

Abstract
The Signal Transducer and Activator of Transcription (STAT)-5 proteins are required in immune regulation and homeostasis and play a crucial role in the development and function of several hematopoietic cells. STAT5b activation is involved in the expression of genes that participate in cell development, proliferation, and survival. STAT5a and STAT5b are paralogs and only human mutations in STAT5B have been identified leading to immune dysregulation and hematopoietic malignant transformation. The inactivating STAT5B mutations cause impaired post-natal growth, recurrent infections and immune dysregulation, whereas gain of function somatic mutations cause dysregulated allergic inflammation. These mutations are rare, and they are associated with a wide spectrum of clinical manifestations which provide a disease model elucidating the biological mechanism of STAT5 by studying the consequences of perturbations in STAT5 activity. Further, the use of Jak inhibitors as therapy for a variety of autoimmune and malignant disorders has increased substantially heading relevant lessons for the consequences of Jak/STAT immunomodulation from the human model. This review summarizes the biology of the STAT5 proteins, human disease associate with molecular defects in STAT5b, and the connection between aberrant activation of STAT5b and the development of certain cancers.
Keywords: STAT5, STAT5b, hematopoiesis, gain of function (GOF), loss of function (LOF)
Introduction
Signal Transducer and Activator of Transcription (STAT) 5 are proteins involved in a variety of critical cellular functions and pathways. STAT5 has two paralogs, STAT5a and STAT5b (1). STAT5 activates in response to a number of cytokines and growth factors. Upon activation, it plays a key role in hematopoiesis, particularly lymphocyte development, proliferation, and survival (2). Mutations in the STAT5B gene are linked to impaired protein signaling and function and are associated with stunted growth, autoimmunity and immunodeficiency (3, 4). Hyperactivation of STAT5b is associated with the development of various blood malignancies and tumors and more recently a syndrome of severe allergic inflammation (5, 6). The mechanisms of aberrant STAT5b function are complicated and not all have been fully elucidated. A more robust understanding of how this crucial protein modulates function in the hematopoietic compartment will aid in development of treatment strategies for patients suffering the effects of STAT5b dysfunction.
This review addresses the biology of STAT5 proteins and we discuss STAT5’s role within the context of human disease.
Biology of STAT proteins
The STAT protein family is named for its dual roles in transducing signal from the cytokine-receptor to the nucleus and activating gene transcription (7). The family includes seven highly related homologous proteins: STAT1, STAT2, STAT3, STAT4, STAT5a and STAT5b (referred to collectively as STAT5), and STAT6 (8–10). Each STAT contains the following conserved domains: an N-terminal region (associated with cooperative DNA binding between dimers) (11, 12), a coiled-coil domain (plays a role during the DNA binding process and provides a site for interactions with other proteins) (10, 13), a DNA binding domain (allows function as transcription factors and targets specific DNA sites) (14), a linker region and SH2 domain (modulates the interaction through phospho-tyrosines) (15, 16), and a C-terminal transactivation domain (17, 18) ( Figure 1 ).
Figure 1.
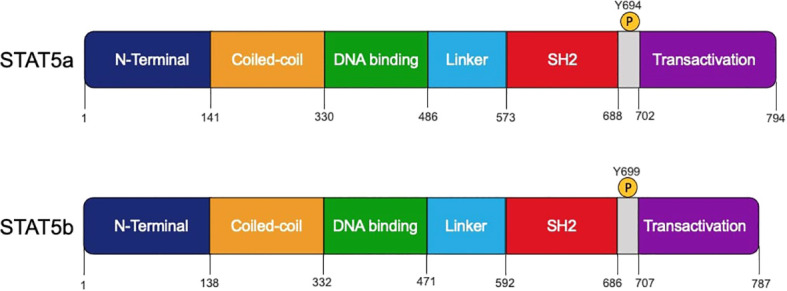
Schematic structures of human STAT5a and STAT5b protein domains. The domains, amino acids residue positions, and the tyrosine 694 and 699 that can be phosphorylated by JAK proteins are shown.
STAT5A/B Gene and Protein
Mammals possess two STAT5 homologs, STAT5A and STAT5B, and both genes are located on chromosome 17 (19). STAT5a and STAT5b are very similar, with 95% homology between their sequences (1) ( Figure 1 ). STAT5a and STAT5b are considered paralogs because they are the protein products of different genes. While they display a high degree of structural similarity, STAT5a differs from STAT5b in several respects. STAT5a contains 12 additional amino acids on the carboxy-terminus and is shortened by five residues on the phosphotyrosyl tail segment (5). The STAT5a and STAT5b DNA binding domains differ by 5 amino acids, which contributes to differences in DNA affinity once they have been activated through phosphorylation and homodimers are formed (20). In addition to these structural differences, STAT5a and STAT5b are phosphorylated at tyrosine at different sites. STAT5a is activated by phosphorylation of a tyrosine found at position 694 while STAT5b is phosphorylated at position 699 (5). Finally, STAT5b is more highly expressed than STAT5a in hematopoietic cell types such as erythrocytes, megakaryocytes, Natural Killer (NK) cells, CD4+ and CD8+ T cells, and B cells. STAT5a is expressed at higher levels only in CD34+ hematopoietic stem cells (20).
STAT5 is activated in response to a variety of cytokines (IL-2, -3, -5, -7, -9, -15, -21) (20, 21). Prior to cytokine-receptor interaction, STAT5 proteins are found in a cell’s cytoplasm as inactive antiparallel dimers (10). The canonical JAK-STAT5 signaling pathway is characterized by three sequential tyrosine phosphorylations that occur in response to cytokine and growth factor stimulations (7, 10). After activation, STAT5 proteins form homodimers, heterodimers, or tetramers (7, 22–24). These complexes then translocate to the nucleus, where they induce the transcription of specific genes (2, 25) ( Figure 2 ). In the nucleus, the functional dimers are dephosphorylated by phosphatases and signaled for export back to the cytoplasm for further rounds of activation (10). One important canonical signaling pathway for STAT5 includes growth hormone (GH). The interaction between GH and its cell surface receptor (GHR) induces the auto-activation of JAK2, followed by phosphorylation of three tyrosines located in the intracellular domain of the GHR. This sequence of events requires the recruitment and docking of cytosolic STAT5b to the intracellular domain of the GHR (26), activation of STAT5b (tyrosine phosphorylation) by JAK2, dimer formation, and the mobilization of the complex to the nucleus to drive transcriptional programs (3, 27, 28). The GH/GHR system can also activate other STAT proteins such as STAT1, STAT3 (3, 28, 29). STAT5 function can also be initiated by other growth factors reviewed here, including granulocyte-macrophage colony-stimulating factor (GM-CSF), erythropoietin (EPO), thrombopoietin (TPO), and Fms-like tyrosine kinase 3 (Flt3) (20, 21) ( Figure 2 ). STAT5 activation downstream of these growth factors is very similar to the pathway initiated by cytokine activation.
Figure 2.

Schematic of STAT5 signaling. The activation of STAT5 requires the binding of cytokines or growth factors to their respective receptors. The cytoplasmic domains of the receptors are trans-phosphorylated by activated JAK proteins. STAT5 proteins then bind to the receptor and become activated by trans-phosphorylation at their specific tyrosine residues. After activation, STAT5 dimerizes and translocates to the nucleus to induce gene transcription.
STAT5 is also tightly regulated. To begin, the suppressor of cytokine signaling (SOCS) proteins act as negative feedback inhibitor dampening specific cytokine signaling and preventing an excessive response (30). The Cytokine-induced SH2-containing protein (CIS), SOCS1, 2 and 3 are all associated with inhibition of the JAK/STAT signaling pathway (10). While SOCS1 is primarily associated with IFN-γ signaling (31), in the context of IL-2 signaling in T cells, SOCS1 shows a high affinity for regulating cytokines that signal through common gamma chain subunit, such as, IL-2, IL-4, IL-7, and IL-15 (32–34). Conversely, SOCS3 regulates various other signaling pathways by interacting with receptors such as granulocyte colony-stimulating receptor (G-CSFR), leptin receptor, and erythropoietin receptor (EpoR) (35). Different studies demonstrated that during GHR/JAK2/STAT5 signaling, SOCS1 and SOCS3 bind JAK2, leading to degradation of the GHR/JAK2 complex by ubiquitination (30, 36). SOCS2 binds to motifs in the GH receptor through its SH2 domain, inhibiting the STAT5 recruitment (37, 38).
