

Abstract
Proteomic analysis on the scale that captures population and biological heterogeneity over hundreds to thousands of samples requires rapid mass spectrometry methods, which maximize instrument utilization (IU) and proteome coverage while maintaining precise and reproducible quantification. To achieve this, a short liquid chromatography gradient paired to rapid mass spectrometry data acquisition can be used to reproducibly quantify a moderate set of analytes. High-throughput profiling at a limited depth is becoming an increasingly utilized strategy for tackling large sample sets but the time spent on loading the sample, flushing the column(s), and re-equilibrating the system reduces the ratio of meaningful data acquired to total operation time and IU. The dual-trap single-column configuration (DTSC) presented here maximizes IU in rapid analysis (15 min per sample) of blood and cell lysates by parallelizing trap column cleaning and sample loading and desalting with the analysis of the previous sample. We achieved 90% IU in low microflow (9.5 μL/min) analysis of blood while reproducibly quantifying 300–400 proteins and over 6000 precursor ions. The same IU was achieved for cell lysates and over 4000 proteins (3000 at CV below 20%) and 40,000 precursor ions were quantified at a rate of 15 min/sample. Thus, DTSC enables high-throughput epidemiological blood-based biomarker cohort studies and cell-based perturbation screening.
INTRODUCTION
Automation of sample preparation and the commercialization of remote sampling devices have overcome two bottlenecks in large-scale mass spectrometry-based proteomic studies. The once cumbersome sample preparation for bottom-up proteomic analysis in which the protein content is extracted and enzymatically digested into peptides1 can now reproducibly prepare hundreds of samples per day through automation.2 Remote sampling devices allow donors to autonomously and reproducibly extract and ship several microliters of blood, thus greatly accelerating collection of specimens from many cohorts across multiple time points. With these developments, the remaining bottleneck is the analysis itself in which the generated peptides are separated by liquid chromatography and quantified by mass spectrometry (LC-MS). While multiplexing through isobaric mass tag labeling significantly improves LC-MS throughput,3–8 limitations of this strategy across large sample sets9–11 have encouraged the use of rapid and individual sample analysis with data-independent acquisition (DIA).12–14
Large-scale epidemiological biomarker investigations enabled by remote sampling require high throughput. However, blood is a challenging matrix for biomarkers due to its dynamic proteome, which varies within an individual depending on the time of day15,16 and is impacted by the collection protocol17,18 and various other factors.19 Blood is also highly heterogeneous between individuals and analysis of thousands of samples is required to appropriately capture the biological diversity of populations.10,17,20 The steep dynamic range of the blood proteome is an added strain, where highly abundant species like hemoglobin, albumin, and immunoglobulins mask the presence of less abundant proteins. It is expected that the coverage of the blood proteome will be limited,21–23 thus there is an impetus to quantify several hundred protein species reproducibly across a vast number of samples. As a proof of concept, the presented platform quantified 318 proteins with 100% data completeness in remotely sampled blood from 87 different donors.
Another application that benefits from high throughput is cell-based perturbation screening. In these studies, individual cell cultures are subjected to various treatments. The effects of these perturbations are then examined by profiling the cell’s molecular species.24–26 Each added variable (e.g., dosages, combinations of treatments, grow conditions, etc.) multiplies the number of samples. With cell culture automation, the number of conditions that can be explored is only limited by the throughput of subsequent analysis. A pace of one 96-well plate/day or 15 min/sample can reasonably match sample generation. In cell lysate thousands of proteins are typically quantified and this platform quantified over 4000 proteins in the AC16 cell lysate at 15 min/sample throughput.
Two mass spectrometry innovations address the challenge of maximizing the number of quantified analytes within a short analysis time: an additional dimension of analyte separation through ion mobility (IMS) and rapid high-resolution mass analysis. IMS obviates the need for long chromatographic separation by providing an orthogonal second dimension that multiplicatively increases peak capacity and detectable analytes.27 Then, a fast mass analyzer performs the necessary selective fragmentations for identification and specific quantification.28
With these innovations, the role of LC becomes rapid and reproducible introduction of separated analytes into the MS via electrospray ionization. The ratio of the time during which analytes are detected to the total time of analysis (instrument utilization, IU) is a critical metric of throughput because it correlates to the number of quantified analytes. However, achieving a high IU during rapid analysis is challenging because the required steps during which useful data is not generated including injection of the sample, the dwell time during which the analytes traverse the analytical column, and the cleaning and equilibration steps cannot be accelerated with the same flexibility as the analytical gradient. For example, the recently commercialized EvoSep29 platform offers standard methods with throughputs of 100 and 200 samples per day (SPD). The 100 SPD (14.4 min/sample) method uses an 11.5 min gradient with 1.5 min of dwell time thus generating 10 min of data and achieving 70% IU. In the 200 (7.2 min/sample) SPD method, there is about 1 min of dwell time and 4.6 min of data is collected over 7.2 min at 64% IU. High flowrate can be used to mitigate dwell time; for example in analysis at 50 μL/min, an IU of 75% is achieved for 20 min sample run times with direct injection onto a 1 mm internal diameter analytical column, but the 5 min required to inject the sample, flush, and equilibrate the column limit IU.30
Parallelization allows substantially higher IU because the steps during which no data is generated can be performed simultaneously with the analysis of the previous sample. In some configurations, two alternating separations are performed with two analytical columns driven by two analytical pumps with and without corresponding trapping columns.31,32 Our platform uses dual trapping columns and a single analytical column (DTSC) and offers several advantages over these more complex setups. First, LCs equipped with a loading pump and a binary analytical pump (e.g., Ultimate 3000 nano-RSLC and Waters Acuity) are widely implemented making DTSC accessible to many researchers. The dual analytical column, also referred to as the double barrel strategy, may use a valve to select which column is connected to the electrospray source during analyte elution at high flowrates,33 but this post-column volume reduces performance at the nano- and low microliter/min flowrates, thus a custom electrospray source with two separate emitters is required.34 Furthermore, a second analytical column is an additional source of variability. By using a single analytical column, DTSC is compatible with standard single-emitter sources, a wide range of conventional liquid chromatography instruments, and low flowrates.
In DTSC, one trap is back-flushed with a highly organic buffer, equilibrated, and loaded with the subsequent sample, while, in parallel, a sample loaded on the other trap is separated on the analytical column and analyzed. With this parallelization, a maximum ratio of instrument time is devoted to the collection of useful data (Figure 1). The presented adaptation of DTSC analyzes blood from remote sampling devices and cell lysates at 15 min/sample while maintaining 90% IU and achieves optimal performance at 1000 ng of injected peptides making it suitable for limited samples.
Figure 1.
