

Abstract
Arrhythmogenic cardiomyopathy is an inheritable heart disease characterized by lethal heart rhythms and abnormal contractile function. Mutations in desmoplakin (DSP), a protein linking the cardiac desmosome with intermediate filaments, are associated with arrhythmogenic cardiomyopathy. Here we generated a human induced pluripotent stem cell (hiPSC) line from a patient with a heterozygous protein-truncating variant in DSP (c.1386del Leu462Serfs*22). This line has a normal karyotype and expression of pluripotency markers, and can differentiate into all three germ layers. This line is well suited for in vitro mechanistic studies of mechanism of DSP protein-truncation mutations in the context of arrhythmogenic cardiomyopathy.
1. Resource utility
The patient carrying the DSP (c.1386 Leu462Serfs*22) mutation showed an arrhythmogenic cardiomyopathy phenotype and died of heart failure at age 47. The hiPSC line described here provides a means for generating in vitro populations expressing the DSP (c.1386 Leu462Serfs*22) protein-truncating variant, enabling study of disease mechanism and potential drug screening.
2. Resource details
Arrhythmogenic cardiomyopathy (AC) is an inherited heart disease estimated to affect 0.02 % to 0.1 % of the population with increased risk of sudden cardiac death and heart failure (Corrado et al., 2017). AC typically presents with ventricular arrhythmia and systolic dysfunction, myocyte depletion, and incursion of fibrotic and fatty tissue. Diagnosis relies on a combination of tools including electrocardiogram, echocardiograph, genetic screening, and biopsy.
Genetic variants associated with AC are predominantly clustered in genes associated with the desmosome, a key intracellular structure in cardiac tissue connecting the cytoskeleton and cell surface across cell junctions (Delmar and McKenna, 2010). Desmoplakin (DSP) is a critical protein in the desmosome, linking intermediate filaments with intercalated discs to maintain structure during contraction. Mutations in DSP are associated with many related diseases including AC, Naxos disease, and Carvajal syndrome; 5–10 % of AC cases exhibit a pathogenic DSP variant (Gerull et al., 2019). A conditional mouse knockout model of DSP recapitulated the AC phenotype, suggesting DSP haploinsufficiency as a possible mechanism (Garcia-Gras et al., 2006). 334 of the reported 488 pathogenic and likely pathogenic DSP variants on ClinVar are protein-truncating variants (as of 17 August 2022). However, no patient-derived induced pluripotent stem cell (iPSC) model with a DSP protein-truncating variant has been described. A missense mutation iPSC model showed reduced DSP expression but failed to exhibit the characteristic arrhythmia or contractile dysfunction in cardiomyocytes (Ng et al., 2019). As such, a DSP protein-truncating variant iPSC model would provide a new tool for studying of disease mechanisms in an in vitro context.
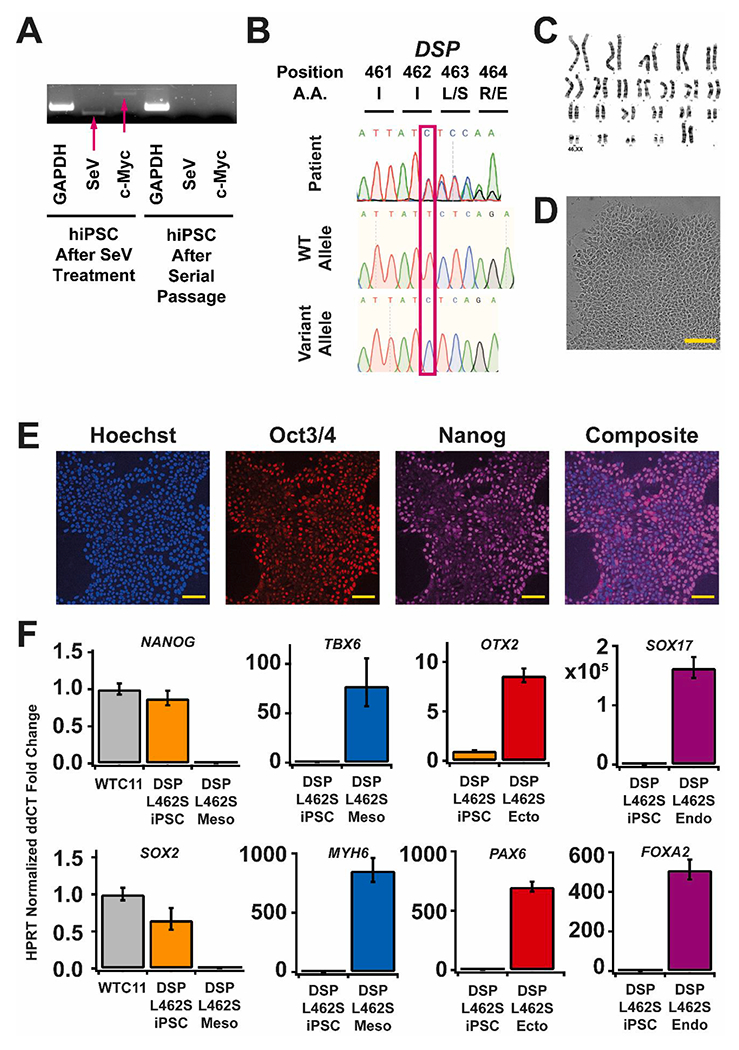
