Broad mutation profiles are insufficient to define patient populations with distinct clinical outcomes.
a, PCA plots displayed as in figure 1d, show PCs along different axes to highlight clusters. b, PCA plots, depicted in 3D, show patients coloured according to mutation status. Broad transcriptional profiling is insufficient to distinguish between the populations of patients categorised by mutation of BAP1 and NF2 in the TCGA (left; n = 86) and Bueno et al (right; 98) cohorts. c, Barplot shows the percentage of patients classified by consensus subtypes as generated in Bueno et al 16, with patients split according to mutation status of BAP1 and NF2. n = 98. d, Barplot, as in (c), highlights the absence of significant association between mutation status and T‐staging across the merged TCGA and Bueno et al datasets (n = 163). e, Kaplan‐Meier curves show overall survival of patients split according to mutation status, with patients categorised as exhibiting either NF2 or BAP1 mutations, as well as mutations in both or neither. No significant difference in survival across the patient populations is observed. P values for (c) and (d) determined via Fisher's exact test, while P values for (e) were calculated via log‐rank test.
Supplementary Figure 2| Analysing TCGA and Bueno cohorts separately highlights consistency between datasets.
a, Violin plots show GSVA scores of YAP/TAZ signature gene expression as in figure 2a, with results split by dataset. A significant collective overexpression of this gene‐set is observed in patients with Hippo kinase cascade inactivating mutations in both TCGA (left; n = 86) and Bueno et al (right; n = 98) cohorts. b, Violin plots as in (a), show signature scores in patients split according to BAP1 mutation status, excluding patients harbouring Hippo pathway inactivating mutations. While there is an association between BAP1 mutation and expression of YAP/TAZ target genes, this is minor and only seen to be significant in the TCGA (left) and not Bueno et al (right) cohort. c, Kaplan‐Meier curves show overall survival of patients split according to YAP/TAZ signature thresholds. Patients classed as YAP/TAZ signature display a pronounced reduction in overall survival in both TCGA (top; HR = 4.4, n = 86, threshold at 74%) and Bueno et al (top; HR = 1.76, n = 211, threshold set at 52%) cohorts. d, Bar‐plots, as in figure 2e, show tumour stage in patients categorised as signature high/low. P values in (a) and (b) were determined by Mann‐Whitney U test, P values and hazard ratios for (c) were calculated via log‐rank test and Cox proportional hazard model respectively, while P values for (d) were calculated via Fisher's exact test. n.s. = Not significant, *P < 0.05, **P < 0.01, and ****P < 0.0001 relative to WT.
Supplementary Figure 3| Validation of NF2 KO mediated activation of YAP.
a, Phos‐tag based western blots (top), as in as in figure 3c, shows phosphorylation status of YAP (left) and TAZ (right) in response to serum starvation across a range of time‐points. Responses in WT MeT‐5A cells are compared to the second NF2 KO clone (#2), with a similar decreased sensitivity to starvation observed on NF2 loss in NF2 KO #1. A standard SDS‐gel based Western blot (bottom) is also shown with the same samples analysed, highlighting levels of phospho‐YAP (S127) and YAP (left), TAZ (right), together with GAPDH. b, Western blots showing NF2 expression levels in WT, NF2 KO, and re‐expression of NF2 in NF2 KO MeT‐5A clones. c‐d, Phos‐tag based western blots, as in (a), with NF2 re‐expressed in NF2 KO MeT‐5As. Response to serum starvation is restored upon NF2 re‐expression, with increased phosphorylation of YAP observed when cells are deprived of serum. This rescue of YAP regulation upon exogenous NF2 expression is observed in both NF2 KO clone #1 (c) and clone #2 (d). e, Violin plot showing levels of nuclear YAP, as determined by immunofluorescence based images of cell monocultures, such as those shown in figure 3e, normalised to levels of cytoplasmic YAP (n = 8). f, Confocal based image acquired on the Opera Phenix Plus. Representative maximum projection images showing difference in YAP nuclear localisation between WT and NF2 KO MeT‐5A cells. Cells were mixed before seeding, with NF2 KO cells stained with CellTracker Red. Individual channels (taken from region within red dashed box) highlight the relative increase in nuclear YAP in NF2 KO cells (diagonal arrows) relative to WT cells (arrowheads). Scale bar = 50 μm. g, Representative western blot (top) showing levels of LATS1/2 in MeT‐5A KO cells of different genotypes, with quantification of LATS2 (n = 3) below, with bars showing mean levels and error bars representing SD across replicates. LATS1 appears unchanged across all genotypes, while LATS2 is generally upregulated in both NF2 and BAP1 KO MeT‐5A. h, Bar‐plot shows qPCR quantification of LATS2 expression relative to WT cells across BAP1 KO and YAP KO MeT‐5A cells. Expression is shown as FC relative to WT MeT‐5A (n = 3). i‐j, Western blots show CRISPR‐mediated knockout of YAP in MeT‐5A cells (i), as well as YAP or TAZ double knockout (dKO) on NF2 KO background MeT‐5A cells (j).