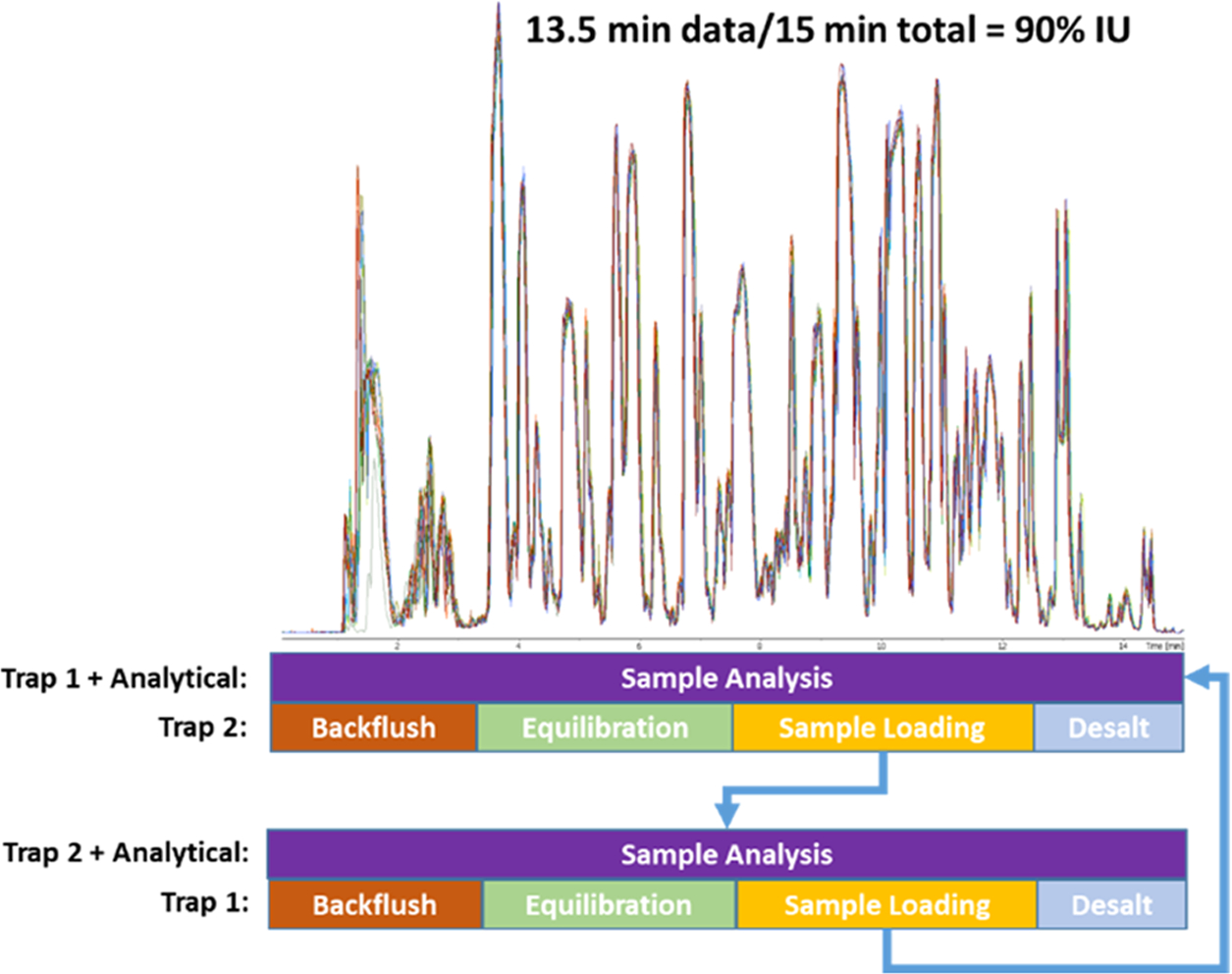
Parallelization of sample analysis on one trap, while the second trap is back-flushed, equilibrated, loaded with the next sample, and desalted. The top is an overlay of 10 injections (5 on each trap) demonstrating the reproducibility of the setup. The dwell time is approximately 1 min, while 30 s is used to flush the analytical column, thus generating 13.5 min of data during a 15 min run time resulting in 90% IU.
MATERIALS AND METHODS
LC Configuration.
The DTSC valve schematic is illustrated in Figure 2 and requires a 10-port 2-position valve, a second 2-position valve with at least 6 ports, an analytical pump, and a loading pump. In these experiments, an Ultimate 3000 nano-RSLC instrument (Thermo) was used, but any instrument with the aforementioned features can implement the DTSC configuration. Two complementary instrument methods operate this configuration. In the first method (Figure 2A,B), the analytical gradient is delivered through Trapping Column 1 and an Analytical Column, while the loading pump connected through the autosampler delivers a highly organic solvent to clean and back-flush (in the reverse direction of the analytical gradient) Trapping Column 2 (Figure 2A). The direction of loading pump flow is switched and the subsequent sample picked up by the autosampler is loaded onto Trapping Column 2 in the analytical direction (forward loaded, Figure 2B) and desalted for several minutes. In the next run, the second method reverses the roles of the traps (Figure 2C,D). The methods are alternated for the entire sample set. A blank is analyzed in the first run since no sample was loaded in the previous run and a buffer is injected in the final run. The loading pump is not intended for gradient delivery, and while it can be used to deliver a slug of organic buffer and equilibrate back to aqueous composition before sample loading at the 60 μL/min flowrate, it is not able to do so at the nanoliter/min flowrate, so instead the autosampler sample loop can be used to inject organic buffer during trap column cleaning.
Figure 2.
