

Summary
The speed and scope of cognitive deterioration in Alzheimer's disease is highly associated with the advancement of tau neurofibrillary lesions across brain networks. We tested whether the rate of tau propagation is a heritable disease trait in a large, well-characterized cohort of genetically divergent mouse strains. Using an AAV-based model system, P301L-mutant human tau (hTau) was introduced into the entorhinal cortex of mice derived from 18 distinct lines. The extent of tau propagation was measured by distinguishing hTau-producing cells from neurons that were recipients of tau transfer. Heritability calculation revealed that 43% of the variability in tau spread was due to genetic variants segregating across background strains. Strain differences in glial markers were also observed, but did not correlate with tau propagation. Identifying unique genetic variants that influence the progression of pathological tau may uncover novel molecular targets to prevent or slow the pace of tau spread and cognitive decline.
Subject areas: Neurogenetics, Molecular neuroscience, Cellular neuroscience
Graphical abstract
Highlights
-
•
Neuron-to-neuron spread of human tau is influenced by mouse genetic background
-
•
Humans and mice share genes with variants associated with tau pathology
-
•
Tau propagation and glial abundance likely have non-overlapping genetic drivers
-
•
Divergent mouse strains can model genetic heterogeneity underlying disease traits
Neurogenetics; Molecular neuroscience; Cellular neuroscience
Introduction
One of the most important risk indicators for developing Alzheimer's disease (AD) is a family history of dementia: having a parent or sibling with a diagnosis of AD confers a 70% increase in disease risk.1 Sequence variants at 75 unique genetic loci have been associated with sporadic AD, and have been implicated in established disease processes, including brain amyloid processing, lipid metabolism, and immune responses.2 Despite growing interest in understanding how AD-associated genetic polymorphisms lead to clinical and pathological outcomes, it remains unclear how polygenic factors contribute to the formation and expansion of tau neurofibrillary inclusions, which are the closest correlate of neural degeneration and clinical symptoms during AD.3,4
That there are genetic drivers of preclinical tau pathology is supported by the observation that genetically identical monozygotic twins exhibit a high degree of concordance in early measures of total-tau (t-tau) and phospho-tau (p-tau) in cerebrospinal fluid (CSF), as well as tau regional distribution as measured by positron emission tomography (PET).5,6 Genome-wide association studies (GWAS) of tau biomarkers and endophenotypes have so far identified 8 novel gene variants associated with elevated levels of CSF t-tau and p-tau, and tau-PET burden.7,8,9,10,11 While clinical data indicate that severity of tau pathology is likely influenced by heritable factors, studying this phenomenon in an experimental setting is especially difficult.
Few studies have used divergent murine lines to investigate heritable contributions to AD. Different rodent strains exhibit dissimilar tau tangle burden and microglial responses following mutant tau transgene expression or injection of AD patient-derived tau.12,13 We have also shown that tau propagation varies considerably among 5 genetically diverse mouse strains, suggesting that discrete cellular phases of tau transfer may be regulated by distinct genetic modifiers.14 We expand on these initial findings by employing a large, well-characterized panel of genetically diverse mouse strains that have been generated by combining the well-known 5XFAD transgenic line with the BXD genetic reference panel. The resulting nontransgenic (Ntg) BXD progeny segregate for almost 6 million sequence variants, including those at genetic loci previously implicated in AD risk.15 Interestingly, BXD mice transgenic for familial AD mutations exhibit strain-dependent susceptibility to amyloid accumulation and age-dependent cognitive decline.16,17,18 We leveraged the genetic diversity among Ntg-BXD animals to investigate whether processes underlying tau propagation may be similarly governed by heritable factors.
Results
Tau propagation is strain dependent and exhibits significant heritability
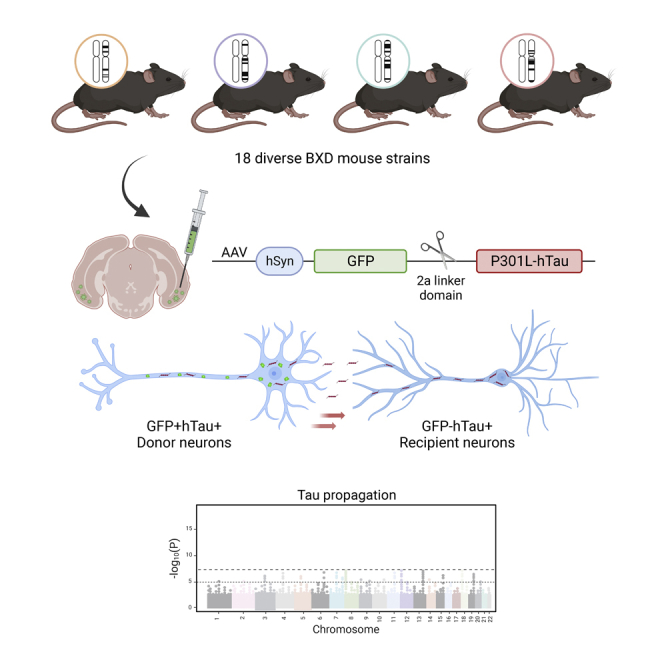
To study the effect of mouse genetic background on tau propagation, we employed an AAV-based method of tau expression and proliferation previously used by our group.14,19,20,21,22 AAV vectors express GFP and P301L-mutant human tau (hTau) separated by a central 2a domain. The 2a sequence encodes a short linker peptide that self-cleaves during protein translation, liberating independent but equimolar GFP and hTau peptides (Figure 1A). As a result, transduced “donor” neurons in the entorhinal cortex and hippocampus generate both GFP and hTau, whereas connected neurons that are “recipients” of tau transfer harbor only hTau peptides (Figures 1A–1C).