Supplementary figure 4| The transcriptional effects of BAP1 loss in vitro consistently and robustly mirrors mutation in patients.
a, Heat‐map shows expression of 1,540 genes included in the combined NanoString PanCancer Progression and Immune Profiling nCounter panels across various KO MeT‐5A cell‐lines. Cells were analysed at steady‐state (‘+’) and after 3 hours of serum starvation (‘‐’). Dendrograms show the clustering of genotypes, highlighting similarities in transcriptional profiles observed in lines bearing the same KO. b, Heat‐map, as in figure 4d, shows the quantification of expression of genes found to be significantly differentially expressed in BAP1 KO relative to WT MeT‐5A cells in patients from both cohorts grouped. No patients were excluded in this analysis, with mutation status for BAP1 and Hippo‐associated genes annotated above. c, Violin plots show the GSVA scores in patients from both cohorts grouped, of genesets consisting of genes found to be dysregulated in BAP1 KO MeT‐5A cells. Genes found to be both up‐regulated (left) and down‐regulated (right) in vitro are similarly dysregulated in patients. d, Heatmap shows dysregulation of YAP/TAZ signature genes within BAP1 KO MeT‐5A cells. Expression is shown across two distinct clones as mean log2FC relative to WT cells across biological replicates (n = 4). e, Scatter‐plots as in figure 4c‐d show correlation between gene dysregulation in NF2 mutant patients from the Bueno et al cohort vs NF2 KO MeT‐5A cells. Noise has been reduced by limiting analysis to those genes found to be significantly dysregulated in vitro. While some significant correlation exists in one NF2 KO clone, this is not consistent, which suggests the same degree of conserved, broad transcriptomic dysregulation observed with BAP1 disruption is not reproduced on loss of NF2. P values for (c) were determined via Kruskal‐Wallis tests, while correlation coefficients and P values for (d) were determined via the Spearman method, *P < 0.05.
Supplementary figure 5| YAP‐TEAD coordinate regulation of expression of NF2 downstream targets.
a, Bar‐plots show expression of putative NF2 target genes, as determined by qPCR. Expression is quantified in both NF2 (red) and YAP (green) KO MeT‐5A cells, with expression plotted as fold‐change relative to WT MeT‐5A cells. In total, 10 dysregulated genes, as inferred from NanoString nCounter analysis, were assessed, comprising 4 up‐regulated (left) and 6 down‐regulated (right) genes. b, Scatter‐plot shows dysregulation of genes included in (a) in NF2 KO cells plotted against YAP KO cells. A clear inverse correlation was observed between FCs relative to WT MeT‐5A in both NF2 KO clone #1 (light red) and #2 (dark red). c, ChIP‐seq tracks, obtained using the Cistrome Data Browser, showing TEAD1 (top) and TEAD4 (bottom) co‐localisation at the most up‐ (KISS1, left) and down‐ (CXCL8, right) regulated NF2 genes. Co‐localisation was assessed in a number of publicly available cancer cell‐lines, comprising a range of different cancer‐types, including PM (MSTO‐211H, top track). TEAD peaks were observed at the promoter region of both genes (dark shading) across most of the cell‐lines included in analysis. Correlation coefficients and P values in (b) were determined by Pearson methods.
Table 1| Lisa analysis of NF2 KO differentially expressed genes.
Results from Lisa analysis103, showing the top 10 most significant P values across corresponding transcriptional regulators (including both transcription factors and chromatin regulators). The top 5 most significant samples, which consist of cell‐lines in which ChIP‐seq data corresponding to that regulator have been deposited in the Cistrome database, are included for each regulator.
ACKNOWLEDGEMENTS
Work ongoing in the Gram Hansen lab is supported by a University of Edinburgh Chancellor's Fellowship, the JHMRF, LifeArc‐CSO as well as by Worldwide Cancer Research (19‐0238). The early stages of this study were supported by the Wellcome Trust—the University of Edinburgh Institutional Strategic Support Fund (ISSF) ISSF2 and the final parts of the project by ISSF3. Siyang Jia holds a scholarship from the Chinese Scholarship Council. Krishna Purohit holds a MRC Precision Medicine DTP Studentship. Ning Sze Hui a Martin Lee Doctoral Scholarship in Stem Cell and Regenerative Medicine. Part of the results published here is formed from data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Genentech is acknowledged for access to datasets EGAD00001001913 and EGAD00001001915 through the (European Genome‐phenome Archive) EGA. Alison Munro from the Host and Tumour Profiling Unit (HTPU), University of Edinburgh is thanked for conducting NanoString nCounter assays. We are grateful for the help provided by Justyna Cholewa‐Waclaw in the High Content Screening Facility at the Centre for Regenerative Medicine, Edinburgh. Single‐cell sorting for clonal cell populations was obtained with excellent support from the QMRI Flow Cytometry and cell sorting facility, at the University of Edinburgh. This research was funded in part, by the Wellcome Trust [Grant number 204804/Z/16/Z]. For the purpose of open access, the author has applied a CC BY public copyright licence to any author‐accepted manuscript version arising from this submission. Figures 1A and 3B were generated using BioRender (https://biorender.com/). This work acknowledges the generous input in the early stages of this project from our friend and collaborator Dr Andy Sims, who passed away in May 2021.
