

Abstract
Macrophages are key cells after tissue damage since they mediate both acute inflammatory phase and regenerative inflammation by shifting from pro‐inflammatory to restorative cells. Glucocorticoids (GCs) are the most potent anti‐inflammatory hormone in clinical use, still their actions on macrophages are not fully understood. We show that the metabolic sensor AMP‐activated protein kinase (AMPK) is required for GCs to induce restorative macrophages. GC Dexamethasone activates AMPK in macrophages and GC receptor (GR) phosphorylation is decreased in AMPK‐deficient macrophages. Loss of AMPK in macrophages abrogates the GC‐induced acquisition of their repair phenotype and impairs GC‐induced resolution of inflammation in vivo during post‐injury muscle regeneration and acute lung injury. Mechanistically, two categories of genes are impacted by GC treatment in macrophages. Firstly, canonical cytokine regulation by GCs is not affected by AMPK loss. Secondly, AMPK‐dependent GC‐induced genes required for the phenotypic transition of macrophages are co‐regulated by the transcription factor FOXO3, an AMPK substrate. Thus, beyond cytokine regulation, GR requires AMPK‐FOXO3 for immunomodulatory actions in macrophages, linking their metabolic status to transcriptional control in regenerative inflammation.
Keywords: acute lung injury, glucocorticoids, macrophages, regenerative inflammation, skeletal muscle regeneration
Subject Categories: Immunology, Signal Transduction, Stem Cells & Regenerative Medicine
Macrophages regulate the resolution of inflammation after tissue damage. The metabolic sensor AMP‐activated protein kinase and the transcription factor FOXO3 are required for glucocorticoids to induce restorative macrophages.

Introduction
Resolution of inflammation is essential for the return to tissue homeostasis after an inflammatory insult, both to limit excessive organ damage and to initiate the regenerative inflammation that sustains the repair process (Watanabe et al, 2019). Resolution involves the suppression of pro‐inflammatory active cells and the acquisition of an anti‐inflammatory/restorative phenotype by immune cells to initiate tissue repair. Macrophages are central to this process, being involved in both the acute inflammatory phase and the regenerative phase (Arnold et al, 2007; Wynn & Vannella, 2016; Watanabe et al, 2019). Inflammatory macrophages rapidly infiltrate the target tissue and release pro‐inflammatory cytokines (Mantovani et al, 2004; Turner et al, 2014). Macrophages then phagocytose apoptotic cells in a process called efferocytosis. Engulfment of apoptotic cells provokes a macrophage phenotypic switch to a repair state, which promotes return to tissue homeostasis (Ortega‐Gomez et al, 2013). A failure of inflammatory resolution leads to chronic inflammation, impaired repair/regeneration, and eventually tissue erosion. Thus, elucidating pathways that induce and stabilize restorative phenotypes of macrophages is of utmost importance.
We and others showed that AMP‐activated protein kinase (AMPK) plays is required for the promotion of repair macrophage phenotype (Sag et al, 2008; Mounier et al, 2013). AMPK controls a wide variety of metabolic functions in cells and is activated by a high AMP:ATP ratio or through diminished cellular glucose concentration (Hardie et al, 2012), making AMPK a sensor of cellular energy deprivation. Upon activation, AMPK phosphorylates various proteins to inhibit cellular anabolism and activate catabolism (Hardie et al, 2012). Of note, the sole catalytic subunit of AMPK being expressed in macrophages is α1.
Having underscored the role of AMPKα1 for inducing restorative macrophage polarization, it is absolutely unclear whether AMPK contributes to the efficacy of anti‐inflammatory drugs. Most prominent anti‐inflammatory agents are glucocorticoids (GCs), with over 1% of the UK, Danish, and American populations, at one point, having received a prescription for GCs (Fardet et al, 2011; Overman et al, 2013; Laugesen et al, 2017). GCs signal through the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily, which acts as a transcription factor to regulate gene expression upon exposure to ligand (Weikum et al, 2017b). GR regulates transcription through transactivation and transrepression (Weikum et al, 2017b). Suppression of pro‐inflammatory cytokines occurs at the transcriptional level through GR tethering to pro‐inflammatory transcription factors such as NFκB and AP‐1, or by direct binding to DNA at, or nearby NFκB and AP‐1 response elements and represses transcription through a tethering independent mechanism (Uhlenhaut et al, 2013; Weikum et al, 2017a; Hudson et al, 2018). Suppression of cytokines was considered as the canonical mechanism of immunosuppression by GCs for decades (Barnes, 2011). However, recent studies implicated that GR‐dependent upregulation of anti‐inflammatory genes plays an essential role in the resolution of inflammation. In particular, the expression of Tsc22d3 (Gilz), Dusp1, Sphk1, Ikkb, and A20 are important for the anti‐inflammatory properties of GCs (Eddleston et al, 2007; Vandevyver et al, 2012; Vettorazzi et al, 2015; Oh et al, 2017). Whether and how GCs interfere with a metabolic sensor such as AMPK in inflammatory cells is still uncompletely understood.
In this study, we demonstrate that AMPK is activated by the GC Dexamethasone in macrophages and that GR S211 phosphorylation is decreased in AMPK‐deficient cells. In vitro, loss of AMPK results in the failure to induce a restorative macrophage phenotype, preventing the GC‐induced repair phenotype. In vivo, mice having a myeloid‐specific deletion of AMPKα1 show impaired GC‐driven resolution of tissue repair (skeletal muscle injury) and resolution of acute lung inflammation (induced by endotoxin shock). The signature of GC‐regulated genes in macrophages revealed two categories of genes. AMPK‐independent genes include the canonical GR‐regulated genes. AMPK‐dependent genes are enriched in FOXO3 target genes, and we identified an AMPK‐dependent loading of FOXO3 together with GR at their regulatory regions. These results demonstrate a new regulatory pathway of GC action in macrophages, that is required for their function in the resolution of inflammation.
Results and Discussion
AMPKα1 is required for a Dex‐induced restorative phenotype in macrophages
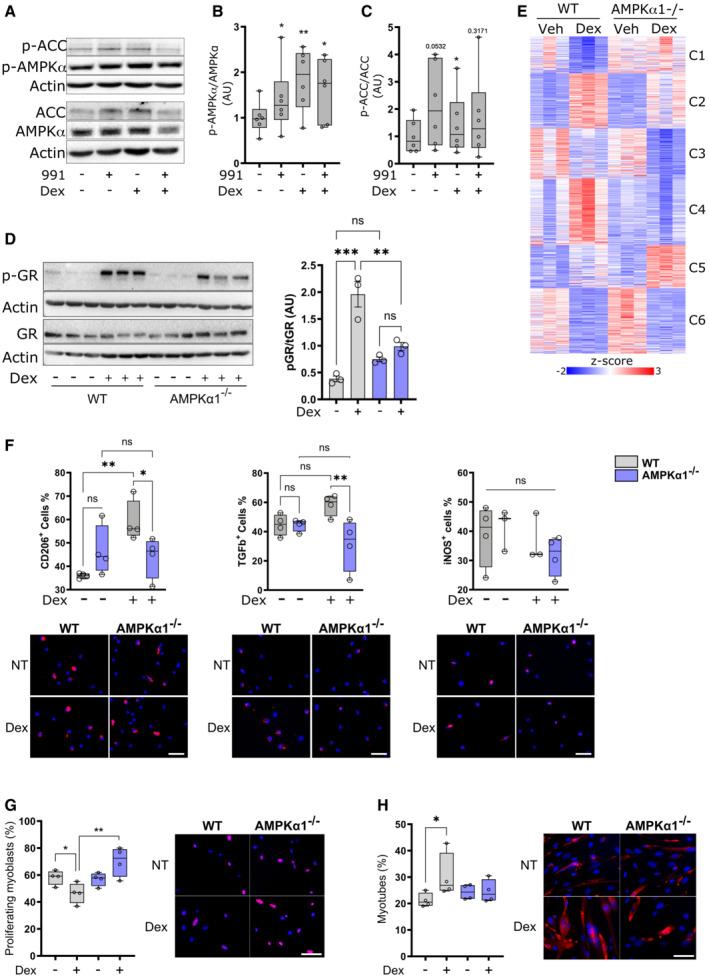
To determine whether GCs affect AMPK activity, we determined the activation of AMPK in bone marrow‐derived macrophages (BMDMs). BMDMs were treated with the AMPK activating compound “991” (Willows et al, 2017), Dexamethasone (Dex) or a combination of both for 1 h and AMPK signaling was assessed. Dex significantly increased the phosphorylation of AMPK at T172 to a similar extent as 991, but with no additive effect of co‐treatment (Fig 1A and B). AMPK phosphorylation resulted in increased activity of AMPK, as the downstream target ACC was also phosphorylated in response to Dex treatment (Fig 1C), indicating a previously unknown role of GCs in stimulating AMPK signaling. Only four studies have reported a link between AMPK signaling and GR signaling in non‐immune cells and organs, through various crosstalks. Indirect AMPK activation was observed in muscle cells due to GC induction of mitochondrial dysfunction (Liu et al, 2016). Dexamethasone‐dependent activation of AMPK in hepatocytes but inhibition in adipocytes was observed, highlighting the cell‐type selectivity of GR‐AMPK cross‐talk (Christ‐Crain et al, 2008). We determined how genetic loss of AMPKα1, the only AMPK catalytic subunit expressed in macrophages, affects GR phosphorylation. Both wildtype (WT) and AMPKα1 knockout (AMPKα1−/−) BMDMs were treated with Dex for 1 h, and phosphorylation of GR at S211, a strong indicator of transcriptional activity (Wang et al, 2002), was assessed by immunoblot (Fig 1D). Upon Dex treatment, GR phosphorylation was increased as expected; however, GR phosphorylation was strongly diminished in AMPKα1−/− macrophages (Fig 1D). This was in agreement with elevated GR‐S211 phosphorylation in the presence of phosphorylated AMPK in hepatocytes (Ratman et al, 2016). AMPK indirectly phosphorylates GR S211 via p38 in preadipocytes and hepatocytes (Nader et al, 2010) but not in macrophages, in both genotypes (Fig EV1A). Finally, a physical link between phospho‐AMPK and phospho‐GR in complex with PPARα was proposed to increase the transcriptional activity of the latter upon starvation (Ratman et al, 2016). The activation of S211 phosphorylation is still poorly understood. Many kinases were described to be involved in different cell types, such as JNKs, CDKs, Erks, Akt and GSK3 (Faus & Haendler, 2006). To which other kinases and phosphatases are involved in GR‐S211 phosphorylation dependent on AMPK is of interest to investigate in the future.
Figure 1. Glucocorticoid signaling through AMPK in macrophages.
-
ABone marrow‐derived macrophages (BMDMs) were treated with either the AMPK activator 991, Dexamethasone (Dex) or a combination for 1 h before analysis by Western blot.
-
B, CPhospho‐AMPK (B) and phospho‐ACC (C) were quantified.
-
DWT and AMPKα1−/− BMDMs were treated with Dex for 1 h before analysis by Western blot and GR phosphorylation at S211 was quantified.
-
EWT and AMPKα1−/− BMDMs were treated with Dex for 24 h and analyzed by RNA‐seq. Differentially expressed genes between any condition were clustered using c‐means clustering.
-
FWT and AMPKα1−/− BMDMs were treated with Dex for 24 h and anti−/pro‐inflammatory markers assessed using immunofluorescence.
-
G, HWT and AMPKα1−/− BMDMs were treated with Dex for 72 h, washed and conditioned medium was recovered after 24 h used to treat muscle stem cells. Proliferation (Ki67 immunostaining) (G) or differentiation (desmin immunostaining and counting the percentage of muscle cells with 2 nuclei or more) (H) were assessed in muscle stem cells.
Data information: results are means ± SEM or z‐score or displayed with boxes and whiskers in which the central band represents the median. N = 6 (B, C), 3 (D), 3–4 (F) of 4 biological replicates (G, H). *P < 0.05, **P < 0.01, ***P < 0.001 statistical analysis using Student's t‐test (B, C) or ANOVA tests (D, G, H). Bar = 25 μm (G, H).
Source data are available online for this figure.