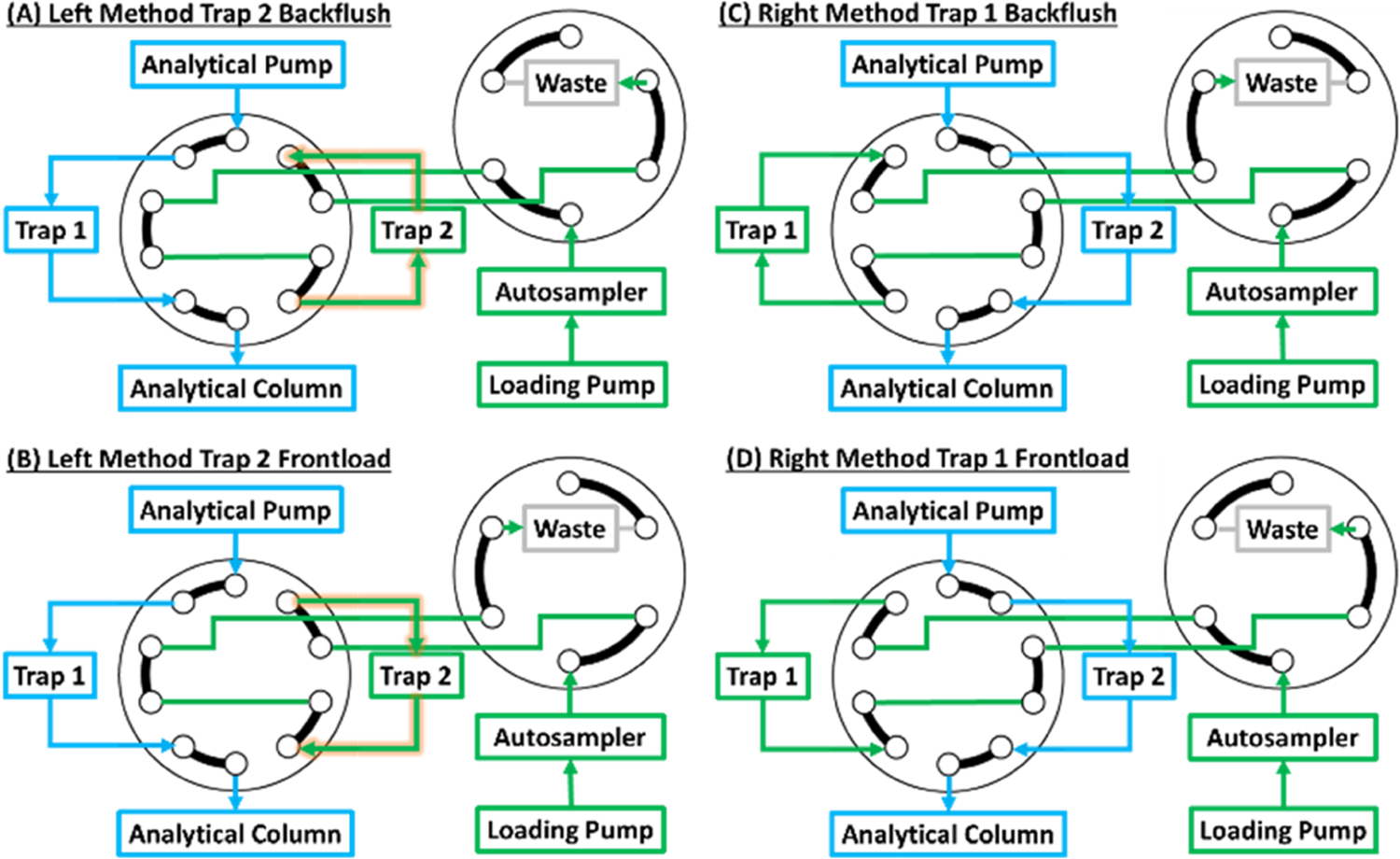
Valve schematic of the dual-trap one-column configuration. The 10-port valve dictates which trap is connected to the analytical pump and column, while the other trap is connected to the loading pump and autosampler. In the Left Method (A and B), the sample loaded on Trap 1 is analyzed, while Trap 2 is back-flushed with high organic buffer (A). After equilibration, the next sample is loaded in the same direction as the analytical separation (B) preventing precipitated contaminants from reaching and clogging the analytical column. In the next run (Right Method), the roles of the traps are reversed (C and D).
LC-MS Analysis.
All samples were analyzed on an Ultimate 3000 nano-RSLC (capillary flow selector) at the 9.5 μL/min analytical flowrate. Peptides were trapped on Phenomenex 0.3 mm internal diameter, 50 mm long columns packed with 5 μm Kinetex C8 beads, and separated on a CapLC 50 cm micropillar array column from Pharmafluidics (now Thermo). An eight-nozzle M3 emitter installed in a MnESI source from Newomics was used to split the flow just prior to electrospray.35,36 The entire assembly was connected with 50 μm internal diameter Viper capillaries (Thermo) and kept at 50 °C in the column oven.
The reversed-phase analytical gradient was delivered as follows: (mobile phase A: 0.1% formic acid in water, mobile phase B: 0.1% formic acid in acetonitrile): start at 7% B, linear ramp to 22% B over 8.5 min, linear ramp to 38% B in 4.3 min; jump to 98% B over 0.2 min, 0.9 min hold at 98% B, drop back to 7% B, and hold for 1 min (15 min total).
The loading pump was connected to three solvents: 0.1% formic acid in water (loading buffer A), 0.2% formic acid in 70% acetonitrile and 30% water with 5 mM ammonium formate (loading buffer B), and 0.2% formic acid in 90% isopropanol with 10% acetonitrile and 5 mM ammonium formate (loading buffer C). This set of buffers was used for convenient switching to a different application, but one organic buffer and one aqueous buffer are sufficient for DTSC implementation. At the start of the method, the loading pump delivered 50% B and 50% C at 60 μL/min for 0.5 min while reducing the flowrate to 55 μL/min. The solvent was switched to 100% A over 0.3 min and held for 8.2 min at 55 μL/min to equilibrate back to aqueous composition. The loading pump flowrate was reduced to 20 μL/min at 9.5 min and held for the remainder of the method for sample loading and desalting.
The autosampler was operated with a user program in which the first command started the run, next the autosampler 20 μL sample loop was filled with acetonitrile and injected to flush the trapping column. Two 25 μL rinses with water equilibrated the needle assembly. After a 3 min pause, 20 μL of the sample was collected into the loop and injected onto the trapping column; this coincided with the drop of the loading pump flowrate to 20 μL/min at 9.5 min run time. Finally, the syringe was emptied and the needle was washed. The autosampler program is presented in Table S1.
Data were acquired on a Bruker TIMS-TOF Pro using PASEF-DIA. Ions were accumulated for 70 ms, separated with 70 ms ramp, and fragmented in 40 m/z windows, which covered the 360 to 1120 m/z and 0.65 to 1.41 1/K0 ion mobility ranges corresponding to the majority of observed multicharged peptide ions. Each ramp cycle contained 1 to 3 DIA scans resulting in a 0.76 s total cycle time. The DIA isolation scheme is presented in Table S2. Electrospray ionization was performed using a Bruker MnESI source and 8-nozzle M3 emitter from Newomics with the capillary voltage set to 4800 V, the endplate offset voltage set to 500 V, the nebulizer set to 2.9 Bar, and the Dry Gas set to 6.0 L/min and 200 °C. Both MS and LC were controlled through Hystar 6.0 with the SII plug-in. The method files can be downloaded from LCMSMethods.com (dx.doi.org/10.17504/protocols.io.5qpvob27dl4o/v1).
Data Analysis.
Data were analyzed in DIA-NN 1.837 using the library-free search feature or with libraries generated by gas phase fractionation (GPF). To generate the GPF libraries, pooled samples were analyzed repeatedly using the aforementioned LC settings with data-dependent acquisition (DDA) to acquire fragmentation spectra for precursors within a restricted mass to charge range (e.g., 300–500 m/z) (Figure S1). Different precursor m/z ranges were analyzed in each run to span the 300–1100 range, and one full range experiment was carried out as the reference for retention time alignment during library assembly. The GPF data were analyzed in FragPipe 17.1.38 In the first search, the open search pipeline with strict tryptic specificity was used to identify the prevalent post-translational modifications (PTMs) and the detectable protein population. The second search was restricted to the identified proteins and most prevalent PTMs but with semitryptic protein specificity and up to 2 allowed missed cleavages. The peptides identified in the second search at <1% FDR were compiled into a spectral library with EasyPQP. A quick library was generated for DIA method optimization using dried blood spots with just nine DDA runs. A more thorough library was generated for remote sampling device blood by collecting each GPF range in duplicate for each of the four pooled cohorts (64 with an additional alignment reference run). Cell lysates were analyzed using the DIA-NN library-free feature in which the spectra, retention times, and ion mobilities were modeled by machine learning for tryptic peptides extracted from the human SwissProt protein database. In all analyses, the match between runs (MBRs) and double pass mode were enabled and MS1 and MS2 mass errors were set to 15 ppm.