Cunningham R, Jia S, Purohit K, et al. YAP/TAZ activation predicts clinical outcomes in mesothelioma and is conserved in in vitro model of driver mutations. Clin Transl Med. 2023;13:e1190. 10.1002/ctm2.1190
REFERENCES
- 1. Curran D, Sahmoud T, Therasse P, Van Meerbeeck J, Postmus PE, Giaccone G. Prognostic factors in patients with pleural mesothelioma: the European Organization for Research and Treatment of Cancer experience. J Clin Oncol. 1998;16:145‐152. [DOI] [PubMed] [Google Scholar]
- 2. Milano MT, Zhang H. Malignant pleural mesothelioma: a population‐based study of survival. J Thorac Oncol. 2010;5:1841‐1848. [DOI] [PubMed] [Google Scholar]
- 3. Zhai Z, Ruan J, Zheng Y, et al. Assessment of global trends in the diagnosis of mesothelioma from 1990 to 2017. JAMA Netw Open. 2021;4:e2120360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collegium Ramazzini . The 18th Collegium Ramazzini statement: the global health dimensions of asbestos and asbestos‐related diseases. Scand J Work Environ Health. 2016;42:86‐90. [DOI] [PubMed] [Google Scholar]
- 5. Tan E, Warren N, Darnton AJ, Hodgson JT. Projection of mesothelioma mortality in Britain using Bayesian methods. Br J Cancer. 2010;103:430‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mazurek JM, Syamlal G, Wood JM, Hendricks SA, Weston A. Malignant mesothelioma mortality ‐ United States, 1999‐2015. MMWR Morb Mortal Wkly Rep. 2017;66:214‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nasser A, Baird A, Saint‐Pierre MD, Amjadi K, Laurie S, Wheatley‐Price P. Three decades of malignant pleural mesothelioma: an academic center experience. Clin Lung Cancer. 2021:441‐448. doi: 10.1016/j.cllc.2021.03.011 [DOI] [PubMed] [Google Scholar]
- 8. Chimed‐Ochir O, Arachi D, Driscoll T, Lin R‐T, Takala J, Takahashi K. Burden of mesothelioma deaths by national income category: current status and future implications. Int J Environ Res Public Health. 2020;17:6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park E‐K, Takahashi K, Hoshuyama T, et al. Global magnitude of reported and unreported mesothelioma. Environ Health Perspect. 2011;119:514‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chernova T, Murphy FA, Galavotti S, et al. Long‐fiber carbon nanotubes replicate asbestos‐induced mesothelioma with disruption of the tumor suppressor gene Cdkn2a (Ink4a/Arf). Curr Biol. 2017;27:3302‐3314. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hylebos M, Van Camp G, Van Meerbeeck JP, Op De Beeck K. The genetic landscape of malignant pleural mesothelioma: results from massively parallel sequencing. J Thorac Oncol. 2016;11:1615‐1626. [DOI] [PubMed] [Google Scholar]
- 12. Hmeljak J, Sanchez‐Vega F, Hoadley KA, et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018;8:1548‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bueno R, Stawiski EW, Goldstein LD, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48:407‐416. [DOI] [PubMed] [Google Scholar]
- 14. Baas P, Scherpereel A, Nowak AK, et al. First‐line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open‐label, phase 3 trial. Lancet. 2021;397:375‐386. [DOI] [PubMed] [Google Scholar]
- 15. Goudar RK. Review of pemetrexed in combination with cisplatin for the treatment of malignant pleural mesothelioma. Ther Clin Risk Manag. 2008;4:205‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Obacz J, Yung H, Shamseddin M, et al. Biological basis for novel mesothelioma therapies. Br J Cancer. 2021:1039‐1055. doi: 10.1038/s41416-021-01462-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yap TA, Aerts JG, Popat S, Fennell DA. Novel insights into mesothelioma biology and implications for therapy. Nat Rev Cancer. 2017;17:475‐488. [DOI] [PubMed] [Google Scholar]
- 18. Guo G, Chmielecki J, Goparaju C, et al. Whole‐exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015;75:264‐269. [DOI] [PubMed] [Google Scholar]
- 19. Sahtoe DD, Van Dijk WJ, Ekkebus R, Ovaa H, Sixma TK. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat Commun. 2016;7:10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conway E, Rossi F, Fernandez‐Perez D, et al. BAP1 enhances Polycomb repression by counteracting widespread H2AK119ub1 deposition and chromatin condensation. Mol Cell. 2021:3526‐3541. doi: 10.1016/j.molcel.2021.06.020. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hong AW, Meng Z, Plouffe SW, Lin Z, Zhang M, Guan K‐L. Critical roles of phosphoinositides and NF2 in Hippo pathway regulation. Genes Dev. 2020;34:511‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamaratoglu F, Willecke M, Kango‐Singh M, et al. The tumour‐suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8:27‐36. [DOI] [PubMed] [Google Scholar]
- 23. Zhao B, Li Li, Tumaneng K, Wang C‐Y, Guan K‐L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta‐TRCP). Genes Dev. 2010;24:72‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao B, Ye X, Yu J, et al. TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev. 2008;22:1962‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747‐2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang S, Li H, Wang G, et al. Yes‐associated protein (YAP) expression is involved in epithelial–mesenchymal transition in hepatocellular carcinoma. Clin Transl Oncol. 2016;18:172‐177. [DOI] [PubMed] [Google Scholar]
- 27. Yuan Y, Li D, Li H, Wang L, Tian G, Dong Y. YAP overexpression promotes the epithelial‐mesenchymal transition and chemoresistance in pancreatic cancer cells. Mol Med Rep. 2016;13:237‐242. [DOI] [PubMed] [Google Scholar]