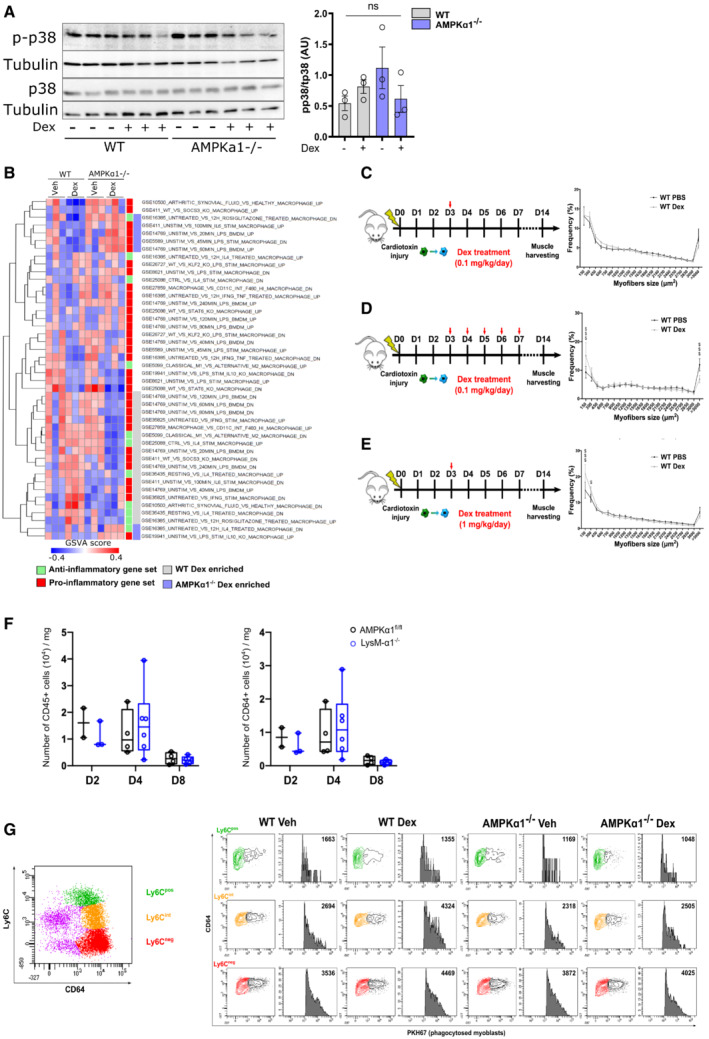
Figure EV1. Glucocorticoid treatment during skeletal muscle regeneration and signaling through AMPK in macrophages is independent of p38.
-
AWT and AMPKα1−/− Bone marrow‐derived macrophages (BMDMs) were treated with Dexamethasone (Dex) for 1 h before analysis by Western blot for p38 phosphorylation and quantified.
-
BRepresentation of GSVA analysis from Fig 1F with gene sets labeled.
-
C–EExperimental procedure of mice treatment with Dex. After cardiotoxin injection to damage the muscle, AMPKα1fl/fl (WT) and LysM‐α1−/− mice were treated with (C) a single dose of Dexamethasone (Dex) intra‐peritoneal (i.p.) (0.1 mg/kg) at day 3 (D3), or (D) multiple doses of Dex i.p. (0.1 mg/kg) on D3 to D7 after CTX, or (E) a single dose of Dex i.p. (1 mg/kg) at D3, and Tibialis Anterior (TA) muscles were harvested at day 8 (D8) and 14 (D14) after injury. Myofiber cross sectional area was then quantified.
-
FThe number of immune cells (CD45pos, left panel) and macrophages (CD45posCD64pos, right panel) per milligram of muscle tissue (TA muscle) was assessed by flow cytometry in AMPKα1fl/fl (WT) and LysM‐α1−/− mice at day 2, 4 and 8 after cardiotoxin injury.
-
GMuscles and cells were treated as described in the legend of Fig 2D. Plot on top shows the whole population of macrophages stained with CD64 and Ly6C. Plots on bottom show proportion of macrophages that have phagocytosed fluorescent dead myoblasts in the Ly6Cpos (green population), Ly6Cint (orange population) and Ly6Cneg (red population) macrophages in the various conditions.
Data information: results are means ± SEM (A) or box and whiskers in which the central band represents the median of three biological replicates (A) or 7 (D), 8–10 (C, E), 2–6 (F–G) animals. $ P < 0.05, $$$ P < 0.001 vs untreated WT, using ANOVA tests.
Source data are available online for this figure.
RNA‐Seq analysis revealed that a substantial fraction of Dex‐regulated genes was differentially expressed between WT and AMPKα1−/− macrophages, supporting the need of AMPK to modulate Dex‐triggered gene expression (Fig 1E). Clustering of genes differentially expressed between all conditions highlighted genes which regulation was independent from AMPK (genes activated [cluster C2] or repressed [clusters C3 & C6] by Dex in both genotypes) and genes regulated in an AMPK‐dependent manner: genes repressed by Dex in WT and not in AMPKα1−/− macrophages (cluster C1), genes activated by Dex in WT and not in AMPKα1−/− cells (cluster C4) and genes that were activated by Dex only in AMPKα1−/− macrophages (cluster C5).
Dex‐regulated AMPK‐independent genes were enriched in GOs related to metabolic processes, mainly catabolic (C2), immune cell response and migration (C3), and cell division (C6; Dataset EV1). Dex‐regulated AMPK‐dependent genes were highly enriched in GOs associated with mitochondria function, respiration, oxidative metabolism, and oxidoreductase activity (C1), with mitochondria biosynthesis, biosynthetic processes, and response to stress (C5; Dataset EV2). Interestingly, C4, which contained genes that were not upregulated upon Dex treatment in AMPKα1−/− macrophages, was strongly enriched in chromatin organization and regulation of transcription, regulation of biosynthetic processes and a series of enzymatic activities (including kinases), as well as, for a lesser extent, microtubule organization, matrix/cell interactions, and organelle lumen formation (Dataset EV2).
To gain more insight into macrophage function, we used gene set variation analysis (GSVA, Hanzelmann et al, 2013) to obtain enrichment of genes in datasets using macrophages upon pro‐inflammatory and anti‐inflammatory conditions (Fig EV1B). As expected, datasets involving genes enriched in classically activated (pro‐inflammatory, red label, Fig EV1B) macrophages were less enriched in Dex‐treated WT macrophages. Genes involved in alternative macrophage activation (e.g., upon IL‐4 treatment, green label, Fig EV1B) were more enriched in Dex‐treated WT than in Dex‐treated AMPKα1−/− macrophages. Dex‐treated AMPKα1−/− macrophages had higher enrichment of genes regulated by pro‐inflammatory stimuli (71% in AMPKα1−/−, as compared with 64% in WT). This analysis indicates that genes regulated in WT by Dex and not in AMPKα1−/− were more associated with the acquisition of the restorative phenotype. This was confirmed at the protein level on naive macrophages. The expression of the anti‐inflammatory marker CD206 was increased in Dex‐treated WT cells, but not in AMPKα1−/− cells (Fig 1F). TGFβ expression was even decreased in AMPKα1−/− cells upon Dex treatment (Fig 1F). Unsurprisingly, Dex had no effect on the expression of the pro‐inflammatory marker iNOS in either genotype since these were not activated macrophages (Fig 1F).
The phenotypic failure of acquiring a restorative profile upon Dex treatment in the absence of AMPK was confirmed at the functional level. Conditioned medium from Dex‐treated WT macrophages decreased myoblast proliferation (Fig 1G) and increased myoblast differentiation (Fig 1H), a cardinal feature of restorative phenotype of macrophages (Mounier et al, 2013; Varga et al, 2016a). However, these properties were lost in AMPKα1−/− macrophages (Fig 1G and H).
Taken together, these results establish that AMPK deficiency substantially impairs the induction of a restorative phenotype of macrophages driven by GCs and that this translates into a failure of Dex‐induced expression of anti‐inflammatory marker proteins and secreted factors associated with the restorative function of macrophages, regulating myogenesis.
AMPKα1 in macrophages is required for the GC‐induced effects on post‐injury muscle regeneration
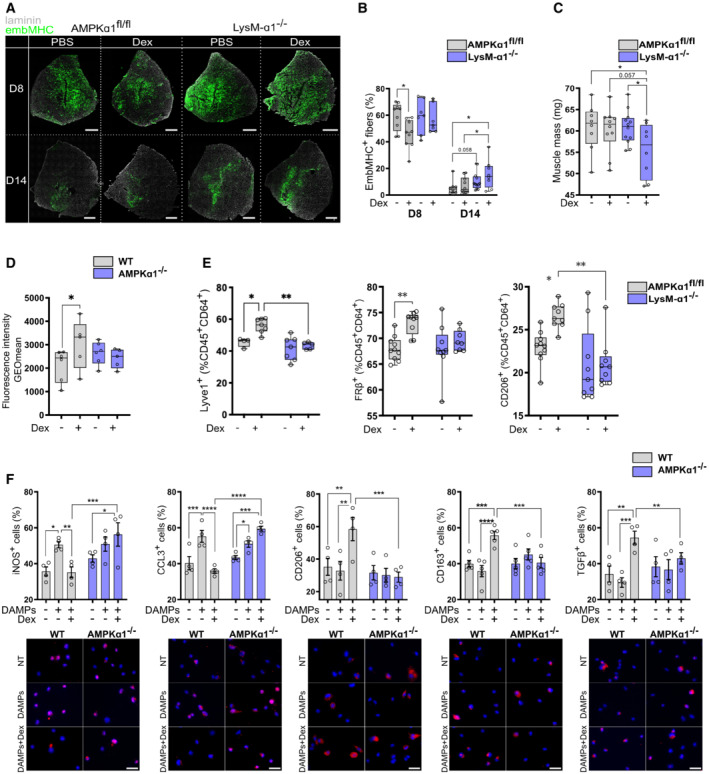
We previously showed that AMPKα1 in macrophages is required for the resolution of inflammation during skeletal muscle regeneration (Mounier et al, 2013). Thus, we aimed to determine whether the anti‐inflammatory actions of GCs could promote muscle regeneration in AMPK‐dependent manner, despite the usual catabolic effects of GCs (Schakman et al, 2009). To induce muscle injury, cardiotoxin (CTX) was injected into the Tibialis Anterior muscle of wildtype (AMPKα1fl/fl) or mice lacking AMPKα1 specifically in the myeloid lineage (LysM‐α1−/−). We optimized the application of Dex to avoid the usual catabolic effects of GCs on muscle (Fig EV1C–E). While multiple daily dosing (Fig EV1D), or a single high dose of 1 mg/kg (Fig EV1E) resulted in muscle atrophy as measured by myofiber cross‐sectional area, a single I.P. dose of 0.1 mg/kg Dex given 3 days after muscle damage, that is, at the time of resolution of inflammation, did not reduce myofiber size (Fig EV1C). This latter application regimen was therefore used for the further experiments. In this model, no differences were observed between WT and LysM‐α1−/− neither on the number of immune cells (CD45+) nor macrophages (CD64+; Fig EV1F). We determined the expression of embryonic myosin heavy chain (EmbMHC), which identifies myofibers undergoing regeneration, to monitor the repair process (Fig 2A). Kinetics of EmbMHC expression in untreated WT muscle indicated a strong decrease (from 59.3 to 5.6%) of myofibers expressing EmbMHC between day 8 and day 14, indicative of the maturation of new regenerating myofibers (Fig 2A and B). Dex accelerated muscle regeneration in an AMPK‐dependent fashion since Dex treatment decreased the number of EmbMHCpos myofibers at day 8 in WT mice (−23%) but not in LysM‐α1−/− muscle (Fig 2A and B). Then, at day 14, when the WT muscle is almost fully recovered (indicated by very low number of embMHCpos myofibers), Dex treatment did not rescue the delay in the maturation of regenerating myofibers observed in LysM‐α1−/− with a 2‐fold increase of EmbMHCpos myofibers (Fig 2A and B). Accordingly, muscle mass was found low in Dex treated LysM‐α1−/− animals (Fig 2C). Altogether, these data show that Dex promoted muscle regeneration in a macrophage AMPKα1‐dependent manner.
Figure 2. AMPKα1 in macrophages is required for glucocorticoid‐dependent resolution of inflammation during cardiotoxin induced skeletal muscle damage.
-
A–CAfter cardiotoxin injection to damage the muscle, AMPKα1fl/fl (WT) and LysM‐α1−/− mice were treated with a single dose of Dexamethasone (Dex) intra‐peritoneal (i.p.) (0.1 mg/kg) at day 3 (D3) and Tibialis Anterior (TA) muscles were harvested at day 8 (D8) and 14 (D14) after injury. (A) Representative embryonic myosin heavy chain (EmbMHC) immunostaining on whole TA muscle section at D8 and D14 after injury. (B) Quantification of (A), results are expressed as percentage of positive myofibers expressing the EmbMHC on the whole muscle section. (C) Muscle mass at D14 after injury.
-
D, ETA muscle from WT and AMPKα1−/− (D) or LysM‐α1−/− (E) mice was damaged with cardiotoxin and Dex (or vehicle) was i.p. injected around 60 h after injury (~ 2.5 days) prior CD45pos cells were sorted with magnetic beads 15 h later (day 3). (D) CD45pos CD64pos Ly6Cpos, Ly6Cint and Ly6Cneg macrophage subsets were cultured with dead fluorescently labeled myoblasts for 6 h. Phagocytosis was evaluated by flow cytometry and expressed as the GeoMean fluorescence Intensity of the phagocyting cells. (E) The proportion of macrophages (CD64pos) expressing anti‐inflammatory markers was assessed by flow cytometry.