Figure 1.

Modeling tau propagation using AAV-GFP-2a-P301LhTau
(A) GFP and P301L-mutated human tau (hTau) were virally expressed in the entorhinal cortex using an AAV construct driven by the neuron-specific synapsin-1 transcriptional promoter (hSyn). Incorporation of a self-cleaving 2a-linker results in the release of separate GFP and hTau peptides, such that GFP-expressing “donor” neurons and tau-bearing “recipient” neurons can be differentiated by double immunofluorescent labeling.
(B) Broad GFP expression (green) was visualized in the entorhinal cortex, hippocampus, and connecting fibers following bilateral injection of AAV-GFP-2a-P301LhTau (scale bar = 5 mm).
(C) AAV-transduced neurons and fibers displayed strong GFP (green) and hTau (red) immunoreactivity, whereas recipient neurons of tau transfer could be discriminated by their lack of GFP expression. Higher magnification photomicrographs (right) allowed for the visualization and subsequent quantification of donor and recipient cells (left scale bar = 200 μm; right scale bar = 50 μm).
AAV vectors expressing GFP-2a-P301LhTau were bilaterally injected into the entorhinal cortex of 6-month-old mice from 18 different strains. Three months post injection, we quantified the number of GFP+hTau+ donor neurons and GFP-hTau+ recipient neurons to evaluate strain differences in tau propagation (Figures 2A and 2B). The number of GFP-hTau+ cells that had acquired hTau through neuron-to-neuron transfer varied considerably among BXD groups, resulting in a significant strain-effect (Figure 2B). Given that the number of donor neurons may be influenced by differences in AAV transduction resulting from surgical variability, we also generated a tau propagation ratio in a secondary analysis (Figure 2C).
Figure 2.

Tau propagation is strain dependent and exhibits significant heritability
(A) GFP+hTau+ donor neurons were quantified to confirm consistent AAV transduction in BXD animals. All strains exhibited strong AAV transduction and substantial donor neurons available for tau propagation (one-way ANOVA, p = 0.160).
(B) The number of GFP-hTau+ recipient neurons varied significantly between 18 BXD mouse lines in a strain-dependent manner (Kruskal Wallis, p = 0.007).
(C) The ratio of GFP-hTau+ recipient neurons to AAV-expressing donor neurons showed significant variability between BXD strains (Kruskal Wallis, p = 0.044). The heritability (h2RIX) of tau propagation ratio was calculated to be 0.435, indicating that ∼43.5% of the variability in tau propagation is accounted for by genetic contributions.
(D) Representative micrographs in the hippocampal dentate gyrus of mice from BXD strains exhibiting varying levels of tau propagation: “low propagators” showed few neurons that were hTau-immunopositive only, whereas “high propagators” displayed strong GFP expression, as well as abundant GFP-hTau+ recipient neurons (scale bar = 100 μm).
(E) Tau propagation ratio was equivalent between male and female animals between all BXD groups (Mann-Whitney, p = 0.592). Data are presented in column bar graphs, where error bars represent the mean ± SEM (∗p ≤ 0.05; ∗∗p ≤ 0.01).
Striking differences in tau propagation were observed across mouse strains. Some genetic backgrounds, such as B6xBXD42, were relatively resistant to tau spread despite strong GFP expression, while others were particularly vulnerable, such as B6xBXD113 (Figure 2D). Importantly, the heritability (h2RIX) of tau propagation ratio was calculated to be 0.435; that is, ∼43.5% of the observed variance in tau propagation could be explained by genetic factors segregating in this cohort of animals. The cohort was sex balanced, and no significant sex differences were detected (Figure 2E).
Translating human GWAS hits to test for an association with tau propagation in BXD mice
We took advantage of the genetic diversity among BXD strains to carry out a preliminary quantitative trait locus (QTL) test for tau propagation (Figure 3A). Although no genome-wide significant QTL was identified in this analysis, future studies applying a larger array of mouse strains may reveal genetic loci associated with tau propagation. Using the BXD Power Calculator tool at GeneNetwork.org (https://power.genenetwork.org/), with 18 strains, an average of 6 replicates per strain, and a power threshold of 0.8, we are powered to detect loci explaining about 60%–65% of the variance in tau propagation. To detect a locus of lower effect size (e.g., a locus contributing to ∼40% of the variance in the trait), we would need 40 BXD strains.
Figure 3.

Translating human GWAS hits to test for an association with tau propagation in BXD mice
(A) Quantitative trait locus (QTL) mapping for tau propagation did not yield genome-wide significant QTL in BXD mice (gray bar, p = 0.30; red bar, p < 0.05).
(B and C) Extent of tau propagation was compared between genotypes of mouse loci with human orthologs found to be associated with tau endophenotypes. Cell-to-cell transfer of tau was more pronounced in BXD mice harboring the D allele of the genetic locus near GMNC/Ostn (p = 0.017). A trend toward increased tau propagation was also observed in mice carrying the B allele of the BIN1 gene. Data are presented in column bar graphs, where error bars represent the mean ± SEM (∗p ≤ 0.05).