- 28. Song S, Ajani JA, Honjo S, et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem‐like properties. Cancer Res. 2014;74:4170‐4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen D, Sun Y, Wei Y, et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo‐YAP pathway and a prognostic marker. Nat Med. 2012;18:1511‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lamar JM, Stern P, Liu H, Schindler JW, Jiang Z‐G, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD‐interaction domain. Proc Natl Acad Sci. 2012;109:E2441‐E2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song S, Honjo S, Jin J, et al. The Hippo coactivator YAP1 mediates EGFR overexpression and confers chemoresistance in esophageal cancer. Clin Cancer Res. 2015;21:2580‐2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang L, Yang S, Chen X, et al. The Hippo pathway effector YAP regulates motility, invasion, and castration‐resistant growth of prostate cancer cells. Mol Cell Biol. 2015;35:1350‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu MZ, Yao T‐J, Lee NPY, et al. Yes‐associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer. 2009;115:4576‐4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y, Dong Q, Zhang Q, Li Z, Wang E, Qiu X. Overexpression of yes‐associated protein contributes to progression and poor prognosis of non‐small‐cell lung cancer. Cancer Sci. 2010;101:1279‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muramatsu T, Imoto I, Matsui T, et al. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis. 2011;32:389‐398. [DOI] [PubMed] [Google Scholar]
- 36. Cho SY, Kim K, Park MS, et al. Expression of Yes‐associated protein 1 and its clinical significance in ovarian serous cystadenocarcinoma. Oncol Rep. 2017;37:2620‐2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941‐D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chapel DB, Hornick JL, Barlow J, Bueno R, Sholl LM. Clinical and molecular validation of BAP1, MTAP, P53, and Merlin immunohistochemistry in diagnosis of pleural mesothelioma. Mod Pathol. 2022:1383‐1397. doi: 10.1038/s41379-022-01081-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miyanaga A, Masuda M, Tsuta K, et al. Hippo pathway gene mutations in malignant mesothelioma: revealed by RNA and targeted exon sequencing. J Thorac Oncol. 2015;10:844‐851. [DOI] [PubMed] [Google Scholar]
- 40. Yang H, Hall SRR, Sun B, et al. NF2 and Canonical Hippo‐YAP pathway define distinct tumor subsets characterized by different immune deficiency and treatment implications in human pleural mesothelioma. Cancers. 2021;13:1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Xu X, Maglic D, et al. Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep. 2018;25:1304‐1317. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoshikawa Y, Sato A, Tsujimura T, et al. Frequent inactivation of the BAP1 gene in epithelioid‐type malignant mesothelioma. Cancer Sci. 2012;103:868‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zauderer MG, Bott M, Mcmillan R, et al. Clinical characteristics of patients with malignant pleural mesothelioma harboring somatic BAP1 mutations. J Thorac Oncol. 2013;8:1430‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cozzi I, Oprescu FA, Rullo E, Ascoli V. Loss of BRCA1‐associated protein 1 (BAP1) expression is useful in diagnostic cytopathology of malignant mesothelioma in effusions. Diagn Cytopathol. 2018;46:9‐14. [DOI] [PubMed] [Google Scholar]
- 45. Louw A, Panou V, Szejniuk WM, et al. BAP1 loss by immunohistochemistry predicts improved survival to first‐line platinum and pemetrexed chemotherapy for patients with pleural mesothelioma: a validation study. J Thorac Oncol. 2022:921‐930. doi: 10.1016/j.jtho.2022.04.008 [DOI] [PubMed] [Google Scholar]
- 46. Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154:1342‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang N, Bai H, David KK, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee H‐J, Pham T, Chang MT, et al. The tumor suppressor BAP1 regulates the Hippo pathway in pancreatic ductal adenocarcinoma. Cancer Res. 2020;1704:1656. doi: 10.1158/0008-5472.CAN-19-1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou X, Wang S, Wang Z, et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest. 2015;125:2123‐2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cottini F, Hideshima T, Xu C, et al. Rescue of Hippo coactivator YAP1 triggers DNA damage–induced apoptosis in hematological cancers. Nat Med. 2014;20:599‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hansen CG, Moroishi T, Guan K‐L. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rausch V, Hansen CG. The Hippo pathway, YAP/TAZ, and the plasma membrane. Trends Cell Biol. 2020;30:32‐48. [DOI] [PubMed] [Google Scholar]
- 53. Cunningham R, Hansen CG. The Hippo pathway in cancer: yAP/TAZ and TEAD as therapeutic targets in cancer. Clin Sci. 2022;136:197‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pearce DA, Nirmal AJ, Freeman TC, Sims AH. Continuous biomarker assessment by exhaustive survival analysis. Biorxiv. 2018:208660. doi: 10.1101/208660. bioRxiv. [DOI] [Google Scholar]
- 55. Gladden AB, Hebert AM, Schneeberger EE, Mcclatchey AI. The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev Cell. 2010;19:727‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cole BK, Curto M, Chan AW, Mcclatchey AI. Localization to the cortical cytoskeleton is necessary for Nf2/merlin‐dependent epidermal growth factor receptor silencing. Mol Cell Biol. 2008;28:1274‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lallemand D, Curto M, Saotome I, Giovannini M, Mcclatchey AI. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 2003;17:1090‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moroishi T, Hansen CG, Guan K‐L. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15:73‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu F‐X, Zhao B, Panupinthu N, et al. Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell. 2012;150:780‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miller E, Yang J, Deran M, et al. Identification of serum‐derived sphingosine‐1‐phosphate as a small molecule regulator of YAP. Chem Biol. 2012;19:955‐962. [DOI] [PubMed] [Google Scholar]
- 61. Plouffe SW, Meng Z, Lin KC, et al. Characterization of Hippo pathway components by gene inactivation. Mol Cell. 2016;64:993‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rausch V, Bostrom JR, Park J, et al. The Hippo pathway regulates caveolae expression and mediates flow response via caveolae. Curr Biol. 2019;29:242‐255. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hansen CG, Ng YLD, Lam W‐LM, Plouffe SW, Guan K‐L. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015;25:1299‐1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rausch V, Hansen CG. Immunofluorescence study of endogenous YAP in mammalian cells. Methods Mol Biol. 2019;1893:97‐106. [DOI] [PubMed] [Google Scholar]