-
FMacrophage polarization was determined by immunofluorescence in vehicle, DAMPs and DAMPs + Dex treated WT and AMPKα1−/− BMDMs.
Data information: results are displayed with box and whiskers in which the central band represents the median (B, C, D, E) or means ± SEM (F). N = 7–12 muscles (B, C), 5–6 biological replicates (D), 4–12 muscles (E) and 4–5 biological replicates (F). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 using ANOVA (B‐F) tests. Bars = 500 μm (A), 25 μm (F).
Efferocytosis of muscle debris is required for the resolution of inflammation during skeletal muscle regeneration (Arnold et al, 2007) and is dependent on AMPK activation (Mounier et al, 2013). WT and LysM‐α1−/− mice were injected with Dex at 2.5 days after injury and CD64+ macrophages were FACS‐isolated and ex vivo tested for their capacity to engulf fluorescent apoptotic myoblasts. Three macrophage subsets were sorted as pro‐inflammatory Ly6Cpos macrophages, Ly6Cneg restorative macrophages, and a Ly6Cint subset of macrophages that are shifting from pro‐inflammatory to restorative profile (Mounier et al, 2013; Varga et al, 2013, 2016a,b; Fig EV1G). In WT mice, Dex treatment 15 h prior to macrophage isolation increased by +42% the efferocytic capacity of Ly6Cint macrophages assessed by the increased GeoMean Fluorescent Intensity of macrophages having engulfed apoptotic fluorescent myoblasts (Figs 2D and EV1G). On the contrary, Ly6Cint macrophages from regenerating LysM‐α1−/− muscle did not modify their efferocytic capacity upon Dex treatment (Figs 2D and EV1G). Moreover, upon Dex treatment, macrophages isolated from regenerating WT muscles showed an increased expression of markers associated with the restorative phenotype (Lyve1, Folate Receptor, CD206), while they did not respond to Dex when lacking AMPK (Fig 2E). In vitro experiments were performed to mimic the damaged muscle environment. Muscle‐derived damage‐associated molecular patterns (DAMPs, from injured muscle, see experimental procedures) induced a pro‐inflammatory profile in both WT and AMPKα1−/− BMDM, as assessed by the increased expression of iNOS and CCL3 proteins (Fig 2F). As expected, Dex treatment of WT DAMP‐activated macrophages induced a decreased expression of those pro‐inflammatory markers and an important increase of the expression of the anti‐inflammatory markers CD206, CD163, and TGFβ (Fig 2F). On the contrary, Dex had no impact on the expression of pro‐ and anti‐inflammatory marker proteins in AMPKα1−/− macrophages (Fig 2F).
Taken together, these results show that the absence of AMPK abrogates Dex‐induced repair phenotype in macrophages in vitro and in vivo and positive effects on muscle regeneration. This is in accordance with previous works showing that AMPKα1 activation in macrophages is triggered by upstream signals delivered early after the injury by myeloid cells, such as the specialized pro‐resolving mediator Annexin A1, which is a mediator of GC‐induced resolution of inflammation (Desgeorges et al, 2019a; McArthur et al, 2020).
AMPKα1 in macrophages is required for GC protection during acute lung inflammation and endotoxin shock
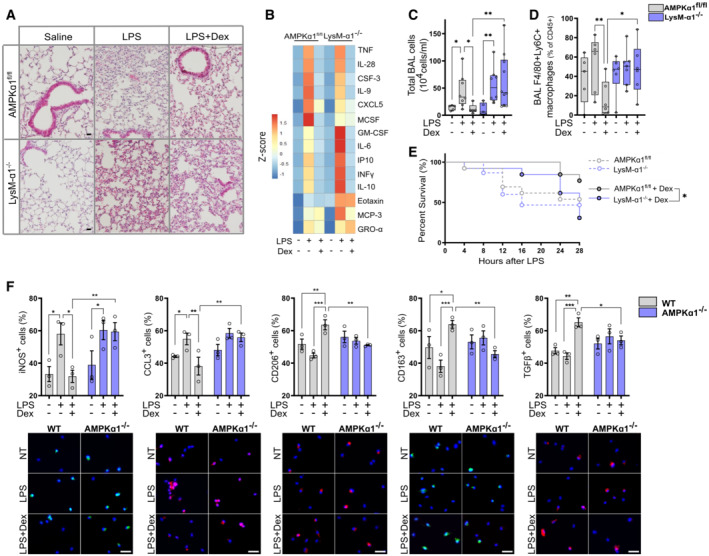
Since AMPKα1 alters GC‐dependent restorative phenotype in macrophages, we suspected that AMPK would also be essential for the resolution of acute inflammation. To this end, we used an LPS‐induced endotoxin shock to model acute lung injury (ALI), where we showed that GR action in macrophages is decisive for the effects of GCs (Vettorazzi et al, 2015). We treated WT and LysM‐α1−/− mice with vehicle, 10 mg/kg LPS or LPS + 1 mg/kg Dex, before intravenous injection of oleic acid to induce lung inflammation and mice were sacrificed 24 h later. LPS induced alveolar collapse in both WT and LysM‐α1−/− mice, which was partially recovered in WT mice treated with Dex, but not in mice with myeloid cells deficient for AMPK (Fig 3A). At the molecular level, no effect on bronchioalveolar lavage (BAL) cytokine content was observed upon AMPKα1 deletion since the levels of TNFα, IL‐6, IFNγ, and IL‐10 (among others) were increased upon ALI and reduced by Dex treatment in both WT and LysM‐α1−/− mice (Figs 3B and EV2A). At the cellular level, LPS increased the number of cells in the BAL fluid of both WT and LysM‐α1−/− mice, but Dex treatment reduced the cell numbers in BAL only in WT, but not in LysM‐α1−/− mice (Fig 3C).
Figure 3. AMPKα1 in macrophages is required for glucorticoid‐dependent suppression of inflammation in endotoxin‐induced lung injury.
-
AHematoxylin–eosin staining of lung tissue after 24 h.
-
BBroncho‐alveolar lavage (BAL) cytokine content was measured by Luminex after 24 h.
-
C, DAfter 24 h, mice were culled and cells infiltrating the lungs were analyzed. (C) The number of cells present in the BAL fluid was counted. (D) The number of pro‐inflammatory macrophages (F4/80pos Ly6Cpos) was evaluated by flow cytometry as a percentage of CD45pos cells.
-
ESurvival curve of mice treated with vehicle or Dex.
-
FMacrophage polarization was determined by immunofluorescence in vehicle, LPS and LPS + Dex treated WT and AMPKα1−/− BMDMs, data are expressed as percentage positive cells.
Data information: Results are displayed with box and whiskers in which the central band represents the median (C, D) or means ± SEM (F). N = 5–10 (C), 5–7 (D), 3–7 (data below detection threshold were excluded from analysis) (B), 13–15 (E) or 3 (F) biological replicates. Statistical analysis by ANOVA (C, D, F) or Gehan–Breslow–Wilcoxon test (E). *P < 0.05, **P < 0.01, ***P < 0.001. Bar = 20 μm (A), 25 μm (F).
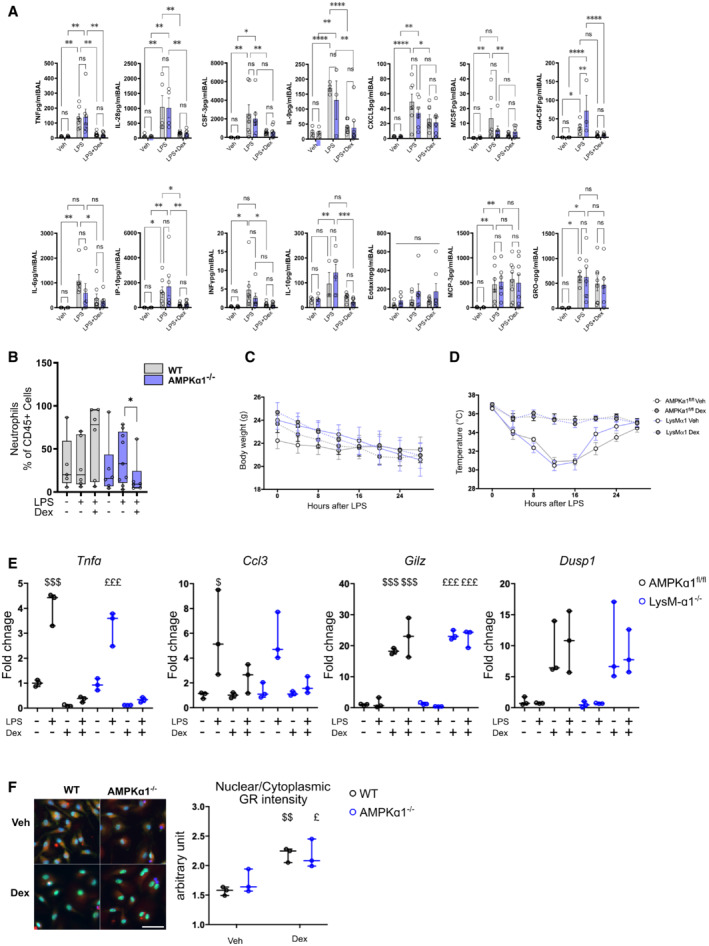
Figure EV2. AMPKα1 in macrophages is not required for glucocorticoid‐dependent suppression of specific classical inflammatory gene expression and glucocorticoid receptor localization.
-
A, BAMPKα1fl/fl (WT) and LysM‐α1−/− mice were exposed for 24 h to vehicle, lipopolysaccharide (LPS) or LPS + Dex treatment and (A) cytokines and (B) neutrophil numbers were quantified in BAL.
-
C, D(C) Body mass and (D) body temperature curves for each group during survival experiment in Fig 3E.
-
ERT–qPCR was performed on AMPKα1fl/fl and LysM‐α1−/− macrophages treated with LPS and Dex for 6 h.
-
FWT and AMPKα1−/− BMDMs were treated with Dex for 1 h and nuclear Glucocorticoid Receptor (GR) intensity determined by immunofluorescence was compared to cytoplasmic GR intensity.
Data information: results are means ± SEM or box and whiskers in which the central band represents the median of 5–9 (B), 13–15 (C, D), 3–7 (A, data below detection threshold were excluded from analysis) animals, and 3 (E, F) biological replicates. Statistical analysis by ANOVA (A, B) or ANOVA with repeated measures (C, D). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. $ P < 0.05, $$ P < 0.01, $$$ P < 0.001 vs untreated WT; £ P < 0.05 vs untreated LysM‐α1−/− or AMPKα1−/−, using one‐way ANOVA. Bar = 70 μm.
No differences were observed in the abundance of neutrophils in WT and LysM‐α1−/− mice, only a small reduction in mutant mice after Dex administration (Fig EV2B), in agreement with our previous data showing that macrophages are the major targets of GC effects in this model (Vettorazzi et al, 2015). Importantly, the number of Ly6Cpos pro‐inflammatory macrophages was dramatically reduced after Dex treatment (to reach 10% of CD45pos cells) while it remained high in LysM‐α1−/− mice, accounting for 50% of the immune cells present in the damaged tissue (Fig 3D). These data indicate a failure of Dex to resolve inflammation when AMPK is absent in macrophages, which is independent of cytokine regulation. The final evaluated outcome was the survival of the mice in both conditions (Stearns‐Kurosawa et al, 2011). ALI induced death similarly in WT and LysM‐α1−/− (Fig 3E). However, Dex treatment allowed the survival of most WT but not LysM‐α1−/− mice, indicating that Dex rescue from ALI was AMPK dependent (Fig 3E). Temperature and weight loss were similar in both genotypes, these parameters being part of the ethical reasons for sacrifice (Fig EV2C and D). These data demonstrate that GCs suppressed inflammation during ALI through AMPK in macrophages to reduce the number of pro‐inflammatory macrophages, promoting the resolution of inflammation leading to improved tissue recovery and survival.
Dex effect on the resolution of inflammation upon LPS activation in macrophages was confirmed in vitro in the same conditions as for ALI in vivo. We found that in WT BMDMs, Dex strongly reduced the LPS‐induced expression of iNOS and CCL3, that was not observed in AMPKα1−/− BMDMs (Fig 3F). Inversely, the expression of the anti‐inflammatory markers CD206, CD163, and TGFβ were induced by Dex in LPS‐exposed WT BMDMs, but not in AMPKα1−/− cells (Fig 3F), showing that AMPK is required for the anti‐inflammatory effects of GCs in LPS‐treated macrophages.
Taken together, these results demonstrate that AMPK is necessary to mediate the anti‐inflammatory effects of Dex in the resolution of an acute lung injury model. These data implicate convergence and interdependence of GR and AMPK signalings in the resolution of inflammation that is independent on the canonical regulation of cytokine expression by GCs.
