

Abstract
The transcription factor EB (TFEB) regulates energy homeostasis and cellular response to a wide variety of stress conditions, including nutrient deprivation, oxidative stress, organelle damage, and pathogens. Here we identify S401 as a novel phosphorylation site within the TFEB proline‐rich domain. Phosphorylation of S401 increases significantly in response to oxidative stress, UVC light, growth factors, and LPS, whereas this increase is prevented by p38 MAPK inhibition or depletion, revealing a new role for p38 MAPK in TFEB regulation. Mutation of S401 in THP1 cells demonstrates that the p38 MAPK/TFEB pathway plays a particularly relevant role during monocyte differentiation into macrophages. TFEB‐S401A monocytes fail to upregulate the expression of multiple immune genes in response to PMA‐induced differentiation, including critical cytokines, chemokines, and growth factors. Polarization of M0 macrophages into M1 inflammatory macrophages is also aberrant in TFEB‐S401A cells. These results indicate that TFEB‐S401 phosphorylation links differentiation signals to the transcriptional control of monocyte differentiation.
Keywords: autophagy, lysosomes, monocytes, p38 MAPK, TFEB
Subject Categories: Immunology, Post-translational Modifications & Proteolysis, Signal Transduction
Stress‐induced phosphorylation of TFEB serine 401 by p38 MAPK promotes the differentiation of monocytes into M1 inflammatory macrophages.
Introduction
The MiT/TFE family of transcription factors consists of four closely related basic helix–loop–helix leucine zipper (bHLH‐ZIP)‐containing proteins named MITF, TFEB, TFE3, and TEFC (Steingrimsson et al, 2004). MiT/TFE proteins have been associated with different cellular processes including cell differentiation, development, energy metabolism, immune response, longevity, and cancer (Raben & Puertollano, 2016; La Spina et al, 2020). One of the best characterized roles of TFEB and its paralogue TFE3 is in the control of cellular homeostasis maintenance by coordinating the expression of autophagy and lysosomal genes in response to environmental clues (Raben & Puertollano, 2016; Slade & Pulinilkunnil, 2017; Yang et al, 2018). Nucleocytoplasmic shuttling of TFEB in response to changes in nutrient status is crucial for the transcriptional control of its target genes. Under nutrient‐rich conditions, TFEB inactivation and retention in the cytoplasm is regulated by the mTORC1‐dependent phosphorylation of several residues, including serine 211, and concomitant binding to the chaperone‐like protein 14‐3‐3. Changes in nutrient availability result in TFEB dissociation from 14‐3‐3, nuclear translocation, and activation, leading to the upregulation of a plethora of genes involved in lysosomal biogenesis, autophagy activation, and metabolic regulation (Martina et al, 2012; Roczniak‐Ferguson et al, 2012; Settembre et al, 2012). In response to different stimuli, TFEB function is also regulated by the action of additional protein kinases, including AKT, CDK4/6, ERK, GSK3β, MAPK3, AMPK, and PKCβ (Settembre et al, 2011; Marchand et al, 2015; Li et al, 2016; Palmieri et al, 2017; Hsu et al, 2018; El‐Houjeiri et al, 2019; Yin et al, 2020), as well as by the activation of protein phosphatases, such as calcineurin and PP2A (Medina et al, 2015; Martina & Puertollano, 2018). Furthermore, additional post‐translational modifications like acetylation, SUMOylation, oxidation, glutathionylation, and ubiquitination (Sha et al, 2017; Puertollano et al, 2018; Goding & Arnheiter, 2019; Wang et al, 2020a, 2020b; Martina et al, 2021) have been reported to contribute to the control of TFEB activation and stability under different stress conditions.
In addition to its role in lysosomal biogenesis and autophagy, TFEB has been described to participate in the control of inflammation and host defense against pathogen infection (Visvikis et al, 2014; Brady et al, 2018; Kim et al, 2021), either directly by regulating expression of pro‐inflammatory cytokine and chemokine genes (Visvikis et al, 2014; Pastore et al, 2016), or indirectly by modulating cellular processes that impact the inflammatory response (Irazoqui, 2020; Rawat & Manjithaya, 2021). However, the contribution of TFEB to these processes seems to be context‐specific, where host effectors induce TFEB activation to restrict the intracellular growth of the pathogens (Rawat & Manjithaya, 2021) whereas some pathogens inhibit TFEB activation to facilitate evasion from the host immune response (Irazoqui, 2020).
Activated macrophages lacking TFEB display an impaired ability to respond to pathogen infection due to a deficient upregulation of anti‐bacterial genes in combination with their failure to activate autophagy and lysosomal biogenesis (Gray et al, 2016; Pastore et al, 2016). In addition, TFEB overexpression enhances the degradative capacity of activated macrophages to reduce atherosclerosis (Sergin et al, 2017) and stimulates the suppression of tumor‐promoting signaling pathways in tumor‐associated macrophages (Li et al, 2020). The activation of TFEB in toll‐like receptor‐induced or bacteria‐exposed macrophages is regulated by a mechanism independent of mTORC1 inactivation (Pastore et al, 2016), suggesting that other protein kinases may participate in the regulation of TFEB function during macrophage activation.
In this study, we identified a novel role of p38 MAPK in TFEB regulation. We showed p38 MAPK‐dependent TFEB phosphorylation at serine 401 (S401) in response to a variety of stress conditions, including oxidative stress, UVC irradiation, growth factors, and lipopolysaccharide (LPS) treatment. Furthermore, inhibition of S401 phosphorylation during PMA‐induced monocyte differentiation prevented TFEB nuclear accumulation and resulted in reduced expression of multiple immune genes. TFEB‐S401A expressing M0 macrophages also failed to efficiently polarize into M1 inflammatory macrophages, showing defective upregulation of cytokines and chemokines, as well as reduced inflammasome activation. We conclude that TFEB is a target of the p38 MAPK signaling pathway that is required for monocyte/macrophage differentiation and function.
Results
Identification of a novel phosphorylation site at TFEB proline‐rich domain
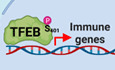
Phosphorylation plays a crucial role in TFEB regulation (Puertollano et al, 2018). In a previous study, we performed mass spectrometry (MS) analysis with the goal to identify changes in TFEB phosphorylation status in response to oxidative stress (Martina & Puertollano, 2018). We found that the phosphorylation levels of several serine residues decreased following treatment with NaAsO2, thus facilitating TFEB translocation from the cytosol to the nucleus (Martina & Puertollano, 2018). However, we also identified a serine residue (S401), whose phosphorylation increased dramatically in response to oxidative stress (Fig 1A). Treatment of U2OS cells expressing TFEB‐FLAG with 150 μM NaAsO2 for 2 h caused over 7‐fold increase in the phosphorylation levels of TFEB‐S401 (Fig 1B and C). Results from an independent experiment are shown in Fig EV1A. An interesting aspect about S401 is that this residue is located within TFEB proline‐rich domain, a region of uncharacterized function but highly conserved during evolution (Fig 1D). Conversely, S401 does not appear to be conserved in TFE3, although the reduced homology between TFEB and TFE3 in this region makes it difficult to rule out the presence of other serine residues with equivalent regulation/function (Fig 1E).
Figure 1. TFEB serine 401 is phosphorylated in response to oxidative stress.
- Schematic representation of TFEB domains, indicating localization of serine 401 (S401) to the C‐terminal proline‐rich domain.
- Table showing mass spectrometry analysis of the abundance ratios of TFEB phosphorylated peptides in S401 from NaAsO2‐treated cells versus control cells.
- Extracted ion chromatograms (XIC) of TFEB phosphorylated peptide in S401 from cells either untreated (Control) or treated with NaAsO2.
- Multi‐sequence alignment of TFEB orthologs showing conserved homology between different species of the proline‐rich region containing TFEB Serine 401 (indicated in red).
- Sequence homology analysis between human TFEB and TFE3 depicting the proline‐rich region containing the S401 in TFEB.
- Immunoblot analysis of protein lysates from ARPE‐19 cells expressing TFEB‐WT‐FLAG or TFEB‐S401A‐FLAG treated with 200 μM NaAsO2 or 250 nM Torin‐1 for 1 h. Two different rabbit polyclonal antibodies raised against a TFEB‐S401 phospho‐specific peptide were tested.
- Immunoblot analysis of protein lysates from ARPE‐19 cells expressing TFEB‐WT‐FLAG, TFEB‐S3A/R4A‐FLAG or TFEB‐R245‐247A‐FLAG treated with 200 μM NaAsO2 for 1 h. Immunoblots are representative of at least three independent experiments.
- Immunofluorescence confocal microscopy of ARPE‐19 cells overexpressing TFEB‐WT‐FLAG, TFEB‐S3A/R4A‐FLAG, and TFEB‐R245‐247A‐FLAG showing the subcellular distribution of recombinant TFEB in response to treatments with 200 μM NaAsO2 for 1 h. Scale bars, 10 μm.
- Immunofluorescence confocal microscopy of ARPE‐19 cells overexpressing TFEB‐WT‐FLAG or TFEB‐S401A‐FLAG showing the subcellular distribution of recombinant TFEB in response to treatments with 250 nM Torin‐1 or 200 μM NaAsO2 for 1 h or EBSS for 4 h. Scale bars, 10 μm.
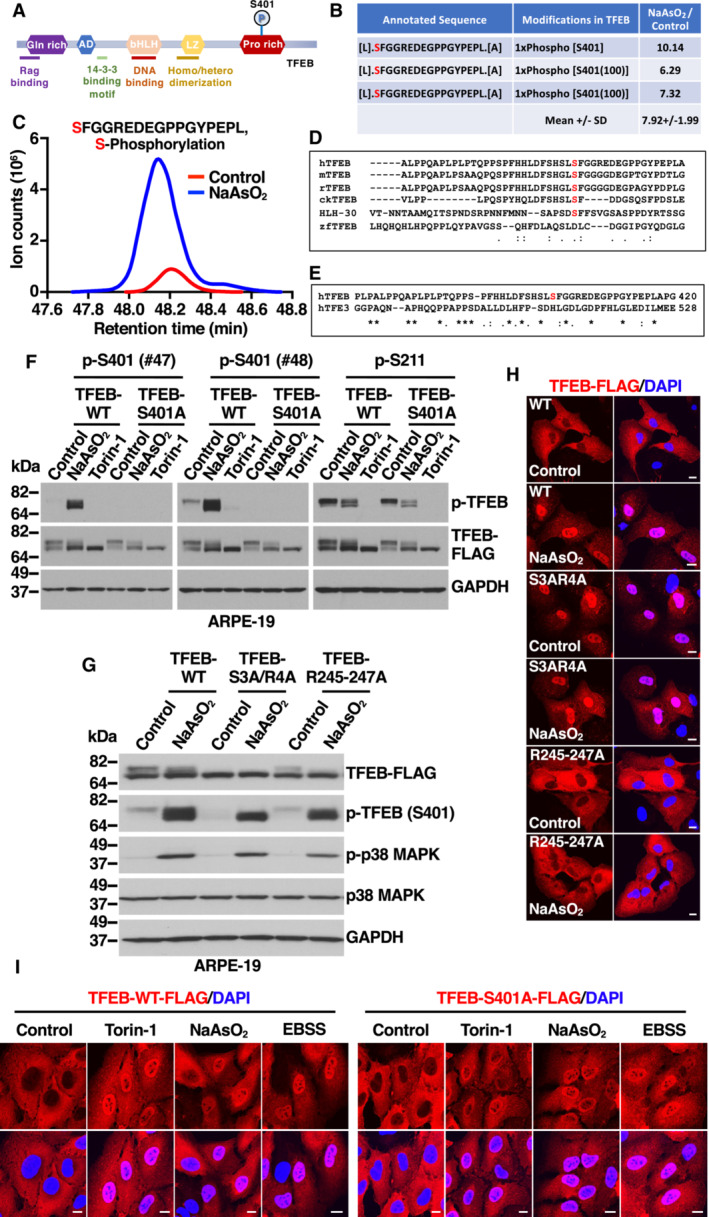
Figure EV1. Phosphorylation of TFEB S401 induced by oxidative stress. Related to Fig 1 .
- Table showing mass spectrometry analysis of the abundance ratios of TFEB phosphorylated peptides in S401 from NaAsO2‐treated cells versus control cells.
- Immunoblot analysis of protein lysates from U2OS cells expressing TFEB‐WT‐FLAG treated with 150 μM NaAsO2 or 250 nM Torin‐1 for 1 h. Two different rabbit polyclonal antibodies raised against a TFEB‐S401 phospho‐specific peptide were tested.
- Quantification of the nuclear localization of recombinant TFEB in ARPE‐19 cells expressing TFEB‐WT‐FLAG or TFEB‐S401A‐FLAG and treated with 250 nM Torin‐1 or 250 μM NaAsO2 for 1 h or EBSS for 4 h. Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant as compared to the same treatment in TFEB‐WT‐FLAG overexpressing cells, with > 300 cells counted per trial from three independent experiments.
- Immunoblot analysis of protein lysates from ARPE‐19 cells treated with 250 μM NaAsO2 for 1 h were subjected to subcellular fractionation. Immunoblots are representative of at least three independent experiments.
- Relative quantitative RT–PCR analysis of the mRNA expression of lysosome‐ (MCOLN1 and ATP6V1C1), autophagy‐ (UVRAG), mitochondria‐ (PGC‐1α), cell cycle‐ (CDKN1a), and heme catabolism‐related (HMOX1) genes in ARPE‐19 cells overexpressing either TFEB‐WT‐FLAG, TFEB‐S401A‐FLAG, or TFEB‐S401D‐FLAG. Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant and *P < 0.01 from three independent experiments.
Data information: n = 3 biological replicates (each dot represents a biological replicate).
Source data are available online for this figure.
To corroborate our MS data, affinity‐purified phospho‐specific peptide antibodies directed against TFEB‐S401 from two different rabbits (#47 and #48) were tested in ARPE‐19 cells infected with adenovirus expressing either TFEB‐WT or TFEB‐S401A. As expected, treatment with NaAsO2 induced a robust increase in S401 phosphorylation, while the signal was not detected in TFEB‐S401A‐expressing cells (Fig 1F). As previously described, phosphorylation of TFEB‐S211 was reduced by treatment with NaAsO2 or the mTORC1 inhibitor Torin‐1 (Fig 1F). Comparable results were obtained in U2OS cells (Fig EV1B).