At least 13 genetic variants have been previously associated with tau-related biomarkers in humans.7,8,9,10 We looked more specifically at whether some of these genes could be mediators of tau propagation in the Ntg-BXD cohort. The mouse ortholog of each gene and its genomic location were identified using the Mouse Genome Informatics database (informatics.jax.org). We then identified the nearest marker SNP for each locus within the BXD genome using the BXD genotypes file published at GeneNetwork.org, and mapped to the mm10/GRCm38 genome build. We segregated the strains by genotype at each allele (BB or BD; denoted as B or D, respectively) and tested for significant differences in tau propagation (Figures 3B and 3C). Surprisingly, the genotype at the GMNC/Ostn locus predicted tau propagation in BXD mice. Animals carrying the B allele of the BIN1 gene, a well-established risk locus for AD, also exhibited increased tau propagation. These findings suggest that, with a larger sample size and more statistical power, this model could be employed to identify additional human-relevant disease modifiers.
Glial abundance exhibits strain-dependent variability, but does not correlate with tau propagation
It has previously been reported that microglial abundance and MHC-II expression are differentially affected by the presence of tauopathy in two rat models bred on distinct genetic backgrounds.12 Given that glia may play a role in tau propagation,23,24,25 we assessed whether strain differences in glial abundance might parallel the degree of tau spread between groups. Total fluorescence and percent area of microglia- and astrocyte-specific markers, Iba1 and GFAP, respectively, varied significantly among BXD groups and were highly heritable traits (h2RIX = 0.82–0.97; Figure 4A). Importantly, there was no correlation between tau propagation ratio and these measures of glial abundance (Figure 4B). While strong genetic factors appear to influence neuronal tau transfer and glial phenotypes, it is likely that independent mechanisms differentially contribute to both. Given the available data, it is unclear whether these measures are reflective of baseline strain differences in cell marker expression or a glial response to propagated tau. We therefore examined Iba1 and GFAP fluorescence intensity in the visual cortex, which does not express GFP or hTau, in these same animals (Figure S1). When strains are organized in rank order of increasing hippocampal Iba1 and GFAP fluorescence intensity, it is evident that these measures are not aligned in unaffected visual cortex areas. Pearson correlation analyses also showed that Iba1 and GFAP fluorescence intensity in the hippocampus is not correlated with those in the visual cortex. This indicates that what is observed in Figure 4A might represent differential reactivity to tau expression, as opposed to baseline biology.
Figure 4.

Glial abundance exhibits strain-dependent variability, but does not correlate with tau propagation
(A) Total fluorescent intensity and image-percent area of Iba1 and GFAP immunoreactivity, which are cell-specific markers of microglia and astrocytes, respectively, were significantly different between BXD strains (one-way ANOVA of Iba1 fluorescence intensity, p < 0.0001; Iba1 %area, p = 0.0002; GFAP fluorescence intensity, p = 0.008; GFAP %area p = 0.0002; scale bar = 100 μm).
(B) Pearson correlation coefficient tests showed that tau propagation ratio in the hippocampus was not associated with Iba1 or GFAP percent-area. Data are presented in column bar graphs, where error bars represent the mean ± SEM.
Discussion
During AD, insoluble tau aggregates arise focally in the medial temporal lobe and progress along a hierarchical trajectory toward connected brain areas. Given that the severity and scope of tau spread is highly correlated with neuronal loss and cognitive deficits, it is likely that the rate of tau propagation impacts the rate of clinical progression in patients with AD.26,27,28 Genetic loci associated with increased risk of sporadic AD also modify tau pathology. The presence of the APOEε4 allele, which is the most significant genetic risk factor for non-autosomal dominant AD, accelerates tau-mediated neurodegeneration and cognitive decline.29,30 Other disease-related variants, including BIN1, CLU, and TREM2, have also been shown to exacerbate tau deposition.31,32,33,34,35 While case-control GWAS can define sequence polymorphisms associated with the presence or absence of a disease, they are limited in their ability to resolve genetic contributions to specific disease traits that exhibit broad heterogeneity within a patient population. We used a novel experimental tool to precisely track neuron-to-neuron tau transfer combined with an extensive panel of genetically diverse mouse strains to determine whether trait heritability facilitates or restrains the advancement of tau pathology. Over 43% of the variability in tau propagation between strains was attributable to mouse genetic background. While this study was underpowered for QTL mapping, we were able to replicate two human GWAS loci associated with tau transmission in this Ntg-BXD model. These results indicate that mechanisms underlying tau spread in humans may be similarly regulated by genetic determinants and warrant further investigation.