FOXO3 coordinates AMPK‐dependent GC‐mediated gene expression and macrophage function
To determine the nature of genes regulated by Dex dependent on AMPK, we re‐visited the RNA‐seq data of Dex exposed wild type and AMPKα1−/− BMDMs shown in Fig 1 to assess genes up and down‐regulated in WT cells. This analysis identified six major clusters, as mentioned previously. Interestingly, canonical GR targets, both upregulated and repressed, were found in the AMPK‐independent clusters 2, 3, and 6 (Fig 1E). The expression of these classically transactivated (Gilz, Dusp1) and transrepressed (Tnfa, Ccl3) genes by GR was similar in WT and AMPKα1−/− macrophages treated with LPS and Dex (Fig EV2E). Accordingly, we saw no effect of loss of AMPK on GR translocation in response to Dex (Fig EV2F), suggesting an alternate mechanism of AMPK‐GR crosstalk than AMPK regulation of translocation.
Then, we investigated the AMPK‐dependent Dex‐regulated genes and subjected genes of clusters 1 (down) and 4 (up) to oPOSSUM analysis for potential transcriptional regulators and the most enriched binding sites are listed in Figs EV3 and EV4. As expected, GR consensus binding site was enriched in genes commonly activated by Dex in WT and AMPKα1−/− macrophages, validating the relevance of this strategy. Interestingly, binding sites for the transcription factors Sry, Nkx2‐5, Foxd1, and Foxo3 were specifically enriched in genes upregulated by Dex only in WT cells (Fig EV3) together with genes down‐regulated by Dex only in AMPKα1−/− cells (Fig EV4). To refine our search for potential AMPK‐dependent regulators of GR activity, we assessed the z‐score of regulation for GR, AR (which has high motif similarity to GR) and three transcription factors known to be regulated by AMPK, FOXO3 (Li et al, 2009), HIF1α (Faubert et al, 2013), and PPARγ (Sozio et al, 2011; Fig 4A). Interestingly, FOXO3 sites were highly enriched in WT‐selective Dex‐upregulated genes, thus AMPKα1‐dependent genes (cluster 4 shown in Fig 1E). As FOXO3 is a well‐established direct target of AMPK activity (Greer et al, 2007b), we focused our further analysis on FOXO3 function in AMPK‐dependent GC‐mediated gene regulation.
Figure EV3. Transcription factor binding sites enriched in Dexamethasone‐activated genes.
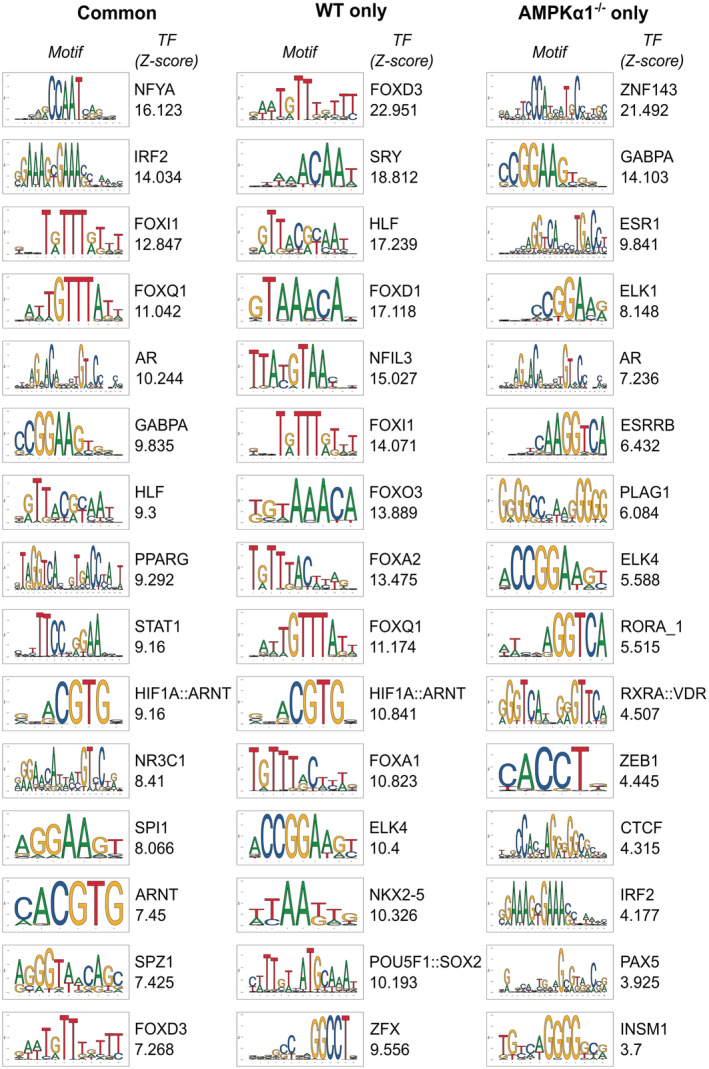
Top 15 transcription factor binding sites enriched in Dexamethasone (Dex)‐activated genes commonly in WT and AMPKα1−/− macrophages (left panel), in WT cells only (middle panel) and in AMPKα1−/− cells only (right panel), ranked by decreasing Z‐score. The consensus binding motif for each transcription factor is shown from the JASPAR database, and its associated Z‐score.
Figure EV4. Transcription factor binding sites enriched in Dexamethasone‐repressed genes.
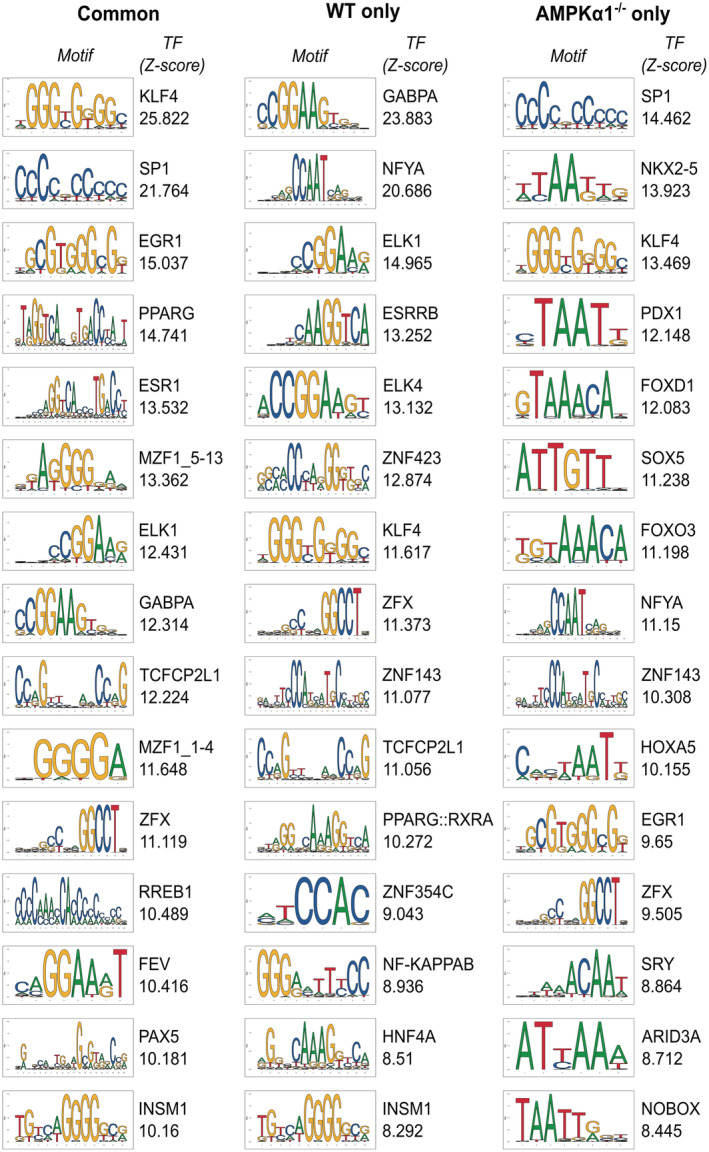
Top 15 transcription factor binding sites enriched in Dexamethasone (Dex)‐repressed genes commonly in WT and AMPKα1−/− macrophages (left panel), in WT cells only (middle panel) and in AMPKα1−/− cells only (right panel), ranked by decreasing Z‐score. The consensus binding motif for each transcription factor is shown from the JASPAR database, and its associated Z‐score.
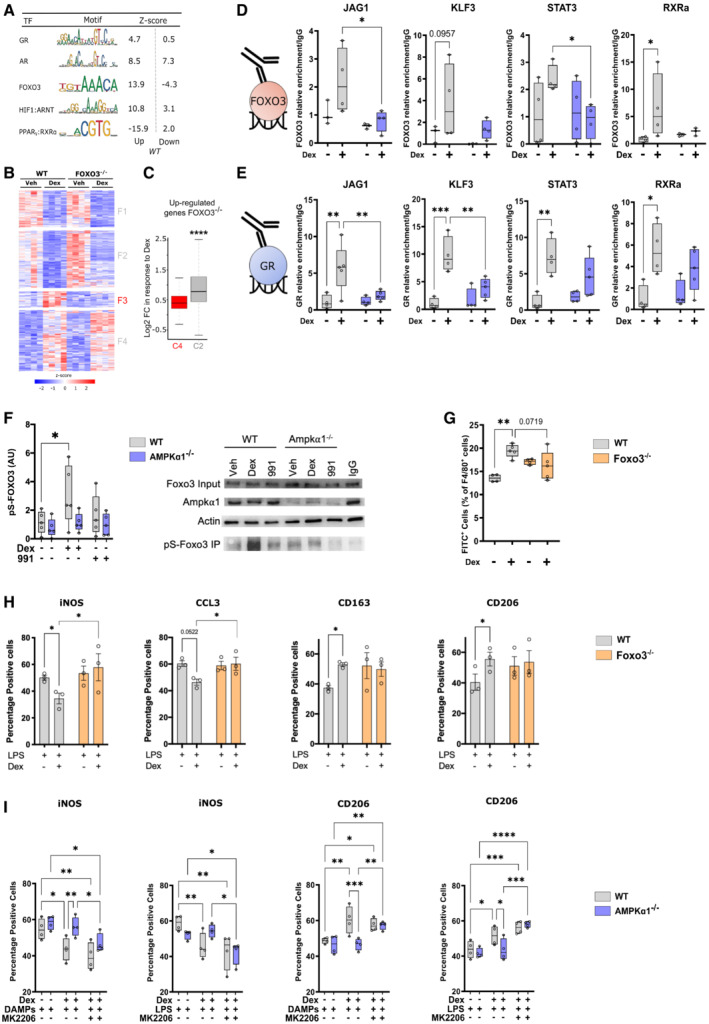
Figure 4. An AMPK‐GR‐FOXO3 trinity regulates glucocorticoid induction of restorative macrophages.
-
ATranscriptional regulators of differentially regulated identified by oPOSSUM were stratified into known interactors with AMPK.
-
BHeat map from RNA‐seq data performed in WT and FOXO3−/− macrophages treated or not with Dex clustered to show genes responsive in WT.
-
CBox plot of log2 fold change of genes upregulated in response to Dexamethasone (Dex) of genes in FOXO3−/− macrophages, stratified by AMPK dependency (clusters C4 and C2 of Fig 1E).
-
D, EFOXO3 (D) and GR (E) enrichment at GR binding sites for 4 AMPK‐dependent GC‐regulated genes.
-
FWT and AMPKα1−/− bone marrow‐derived macrophages (BMDMs) were treated with either Dex or the AMPK activator 991 and protein phosphorylated on serine were immunoprecipitated. Western blot for FOXO3, AMPKα1, actin and the phosphorylated form of FOXO3 were done. Quantification of the band intensity is provided normalized to those of actin and total FOXO3, a representative example is shown.
-
GFOXO3−/− BMDMs were treated or not with Dex and cultured with CSFE‐fluorescently labeled apoptotic thymocytes for 2.5 h. The number of macrophages with ingested apoptotic cells was evaluated by flow cytometry. Results are expressed as percentages of positive cells.
-
HWT and FOXO3−/− BMDMs were treated with LPS for 24 h prior addition of Dex for 6 h; immunofluorescence for pro‐ (iNOS, CCL3) and anti‐ (CD163, CD206) inflammatory markers was performed.
-
IWT and AMPKα1−/− BMDMs were treated with either lipopolysaccharide (LPS) or muscle Damaged Associated Molecular Patterns (DAMPs) for 24 h and further treated with the AKT inhibitor MK2206 for 1 h prior addition of Dex for 6 h; before immunofluorescence for pro‐ (iNOS) and anti‐ (CD206) inflammatory markers was performed. Results are expressed as percentages of positive cells.