Next, we addressed whether phosphorylation of S401 occurs in the nucleus or the cytosol. Mutation of TFEB nuclear import signal (R245‐247>A) (Roczniak‐Ferguson et al, 2012) prevented its translocation to the nucleus in response to oxidative stress but did not affect S401 phosphorylation, indicating that modification of this residue is a cytosolic event (Fig 1G and H). Increased S401 phosphorylation in response to NaAsO2 was also observed in the TFEB S3A/R4A mutant (Fig 1G). We have previously described that mutation of TFEB at S3 and R4 blocks its interaction with Rag GTPases, preventing mTORC1‐dependent phosphorylation of S211 and TFEB interaction with 14‐3‐3 (Martina & Puertollano, 2013). As a result, TFEB‐S3A/R4A cannot be retained in the cytosol and mainly accumulates in the nucleus, even under control (non‐stress) conditions (Fig 1H). Our data showed that nuclear accumulation was not sufficient to cause S401 phosphorylation, since the TFEB‐S3A/R4A mutant was not phosphorylated under control conditions (Fig 1G). Considering that the mutant is still able to rapidly recycle between nucleus and cytosol, it is likely that its phosphorylation at S401 in response to oxidative stress occurs in the cytosol. Overall, these results indicate that TFEB can translocate and be present in the nucleus while phosphorylated at S401. This is in clear contrast with phosphorylation of S211, which is never observed in nuclear TFEB (Martina et al, 2016).
Next, we investigated whether the phosphorylation status of S401 may influence phosphorylation of S211 and, consequently, TFEB intracellular distribution and activation. We found that dephosphorylations of S211 by treatment with NaAsO2 was not affected by mutation of S401 to either alanine (TFEB‐S401A) or aspartic acid (TFEB‐S401D) (Appendix Fig S1A and B). Furthermore, mutation of S211 did not prevent phosphorylation of S401 in response to NaAsO2 (Appendix Fig S1A), while phosphomimetic mutation of S401 did not prevent the constitutive nuclear accumulation of the S211A mutant (Appendix Fig S1C and D). These data indicate that phosphorylation of S211 and S401 are independent of each other.
Accordingly, TFEB‐WT, TFEB‐S401A, and TFEB‐S401D translocated to the nucleus with similar efficiency in response to a variety of stress conditions, including treatment with Torin‐1, starvation medium (EBSS), and NaAsO2 (Figs 1I and EV1C; Appendix Fig S1E and F). Activation of TFEB‐S401A and presence of phospho‐S401 TFEB in the nucleus were also corroborated by subcellular fractionation experiments (Fig EV1D).
Furthermore, mutation of S401 to either alanine or aspartic acid did not affect TFEB transcriptional activity. Over‐expression of TFEB‐WT, TFEB‐S401A, or TFEB‐S401D in ARPE‐19 cells resulted in a comparable upregulation of TFEB targets implicated in different cellular pathways, including lysosomal function (MCOLN1 and ATP6V1C1), autophagy (UVRAG), metabolic regulation (PGCα), cell cycle (CDKN1a), and oxidative stress (HMOX1) (Fig EV1E).
p38 MAPK phosphorylates TFEB at S401
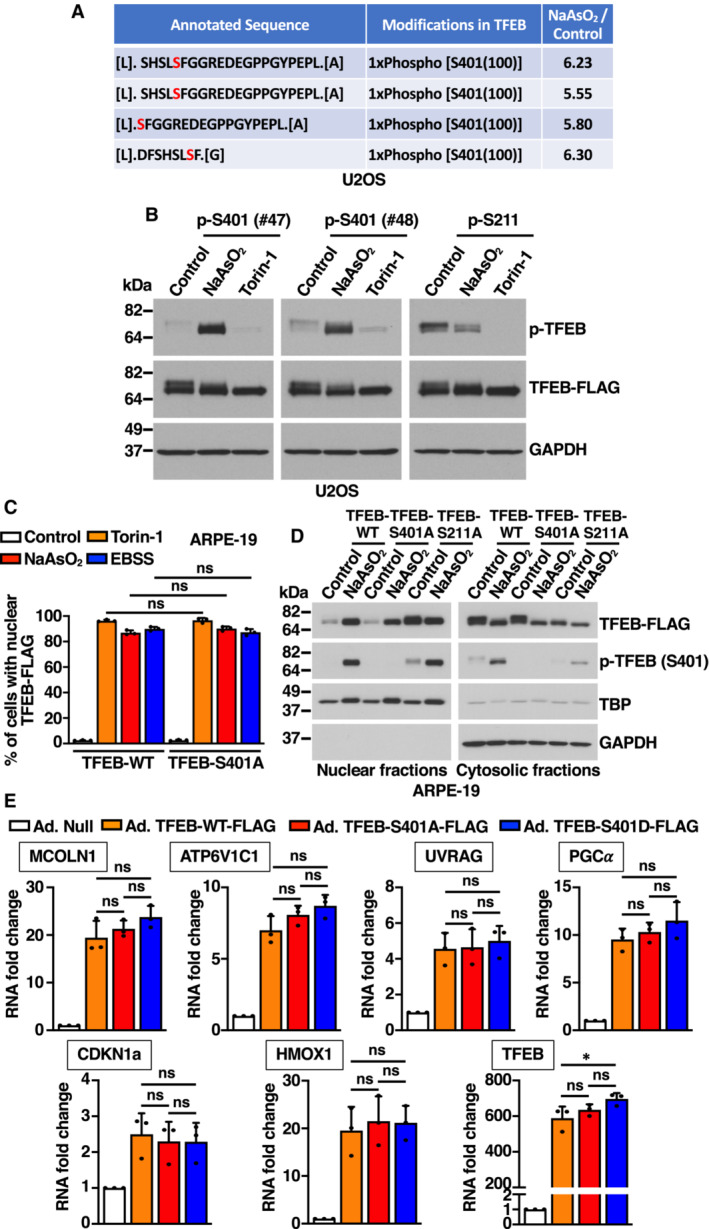
Mitogen‐activated protein kinases (MAPK), a family of serine/threonine/tyrosine‐specific protein kinases involved in directing cellular responses to diverse stimuli, consists of three members: p38 MAPK, c‐Jun NH2‐terminal kinase (JNK), and extracellular signal‐regulated kinase (ERK; Wada & Penninger, 2004). Much evidence has demonstrated that MAPKs strongly activate in response to oxidative stress (Foo et al, 2020); therefore, we assessed if they might contribute to S401 phosphorylation. Importantly, we found that incubation with SB203580, a selective inhibitor of p38 MAPK, entirely prevented increased TFEB‐S401 phosphorylation in NaAsO2‐treated cells (Fig 2A and B). In contrast, incubation with JNK (JNK inhibitor VIII) or ERK1/2 (U0126) inhibitors did not have any measurable effect on S401 phosphorylation (Fig 2A and B). The efficiency and selectivity of the inhibitors was assessed by monitoring the phosphorylation status of specific p38 (MAPKAPK2) and JNK (c‐Jun) effectors, as well as ERK1/2 phosphorylation (Fig 2A).
Figure 2. p38 MAPK‐dependent phosphorylation of TFEB‐S401.
- Immunoblot analysis of protein lysates from HeLa cells stably expressing TFEB‐WT‐FLAG incubated with the indicated kinase inhibitors for 1 h prior to the addition of 200 μM NaAsO2 for 1 h.
- Quantification of immunoblot data shown in (A). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant, and ****P < 0.0001 from three independent experiments.
- Immunoblot analysis of protein lysates from HeLa cells stably expressing TFEB‐WT‐FLAG incubated with either 200 μM NaAsO2 for 1 h, EBSS for 4 h, 100 ng/ml EGF for the indicated times or 37 μM Anisomycin for 1 h. Before the addition of EGF or Anisomycin, cells were serum starved for 8 h.
- Immunoblot analysis of protein lysates from HeLa cells stably expressing TFEB‐WT‐FLAG exposed to 30 J/m2 of UV‐C and allowed to recover in complete medium for the indicated times. Cells were incubated with p38 MAPK inhibitor (20 μM, SB203580) for 1 h before UV‐C irradiation and allowed to recover for 30 min in the presence of the inhibitor.
- Immunoblot analysis of protein lysates from HeLa cells stably expressing TFEB‐WT‐FLAG depleted of either p38 MAPK (α + β) or JNK1 or JNK2 or JNK(1 + 2) and incubated with 200 μM NaAsO2 for 1 h. Immunoblots are representative of at least three independent experiments.
- Immunoblot analysis of in vitro p38 MAPK kinase assay using GST‐TFEB‐PRD as substrate in the presence or absence of either recombinant human active p38α MAPK or ATP.
- Quantification of immunoblot data shown in (F). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, *P < 0.05 from three independent experiments.
Data information: n = 3 biological replicates (each dot represents a biological replicate).
Source data are available online for this figure.
mTORC1 plays a critical role in TFEB regulation by phosphorylating several residues, including S211, so we asked whether this kinase may also contribute to the phosphorylation of S401. As seen in Fig EV2A, increased TFEB‐S401 phosphorylation in response to oxidative stress was inhibited by p38 MAPK inhibitors or the antioxidant NAC. Conversely, inhibition of mTORC1 by either starvation (EBSS) or Torin‐1 did not prevent increased S401 phosphorylation. Efficient mTORC1 inactivation was confirmed by reduced phosphorylation of TFEB‐S211 and 4EBP1 (Fig EV2A).
Figure EV2. Phosphorylation of TFEB S401 by p38 MAPK. Related to Fig 2 .
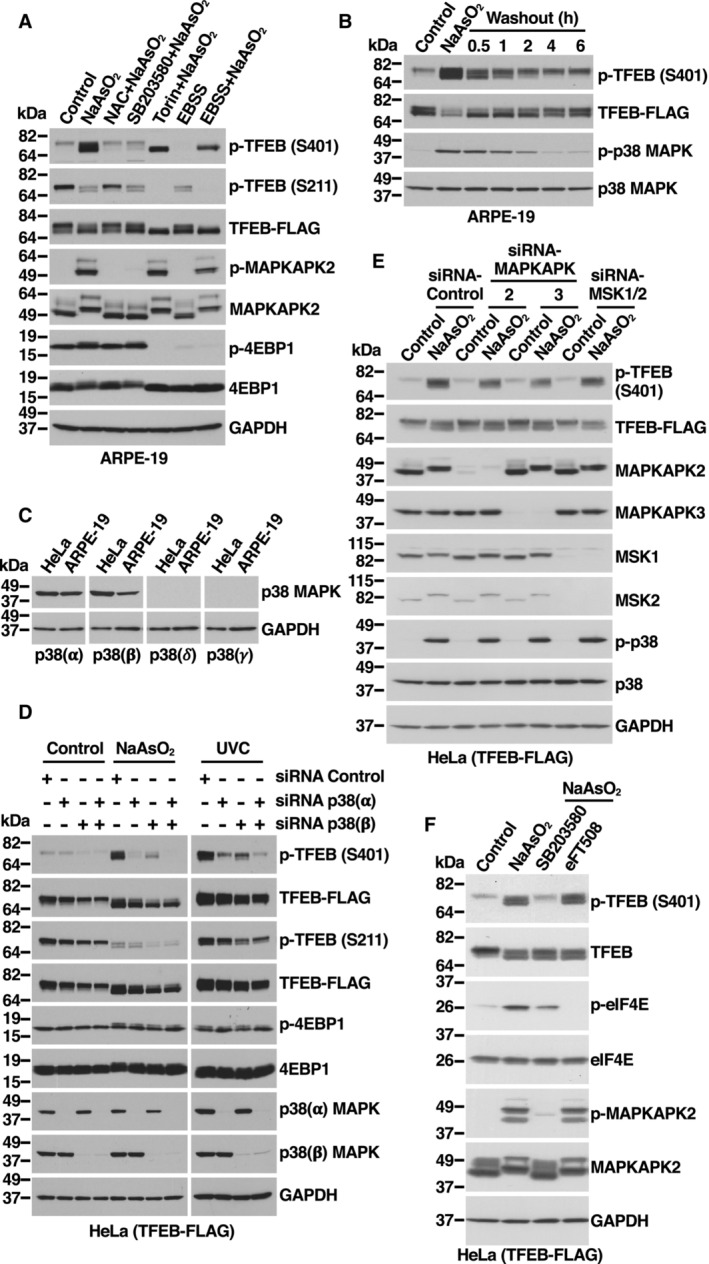
- Immunoblot analysis of protein lysates from ARPE‐19 cells expressing TFEB‐WT‐FLAG incubated with 15 mM NAC, 20 μM SB203580, 250 nM Torin‐1 for 1 h or EBSS for 4 h prior to the addition of 250 μM NaAsO2 for 1 h.
- Immunoblot analysis of protein lysates from ARPE‐19 cells expressing TFEB‐WT‐FLAG incubated with 250 μM NaAsO2 for 1 h. Then, cells were washed with HBSS and incubated in fresh medium without NaAsO2 for the indicated times.
- Immunoblot analysis of protein lysates from HeLa and ARPE‐19 cells.
- Immunoblot analysis of protein lysates from HeLa cells stably expressing TFEB‐WT‐FLAG depleted of p38 MAPK (α), p38 MAPK (β), or p38 MAPK (α + β) and incubated with 200 μM NaAsO2 for 1 h or irradiated with UV‐C light and recovered for 30 min. Immunoblots are representative of at least three independent experiments.
- Immunoblot analysis of protein lysates from HeLa cells stably expressing TFEB‐WT‐FLAG depleted of MAPKAPK2, MAPKAPK3, MSK1, and MSK2 and incubated with 200 μM NaAsO2 for 1 h. Immunoblots are representative of at least three independent experiments.
- Immunoblot analysis of protein lysates from HeLa cells stably expressing TFEB‐WT‐FLAG incubated with the indicated kinase inhibitors for 1 h prior to the addition of 200 μM NaAsO2 for 1 h.
Source data are available online for this figure.
p38 MAPK is activated by a variety of cellular stresses including osmotic shock, inflammatory cytokines, lipopolysaccharide (LPS), UVC light, and growth factors (Chang & Karin, 2001; Kyriakis & Avruch, 2001). We hypothesized that stress conditions that cause p38 MAPK activation may also result in increased TFEB‐S401 phosphorylation. As seen in Fig 2C and D, oxidative stress, UVC light, and, to a lesser extent, growth factors induced p38 MAPK activation, as well as a proportional increase in TFEB‐S401 phosphorylation. Anisomycin, a well characterized p38 MAPK activator (Hazzalin et al, 1998), also induced TFEB‐S401 phosphorylation with high efficiency (Fig 2C). As expected, increased S401 phosphorylation in response to UV light was blocked by p38 MAPK inhibitors (Fig 2D). Furthermore, when cells were treated with NaAsO2 and then placed in normal medium (washout), the progressive inactivation of p38 MAPK correlated with a gradual reduction in S401 phosphorylation (Fig EV2B). In contrast, induction of ER stress by treatment with either Tunicamycin or Thapsigargin did not cause p38 MAPK activation or TFEB‐S401 phosphorylation (Appendix Fig S2A and B).
