

ABSTRACT
Background
Autosomal dominant polycystic kidney disease (ADPKD) is the most common renal monogenic disease, characterized by bilateral accumulation of renal fluid-filled cysts leading to progressive renal volume enlargement and gradual impairment of kidney function, often resulting in end-stage renal disease. Kuwait could provide valuable genetic insights about ADPKD, including intrafamilial phenotypic variation, given its large household size. This study aims to provide a comprehensive description of the pathogenic variants linked to ADPKD in the Kuwaiti population using multiple genetic analysis modalities and to describe and analyse the ADPKD phenotypic spectrum in terms of kidney function, kidney volume and renal survival.
Methods
A total of 126 ADPKD patients from 11 multiplex families and 25 singletons were recruited into the study. A combination of targeted next-generation sequencing (tNGS), long-range polymerase chain reaction, Sanger sequencing and multiplex ligation-dependent probe amplification were utilized for genetic diagnosis. Clinical evaluation was conducted through renal function testing and ultrasonographic kidney volume analysis.
Results
We identified 29 ADPKD pathogenic mutations from 36 families achieving an overall molecular genetic diagnostic rate of 112/126 (88.9%), including 29/36 (80.6%) in families. A total of 28/36 (77.8%) families had pathogenic mutations in PKD1, of which 17/28 (60.7%) were truncating, and 1/36 (2.8%) had a pathogenic variant in the IFT140 gene. A total of 20/29 (69%) of the identified ADPKD mutations were novel and described for the first time, including a TSC2-PKD1 contiguous syndrome. Clinical analysis indicated that genetically unresolved ADPKD cases had no apparent association between kidney volume and age.
Conclusion
We describe for the first time the genetic landscape of ADPKD in Kuwait. The observed genetic heterogeneity underlining ADPKD along with the wide phenotypic spectrum reveal the level of complexity in disease pathophysiology. ADPKD genetic testing could improve the care of patients through improved disease prognostication, guided treatment and genetic counselling. However, to fulfil the potential of genetic testing, it is important to overcome the hurdle of genetically unresolved ADPKD cases.
Keywords: eGFR, htTKV, IFT140, polycystic kidney disease, TSC2/PKD1 contiguous gene syndrome
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is the most common renal monogenic disease, affecting ≈1/1000 individuals, and is the fourth leading cause of end-stage renal disease (ESRD) in adults [1, 2]. It is a systemic disease characterized by bilateral accumulation of renal fluid-filled cysts, leading to progressive renal volume enlargement and gradual impairment of kidney function, often resulting in ESRD [3]. ADPKD patients can also develop extrarenal manifestations such as polycystic liver disease (PLD), cerebral aneurysms and cardiovascular abnormalities, including early-onset hypertension [4]. Around 78% of ADPKD cases are linked to pathogenic variants in PKD1 and ≈15% to pathogenic variants in PKD2, while 7% of ADPKD cases remain genetically unresolved or are due to pathogenic variants in other genes [5, 6]. PKD1 and PKD2 encode polycystin-1 (PC1) and polycycstin-2 (PC2), respectively, which are membrane glycoproteins forming a 1:3 heteromeric complex and function, most likely, in the primary cilium [7–9]. ADPKD severity shows some degree of complexity, where mutations in PKD1 have shown a more severe disease phenotype than mutations in PKD2 in terms of total kidney volume and onset of ESRD (54.3 years for PKD1 and 79.7 years for PKD2) [10–14]. Furthermore, truncating mutations in PKD1 are associated with a shorter time to ESRD when compared with PKD1 non-truncating mutations (55.6 and 67.9 years, respectively) [15].
Recent advances in genetic sequencing technologies have allowed wider utilization of next-generation sequencing (NGS) platforms, including whole genome sequencing (WGS), whole exome sequencing (WES) and targeted sequencing (tNGS), to explore the genetic basis of the heterogeneous spectrum of ADPKD-like phenotypes that is unrelated to mutations in either of PKD1 or PKD2. A recent report has shown that IFT140 is the third most common ADPKD spectrum gene after PKD1 and PKD2. Monoallelic pathogenic variants in this gene, which encodes a component of the intraflagellar transport (IFT) complex that is involved in the formation and maintenance of cilia, have been associated with a milder spectrum of the ADPKD phenotype with limited renal insufficiency [16]. Other genes causative of rarer forms of the disease include GANAB, DNAJB11 and ALG9. Monoallelic mutations in GANAB, which encodes the glucosidase IIα subunit, have been associated with a milder form of ADPKD and autosomal dominant polycystic liver disease (ADPLD) [17]. Monoallelic mutations in DNAJB11, which encodes a soluble glycoprotein that is involved in protein processing in the endoplasmic reticulum, have been shown to cause an atypical presentation of ADPKD with phenotypic overlap with a milder form of ADPKD and autosomal dominant tubulointerstitial diseases (ADTKDs) characterized by cystic kidneys with normal size and progressive interstitial fibrosis [18]. Other reports have shown that mutations in ALG9, which encodes alpha-1,2-mannosyltransferase enzyme, are associated with a milder form of ADPKD [19].