Data information: results are z‐score of 4 (B), or box and whiskers in which the central band represents the median (D–G, I) or means ± SEM (I). N = 4–6 (D, E), 4–5 (F, G), 3 (H), 4 (I) biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 ****P < 0.0001 using Mann–Whitney–Wilcoxon test (C) or ANOVA and Kruskal–Wallis tests (D–I).
Source data are available online for this figure.
First, we performed Gene Set Enrichment Analysis on Dex‐regulated genes in WT and AMPKα1−/− macrophages combined to a gene set of Dex‐regulated and FOXO3‐dependent genes defined in FOXO3−/− macrophages (Litvak et al, 2012; Fig EV5A). We found that WT cells had a significant enrichment for FOXO3‐dependent genes upon Dex treatment, while Dex‐regulated genes in AMPKα1−/− macrophages did not. We thus performed an RNAseq analysis of WT and FOXO3−/− macrophages treated or not with Dex. We found a subset of FOXO3‐dependent genes, cluster F3, which were no longer upregulated by GC treatment in the FOXO3−/− macrophages (Fig 4B). These data suggest a co‐regulatory role of FOXO3 and GR, requiring AMPK for gene expression. To determine whether genetic deletion of Foxo3 may affect AMPK‐dependent Dex‐regulated genes, we compared the log2 fold change of upregulated genes in response to Dex in the FOXO3−/− cells between AMPK‐dependent (cluster 4 shown in Fig 1E) and AMPK‐independent genes (cluster 2 shown in Fig 1E). We observed that in FOXO3−/− macrophages, AMPK‐dependent genes had a strongly reduced response to Dex, as compared to AMPK‐independent genes (Fig 4C), suggesting a common regulatory mechanism of AMPK and FOXO3 on GR action. Therefore, we selected four genes involved in macrophage functions, which are upregulated by Dex and are dependent on AMPK and FOXO3 (Fig EV5B from Figs 1E and 4B), and assessed by ChIP‐PCR whether GR and FOXO3 DNA loading was regulated by Dex in both WT and AMPKα1−/− cells (Fig 4D and E). We used publicly available ChIP‐seq data to choose sites where GR binds to determine whether FOXO3 binds at similar loci (Fig EV5C). Strikingly, FOXO3 was recruited to similar loci as GR in response to Dex at Jag1, Klf3, Rxra and Stat3 genes, but FOXO3 loading was diminished in AMPKα1−/− macrophages (Fig 4D). Along with diminished FOXO3 binding at these sites, we identified reduced GR loading in the AMPKα1−/− cells (Fig 4E), suggesting that a co‐regulatory even on the DNA between GR and FOXO3 promotes gene expression, dependent on AMPKα1. As for GR localization, FOXO3 cellular localization was not affected by AMPK deficiency (Fig EV5D). However, phosphorylation of FOXO3 was increased upon Dex treatment in WT macrophages but not in AMPKα1−/− cells, indicating the need of AMPK for Foxo3 phosphorylation upon GC treatment (Fig 4F). Of note, AMPKα1 activator 991 did not induce Foxo3 phosphorylation (Fig 4F).
Figure EV5. AMPK‐GC signaling required FOXO3.
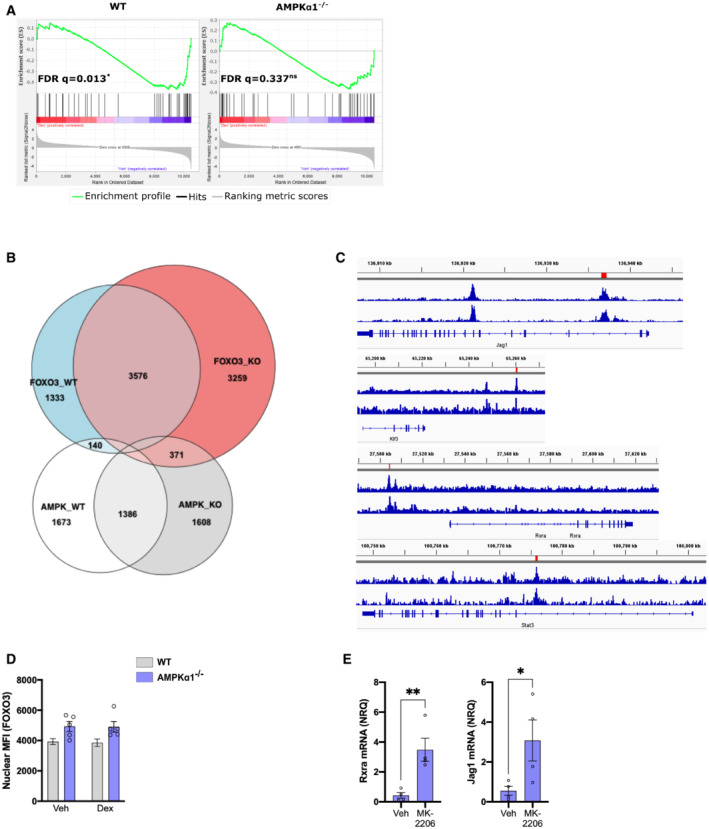
- GSEA analysis of Dexamethasone (Dex)‐regulated genes in WT and AMPKα1−/− Bone marrow‐derived macrophages (BMDMs) for FOXO3‐dependent targets.
- Venn diagram corresponding to the heatmap shown in Fig 4B.
- WT and AMPKα1−/− BMDMs were treated with Dex for 1 h and nuclear FOXO3 intensity was determined by immunofluorescence (results are means ± SEMs of five biological replicates, ANOVA test, no statistical differences).
- AMPKα1−/− BMDMs were treated with lipopolysaccharide (LPS) for 24 h and then incubated with Dex for 6 h in the presence or not of the AKT inhibitor MK‐2206. Rxra and Jag1 mRNA levels were analyzed by RT–qPCR (results are means ± SEMs of four biological replicates, Student's t‐test with *P < 0.05, **P < 0.01).
Next, we performed in vitro loss‐ and gain‐of‐function for FOXO3 in macrophages. Efferocytosis assay using FOXO3−/− BMDMs showed that deletion of Foxo3 in macrophages blunted GC‐induced efferocytosis (Fig 4G). Accordingly, pro‐inflammatory FOXO3−/− macrophages did not acquire the restorative phenotype upon Dex treatment (Fig 4H). Gain‐of‐function included the indirect activation of FOXO3 using the ATK inhibitor MK2206 (Brunet et al, 1999; and shown by increased expression of FOXO3 target genes Rxra and Jag1 in Fig EV5E), after which the evaluation of the macrophage inflammatory state was done in WT and AMPKα1−/− macrophages activated to be pro‐inflammatory cells with LPS or DAMPs, then treated with Dex. Activation of FOXO3 restored Dex sensitivity of AMPKα1−/− macrophages, as assessed by the decreased expression of the pro‐inflammatory marker iNOS (Fig 4I) and the increased expression of the anti‐inflammatory marker CD206 (Fig 4I) upon MK2206 treatment.
Taken together, these data demonstrate that FOXO3 regulates with GR the expression of a subset of AMPKα1‐dependent genes that are required for anti‐inflammatory actions and the resolution of inflammation by GCs. FOXO3 is activated upon metabolic stress (high cortisol and high AMP), suggesting that it is regulated by both GR and AMPK. Indeed, FOXO3 is a direct target of AMPK‐dependent phosphorylation (Greer et al, 2007a; Lutzner et al, 2012) and FOXO3 is activated by GR, indirectly through the expression of SGK1, which alleviates the inhibition of FOXO3 transcriptional activity. FOXO3 was previously highlighted as an important factor in macrophages and inflammatory responses: FOXO3 is required for the differentiation of monocytes into macrophages via AMPKα1 activation and autophagy (Zhang et al, 2017) and promotes anti‐viral response in macrophages (Zhang et al, 2019). Strikingly, FOXO3 is involved in the GC‐driven anti‐inflammatory response in Systemic Lupus Erythematosus through the reinforcement of the blockage of NF‐ΚB activity (Lu et al, 2016). Of note, GR and FOXO1, a related Forkhead box transcription factor, are found at similar loci in the liver, suggesting that GR‐FOXO cross‐talk occurs across multiple transcription factors, potentially in a cell‐type specific manner (Kalvisa et al, 2018). These observations open the possibility that GR or FOXO3 are assisting one another in binding the DNA and regulating macrophage gene expression. Our data did not define a hierarchy in AMPK‐GR‐FOXO3 signaling, partly due to the complex molecular links between GC activation of both GR and AMPK, and AMPK‐dependent activation of FOXO3. However, as GR has the capacity to promote the binding of other transcription factors to DNA, via chromatin remodeling (Miranda et al, 2013; Goldstein et al, 2017), it is likely that GR would promote FOXO3 binding at regulatory elements, as FOXO3 requires pre‐programmed chromatin to bind, and it does not often work as a pioneer factor (Eijkelenboom et al, 2013a,b). Cortisol and insulin, which activate GR and inhibit FOXO3 respectively, along with changes in cellular AMP levels may therefore modulate inflammation together, depending on the metabolic status of the cell and organism. Supporting the notion that FOXOs are important for the resolution of inflammation, insulin treatment has been shown to exacerbate sepsis‐induced lung injury in diabetic rats (Filgueiras et al, 2014). Moreover, GCs control skeletal muscle metabolism, through the induction of the transcription factor KLF15, which activity is a key component for repair in a mouse model of Duchenne muscular dystrophy (Morrison‐Nozik et al, 2015). The metabolic actions of GCs may therefore be highly unappreciated in the regulation of inflammatory diseases, but our data further support the necessity for metabolic modulation as a core aspect of GCs in the resolution of inflammation. However, the clear implication of an AMPK‐GR‐FOXO3 regulatory trinity is that multiple hormonal signals are integrated to fine‐tune gene expression in immune cells. We demonstrate that Foxo3 deletion recapitulates the effects of AMPKα1 loss, also reducing the effect of GCs on efferocytosis, linking these three pathways together.
To conclude, we demonstrate that GCs activate and require AMPKα1 to efficiently polarize macrophages to a restorative phenotype and promote the resolution of inflammation. This effect requires the recruitment of FOXO3 at loci targeted by GR in an AMPKα1‐dependent way. Intriguingly, a part of the GC response was maintained in AMPKα1‐deficient macrophages and another part was strictly dependent on AMPK activity. Indeed, the expression of the classical GR target genes was not affected in AMPKα1‐deficient macrophages, despite a reduced phosphorylation of the GR, leading to unaltered cytokine regulation. On the other hand, AMPKα1 activation appeared to control the gene networks involved in the metabolic‐dependent change required for the shift of macrophage phenotype (Varga et al, 2016a). This has implications for the role of GR signaling on macrophage function depending on the energy status of the cell and could help identification of new small molecule strategies to enhance GC action in a clinical setting.
Materials and Methods
Animal models
C57BL/6, LysMcre/+;AMPKɑ1fl/fl (B6‐Prkaa1flox/flox/J; B6‐Lyz2tm1(cre)ifo, designated as LysM‐α1−/−) and 129;B6‐Prkaa1tm (AMPKα1−/−) adult male mice (8–14 weeks old), Foxo3−/− mixed sex between 9 and 21 weeks old were bred, FVB/N mixed sex mice between 8 and 21 weeks old were purchased from Charles River and used according to French and German legislations and protocols were approved by ethical committees. Mice received water and food ad libitum. For muscle injury experiments, mice were anesthetized with isoflurane and Tibialis Anterior (TA) muscle was injected with cardiotoxin (Latoxan) at 12 μM (50 μl per muscle) as previously described (Mounier et al, 2013). Mice were treated by intra‐peritoneal (i.p.) injection of either Dexamethasone (Dex; concentrations indicated in the figures, Sigma‐Aldrich, D2915) or PBS (Vehicle) at various time points after injury. Muscles were harvested at different time points after injury. For lung inflammation experiments, male and female mice (8–14 weeks) were injected i.p. with either vehicle (PBS) or LPS (Sigma‐Aldrich, L2880) at a concentration of 10 mg/kg. Mice were then injected with either Dex (Sigma‐Aldrich, D2915) 1 mg/kg or cyclodextrin (1 mg/kg) vehicle control. After 30 min, mice were injected intra‐venous (i.v.) with 2.6 μl/g oleic acid (Sigma‐Aldrich, O1008) as a solution of 40% oleic acid in 1% bovine serum albumin (Sigma‐Aldrich, A3912) as previously described (Vettorazzi et al, 2015). Mice were killed 24 h later and bronchoalveolar lavage (BAL) was performed by instilling the lungs with 1 ml of BAL fluid (PBS, 10 mM EDTA, 1% BSA) three times. Survival after lung inflammation was measured for 28 h. Mice body temperature and body mass were measured each 4 h after induction of lung injury. Mice were sacrificed when limit points were reached, body temperature lower than 29°C and 15% of total body weight loss. General aspect of mice was also considered.