We generated an iPSC line from a 47-year old female carrying DSP (c.1386del Leu462Serfs*22) protein-truncation variant and exhibiting severe biventricular dysfunction (Table 1). With family consent, peripheral mononuclear blood cells (PBMCs) were reprogrammed into iPSCs using Yamanaka factors Oct3/4, Sox2, Klf4, and c-Myc and Sendaivirus transfection. Transgene and vector removal were induced with four days of culture at 38° C and confirmed via PCR (Fig. 1A). The resulting iPSC clone was confirmed to have the DSP protein-truncating variant by Sanger sequencing of the genomic DNA and of the individual alleles via standard subcloning methods (Fig. 1B). Short tandem repeat (STR) analysis verified these hiPSCs genetically matched the donor PBMCs (submitted in the archive with journal). The karyotype of the iPSC clone was assessed as normal at passage 9 (Fig. 1C), and the culture was assessed as free of mycoplasma contamination (Supp Fig. A). The iPSC clone exhibited normal morphology (Fig. 1D, scale 100 μm) and nuclear expression of pluripotency markers Nanog and Oct3/4 at passage 11 (Fig. 1E–F, scale 25 μm). Expression of NANOG and SOX2 was confirmed by RT-qPCR as compared to WTC-11 hiPSCs (Fig. 1F). Trilineage differentiation was confirmed by RT-qPCR detection of expression of markers for ectoderm (PAX6, OTX2), mesoderm (TBX6, MYH6), and endoderm (SOX17, FOXA2). In sum, this DSP variant line exhibits all characteristics of pluripotency and is well suited for modelling the effects of DSP protein-truncating variants in the context of AC and in other desmosomal diseases.
Table 1.
Characterization and validation.
| classification | Test | Result | Data |
|---|---|---|---|
| Morphology | Photography Bright field | Normal | Fig. 1D |
| Quantitative analysis (RT-qPCR) | mRNA expression of NANOG and SOX2 | Fig. 1G | |
| Genotype | Karyotype (G-banding) and resolution [mandatory] | Normal Karyotype 46XX Resolution 5 Mb | Fig. 1C |
| Identity | Microsatellite PCR (mPCR) OR | N/A | N/A |
| STR analysis [mandatory] | Matched 27 allelic polymorphisms across 15 STR loci | Available with the authors | |
| Mutation analysis (IF APPLICABLE) | Sequencing | Heterozygous | Fig. 1B |
| Southern Blot OR WGS | N/A | N/A | |
| Microbiology and virology | Mycoplasma | Luminescence:Negative | Supplementary |
| Donor screening (OPTIONAL) | HIV 1 + 2 Hepatitis B, Hepatitis C | N/A | N/A |
| Genotype additional info (OPTIONAL) | Blood group genotyping | N/A | N/A |
| HLA tissue typing | N/A | N/A |
3. Materials and methods
3.1. Reprogramming
Patient PBMCs were isolated from blood with familial consent. PBMCs were plated at 3x105 cells/well and transformed using CytoTune-iPS 2.0 Sendai Reprogramming Kit (Invitrogen #A16517, Lot #L2170059) with recommended titers at MOI 5:5:3 (Klf4-Oct4-Sox2:c-Myc:Klf4). Cells were spinfected 45 min at 1000 × g with virus and 4 μg/mL polybrene. Post-reprogramming, cells were maintained on MEF-feeder cells in StemPro-34 SFM (Gibco #10639) and transitioned to mTeSR+ (STEMCELL Technologies #100-0276) before colony picking. Transgene removal was confirmed via RNA isolation, cDNA generation, and PCR (Table 2).
Table 2.