For cohort comparison of remote sampling device blood, the DIA-NN generated protein intensities were log base 2-transformed and standardized across each sample so that the protein quantity distribution in each sample had a mean of zero and standard deviation of one. UMAP (Uniform Manifold Approximation and Projection for Dimension)39 was used to project the protein quantities onto two dimensions to allow for visualization of protein clustering by cohort. Proteins quantified in every sample (318 proteins) were compared between the control group and the three hypertension cohorts as a combined second group using t-tests with Benjamini–Hochberg correction. The proteins with significantly different means (adjusted p-values < 0.05) were used as inputs to train five classification models (Gradient Boosting Decision Trees [GB], Support Vector Classification [SVC], Random Forest [RF], Extra Trees [ET], and Logistic Regression [LR])40–44 using 70% of the data. The remaining 30% of the data was used as the final test set and the model with the highest precision recall area under the curve (PR AUC) was selected as the classifier for hypertension prediction. This data normalization, analysis, and model training were performed in Python version 3.7.11.
Dried Blood Spot (DBS) Sample Preparation.
Pooled, mixed-gender human blood sample (Golden West Biosolutions, LLC., Lot# PS1000.104) was spotted on Whatman cards. The discs with dried blood spots were transferred to 96-well plates (1.1 mL well volume, Thermo). The proteins were solubilized in 9 M Urea, 30 mM tris(2-carboxyethyl)phosphine (TCEP; Thermo Fisher Scientific), and 200 mM Trizma (Sigma-Aldrich) by shaking at 1200 rpm for 60 min at 37 °C. The remainder of the protocol was automated on a Biomek i7 workstation (Beckman Coulter). The samples were alkylated in 50 mM iodoacetamide (IAA) and then diluted with 200 mM Trizma to a final volume of 567 μL, and trypsin (Promega) was added to a 1:25 protease to substrate ratio. The plate was incubated at 42 °C for 4 h with slight shaking at 120 rpm and digestion was quenched by acidification to 1% formic acid. Digested samples were automatically desalted on an Oasis 30 μm HLB 96-well plate (Waters) following the manufacturer’s protocol adapted for the Biomek i7 with the positive pressure module.45
Mitra Device Dried Blood Sample Preparation.
Blood was obtained from 87 patients using Mitra devices (Neoteryx). The protein content was extracted by 60 min incubation at 60 °C in a buffer of 35% trifluoroethanol (TFE), 40 mM dithiothreitol (DTT; Sigma), and 50 mM ammonium bicarbonate (ABC; Sigma). The rest of the sample processing was performed automatically on the Biomek i7. Samples were alkylated with 10 mM IAA at 25 °C in the dark for 30 min. An addition of 5 mM DTT quenched alkylation and addition of 50 mM ABC diluted TFE to 5% final concentration. Samples were digested for 4 h at 43 °C with Trypsin (1:25 protease to enzyme, Promega). Digestion was quenched with addition of formic acid to 1% final concentration. Samples were diluted to 50 ng/μL concentration before LC-MS analysis.46
Cell Lysate Preparation.
The AC16 cell line was grown in-house in high-glucose Dulbecco’s modified Eagle’s medium: nutrient mixture F-12 (DMEM/F-12; Gibco), with 12.5% fetal bovine serum (FBS; Gibco) supplemented with antibiotic–antimycotic (100×; Gibco) at 37 °C (5% CO2). At 1.44 × 107 cells/mL concentration, the cells were washed, trypsinized, and pelleted at 180g. The pellet was washed twice and aliquots of 3.7 × 104 cells suspended in 20 μL of 100 mM ammonium bicarbonate (MilliporeSigma) buffer were loaded into individual wells on three 96-well AFA-Tube TPX plates (COVARIS). Cells were lysed by sonication for 5 min on the LE220-Plus focused-ultrasonicator (COVARIS) using 350 power peak intensity, 25% duty factor, and 200 cycles per burst. Each sample was reduced with 20 mM DTT for 10 min at 60 °C and alkylated with 40 mM IAA for 30 min at room temperature in the dark. The sample volume was adjusted to 150 μL and 10% acetonitrile. The samples were digested with trypsin at a ratio of 1:20 protease to substrate ratio for 2 h at 42 °C. The samples were acidified to 0.6% trifluoroacetic acid (TFA; Fisher Scientific) and dried under vacuum. Prior to analysis, the dried peptides were reconstituted in 80 μL of 0.1% formic acid to load at 50 ng/μL concentration.
RESULTS AND DISCUSSION
In DTSC, one trapping column is cleaned and loaded with the following sample during analysis of the sample loaded on the second trap. A typical autosampler injection requires 2.5 min and the system cleaning and equilibration is reduced by 0.5 min. The 3 min recovered per run amounts to a 20% increase in throughput at 15 min per sample. An important consideration for DTSC implementation is a delay due to instrument warm-up; the TIMS-TOF Pro required 30 s to prepare for the run. Under normal circumstances, we would compensate for this delay by shortening the run time by 30 s, but we elected to keep the final 30 s of data acquisition to ensure that no detectable peptides were eluting. Peptides were quantified for 13.5 min of the 15 min method thus achieving a 90% IU.
The DIA data acquisition scheme was optimized using pooled DBS, and the isolation windows were selected to cover the majority of detectable multicharged peptide ions. Data acquisition schemes with varying ramp times and isolation window widths were evaluated by assessing the peptide level quantitative reproducibility from 10 injections of 1000 ng of DBS (Figure S2). The highest number of identified precursors (3985 total, 3697 on average) and the lowest CV (23% median, 26% average) correlated with the highest average number of points across the elution peak, which DIA-NN reported as an average of 3.1 MS2 scans across the full-width half max (FWHM). The isolation windows were further optimized to increase the points across the peak, and the finalized method averaged 3.3–4 MS2 scans at FWHM.
A loading curve spanning 10–1500 ng of the DBS standard was generated to establish optimal loading quantity and evaluate quantitative variance at each level. Each quantity was injected 4 times, except for the 1000 and 1500 ng levels, which were injected 10 times to serve as the first-day reproducibility set. The middle 4 injections (injections 3–7) were used to represent the variance for the 1000 and 1500 ng loads. One of the 25 ng injections was not picked up by the autosampler and was excluded from analysis. The number of precursors and protein identifications and the %CV at each level are presented in Figure 3A,B. The most reproducible identifications were achieved at 1000 ng. Although the LC columns could accommodate up to 4 micrograms of peptides, such a high ion current overloads the trapped ion mobility cell and reduces performance as reflected at the 1500 ng load.
Figure 3.