To further corroborate our results, we depleted p38 MAPK and JNK with specific siRNAs. Four isoforms of p38 MAPK (p38α, β, γ, and δ) and two isoforms of JNK (JNK1 and JNK2) have been identified, although p38α and p38β were the only p38 MAPK isoforms detected in HeLa and ARPE‐19 cells (Fig EV2C). In agreement with our data using p38 MAPK inhibitors, we found that simultaneous depletion of p38α and p38β completely prevented increased S401 phosphorylation in response to NaAsO2 (Fig 2E). In contrast, depletion of JNK1 and JNK2 did not have a noticeable effect (Fig 2E). Depletion of p38α+β also inhibited phosphorylation of S401 in response to UVC light (Fig EV2D). Individual depletion of p38α and p38β demonstrated that while both isoforms contribute to an efficient S401 phosphorylation, the role of p38α seems to be more significant (Fig EV2D).
It is well established that p38 MAPK can phosphorylate and activate several kinases, which in turn phosphorylate additional proteins to regulate a wide variety of processes (Canovas & Nebreda, 2021). Of particular relevance are MSK1 and MSK2, which phosphorylate transcription factors implicated in immune response and differentiation. Additional kinases that are regulated by p38 include MAPKAPK2, MAPKAPK3, MNK1, and MNK2. We used specific siRNAs and inhibitors to assess the potential contribution of p38‐regulated kinases to TFEB‐S401 phosphorylation. As seen in Fig EV2E and F, neither depletion of MSK1, MSK2, MAPKAPK2, and MAPKAPK3 nor inhibition of MNK1 and MNK2 with eFT508 prevented increased S401 phosphorylation in response to NaAsO2, suggesting that p38 likely phosphorylate TFEB directly. To further confirm this, we performed in vitro kinase assays. TFEB C‐terminal region (Ala364 to Leu476), which includes the proline‐rich domain (PRD), was fused to GST, purified, and incubated with recombinant active p38α MAPK. As expected, phosphorylation of TFEB‐S401 was detected only when both p38 MAPK and ATP were present (Fig 2F and G). These results provide strong evidence that p38 MAPK directly phosphorylates TFEB at S401.
p38 MAPK phosphorylates TFEB‐S401 in macrophages following LPS stimulation
It is well established that p38 MAPK plays a crucial role in macrophage‐mediated inflammatory responses. Following pathogen or LPS stimulation, p38 MAPK is rapidly phosphorylated and activated, contributing to the expression of multiple proinflammatory mediators (Garcia et al, 1998; Byeon et al, 2011; Yang et al, 2012). In agreement with previous studies, we observed p38 MAPK and MAPKAPK2 phosphorylation in Raw 264.7 mouse macrophages within 5 min of LPS stimulation (Fig 3A and B). We reasoned that macrophage might be a useful model system to address TFEB‐S401 phosphorylation under physiologically relevant conditions. Since our phospho‐S401 antibody does not efficiently recognize mouse TFEB, we generated a stable clone in Raw 264.7 cells expressing human TFEB‐FLAG. Interestingly, p38 MAPK activation correlated with a robust increase in TFEB‐S401 phosphorylation following LPS stimulation (Fig 3A and B). Furthermore, S401 phosphorylation was abolished by p38 MAPK inhibitors but not by Torin‐1 (Fig 3B and C) further supporting a role of p38 MAPK in LPS‐mediated S401 phosphorylation.
Figure 3. LPS stimulation induces p38 MAPK‐dependent phosphorylation of TFEB‐S401 in macrophages.
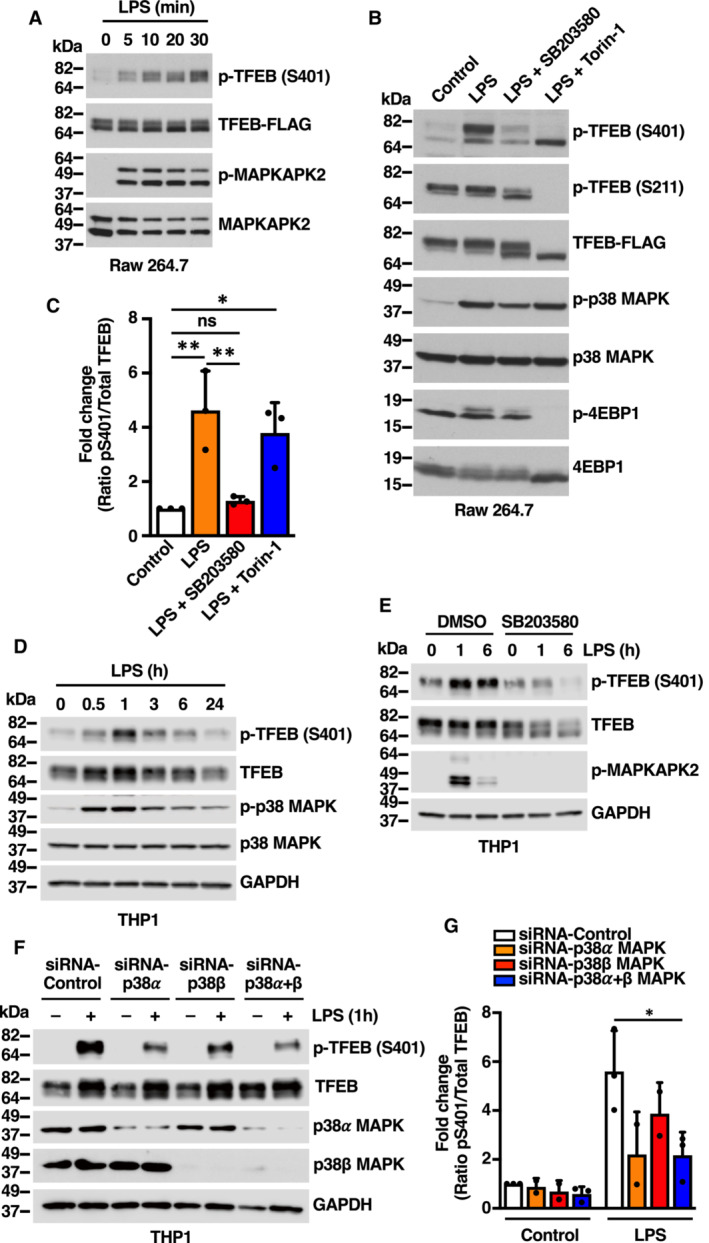
- Immunoblot analysis of protein lysates from Raw 264.7 cells stably expressing TFEB‐WT‐FLAG incubated with 1 μg/ml LPS for the indicated times.
- Immunoblot analysis of protein lysates from Raw 264.7 cells stably expressing TFEB‐WT‐FLAG incubated with either 20 μM SB203580 or 250 nM Torin‐1 for 1 h prior to the addition of 1 μg/ml LPS for 30 min.
- Quantification of immunoblot data shown in (B). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant, *P < 0.05 and **P < 0.01 from three independent experiments.
- Immunoblot analysis of protein lysates from THP1 cells incubated with 1 μg/ml LPS for the indicated times.
- Immunoblot analysis of protein lysates from THP1 cells incubated with either vehicle (DMSO) or 20 μM SB203580 for 1 h prior to the addition of 1 μg/ml LPS for the indicated times.
- Immunoblot analysis of protein lysates from THP1 cells depleted of p38 MAPK (α), p38 MAPK (β) or p38 MAPK (α + β) and incubated with 1 μg/ml LPS for 1 h. Immunoblots are representative of at least three independent experiments.
- Quantification of immunoblot data shown in (F). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, *P < 0.05 from three independent experiments.
Data information: n = 3 biological replicates (each dot represents a biological replicate).
Source data are available online for this figure.
To confirm that endogenous TFEB also undergoes phosphorylation at S401, we used human differentiated THP1 cells. As seen in Fig 3D, endogenous TFEB was rapidly phosphorylated at S401 in response to LPS, reaching maximum phosphorylation levels at around 1 h of LPS stimulation. Once again, S401 phosphorylation was inhibited by either p38 MAPK inhibition or depletion (Fig 3E and F). In agreement with our results in HeLa cells, we found that p38 MAPK alpha plays a primary role in S401 phosphorylation (Fig 3G).
To further corroborate our data in primary cells, human monocytes were isolated from blood and differentiated into macrophages by treatment with GM‐CSF for 6 days (Fig EV3A and B). As expected, incubation with LPS resulted in p38 MAPK activation and increased S401 phosphorylation (Fig EV3C and D).
Figure EV3. TFEB‐S401 phosphorylation induced by NaAsO2 and LPS treatments is absent in S401A knock‐in THP1 cells. Related to Fig 3 .
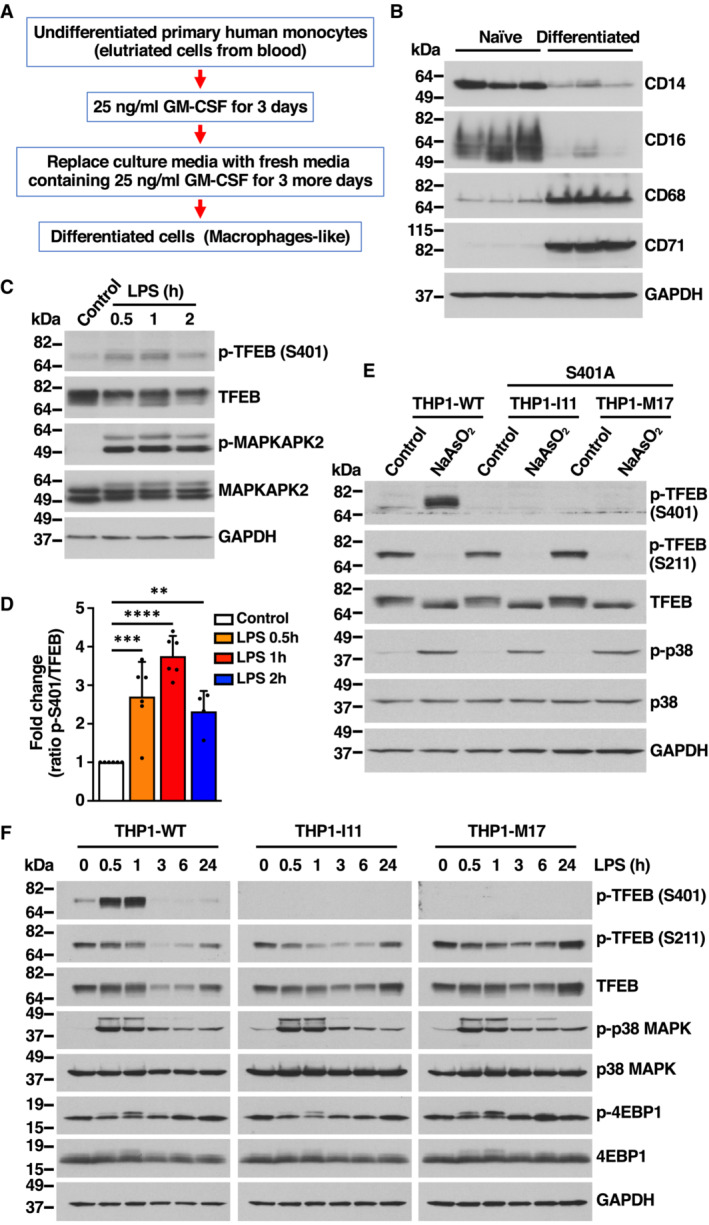
- Flowchart indicating the different steps followed to differentiate primary human monocytes into macrophage cells.
- Immunoblot analysis of protein lysates from primary human monocytes undifferentiated (Naïve) or GM‐CSF‐differentiated primary macrophages. Samples are from three independent experiments.
- Immunoblot analysis of protein lysates from primary human macrophages incubated with 1 μg/ml LPS for the indicated times. Immunoblots are representative of at least four independent experiments.
- Quantification of immunoblot data shown in (C). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Dunnett's multiple comparisons test, **P < 0.01, ***P < 0.001 and ****P < 0.0001 as compared to untreated cells from at least four independent experiments.
- Immunoblot analysis of protein lysates from THP1 cells WT or TFEB‐S401A knock‐ins (clones I11 and M17) incubated with 100 μM NaAsO2 for 1 h.
- Immunoblot analysis of protein lysates from PMA‐differentiated THP1 cells WT or TFEB‐S401A knock‐ins (clones I11 and M17) incubated with 1 μg/ml LPS for the indicated times. Immunoblots are representative of at least three independent experiments.
Data information: n ≥ 3 biological replicates (each dot represents a biological replicate).
Source data are available online for this figure.
Next, we used CRISPR/Cas9 knock‐in technology to substitute S401 by alanine within endogenous TFEB in THP1 cells. We were able to identify two independent single cell clones I11 and M17 (subsequently termed THP1‐I11 and THP1‐M17) displaying homozygous introduction of S401A into the TFEB locus of THP1 cells. Expression of TFEB S401A mutants was comparable to TFEB‐WT (Fig EV3E and F). However, phosphorylation of S401 in response to NaAsO2 or LPS was completely abrogated in the mutant clones (Fig EV3E and F). Altogether, our data indicate that THP1 cells constitute a suitable model to investigate the role of S401 phosphorylation.