Western blot
AMPK/ACC: Macrophages were seeded at 208,000 cells/cm2 in 6‐well plates. Cells were treated with Dex (Sigma‐Aldrich, D4902, 1 × 10−7 M or 1 × 10−8 M) and 991 (Spirochem, 129739‐36‐2, 1 μM) for 1 h in DMEM containing 10% of charcoal stripped serum (Gibco, 12676029). Total protein extract was obtained in cell lysis buffer containing 50 mM Tris–HCL (pH 7.5), 1 mM EDTA, 1 mM EGTA, 0.27 M sucrose, 1% triton, 20 mM glycerol‐2‐phosphate disodium, 50 mM NaF, 0.5 mM PMSF, 1 mM benzamidine, 1 mM Na3VO4, and 1% cocktail phosphatase inhibitor 3 (Sigma‐Aldrich, P0044). Thirty micrograms of protein were subjected to SDS–PAGE (100 V) and transferred onto a nitrocellulose membrane overnight at 4°C, 30 V. Blots were probed with antibodies against p‐AMPKɑ (Cell Signaling, #2535, 1:1,000), AMPKɑ (Cell Signaling, #2532, 1:1,000), p‐ACC (Cell Signaling, #3661, 1:1,000), ACC (Cell Signaling, #3662, 1:1,000), with β‐actin (Sigma‐Aldrich, A5316, 1:2,000) as a loading control. p‐AMPK1α immunoblots and AMPKα1ɑ immunoblots were performed on separated blot and each independent experiment was also performed on independent Western blot. Blots were quantified using ImageJ software and results represent (p‐AMPKα/β‐actin) ratio on (AMPKα/β‐actin) ratio for each experiment and they are also normalized to the control condition.
GR/p38: Macrophages were seeded at confluency in 10 cm plates, and changed to DMEM containing charcoal stripped serum (Sigma‐Aldrich, F7524) overnight. The following day, cells were treated with vehicle (DMSO) or Dex (Sigma‐Aldrich, D4902, 1 × 10−7 M) for 1 h. Cells were then washed twice in ice cold PBS and total protein was extracted using RadioImmunoprecipitation Assay (RIPA) buffer (50 mM Tris–HCl pH 7.4, 1% NP40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA) containing protease inhibitors (Roche, 04694124001), phosphatase inhibitor cocktail 2 (Sigma‐Aldrich, P5726) and phosphatase inhibitor cocktail 3 (Sigma‐Aldrich, P0044). 25 μg of protein was subjected to SDS‐PAGE (100 V) and transferred to a nitrocellulose membrane for 1 h at 4°C, 100 V. Blots were blocked with 5% BSA (Sigma‐Aldrich) and probed with antibodies against p‐GR (Cell Signaling, #4161, 1:1,000), GR (Cell Signaling, #3660, 1:1,000), p‐p38 (Cell Signaling #4511, 1:1,000), p38 (Cell Signaling, #8690, 1:1,000) with β‐actin (Sigma‐Aldrich, A5316, 1:2,000) or tubulin (Santa Cruz, sc‐8035, 1:1,000) as a loading control. Blots were quantified using ImageJ.
Immunoprecipitation
BMDMs were treated with Dex or 911 as described above and washed with PBS containing pStop (Roche) and Complete Protease inhibitor (Roche). Cells were lysed using RIPA buffer and protein content was measured using BCA. Immunoprecipitation was performed with 300 μg of proteins using Pierce Protein A Magnetic Beads (Thermo Fisher Scientific). Protein lysates were precleared by incubation with protein A beads followed by incubation with 1:100 anti‐pSerine AB (Novus NB100‐1953) or 3 μg of IgG control antibodies (Cell Signaling Technology) overnight on a rotator at 4°C. Beads were washed with PBS and resuspended in 1× SDS buffer, which was then boiled for 5 min. Leftover beads were removed using a magnet. Western blot was then performed as described above. IgG band intensity was removed from calculations.
Analysis of lung immune cells, total cell counts and FACS
Cells isolated by BAL were centrifuged, washed with BAL fluid and red blood cells lysed. Resulting cells were counted using a hemocytometer and normalized to the total amount of BAL fluid recovered. For FACS, cells were suspended in BAL fluid containing FcɣRII/III block (eBiosciences, 1:300), washed then incubated with antibodies against CD45 (APC, Invitrogen 30‐F11, 1:300), CD11b (FITC, Invitrogen M1/70, 1:300), F4/80 (AF700, Invitrogen BM8, 1:300), Ly6G (Pe‐Cy7, Invitrogen RB6‐8C5, 1:300), Ly6C (PerCP‐Cy5.5, eBiosciences HK1.4, 1:300). After washing, cells were analyzed immediately using a flow cytometer (LSRII, BD). Data were analyzed using FlowJo software.
Cytokine multiplex
Cytokines were determined in BAL fluid using a Cytokine & Chemokine Convenience 36‐Plex Mouse ProcartaPlex Panel 1A (ThermoFisher) using a Bio‐plex 200 System (Bio‐Rad). Samples below the detection threshold were excluded from analysis.
Histological analysis and in vivo immunostaining
TA muscles were mounted on piece of cork and fixed with tragacanth gum (VWR, 24437.260) before being frozen in isopentane cooled by liquid nitrogen. Muscles were stored at −80°C. TA muscles were sectioned with cryomicrotome (NX50, Microm) in 10 μm thick sections and slides were kept at −80°C. Slides were first stain with hematoxylin and eosin to check cardiotoxin injection and immunofluorescence was performed only on muscles that showed at least 90% of their area occupied by regenerating myofibers (i.e., exhibiting central nuclei). For immunofluorescence experiments, slides were incubated with Triton ×100 0.5%, saturated with bovine serum albumin (BSA) 2% and were incubated overnight with primary anti‐body: anti‐embMHC (Santa Cruz, sc‐53091, 1:100) or anti‐laminin (Sigma Aldrich, L9393, 1:200). Slides were washed with PBS and incubated with secondary antibodies: Alexa Fluor 488 anti‐mouse IgG1 (Jackson Immuno research, 115‐545‐205, 1:200) or Cy5 Donkey anti‐Rabbit IgG (H + L; Jackson Immuno research, 711‐175‐152, 1:200). Sections were soaked for 10 s in Hoechst solution H 33342 (Sigma‐Aldrich, B2261, 1:1,000) and washed once with PBS before mounting with antifading Fluoromount G medium (Interchim, FP‐483331). Slides were stored at 4°C protected from light until picture acquisition. For adult myosin immunostaining, triton was not used and slides were blocked with Mouse on Mouse (M.O.M™) Blocking Reagent (Vector Laboratories, MKB‐2213). All samples of the same experiment were recorded in similar conditions (microscope, magnification, exposure time, and binning). For whole section analysis, slides were automatically scanned at ×10 or ×20 of magnification using an Axio Observer.Z1 (Zeiss) connected to a CoolSNAP HQ2 CCD Camera (Photometrics) using Metavue software. The whole muscle section was automatically reconstituted by Metavue software. Cross‐sectional analysis was performed using Open‐CSAM ImageJ macro (Desgeorges et al, 2019b).
After BAL, lungs were fixed in 4% PFA overnight, then transferred to 70% ethanol. Fixed lungs were processed and embedded into paraffin, then cut to 5 μm sections using a microtome (Leica RM2255). Slides were then stained with hematoxylin and eosin and imaged using an Olympus BX41 microscope at 20× magnification.
Bone marrow‐derived macrophages
Macrophages were obtained from bone marrow precursors as previously described (Mounier et al, 2013). Total bone marrow was isolated from mice by flushing the femur and tibiae bone marrow with culture medium. Extracted bone marrow was cultured in DMEM high glucose high pyruvate (Gibco, 11995065) containing 20% of FBS, 1% of penicillin/streptomycin, 1% of fungizone and 30% of L929 conditioned medium for 5–7 days. L929 secrete high levels of macrophage colony‐stimulating factor (MCSF) that triggers macrophage differentiation. L929 conditioned medium was obtained after 10 days of L929 cells culture with DMEM containing 10% FBS.
DAMPs—Damaged associated molecular patterns from muscle
DAMPs were obtained from ischemia‐injured muscles. Briefly, mice hindlimb underwent an ischemia for 2 h. After 20 h of reperfusion, all hindlimb muscles were harvested. Three mice were pooled for each substrate. Once harvested, muscles were digested with Mammalian cell lysis buffer (Sigma‐Aldrich, MCL1). Two tungsten beads were added in samples to homogenize the muscle extract with a tissue lyser. After centrifugation, samples were sonicated and supernatant was recovered, pooled, and stored at −80°C.
Macrophage inflammatory status—in vitro
Macrophages were seeded at 54,000 cells/cm2 in DMEM containing 10% of FBS on glass coverslips in 12‐well plates. Cells were treated with muscle DAMPs (1 μg/ml) or LPS (100 ng/ml for 24 h) and Dex (Sigma‐Aldrich, D4902, 1 × 10−8 M) was further directly added in the well for 6 h. In the AKT inhibition experiments, after the 24 h incubation with muscle DAMPs or LPS, cells were treated with MK‐2206 (Adooq Bioscience, A10003, 1 μM; or 0.01% of DMSO for controls) for 1 h prior addition of Dex.
Cells were fixed with PFA 4%, permeabilized with Triton ×100 0.5% and incubated for 1 h with BSA 4%. Staining for CCL3 (Santa Cruz, sc‐1383, 1:50), iNOS (Abcam, ab3523, 1:50), CD206 (Santa Cruz, sc‐58987, 1:50), CD163 (Santa Cruz, sc‐33560, 1:50 or BIOSS, bs‐2527R, 1:100), TGFβ1 (Abcam, ab64715, 1:50) was performed as in (Mounier et al, 2013). All samples of the same experiment were recorded in similar conditions (microscope, magnification, exposure time and binning). At least 10 pictures per condition were acquired at ×20 magnification on an Axio Imager.Z1 microscope (Zeiss) connected to a CoolSNAP MYO CCD camera (photometrics) using MetaMorph Software. Quantification of macrophage markers was done manually using ImageJ software.
RNA‐sequencing and RT–qPCR
Macrophages were seeded and allowed to adhere for 16 h. Cells were then washed twice with PBS and medium was changed to Macrophage‐SFM (Thermo Fisher, 12065074) with 1% penicillin/streptomycin, 1% L‐glutamine, 1% sodium pyruvate, 1% fungizone for 16 h. Cells were then treated with either vehicle (DMSO) or 100 nM Dex (Sigma, D4902), for 24 h. Cells were then washed twice with PBS and total RNA extracted using TRIzol (Invitrogen, 15596018), according to the manufacturer's protocol. For qRT–PCR, RNA was measured using a Nanodrop 1 μg of RNA was then reverse transcribed into cDNA (Applied Biosystems, High Capacity cDNA kit, 4368813) with additional RNase inhibitor (Invitrogen RNase OUT, 10777019). cDNA was quantified using a ViiA 7 (Applied Biosystems) and analyzed using the delta–delta CT method, relative to the average of WT vehicle treated samples. For Rxra and Jag1 expression analysis, the normalized relative quantity (NRQ) method was used as previously described (Juban et al, 2018) with Ppia as the housekeeping gene (list of primers in Table 1). RNA integrity was assessed using a 2100 Bioanalyzer (Agilent) and samples with a RIN value of > 6 were taken forward for library prep. Library prep and sequencing was performed using the Novaseq 6000 S4 platform at Novogene (Novogene, Beijing, China). Differential expression analysis was performed using DESeq2. Gene ontology and pathway analyses were performed using DAVID (Huang da et al, 2009), and ENRICHR (Kuleshov et al, 2016).
Table 1.
Primer list for RT–qPCR.
| Target | Forward | Reverse |
|---|---|---|
| Rpl | CCTGCTGCTCTCAAGGTT | TGGCTGTCACTGCCTGGTACTT |
| Tnf | AGGGGCCACCACGCTCTTCT | TGAGTGTGAGGGTCTGGGCCAT |
| Gilz | GACCTCGTGAAGAACCACCT | AGCCACTTACACCGCAGAAC |
| Dusp1 | GTGCCTGACAGTGCAGAATC | CACTGCCCAGGTACAGGAAG |
| Ccl3 | TGCCCTTGCTGTTCTTCTCTG | CAACGATGAATTGGCGTGG |
| Il6 | AAACCGCTATGAAGTTCCTCTCT | AGCCTCCGACTTGTGAAGTGGT |
| Ppia | GTGACTTTACACGCCATAATG | ACAAGATGCCAGGACCTGTAT |
| Rxra | CAAACATTTCCTGCCGCTCG | CAGTGGAGAGCCGATTCCC |
| Jag1 | GGGTCTACGCCTGTCATCG | TCCACCAGCAAAGTGTAGGAC |
Transcription factor binding site analysis
Genes up‐ or down‐regulated by Dex were analyzed for over‐represented transcription factor binding sites using the Single Site Analysis function from oPOSSUM‐3 software (Kwon et al, 2012). Sequences from 2000 bp upstream to 2000 bp downstream of the transcription start sites were analyzed against all the 29,347 genes in the oPOSSUM database using a 0.4 conservation cut‐off and a 85% matrix score threshold. Enriched transcription factor binding sites were ranked by Z‐score and only the 15 highest scores were considered.