Recent shortfalls in AD clinical trials have prompted a re-evaluation of the true translational potential of murine models as they are currently applied.36,37 The results of this study and others underscore the importance of considering animal genetic background when modeling complex neurological conditions using laboratory rodents.16 As an example, moving the transgenes of two mouse models of tauopathy (rTg4510 and TPR50 mice) onto the common C57BL/6J background has been shown to modify tau accumulation.38,39,40 Of note, the majority of AD transgenic mice are bred on a C57BL/6J genetic background, which is known to be relatively resilient to amyloid deposition.16 The incomplete development of disease phenotypes in murine models may be due to an inherent resistance to human AD neuropathologies; this phenomenon could also provide insight into the rare human condition of resilience to AD.41
In addition to strain-dependent differences in Aβ- and tau neuropathologies, it is becoming clear that mouse strains may exhibit distinct neuroinflammatory responses to brain insult. In the present study, glial percent area and cell marker expression were strongly impacted by heritable factors. Our findings indicate that distinct molecular and genetic networks are involved in cell-type-specific disease processes. Indeed, using the same BXD mouse genetic reference panel, we have identified cell-type-specific mechanisms of resilience and susceptibility to AD pathology.18 As a result, different mouse strains may be more or less resistant to neuronal versus glial features of disease. While we cannot dissect-out the influence of individual genetic loci, it is likely that those genetic factors that contribute to glial phenotypes are at least in part distinct from those that contribute to tau propagation. Several AD risk polymorphisms are located within or near genetic loci of receptors that are exclusively or highly expressed in brain-resident macrophages and astroglia, including APOE, TREM2, CD33, and CR1. Convincing evidence suggests that these disease variants promote maladaptive glial responses to AD pathological changes. Identifying heritable factors that are detrimental or beneficial in directing tau propagation and glial functions may help elucidate multiple molecular networks that are amenable to personalized therapeutic targeting.
Limitations of the study
Using mice as a model system to study human genetic contributions to disease susceptibility is of course limited by the extent of overlap between the mouse and human genomes, and their effects on downstream biology. While variants at the GMNC locus are associated with tau biomarkers in humans, murine ortholog alleles may have distinct effects on protein translation or function. However, the major advantage of using mice to study AD phenotypes is the ability to measure dynamic cellular and molecular events in a physiological milieu that cannot be studied in living patients. Using diverse mouse strains to study these discrete pathological mechanisms may help resolve genetic modifiers of specific disease processes that would otherwise not be detected in classic case-control GWAS.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-GFP | Aves Labs | Cat# GFP-1020 |
| Mouse anti-tau15-25 (Tau13) | BioLegend | Cat# MMS-520R |
| Rabbit anti-Iba1 | Fujifilm Wako Chemicals USA Corporation | Cat# 012–19741 |
| Cy3-conjugated mouse anti-GFAP | Millipore Sigma | Cat# C9205 |
| Alexa Fluor 488-conjugated goat anti-chicken | Invitrogen | Cat# A-11039 |
| Alexa Fluor 594-conjugated goat anti-mouse | Invitrogen | Cat# A-11005 |
| Alexa Fluor 568-conjugated goat anti-rabbit | Invitrogen | Car# A-11011 |
| Bacterial and virus strains | ||
| AAV2/8 GFP-2a-P301LhTau | Wegmann et al.17 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse B6xBXD75 | The Jackson Laboratory | Stock# 033260 |
| Mouse B6xBXD12 | The Jackson Laboratory | Stock# 035618 |
| Mouse B6xBXD42 | The Jackson Laboratory | Stock# 033248 |
| Mouse B6xBXD169 | The Jackson Laboratory | Stock# 035618 |
| Mouse B6xBXD50 | The Jackson Laboratory | Stock# 035630 |
| Mouse B6xBXD62 | The Jackson Laboratory | Stock# 033255 |
| Mouse B6xBXD74 | The Jackson Laboratory | Stock# 035642 |
| Mouse B6xBXD68 | The Jackson Laboratory | Stock# 035638 |
| Mouse B6xBXD154 | The Jackson Laboratory | Stock# 035655 |
| Mouse B6xBXD73 | The Jackson Laboratory | Stock# 035641 |
| Mouse B6xBXD14 | The Jackson Laboratory | Stock# 032332 |
| Mouse B6xBXD31 | The Jackson Laboratory | Stock# 035622 |
| Mouse B6xBXD152 | The Jackson Laboratory | Stock# 035654 |
| Mouse B6xBXD77 | The Jackson Laboratory | Stock# 033261 |
| Mouse B6xBXD55 | The Jackson Laboratory | Stock# 035633 |
| Mouse B6xBXD113 | The Jackson Laboratory | Stock# 035651 |
| Mouse B6 | The Jackson Laboratory | Stock# 032883 |
| Mouse B6xBD2Gpnmb | The Jackson Laboratory | Stock# 032881 |
| Software and algorithms | ||
| CellSens Software | Olympus | https://www.olympus-lifescience.com/en/software/cellsens/ |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead contacts, Catherine C. Kaczorowski (catherine.kaczorowski@jax.org) and Bradley T. Hyman (bhyman@mgh.harvard.edu).
Materials availability
This study did not generate new unique reagents.
The plasmid encoding reported 2A tau constructs can be provided by Bradley T. Hyman pending scientific review and a completed material transfer agreement.
All mouse lines are available for purchase from The Jackson Laboratory.