Phosphorylation of S401 is required for efficient TFEB activation during monocyte differentiation
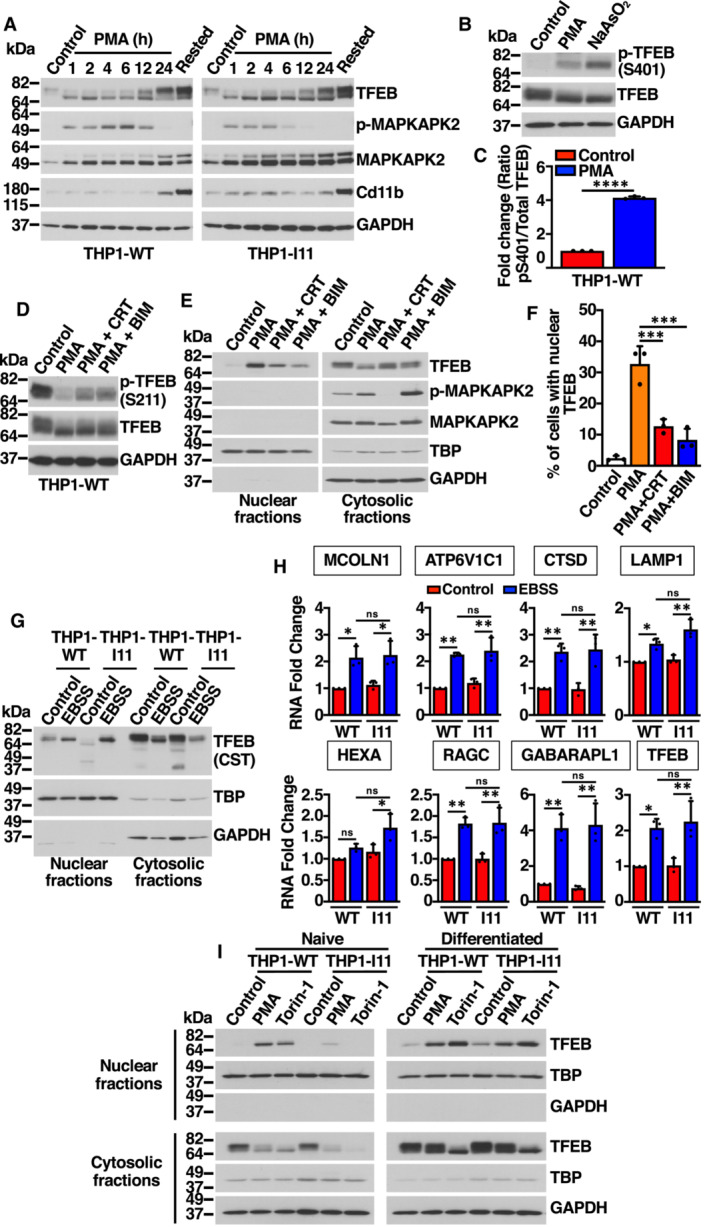
THP1 is a human leukemia monocytic cell line that has been extensively used to characterize monocyte and macrophage activation and differentiation. THP1 monocytes can be differentiated into M0 macrophages by incubation with Phorbol 12‐myristate 13‐acetate (PMA) for 24 h, followed by 24 h rest (Fig 4A). Interestingly, we observed rapid p38 MAPK activation in THP1 monocytes treated with PMA, as assessed by increased levels of phospho‐p38 MAPK and phospho‐MAPKAPK2 (Figs 4B and EV4A), as well as increased TFEB‐S401 phosphorylation (Fig EV4B and C). Incubation with PMA also caused a very rapid and noticeable change in TFEB electrophoretic mobility, which is usually indicative of TFEB activation (Figs 4B and EV4A). Accordingly, phosphorylation of TFEB‐S211 was dramatically reduced following PMA treatment but recovered once PMA was removed (rested) (Fig 4B).
Figure 4. Activation of TFEB depends on S401 phosphorylation during macrophage differentiation.
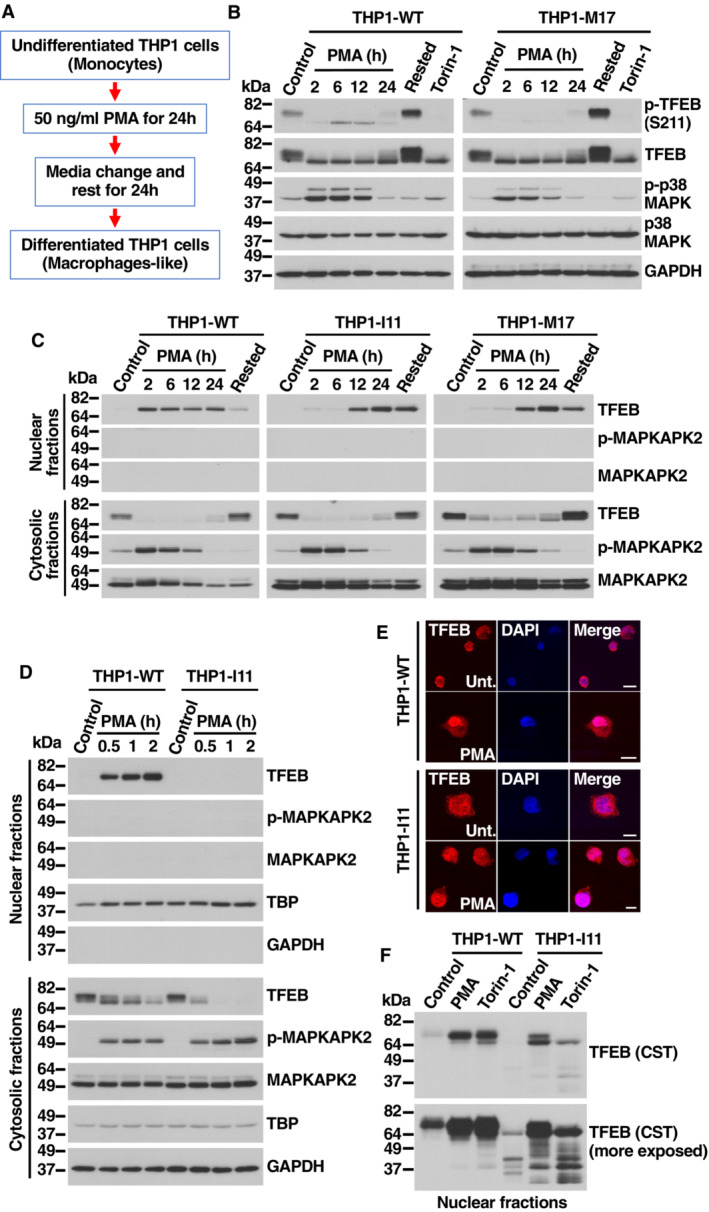
- Flowchart indicating the different steps followed to differentiate Naïve into macrophage‐like THP1 cells.
- Immunoblot analysis of protein lysates from naïve THP1‐WT or TFEB‐S401A knock‐in (clone M17) cells treated with 50 ng/ml PMA for the indicated times, PMA‐differentiated THP1 (Rested) cells treated without or with 250 nM Torin‐1 for 1 h.
- Immunoblot analysis of proteins from nuclear and cytosolic fractions from naïve THP1‐WT or TFEB‐S401A knock‐in (clones I11 and M17) cells treated with 50 ng/ml PMA for the indicated times and PMA‐differentiated THP1 (Rested) cells.
- Immunoblot analysis of proteins from nuclear and cytosolic fractions from naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells treated with 50 ng/ml PMA for the indicated times.
- Immunofluorescence confocal microscopy of naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells treated with 50 ng/ml PMA for 1 h. Scale bars, 10 μm.
- Immunoblot analysis of proteins from nuclear fractions from naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells treated with 50 ng/ml PMA or 250 nM Torin‐1 for 1 h. Immunoblots are representative of at least three independent experiments. The antibody directed against the central region of TFEB was obtained from Cell Signaling Technology (CST).
Source data are available online for this figure.
Figure EV4. PMA‐dependent activation of TFEB in THP1 cells. Related to Fig 4 .
- Immunoblot analysis of protein lysates from naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells treated with 50 ng/ml PMA for the indicated times, and PMA‐differentiated THP1 (Rested) cells.
- Immunoblot analysis of protein lysates from naïve THP1‐WT cells treated with 50 ng/ml PMA for 30 min and with 100 μM NaAsO2 for 1 h.
- Quantification of immunoblot data shown in (B). Data are presented as mean ± SD using unpaired Student's t‐test, ****P < 0.0001 from three independent experiments.
- Immunoblot analysis of protein lysates from naïve THP1‐WT cells treated with 5 μM CRT0066101(CRT, PKD inhibitor) or 5 μM Bisindolylmaleimide IV (BIS, PKC inhibitor) for 1 h prior to the addition of 50 ng/ml PMA for 1 h.
- Immunoblot analysis of proteins from nuclear and cytosolic fractions from naïve THP1‐WT treated with 5 μM CRT0066101(CRT, PKD inhibitor) or 5 μM Bisindolylmaleimide IV (BIS, PKC inhibitor) for 1 h prior to the addition of 50 ng/ml PMA for 1 h.
- Quantification of the nuclear localization of recombinant TFEB in HeLa cells expressing TFEB‐WT‐FLAG treated with 5 μM CRT0066101(CRT, PKD inhibitor) or 5 μM Bisindolylmaleimide IV (BIS, PKC inhibitor) for 1 h prior to the addition of 500 ng/ml PMA for 45 min. Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, ***P < 0.001 as compared to PMA‐treated cells, with > 200 cells counted per trial from three independent experiments.
- Immunoblot analysis of proteins from nuclear and cytosolic fractions from naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells nutrients starved with EBSS for 12 h. The antibody directed against the central region of TFEB was obtained from Cell Signaling Technology (CST).
- Relative quantitative RT–PCR analysis of the mRNA expression of lysosome‐ (MCOLN1, ATP6V1C1, CTSD, LAMP1, HEXA, RAGC) and autophagy‐related (GABARAPL1) genes in naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells nutrients starved with EBSS for 12 h. Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, *P < 0.05 and **P < 0.01 from three independent experiments.
- Immunoblot analysis of proteins from nuclear and cytosolic fractions from naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells or PMA‐differentiated THP1 (Rested) cells treated with either 50 ng/ml PMA or 250 nM Torin‐1 for 1 h.
Data information: n = 3 biological replicates (each dot represents a biological replicate).
Source data are available online for this figure.
Previous studies have suggested that PMA‐mediated activation of recombinant TFEB in Raw 264.7 cells requires PKC and PKD activation (Najibi et al, 2016). In agreement with these observations, we found that Bisindolylmaleimide IV (BIM) and CRT 0066101, two specific inhibitors of PKC and PKD, respectively, reduced S211 dephosphorylation (Fig EV4D) and TFEB translocation to the nucleus (Fig EV4E). BIM and CRT also significantly decreased PMA‐induced TFEB nuclear translocation in HeLa cells (Fig EV4F).
To further assess TFEB activation in response to PMA, we performed subcellular fractionation. As expected, TFEB rapidly translocated to the nucleus in THP1‐WT cells and remained there while PMA was present (Fig 4C). Notably, the situation was different in the two clones in which S401 was mutated to alanine. Despite showing electrophoretic mobility changes and S211 dephosphorylation comparable to TFEB‐WT (Figs 4B and EV4A), the TFEB‐S401 mutant showed a delayed activation kinetic, with very reduced amounts of TFEB‐S401A present in nuclear fractions before 12 h treatment with PMA (Fig 4C). It is important to note that p38 MAPK activation was comparable in WT and mutant clones (Fig 4B and C).
Further experiments showed that the accumulation of TFEB‐WT into the nucleus following PMA treatment was very rapid and could be detected after only 30 min of incubation with PMA (Fig 4D). In contrast, almost no TFEB‐S401 mutant was detected in the nucleus at early PMA incubation times (Fig 4D). Decreased TFEB‐S401A nuclear accumulation was also observed by immunofluorescence (Fig 4E). These results suggest that phosphorylation of S401 is required for efficient TFEB nuclear accumulation in response to PMA. While we cannot rule out that S401 phosphorylation regulates transport to the nucleus, an alternative possibility is that the S401 mutant displays reduced stability and gets quickly degraded either in the cytosol or the nucleus following activation. In agreement with this idea, we found that when we used a different anti‐TFEB antibody directed against the central part of the protein, instead of the C‐terminal residues, we were able to detect multiple proteolytic fragments in the THP1‐I11 clone, further suggesting increased degradation of the TFEB‐S401A mutant (Fig 4F). The levels of nuclear TFEB‐S401A remained barely detectable even in the presence of proteosome (MG132) or lysosome (Leupeptin+E64d) inhibitors, hinting at the possibility that TFEB‐S401 undergoes protease‐specific cleavage (Appendix Fig S3).
As control that our knock‐in clones do not present an unrelated defect that might affect TFEB transport to the nucleus, we incubated THP1‐WT and THP1‐I11 cells in starvation medium (EBSS). As shown in Fig EV4G and H, both TFEB‐WT and TFEB‐S401A efficiently translocated to the nucleus following nutrient deprivation and induced expression of lysosomal and autophagic genes with comparable efficiency. Furthermore, we did not observe reduced nuclear accumulation of the S401A mutant in differentiated THP1 cells treated with PMA (Fig EV4I). These results suggest that the role of S401 phosphorylation in promoting TFEB nuclear accumulation is highly specific of monocytes at early times of PMA‐induced differentiation.
Expression of immune genes is severely altered in TFEB‐S401A mutant cells
Next, we seek to investigate how the reduced nuclear accumulation of the TFEB‐S401 mutant may impact monocyte differentiation. For this we treated THP1‐WT and THP1‐I11 cells with PMA for 6 h and processed the samples for RNA‐seq analysis. In parallel we also analyzed the cells by subcellular fractionation, confirming reduced amount of TFEB‐S401A in the nucleus when compared with TFEB‐WT (Fig 5A). It is important to note that TFE3 activation was normal in THP1‐I11 cells, once again arguing against a general defect in nuclear import in our knock‐in clones (Fig 5A).
Figure 5. Immune genes expression is affected in TFEB‐S401A mutant cells.