Reagents details.
| |
Antibodies used for immunocytochemistry/flow-cytometry |
|||
|---|---|---|---|---|
| Antibody | Dilution | Company Cat # | RRID | |
|
| ||||
| Pluripotency Markers | Mouse anti-OCT3/4 | 1:30 | Santa Cruz Cat# sc-5279 | RRID: AB_628051 |
| Rabbit anti-NANOG | 1:100 | Proteintech Cat# 14295-1-AP | RRID: AB_1607719 | |
| Secondary Antibodies | Alexa Fluor Plus 647 Goat anti-Mouse IgG | 1:1000 | Invitrogen Cat# A32728 | RRID: AB_2633277 |
| Alexa Fluor Plus 647 Goat anti-Rabbit IgG | 1:1000 | Invitrogen Cat# A32733 | RRID: AB_2633282 | |
| Primers | ||||
|
| ||||
| Target | Size of band | Forward/Reverse primer (5′-3′) | ||
|
| ||||
| Sendai Virus (PCR) | Sendai Virus Genome | 181 bp | GGATCACTAGGTGATATCGAGC/ACCAGACAAGAGTTTAAGAGATATGTATC | |
| Transgene (PCR) | c-Myc | 532 bp | TAACTGACTAGCAGGCTTGTCG/TCCACATACAGTCCTGGATGATGATG | |
| House-Keeping Gene (PCR) | GAPDH | 238 bp | GAGTCAACGGATTTGGTCGT/TTGATTTTGGAGGGATCTCG | |
| Pluripotency Markers (qPCR) |
NANOG
SOX2 |
237 bp 155 bp |
AAGGTCCCGGTCAAGAAACAG/CTTCTGCGTCACACCATTGC GCCGAGTGGAAACTTTTGTCG/GGCAGCGTGTACTTATCCTTCT |
|
| Ectoderm Markers (qPCR) | PAX6 | 111 bp | TGGGCAGGTATTACGAGACTG/ACTCCCGCTTATACTGGGCTA | |
| OTX2 | 179 bp | CAAAGTGAGACCTGCCAAAAAGA/TGGACAAGGGATCTGACAGTG | ||
| Endoderm Markers (qPCR) | SOX17 | 94 bp | GTGGACCGCACGGAATTTG/GGAGATTCACACCGGAGTCA | |
| FOXA2 | 83 bp | GGAGCAGCTACTATGCAGAGC/CGTGTTCATGCCGTTCATCC | ||
| Mesoderm Markers (qPCR) | TBX6 | 139 bp | CATCCACGAGAATTGTACCCG/AGCAATCCAGTTTAGGGGTGT | |
| MYH6 | 187 bp | CAAGTTGGAAGACGAGTGCT/ATGGGCCTCTTGTAGAGCTT | ||
| House-Keeping Gene (qPCR) | HPRT | 94 bp | TGACACTGGCAAAACAATGCA/GGTCCTTTTCACCAGCAAGCT | |
| DSP Amplification for Sequencing (PCR) | DSP | 1406 bp | GGGGCTCCAGGACTCCATCAG/GTCCTTCAGGATACACTCATCCCCC | |
| DSP Sequencing (Sanger) | DSP | GCGTCAGGTGCAGAACTTGG | ||
3.2. Cell culture
iPSCs were cultured in mTeSR + on 80 μg/mL Matrigel (Corning #356231, Lot #1242001) coated plates, 5 % CO2, 37 °C. Cells were fed every other day and passaged with versene before differentiation was morphologically evident. Media was supplemented with 10 μM ROCK inhibitor (SelleckChem #Y27632) for 24 h post-passage. WTC-11 hiPSC line was a generous gift from the Allen Institute (Coriell #GM25256).
3.3. Trilineage differentiation
Passage 10 iPSCs were seeded in Matrigel-coated 24-well plates at 4x105, 1x105, and 4x105 cells/well for ectoderm, mesoderm, and endoderm differentiations, respectively. Cells were fed daily per manufacturer’s protocol (STEMCELL Technologies #5230). Cells were lysed for RNA isolation at day 5 (ectoderm and mesoderm) or 7 (endoderm).
3.4. Immunofluorescence
Passage 11 iPSCs were fixed in 4 % paraformaldehyde 10 min RT. Cells were permeabilized with 0.2 % Triton X-100 in PBS 5 min RT and blocked overnight with 3 % BSA (Sigma #A7030) in PBS. Cells were incubated overnight 4 °C with primary antibody (Table 2) in blocking buffer. Cells were washed and incubated with secondary antibody (Table 2) in PBS2 hours RT. Nuclei were stained with 1:2000 Hoechst 33,342 (Invitrogen H3570) 10 min RT.