Experimental model and subject details
Animals
Mice were housed under 12-hour light/dark cycle with food and water ad libitum. F1 Ntg-BXD mice were generated as previously described.16,17,42 Briefly, female hemizygote 5XFAD mice on a congenic C57BL/6J background were bred with males from the BXD mouse genetic reference panel that originated from C57BL/6J and DBA/2J founder strains. Half of the F1 offspring were transgenic for 5 familial Alzheimer’s disease (FAD) mutations, and the other half did not inherit the 5XFAD transgenes: tau propagation experiments were conducted using nontransgenic controls from 18 strains. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at The Jackson Laboratory in accordance with the standards of the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Method details
AAV vector design and production
Cloning of GFP-2a-P301LhTau under the control of the neuron-specific synapsin-1 promoter was carried out as previously described.19 Plasmid DNA was assessed for inverted terminal repeat integrity by Sma1 restriction enzyme digestion and packaged into an adeno-associated virus 2/8 (AAV2/8) capsid at a titre of 0.6 × 1013 viral particles/mL (Massachusetts Eye and Ear Institute Vector Core). The final AAV stock was aliquoted and preserved at −80°C until use.
Stereotactic injections
AAVs expressing GFP-2a-P301LhTau were bilaterally injected into the entorhinal cortex of 3-month-old BXD mice. Animals were anesthetized using 3% isoflurane and maintained at 2% isoflurane for the duration of the surgery. A 10 μL Hamilton syringe and 30-gauge beveled needle coupled to an injector pump were used to inject 2 μL of AAV vector solution (1.2 × 1010 viral particles) at the following stereotactic coordinates: right side, 18-degree angle; AP -4.7 mm; ML -4.5 mm; DV -2.0 mm from the surface of the brain; left side, AP -4.7 mm; ML +3.2 mm; DV -4.0 mm from the surface of the brain. The needle was held in place for 2min to allow for sufficient diffusion of the injected material. After slowly withdrawing the needle, the skin was sutured, and the animal was allowed to recover from anesthesia for at least 1h on a 37°C warming pad before being returned to their home cage. 0.05 mg/kg buprenorphine was administered immediately after surgery, and each cage was supplied with 3 days of normal drinking water mixed with acetaminophen. All surgeries were performed at The Jackson Laboratory.
Tissue preparation and fluorescent immunolabeling
Two months after surgery, mice were transcardially perfused with ice-cold phosphate buffered saline (PBS) at a flow rate of 5 mL/min for 5min, followed by 4% paraformaldehyde (PFA) in PBS. Whole brains were isolated and post-fixed in 4% PFA for 48h before being transferred to a large volume of PBS. Tissue was shipped at room temperature to NeuroScience Associates Inc. (USA) and embedded in a non-infiltrating gelatin matrix before sectioning on a sliding microtome. 35 μm-thick free-floating coronal tissue sections were shipped back to Massachusetts General Hospital in cryoprotectant and stored at −20°C.
Sections were washed of cryoprotectant using large volumes of PBS, and permeabilized using 0.2% TritonX-100 in Tris buffered saline (TBS) for 20 min at room temperature. Tissue was blocked using 5% normal goat serum (NGS) in PBS for 1h at room temperature before incubating with the following primary antibody solutions overnight at 4°C: chicken anti-eGFP (GFP-1020, 1:1000; Aves Labs, USA); mouse anti-human tau (Tau13, MMS-520R, 1:1000; BioLegend, USA); rabbit anti-Iba1 (019–19741, 1:500; Fujifilm Wako Chemicals USA Corporation, USA); Cy3-conjugated mouse anti-GFAP (C9205, 1:500; Millipore Sigma, Germany). Sections were washed 3 times with PBS and incubated with fluorophore-conjugated secondary antibodies for 1h at room temperature (1:1000; Thermo Fisher Scientific, USA). Tissue was treated with 4′6-diamidino-2-phenylindole (DAPI), mounted onto charged glass microscope slides, and cover-slipped with Fluoromount mount media. Fluorescent imaging was performed using an Olympus VS120 slide scanner with fixed image acquisition settings across all images. The mean intensity of each image was automatically measured and used for subsequent statistical analyses.
Stereological cell counts
The number of GFP- and hTau-immunopositive cells in the entorhinal cortex, hippocampus, and isocortex was counted using CellSens Software (Olympus, Japan). Neurons transduced with AAV-GFP-2a-P301LhTau expressed both GFP and hTau and were considered potential donor cells for inter-neuronal tau transfer. GFP−/hTau+ neurons were considered recipient cells of propagated tau. Both values were used to calculate the tau propagation ratio, which represents the extent of cell-to-cell tau transfer corrected for variability in AAV transduction and/or hTau expression.
Heritability calculation
Heritability of tau propagation ratio was calculated as a ratio of genetic variance to total variance (genetic + environmental variance) normalized by the number of biological replicates per strain. Heritability scores range between 0 and 1.0, with a score of 1.0 indicating that 100% of the variance observed in a given trait is controlled by genetic factors.43
Quantitative trait locus (QTL) mapping
Genotypes for BXD strains were obtained from GeneNetwork.org. QTL mapping was carried our using R package, qtl2, using the LOCO method for kinship correction.44 Permutation tests (1000) were applied to determine statistical significance.