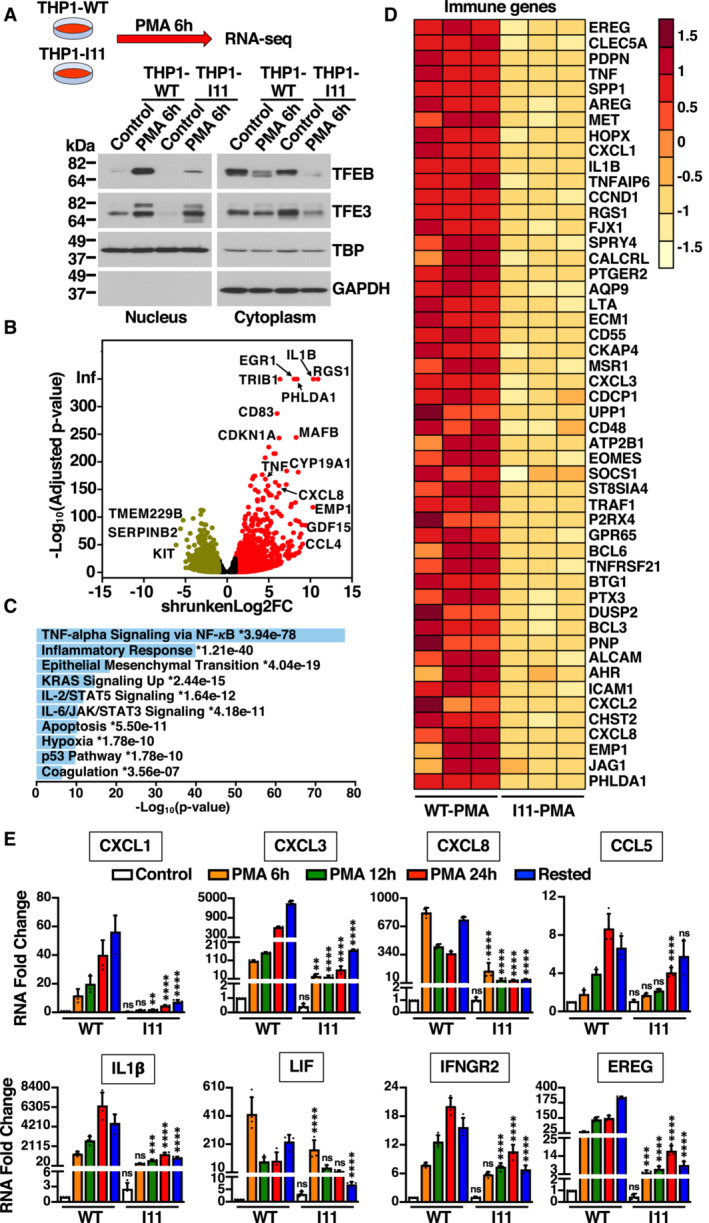
- Immunoblot analysis of proteins from nuclear and cytosolic fractions from naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells treated with 50 ng/ml PMA for 6 h. This condition was used to perform RNA‐Seq analysis.
- Volcano plot indicating distribution of genes up‐ (red, shrunkenLog2FC ≥1) and down‐regulated (green, shrunkenLog2FC ≤ −1) in naïve THP1‐WT cells (Control) versus naïve THP1‐WT cells incubated with 50 ng/ml PMA for 6 h. Black symbols represent genes with a shrunkenLog2FC between 1 and −1.
- MSigDB Hallmark 2020 bar chart showing the top 10 enriched terms from RNA‐Seq analysis of naïve THP1‐WT cells (Control) versus naïve THP1‐WT cells incubated with 50 ng/ml PMA for 6 h. The corresponding significant P‐values (< 0.05) are included with an asterisk (*) indicating that the terms also have significant q‐value (< 0.05).
- Heat map of 50 differentially expressed immune genes from RNA‐Seq analysis of naïve THP1‐WT cells (WT‐PMA) versus naïve TFEB‐S401A knock‐in (I11‐PMA) cells incubated with 50 ng/ml PMA for 6 h.
- Relative quantitative RT–PCR analysis of the mRNA expression of chemokines (CXCL1, CXCL3, CXCL8, and CCL5), cytokines (IL1β and LIF), and key immune regulators (IFNGR2 and EREG) genes in naïve THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells incubated with 50 ng/ml PMA for the indicated times. Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant, **P < 0.01, ***P < 0.001 and ****P < 0.0001 as compared to the same treatment condition in THP1‐WT cells from three independent experiments.
Data information: n ≥ 3 biological replicates (each dot represents a biological replicate).
Source data are available online for this figure.
The comparative transcriptome analysis between control and PMA‐treated THP1‐WT cells indicated that over 7,000 genes were differentially expressed between the two conditions, with a clear upregulation of multiple genes implicated in proliferation regulation and differentiation (Fig 5B; Dataset EV1). MSigDM Hallmark Pathway analysis revealed that among the most upregulated categories were many related to immune and inflammatory response, which is consistent with the acquisition of macrophage functions (Fig 5C). Importantly, the upregulation of many of those immune genes was significantly reduced in THP1‐I11 cells (Fig 5D). Comparison of gene expression between THP1‐WT and THP1‐I11 cells after treatment with PMA for 6 h revealed that the most significant differences were found in genes belonging to the “TNF‐alpha Signaling via NF‐KB,” “Inflammatory Response,” and “IL‐2/STAT5 Signaling” categories (Dataset EV2 and Appendix Table S1).
To confirm our RNA‐seq data, we analyzed expression of several immune genes at different times of PMA treatment by q‐PCR. As shown in Fig 5E, PMA efficiently increased the transcriptional expression of multiple chemokines (CXCL1, CXCL3, CXCL8, and CCL5), cytokines (IL1β and LIF), and key immune regulators (IFNGR2 and EREG) in THP1‐WT cells. In contrast, the expression of these genes was severely reduced in THP1‐I11 cells, even at late PMA incubation times (Fig 5E). As previously described, TFEB binds to the promoter of multiple immune genes (Pastore et al, 2016). However, we did not detect noticeable differences in promoter binding affinity between TFEB‐WT and TFEB‐S401A (Appendix Fig S4).
Given the well‐established role of TFEB as master regulator of lysosomal biogenesis, we analyzed our RNA‐seq data to compare expression of lysosomal genes between THP1‐WT and THP1‐I11 cells. Our data showed that there was a modest but significant increase in the expression of several lysosomal genes after 6 h treatment with PMA; however, this upregulation was comparable between THP1‐WT and THP1‐I11 cells (Appendix Fig S5A). Similar lysosomal genes expression was also observed when WT and mutant cells were incubated with PMA for longer periods of time (24 h) or were fully differentiated (24 h PMA+ 24 h rested) (Appendix Fig S5B). Immunoblot analysis showed similar protein levels for several lysosomal, autophagic, oxidative stress, and mitochondrial markers in THP1‐WT, THP1‐I11, and THP1‐M17 cells (Appendix Fig S5C). Furthermore, in response to PMA, both WT and mutant cells showed increased cell adhesion, spread morphology, increased granularity, and acquisition macrophage surface markers, such as CD11b, CD36, and CD14 (although the upregulation of CD14 was less efficient in mutant cells; Appendix Fig S5D–F).
These results suggest that preventing TFEB phosphorylation at S401 has a significant and specific impact in the expression of immune genes during the differentiation of THP1 monocytes into macrophages.
TFEB‐S401A mutant macrophages fail to properly activate in response to LPS stimulation
M0 macrophages were polarized into M1 inflammatory macrophages by incubation with 1 μg/ml LPS for 1.5 and 4 h, and the expression of several immune mediators was measured by qRT–PCR. As seen in Fig 6A, LPS‐mediated upregulation of most of the tested cytokines (IL1β, IL33, IL23A, IL1R) and chemokines (CXCL3, CXCL5, CXCL6) was very significantly reduced in THP1‐I11 and THP1‐M17 mutant clones when compared to THP1‐WT cells. The only exception was IL18, which remained elevated before and during LPS stimulation (Fig 6A–C). Since constitutive expression of pro‐IL18 is high in monocytes (Puren et al, 1999; Gritsenko et al, 2020), the increased pro‐IL18 levels in mutant cells might constitute an additional indication of defective differentiation. We also observed a very significant reduction in the expression of TNFα, both at the mRNA and protein levels (Figs 6A and EV5A and B). In contrast, upregulation of IL10, an anti‐inflammatory cytokine that plays an important role by limiting the immune response, was significantly increased in mutant cells (Fig 6A). These results suggest that phosphorylation of S401 is required for efficient polarization of M1 inflammatory macrophages.
Figure 6. Defect in activation of TFEB‐S401A mutant macrophages upon LPS treatment.
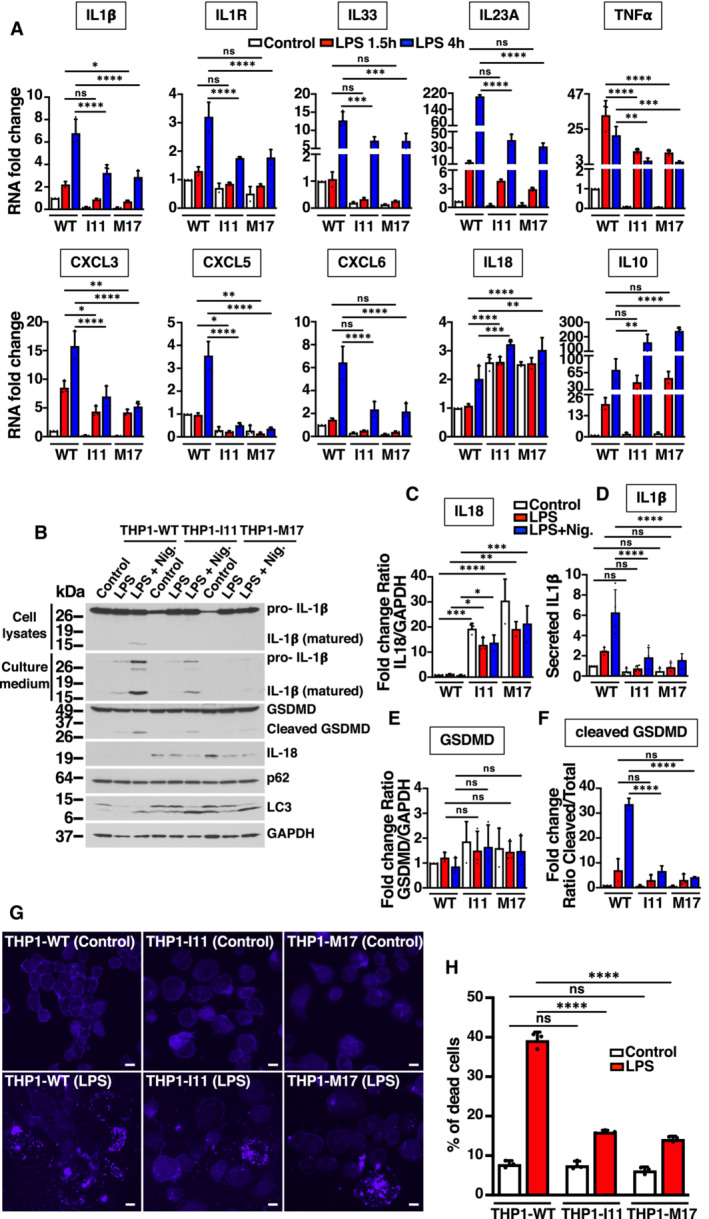
-
ARelative quantitative RT–PCR analysis of the mRNA expression of cytokine‐ (IL1β, IL10, IL18, IL33, IL23A, IL1R, and TNFα) and chemokine‐related (CXCL3, CXCL5, and CXCL6) genes in PMA‐differentiated (Rested) THP1‐WT or TFEB‐S401A knock‐ins (clones I11 and M17) incubated with 1 μg/ml LPS for the indicated times. Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 as compared to the same treatment condition in THP1‐WT cells from three independent experiments.
-
BImmunoblot analysis of protein lysates and cell culture medium from PMA‐differentiated (Rested) THP1‐WT or TFEB‐S401A knock‐in (clones I11 and M17) cells incubated with 0.1 μg/ml LPS for 4 h prior to the addition of 15 μM Nigericin for 45 min.
-
C–FQuantification of immunoblot data shown in (B). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 from three independent experiments.
-
GFluorescence confocal microscopy of PMA‐differentiated (Rested) THP1‐WT or TFEB‐S401A knock‐in (clones I11 and M17) cells incubated with 1 μg/ml LPS for 6 h prior to the detection of dead cells with LIVE/DEAD fixable blue dead cell stain. Scale bars, 10 μm.
-
HQuantification of the percentage of dead cells shown in (G). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant and ****P < 0.0001 as compared to their corresponding untreated (Control) cells, with > 200 cells counted per trial from three independent experiments.
Data information: n ≥ 3 biological replicates (each dot represents a biological replicate).
Source data are available online for this figure.
Figure EV5. Macrophage polarization is affected in THP1 TFEB‐S401A mutant cells in response to LPS. Related to Fig 6 .
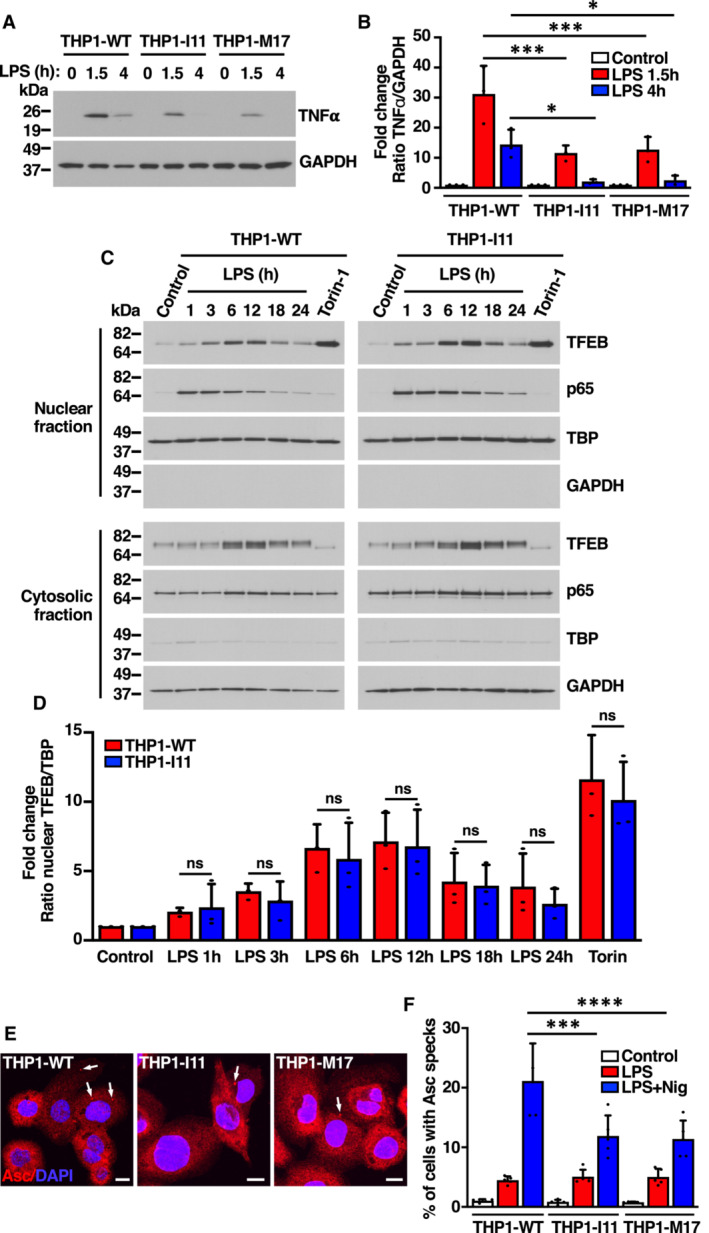
- Immunoblot analysis of protein lysates from PMA‐differentiated (Rested) THP1‐WT or TFEB‐S401A knock‐ins (clones I11 and M17) cells incubated with 1 μg/ml LPS for the indicated times.