The binding of cytokines to their receptors triggers an intracellular tyrosine phosphorylation cascade, that is also regulated by two other proteins: phosphotyrosines phosphatases (PTPs) and protein inhibitors of activated STATs (PIAS). The PTP protein family includes six members found in both the cytoplasm and nucleus that regulate the intracellular tyrosine phosphorylation cascade. These proteins are constitutively expressed, and therefore are not feedback-inhibitors (30, 39). Finally, the PIAS proteins modulate STAT5 activity in the nucleus by inhibiting the interaction between STAT5-dimers and their DNA promoter regions through SUMOylation of STAT5 (30, 40, 41). STAT5 activation is tightly regulated, importantly so, because the downstream activity of STAT5 has implications on multiple cell types in tissues and the immune system.
Role of STAT5 proteins during hematopoiesis
STAT5 plays a critical role in the development of many hematopoietic lineages, including B cells, T cells, NK cells, erythroid cells, and megakaryocytic cells ( Figure 3 ). In regards to hematopoiesis, STAT5b is expressed at higher levels than STAT5a in all of these cell types (20).
Figure 3.

STAT5 and hematopoiesis. The genes and transcription factors involved in hematopoiesis following activation of STAT5.
STAT5 activation that occurs in conjunction with IL-7R signaling regulates B cell lymphopoiesis by promoting cell survival and regulating the immunoglobulin gene rearrangement in pro-B cells (42). STAT5 also regulates the expression of transcription factors critical during early B cell stages such as E2A, EBF1, and Pax5 (43–46). In Stat5a/bnull/null mice there is a block of B cell development between the pro- and pre- B cell stages (47). However, mice that expressed an N-terminal truncated Stat5a/b protein (Stat5a/b ΔN/ΔN mice) showed far less impairment in B cell development than the knockout mice leading to a more mild phenotype. These results suggest that the N-terminal truncated STAT5a/b may partially rescue B cell development, but the mechanism behind this is unclear (47). The activation of STAT5 in response to other cytokines can have distinct effects on B cell differentiation as well. Recently, Pelham and colleagues demonstrated that STAT5b can induce the expression of SOCS3 in response to IL-21 regulating naïve B cell differentiation to memory B cells and plasma cells (48). So, when STAT5b is decreased or absent, as in STAT5b deficient patients, there is a skewing towards terminally differentiated memory and plasma cells compared to wild type (48). Studies such as this demonstrate the ubiquitous function of STAT5b in immune cells and highlight the importance in regulating lymphocyte differentiation ( Figure 3 ).
STAT5 plays an important role in the development, differentiation, and survival of T cells (49). To start with data surrounding development, Stat5a/b-/- mice show severe impairment of lymphocyte development which mimic the findings observed in Il7r-/- mice (50, 51). The IL-7/STAT5 signaling plays a critical role driving the expression of important transcription factors, such as Runx3, required for CD8+ T cell differentiation (52). Moreover, Lin and colleagues demonstrated that STAT5-tetramers are essential for the expansion of antigen-specific activated CD8+ T cell (24).
As STAT5 has been shown to have a role in the development and differentiation of CD8+ T cells, it is similarly involved with the development of the various CD4+ T cell subsets (49). Th1 polarization is induced by IL-12 and expression of Tbet leads to IFN-γ production along with other Th1 cytokines. Liao and colleagues demonstrated that while naïve T cells are unable to respond to IL-12 because they do not express the IL-12 receptor β2 (IL-12Rβ2), IL-2 potentiates Th1 differentiation via STAT5 activation by inducing IL-12Rβ2 and Tbet expression (53). Similarly, Th2 polarization requires IL-4 signaling and GATA3 expression (49, 54). Importantly, the activation of STAT5 by IL-2 is first required to induce the expression of IL-4 receptor α expression leading to Th2 differentiation (54–57).
The activation of STAT5 in response to IL-2 is also associated with the development and function of CD4+FoxP3+ regulatory T cells (Tregs) (58–61). In mature Treg cells, the activation of STAT5b drives and maintains FOXP3, CD25, and Bcl-2 gene expression (21, 61, 62). Combined with studies using Il2rb-deficient mice (63, 64), these findings demonstrate that STAT5 acts as a central effector during the development and function of Treg cells. Reports involving patients with STAT5b deficiency show impaired Treg homeostasis and decreased numbers of CD4+ and CD8+ T cell subsets (61, 65).
NK cells are the third subset of lymphocytes in the peripheral blood whose development is dependent on STAT5 activation (66, 67). IL-15 is required for the generation of fully functional NK cells, and the IL-15 trans-presentation by dendritic cells is vital to prime the terminal NK cell maturation (68–74). NK cell activation through IL-15 receptors induces the recruitment and activation of STAT5 (75, 76). Compared to Stat5a−/− mice, Stat5b−/− mice exhibit a profound reduction of NK cell frequencies in peripheral blood with a reduction in their activity in response to IL-2 and IL-15 (77, 78). STAT5 drives the expression of essential transcription factors for NK cell development such as ID2, EOMES, and T-bet, and acts as a regulator of the expression of important effector molecules (perforin, granzymes, and IFN-γ) (79). STAT5 can also form tetramers, and Lin and colleagues showed that STAT5 tetramer-deficient mice (DKI mice) had lower numbers of NK cells and that the absence of tetramers was associated with abnormal maturation and with decreased expression of maturation-associated genes (80). Importantly, owing to STAT5b’s ubiquitous role in the development of multiple lymphocyte cell subsets, these DKI mice presented with decreased and defective proliferation of CD8+ T cells along with reduced Treg cell presence and function (80).
Similar to IL-15, STAT5 is the principal downstream effector after IL-2 stimulation. NK cells are activated in response to IL-2, enhancing their cytotoxicity capacity, activating receptor expression and cytokine production (81–83) which is severely impaired in STAT5b deficient patients (25). Taken together, the abnormal NK cell maturation observed in humans mimics the abrogated NK cell terminal maturation observed in Stat5b -/- mice (25, 84).
NK cells are closely related to innate lymphoid cells (ILC). While NK cells are more closely related to group 1 ILC, all groups of ILCs deserve mention in regards to STAT5b biology. The ILC family includes four subsets: ILC1, ILC2, ILC3, and lymphoid tissue inducer cells (85, 86). According to their functional and phenotypic characteristics, they are classified into Group 1 ILCs characterized by T-bet expression and IFN-γ production (87), Group 2 ILCs characterized by IL-5 and IL-13 production along with the expression of the transcription factors GATA3 and RORα, and Group 3 ILCs which are characterized by IL-17 and IL-22 production and are dependent on AHR and RORγt (85). Using a mouse model with decreased numbers of STAT5 alleles, Villarino and colleagues demonstrated that STAT5 is necessary for accumulation of all ILC subsets in lymphoid and nonlymphoid tissues. In this model, mice retaining Stat5a alleles rather than Stat5b alleles seemed to have a larger impact on ILC development. Early developmental precursors were present and consistent with γc and IL-7 deficient mice important because γc cytokines involved with lymphocyte development signal through STAT5b (78). As expected ILC2 progenitors were also present but NK cell and IL-1 progenitors were severely affected; an important finding since it corroborates the notion that STAT5 is a critical multilineage transcription factor (MLTF) that induces ILC development, homeostasis and function (78). These findings show the relevance of STAT5 beyond classic lymphocyte biology since ILCs are typically known for their surveillance and defense in tissues like lungs, skin, and intestines.
STAT5 is a regulator that determines lineage commitment between erythroid and megakaryocytic cell fates (88). Starting with erythroid development, EPO induces the activation of the STAT5/JAK2 pathway (89–93). After activation, STAT5 interacts with the transcription factors TAL1, GATA1, and KLF1 (called “master regulators”), and the resulting complex binds enhancers of specifics genes that induce erythroid differentiation (93–96) ( Figure 3 ). In mice, the absence of any one of these transcription factors important to the development of erythroid cells results in severe anemia and death (97–100). Studies with Stat5a/b -/- mice demonstrate that both STAT5a and STAT5b are critical regulators of iron uptake during erythroid development, as the absence of both STAT5 proteins resulted in severe anemia and reduction of Tfr1 expression (transferrin receptor) (101). Finally, chronic anemia was reported in STAT5b deficient patients, further highlighting the role of STAT5b during erythropoiesis (25, 48).
While erythroid cell development is initiated by EPO, megakaryocytic differentiation is activated by TPO via JAK/STAT5a/b signaling. TPO signaling activation induces the expression of Bcl-xL and megakaryocytic cell survival (102) ( Figure 3 ). Olthof and colleagues reported that the overexpression of constitutively activated Stat5a mutant in CD34+ cells in mice induces erythroid differentiation at the expense of megakaryocyte development (88). Furthermore, studies in hematopoietic stem/progenitor cells demonstrated that in the absence of TPO, tyrosine-unphosphorylated STAT5 represses megakaryocytic differentiation by blocking the megakaryocytic transcriptional program (103). The role STAT5 plays in erythroid and megakaryocyte development comes into play when assessing the side effects of suppressing STAT5 through various JAK inhibitor therapies now used for hematopoietic and autoimmune disorders.