Gene set variation analysis
For GSVA (Hanzelmann et al, 2013), human analogues of differentially expressed mouse genes were determined before analysis using selected macrophage datasets from the Molecular Signatures Database using C7 – Immunologic signature gene sets (Subramanian et al, 2005; Liberzon et al, 2011). A FOXO3‐dependent gene set was defined using GSE37051, comparing unstimulated wildtype BMDMs to unstimulated FOXO3 deficient BMDMs, analyzed by GEO2R. Gene set enrichment was performed using GSEA 4.0.3 (Mootha et al, 2003; Subramanian et al, 2005).
Myoblast—macrophage coculture
Macrophages were seeded at 54,000 cells/cm2 in 12‐well plates in DMEM 10% FBS. They were treated for 3 days with 1 × 10−7 or 1 × 10−8 M of Dex and washed. DMEM free serum was added for 24 h to obtain conditioned medium. Myoblasts were obtained from TA muscles and were cultured in DMEM/F12 containing 20% FBS and 2% UltroserG (Pall Inc, 15950‐017) as previously described (Mounier et al, 2013). For proliferation studies, myoblasts were seeded at 10,000 cells/cm2 on Matrigel (Corning, 356231, 1:10) and incubated for 1 day with macrophage‐conditioned medium +2.5% FBS. Myoblasts were then washed, fixed and incubated with anti‐Ki67 antibody (Abcam, ab15580, 1:100) visualized using cy3‐conjugated secondary antibody (Jackson Immunoresearch, 711‐165‐152, 1:200). For differentiation experiments, myoblasts were seeded at 30,000 cells/cm2 on Matrigel (Corning, 356231, 1:10) for 3 days with macrophage‐conditioned medium containing 2% Horse Serum (Gibco). Cells were washed, fixed, and stained with anti‐desmin antibody (Abcam, ab32362, 1:200) and visualized by cy3‐conjugated secondary antibody (Jackson immunoresearch, 711‐165‐152, 1:200). At least 9 ± 1 pictures per condition were acquired at ×20 magnification on an Axio Imager.Z1 microscope (Zeiss) connected to a CoolSNAP MYO CCD camera (Photometrics) using MetaMorph Software. Quantification of proliferation (Ki67pos cells) and of differentiation (percentage of cells with 2 nuclei or more) was done manually using ImageJ software.
Glucocorticoid receptor and Foxo3a subcellular localization
BMDMs were seeded at a density of 20,000 cells in 96‐well plates. Medium was changed to Macrophage‐SFM (Thermo Fisher, 12065074) with 1% penicillin/streptomycin, 1% L‐glutamine, 1% sodium pyruvate, 1% fungizone overnight. The following day, cells were treated with 100 nM Dex (Sigma‐Aldrich, D4902) for 1 h, and fixed with 4% PFA for 15 min at 4°C. PFA was removed, and samples were washed with PBS 3 times. Cells were permeabilized with permeabilization buffer (PBS, 0.1% TritonX‐100) for 30 min at 4°C, then non‐specific binding sites blocked with blocking buffer (PBS, 0.1% TritonX‐100, 1% fetal calf serum) for 1 h at room temperature, then overnight at 4°C. Cells were incubated with anti‐GR antibody (Cell Signaling D8H2 3660, 1:200) or with anti‐Foxo3a antibody (Cell Signaling 75D8 9467, 1:250) overnight at 4°C. After washing with PBS 5 times, cells were incubated with a fluorescently labeled secondary antibody (Thermo Fisher, A11037 anti‐rabbit IgG Alexafluor 594) for 2 h at room temperature. Cells were washed five more times and incubated with 1 μg/ml DAPI (Sigma‐Aldrich, D9542) for 10 min at room temperature. Cells were washed twice more and retained in PBS for immediate imaging. Images were acquired using the ImageXpress MicroConfocal (Molecular Devices), at least 12 images were acquired per biological sample. Nuclear GR intensity was determined using ImageJ.
Ex vivo phagocytosis assay
TA muscle was damaged as described above. Two and a half days after injury, mice were treated with i.p. injection of Dex (Sigma‐Aldrich, D2915, 1 mg/kg). At day 3, TA muscles were harvested, digested for 1 h with collagenase (Roche, 11088831001, 1 mg/ml) in RPMI medium at 37°C. Digested muscles were filtered through 30 μm filter with DMEM containing 50% FBS. CD45pos cells were isolated using magnetic sorting (Milteny Biotec, 130‐042‐201 and 130‐052‐301) and were seeded at 190,000 cells per wells in 12‐well plates in DMEM serum free. Primary myoblasts previously stained with PKH67 (Sigma‐Aldrich, MINI67) were first submitted to 20 mM of H2O2 for 20 min to induce necrosis before being added to wells at 1:3 ratio (570,000 myoblasts per condition). After 6 h of phagocytosis, cells were harvested, blocked with FcR Blocking Reagent (Miltenyi Biotec, 130‐059‐901) and thereafter stained with anti‐CD45 antibody (eBioscience, 25‐0451‐82, 1:200), anti‐Ly6C antibody (eBioscience, 12‐5932‐82, 1:400) and anti‐CD64 antibody (BD Pharmingen, 558539, 1:100) for flow cytometry analysis.
Macrophage inflammatory status—in vivo
TA muscle was damaged and treated as described above for the ex vivo phagocytosis. After muscle digestion, non‐viable cells were labeled with Ghost Dye Red 780 (Tonbo Biosciences, 13‐0865‐T100, 1:1,000). Then, the cell suspension was incubated with anti‐mouse FcR Blocking Reagent and further stained with BV510‐conjugated anti‐CD45 (Biolegend, 103138, 1:100), AF647‐conjugated anti‐CD64 (Biolegend, 139322, 1:50), PE‐Cy7‐conjugated anti‐Lyve1 (Novus Biologicals, NBP1‐43411PECY7, 1:100), PE‐conjugated anti‐Folate receptor (Biolegend, 153304, 1:100) or AF488‐conjugated anti‐CD206 (eBioscience, 53‐2061‐82, 1:100) antibodies for 20 min at 4°C. Macrophages were analyzed with a FACSCanto II flow cytometer (BD Biosciences).
In vitro phagocytosis assay
BMDMs were isolated as described above. After differentiation, 100,000 BMDMs per well were seeded into 12 well plates, allowed to adhere and medium changed to L929 conditioned medium with charcoal stripped serum. Simultaneously, thymuses were isolated from two mice, and treated over‐night with Dex (Sigma‐Aldrich, D4902, 1 × 10−6M) in RPMI‐1640 (Sigma‐Aldrich, R8758) to induce apoptosis. The next day, apoptotic thymocytes were washed with cold PBS and FCS to remove excess Dex, then 7 × 106 cells were incubated with CSFE (Thermofisher, C34554, 1:25,000) for 20 min at 37°C. Labeled thymocytes were then added to BMDMs in a ratio of 5:1 and incubated at 37°C for 2.5 h to allow phagocytosis. A negative control was maintained at 4°C. Cells were then washed twice with PBS, and scraped into FACS buffer (PBS, 10 mM EDTA, 1% BSA). The cell mixture was first blocked with anti‐FcɣRII/III (eBiosciences, 14‐0161‐82) then stained with anti‐F4/80 (Invitrogen, BM8, 1:300) and analyzed on a flow cytometer (LSRII, BD). Data were analyzed with FlowJo, with signal higher than the 4°C control considered a positive phagocytosis event.
Chromatin immunoprecipitation
ChIP was performed as described elsewhere (Nelson et al, 2006), with some modifications. 20 × 106 BMDMs were changed to macrophage serum free medium (Gibco) overnight before treatment with vehicle (DMSO) or Dex (100 nM; Sigma) for 2 h. Cells were washed twice with ice cold PBS then fixed for 15 min with 1% formaldehyde (ThermoScientific 28908). Formaldehyde was quenched using 1 M Glycine (Sigma, 33226) for 5 min at room temperature. Cells were then washed twice more with ice‐cold PBS before homoengization with a Dounce homogenizer (Active Motif, 40415) in Fast IP buffer (150 mM NaCl, 50 mM Tris–HCl (pH7.5), 5 mM EDTA, 0.5%v/v NP‐40, 1%v/v Triton X‐100) with protease inhibitors (cOmplete Tablets EDTA‐free, EASYpack, Roche, 04693132001). Resulting nuclei were centrifuged at 4,000 g and resuspended in shearing buffer (1% SDS, 10 mM EDTA pH 8, 50 mM Tris pH8) and sheared in 1.5 ml Bioruptor Microtubes (Diagenode, C30010016) in a Bioruptor (Diagenode) for 8 cycles 30 s on, 30 s off. Resulting sheared chromatin was cleared by centrifugation for 10 min at 13,500 g, 4°C before diluting 1/10. 1 ml of diluted chromatin was incubated with 3 μg anti‐FOXO3 antibody (Santa Cruz D‐12, sc‐48348), or 3 μg of isotype control antibody (Rabbit DA1E mAb IgG XP Isotype Control, 3900, Cell Signaling Technologies) rotating overnight at 4°C. Chromatin‐antibody mixes were then incubated with 20 μl Protein A Dynabeads (Invitrogen, 10001D) for 3 h, rotating at 4°C. After washing with Fast IP buffer, beads were eluted, and DNA precipitated. Chromatin was then analyzed by qPCR and calculated as fold enrichment over IgG control (list of primers in Table 2).
Table 2.
Primer list for ChIP‐PCR.
| Target | Forward | Reverse |
|---|---|---|
| Jag1 ChIP | ACCTTGGGCTAGTTAGGGGT | GAGCCAGATTCGGAGTAGGC |
| Stat3 ChIP | CTGACAGCTGGGCGTTTAGA | GCAATGTCCCCAGCCTCTTA |
| Rxra ChIP | CTCCCATCTCCCAGTTAGCG | TTAGAGCCTGGAGTGAGGCA |
| Klf3 ChIP | CCCACCACAATGTACTGCCT | CGCTTGGCATCTCTGAAGC |
Statistical analysis
Whenever possible, counting and analyses were done in a blind way. All experiments were performed at least using three independent primary cultures. For in vivo experiments, at least four mice were analyzed from three independent experiments for each group. Statistic tests were performed using GraphPad Prism software. Normality was tested with Shapiro–Wilk test. Depending on the experiment and the normality, parametric (Student t‐test, ANOVA) or non‐parametric (Wilcoxon–Mann–Whitney, Kruskal–Wallis with post hoc comparisons) tests (two‐sided) were used. Gehan Breslow Wilcoxon test was used for survival experiments. P < 0.05 was considered significant.
Author contributions
Giorgio Caratti: Conceptualization; data curation; formal analysis; validation; visualization; methodology; writing – original draft; writing – review and editing. Thibaut Desgeorges: Conceptualization; data curation; formal analysis; validation; visualization; methodology; writing – original draft; writing – review and editing. Gaëtan Juban: Data curation; validation; visualization; methodology; writing – review and editing. Ulrich Stifel: Methodology. Aurélie Fessard: Investigation; methodology. Mascha Koenen: Validation; investigation; methodology. Bozhena Caratti: Investigation; methodology. Marine Théret: Data curation; validation; investigation; methodology. Carsten Skurk: Resources; writing – review and editing. Bénédicte Chazaud: Conceptualization; supervision; funding acquisition; validation; investigation; visualization; writing – original draft; project administration; writing – review and editing. Jan P Tuckermann: Conceptualization; supervision; funding acquisition; validation; investigation; visualization; writing – original draft; project administration; writing – review and editing. Rémi Mounier: Conceptualization; supervision; funding acquisition; validation; investigation; visualization; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 4
Acknowledgements
This work was supported by French‐German Collaboration for Joint Projects of the French Agence Nationale de Recherche (ANR) and the Deutsche Forschungsgemeinschaft (DFG) to BC, RM (15‐CE15‐0024‐01) and JT (Tu220/13), Campus France‐DAAD to BC and JPT. BC and GJ were supported by Fondation pour la Recherche Médicale (Equipe FRM DEQ20140329495). BC and RM were supported by CNRS (Centre National de la Recherche Scientifique), Inserm and Société Française de Myologie, GJ by AFM‐Telethon. JPT was supported by FDG Collaborative Research Centers CRC1149 Trauma Project‐ID 251293561 and GC was supported by Baustein Grant from the Medical Faculty of Ulm University and ProtrainU grant from Ulm University. Excellent support was supplied by the animal facilities at University of Ulm and the Université Claude Bernard Lyon 1.