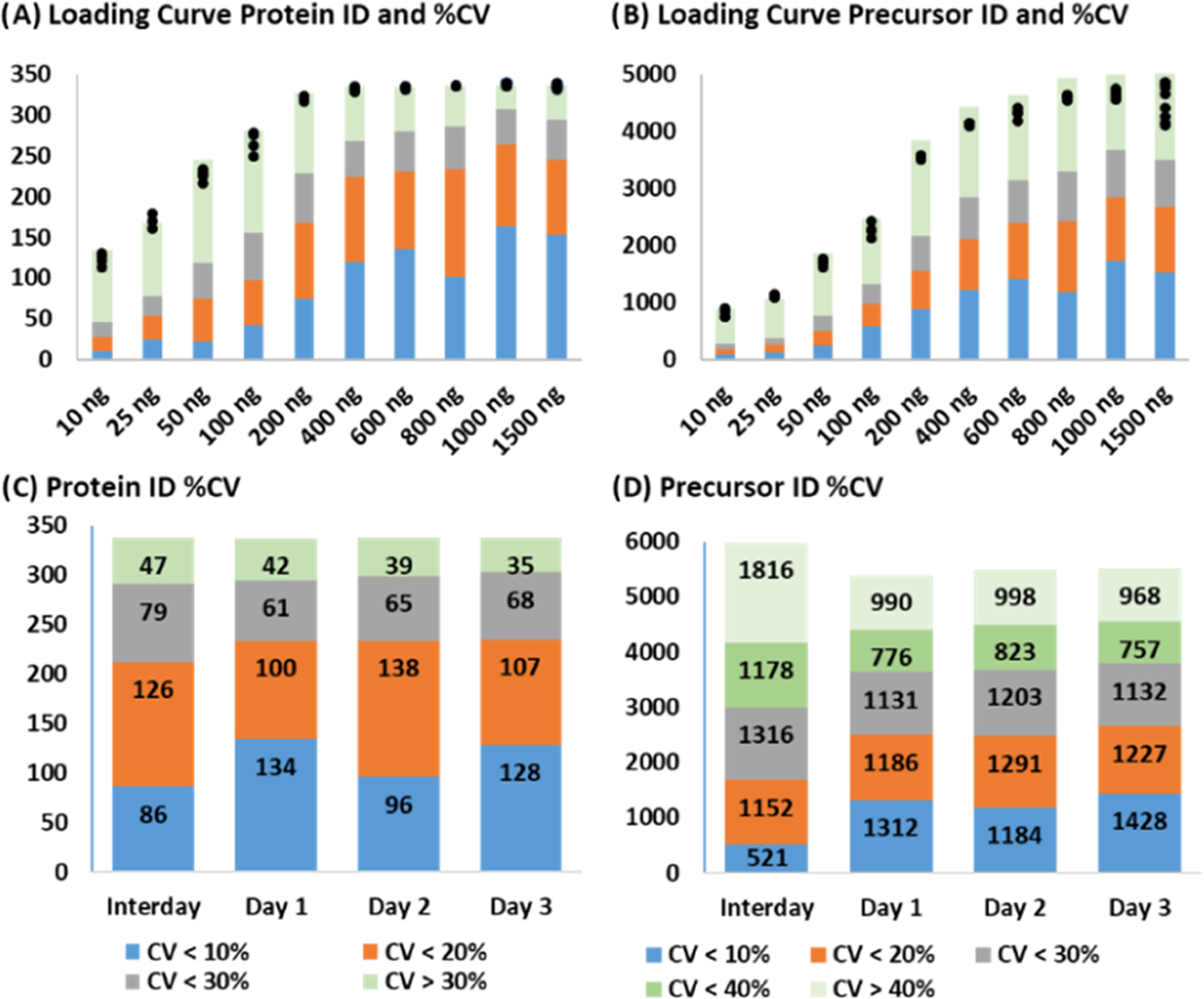
Loading curve and reproducibility. (A, B) Protein and precursor ion identifications, respectively, at each DBS sample load with identifications in individual replicates marked by black circles. The colors represent %CV criteria: blue < 10%; orange 10–20%; gray 20–30%; and green > 30%. (C, D) Protein and precursor ion reproducibility on 3 different days (10 replicate injections) and interday reproducibility. The same color code is repeated from Panels A and B, except in D: green 30–40% (green) and pale green > 40%.
The intra- and interday reproducibility was tested with ten 1000 ng injections on 2 additional days, and the %CV distribution is presented in Figure 3C,D. On Days 1 and 3, 134 and 128 proteins had a CV of less than 10%, but on the second day, which reduced the interday reproducibility, only 96 proteins were under this threshold. Despite this, 234 proteins were quantified below 20% CV on all 3 days and 212 met this threshold interday. If an interday variance of 30% CV is acceptable, then 291 proteins and 2999 precursors (or 4177 at 40% CV) are reproducibly quantifiable with this platform.
Next, we applied the platform to blood samples collected through remote sampling. The specimens were grouped by blood pressure (BP): Cohort 1 had normal BP (n = 37), Cohort 2 had BP between 120/80 and 130/90 (n = 29), Cohort 3 had a BP above 130/90 (n = 11), and Cohort 4 had high BP that was treated with medication (n = 10). On average, 366 proteins were quantified in each cohort with 318 proteins quantified in every sample and 362 proteins quantified in 80% of the samples (Figure 4A,B). At the precursor level, 2166 were quantified in every sample, while 4483 were quantified in 80% of the samples with a total of 5500 to 6000 precursors quantified in most samples (Figure 4). The reproducibility was excellent based on uncorrected log2-transformed protein quantity distributions across all 87 samples. After standardization, UMAP was used for dimension reduction to examine the unbiased relationship between the 4 cohorts (Figure 4C). Cohorts 3 and 4 formed separate clusters, while the control and lower hypertension cohorts were intermixed. To separate samples based on the hypertension phenotype, machine learning classification models were trained based on proteins that were quantified in every sample and significantly differentiated Cohort 1 from the combined hypertension cohorts. Data were split into 70% training and 30% test sets, and various models were optimized using random hyperparameter searches before assessing performance on the test set. Of all of the models, Extra Trees had the highest PR AUC score of 0.91 (Table S3), which was substantially better than the no-skill model (Figure 4D). Altogether, this analysis demonstrates that data generated from difficult samples at high throughput can be used to distinguish patient groups even when the underlying biology is subtle.
Figure 4.