- 65. Morrison H, Sherman LS, Legg J, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001;15:968‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim N‐G, Gumbiner BM. Cell contact and Nf2/Merlin‐dependent regulation of TEAD palmitoylation and activity. Proc Natl Acad Sci. 2019;116:9877‐9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Machida YJ, Machida Y, Vashisht AA, Wohlschlegel JA, Dutta A. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF‐1. J Biol Chem. 2009;284:34179‐34188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zarrizi R, Menard JA, Belting M, Massoumi R. Deubiquitination of γ‐Tubulin by BAP1 prevents chromosome instability in breast cancer cells. Cancer Res. 2014;74:6499‐6508. [DOI] [PubMed] [Google Scholar]
- 69. Deng R, Guo Y, Li L, et al. BAP1 suppresses prostate cancer progression by deubiquitinating and stabilizing PTEN. Mol Oncol. 2021;15:279‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Scheuermann JC, De Ayala Alonso AG, Oktaba K, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR‐DUB. Nature. 2010;465:243‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dai X, Liu H, Shen S, et al. YAP activates the Hippo pathway in a negative feedback loop. Cell Res. 2015;25:1175‐1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moroishi T, Park HW, Qin B, et al. A YAP/TAZ‐induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015;29:1271‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Park J, Hansen CG. Cellular feedback dynamics and multilevel regulation driven by the hippo pathway. Biochem Soc Trans. 2021;49:1515‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kuznetsov JN, Aguero TH, Owens DA, et al. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1‐mutant cancers. Sci Adv. 2019;5:eaax1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Caporali S, Butera A, Amelio I. BAP1 in cancer: epigenetic stability and genome integrity. Discov Oncol. 2022;13:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Abdel‐Rahman MH, Pilarski R, Cebulla CM, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48:856‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Figueiredo CR, Kalirai H, Sacco JJ, et al. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J Pathol. 2020;250:420‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Matatall KA, Agapova OA, Onken MD, Worley LA, Bowcock AM, Harbour JW. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer. 2013;13:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Carbon S, Douglass E, Good BM, et al. The gene ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325‐D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang Y, Shi J, Chai K, Ying X, Zhou B. The role of snail in EMT and tumorigenesis. Curr Cancer Drug Targets. 2013;13:963‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ota I, Masui T, Kurihara M, et al. Snail‐induced EMT promotes cancer stem cell‐like properties in head and neck cancer cells. Oncol Rep. 2016;35:261‐266. [DOI] [PubMed] [Google Scholar]
- 83. Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors Snail and Slug activate the transforming growth factor‐beta signaling pathway in breast cancer. PLoS One. 2011;6:e26514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nakamura R, Ishii H, Endo K, et al. Reciprocal expression of Slug and Snail in human oral cancer cells. PLoS One. 2018;13:e0199442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kurrey NK, K A, Bapat SA. Snail and Slug are major determinants of ovarian cancer invasiveness at the transcription level. Gynecol Oncol. 2005;97:155‐165. [DOI] [PubMed] [Google Scholar]
- 86. CôMe C, Magnino F, Bibeau F, et al. Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res. 2006;12:5395‐5402. [DOI] [PubMed] [Google Scholar]
- 87. Ganesan R, Mallets E, Gomez‐Cambronero J. The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Mol Oncol. 2016;10:663‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sivertsen S, Hadar R, Elloul S, et al. Expression of Snail, Slug and Sip1 in malignant mesothelioma effusions is associated with matrix metalloproteinase, but not with cadherin expression. Lung Cancer. 2006;54:309‐317. [DOI] [PubMed] [Google Scholar]
- 89. Wu Q, You Li, Nepovimova E, et al. Hypoxia‐inducible factors: master regulators of hypoxic tumor immune escape. J Hematol Oncol. 2022;15:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Downes NL, Laham‐Karam N, Kaikkonen MU, Ylä‐Herttuala S. Differential but complementary HIF1α and HIF2α transcriptional regulation. Mol Ther. 2018;26:1735‐1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Forristal CE, Wright KL, Hanley NA, Oreffo ROC, Houghton FD. Hypoxia inducible factors regulate pluripotency and proliferation in human embryonic stem cells cultured at reduced oxygen tensions. Reproduction. 2010;139:85‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Park JH, Shin JE, Park HW. The role of Hippo pathway in cancer stem cell biology. Mol Cells. 2018;41:83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Li J, Williams MJ, Park HJ, et al. STAT1 is essential for HSC function and maintains MHCIIhi stem cells that resist myeloablation and neoplastic expansion. Blood. 2022;140:1592‐1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kroon P, Berry PA, Stower MJ, et al. JAK‐STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem‐like cells. Cancer Res. 2013;73:5288‐5298. [DOI] [PubMed] [Google Scholar]
- 95. Lapidot M, Case AE, Larios D, et al. Inhibitors of the transcription factor STAT3 decrease growth and induce immune response genes in models of malignant pleural mesothelioma (MPM). Cancers. 2020;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Heldin C‐H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal. 2013;11:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Uner OE, See TRO, Szalai E, Grossniklaus HE, Stålhammar G. Estimation of the timing of BAP1 mutation in uveal melanoma progression. Sci Rep. 2021;11:8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lee JH, Welch DR. Suppression of metastasis in human breast carcinoma MDA‐MB‐435 cells after transfection with the metastasis suppressor gene, KiSS‐1. Cancer Res. 1997;57:2384‐2387. [PubMed] [Google Scholar]