- Quantification of immunoblot data shown in (A). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, *P < 0.05 and ***P < 0.001 as compared to the same treatment condition in THP1‐WT cells from three independent experiments.
- Immunoblot analysis of proteins from nuclear and cytosolic fractions from PMA‐differentiated (Rested) THP1‐WT or TFEB‐S401A knock‐in (clone I11) cells incubated with 1 μg/ml LPS for the indicated times or 250 nM Torin‐1 for 1 h.
- Quantification of immunoblot data shown in (C). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, (ns) not significant as compared to the same treatment condition in THP1‐WT cells from three independent experiments.
- Immunofluorescence confocal microscopy of PMA‐differentiated (Rested) THP1‐WT or TFEB‐S401A knock‐in (clones I11 and M17) cells incubated with 0.1 μg/ml LPS for 4 h prior to the addition of 15 μM Nigericin for 45 min. Arrows point to the position of ASC specks indicative of inflammasome activation. Scale bars, 10 μm.
- Quantification of the percentage of cells with ASC specks shown in (E). Data are presented as mean ± SD using one‐way ANOVA (unpaired) followed by Tukey's multiple comparisons test, ***P < 0.001 and ****P < 0.0001 as compared to THP1‐WT cells treated with LPS + Nigericin with > 200 cells counted per trial from three independent experiments.
Data information: n ≥ 3 biological replicates (each dot represents a biological replicate).
Source data are available online for this figure.
We found that THP1‐WT and THP1‐I11 macrophages showed early and comparable activation of NF‐КB in response to LPS stimulation (Fig EV5C; Appendix Fig S6A and B), whereas the activation of TFEB was a late event, with maximum levels of nuclear TFEB at 6–12 h post‐stimulation (Fig EV5C). However, we did not find significant differences in the ability of TFEB‐WT and TFEB‐S401A to translocate to the nucleus (Fig EV5D). This observation, together with the fact that reduced cytokine expression in mutant cells was observed as early as 1.5 h following LPS treatment (Fig 6A), suggests that the polarization defects seen in THP1‐I11 macrophages are likely a consequence of the aberrant differentiation of mutant monocytes into M0 macrophages.
Inflammasome is a cytosolic multiprotein complex that activates caspase‐1 upon assembly (Kelley et al, 2019). Active caspase‐1 then cleaves the biologically inactive pro‐peptide pro‐IL1β and pro‐IL18 into mature cytokines, which are secreted by the cell. We found that inflammasome function was impaired in THP1‐I11 and THP1‐M17 cells. Assembly of apoptosis‐associated speck‐like protein containing a CARD (ASC) complexes (Fig EV5E and F), as well as IL1β cleavage and secretion (Fig 6B and D) were significantly decreased in mutant cells. Inflammasome activation also induces a pro‐inflammatory form of cell death known as pyroptosis. Cleavage of the amino‐terminal domain of gasdermin D (GSDMD) causes its oligomerization and subsequent incorporation at the plasma membrane, forming pores that lead to cell swelling and osmotic lysis (Kesavardhana et al, 2020). Consistently, we found a very significant decrease in GSDMD cleavage (Fig 6B, E and F) and reduced cell death upon mutation of TFEB‐S401 (Fig 6G and H).
Altogether our results show that TFEB controls transcriptional programs that are essential for differentiation of monocytes into naïve M0 macrophages, as well as polarization of macrophages to pro‐inflammatory phenotypes. Furthermore, phosphorylation of TFEB at S401 by p38 MAPK constitutes a key checkpoint in the interface between signaling networks and transcriptional control.
Discussion
TFEB coordinates multiple cellular pathways, including lysosomal biogenesis, autophagy, metabolism, survival, cell‐cycle progression, and differentiation. However, how this transcription factor integrates the output from complex microenvironmental cues to regulate vastly different transcriptional programs in a wide variety of cell types is poorly understood.
In this study, we identify a novel mechanism of TFEB regulation through phosphorylation of TFEB‐S401 by p38 MAPK. We found that multiple inputs that induced p38 MAPK activation, including oxidative stress, UVC light, growth factors, LPS, and anisomycin, dramatically increased S401 phosphorylation levels, while modification of this residue was prevented by p38 MAPK inhibition or depletion. Generation of THP1 knock‐in clones in which endogenous TFEB‐S401 was mutated to alanine demonstrated that the p38 MAPK/TFEB pathway plays a particularly relevant role during monocyte differentiation into macrophages. THP1 monocytes expressing TFEB‐S401A failed to efficiently upregulate expression of multiple immune genes in response to PMA, including critical cytokines, chemokines, and growth factors. Polarization of M0 macrophages into M1 inflammatory macrophages was also aberrant in TFEB‐S401A cells in terms of gene expression, cytokine and chemokine secretion, and inflammasome activation.
One important question is how S401 phosphorylation modulates monocyte differentiation. We found that the amount of nuclear TFEB was dramatically reduced in mutant cells at early times of PMA treatment. In fact, TFEB‐S401A was undetected in nuclear fractions when we used an anti‐TFEB antibody directed against the protein C‐terminal region. In contrast, an antibody generated against TFEB central region detected several proteolytic fragments. This suggests that prevention of TFEB phosphorylation at S401 at early times of PMA‐induced monocyte differentiation causes TFEB instability and degradation. Given a recent report showing that TFEB mutants harboring C‐terminal truncations lack transcriptional activity (Paquette et al, 2021), it is likely that the TFEB‐S401A proteolytic fragments detected in the nuclear fraction are inactive. It is very intriguing that the reduced nuclear accumulation of the TFEB‐S401A mutant was only observed in undifferentiated monocytes. Treatment of differentiated macrophages with PMA or LPS did not cause noticeable differences between TFEB‐WT and TFEB‐S401A in terms of stability or nuclear accumulation. Likewise, over‐expressed TFEB‐WT and TFEB‐S401A translocated to the nucleus with comparable efficiency in ARPE‐19 cells treated with NaAsO2, Torin‐1, or EBSS. This suggests that p38 MAPK‐dependent phosphorylation of S401 may play a key role during early stages of monocyte differentiation by linking extracellular differentiation signals to TFEB stability and activation.
Since TFEB‐S401 localizes within a proline‐rich domain, which is usually implicated in protein–protein interactions, it is also possible that phosphorylation promotes TFEB interaction with unknown transcriptional co‐activators or lowers its affinity for specific co‐repressors. For example, in RANK‐stimulated osteoclast precursors, activation of p38 MAPK results in phosphorylation of the TFEB family member MITF at S307 (Mansky et al, 2002). This phosphorylation allows the recruitment of transcriptional co‐activators FUS and BRG1, leading to the expression of the MITF targets TRAP, OSCAR, CLCN7, and cathepsin K, which are necessary for osteoclasts function (Sharma et al, 2007; Bronisz et al, 2014). At the same time, RANK downstream signaling promotes dissociation of a MITF repressor complex consisting of EOS, HDAC, Sin3A, and CtBP, further enhancing MITF‐dependent transcription (Hu et al, 2007). Importantly, loss of the p38 MAPK phosphorylation site prevents osteoclasts differentiation (Carey et al, 2016). In addition, a previous study suggested that the proline‐rich region of TFE3 may function as an activation domain that acts synergistically with the acidic domain to ensure efficient transcription of targets genes (Artandi et al, 1995). If the TFEB proline‐rich domain were to play a similar role, one may envision S401 phosphorylation functioning as a rheostat to adjust the strength or selectivity of the transcriptional response.
Another interesting aspect is that while the expression of numerous immune genes was significantly reduced in mutant cells as result of the deficient TFEB‐S401A nuclear accumulation, the levels of lysosomal and autophagic genes were not affected. This is probably due to the fact that TFE3 activation was normal in mutant cells. However, these results raise the interesting possibility that TFEB and TFE3 may be at least partially redundant in regulation of lysosomal biogenesis but not in expression of lineage determination genes.
A final consideration is whether TFEB directly binds to the promoter of multiple immune genes to regulate their expression or if it plays a more indirect role by altering the transcriptional landscape of differentiating monocytes. We previously performed ChIP‐seq analysis in LPS‐stimulated RAW 264.7 mouse macrophages and showed TFE3 binding to the promoter of several genes implicated in immune activation (Pastore et al, 2016), indicating the possibility of a direct regulation. However, a recent study has also suggested that TFEB may participate in granulocyte and monocyte differentiation and survival through modulation of epigenetic programs. TFEB directly induces transcription of IDH1 and IDH2, two enzymes that catalyze the production of α‐ketoglutarate (α‐KG), a substrate of the TET family of dioxygenases that convert 5‐methylcytosine (5mC) to 5‐hydroxylmethylcytosine (5hmC) to initiate DNA demethylation (Yun et al, 2021). It is, therefore, plausible that the p38 MAPK/TFEB pathway contributes to monocyte cell fate, at least in part, via epigenetic regulation of genome methylation. This might explain why the expression of many immune genes remains deficient in mutant cells during polarization to M1 macrophages.
Understanding the regulatory mechanisms involved in monocyte differentiation is essential in comprehending the pathogenesis and in developing therapies for the treatment of hematologic disorders, such as acute myeloid leukemia. Our work reveals a critical role of TFEB in the transcriptional control of monocyte differentiation and identifies phosphorylation of S401 as a novel post‐translational modification that enables coordination of signaling pathways, gene expression, and lineage determination.
Materials and Methods
Cell line cultures and treatments
ARPE‐19 cells (CRL‐2302, American Type Culture Collection) were grown at 37°C in a 1:1 mixture of DMEM and Ham's F12 media supplemented with 10% fetal bovine serum (Invitrogen), 2 mM Glutamax™, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco) in a humidified 5% CO2 atmosphere. RAW 264.7 cells (TIB‐71, American Type Culture Collection), U2OS cells (HTB‐96 American Type Culture Collection), and HeLa (TFEB‐FLAG) cells stably expressing TFEB‐FLAG (previously described (Martina et al, 2012) were grown in DMEM media supplemented with fetal bovine serum), Glutamax™, and antibiotics as indicated for ARPE‐19 cells media. THP1 cells (TIB‐202, American Type Culture Collection) and primary human monocytes were grown in RPMI 1640 media supplemented with fetal bovine serum, Glutamax™, and antibiotics as indicated for ARPE‐19 cells media. All cell lines are free of mycoplasma contamination. For transient expression, ARPE19 cells and RAW 264.7 cells were nucleofected using Cell Line Nucleofector® Kit V and (Lonza) following manufacturer's recommendations. Cells were analyzed 12–24 h post‐nucleofection. For drug treatment experiments, cells were incubated the indicated time at 37°C in medium containing one of the following reagents: DMSO (Sigma‐Aldrich), 250 nM Torin‐1 (TOCRIS), 15 mM N‐Acetyl‐L‐cysteine (NAC, Sigma‐Aldrich), 100–200 μM NaAsO2 (Sodium Arsenite, Santa Cruz Biotechnology), 20 μM SB203580 (p38 MAPK inhibitor, Cayman Chemical), 20 μM JNK VIII (c‐Jun amino terminal kinases inhibitor, Cayman Chemical), 20 μM U0126 (Dual specificity mitogen‐activated protein kinase 1 and 2 inhibitor, Cayman Chemical), 37 μM Anisomycin (p38 MAPK and JNK activator, Cayman Chemical), 5 μM Bisindolylmaleimide IV (PKC inhibitor, Cayman Chemical), 5 μM CRT0066101 (PKD inhibitor, Cayman Chemical), 10 μM eFT508 (MNK1 and 2 inhibitor, Selleck Chemicals), 10 μM E‐64d (lysosomal and cytosolic cysteine proteases inhibitor, Cayman Chemical), 100 μg/ml Leupeptin (cysteine, serine, and threonine proteases inhibitor, Cayman Chemical), 10 μM MG132 (proteasomal inhibitor, Cell Signaling Tech.), 100 nM Thapsigargin (ER stress inducer, Cell Signaling Tech.), and 5 μg/ml Tunicamycin (ER stress inducer, Cell Signaling Tech.). For starvation experiments, cells were washed three times in Hank's balances salt solution (ThermoFisher Scientific) and incubated for 4–12 h at 37°C in Earle's balanced salt solution (Sigma‐Aldrich). For EGF treatment, cells were serum starved for 8 h and then incubated with 100 ng/ml of recombinant human Epidermal Growth Factor (ThermoFisher Scientific) for the indicated times. For macrophage differentiation, naïve THP1 cells were cultured in RPMI medium containing 50 ng/ml PMA (Phorbol 12‐myristate 13‐acetate, InvivoGen) for 24 h. Then, cells were rested in fresh medium without PMA for additional 24 h. For LPS treatment, differentiated THP1 cells were cultured in RPMI medium containing 1 μg/ml LPS (Lipopolysaccharide, InvivoGen) for the indicated times. For inflammasome activation, differentiated THP1 cells were primed in RPMI medium containing 0.1 μg/ml LPS for 4 h, then cells were incubated in the presence of 15 μM Nigericin (Cell Signaling Technology) for 45 min. For primary macrophage differentiation, human monocytes were obtained by elutriation from blood of anonymized healthy human donors provided by the National Institutes of Health blood bank. Volunteers provided written informed consent for the collection of samples and subsequent analysis. Monocytes were cultured in RPMI medium containing 25 ng/ml human recombinant GM‐CSF (Granulocyte‐macrophage colony‐stimulating factor, R&D Systems) for 3 days. Then, medium was replaced with fresh medium containing 25 ng/ml GM‐CSF for additional 3 days.
Recombinant DNA plasmid
TFEB‐WT‐FLAG, TFEB‐S3A/R4A‐FLAG, TFEB‐ΔNLS‐FLAG (TFEB‐R245‐247A‐FLAG) expression vectors have been previously described (Martina & Puertollano, 2013). GST‐TFEB‐PRD bacterial expression vector was generated by cloning the cDNA fragment coding the amino acid sequence corresponding to the proline‐rich domain (Ala364 to Leu476) of human TFEB obtained by PCR amplification from p3xFLAG‐CMV‐14‐TFEB‐FLAG‐WT (Martina et al, 2021). Followed by in‐frame cloning into EcoRI site of pGST‐Parallel‐1 (Sheffield et al, 1999) with a triple FLAG tag fused to the carboxy‐termini of TFEB‐PRD using In‐fusion HD EcoDry system (Takara Bio USA, Inc.). Amino acid substitutions in TFEB were made using the QuikChange Lightning site‐directed mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions.