Quantification and statistical analysis
Comparisons were performed between genetic strains within each brain region analyzed. One-way ANOVA was performed for parametric analyses. Mann-Whitney and Kruskal Wallis nonparametric tests were performed when normal distribution could not be assumed, as determined by the Shapiro-Wilk normality test. Outliers were identified using ROUT and Grubb’s methods for parametric and non-parametric groups respectively and removed from all subsequent datasets. Data are presented in column bar graphs, where error bars represent the mean ± SEM. To assess the relationship between tau propagation and glial changes, Pearson correlation coefficients (r) were measured. The sample size, n, for each experiment represents the number of brain hemispheres transduced with AAV-GFP-2a-P301LhTau. Each data point (n = 4–8 per group) represents the average value of all technical replicates for each hemisphere. In systems genetics analyses using recombinant inbred strains, each strain is not genetically independent: replication occurs at the level of the allele, and each strain shares approximately 50% of its variant alleles with any other strain. Increasing the number of strains, rather than the number of individuals per strain, increases the statistical power of our analyses. Using BXD-mice, we determined that 2–4 replicates per strain was sufficient for our analyses.15,43,45 The final sample size is represented in all figures. Statistical significance was set at p < 0.05 for all tests (∗p ≤ 0.05; ∗∗p ≤ 0.01).
Acknowledgments
This research is supported by the National Institutes of Health (R01AG057914, R01AG054180, and RF1AG063755 to C.C.K.; 1RF1AG058674-01 to B.T.H.); the BrightFocus Foundation (A2016397S to C.C.K.); the Alzheimer's Association (ZEN-21-846037 to C.C.K.); and the JCB Foundation. L.A.W. is the recipient of Postdoctoral Fellowships from the Canadian Institutes of Health Research and the Fonds de Recherche du Québec - Santé.
Author contributions
Conceptualization, S.D. and B.T.H.; Methodology, L.A.W., S.D., A.R.D., C.C.K., and B.T.H.; Formal analysis, L.A.W., S.D., A.R.D., and L.B.C.; Investigation, L.A.W., S.D., A.R.D., A.R.F., and A.K.; Writing – Original Draft, L.A.W.; Writing – Review & Editing, L.A.W., S.D., A.R.D., A.R.F., A.K., L.B.C., C.C.K., and B.T.H.; Supervision, C.C.K. and B.T.H.
Declaration of interests
B.T.H. has a family member who works at Novartis and owns stock in Novartis. He serves on the SAB of Dewpoint and owns stock, and he serves on a scientific advisory board or is a consultant for AbbVie, Arvinas, Avrobio, Axon, Biogen, BMS Cell Signaling, Eisai, Genentech, Ionis, Novartis, Sangamo, Takeda, the US Dept of Justice, Vigil, Voyager. His laboratory is supported by Sponsored research agreements with Abbvie, F Prime, and research grants from the National Institutes of Health, Cure Alzheimer’s Fund, Tau Consortium, and the JPB Foundation, and a sponsored research agreement from Abbvie. S.D. is a current employee of Sanofi. A.R.F. is currently affiliated with the Mayo Clinic Graduate School of Biomedical Sciences.
Published: February 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.105983.
Contributor Information
Catherine C. Kaczorowski, Email: catherine.kaczorowski@jax.org.
Bradley T. Hyman, Email: bhyman@mgh.harvard.edu.
Supplemental information
Data and code availability
-
•
Data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This study does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
References
- 1.Cannon-Albright L.A., Foster N.L., Schliep K., Farnham J.M., Teerlink C.C., Kaddas H., Tschanz J., Corcoran C., Kauwe J.S.K. Relative risk for Alzheimer disease based on complete family history. Neurology. 2019;92:e1745–e1753. doi: 10.1212/WNL.0000000000007231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellenguez C., Küçükali F., Jansen I.E., Kleineidam L., Moreno-Grau S., Amin N., Naj A.C., Campos-Martin R., Grenier-Boley B., Andrade V., et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat. Genet. 2022;54:412–436. doi: 10.1038/s41588-022-01024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arriagada P.V., Growdon J.H., Hedley-Whyte E.T., Hyman B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 4.Gómez-Isla T., Hollister R., West H., Mui S., Growdon J.H., Petersen R.C., Parisi J.E., Hyman B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 5.Konijnenberg E., Tomassen J., den Braber A., Ten Kate M., Yaqub M., Mulder S.D., Nivard M.G., Vanderstichele H., Lammertsma A.A., Teunissen C.E., et al. Onset of preclinical alzheimer disease in monozygotic twins. Ann. Neurol. 2021;89:987–1000. doi: 10.1002/ana.26048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coomans E.M., Tomassen J., Ossenkoppele R., Golla S.S.V., den Hollander M., Collij L.E., Weltings E., van der Landen S.M., Wolters E.E., Windhorst A.D., et al. Genetically identical twins show comparable tau PET load and spatial distribution. Brain. 2022;145:3571–3581. doi: 10.1093/brain/awac004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruchaga C., Kauwe J.S.K., Harari O., Jin S.C., Cai Y., Karch C.M., Benitez B.A., Jeng A.T., Skorupa T., Carrell D., et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron. 2013;78:256–268. doi: 10.1016/j.neuron.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deming Y., Li Z., Kapoor M., Harari O., Del-Aguila J.L., Black K., Carrell D., Cai Y., Fernandez M.V., Budde J., et al. Genome-wide association study identifies four novel loci associated with Alzheimer's endophenotypes and disease modifiers. Acta Neuropathol. 