3.5. RT-qPCR
RNA from passage 11 iPSCs and passage 10 trilineage differentiated cells was isolated with PureLink RNA minipreps (Invitrogen #12183018A) with PureLink Dnase treatment (Invitrogen #12185010). cDNA was reverse transcription generated using SensiFast cDNA Synthesis Kit (Thomas Scientific #C755H65). qPCR was performed with 10 μL technical triplicate reactions, 40 cycles, with SYBR Select Master Mix (Invitrogen #4472919), 5 ng cDNA, 100 nM forward/reverse primer (Table 2).
3.6. PCR and Sequencing
Genomic DNA was isolated from PBMC passage 1 and iPSC passage 11 using Dneasy Blood and Tissue Kit (Qiagen #69506). DSP fragment containing mutation was PCR amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs #M0491L), 40 cycles: 95 °C 30 s, 55 °C 30 s, 72 °C 30 s, using Bio Rad T100 Thermocycler. PCR products were run on 2 % agarose gels and extracted using Fermentas Gel Extraction Kit (Invitrogen #K0692). Alleles were cloned into plasmids using CloneJET PCR Cloning Kit (Thermo Scientific #K1321) and Stbl3 chemically competent E. coli (Invitrogen #C737303). Sanger sequencing was performed by Eurofins Genomics.
3.7. Karyotyping
G-banded chromosome analysis of twenty metaphase cells at passage 9 was performed by Diagnostic Cytogenetics Incorporated.
3.8. STR analysis
STR analysis was performed by WiCell from 500 ng DNA isolated from PBMC passage 1 and iPSC passage 11.
3.9. Mycoplasma detection
Supernatant media from iPSCs at passage 10 was tested for mycoplasma per manufacturer’s protocol with MycoAlert Mycoplasma Detection Kit (Lonza #LT07-118) and MycoAlert Assay Control Set (Lonza #LT07-518).
Supplementary Material
| Unique stem cell line identifier | ISCRMi001-A |
|---|---|
| Alternative name(s) of stem cell line | 3-0062; DSP Leu462Serfs*22 |
| Institution | University of Washington Department of Medicine |
| Contact information of distributor | Kai-Chun Yang, kcyang@uw.edu |
| Type of cell line | iPSC |
| Origin | Human |
| Additional origin info required for human ESC or iPSC | Age: 47 Sex: Female Ethnicity if known: White |
| Cell Source | Peripheral blood mononuclear cells |
| Clonality | Clonal |
| Method of reprogramming | Integration-free Sendai virus expressing human OCT4, SOX2, KLF4, c-MYC |
| Genetic Modification | N/A |
| Type of Genetic Modification | N/A |
| Evidence of the reprogramming transgene loss (including genomic copy if applicable) | PCR |
| Associated disease | Arrhythmogenic cardiomyopathy |
| Gene/locus | GRCh38 6: 7,568,555NM_004415.4 (DSP):c.1386 (3′ rule) (p. Leu462Serfs*22) |
| Date archived/stock date | Aliquots frozen 11 Aug 2022 Currently under application for archival at EBiSC |
| Cell line repository/bank | https://hpscreg.eu/cell-line/ISCRMi001-A |
| Ethical approval | IRB Consent STUDY00002544 |
Funding
This work was supported in part by a Career Development Award (IK2 BX004642) from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory R&D (BLRD) Service and the John L. Locke Jr. Charitable Trust to K-C.Y. Additional support comes from the F32 fellowship (1F32HL164108-01 NIH NHLBI) to A.L and the CMAP postdoctoral fellowship (to C.E.F.).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Veterans Affairs or the United States Government.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.scr.2022.102987.
References
- Corrado D, Basso C, Judge DP, 2017. Arrhythmogenic cardiomyopathy. Circulation Res. 121 (7), 784–802. 10.1161/CIRCRESAHA.117.309345. [DOI] [PubMed] [Google Scholar]
- Delmar M, McKenna WJ, 2010. The cardiac desmosome and arrhythmogenic cardiomyopathies. Circulation Res. 107, 700–714. 10.1161/CIRCRESAHA.l10.223412. [DOI] [PubMed] [Google Scholar]
- Garcia-Gras E, et al. , 2006. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest 116, 2012–2021. 10.1172/jci27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerull B, Klaasen S, Brodehl A (2019) The genetic landscape of cardiomyopathies. Genetic causes of cardiac disease, eds Edrmann J, Moretti A (Cham: Springer; ), pp. 45–91. 10.1007/978-3-030-27371-22. [DOI] [Google Scholar]
- Ng R, et al. , 2019. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. JCI Insight 4 (14), e128643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.