Finally, the canonical signaling for the phagocytic growth factor GM-CSF occurs via STAT5. While GM-CSF was initially described for its ability to initiate the production of neutrophils and macrophages, it can also participate in the production of eosinophils, erythrocytes, megakaryocytes, and dendritic cells (104–106). Once GM-CSF binds to its receptor (GM-CSFR), the JAK2/STAT5 pathway is initiated leading to downstream gene expression (107). This GM-CSF/STAT5 signaling pathway regulates target genes such as cytokine-inducible SH2-containing protein (CIS), a regulator of STAT proteins (10, 107–109). In addition to its role in phagocytic cell proliferation, GM-CSF/STAT5 signaling is also involved in various functions of alveolar macrophages and the growth of alveolar epithelium in lung tissue (105, 110, 111). These critical alveolar macrophage functions include surfactant and cholesterol clearance (104). Impairment in this signaling pathway can lead to the inability to initiate this protein clearance causing pulmonary alveolar proteinosis (PAP), a known complication in STAT5b deficient patients (112).
Human defects caused by STAT5B mutations
In humans, the identification of deleterious STAT5B mutations demonstrates that STAT5a is unable to compensate for the absence of STAT5b, despite their high amino acid similarity, and mutations causing human disease in STAT5A have not yet been identified. Pathologic mutations affecting its function and signaling have been described in all domains of STAT5b (3, 4). Thus far, different molecular mechanisms of STAT5B mutations have been identified. Autosomal recessive loss-of-function (LOF) mutations and heterozygous dominant negative mutations as well as recently, somatic gain-of-function (GOF) mutations, associated with severe allergic inflammation and hematological malignancies, have been reported.
Loss of Function Mutations
Nine homozygous LOF STAT5B mutations have seen been reported: three missense mutations (p.A630P, p.F646S, p.L151P) (27, 113, 114); two nonsense mutation (p. Q41X, p.R152X) (48, 115); a deletion of four nucleotides (c.424-427del) (116); two single nucleotide insertions (c.1102insC, c.1191insG) (117, 118); and a single nucleotide deletion (c.1680delG) (119) ( Figure 4 ). Nonsense and frameshift mutations predict a truncated STAT5b protein, and the lack of detectable protein in primary cells from these patients suggests that the truncated STAT5b protein is not stably expressed (25, 27, 115, 117, 118). The missense mutation p.A630P located in SH2 domain, was the first mutation described as responsible for STAT5b deficiency (27). Overexpression studies demonstrated that this variant can be expressed, but it cannot be activated by GH or IFN-γ stimulation (27). Furthermore, studies in silico, showed that the autosomal recessive p.A630P mutation disrupts the β-sheet core, affecting protein solubility leading to misfolding (120). In addition, two other missense mutations have been described with abnormal functionality and correlate with the clinical STAT5b phenotype (113, 114). Using a reconstitution system, Hwa and colleagues demonstrated that the c.1680delG frameshift mutation was not stably expressed (119).
Figure 4.

Pathologic STAT5B mutations identified in patients. The STAT5b domains and amino acids residue positions are indicated. Red, homozygous LOF mutations; Purple, dominant-negative LOF mutations with growth hormone insensitivity and eczema; Blue, dominant-negative LOF mutation associated with immune dysregulation lacking growth hormone insensitivity; Green, severe allergic somatic GOF mutation; Black, somatic GOF mutations.
Loss-of-function mutations are associated with severe forms of pulmonary disease, eczema, combined immunodeficiency, autoimmune disease, bacterial and/or viral infections and post-natal growth delay ( Table 1 ). As previously mentioned, STAT5b plays an important role in lymphocyte development, function, and survival; therefore, it would be expected that these patients have recurrent and severe viral infections in addition to bacterial and some reported fungal infections putting these patients at risk for opportunistic infections (21, 61, 84). Autoimmune disease has been reported such as juvenile idiopathic arthritis, interstitial lung disease (ILD), inflammatory bowel disease, Hashimoto’s thyroiditis, and type 1 diabetes (25, 75). The proposed mechanism of autoimmunity in STAT5b deficient patients was reviewed nicely by Kanai et al. where they suggested that STAT5b’s role in driving FOXP3 led to lack of Tregs in these patients. In addition, they suggested that cells are more likely to be susceptible to apoptosis due to lack of STAT5b dependent Bcl2; a protein important for regulation of apoptosis (75).
Table 1.
Human Disease Manifestations.
| Human Disease Manifestations | ||
|---|---|---|
| Gain of Function | Loss of Function | Dominant Negative |
| ◼ Non-clonal eosinophilia ◼ Urticaria ◼ Dermatitis ◼ Diarrhea ◼ LGL leukemia |
◼ Impaired post-natal growth ◼ Low expression of IGF-I, IGFBP-3 and ALS ◼ Elevated prolactin levels ◼ Severe pulmonary disease, eczema, autoimmune disease (juvenile idiopathic arthritis), lung disease, and bacterial and/or viral infections ◼ Low NK cell counts and cytotoxic capabilities ◼ T-lymphopenia (CD4+ and CD8+ T cells) ◼ Reduction of T-regulatory cells ◼ Hypergammaglobulinemia ◼ Chronic Anemia |
◼ Impaired post-natal growth ◼ Eczema ◼ Hyper IgE ◼ Growth hormone insensitivity syndrome (GHIS) ◼ Mild immune dysregulation ◼ Low expression of IGF-I, IGFBP-3 and ALS |
The clinical manifestations found in patients with GOF, LOF, and dominant negative STAT5B variants.
These deleterious loss-of-function STAT5B mutations also lead to the disruption of GM-CSF signaling, which is associated with pulmonary alveolar proteinosis (PAP) (104). GM-CSF binds with its heterodimeric receptor, causing activation of JAK2 and initiating multiple pathways. One of these pathways involves STAT5 is important for the development and function of alveolar macrophages (104, 107). Recently, Krone and colleagues described impaired GM-CSF signaling in a patient with STAT5b deficiency (112).
Extra immune manifestations of STAT5b deficiency include growth hormone related abnormalities. Since STAT5b is activated by growth hormone through the GHR, patients with STAT5b deficiency present with normal intrauterine growth but impaired post-natal growth, similar to patients with GHR defects (4). In most cases involving LOF mutations, the endocrine profile reveals a normal basal level of GH, low expression of IGF-1, IGFBP-3 and ALS with elevated prolactin levels, suggesting an altered negative feedback loop for GH signaling (4, 21) ( Table 1 ). GH treatment is somewhat challenging because, like GHR deficient patients, there is poor response to endogenous and exogenous GH (27, 117–119). Recently, Bang and colleagues reported a study using recombinant human IGF1 supplementation in patients with GHR defects. They saw a positive gain in linear growth suggesting IGF1 supplementation might be a plausible therapy to promote growth in patients with aberrant growth hormone signaling such as STAT5b deficiency (121).
Heterozygous dominant negative LOF STAT5B mutations were recently described in three unrelated patients and are extremely rare (122). These patients only present with impaired post-natal growth and eczema (122). Their characterization provides information about STAT5b as a critical effector for GH signaling and normal human hematopoiesis. Their clinical profile confirmed growth hormone insensitivity syndrome (GHIS), with postnatal growth failure and IGF-I deficiency (122). Two patients developed eczema in the neonatal period and higher levels of IgE ( Table 1 ). None experienced severe immune or pulmonary symptoms (122). These missense mutations retain their capacity to become phosphorylated in response to GH and their heterodimer formation capacity is not affected. However, the mutations interfere with the transcriptional activity of the STAT5b wild type. Studies in vitro showed that the p.Q177P variant has an abnormal mobilization to the nucleus with an accumulation at the nuclear membrane. The de novo mutation is located within an α-helix and studies suggest that it disrupts the secondary structure (122). The p.Q474R and p.A478V variants were characterized with a normal capacity to translocate to nucleus, but their DNA-binding capacity was impaired (122) ( Figure 4 ). A new variant, p.Q206R, was discovered in a patient with autoimmunity, chronic lymphadenopathy, and splenomegaly; however, growth hormone insensitivity was not reported (123). This variant, located in the CCD, showed normal expression and similar mobilization to nucleus compared to healthy donors. The authors performed a luciferase reporter assay showing the mutation has a dominant negative effect and correlated this finding with an impaired STAT5 phosphorylation in response to IL-2, IL-7 and IL-15 (123).