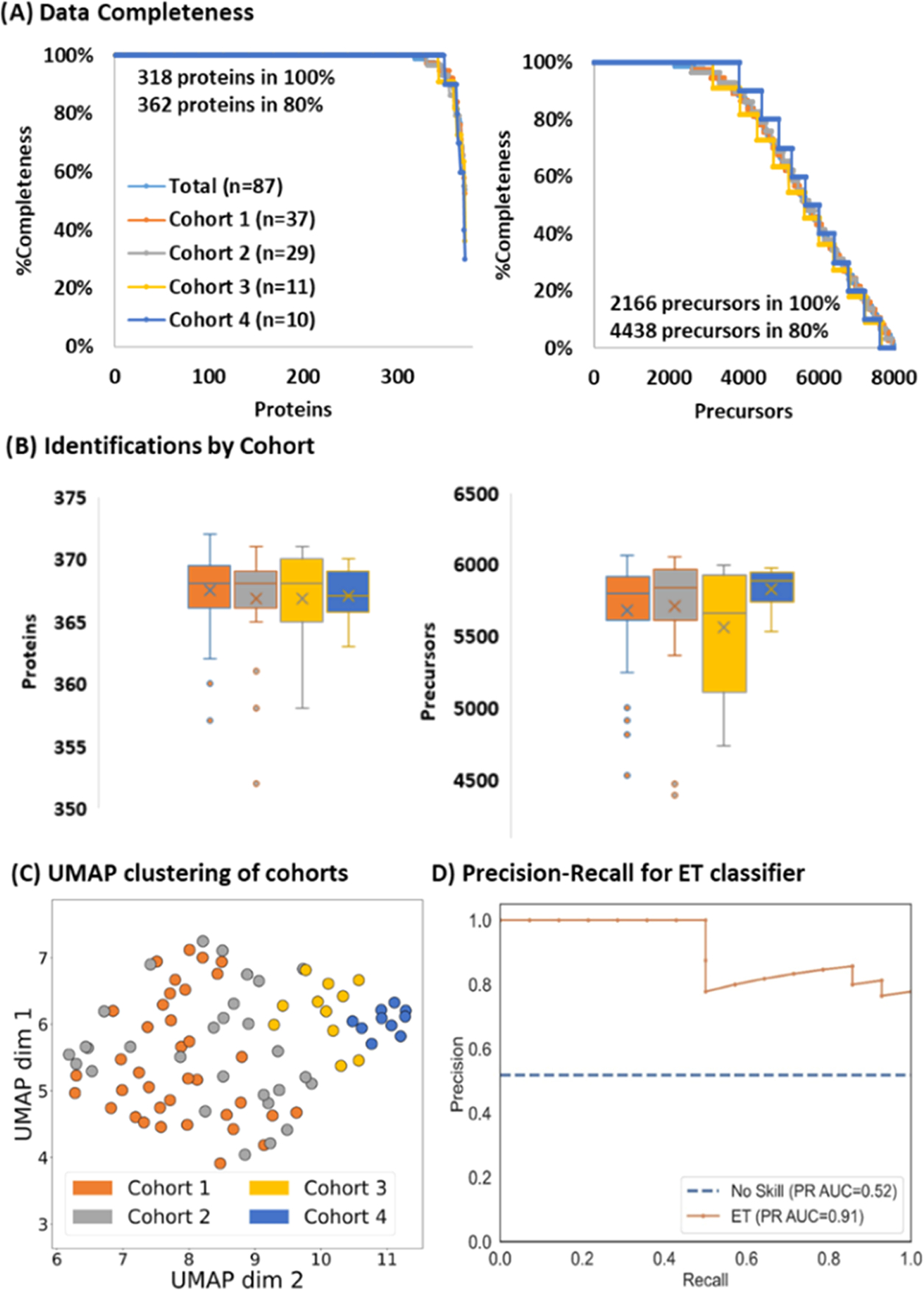
Hypertension Cohort Analysis (A) Data completeness curves for total (light blue), Cohort 1 (orange), Cohort 2 (gray), Cohort 3 (yellow), and Cohort 4 (dark blue). (B) Identification distributions at the protein (left) and precursor ion (right). (C) UMAP clustering of the different cohorts. (D) Precision recall curve of the best classifier (Extra Trees) trained to distinguish Cohort 1 (the control group) from the remaining hypertension cohorts achieves a PR AUC of 0.91.
The platform was next evaluated for rapid analysis of cell lysates in perturbation studies. Three identical 96-well plates containing untreated AC16 cell lysates were prepared. This experiment evaluated the depth of analysis, which can be accomplished in 15 min, and the combined reproducibility of the sample preparation protocol and analytical setup. The acquired data were analyzed against the human SwissProt database using the library-free search option of DIA-NN. Out of 288 samples, 9 injections failed because the autosampler did not pick up the sample, establishing a 3% re-injection rate that can be improved with a more robust autosampler. On average, 37.5 k precursor ions and 4534 proteins were identified across the 279 successful injections. The distribution of identifications by the plate is presented in Figure 5A. The dataset completeness and reproducibility both at the protein and precursor level are presented in Figure 5B; 13,165 precursors and 3515 proteins were identified in all 279 samples. If a rate of 5% missing values is acceptable, then these numbers increase to 22,621 and 3977 for precursors and proteins, respectively. For the core proteins and precursors (those detected in all successful injections), the average %CV was 11 and 19%, respectively.
Figure 5.
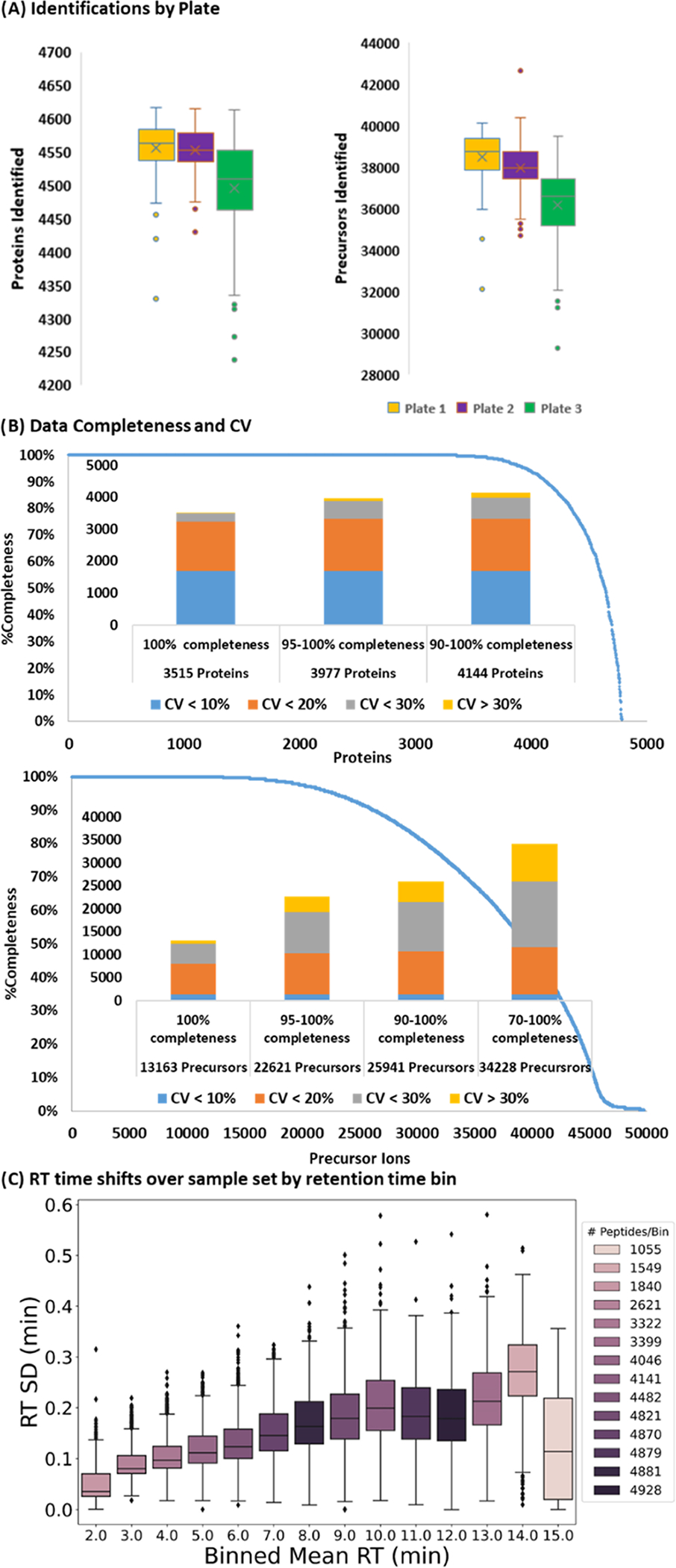
Cell lysate analysis (A) protein (left) and precursor ion (right) identification distribution on each plate. (B) Data completeness curve for proteins (top) and precursor ions (bottom) over all samples. The bar graphs represent the %CV distributions at different completeness levels: blue < 10 %; orange 10–20%; gray 20–30%; and yellow > 30%. (C) Standard deviation of peptide RT binned by average retention time.