- 99. Cho S‐G, Wang Y, Rodriguez M, et al. Haploinsufficiency in the prometastasis Kiss1 receptor Gpr54 delays breast tumor initiation, progression, and lung metastasis. Cancer Res. 2011;71:6535‐6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ciaramella V, Della Corte CM, Ciardiello F, Morgillo F. Kisspeptin and cancer: molecular interaction, biological functions, and future perspectives. Front Endocrinol. 2018;9:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhao B, Ye X, Yu J, et al. TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev. 2008;22:1962‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zheng R, Wan C, Mei S, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47:D729‐D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Qin Q, Fan J, Zheng R, et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP‐seq data. Genome Biol. 2020;21:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shen Y, Wang X, Liu Y, et al. STAT3‐YAP/TAZ signaling in endothelial cells promotes tumor angiogenesis. Sci Signal. 2021:14. [DOI] [PubMed] [Google Scholar]
- 105. Li J, Shi C, Zhou R, et al. The crosstalk between AXL and YAP promotes tumor progression through STAT3 activation in head and neck squamous cell carcinoma. Cancer Sci. 2020;111:3222‐3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zanconato F, Forcato M, Battilana G, et al. Genome‐wide association between YAP/TAZ/TEAD and AP‐1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. He L, Pratt H, Gao M, Wei F, Weng Z, Struhl K. YAP and TAZ are transcriptional co‐activators of AP‐1 proteins and STAT3 during breast cellular transformation. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Irianto J, Pfeifer CR, Xia Y, Discher DE. SnapShot: mechanosensing matrix. Cell. 2016;165:1820‐1820.e1. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gilbert PM, Havenstrite KL, Magnusson KEG, et al. Substrate elasticity regulates skeletal muscle stem cell self‐renewal in culture. Science. 2010;329:1078‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. White ES. Lung extracellular matrix and fibroblast function. Ann Am Thorac Soc. 2015;12(1):S30‐3. Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gouldstone A, Brown RE, Butler JP, Loring SH. Stiffness of the pleural surface of the chest wall is similar to that of the lung. J Appl Physiol. 2003;95:2345‐2349. [DOI] [PubMed] [Google Scholar]
- 112. Ansardamavandi A, Tafazzoli‐Shadpour M, Shokrgozar MA. Behavioral remodeling of normal and cancerous epithelial cell lines with differing invasion potential induced by substrate elastic modulus. Cell Adh Migr. 2018;12:472‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Abidine Y, Constantinescu A, Laurent VM, et al. Mechanosensitivity of cancer cells in contact with soft substrates using AFM. Biophys J. 2018;114:1165‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Manickam K, Machireddy RR, Seshadri S. Characterization of biomechanical properties of agar based tissue mimicking phantoms for ultrasound stiffness imaging techniques. J Mech Behav Biomed Mater. 2014;35:132‐143. [DOI] [PubMed] [Google Scholar]
- 115. Nayar VT, Weiland JD, Nelson CS, Hodge AM. Elastic and viscoelastic characterization of agar. J Mech Behav Biomed Mater. 2012;7:60‐68. [DOI] [PubMed] [Google Scholar]
- 116. Zanconato F, Battilana G, Forcato M, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med. 2018;24:1599‐1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Han H, Yang B, Nakaoka HJ, et al. Hippo signaling dysfunction induces cancer cell addiction to YAP. Oncogene. 2018;37:6414‐6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Carbone M, Harbour JW, Brugarolas J, et al. Biological Mechanisms and clinical significance of BAP1 mutations in human cancer. Cancer Discov. 2020;10:1103‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179‐183. [DOI] [PubMed] [Google Scholar]
- 120. Xu D, Liang S‐Q, Yang Z, et al. Malignant pleural mesothelioma co‐opts BCL‐XL and autophagy to escape apoptosis. Cell Death Dis. 2021;12:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhang M, Luo J‐L, Sun Q, et al. Clonal architecture in mesothelioma is prognostic and shapes the tumour microenvironment. Nat Commun. 2021;12:1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Finicle BT, Jayashankar V, Edinger AL. Nutrient scavenging in cancer. Nat Rev Cancer. 2018;18:619‐633. [DOI] [PubMed] [Google Scholar]
- 123. Klabatsa A, Sheaff MT, Steele JPC, Evans MT, Rudd RM, Fennell DA. Expression and prognostic significance of hypoxia‐inducible factor 1α (HIF‐1α) in malignant pleural mesothelioma (MPM). Lung Cancer. 2006;51:53‐59. [DOI] [PubMed] [Google Scholar]
- 124. Francis RJ, Segard T, Morandeau L, et al. Characterization of hypoxia in malignant pleural mesothelioma with FMISO PET‐CT. Lung Cancer. 2015;90:55‐60. [DOI] [PubMed] [Google Scholar]
- 125. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31‐46. [DOI] [PubMed] [Google Scholar]
- 126. Navarro P, Gómez M, Pizarro A, Gamallo C, Quintanilla M, Cano A. A role for the E‐cadherin cell‐cell adhesion molecule during tumor progression of mouse epidermal carcinogenesis. J Cell Biol. 1991;115:517‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Astin JW, Batson J, Kadir S, et al. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat Cell Biol. 2010;12:1194‐1204. [DOI] [PubMed] [Google Scholar]
- 128. Wei SC, Yang J. Forcing through tumor metastasis: the interplay between tissue rigidity and epithelial‐mesenchymal transition. Trends Cell Biol. 2016;26:111‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Plodinec M, Loparic M, Monnier CA, et al. The nanomechanical signature of breast cancer. Nat Nanotechnol. 2012;7:757‐765. [DOI] [PubMed] [Google Scholar]