Gain of Function Mutations
Studies on STAT5 GOF mutations shed additional light on pivotal STAT5 functions. The reported N642H variant shows increased STAT5b activity compared to the wild type and promotes upregulation of genes that are required for cell proliferation and survival (124). In silico studies show that the STAT5BN642H variant adopts a conformation that enhances the affinity for self-dimerization and is resistant to dephosphorylation (125). In addition, Ma and colleagues reported two patients with the N642H variant that developed non-clonal eosinophilia, urticaria, dermatitis, and diarrhea (6) ( Table 1 ). So far, only somatic STAT5 GOF mutations have been reported, located in the SH2 domain and in the transactivation domain (124, 126–130) ( Figure 4 ).
Somatic GOF mutations can also lead to the development of large granular cell (LGL) leukemia in addition to dysregulated activation of STAT5 associated with other types of cancers.
STAT5 and cancer
LGL leukemia is a chronic proliferation of clonal cytotoxic lymphocytes, frequently complicated with cytopenia and autoimmunity (131). The disease is classified in two subtypes: T cell large granular lymphocyte leukemia (T-LGL) (which represents about 85% of cases) and chronic lymphoproliferative disease of NK cells (NK-LGL) (which represents 10% of cases) (132). Mutations in STAT5B are more frequently associated with T-LGL leukemia, more specifically, CD3+/CD56+ and CD4+ subsets of T-LGL leukemia (133). Along with mutations in STAT5B, mutated STAT3 leads to constitutive activation and plays a fundamental role in the pathogenesis of LGL leukemia (133). When evaluating the transcriptome of LGL leukemia patients, many of the genes are regulated by STAT3 (133). There are a couple key differences to note between STAT3 and STAT5b mediated LGL leukemia. One is that CD4+ T-LGL leukemia is characterized only by STAT5B mutations and not STAT3 mutations (134). Additionally, STAT5B mutations often lead to a more aggressive presentation of the disease (133).
Hyperactivation of STAT5 is also associated with other blood malignancies such as acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia (CML), myeloproliferative neoplasms (MPNs), in addition to human tumors (breast, prostate, liver, etc.), and other cancers (5). Because the dysregulated activation of STAT5 leads to the development of leukemias and myeloproliferative disease, it is important to evaluate the role of STAT5 activators such as tyrosine kinases in these diseases. The TEL-Jak2 fusion protein has been found in patients with pre-B cell and T cell ALL and CML. The fusion of TEL and Jak2 leads to the constitutive activation of its tyrosine activity, followed by constitutive activation of STAT5 (135, 136). Other tyrosine kinase translocation products are also important drivers of cellular transformation and leukemias (137). The relationship between STAT5 and an upstream oncogene plays a critical role in the development of CML. Transformation of cells by BCR-ABL, the oncogene resulting from the fusion of BCR to ABL kinase, is the most common cause of CML. Following translocation and the merging of these genes, the tyrosine kinase activity of ABL increases, over-activating downstream STAT5. The constitutive activation of STAT5 by BCR-ABL then leads to the upregulation of genes associated with CML such as Bcl-x, Mcl-1, and D ½, and this hyperactivation leads to the dysregulated proliferation of cells (136, 138)
Because STAT5 is involved in the development and survival of lymphoid cells, its activation is also correlated with lymphocyte transformation. In a study by Heltemes-Harris and Farrar, a sample of 128 patients with ALL showed elevated levels of STAT5 phosphorylation, demonstrating that the protein plays a key role in the transformation of progenitor B cells. High levels of phosphorylated STAT5 were negatively correlated with survival. In addition, cooperation between STAT5 and mutated forms of EBF1 and PAX5 (important transcription factors for the development of B cells) was linked to the initiation of B cell transformation (1, 139).
Hyperactivation of the JAK2-STAT5 pathway can also lead to myeloproliferative neoplasms (MPNs)—a disease of hematopoietic stem cells (140). One of the most common mutations, V617F, in JAK2 has been directly linked to the constitutive activation of STAT5 (141). This mutated JAK2 requires the presence of thrombopoietin receptor (TPO-R) to promote the cytokine-independent activation of STAT5 and the MPN phenotype (141). Understanding the relationship between JAK2-STAT5 activation in MPNs can help expand treatment options for patients including ruxolitinib and other JAK inhibitors (142).
STAT5 also regulates apoptosis in cancer cells, and when it is constitutively active, mutant cells have properties of transformed cancer cells (143, 144). STAT5 activation has been shown to turn on Bcl-xL, a gene with an anti-apoptotic role, leading to the development of aggressive solid tumors (145). The link between activation of STAT5a and STAT5b to the progression of solid tumors in breast and prostate cancer, respectively, is clear but the mechanism behind the developments of these cancers requires further elucidation. The regulatory role of STAT5a during mammary gland remodeling and cell death has been well documented. Additionally, mouse models involving constitutively activated STAT5a further emphasize the transcription factor’s involvement in tumorigenesis (146). This constitutively active state of STAT5a has also been detected in humor breast cancer cell lines (147). STAT5b, on the other hand, is directly linked to the development and growth of prostate cancer. Along with being critical for the survival of prostate cancer cells, high levels of active STAT5b are also linked to early recurrence of the disease. Substantial cell death occurs in prostate cancer cells when the activation of STAT5b is inhibited (148–150). Being able to directly link this key transcription factor to the development of breast and prostate cancers has opened new doors for individualized treatment plans with an opening for the expanded landscape of biologic precision therapy. For this reason, it is critical to continue expanding our understanding of how the activation of STAT5 can promote tumorigenesis.
Evidence also suggests that STAT5 is involved in epithelial-mesenchymal transition (EMT), an important step in the process of tumor invasion. Because EMT plays such a significant role in the development of aggressive hepatocellular carcinoma (HCC), studies have been conducted linking STAT5b to the invasive properties of HCC (145). Along with solid breast and prostate tumors, dysregulation in the JAK/STAT5 pathway has been directly linked to the development of osteosarcoma (OS), a common primary bone tumor. In a study by Subramaniam and colleagues, they showed that treatment of cells with pimozide, a STAT5 inhibitor, lead to suppressed growth of OS cells. These results emphasized STAT5’s role in the development of OS and offer promising treatment options (151).
Continued research into the mechanism by which STAT5 activation contributes to the development of cancers and a better understanding of the downstream effects of dysregulated STAT5 activation may lead to the development of more accurate and effective therapeutic targets.
Conclusions
STAT5b is a master regulator in the hemopoietic compartment as well as an essential signaling partner in extra-hematopoietic tissue regulation and growth. The critical role of STAT5 in lymphocyte development, GM-CSF signaling, and growth highlights the importance of studying these mechanisms in correlation with human diseases. Enhanced knowledge on STAT5 activation and regulation will guide genomic sequencing, improve early diagnosis and targeted therapeutic interventions.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Acknowledgments
We would like to acknowledge the Texas Children’s Hospital William T. Shearer Center for Human Immunobiology for their generous support for this research. Figures 2 , 3 were made using BioRender.
Funding Statement
Texas Children’s Hospital Pediatric Pilot Award, Jeffery Model Foundation.
Abbreviations
STAT5, Signal Transducer of Activation of Transcription Factor 5; LOF, loss-of-function; GOF, gain-of-function; NK cells, Natural Killer cells; GM-CSF, granulocyte-macrophage colony-stimulating factor; EPO, erythropoietin; TPO, thrombopoietin; GH, growth hormone; Flt, Fms-like tyrosine kinase 3; GHR, growth hormone receptor; PTPs, protein tyrosine phosphatases; SOCS, suppressor of cytokine signaling; PIAS, protein inhibitors of activated STATs; Tregs, regulatory T cells; DKI mice, STAT5 tetramer-deficient mice; PAP, pulmonary alveolar proteinosis; CMV, cytomegalovirus; ALL, acute lymphoblastic leukemia; CML, chronic myelogenous leukemia.