Adenovirus production
Adenovirus expressing TFEB‐FLAG‐S401A and TFEB‐FLAG‐S401D were prepared, amplified, and purified by Welgen, Inc. Adenovirus expressing TFEB‐FLAG‐WT has been previously described (Martina et al, 2014).
RNA interference (RNAi)
HeLa (TFEB‐FLAG) or THP1‐WT cells (treated for 24 h with PMA before transfection) were transfected using Lipofectamine RNAimax (ThermoFisher, 13778075) or Viromer (Viromer Transfection) transfection reagents respectively, according to the manufacturer's instructions. ON‐TARGETplus non‐targeting pool siRNA duplexes (Horizon Discovery, D‐001810‐10) or ON‐TARGETplus smart pool siRNA duplexes targeted against human p38α (MAPK14) (Horizon Discovery, L‐003512‐00), human p38β (MAPK11) (Horizon Discovery, L‐003972‐00), human JNK1 (MAPK8) (Horizon Discovery, L‐003514‐00), human JNK2 (MAPK9) (Horizon Discovery, L‐003505‐00), human MSK1 (RPS6KA5) (Horizon Discovery, L‐004665‐00), human MSK2 (RPS6KA4) (Horizon Discovery, L‐004664‐00), human MAPKAPK2 (Horizon Discovery, L‐003516‐00), or human MAPKAPK3 (Horizon Discovery, L‐005014‐00) were used at 100 nM. Seventy‐two hours after transfection cells were treated and processed as indicated in the corresponding figure legends.
Generation of CRISPR knock‐in TFEB‐S401A in THP1 cells
CRISPR Cas9‐mediated knock‐ins (clones I11 and M17) of TFEB‐S401 in THP1 cells were generated by Synthego Corporation (Redwood City, CA, USA).
To generate these cells, ribonucleoproteins containing the Cas9 protein and synthetic chemically modified sgRNA (UCAGCCACAGCCUGAGCUUU) and donor templates as single‐stranded oligo DNA nucleotides (CCACCCAGCCACCATCCCCATTCCATCACCTGGACTTCAGC CACTCTCTGGCCTTTGGGGGCAGGGAGGACGAGGGTCCCCCGGGCTACCCCGAACCCCTGGC) produced at Synthego were electroporated into the cells using Synthego's optimized protocol.
Editing efficiency was assessed upon recovery, 48 h post electroporation. Genomic DNA was extracted from a portion of the cells, PCR amplified, and sequenced using Sanger sequencing. The resulting chromatograms were processed using Synthego Inference of CRISPR edits software (ice.synthego.com).
To create monoclonal cell populations, edited cell pools were seeded at < 1 cell/well using a single cell printer into 96 well plates. All wells were imaged every 3 days to ensure expansion from a single‐cell clone. Clonal populations were screened and identified using the PCR‐Sanger‐ICE genotyping strategy described above.
Production of anti‐phospho‐TFEB (S401) antibody
For antibody production, the synthesis and purification of a phospho‐specific TFEB peptide (DFSHSL‐pS‐FGGREDE; amino acids 395–408), and a non‐phosphorylated peptide counterpart (DFSHSLSFGGREDE) as well as the production of polyclonal antisera was performed by YenZym Antibodies (South San Francisco, CA). Two New Zealand white rabbits were immunized with the phosphopeptide following a 90 days immunization protocol. The antisera were further purified by affinity chromatography against the same phosphopeptide used for immunization. The purified antibody was then affinity‐absorbed against the non‐phosphorylated peptide counterpart, to separate the phosphopeptide‐specific antibody from the cross‐reactive population. The specificity of anti‐phospho‐TFEB antibody was examined by immunoblotting.
Immunofluorescence confocal microscopy
For immunofluorescence, cells grown on coverslips were washed with PBS and fixed with 4% formaldehyde for 15 min at room temperature. For monitoring nuclear localization of TFEB or TFEB‐FLAG, cells were permeabilized in PBS containing 0.2% Triton X100 for 10 min at room temperature. Cells were then incubated with the indicated primary antibodies in PBS containing 10% Fetal Bovine Serum, and 0.1% (wt/v) saponin for 1 h at room temperature, followed by incubation with the corresponding Alexa Fluor 568‐conjugated secondary antibodies. For a list of antibodies and their dilutions, see Appendix Table S2. After staining, the coverslips were mounted onto glass slides with Dapi‐Fluoromount‐G (Electron Microscopy Sciences, 17984‐24). Images were acquired on a Zeiss LSM 510 confocal system equipped with filter sets for Rhodamine and DAPI, 543 nm and 405 laser excitations respectively, an AxioCam camera, a 63× NA 1.4 oil immersion objective, and LSM 510 operating software (Carl Zeiss). Confocal images taken with the same acquisition parameters were processed with ImageJ software (NIH). Photoshop CC 2022 software was used to produce the figures.
Cell viability assay
PMA‐differentiated THP1 cells grown on glass coverslips were incubated in medium containing 1 μg/ml LPS for 6 h at 37°C. Then, cells were washed with Hanks' Balanced Salt Solution (HBSS) and incubated with LIVE/DEAD fixable blue dead cell stain (ThermoFisher Scientific) in HBSS for 30 min at room temperature. Cells were washed with HBSS and fixed with 4% formaldehyde for 15 min at room temperature. Fixed cells were analyzed, and images acquired as detailed in “immunofluorescence confocal microscopy” section.
Subcellular fractionation
Cells were resuspended in buffer A (10 mM HEPES, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1.5 mM dithiothreitol) supplemented with protease and phosphatase inhibitors at a concentration of 7.5 × 106 cells/ml. Cells were gently mixed and kept on ice for 10 min. Then, cells were lysed by the addition of 0.05% NP‐40 and gently mixing for 3 s. The lysate was then centrifuged at 800 g for 10 min at 4°C. The supernatant was centrifuged at 13,500 g for 15 min at 4°C. The resulting supernatant represents the cytosolic fraction. The 800 g pellet was resuspended in buffer A containing NP‐40 followed by centrifugation at 800 g for 10 min at 4°C. The resulting pellet was resuspended in buffer B (20 mM HEPES, pH 7.4, 400 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 10% glycerol, 0.2% NP‐40) supplemented with protease and phosphatase inhibitors and kept on ice for 30 min with intermittent vigorous vortexing. The supernatant obtained after centrifugation at 18,500 g for 30 min at 4°C contained the nuclear fraction.
Immunoprecipitation, electrophoresis, and immunoblotting
Cells washed with ice‐cold PBS were lysed in lysis buffer containing 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, and 1% Triton X‐100 (wt/v) and supplemented with protease and phosphatase inhibitors cocktail (Sigma‐Aldrich). Cell lysates were incubated on ice for 30 min and then were passed 10 times through a 25‐gauge needle. Cell lysates were centrifuged at 16,000 g for 10 min at 4°C. For immunoprecipitation, the soluble fractions were incubated with 20 μl of anti‐FLAG M2 affinity gel beads (Sigma‐Aldrich) for 4 h at 4°C. The immunoprecipitates were collected, washed three times with lysis buffer, and proteins were eluted with Laemmli sample buffer.
Samples were analyzed by SDS–PAGE (4–20% gradient, ThermoFisher) under reducing conditions and transferred to nitrocellulose membranes. Membranes were immunoblotted using the indicated antibodies. For a list of antibodies and their dilutions, see Appendix Table S2. HRP‐chemiluminescence was developed using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences). The exposed films were scanned, and the protein band intensities quantified using ImageJ software (NIH). Photoshop CC 2022 software was used to produce the figures.
Expression and purification of GTS‐TFEB‐PRD
BL21(DE3) competent cells were transformed with plasmid pGST‐Parallel‐TFEB‐PRD following manufacturer instructions (New England Biolabs). Expression of GST‐TFEB‐PRD fusion protein was done in 1 liter culture of LB (KD medical) and incubated at 37°C until an OD600 of 0.8–0.9. Then the expression was induced by the addition of 1 mM IPTG (ThermoFisher Scientific) and the culture was incubated for 4 h at 37°C. Cells were harvested by centrifugation at 8,000 g for 30 min at 4°C. Bacterial pellet was resuspended in GST lysis buffer (PBS, 2 mM EDTA) containing protease inhibitors cocktail (Sigma‐Aldrich), and lysed by high pressure homogenization. The bacterial lysate was incubated with 1% Triton X‐100 (Sigma‐Aldrich) and 100 U of DNAse solution (Fisher Scientific) for 30 min at 4°C with rotation. The soluble protein fraction was recovered by centrifugation at 22,000 g for 30 min at 4°C and then incubated with 0.7 ml of glutathione–Sepharose beads (Cytiva). The GST‐fusion protein was allowed to bind to the beads for 2 h at 4°C with rotation. Beads were washed twice with 50 ml of ice‐cold GST lysis buffer containing 1% Triton X‐100, 50 ml of GST lysis buffer containing 150 mM NaCl, and 1% Triton X‐100 and twice with PBS before eluting the GST‐fusion protein with 0.6 ml (4 times) of 0.1 M NaCl and 0.1 M Tris–HCl (pH 8) containing 20 mM reduced glutathione at room temperature for 10 min each with rotation. GST‐TFEB‐PRD protein purity was confirmed by SDS–PAGE and Coomassie blue staining and concentration was determined by Bradford protein assay (Bio‐Rad).
In vitro protein kinase assay
p38 MAPK assay was performed at 30°C for 1 h in kinase buffer (60 mM HEPES (pH 7.5), 3 mM MgCl2, 3 mM MnCl2, 3 μM Na‐orthovanadate, 1.2 mM DTT and 10% glycerol) containing 40 μM ATP, 10 μg of GST‐TFEB‐PRD and 600–800 ng of recombinant human active p38α MAPK (Reaction Biology or Sino Biological) in a final volume of 30 μl. One microliter of the kinase reaction was analyzed by SDS–PAGE under reducing conditions and immunoblotting using the indicated antibodies.
Mass spectrometry
Immunoprecipitated TFEB‐FLAG from HeLa (TFEB‐FLAG) and U2O2 (Ad. TFEB‐FLAG) cells treated with DMSO or 150 μM NaAsO2 for 1 h were subjected to SDS–PAGE, and gel bands from corresponding molecular weights were excised for enzymatic digestion. Briefly, the samples were first reduced with TCEP (tris(2‐carboxyethyl)phosphine, Sigma‐Aldrich) and alkylated with CAA (chloroacetamide, Sigma‐Aldrich), and then digested with chymotrypsin (Promega). The resulting peptide mixtures were analyzed with an Orbitrap Fusion Lumos that is equipped with a Dionex Ultimate 3000 nanoLC system (ThermoFisher Scientific). Peptide IDs and phosphorylation sites were assigned with Mascot V2.5 (Matrix Science). The confidence of phosphorylation site localization is assessed with ptmRS node in Proteome Discoverer 2.2 platform (ThermoFisher Scientific). All peptides were filtered out at 1% false discovery rate (FDR) and their relative abundances were compared based on the areas under curve (AUC) of their corresponding chromatographic peaks.
RNA isolation and relative quantitative Real‐Time PCR
RNA was isolated from samples with the PureLink RNA Mini Kit (ThermoFisher, 12183018A) and reverse transcribed using SuperScript III First‐Strand Synthesis SuperMix kit (ThermoFisher, 11752). Quantitative real‐time PCR reactions were set up in triplicate with 50 ng cDNA per reaction and 200 nM gene specific primers mix (QuantiTect primer Assays, Qiagen) along with PowerUp SYBR Green Master Mix (Applied Biosystems, A25741). For a list of primers, see Appendix Table S3. Reactions were run and analyzed using a QuantStudio 12 K Flex Real‐Time PCR system (Applied Biosystems, Life Technologies). The values were expressed as a fold change relative to RNA from cells infected with control adenovirus (Ad. Null) or WT non‐treated cells, normalized against GAPDH using the ΔΔCT methods.
RNA‐seq
Naïve THP1 cells (4 × 106) were treated with 50 ng/ml PMA for 6 h and transferred to a 15 ml conical tube and then centrifuged at 800 g for 5 min at 4°C. Cell pellets were resuspended with ice‐cold PBS and centrifuged again at 800 g for 5 min. Washed cell pellets were snap‐frozen on dry ice and then processed for RNA‐seq assay by Active Motif, Inc.
RNA‐Seq sample processing
Total RNA was isolated from cells using the Qiagen RNeasy Mini Kit (Qiagen, cat# 74104). For each sample, 0.5 ng of total RNA was then used in Illumina's TruSeq Stranded mRNA Library kit (Cat# 20020594). Libraries were sequenced on Illumina NextSeq 500 as paired‐end 42‐nt reads. Sequence reads were analyzed with the STAR alignment—DESeq2 software pipeline.
RNA‐Seq analysis
Read mapping
The paired‐end 42 bp sequencing reads (PE42) generated by Illumina sequencing (using NextSeq 500) were mapped to the genome using the STAR algorithm with default settings. Alignment information for each read was stored in the BAM format.
Fragment assignment
The number of fragments overlapping predefined genomic features of interest (e.g., genes) were counted. Only read pairs that have both ends aligned were counted. Read pairs that have their two ends mapping to different chromosomes or mapping to same chromosome but on different strands are discarded. The gene annotations were obtained from Subread package. These annotations were originally from NCBI RefSeq database and then adapted by merging overlapping exons from the same gene to form a set of disjoint exons for each gene. Genes with the same Entrez gene identifiers were also merged into one gene.
Differential analysis
After obtaining the gene table containing the fragment counts of genes, differential analyses were performed to identify statistically significant differential genes using DESeq2. The following lists the pre‐processing steps before differential calling:
Data normalization: DESeq2 expects un‐normalized count matrix of sequencing fragments. The DESeq2 model internally corrects for library size using their median‐of‐ratios method. The gene table obtained from “Fragment Assignment” was used as input to perform the DESeq2's differential test.
Filtering before multiple testing adjustment: After a differential test has been applied to each gene except the ones with zero counts, the P‐value of each gene was calculated and adjusted to control the number of false positives among all discoveries at a proper level.