2017;133:839–856. doi: 10.1007/s00401-017-1685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramanan V.K., Wang X., Przybelski S.A., Raghavan S., Heckman M.G., Batzler A., Kosel M.L., Hohman T.J., Knopman D.S., Graff-Radford J., et al. Variants in PPP2R2B and IGF2BP3 are associated with higher tau deposition. Brain Commun. 2020;2:fcaa159. doi: 10.1093/braincomms/fcaa159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jasen I.E., van der Lee S.J., Gomez-Fonseca D., de Rojas I., Dalmasso M.C., Grenier-Boley B., Zettergren A., Mishra A., Ali M., Andrade V., et al. Genome-wide meta-analysis for Alzheimer’s disease cerebrospinal fluid biomarkers. bioRxiv. 2022 doi: 10.1101/2022.03.08.22271043. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montal V., Diez I., Kim C.M., Orwig W., Bueichekú E., Gutiérrez-Zúñiga R., Bejanin A., Pegueroles J., Dols-Icardo O., Vannini P., et al. Network Tau spreading is vulnerable to the expression gradients of APOE and glutamatergic-related genes. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abn7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stozicka Z., Zilka N., Novak P., Kovacech B., Bugos O., Novak M. Genetic background modifies neurodegeneration and neuroinflammation driven by misfolded human tau protein in rat model of tauopathy: implication for immunomodulatory approach to Alzheimer's disease. J. Neuroinflammation. 2010;7:64. doi: 10.1186/1742-2094-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smolek T., Cubinkova V., Brezovakova V., Valachova B., Szalay P., Zilka N., Jadhav S. Genetic background influences the propagation of tau pathology in transgenic rodent models of tauopathy. Front. Aging Neurosci. 2019;11:343. doi: 10.3389/fnagi.2019.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dujardin S., Fernandes A., Bannon R., Commins C., De Los Santos M., Kamath T.V., Hayashi M., Hyman B.T. Tau propagation is dependent on the genetic background of mouse strains. Brain Commun. 2022;4:fcac048. doi: 10.1093/braincomms/fcac048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashbrook D.G., Arends D., Prins P., Mulligan M.K., Roy S., Williams E.G., Lutz C.M., Valenzuela A., Bohl C.J., Ingels J.F., et al. A platform for experimental precision medicine: the extended BXD mouse family. Cell Syst. 2021;12:235–247.e9. doi: 10.1016/j.cels.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neuner S.M., Heuer S.E., Huentelman M.J., O'Connell K.M.S., Kaczorowski C.C. Harnessing genetic complexity to enhance translatability of alzheimer's disease mouse models: a path toward precision medicine. Neuron. 2019;101:399–411.e5. doi: 10.1016/j.neuron.2018.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heuer S.E., Neuner S.M., Hadad N., O'Connell K.M.S., Williams R.W., Philip V.M., Gaiteri C., Kaczorowski C.C. Identifying the molecular systems that influence cognitive resilience to Alzheimer's disease in genetically diverse mice. Learn. Mem. 2020;27:355–371. doi: 10.1101/lm.051839.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Article on a preprint server or other repository. Telpoukhovskaia M.A., Hadad N., Gurdon B., Dai M., Ouellette A.R., Neuner S.M., Dunn A.R., Hansen S., Wu Y., Dumitrescu L., et al. Conserved cell-type specific signature of resilience to Alzheimer’s disease nominates role for excitatory cortical neurons. bioRxiv. 2022 doi: 10.1101/2022.04.12.487877. Preprint at. [DOI] [Google Scholar]
- 19.Wegmann S., Bennett R.E., Amaral A.S., Hyman B.T. Studying tau protein propagation and pathology in the mouse brain using adeno-associated viruses. Methods Cell Biol. 2017;141:307–322. doi: 10.1016/bs.mcb.2017.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Wegmann S., Bennett R.E., Delorme L., Robbins A.B., Hu M., McKenzie D., Kirk M.J., Schiantarelli J., Tunio N., Amaral A.C., et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aaw6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rauch J.N., Luna G., Guzman E., Audouard M., Challis C., Sibih Y.E., Leshuk C., Hernandez I., Wegmann S., Hyman B.T., et al. LRP1 is a master regulator of tau uptake and spread. Nature. 2020;580:381–385. doi: 10.1038/s41586-020-2156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caballero B., Bourdenx M., Luengo E., Diaz A., Sohn P.D., Chen X., Wang C., Juste Y.R., Wegmann S., Patel B., et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 2021;12:2238. doi: 10.1038/s41467-021-22501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asai H., Ikezu S., Tsunoda S., Medalla M., Luebke J., Haydar T., Wolozin B., Butovsky O., Kügler S., Ikezu T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015;18:1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopp S.C., Lin Y., Oakley D., Roe A.D., DeVos S.L., Hanlon D., Hyman B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J. Neuroinflammation. 2018;15:269. doi: 10.1186/s12974-018-1309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brelstaff J.H., Mason M., Katsinelos T., McEwan W.A., Ghetti B., Tolkovsky A.M., Spillantini M.G. Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abg4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sepulveda-Falla D., Chavez-Gutierrez L., Portelius E., Vélez J.I., Dujardin S., Barrera-Ocampo A., Dinkel F., Hagel C., Puig B., Mastronardi C., et al. A multifactorial model of pathology for age of onset heterogeneity in familial Alzheimer's disease. Acta Neuropathol. 