Conflict of interest
LS consultant for ADMA, Enzyvant, Grifols, Incyte, Orchard, and Takeda.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1. Heltemes-Harris LM, Farrar MA. The role of STAT5 in lymphocyte development and transformation. Curr Opin Immunol (2012) 24(2):146–52. doi: 10.1016/j.coi.2012.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vargas-Hernandez A, Forbes LR. JAK/STAT proteins and their biological impact on NK cell development and function. Mol Immunol (2019) 115:21–30. doi: 10.1016/j.molimm.2018.12.005 [DOI] [PubMed] [Google Scholar]
- 3. Hwa V, Nadeau K, Wit JM, Rosenfeld RG. STAT5b deficiency: Lessons from STAT5b gene mutations. Best Pract Res Clin Endocrinol Metab (2011) 25(1):61–75. doi: 10.1016/j.beem.2010.09.003 [DOI] [PubMed] [Google Scholar]
- 4. Hwa V. Human growth disorders associated with impaired GH action: Defects in STAT5B and JAK2. Mol Cell Endocrinol (2021) 519:111063. doi: 10.1016/j.mce.2020.111063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rani A, Murphy JJ. STAT5 in cancer and immunity. J Interferon Cytokine Res (2016) 36(4):226–37. doi: 10.1089/jir.2015.0054 [DOI] [PubMed] [Google Scholar]
- 6. Ma CA, Xi L, Cauff B, DeZure A, Freeman AF, Hambleton S, et al. Somatic STAT5b gain-of-function mutations in early onset nonclonal eosinophilia, urticaria, dermatitis, and diarrhea. Blood (2017) 129(5):650–3. doi: 10.1182/blood-2016-09-737817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levy DE, Darnell JE, Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 3(9):651–62. doi: 10.1038/nrm909 [DOI] [PubMed] [Google Scholar]
- 8. Villarino AV, Kanno Y, Ferdinand JR, O'Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol (2015) 194(1):21–7. doi: 10.4049/jimmunol.1401867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reich NC. STATs get their move on. JAKSTAT (2013) 2(4):e27080. doi: 10.4161/jkst.27080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci (2018) 27(12):1984–2009. doi: 10.1002/pro.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vinkemeier U, Moarefi I, Darnell JE, Jr., Kuriyan J. Structure of the amino-terminal protein interaction domain of STAT-4. Science (1998) 279(5353):1048–52. doi: 10.1126/science.279.5353.1048 [DOI] [PubMed] [Google Scholar]
- 12. Hu T, Yeh JE, Pinello L, Jacob J, Chakravarthy S, Yuan GC, et al. Impact of the n-terminal domain of STAT3 in STAT3-dependent transcriptional activity. Mol Cell Biol (2015) 35(19):3284–300. doi: 10.1128/MCB.00060-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr., Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell (1998) 93(5):827–39. doi: 10.1016/S0092-8674(00)81443-9 [DOI] [PubMed] [Google Scholar]
- 14. Vinkemeier U, Cohen SL, Moarefi I, Chait BT, Kuriyan J, Darnell JE, Jr. DNA Binding of in vitro activated Stat1 alpha, Stat1 beta and truncated Stat1: Interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. EMBO J (1996) 15(20):5616–26. doi: 10.1002/j.1460-2075.1996.tb00946.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heim MH, Kerr IM, Stark GR, Darnell JE, Jr. Contribution of STAT SH2 groups to specific interferon signaling by the jak-STAT pathway. Science (1995) 267(5202):1347–9. doi: 10.1126/science.7871432 [DOI] [PubMed] [Google Scholar]
- 16. Shuai K, Horvath CM, Huang LH, Qureshi SA, Cowburn D, Darnell JE, Jr. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell (1994) 76(5):821–8. doi: 10.1016/0092-8674(94)90357-3 [DOI] [PubMed] [Google Scholar]
- 17. Neculai D, Neculai AM, Verrier S, Straub K, Klumpp K, Pfitzner E, et al. Structure of the unphosphorylated STAT5a dimer. J Biol Chem (2005) 280(49):40782–7. doi: 10.1074/jbc.M507682200 [DOI] [PubMed] [Google Scholar]
- 18. Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, et al. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell (2005) 17(6):761–71. doi: 10.1016/j.molcel.2005.02.021 [DOI] [PubMed] [Google Scholar]
- 19. Crispi S, Sanzari E, Monfregola J, De Felice N, Fimiani G, Ambrosio R, et al. Characterization of the human STAT5A and STAT5B promoters: Evidence of a positive and negative mechanism of transcriptional regulation. FEBS Lett (2004) 562(1-3):27–34. doi: 10.1016/S0014-5793(04)00166-8 [DOI] [PubMed] [Google Scholar]
- 20. Maurer B, Kollmann S, Pickem J, Hoelbl-Kovacic A, Sexl V. STAT5A and STAT5B-twins with different personalities in hematopoiesis and leukemia. Cancers (Basel) (2019) 11(11). doi: 10.3390/cancers11111726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nadeau K, Hwa V, Rosenfeld RG. STAT5b deficiency: an unsuspected cause of growth failure, immunodeficiency, and severe pulmonary disease. J Pediatr (2011) 158(5):701–8. doi: 10.1016/j.jpeds.2010.12.042 [DOI] [PubMed] [Google Scholar]
- 22. Levy DE, Marie IJ. STATus report on tetramers. Immunity (2012) 36(4):553–5. doi: 10.1016/j.immuni.2012.04.003 [DOI] [PubMed] [Google Scholar]
- 23. Fink K, Grandvaux N. STAT2 and IRF9: Beyond ISGF3. JAKSTAT (2013) 2(4):e27521. doi: 10.4161/jkst.27521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin JX, Li P, Liu D, Jin HT, He J, Ata Ur Rasheed M, et al. Critical role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity (2012) 36(4):586–99. doi: 10.1016/j.immuni.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vargas-Hernandez A, Witalisz-Siepracka A, Prchal-Murphy M, Klein K, Mahapatra S, Al-Herz W, et al. Human signal transducer and activator of transcription 5b (STAT5b) mutation causes dysregulated human natural killer cell maturation and impaired lytic function. J Allergy Clin Immunol (2020) 145(1):345–57 e9. doi: 10.1016/j.jaci.2019.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, et al. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat Struct Mol Biol (2005) 12(9):814–21. doi: 10.1038/nsmb977 [DOI] [PubMed] [Google Scholar]
- 27. Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med (2003) 349(12):1139–47. doi: 10.1056/NEJMoa022926 [DOI] [PubMed] [Google Scholar]
- 28. Rosenfeld RG, Belgorosky A, Camacho-Hubner C, Savage MO, Wit JM, Hwa V. Defects in growth hormone receptor signaling. Trends Endocrinol Metab (2007) 18(4):134–41. doi: 10.1016/j.tem.2007.03.004 [DOI] [PubMed] [Google Scholar]
- 29. Derr MA, Fang P, Sinha SK, Ten S, Hwa V, Rosenfeld RG. A novel Y332C missense mutation in the intracellular domain of the human growth hormone receptor does not alter STAT5b signaling: Redundancy of GHR intracellular tyrosines involved in STAT5b signaling. Horm Res Paediatr (2011) 75(3):187–99. doi: 10.1159/000320461 [DOI] [PubMed] [Google Scholar]
- 30. Wojcik M, Krawczynska A, Antushevich H, Herman AP. Post-receptor inhibitors of the GHR-JAK2-STAT pathway in the growth hormone signal transduction. Int J Mol Sci (2018) 19(7). doi: 10.3390/ijms19071843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DiGiandomenico A, Wylezinski LS, Hawiger J. Intracellular delivery of a cell-penetrating SOCS1 that targets IFN-gamma signaling. Sci Signal (2009) 2(80):ra37. doi: 10.1126/scisignal.1162191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chong MM, Cornish AL, Darwiche R, Stanley EG, Purton JF, Godfrey DI, et al. Suppressor of cytokine signaling-1 is a critical regulator of interleukin-7-dependent CD8+ T cell differentiation. Immunity (2003) 18(4):475–87. doi: 10.1016/S1074-7613(03)00078-5 [DOI] [PubMed] [Google Scholar]
- 33. Cornish AL, Chong MM, Davey GM, Darwiche R, Nicola NA, Hilton DJ, et al. Suppressor of cytokine signaling-1 regulates signaling in response to interleukin-2 and other gamma c-dependent cytokines in peripheral T cells. J Biol Chem (2003) 278(25):22755–61. doi: 10.1074/jbc.M303021200 [DOI] [PubMed] [Google Scholar]
- 34. Fujimoto M, Tsutsui H, Yumikura-Futatsugi S, Ueda H, Xingshou O, Abe T, et al. A regulatory role for suppressor of cytokine signaling-1 in t(h) polarization in vivo . Int Immunol (2002) 14(11):1343–50. doi: 10.1093/intimm/dxf094 [DOI] [PubMed] [Google Scholar]
- 35. Linossi EM, Calleja DJ, Nicholson SE. Understanding SOCS protein specificity. Growth Factors (2018) 36(3-4):104–17. doi: 10.1080/08977194.2018.1518324 [DOI] [PubMed] [Google Scholar]