Differential calling: Differential genes were detected by DESeq2 at 0.1 (or 10%) FDR (i.e., adjusted P‐value).
Gene set enrichment analysis and heatmap representation
The gene set enrichment analysis (MSigDB Hallmark 2020) was performed using the web‐based enrichment tool Enrichr (Xie et al, 2021), and heatmaps of immune‐ and lysosomal‐related genes were analyzed using pheatmap package in R software (The R Project for Statistical Computing).
ChIP‐seq
Chromatin immunoprecipitation
Chromatin was isolated by adding lysis buffer (1% SDS, 10 mM EDTA and 50 mM Tris–HCl, pH 8.1 containing protease inhibitors), followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp with Active Motif's EpiShear probe sonicator (cat# 53051). Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K, and heat for de‐crosslinking, followed by SPRI beads clean up (Beckman Coulter) and quantitation by Clariostar (BMG Labtech). Extrapolation to the original chromatin volume allowed determination of the total chromatin yield. An aliquot of chromatin (40 μg) was precleared with protein A agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using 40 μl of rabbit monoclonal antibody against TFEB (clone D2O7D, Cell Signaling Technology, cat# 37785). Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65°C, and ChIP DNA was purified by phenol‐chloroform extraction and ethanol precipitation.
ChIP sequencing (Illumina)
Illumina sequencing libraries were prepared from the ChIP and Input DNAs by the standard consecutive enzymatic steps of end‐polishing, dA‐addition, and adaptor ligation. Steps were performed on an automated system (Apollo 342, Wafergen Biosystems/Takara). After a final PCR amplification step, the resulting DNA libraries were quantified and sequenced on Illumina's NextSeq 500 (75 nt reads, single end). Reads were aligned to the human genome using the BWA algorithm (default settings; Li & Durbin, 2009). Duplicate reads were removed, and only uniquely mapped reads (mapping quality ≥ 25) were used for further analysis. Alignments were extended in silico at their 3′‐ends to a length of 200 bp, which is the average genomic fragment length in the size‐selected library and assigned to 32‐nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. Peak locations were determined using the MACS algorithm (v2.1.0) with a cutoff of P‐value = 1 e‐7 (Zhang et al, 2008). Peaks that were on the ENCODE blacklist of known false ChIP‐Seq peaks were removed. Signal maps and peak locations were used as input data to Active Motifs proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations, and gene annotations.
Software
bcl2fastq2 (v2.20) was used for processing of Illumina base‐call data and demultiplexing, Samtools (v0.1.19) was used for processing of BAM files, BEDtools (v2.25.0) was used for processing of BED files, and wigToBigWig (v4) was used for the generation of bigWIG files.
Statistical analysis
Data were processed in Excel (Microsoft Corporation) then Prism (GraphPad Software) to generate curve and bar charts and perform statistical analyses. Student's t‐test, One‐way ANOVA, Two‐way ANOVA, and pairwise post‐tests were performed for each dependent variable, as specified in each figure legend. All data are presented as mean ± SD. P < 0.05 was considered statistically significant (*), P < 0.01 very significant (**), P < 0.001 extremely significant (***), and P < 0.0001 extremely significant (****). P > 0.05 was considered not significant (ns).
Author contributions
Rosa Puertollano: Conceptualization; formal analysis; supervision; funding acquisition; writing—original draft; writing—review and editing. José A Martina: Conceptualization; data curation; formal analysis; writing—review and editing. Eutteum Jeong: Data curation; formal analysis.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
J.A.M., E.J., and R.P. were supported by the Intramural Research Program of the NIH, National Heart, Lung, and Blood Institute (NHLBI) (ZIA HL006151). We thank Dr. Yong Chen and Dr. Marjan Gucek, NHLBI Proteomics Core Facility, for help to perform and interpret the mass spectrometry analysis.
EMBO reports (2023) 24: e55472
Data availability
All source and supporting data are available from the corresponding authors on reasonable request. The datasets produced in this study are available in the following database: RNA‐Seq data: Gene Expression Omnibus GSE201826 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE201826). ChIP‐Seq data: Gene Expression Omnibus GSE217608 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE217608).
References
- Artandi SE, Merrell K, Avitahl N, Wong KK, Calame K (1995) TFE3 contains two activation domains, one acidic and the other proline‐rich, that synergistically activate transcription. Nucleic Acids Res 23: 3865–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Martina JA, Puertollano R (2018) Emerging roles for TFEB in the immune response and inflammation. Autophagy 14: 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronisz A, Carey HA, Godlewski J, Sif S, Ostrowski MC, Sharma SM (2014) The multifunctional protein fused in sarcoma (FUS) is a coactivator of microphthalmia‐associated transcription factor (MITF). J Biol Chem 289: 326–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon SE, Lee J, Yoo BC, Sung GH, Kim TW, Park HJ, Cho JY (2011) p38‐targeted inhibition of interleukin‐12 expression by ethanol extract from cordyceps bassiana in lipopolysaccharide‐activated macrophages. Immunopharmacol Immunotoxicol 33: 90–96 [DOI] [PubMed] [Google Scholar]
- Canovas B, Nebreda AR (2021) Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol 22: 346–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey HA, Bronisz A, Cabrera J, Hildreth BE 3rd, Cuitino M, Fu Q, Ahmad A, Toribio RE, Ostrowski MC, Sharma SM (2016) Failure to target RANKL signaling through p38‐MAPK results in defective Osteoclastogenesis in the microphthalmia cloudy‐eyed mutant. J Cell Physiol 231: 630–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410: 37–40 [DOI] [PubMed] [Google Scholar]
- El‐Houjeiri L, Possik E, Vijayaraghavan T, Paquette M, Martina JA, Kazan JM, Ma EH, Jones R, Blanchette P, Puertollano R et al (2019) The transcription factors TFEB and TFE3 link the FLCN‐AMPK signaling Axis to innate immune response and pathogen resistance. Cell Rep 26: 3613–3628.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo NP, Ko CL, Chu CY, Wang CY, So EC, Huang BM (2020) Arsenic compounds activate the MAPK and caspase pathways to induce apoptosis in OECM1 gingival epidermal carcinoma. Oncol Rep 44: 2701–2714 [DOI] [PubMed] [Google Scholar]
- Garcia J, Lemercier B, Roman‐Roman S, Rawadi G (1998) A mycoplasma fermentans‐derived synthetic lipopeptide induces AP‐1 and NF‐kappaB activity and cytokine secretion in macrophages via the activation of mitogen‐activated protein kinase pathways. J Biol Chem 273: 34391–34398 [DOI] [PubMed] [Google Scholar]
- Goding CR, Arnheiter H (2019) MITF‐the first 25 years. Genes Dev 33: 983–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MA, Choy CH, Dayam RM, Ospina‐Escobar E, Somerville A, Xiao X, Ferguson SM, Botelho RJ (2016) Phagocytosis enhances lysosomal and bactericidal properties by activating the transcription factor TFEB. Curr Biol 26: 1955–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsenko A, Yu S, Martin‐Sanchez F, Diaz‐Del‐Olmo I, Nichols EM, Davis DM, Brough D, Lopez‐Castejon G (2020) Priming is dispensable for NLRP3 inflammasome activation in human monocytes In vitro. Front Immunol 11: 565924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazzalin CA, Le Panse R, Cano E, Mahadevan LC (1998) Anisomycin selectively desensitizes signalling components involved in stress kinase activation and fos and Jun induction. Mol Cell Biol 18: 1844–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CL, Lee EX, Gordon KL, Paz EA, Shen WC, Ohnishi K, Meisenhelder J, Hunter T, La Spada AR (2018) MAP4K3 mediates amino acid‐dependent regulation of autophagy via phosphorylation of TFEB. Nat Commun 9: 942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Sharma SM, Bronisz A, Srinivasan R, Sankar U, Ostrowski MC (2007) Eos, MITF, and PU.1 recruit corepressors to osteoclast‐specific genes in committed myeloid progenitors. Mol Cell Biol 27: 4018–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irazoqui JE (2020) Key roles of MiT transcription factors in innate immunity and inflammation. Trends Immunol 41: 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20: 3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavardhana S, Malireddi RKS, Kanneganti TD (2020) Caspases in cell death, inflammation, and Pyroptosis. Annu Rev Immunol 38: 567–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Song HS, Yu J, Kim YM (2021) MiT family transcriptional factors in immune cell functions. Mol Cells 44: 342–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J (2001) Mammalian mitogen‐activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81: 807–869 [DOI] [PubMed] [Google Scholar]
- La Spina M, Contreras PS, Rissone A, Meena NK, Jeong E, Martina JA (2020) MiT/TFE family of transcription factors: an evolutionary perspective. Front Cell Dev Biol 8: 609–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with burrows‐wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu M, Ding X, Yan C, Song Z, Chen L, Huang X, Wang X, Jian Y, Tang G et al (2016) Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol 18: 1065–1077 [DOI] [PubMed] [Google Scholar]
- Li Y, Hodge J, Liu Q, Wang J, Wang Y, Evans TD, Altomare D, Yao Y, Murphy EA, Razani B et al (2020) TFEB is a master regulator of tumor‐associated macrophages in breast cancer. J Immunother Cancer 8: e000543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansky KC, Sankar U, Han J, Ostrowski MC (2002) Microphthalmia transcription factor is a target of the p38 MAPK pathway in response to receptor activator of NF‐kappa B ligand signaling. J Biol Chem 277: 11077–11083 [DOI] [PubMed] [Google Scholar]
- Marchand B, Arsenault D, Raymond‐Fleury A, Boisvert FM, Boucher MJ (2015) Glycogen synthase kinase‐3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J Biol Chem 290: 5592–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Puertollano R (2013) Rag GTPases mediate amino acid‐dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol 200: 475–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Puertollano R (2018) Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J Biol Chem 293: 12525–12534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, Puertollano R (2014) The nutrient‐responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 7: ra9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Brady OA, Puertollano R (2016) TFEB and TFE3 are novel components of the integrated stress response. EMBO J 35: 479–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Guerrero‐Gomez D, Gomez‐Orte E, Antonio Barcena J, Cabello J, Miranda‐Vizuete A, Puertollano R (2021) A conserved cysteine‐based redox mechanism sustains TFEB/HLH‐30 activity under persistent stress. EMBO J 40: e105793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto‐Rosato A, Prezioso C, Forrester A et al (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17: 288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najibi M, Labed SA, Visvikis O, Irazoqui JE (2016) An evolutionarily conserved PLC‐PKD‐TFEB pathway for host defense. Cell Rep 15: 1728–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, Chaudhury A, Bajaj L, Bondar VV, Bremner L et al (2017) mTORC1‐independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun 8: 14338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette M, El‐Houjeiri L, Zirden LC, Puustinen P, Blanchette P, Jeong H, Dejgaard K, Siegel PM, Pause A (2021) AMPK‐dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy 17: 3957–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore N, Brady OA, Diab HI, Martina JA, Sun L, Huynh T, Lim JA, Zare H, Raben N, Ballabio A et al (2016) TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12: 1240–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Ferguson SM, Brugarolas J, Ballabio A (2018) The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J 37: e98804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puren AJ, Fantuzzi G, Dinarello CA (1999) Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci U S A 96: 2256–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Puertollano R (2016) TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol 32: 255–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat KPA, Manjithaya R (2021) TFEB – at the crossroads of host–pathogen interactions. J Cell Sci 134: jcs252981 [DOI] [PubMed] [Google Scholar]
- Roczniak‐Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5: ra42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, Ali S, Dehestani B, Holloway KB, Micevych PS et al (2017) Exploiting macrophage autophagy‐lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun 8: 15750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC et al (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha Y, Rao L, Settembre C, Ballabio A, Eissa NT (2017) STUB1 regulates TFEB‐induced autophagy‐lysosome pathway. EMBO J 36: 2544–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SM, Bronisz A, Hu R, Patel K, Mansky KC, Sif S, Ostrowski MC (2007) MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J Biol Chem 282: 15921–15929 [DOI] [PubMed] [Google Scholar]
- Sheffield P, Garrard S, Derewenda Z (1999) Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr Purif 15: 34–39 [DOI] [PubMed] [Google Scholar]
- Slade L, Pulinilkunnil T (2017) The MiTF/TFE family of transcription factors: master regulators of organelle signaling, metabolism, and stress adaptation. Mol Cancer Res 15: 1637–1643 [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Copeland NG, Jenkins NA (2004) Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet 38: 365–411 [DOI] [PubMed] [Google Scholar]
- Visvikis O, Ihuegbu N, Labed SA, Luhachack LG, Alves AF, Wollenberg AC, Stuart LM, Stormo GD, Irazoqui JE (2014) Innate host defense requires TFEB‐mediated transcription of cytoprotective and antimicrobial genes. Immunity 40: 896–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Penninger JM (2004) Mitogen‐activated protein kinases in apoptosis regulation. Oncogene 23: 2838–2849 [DOI] [PubMed] [Google Scholar]
- Wang H, Wang N, Xu D, Ma Q, Chen Y, Xu S, Xia Q, Zhang Y, Prehn JHM, Wang G et al (2020a) Oxidation of multiple MiT/TFE transcription factors links oxidative stress to transcriptional control of autophagy and lysosome biogenesis. Autophagy 16: 1683–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang Y, Liu J, Zhang J, Xu M, You Z, Peng C, Gong Z, Liu W (2020b) Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep 21: e48335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM et al (2021) Gene set knowledge discovery with Enrichr. Curr Protoc 1: e90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XL, Guo TK, Wang YH, Huang YH, Liu X, Wang XX, Li W, Zhao X, Wang LP, Yan S et al (2012) Ginsenoside Rd attenuates the inflammatory response via modulating p38 and JNK signaling pathways in rats with TNBS‐induced relapsing colitis. Int Immunopharmacol 12: 408–414 [DOI] [PubMed] [Google Scholar]
- Yang M, Liu E, Tang L, Lei Y, Sun X, Hu J, Dong H, Yang SM, Gao M, Tang B (2018) Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun Signal 16: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, Jian Y, Xu M, Huang X, Wang N, Liu Z, Li Q, Li J, Zhou H, Xu L et al (2020) CDK4/6 regulate lysosome biogenesis through TFEB/TFE3. J Cell Biol 219: e201911036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S, Vincelette ND, Yu X, Watson GW, Fernandez MR, Yang C, Hitosugi T, Cheng CH, Freischel AR, Zhang L et al (2021) TFEB links MYC signaling to epigenetic control of myeloid differentiation and acute myeloid leukemia. Blood Cancer Discov 2: 162–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]