We investigated the slightly reduced identifications in Plate 3 by plotting the number of precursor identifications for each sample in chronological order and across Plate 3 (Figure S3). The dip in identifications appears to be localized to columns 8 and 9 and returns to the total average in columns 11 and 12. The MS1 and MS2 intensities are consistent over the whole dataset (Figure S3), and the low deviation in peptide retention time, generally under 12 s, shows that the LC operated robustly across this cohort (Figure 4C). Most likely, this dip in performance is due to reduced sample quality in the aforementioned region of Plate 3.
The presented assembly robustly and rapidly analyzes challenging physiological samples like blood and achieves deep profiling in cell lysates. A key consideration for developing this platform is the ability to perform with minimal down time. Relatively large traps were selected in the expectation that they would not clog and will be able to filter out any contaminants that may compromise analysis. Additionally, identical performance is required between the two traps, and irregularities in manufacturing would be negligible in larger columns. We routinely perform trap synchronization by overlaying the base-peak chromatograms from several runs on each trap to ensure that the retention times and peak widths are identical on both traps, as shown in Figure 1. A failure in this alignment indicates that there is possibly a leak in one of the connections, some contamination is influencing trap retention, or the trap needs to be replaced. In one of the Figure 1 runs, the earliest eluting peptide peak intensity is reduced due to incomplete equilibration of the loading pump buffer back to aqueous, which reduced binding and increased the loss of hydrophilic peptides in the slightly organic buffer. These trapping columns reached a backpressure of ~300 bar when flushed at the 55 μL/min flowrate. Accepting a higher backpressure or using more permeable columns would allow a faster and more efficient organic flush and equilibration.
The analytical flowrate was set at 9.5 μL/min because the Ultimate 3000 operates at a maximum of 10 μL/min with the capillary flow module. The CapLC column was selected because it can robustly process 1–3 thousand samples, but it does not tolerate a pressure above 350 bar. At the analytical flowrate, the backpressure was 200–250 bar, so it is possible to increase the flowrate by several microliters/min and further reduce the dwell time and increase IU. The IU can also be increased by reducing the dwell volume with smaller trapping columns or a different analytical column, but selection of trapping columns that have identical performance and can operate robustly requires thorough testing. It is important to note that while the focus of this publication was tryptic peptides, the described configuration can be used to rapidly profile other sets of analytes such as metabolites, lipids, intact proteins, and RNA with the appropriate chromatographic columns and mobile phases. The manuscript data presented in this manuscript is accessible through the MASSIVE repository by FTP: MSV000089675 and password: dualtrap PXD034611; doi:10.25345/C5PV6BB5N.
CONCLUSIONS
The authors demonstrated that DTSC achieves 90% IU at 15 min per sample throughput in two key LC-MS applications: analysis of cell lysates in a perturbation study and analysis of clinical specimens collected using remote blood sampling devices. The authors envision implementing this platform synchronously with 24 h incubation of cells cultured on 96-well plates with different conditions applied across the plate. This will allow continuous perturbation studies as samples are incubated for 24 h, digested, and analyzed the following day while the subsequent plates are prepared in parallel. Their platform quantified over 4000 proteins in a human cardiomyocyte cell line (AC16) lysate, which corresponds to approximately 20% of the total genome in 15 min. The platform is versatile as demonstrated in the analysis of blood from remote sampling devices. In combination with remote sampling, this platform can be used to execute ambitious surveys of large cohorts at multiple (even unlimited) time points. At the cursory depth of 300–400 blood proteins, they were able to distinguish samples based on the blood pressure phenotype from just 15 min of instrument time using machine learning. Similar algorithms can be generated for other phenotypes and disease states and greater depth can be achieved through longer methods or fractionation. DTSC provides a significant boost in throughput without sacrificing the quality of the generated data thus empowering precise and reproducible quantitation across large sample sets and allowing proteomics to capture population and biological heterogeneity.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank their collaborators at Bruker for instrument support especially Michael Krawitzky, Francesco Pingitore, and Christopher Adams and their collaborators from Newomics for help in optimization of electrospray ionization settings especially Daojing Wang and Mark J. Schroeder. Funding: BIRCHW: NIH K12HD043441, R35: GM142502, 5R01HL155346-02.
Footnotes
Notes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.2c02609
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c02609.
Autosampler control program (Table S1); DIA isolation windows (Table S2); GPF library for remote sampled blood (Figure S1); PASEF-DIA scheme optimization (Figure S2); scores of classifiers for the hypertension phenotype (Table S3); performance across the cell lysate analysis (Figure S3) (PDF)
Contributor Information
Simion Kreimer, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States.
Ali Haghani, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States.
Aleksandra Binek, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States.
Alisse Hauspurg, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15213, United States.
Saeed Seyedmohammad, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States.
Alejandro Rivas, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States.
Amanda Momenzadeh, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States.
Jesse G. Meyer, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States
Koen Raedschelders, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States.