- 36. Babon JJ, Sabo JK, Zhang JG, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: Implications for ubiquitin ligase formation and cytokine signalling suppression. J Mol Biol (2009) 387(1):162–74. doi: 10.1016/j.jmb.2009.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Greenhalgh CJ, Rico-Bautista E, Lorentzon M, Thaus AL, Morgan PO, Willson TA, et al. SOCS2 negatively regulates growth hormone action in vitro and in vivo . J Clin Invest (2005) 115(2):397–406. doi: 10.1172/JCI200522710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Favre H, Benhamou A, Finidori J, Kelly PA, Edery M. Dual effects of suppressor of cytokine signaling (SOCS-2) on growth hormone signal transduction. FEBS Lett (1999) 453(1-2):63–6. doi: 10.1016/S0014-5793(99)00681-X [DOI] [PubMed] [Google Scholar]
- 39. Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr Opin Cell Biol (2001) 13(2):182–95. doi: 10.1016/S0955-0674(00)00196-4 [DOI] [PubMed] [Google Scholar]
- 40. Rytinki MM, Kaikkonen S, Pehkonen P, Jaaskelainen T, Palvimo JJ. PIAS proteins: pleiotropic interactors associated with SUMO. Cell Mol Life Sci (2009) 66(18):3029–41. doi: 10.1007/s00018-009-0061-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sharrocks AD. PIAS proteins and transcriptional regulation–more than just SUMO E3 ligases? Genes Dev (2006) 20(7):754–8. doi: 10.1101/gad.1421006 [DOI] [PubMed] [Google Scholar]
- 42. Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, et al. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-b cell development. Nat Immunol (2010) 11(2):171–9. doi: 10.1038/ni.1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nutt SL, Kee BL. The transcriptional regulation of b cell lineage commitment. Immunity (2007) 26(6):715–25. doi: 10.1016/j.immuni.2007.05.010 [DOI] [PubMed] [Google Scholar]
- 44. Kikuchi K, Lai AY, Hsu CL, Kondo M. IL-7 receptor signaling is necessary for stage transition in adult b cell development through up-regulation of EBF. J Exp Med (2005) 201(8):1197–203. doi: 10.1084/jem.20050158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hirokawa S, Sato H, Kato I, Kudo A. EBF-regulating Pax5 transcription is enhanced by STAT5 in the early stage of b cells. Eur J Immunol (2003) 33(7):1824–9. doi: 10.1002/eji.200323974 [DOI] [PubMed] [Google Scholar]
- 46. Dias S, Silva H, Jr., Cumano A, Vieira P. Interleukin-7 is necessary to maintain the b cell potential in common lymphoid progenitors. J Exp Med (2005) 201(6):971–9. doi: 10.1084/jem.20042393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, et al. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood (2006) 107(12):4898–906. doi: 10.1182/blood-2005-09-3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pelham SJ, Caldirola MS, Avery DT, Mackie J, Rao G, Gothe F, et al. STAT5B restrains human b-cell differentiation to maintain humoral immune homeostasis. J Allergy Clin Immunol (2022) 150(4):931–46. doi: 10.1016/j.jaci.2022.04.011 [DOI] [PubMed] [Google Scholar]
- 49. Owen DL, Farrar MA. STAT5 and CD4 (+) T cell immunity. F1000Res (2017) 6:32. doi: 10.12688/f1000research.9838.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol (2004) 24(18):8037–47. doi: 10.1128/MCB.24.18.8037-8047.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA (2006) 103(4):1000–5. doi: 10.1073/pnas.0507350103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Park JH, Adoro S, Guinter T, Erman B, Alag AS, Catalfamo M, et al. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat Immunol (2010) 11(3):257–64. doi: 10.1038/ni.1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol (2011) 12(6):551–9. doi: 10.1038/ni.2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nat Immunol (2008) 9(11):1288–96. doi: 10.1038/ni.1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity (2003) 19(5):739–48. doi: 10.1016/S1074-7613(03)00292-9 [DOI] [PubMed] [Google Scholar]
- 56. Yamane H, Zhu J, Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med (2005) 202(6):793–804. doi: 10.1084/jem.20051304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, et al. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci U S A. (2004) 101(11):3880–5. doi: 10.1073/pnas.0400339101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. D'Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol (2005) 6(11):1152–9. doi: 10.1038/ni1264 [DOI] [PubMed] [Google Scholar]
- 59. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol (2005) 6(11):1142–51. doi: 10.1038/ni1263 [DOI] [PubMed] [Google Scholar]
- 60. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. implications for the nonredundant function of IL-2. Immunity (2002) 17(2):167–78. doi: 10.1016/s1074-7613(02)00367-9 [DOI] [PubMed] [Google Scholar]
- 61. Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol (2006) 177(5):2770–4. doi: 10.4049/jimmunol.177.5.2770 [DOI] [PubMed] [Google Scholar]
- 62. Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo . J Immunol (2011) 186(11):6329–37. doi: 10.4049/jimmunol.1100061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in treg cell function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science (1995) 268(5216):1472–6. doi: 10.1126/science.7770771 [DOI] [PubMed] [Google Scholar]
- 65. Villarino A, Laurence A, Robinson GW, Bonelli M, Dema B, Afzali B, et al. Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. Elife (2016) 5. doi: 10.7554/eLife.08384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22(11):633–40. doi: 10.1016/S1471-4906(01)02060-9 [DOI] [PubMed] [Google Scholar]
- 67. Angelo LS, Banerjee PP, Monaco-Shawver L, Rosen JB, Makedonas G, Forbes LR, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res (2015) 62(3):341–56. doi: 10.1007/s12026-015-8664-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity (1998) 9(5):669–76. doi: 10.1016/S1074-7613(00)80664-0 [DOI] [PubMed] [Google Scholar]
- 69. Ranson T, Vosshenrich CA, Corcuff E, Richard O, Laloux V, Lehuen A, et al. IL-15 availability conditions homeostasis of peripheral natural killer T cells. Proc Natl Acad Sci USA (2003) 100(5):2663–8. doi: 10.1073/pnas.0535482100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ranson T, Vosshenrich CA, Corcuff E, Richard O, Muller W, Di Santo JP. IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood (2003) 101(12):4887–93. doi: 10.1182/blood-2002-11-3392 [DOI] [PubMed] [Google Scholar]
- 71. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26(4):503–17. doi: 10.1016/j.immuni.2007.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tamzalit F, Barbieux I, Plet A, Heim J, Nedellec S, Morisseau S, et al. IL-15.IL-15Ralpha complex shedding following trans-presentation is essential for the survival of IL-15 responding NK and T cells. Proc Natl Acad Sci USA (2014) 111(23):8565–70. doi: 10.1073/pnas.1405514111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mrozek E, Anderson P, Caligiuri MA. Role of interleukin-15 in the development of human CD56+ natural killer cells from CD34+ hematopoietic progenitor cells. Blood (1996) 87(7):2632–40. doi: 10.1182/blood.V87.7.2632.bloodjournal8772632 [DOI] [PubMed] [Google Scholar]
- 74. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo . J Exp Med (2009) 206(1):25–34. doi: 10.1084/jem.20082013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kanai T, Jenks J, Nadeau KC. The STAT5b pathway defect and autoimmunity. Front Immunol (2012) 3:234. doi: 10.3389/fimmu.2012.00234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lin JX, Leonard WJ. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene (2000) 19(21):2566–76. doi: 10.1038/sj.onc.1203523 [DOI] [PubMed] [Google Scholar]
- 77. Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med (1998) 188(11):2067–74. doi: 10.1084/jem.188.11.2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Villarino AV, Sciume G, Davis FP, Iwata S, Zitti B, Robinson GW, et al. Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J Exp Med (2017) 214(10):2999–3014. doi: 10.1084/jem.20150907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discovery (2016) 6(4):414–29. doi: 10.1158/2159-8290.CD-15-0732 [DOI] [PubMed] [Google Scholar]