- 130. Song Q, Mao B, Cheng J, et al. YAP enhances autophagic flux to promote breast cancer cell survival in response to nutrient deprivation. PLoS One. 2015;10:e0120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Mason DE, Collins JM, Dawahare JH, et al. YAP and TAZ limit cytoskeletal and focal adhesion maturation to enable persistent cell motility. J Cell Biol. 2019;218:1369‐1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Aikawa T, Gunn J, Spong SM, Klaus SJ, Korc M. Connective tissue growth factor‐specific antibody attenuates tumor growth, metastasis, and angiogenesis in an orthotopic mouse model of pancreatic cancer. Mol Cancer Ther. 2006;5:1108‐1116. [DOI] [PubMed] [Google Scholar]
- 133. Gill MK, Christova T, Zhang YY, et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat Commun. 2018;9:3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Krebs M, Carter L, Villa S, et al. P2.06‐09 MiST3: a phase II study of oral selective AXL inhibitor bemcentinib (BGB324) in combination with pembrolizumab in pts with malignant mesothelioma. J Thorac Oncol. 2018;13:S745. [Google Scholar]
- 135. Liu‐Chittenden Yi, Huang Bo, Shim JS, et al. Genetic and pharmacological disruption of the TEAD‐YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wang C, Zhu X, Feng W, et al. Verteporfin inhibits YAP function through up‐regulating 14‐3‐3σ sequestering YAP in the cytoplasm. Am J Cancer Res. 2016;6:27‐37. [PMC free article] [PubMed] [Google Scholar]
- 137. Dasari VR, Mazack V, Feng W, Nash J, Carey DJ, Gogoi R. Verteporfin exhibits YAP‐independent anti‐proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget. 2017;8:28628‐28640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Song S, Xie M, Scott AW, et al. A novel YAP1 inhibitor targets CSC‐Enriched radiation‐resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol Cancer Ther. 2018;17:443‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Nouri K, Azad T, Ling M, et al. Identification of celastrol as a novel YAP‐TEAD Inhibitor for cancer therapy by high throughput screening with ultrasensitive YAP/TAZ–TEAD Biosensors. Cancers. 2019;11:1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Holden JK, Crawford JJ, Noland CL, et al. Small molecule dysregulation of TEAD lipidation induces a dominant‐negative inhibition of Hippo pathway signaling. Cell Rep. 2020;31:107809. [DOI] [PubMed] [Google Scholar]
- 141. Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016;22:128‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhang Y, Shi J, Liu X, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Xu Y‐Y, Ren Z‐L, Liu X‐L, et al. BAP1 loss augments sensitivity to BET inhibitors in cancer cells. Acta Pharmacol Sin. 2022;43:1803‐1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Farzin M, Toon CW, Clarkson A, et al. Loss of expression of BAP1 predicts longer survival in mesothelioma. Pathology. 2015;47:302‐307. [DOI] [PubMed] [Google Scholar]
- 145. Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792‐D799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics. 2015;31:166‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Robinson MD, Mccarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high‐throughput experiments. Bioinformatics. 2012;28:882‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1| Broad mutation profiles are insufficient to define patient populations with distinct clinical outcomes.
a, PCA plots displayed as in figure 1d, show PCs along different axes to highlight clusters. b, PCA plots, depicted in 3D, show patients coloured according to mutation status. Broad transcriptional profiling is insufficient to distinguish between the populations of patients categorised by mutation of BAP1 and NF2 in the TCGA (left; n = 86) and Bueno et al (right; 98) cohorts. c, Barplot shows the percentage of patients classified by consensus subtypes as generated in Bueno et al 16, with patients split according to mutation status of BAP1 and NF2. n = 98. d, Barplot, as in (c), highlights the absence of significant association between mutation status and T‐staging across the merged TCGA and Bueno et al datasets (n = 163). e, Kaplan‐Meier curves show overall survival of patients split according to mutation status, with patients categorised as exhibiting either NF2 or BAP1 mutations, as well as mutations in both or neither. No significant difference in survival across the patient populations is observed. P values for (c) and (d) determined via Fisher's exact test, while P values for (e) were calculated via log‐rank test.
Supplementary Figure 2| Analysing TCGA and Bueno cohorts separately highlights consistency between datasets.
a, Violin plots show GSVA scores of YAP/TAZ signature gene expression as in figure 2a, with results split by dataset. A significant collective overexpression of this gene‐set is observed in patients with Hippo kinase cascade inactivating mutations in both TCGA (left; n = 86) and Bueno et al (right; n = 98) cohorts. b, Violin plots as in (a), show signature scores in patients split according to BAP1 mutation status, excluding patients harbouring Hippo pathway inactivating mutations. While there is an association between BAP1 mutation and expression of YAP/TAZ target genes, this is minor and only seen to be significant in the TCGA (left) and not Bueno et al (right) cohort. c, Kaplan‐Meier curves show overall survival of patients split according to YAP/TAZ signature thresholds. Patients classed as YAP/TAZ signature display a pronounced reduction in overall survival in both TCGA (top; HR = 4.4, n = 86, threshold at 74%) and Bueno et al (top; HR = 1.76, n = 211, threshold set at 52%) cohorts. d, Bar‐plots, as in figure 2e, show tumour stage in patients categorised as signature high/low. P values in (a) and (b) were determined by Mann‐Whitney U test, P values and hazard ratios for (c) were calculated via log‐rank test and Cox proportional hazard model respectively, while P values for (d) were calculated via Fisher's exact test. n.s. = Not significant, *P < 0.05, **P < 0.01, and ****P < 0.0001 relative to WT.