Jennifer E. Van Eyk, Cedars-Sinai Medical Center, Beverly Hills, California 90211, United States
REFERENCES
- (1).Rogers JC; Bomgarden RD Adv. Exp. Med. Biol. 2016, 919, 43–62. [DOI] [PubMed] [Google Scholar]
- (2).Fu Q; Murray CI; Karpov OA; Van Eyk JE Mass Spectrom. Rev. 2021, No. e21750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mertins P; Udeshi ND; Clauser KR; Mani DR; Patel J; Ong SE; Jaffe JD; Carr SA Mol. Cell. Proteomics 2012, 11, No. M111.014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).O’Connell JD; Paulo JA; O’Brien JJ; Gygi SP J. Proteome Res. 2018, 17, 1934–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sonnett M; Yeung E; Wuhr M Anal. Chem. 2018, 90, 5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Thompson A; Schafer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Anal. Chem. 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (7).Edwards A; Haas W Methods Mol. Biol. 2016, 1394, 1–13. [DOI] [PubMed] [Google Scholar]
- (8).Ruprecht B; Zecha J; Zolg DP; Kuster B Methods Mol. Biol. 2017, 1550, 83–98. [DOI] [PubMed] [Google Scholar]
- (9).Wenger CD; Lee MV; Hebert AS; McAlister GC; Phanstiel DH; Westphall MS; Coon JJ Nat. Methods 2011, 8, 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Anderson NL Clin. Chem. 2010, 56, 177–185. [DOI] [PubMed] [Google Scholar]
- (11).Karp NA; Huber W; Sadowski PG; Charles PD; Hester SV; Lilley KS Mol. Cell. Proteomics 2010, 9, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pappireddi N; Martin L; Wuhr M ChemBioChem 2019, 20, 1210–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bekker-Jensen DB; Bernhardt OM; Hogrebe A; Martinez-Val A; Verbeke L; Gandhi T; Kelstrup CD; Reiter L; Olsen JV Nat. Commun. 2020, 11, No. 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Meyer JG; Schilling B Expert Rev. Proteomics 2017, 14, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Anderson NL; Anderson NG Mol. Cell. Proteomics 2002, 1, 845–867. [DOI] [PubMed] [Google Scholar]
- (16).Rifai N; Gillette MA; Carr SA Nat. Biotechnol. 2006, 24, 971–983. [DOI] [PubMed] [Google Scholar]
- (17).Geyer PE; Holdt LM; Teupser D; Mann M Mol. Syst. Biol. 2017, 13, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lehmann S; Guadagni F; Moore H; et al. Biopreserv. Biobanking 2012, 10, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ignjatovic V; Geyer PE; Palaniappan KK; Chaaban JE; Omenn GS; Baker MS; Deutsch EW; Schwenk JM J. Proteome Res. 2019, 18, 4085–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Price ND; Magis AT; Earls JC; Glusman G; Levy R; Lausted C; McDonald DT; Kusebauch U; Moss CL; Zhou Y; Qin S; Moritz RL; Brogaard K; Omenn G; Lovejoy JC; Hood L Nat. Biotechnol. 2017, 35, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Huang L; Shao D; Wang Y; Cui X; Li Y; Chen Q; Cui J Briefings Bioinf. 2021, 22, 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tirumalai RS; Chan KC; Prieto DA; Issaq HJ; Conrads TP; Veenstra TD Mol. Cell. Proteomics 2003, 2, 1096–1103. [DOI] [PubMed] [Google Scholar]
- (23).Villanueva J; Shaffer DR; Philip J; Chaparro CA; Erdjument-Bromage H; Olshen AB; Fleisher M; Lilja H; Brogi E; Boyd J; Sanchez-Carbayo M; Holland EC; Cordon-Cardo C; Scher HI; Tempst PJ Clin. Invest. 2005, 116, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Caldera M; Muller F; Kaltenbrunner I; Licciardello MP; Lardeau CH; Kubicek S; Menche J Nat. Commun. 2019, 10, No. 5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Molinelli EJ; Korkut A; Wang W; Miller ML; Gauthier NP; Jing X; Kaushik P; He Q; Mills G; Solit DB; Pratilas CA; Weigt M; Braunstein A; Pagnani A; Zecchina R; Sander C PLoS Comput. Biol. 2013, 9, No. e1003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Keenan AB; Jenkins SL; Jagodnik KM; et al. Cell Sys. 2018, 6, 13–24. [Google Scholar]
- (27).Meier F; Park MA; Mann M Mol. Cell. Proteomics 2021, 20, No. 100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Meier F; Brunner AD; Frank M; Ha A; Bludau I; Voytik E; Kaspar-Schoenefeld S; Lubeck M; Raether O; Bache N; Aebersold R; Collins BC; Röst HL; Mann M Nat. Methods 2020, 17, 1229–1236. [DOI] [PubMed] [Google Scholar]
- (29).Bache N; Geyer PE; Bekker-Jensen DB; Hoerning O; Falkenby L; Treit PV; Doll S; Paron I; Muller JB; Meier F; Olsen JV; Vorm O; Mann M Mol. Cell. Proteomics 2018, 17, 2284–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bian Y; Zheng R; Bayer FP; Wong C; Chang YC; Meng C; Zolg DP; Reinecke M; Zecha J; Wiechmann S; Heinzlmeiir S; Scherr J; Hemmer B; Baynham M; Gingras A; Boychenko O; Kuster B Nat. Commun. 2020, 11, No. 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hosp F; Scheltema RA; Eberl HC; Kulak NA; Keilhauer EC; Mayr K; Mann M Mol. Cell. Proteomics 2015, 14, 2030–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Orton DJ; Wall MJ; Doucette AA J. Proteome Res. 2013, 12, 5963–5970. [DOI] [PubMed] [Google Scholar]
- (33).Yin L; Shi MY; Wang TT; Zhang MM; Zhao XJ; Zhang Y; Gu JK Chromatographia 2017, 80, 137–143. [Google Scholar]
- (34).Webber KGI; Truong T; Johnston SM; Zapata SE; Liang Y; Davis JM; Buttars AD; Smith FB; Jones HE; Mahoney AC; Carson RH; Nwosu AJ; Heninger J; Liyu AV; Nordin GP; Zhu Y; Kelly RT Anal. Chem. 2022, 94, 6017–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen Y; Mao P; Wang D Anal. Chem. 2018, 90, 10650–10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mao P; Gomez-Sjoberg R; Wang D Anal. Chem. 2013, 85, 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Demichev V; Messner CB; Vernardis SI; Lilley KS; Ralser M Nat. Methods 2020, 17, 41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yu F; Haynes SE; Nesvizhskii AI Mol. Cell. Proteomics 2021, 20, No. 100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mclnnes L; H ,J; Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. 2020, arXiv:1802.03426. arXiv.org e-Print archive. https://arxiv.org/abs/1802.03426. [Google Scholar]
- (40).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion BJ Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- (41).Buitinck L; Louppe G; Blondel M; Pedregosa F; Muller C; Grisel O; Niculae V; Prettenhofer P; Gramfort A; Grobler J; Layton R; Vanderplas J; Joly A; Holt B; Varoquaux G API Design for Machine Learning Software: Experiences from the Scikit-learn Project, 2013, arXiv:1309.0238. arXiv.org e-Print archive. https://arxiv.org/abs/1309.0238. [Google Scholar]
- (42).Breiman L Mach. Learn. 2001, 45, 5–32. [Google Scholar]
- (43).Geurts P; Ernst D; Wehenkel L Mach. Learn. 2006, 63, 3–42. [Google Scholar]
- (44).Yu H; Huang FL; Jen H Mach. Learn. 2011, 85, 41–75. [Google Scholar]
- (45).Fu Q; Johnson CW; Wijayawardena BK; Kowalski MP; Kheradmand M; Van Eyk JE J. Visualized Exp. 2020, e59842. [DOI] [PubMed] [Google Scholar]
- (46).Mc Ardle A; Binek A; Moradian A; Chazarin Orgel B; Rivas A; Washington KE; Phebus C; Manalo DM; Go J; Venkatraman V; Coutelin Johnson CW; Johnson CWC; Fu Q; Cheng S; Cheng S; Raedschelders K; Raedschelders K; Fert-Bober J; Fert-Bober J; Pennington SR; Pennington SR; Murray CI; Murray CI; Van Eyk JE Clin. Chem. 2022, 68, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.