- 80. Lin JX, Du N, Li P, Kazemian M, Gebregiorgis T, Spolski R, et al. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat Commun (2017) 8(1):1320. doi: 10.1038/s41467-017-01477-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. James AM, Hsu HT, Dongre P, Uzel G, Mace EM, Banerjee PP, et al. Rapid activation receptor- or IL-2-induced lytic granule convergence in human natural killer cells requires src, but not downstream signaling. Blood (2013) 121(14):2627–37. doi: 10.1182/blood-2012-06-437012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bonnema JD, Rivlin KA, Ting AT, Schoon RA, Abraham RT, Leibson PJ. Cytokine-enhanced NK cell-mediated cytotoxicity. Positive modulatory effects of IL-2 and IL-12 on stimulus-dependent granule exocytosis. J Immunol (1994) 152(5):2098–104. [PubMed] [Google Scholar]
- 83. Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M, et al. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood (2003) 101(8):3052–7. doi: 10.1182/blood-2002-09-2876 [DOI] [PubMed] [Google Scholar]
- 84. Vargas-Hernandez A, Forbes LR. The impact of immunodeficiency on NK cell maturation and function. Curr Allergy Asthma Rep (2019) 19(1):2. doi: 10.1007/s11882-019-0836-8 [DOI] [PubMed] [Google Scholar]
- 85. Scoville SD, Freud AG, Caligiuri MA. Modeling human natural killer cell development in the era of innate lymphoid cells. Front Immunol (2017) 8:360. doi: 10.3389/fimmu.2017.00360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cording S, Medvedovic J, Aychek T, Eberl G. Innate lymphoid cells in defense, immunopathology and immunotherapy. Nat Immunol (2016) 17(7):755–7. doi: 10.1038/ni.3448 [DOI] [PubMed] [Google Scholar]
- 87. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat Immunol (2016) 17(7):758–64. doi: 10.1038/ni.3482 [DOI] [PubMed] [Google Scholar]
- 88. Olthof SG, Fatrai S, Drayer AL, Tyl MR, Vellenga E, Schuringa JJ. Downregulation of signal transducer and activator of transcription 5 (STAT5) in CD34+ cells promotes megakaryocytic development, whereas activation of STAT5 drives erythropoiesis. Stem Cells (2008) 26(7):1732–42. doi: 10.1634/stemcells.2007-0899 [DOI] [PubMed] [Google Scholar]
- 89. Gillinder KR, Tuckey H, Bell CC, Magor GW, Huang S, Ilsley MD, et al. Direct targets of pSTAT5 signalling in erythropoiesis. PloS One (2017) 12(7):e0180922. doi: 10.1371/journal.pone.0180922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Perreault AA, Venters BJ. Integrative view on how erythropoietin signaling controls transcription patterns in erythroid cells. Curr Opin Hematol (2018) 25(3):189–95. doi: 10.1097/MOH.0000000000000415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tothova Z, Tomc J, Debeljak N, Solar P. STAT5 as a key protein of erythropoietin signalization. Int J Mol Sci (2021) 22(13). doi: 10.3390/ijms22137109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Grebien F, Kerenyi MA, Kovacic B, Kolbe T, Becker V, Dolznig H, et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood (2008) 111(9):4511–22. doi: 10.1182/blood-2007-07-102848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Koulnis M, Porpiglia E, Hidalgo D, Socolovsky M. Erythropoiesis: From molecular pathways to system properties. Adv Exp Med Biol (2014) 844:37–58. doi: 10.1007/978-1-4939-2095-2_3 [DOI] [PubMed] [Google Scholar]
- 94. Love PE, Warzecha C, Li L. Ldb1 complexes: The new master regulators of erythroid gene transcription. Trends Genet (2014) 30(1):1–9. doi: 10.1016/j.tig.2013.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Perkins AC, Gaensler KM, Orkin SH. Silencing of human fetal globin expression is impaired in the absence of the adult beta-globin gene activator protein EKLF. Proc Natl Acad Sci USA (1996) 93(22):12267–71. doi: 10.1073/pnas.93.22.12267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wijgerde M, Gribnau J, Trimborn T, Nuez B, Philipsen S, Grosveld F, et al. The role of EKLF in human beta-globin gene competition. Genes Dev (1996) 10(22):2894–902. doi: 10.1101/gad.10.22.2894 [DOI] [PubMed] [Google Scholar]
- 97. Pevny L, Simon MC, Robertson E, Klein WH, Tsai SF, D'Agati V, et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature (1991) 349(6306):257–60. doi: 10.1038/349257a0 [DOI] [PubMed] [Google Scholar]
- 98. Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature (1995) 375(6529):316–8. doi: 10.1038/375316a0 [DOI] [PubMed] [Google Scholar]
- 99. Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: A direct role for Stat5 in bcl-X(L) induction. Cell (1999) 98(2):181–91. doi: 10.1016/S0092-8674(00)81013-2 [DOI] [PubMed] [Google Scholar]
- 100. Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood (2001) 98(12):3261–73. doi: 10.1182/blood.V98.12.3261 [DOI] [PubMed] [Google Scholar]
- 101. Zhu BM, McLaughlin SK, Na R, Liu J, Cui Y, Martin C, et al. Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood (2008) 112(5):2071–80. doi: 10.1182/blood-2007-12-127480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kirito K, Watanabe T, Sawada K, Endo H, Ozawa K, Komatsu N. Thrombopoietin regulates bcl-xL gene expression through Stat5 and phosphatidylinositol 3-kinase activation pathways. J Biol Chem (2002) 277(10):8329–37. doi: 10.1074/jbc.M109824200 [DOI] [PubMed] [Google Scholar]
- 103. Park HJ, Li J, Hannah R, Biddie S, Leal-Cervantes AI, Kirschner K, et al. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J (2016) 35(6):580–94. doi: 10.15252/embj.201592383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, et al. Pulmonary alveolar proteinosis. Nat Rev Dis Primers (2019) 5(1):16. doi: 10.1038/s41572-019-0066-3 [DOI] [PubMed] [Google Scholar]
- 105. Trapnell BC, Whitsett JA. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol (2002) 64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847 [DOI] [PubMed] [Google Scholar]
- 106. Metcalf D, Johnson GR, Burgess AW. Direct stimulation by purified GM-CSF of the proliferation of multipotential and erythroid precursor cells. Blood (1980) 55(1):138–47. doi: 10.1182/blood.V55.1.138.138 [DOI] [PubMed] [Google Scholar]
- 107. Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J Leukoc Biol (2002) 71(3):511–9. doi: 10.1189/jlb.71.3.511 [DOI] [PubMed] [Google Scholar]
- 108. Matsumoto A, Masuhara M, Mitsui K, Yokouchi M, Ohtsubo M, Misawa H, et al. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood (1997) 89(9):3148–54. doi: 10.1182/blood.V89.9.3148 [DOI] [PubMed] [Google Scholar]
- 109. Lilly M, Le T, Holland P, Hendrickson SL. Sustained expression of the pim-1 kinase is specifically induced in myeloid cells by cytokines whose receptors are structurally related. Oncogene (1992) 7(4):727–32. [PubMed] [Google Scholar]
- 110. Nakata K, Akagawa KS, Fukayama M, Hayashi Y, Kadokura M, Tokunaga T. Granulocyte-macrophage colony-stimulating factor promotes the proliferation of human alveolar macrophages in vitro . J Immunol (1991) 147(4):1266–72. doi: 10.4049/jimmunol.147.4.1266 [DOI] [PubMed] [Google Scholar]
- 111. Huffman Reed JA, Rice WR, Zsengeller ZK, Wert SE, Dranoff G, Whitsett JA. GM-CSF enhances lung growth and causes alveolar type II epithelial cell hyperplasia in transgenic mice. Am J Physiol (1997) 273(4):L715–25. doi: 10.1152/ajplung.1997.273.4.L715 [DOI] [PubMed] [Google Scholar]
- 112. Krone KA, Foley CL, Fishman MP, Vargas SO, Forbes LR, Vece TJ, et al. Signal transducer and activator of transcription 5B deficiency-associated lung disease. Am J Respir Crit Care Med (2022) 205(10):1245–50. doi: 10.1164/rccm.202111-2527LE [DOI] [PubMed] [Google Scholar]
- 113. Scaglia PA, Martinez AS, Feigerlova E, Bezrodnik L, Gaillard MI, Di Giovanni D, et al. A novel missense mutation in the SH2 domain of the STAT5B gene results in a transcriptionally inactive STAT5b associated with severe IGF-I deficiency, immune dysfunction, and lack of pulmonary disease. J Clin Endocrinol Metab (2012) 97(5):E830–9. doi: 10.1210/jc.2011-2554 [DOI] [PubMed] [Google Scholar]
- 114. Acres MJ, Gothe F, Grainger A, Skelton AJ, Swan DJ, Willet JDP, et al. Signal transducer and activator of transcription 5B deficiency due to a novel missense mutation in the coiled-coil domain. J Allergy Clin Immunol (2019) 143(1):413–6 e4. doi: 10.1016/j.jaci.2018.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M, et al. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics (2006) 118(5):e1584–92. doi: 10.1542/peds.2005-2882 [DOI] [PubMed] [Google Scholar]
- 116. Pugliese-Pires PN, Tonelli CA, Dora JM, Silva PC, Czepielewski M, Simoni G, et al. A novel STAT5B mutation causing GH insensitivity syndrome associated with hyperprolactinemia and immune dysfunction in two male siblings. Eur J Endocrinol (2010) 163(2):349–55. doi: 10.1530/EJE-10-0272 [DOI] [PubMed] [Google Scholar]