EMBO reports (2023) 24: e55363
Contributor Information
Bénédicte Chazaud, Email: benedicte.chazaud@inserm.fr.
Jan P Tuckermann, Email: jan.tuckermann@uni-ulm.de.
Rémi Mounier, Email: remi.mounier@univ-lyon1.fr.
Data availability
The datasets produced in this study are available in the following databases:
RNA‐Seq data for AMPKα1KO dataset: Gene Expression Omnibus 167382 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167382).
RNA‐Seq data for Foxo3aKO dataset: Gene Expression Omnibus 190235 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190235).
References
- Arnold L, Henry A, Poron F, Baba‐Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204: 1057–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (2011) Glucocorticosteroids: current and future directions. Br J Pharmacol 163: 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Christ‐Crain M, Kola B, Lolli F, Fekete C, Seboek D, Wittmann G, Feltrin D, Igreja SC, Ajodha S, Harvey‐White J et al (2008) AMP‐activated protein kinase mediates glucocorticoid‐induced metabolic changes: a novel mechanism in Cushing's syndrome. FASEB J 22: 1672–1683 [DOI] [PubMed] [Google Scholar]
- Desgeorges T, Caratti G, Mounier R, Tuckermann J, Chazaud B (2019a) Glucocorticoids shape macrophage phenotype for tissue repair. Front Immunol 10: 1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgeorges T, Liot S, Lyon S, Bouviere J, Kemmel A, Trignol A, Rousseau D, Chapuis B, Gondin J, Mounier R et al (2019b) Open‐CSAM, a new tool for semi‐automated analysis of myofiber cross‐sectional area in regenerating adult skeletal muscle. Skelet Muscle 9: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston J, Herschbach J, Wagelie‐Steffen AL, Christiansen SC, Zuraw BL (2007) The anti‐inflammatory effect of glucocorticoids is mediated by glucocorticoid‐induced leucine zipper in epithelial cells. J Allergy Clin Immunol 119: 115–122 [DOI] [PubMed] [Google Scholar]
- Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, van Triest MH, van Boxtel R, Schulze A, de Laat W, Cuppen E et al (2013a) Genome‐wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol Syst Biol 9: 638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelenboom A, Mokry M, Smits LM, Nieuwenhuis EE, Burgering BM (2013b) FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell Rep 5: 1664–1678 [DOI] [PubMed] [Google Scholar]
- Fardet L, Petersen I, Nazareth I (2011) Prevalence of long‐term oral glucocorticoid prescriptions in the UK over the past 20 years. Rheumatology (Oxford) 50: 1982–1990 [DOI] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B et al (2013) AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo . Cell Metab 17: 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faus H, Haendler B (2006) Post‐translational modifications of steroid receptors. Biomed Pharmacother 60: 520–528 [DOI] [PubMed] [Google Scholar]
- Filgueiras LR, Capelozzi VL, Martins JO, Jancar S (2014) Sepsis‐induced lung inflammation is modulated by insulin. BMC Pulm Med 14: 177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I, Baek S, Presman DM, Paakinaho V, Swinstead EE, Hager GL (2017) Transcription factor assisted loading and enhancer dynamics dictate the hepatic fasting response. Genome Res 27: 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A (2007a) An AMPK‐FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans . Curr Biol 17: 1646–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A (2007b) The energy sensor AMP‐activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem 282: 30107–30119 [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics 14: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Hudson WH, Vera IMS, Nwachukwu JC, Weikum ER, Herbst AG, Yang Q, Bain DL, Nettles KW, Kojetin DJ, Ortlund EA (2018) Cryptic glucocorticoid receptor‐binding sites pervade genomic NF‐kappaB response elements. Nat Commun 9: 1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juban G, Saclier M, Yacoub‐Youssef H, Kernou A, Arnold L, Boisson C, Ben Larbi S, Magnan M, Cuvellier S, Theret M et al (2018) AMPK activation regulates LTBP4‐dependent TGF‐beta1 secretion by pro‐inflammatory macrophages and controls fibrosis in Duchenne muscular dystrophy. Cell Rep 25: 2163–2176 [DOI] [PubMed] [Google Scholar]
- Kalvisa A, Siersbaek MS, Praestholm SM, Christensen LJL, Nielsen R, Stohr O, Vettorazzi S, Tuckermann J, White M, Mandrup S et al (2018) Insulin signaling and reduced glucocorticoid receptor activity attenuate postprandial gene expression in liver. PLoS Biol 16: e2006249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon AT, Arenillas DJ, Worsley Hunt R, Wasserman WW (2012) oPOSSUM‐3: advanced analysis of regulatory motif over‐representation across genes or ChIP‐Seq datasets. G3 (Bethesda) 2: 987–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugesen K, Jorgensen JOL, Sorensen HT, Petersen I (2017) Systemic glucocorticoid use in Denmark: a population‐based prevalence study. BMJ Open 7: e015237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XN, Song J, Zhang L, LeMaire SA, Hou X, Zhang C, Coselli JS, Chen L, Wang XL, Zhang Y et al (2009) Activation of the AMPK‐FOXO3 pathway reduces fatty acid‐induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 58: 2246–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvak V, Ratushny AV, Lampano AE, Schmitz F, Huang AC, Raman A, Rust AG, Bergthaler A, Aitchison JD, Aderem A (2012) A FOXO3‐IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature 490: 421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Peng Y, Wang X, Fan Y, Qin C, Shi L, Tang Y, Cao K, Li H, Long J et al (2016) Mitochondrial dysfunction launches dexamethasone‐induced skeletal muscle atrophy via AMPK/FOXO3 signaling. Mol Pharm 13: 73–84 [DOI] [PubMed] [Google Scholar]
- Lu M, Xu W, Gao B, Xiong S (2016) Blunting autoantigen‐induced FOXO3a protein phosphorylation and degradation is a novel pathway of glucocorticoids for the treatment of systemic lupus erythematosus. J Biol Chem 291: 19900–19912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzner N, Kalbacher H, Krones‐Herzig A, Rosl F (2012) FOXO3 is a glucocorticoid receptor target and regulates LKB1 and its own expression based on cellular AMP levels via a positive autoregulatory loop. PLoS One 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25: 677–686 [DOI] [PubMed] [Google Scholar]
- McArthur S, Juban G, Gobbetti T, Desgeorges T, Theret M, Gondin J, Toller‐Kawahisa JE, Reutelingsperger CP, Chazaud B, Perretti M et al (2020) Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J Clin Invest 130: 1156–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Voss TC, Sung MH, Baek S, John S, Hawkins M, Grontved L, Schiltz RL, Hager GL (2013) Reprogramming the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res 73: 5130–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E et al (2003) PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 [DOI] [PubMed] [Google Scholar]
- Morrison‐Nozik A, Anand P, Zhu H, Duan Q, Sabeh M, Prosdocimo DA, Lemieux ME, Nordsborg N, Russell AP, MacRae CA et al (2015) Glucocorticoids enhance muscle endurance and ameliorate Duchenne muscular dystrophy through a defined metabolic program. Proc Natl Acad Sci U S A 112: E6780–E6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, Sanz N, Ferry A, Sakamoto K, Foretz M et al (2013) AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab 18: 251–264 [DOI] [PubMed] [Google Scholar]
- Nader N, Ng SS, Lambrou GI, Pervanidou P, Wang Y, Chrousos GP, Kino T (2010) AMPK regulates metabolic actions of glucocorticoids by phosphorylating the glucocorticoid receptor through p38 MAPK. Mol Endocrinol 24: 1748–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1: 179–185 [DOI] [PubMed] [Google Scholar]
- Oh KS, Patel H, Gottschalk RA, Lee WS, Baek S, Fraser IDC, Hager GL, Sung MH (2017) Anti‐inflammatory chromatinscape suggests alternative mechanisms of glucocorticoid receptor action. Immunity 47: 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega‐Gomez A, Perretti M, Soehnlein O (2013) Resolution of inflammation: an integrated view. EMBO Mol Med 5: 661–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman RA, Yeh JY, Deal CL (2013) Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res (Hoboken) 65: 294–298 [DOI] [PubMed] [Google Scholar]
- Ratman D, Mylka V, Bougarne N, Pawlak M, Caron S, Hennuyer N, Paumelle R, De Cauwer L, Thommis J, Rider MH et al (2016) Chromatin recruitment of activated AMPK drives fasting response genes co‐controlled by GR and PPARalpha. Nucleic Acids Res 44: 10539–10553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacta MA, Tharmalingam B, Coppo M, Rollins DA, Deochand DK, Benjamin B, Yu L, Zhang B, Hu X, Li R et al (2018) Gene‐specific mechanisms direct glucocorticoid‐receptor‐driven repression of inflammatory response genes in macrophages. Elife 7: e34864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sag D, Carling D, Stout RD, Suttles J (2008) Adenosine 5′‐monophosphate‐activated protein kinase promotes macrophage polarization to an anti‐inflammatory functional phenotype. J Immunol 181: 8633–8641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schakman O, Gilson H, Kalista S, Thissen JP (2009) Mechanisms of muscle atrophy induced by glucocorticoids. Horm Res 72: 36–41 [DOI] [PubMed] [Google Scholar]
- Sozio MS, Lu C, Zeng Y, Liangpunsakul S, Crabb DW (2011) Activated AMPK inhibits PPAR‐{alpha} and PPAR‐{gamma} transcriptional activity in hepatoma cells. Am J Physiol Gastrointest Liver Physiol 301: G739–G747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns‐Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG (2011) The pathogenesis of sepsis. Annu Rev Pathol 6: 19–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MD, Nedjai B, Hurst T, Pennington DJ (2014) Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 1843: 2563–2582 [DOI] [PubMed] [Google Scholar]
- Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, Schwalie P, Hubner N, Evans RM (2013) Insights into negative regulation by the glucocorticoid receptor from genome‐wide profiling of inflammatory cistromes. Mol Cell 49: 158–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandevyver S, Dejager L, Van Bogaert T, Kleyman A, Liu Y, Tuckermann J, Libert C (2012) Glucocorticoid receptor dimerization induces MKP1 to protect against TNF‐induced inflammation. J Clin Invest 122: 2130–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga T, Mounier R, Gogolak P, Poliska S, Chazaud B, Nagy L (2013) Tissue LyC6‐ macrophages are generated in the absence of circulating LyC6‐ monocytes and Nur77 in a model of muscle regeneration. J Immunol 191: 5695–5701 [DOI] [PubMed] [Google Scholar]
- Varga T, Mounier R, Horvath A, Cuvellier S, Dumont F, Poliska S, Ardjoune H, Juban G, Nagy L, Chazaud B (2016a) Highly dynamic transcriptional signature of distinct macrophage subsets during sterile inflammation, resolution, and tissue repair. J Immunol 196: 4771–4782 [DOI] [PubMed] [Google Scholar]
- Varga T, Mounier R, Patsalos A, Gogolak P, Peloquin M, Horvath A, Pap A, Daniel B, Nagy G, Pintye E et al (2016b) Macrophage PPARgamma, a lipid activated transcription factor controls the growth factor GDF3 and skeletal muscle regeneration. Immunity 45: 1038–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vettorazzi S, Bode C, Dejager L, Frappart L, Shelest E, Klassen C, Tasdogan A, Reichardt HM, Libert C, Schneider M et al (2015) Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat Commun 6: 7796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Frederick J, Garabedian MJ (2002) Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem 277: 26573–26580 [DOI] [PubMed] [Google Scholar]
- Watanabe S, Alexander M, Misharin AV, Budinger GRS (2019) The role of macrophages in the resolution of inflammation. J Clin Invest 129: 2619–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weikum ER, de Vera IMS, Nwachukwu JC, Hudson WH, Nettles KW, Kojetin DJ, Ortlund EA (2017a) Tethering not required: the glucocorticoid receptor binds directly to activator protein‐1 recognition motifs to repress inflammatory genes. Nucleic Acids Res 45: 8596–8608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR (2017b) Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol 18: 159–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willows R, Navaratnam N, Lima A, Read J, Carling D (2017) Effect of different gamma‐subunit isoforms on the regulation of AMPK. Biochem J 474: 1741–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Vannella KM (2016) Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44: 450–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhu H, Ding Y, Liu Z, Cai Z, Zou MH (2017) AMP‐activated protein kinase alpha1 promotes atherogenesis by increasing monocyte‐to‐macrophage differentiation. J Biol Chem 292: 7888–7903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang X, Zhang X, Wang J, Ma Y, Zhang L, Cao X (2019) RNA‐binding protein YTHDF3 suppresses interferon‐dependent antiviral responses by promoting FOXO3 translation. Proc Natl Acad Sci U S A 116: 976–981 [DOI] [PMC free article] [PubMed] [Google Scholar]