Supplementary Figure 3| Validation of NF2 KO mediated activation of YAP.
a, Phos‐tag based western blots (top), as in as in figure 3c, shows phosphorylation status of YAP (left) and TAZ (right) in response to serum starvation across a range of time‐points. Responses in WT MeT‐5A cells are compared to the second NF2 KO clone (#2), with a similar decreased sensitivity to starvation observed on NF2 loss in NF2 KO #1. A standard SDS‐gel based Western blot (bottom) is also shown with the same samples analysed, highlighting levels of phospho‐YAP (S127) and YAP (left), TAZ (right), together with GAPDH. b, Western blots showing NF2 expression levels in WT, NF2 KO, and re‐expression of NF2 in NF2 KO MeT‐5A clones. c‐d, Phos‐tag based western blots, as in (a), with NF2 re‐expressed in NF2 KO MeT‐5As. Response to serum starvation is restored upon NF2 re‐expression, with increased phosphorylation of YAP observed when cells are deprived of serum. This rescue of YAP regulation upon exogenous NF2 expression is observed in both NF2 KO clone #1 (c) and clone #2 (d). e, Violin plot showing levels of nuclear YAP, as determined by immunofluorescence based images of cell monocultures, such as those shown in figure 3e, normalised to levels of cytoplasmic YAP (n = 8). f, Confocal based image acquired on the Opera Phenix Plus. Representative maximum projection images showing difference in YAP nuclear localisation between WT and NF2 KO MeT‐5A cells. Cells were mixed before seeding, with NF2 KO cells stained with CellTracker Red. Individual channels (taken from region within red dashed box) highlight the relative increase in nuclear YAP in NF2 KO cells (diagonal arrows) relative to WT cells (arrowheads). Scale bar = 50 μm. g, Representative western blot (top) showing levels of LATS1/2 in MeT‐5A KO cells of different genotypes, with quantification of LATS2 (n = 3) below, with bars showing mean levels and error bars representing SD across replicates. LATS1 appears unchanged across all genotypes, while LATS2 is generally upregulated in both NF2 and BAP1 KO MeT‐5A. h, Bar‐plot shows qPCR quantification of LATS2 expression relative to WT cells across BAP1 KO and YAP KO MeT‐5A cells. Expression is shown as FC relative to WT MeT‐5A (n = 3). i‐j, Western blots show CRISPR‐mediated knockout of YAP in MeT‐5A cells (i), as well as YAP or TAZ double knockout (dKO) on NF2 KO background MeT‐5A cells (j).
Supplementary figure 4| The transcriptional effects of BAP1 loss in vitro consistently and robustly mirrors mutation in patients.
a, Heat‐map shows expression of 1,540 genes included in the combined NanoString PanCancer Progression and Immune Profiling nCounter panels across various KO MeT‐5A cell‐lines. Cells were analysed at steady‐state (‘+’) and after 3 hours of serum starvation (‘‐’). Dendrograms show the clustering of genotypes, highlighting similarities in transcriptional profiles observed in lines bearing the same KO. b, Heat‐map, as in figure 4d, shows the quantification of expression of genes found to be significantly differentially expressed in BAP1 KO relative to WT MeT‐5A cells in patients from both cohorts grouped. No patients were excluded in this analysis, with mutation status for BAP1 and Hippo‐associated genes annotated above. c, Violin plots show the GSVA scores in patients from both cohorts grouped, of genesets consisting of genes found to be dysregulated in BAP1 KO MeT‐5A cells. Genes found to be both up‐regulated (left) and down‐regulated (right) in vitro are similarly dysregulated in patients. d, Heatmap shows dysregulation of YAP/TAZ signature genes within BAP1 KO MeT‐5A cells. Expression is shown across two distinct clones as mean log2FC relative to WT cells across biological replicates (n = 4). e, Scatter‐plots as in figure 4c‐d show correlation between gene dysregulation in NF2 mutant patients from the Bueno et al cohort vs NF2 KO MeT‐5A cells. Noise has been reduced by limiting analysis to those genes found to be significantly dysregulated in vitro. While some significant correlation exists in one NF2 KO clone, this is not consistent, which suggests the same degree of conserved, broad transcriptomic dysregulation observed with BAP1 disruption is not reproduced on loss of NF2. P values for (c) were determined via Kruskal‐Wallis tests, while correlation coefficients and P values for (d) were determined via the Spearman method, *P < 0.05.
Supplementary figure 5| YAP‐TEAD coordinate regulation of expression of NF2 downstream targets.
a, Bar‐plots show expression of putative NF2 target genes, as determined by qPCR. Expression is quantified in both NF2 (red) and YAP (green) KO MeT‐5A cells, with expression plotted as fold‐change relative to WT MeT‐5A cells. In total, 10 dysregulated genes, as inferred from NanoString nCounter analysis, were assessed, comprising 4 up‐regulated (left) and 6 down‐regulated (right) genes. b, Scatter‐plot shows dysregulation of genes included in (a) in NF2 KO cells plotted against YAP KO cells. A clear inverse correlation was observed between FCs relative to WT MeT‐5A in both NF2 KO clone #1 (light red) and #2 (dark red). c, ChIP‐seq tracks, obtained using the Cistrome Data Browser, showing TEAD1 (top) and TEAD4 (bottom) co‐localisation at the most up‐ (KISS1, left) and down‐ (CXCL8, right) regulated NF2 genes. Co‐localisation was assessed in a number of publicly available cancer cell‐lines, comprising a range of different cancer‐types, including PM (MSTO‐211H, top track). TEAD peaks were observed at the promoter region of both genes (dark shading) across most of the cell‐lines included in analysis. Correlation coefficients and P values in (b) were determined by Pearson methods.
Table 1| Lisa analysis of NF2 KO differentially expressed genes.
Results from Lisa analysis103, showing the top 10 most significant P values across corresponding transcriptional regulators (including both transcription factors and chromatin regulators). The top 5 most significant samples, which consist of cell‐lines in which ChIP‐seq data corresponding to that regulator have been deposited in the Cistrome database, are included for each regulator.