- 117. Vidarsdottir S, Walenkamp MJ, Pereira AM, Karperien M, van Doorn J, van Duyvenvoorde HA, et al. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. J Clin Endocrinol Metab (2006) 91(9):3482–5. doi: 10.1210/jc.2006-0368 [DOI] [PubMed] [Google Scholar]
- 118. Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, et al. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab (2005) 90(7):4260–6. doi: 10.1210/jc.2005-0515 [DOI] [PubMed] [Google Scholar]
- 119. Hwa V, Camacho-Hubner C, Little BM, David A, Metherell LA, El-Khatib N, et al. Growth hormone insensitivity and severe short stature in siblings: A novel mutation at the exon 13-intron 13 junction of the STAT5b gene. Horm Res (2007) 68(5):218–24. doi: 10.1159/000101334 [DOI] [PubMed] [Google Scholar]
- 120. de Araujo ED, Orlova A, Neubauer HA, Bajusz D, Seo HS, Dhe-Paganon S, et al. Structural implications of STAT3 and STAT5 SH2 domain mutations. Cancers (Basel) (2019) 11(11). doi: 10.3390/cancers11111757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bang P, Woelfle J, Perrot V, Sert C, Polak M. Effectiveness and safety of rhIGF1 therapy in patients with or without laron syndrome. Eur J Endocrinol (2021) 184(2):267–76. doi: 10.1530/EJE-20-0325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun (2018) 9(1):2105. doi: 10.1038/s41467-018-04521-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Majri SS, Fritz JM, Villarino AV, Zheng L, Kanellopoulou C, Chaigne-Delalande B, et al. STAT5B: A differential regulator of the life and death of CD4(+) effector memory T cells. J Immunol (2018) 200(1):110–8. doi: 10.4049/jimmunol.1701133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rajala HL, Eldfors S, Kuusanmaki H, van Adrichem AJ, Olson T, Lagstrom S, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood (2013) 121(22):4541–50. doi: 10.1182/blood-2012-12-474577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. de Araujo ED, Erdogan F, Neubauer HA, Meneksedag-Erol D, Manaswiyoungkul P, Eram MS, et al. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat Commun (2019) 10(1):2517. doi: 10.1038/s41467-019-10422-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kiel MJ, Sahasrabuddhe AA, Rolland DCM, Velusamy T, Chung F, Schaller M, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in sezary syndrome. Nat Commun (2015) 6:8470. doi: 10.1038/ncomms9470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kucuk C, Jiang B, Hu X, Zhang W, Chan JK, Xiao W, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun (2015) 6:6025. doi: 10.1038/ncomms7025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet (2015) 47(9):1061–6. doi: 10.1038/ng.3358 [DOI] [PubMed] [Google Scholar]
- 129. Andersson EI, Tanahashi T, Sekiguchi N, Gasparini VR, Bortoluzzi S, Kawakami T, et al. High incidence of activating STAT5B mutations in CD4-positive T-cell large granular lymphocyte leukemia. Blood (2016) 128(20):2465–8. doi: 10.1182/blood-2016-06-724856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Bhattacharya D, Teramo A, Gasparini VR, Huuhtanen J, Kim D, Theodoropoulos J, et al. Identification of novel STAT5B mutations and characterization of TCRbeta signatures in CD4+ T-cell large granular lymphocyte leukemia. Blood Cancer J (2022) 12(2):31. doi: 10.1038/s41408-022-00630-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Teramo A, Barila G, Calabretto G, Vicenzetto C, Gasparini VR, Semenzato G, et al. Insights into genetic landscape of Large granular lymphocyte leukemia. Front Oncol (2020) 10:152. doi: 10.3389/fonc.2020.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Munoz-Garcia N, Jara-Acevedo M, Caldas C, Barcena P, Lopez A, Puig N, et al. STAT3 and STAT5B mutations in T/NK-cell chronic lymphoproliferative disorders of Large granular lymphocytes (LGL): Association with disease features. Cancers (Basel) (2020) 12(12). doi: 10.3390/cancers12123508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lamy T, Moignet A, Loughran TP, Jr. LGL leukemia: From pathogenesis to treatment. Blood (2017) 129(9):1082–94. doi: 10.1182/blood-2016-08-692590 [DOI] [PubMed] [Google Scholar]
- 134. Teramo A, Barila G, Calabretto G, Ercolin C, Lamy T, Moignet A, et al. STAT3 mutation impacts biological and clinical features of T-LGL leukemia. Oncotarget (2017) 8(37):61876–89. doi: 10.18632/oncotarget.18711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science (1997) 278(5341):1309–12. doi: 10.1126/science.278.5341.1309 [DOI] [PubMed] [Google Scholar]
- 136. Lewis RS, Ward AC. Stat5 as a diagnostic marker for leukemia. Expert Rev Mol Diagn (2008) 8(1):73–82. doi: 10.1586/14737159.8.1.73 [DOI] [PubMed] [Google Scholar]
- 137. Ferbeyre G, Moriggl R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim Biophys Acta (2011) 1815(1):104–14. doi: 10.1016/j.bbcan.2010.10.004 [DOI] [PubMed] [Google Scholar]
- 138. Nam S, Williams A, Vultur A, List A, Bhalla K, Smith D, et al. Dasatinib (BMS-354825) inhibits Stat5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol Cancer Ther (2007) 6(4):1400–5. doi: 10.1158/1535-7163.MCT-06-0446 [DOI] [PubMed] [Google Scholar]
- 139. Heltemes-Harris LM, Willette MJ, Ramsey LB, Qiu YH, Neeley ES, Zhang N, et al. Ebf1 or Pax5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia. J Exp Med (2011) 208(6):1135–49. doi: 10.1084/jem.20101947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Constantinescu SN, Vainchenker W, Levy G, Papadopoulos N. Functional consequences of mutations in myeloproliferative neoplasms. Hemasphere (2021) 5(6):e578. doi: 10.1097/HS9.0000000000000578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Cahu X, Constantinescu SN. Oncogenic drivers in myeloproliferative neoplasms: From JAK2 to calreticulin mutations. Curr Hematol Malig Rep (2015) 10(4):335–43. doi: 10.1007/s11899-015-0278-x [DOI] [PubMed] [Google Scholar]
- 142. Talpaz M, Kiladjian JJ. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia (2021) 35(1):1–17. doi: 10.1038/s41375-020-0954-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Xi S, Zhang Q, Gooding WE, Smithgall TE, Grandis JR. Constitutive activation of Stat5b contributes to carcinogenesis in vivo . Cancer Res (2003) 63(20):6763–71. [PubMed] [Google Scholar]
- 144. Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol (1998) 18(7):3871–9. doi: 10.1128/MCB.18.7.3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lee TK, Man K, Poon RT, Lo CM, Yuen AP, Ng IO, et al. Signal transducers and activators of transcription 5b activation enhances hepatocellular carcinoma aggressiveness through induction of epithelial-mesenchymal transition. Cancer Res (2006) 66(20):9948–56. doi: 10.1158/0008-5472.CAN-06-1092 [DOI] [PubMed] [Google Scholar]
- 146. Humphreys RC, Hennighausen L. Signal transducer and activator of transcription 5a influences mammary epithelial cell survival and tumorigenesis. Cell Growth Differ (1999) 10(10):685–94. [PubMed] [Google Scholar]
- 147. Yamashita H, Iwase H, Toyama T, Fujii Y. Naturally occurring dominant-negative Stat5 suppresses transcriptional activity of estrogen receptors and induces apoptosis in T47D breast cancer cells. Oncogene (2003) 22(11):1638–52. doi: 10.1038/sj.onc.1206277 [DOI] [PubMed] [Google Scholar]
- 148. Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi M, Alanen K, et al. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. J Biol Chem (2003) 278(29):27287–92. doi: 10.1074/jbc.M304307200 [DOI] [PubMed] [Google Scholar]
- 149. Li H, Zhang Y, Glass A, Zellweger T, Gehan E, Bubendorf L, et al. Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clin Cancer Res (2005) 11(16):5863–8. doi: 10.1158/1078-0432.CCR-05-0562 [DOI] [PubMed] [Google Scholar]
- 150. Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, et al. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. Am J Pathol (2010) 176(4):1959–72. doi: 10.2353/ajpath.2010.090653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Subramaniam D, Angulo P, Ponnurangam S, Dandawate P, Ramamoorthy P, Srinivasan P, et al. Suppressing STAT5 signaling affects osteosarcoma growth and stemness. Cell Death Dis (2020) 11(2):149. doi: 10.1038/s41419-020-2335-1 [DOI] [PMC free article] [PubMed] [Google Scholar]