2021;141:217–233. doi: 10.1007/s00401-020-02249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dujardin S., Commins C., Lathuiliere A., Beerepoot P., Fernandes A.R., Kamath T.V., De Los Santos M.B., Klickstein N., Corjuc D.L., Corjuc B.T., et al. Author Correction: tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat. Med. 2021;27:356. doi: 10.1038/s41591-021-01251-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim C., Haldiman T., Kang S.G., Hromadkova L., Han Z.Z., Chen W., Lissemore F., Lerner A., de Silva R., Cohen M.L., et al. Distinct populations of highly potent TAU seed conformers in rapidly progressing Alzheimer's disease. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abg0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi Y., Yamada K., Liddelow S.A., Smith S.T., Zhao L., Luo W., Tsai R.M., Spina S., Grinberg L.T., Rojas J.C., et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–527. doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian J., Betensky R.A., Hyman B.T., Serrano-Pozo A. Association of APOE genotype with heterogeneity of cognitive decline rate in alzheimer disease. Neurology. 2021;96:e2414–e2428. doi: 10.1212/WNL.0000000000011883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang T., Tan L., Zhu X.C., Zhou J.S., Cao L., Tan M.S., Wang H.F., Chen Q., Zhang Y.D., Yu J.T. Silencing of TREM2 exacerbates tau pathology, neurodegenerative changes, and spatial learning deficits in P301S tau transgenic mice. Neurobiol. Aging. 2015;36:3176–3186. doi: 10.1016/j.neurobiolaging.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 32.Calafate S., Flavin W., Verstreken P., Moechars D. Loss of Bin1 promotes the propagation of tau pathology. Cell Rep. 2016;17:931–940. doi: 10.1016/j.celrep.2016.09.063. [DOI] [PubMed] [Google Scholar]
- 33.Bemiller S.M., McCray T.J., Allan K., Formica S.V., Xu G., Wilson G., Kokiko-Cochran O.N., Crish S.D., Lasagna-Reeves C.A., Ransohoff R.M., et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 2017;12:74. doi: 10.1186/s13024-017-0216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leyns C.E.G., Ulrich J.D., Finn M.B., Stewart F.R., Koscal L.J., Remolina Serrano J., Robinson G.O., Anderson E., Colonna M., Holtzman D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA. 2017;114:11524–11529. doi: 10.1073/pnas.1710311114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wojtas A.M., Carlomagno Y., Sens J.P., Kang S.S., Jensen T.D., Kurti A., Baker K.E., Berry T.J., Phillips V.R., Castanedes M.C., et al. Clusterin ameliorates tau pathology in vivo by inhibiting fibril formation. Acta Neuropathol. Commun. 2020;8:210. doi: 10.1186/s40478-020-01079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sierksma A., Escott-Price V., De Strooper B. Translating genetic risk of Alzheimer's disease into mechanistic insight and drug targets. Science. 2020;370:61–66. doi: 10.1126/science.abb8575. [DOI] [PubMed] [Google Scholar]
- 37.Oblak A.L., Lin P.B., Kotredes K.P., Pandey R.S., Garceau D., Williams H.M., Uyar A., O'Rourke R., O'Rourke S., Ingraham C., et al. Comprehensive evaluation of the 5XFAD mouse model for preclinical testing applications: a MODEL-AD study. Front. Aging Neurosci. 2021;13 doi: 10.3389/fnagi.2021.713726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey R.M., Howard J., Knight J., Sahara N., Dickson D.W., Lewis J. Effects of the C57BL/6 strain background on tauopathy progression in the rTg4510 mouse model. Mol. Neurodegener. 2014;9:8. doi: 10.1186/1750-1326-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swarup V., Hinz F.I., Rexach J.E., Noguchi K.I., Toyoshiba H., Oda A., Hirai K., Sarkar A., Seyfried N.T., Cheng C., et al. Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat. Med. 2019;25:152–164. doi: 10.1038/s41591-018-0223-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yanagisawa D., Hamezah H.S., Pahrudin Arrozi A., Tooyama I. Differential accumulation of tau pathology between reciprocal F1 hybrids of rTg4510 mice. Sci. Rep. 2021;11:9623. doi: 10.1038/s41598-021-89142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gómez-Isla T., Frosch M.P. Lesions without symptoms: understanding resilience to Alzheimer disease neuropathological changes. Nat. Rev. Neurol. 2022;18:323–332. doi: 10.1038/s41582-022-00642-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunn A.R., Hadad N., Neuner S.M., Zhang J.G., Philip V.M., Dumitrescu L., Hohman T.J., Herskowitz J.H., O'Connell K.M.S., Kaczorowski C.C. Identifying mechanisms of normal cognitive aging using a novel mouse genetic reference panel. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.562662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belknap J.K. Effect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behav. Genet. 1998;28:29–38. doi: 10.1023/a:1021404714631. [DOI] [PubMed] [Google Scholar]
- 44.Broman K.W., Gatti D.M., Simecek P., Furlotte N.A., Prins P., Sen Ś., Yandell B.S., Churchill G.A. R/qtl2: software for mapping quantitative trait loci with high-dimensional data and multiparent populations. Genetics. 2019;211:495–502. doi: 10.1534/genetics.118.301595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andreux P.A., Williams E.G., Koutnikova H., Houtkooper R.H., Champy M.F., Henry H., Schoonjans K., Williams R.W., Auwerx J. Systems genetics of metabolism: the use of the BXD murine reference panel for multiscalar integration of traits. Cell. 2012;150:1287–1299. doi: 10.1016/j.cell.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This study does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.