The observed genetic heterogeneity underlining ADPKD along with the wide phenotypic spectrum reveal the level of complexity in disease pathophysiology. A recent ‘threshold model’ indicated that renal cyst formation is likely to be triggered by the reduction in functional levels of polycystin in tubular epithelial cells beyond a critical threshold. This could be a result of either mutations in PKD1 and/or PKD2 or mutations in other genes, including GANAB, DNAJB11 and ALG9, that affect the endoplasmic reticulum protein biosynthesis, which in turn have a negative functional impact on the polycystins [20].
Currently ADPKD genetic testing is clinically indicated in limited scenarios, including to confirm or exclude an ADPKD diagnosis in suspected cases with no apparent family history or with equivocal imaging findings. It is also used to confirm or exclude an ADPKD diagnosis in young patients for kidney donation decisions. As it is the most common inherited kidney disease, genetic testing for ADPKD is a rapidly increasing area [21]. Applying ADPKD genetic testing in the clinical context could improve the care of patients through improved disease prognostication, treatment guidance, support of clinical trials and as an aid in genetic counselling and pre-implementation genetic diagnosis applications [21–23].
The genetic landscape of ADPKD in Kuwait has not been explored previously. As a small country with an average household size of seven people [24], Kuwait could provide valuable genetic insights about ADPKD, including intrafamilial phenotypic variation. In this study we provide a comprehensive description of the pathogenic variants linked to ADPKD in the Kuwaiti population using multiple genetic analysis modalities. We further describe and analyse the phenotypic spectrum of ADPKD in the studied population in terms of kidney function, kidney volume and renal survival.
MATERIALS AND METHODS
Patient recruitment
Participants were recruited from the outpatient nephrology clinic, inpatients and dialysis clinic and kidney transplant outpatients at Mubarak Al-Kabeer Hospital. Patients were recruited into the study if they satisfied the inclusion criteria. All patients are ≥18 years of age, able to provide informed consent and without previous genetic results related to ADPKD. Patients included had a positive family history of ADPKD and met the unified criteria for ultrasonographic diagnosis of ADPKD [25] or were without a family history of ADPKD but with confirmed renal cysts on imaging as per the unified criteria for ultrasonographic diagnosis of ADPKD [25].
The study was reviewed and approved the Ministry of Health (MOH) Research Ethics Committee (reference 1139/2019) and the Joint Committee for the Protection of Human Subjects in Research of the Health Sciences Center at Kuwait University and the Kuwait Institute for Medical Specialization (reference VDR/JC/690). Written informed consent was obtained from all enrolled patients in accordance with the guidelines of the MOH Research Ethics Committee.
Clinical evaluation
Enrolled patients were evaluated using ultrasonographic analysis and renal function tests to confirm their disease status. All enrolled patients, except for those with ESRD or kidney transplant, underwent abdominal sonographic examination using a Logic 7 ultrasound with a curvilinear 3.5-MHZ probe (GE Healthcare, Chicago, IL, USA). Additional imaging was performed by both non-enhanced and contrast-enhanced multidetector computed tomography with BrightSpeed computed tomography (CT) scanners (GE Healthcare) with multiplanar reformation. Magnetic resonance imaging (MRI) was performed as multisequence multiplanar non-enhanced using a 1.5-Tesla machine (GE Healthcare). The scan was performed to examine the kidneys, liver and pancreas, as well as the ovaries in female subjects. Each kidney was examined in multiple views. Renal cysts were examined and counted for diagnostic purposes according to the unified criteria for ultrasonographic diagnosis [25]. The total kidney volume (TKV) for each kidney was calculated using the ellipsoid formula: volume = length × lateral diameter × anterior–posterior diameter × π/6. The machine calculated the TKV automatically in cubic centimetres (cc) and then it was adjusted for height (htTKV, expressed as cc/m). The liver and pancreas of each patient were screened for the presence of cysts. For the renal function test (RFT), a 5-ml blood sample was taken from each enrolled patient, except patients with documented ESRD or kidney transplant. All RFTs were performed at the Mubarak Al-Kabeer Hospital, Jabriya, Kuwait. Estimated glomerular filtration rates (eGFRs) were calculated using the Chronic Kidney Disease Epidemiology Collaboration creatinine equation [26].
DNA isolation
A 10-ml blood sample from each enrolled patient was collected in ethylenediaminetetraacetic acid tubes. Genomic DNA was isolated from each blood sample using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany; catalogue no. 158467) following the manufacturer's protocol.
tNGS
DNA libraries were prepared for hybridization-based targeted capture of a 65-gene, 136-gene or 357-gene panel as previously described [6, 16, 18]. Captured libraries were sequenced on an HiSeq4000 (Illumina, San Diego, CA, USA) with 2 × 150 paired-end reads. Read alignment and variant calling were performed in the Genome Analysis Toolkit and variant mining completed in the Sequence and Variation Suite (Golden Helix, Bozeman, MT, USA) as previously described [16, 18]. Samples were evaluated for copy number variants utilizing the log ratio of observed read depth over expected read depth, based on normalized sample coverage in the target region [6, 16].
Long-range polymerase chain reaction (LR-PCR), Sanger sequencing and multiplex ligation-dependent probeamplification (MLPA)
Variants of interest (VOIs) identified on the panel were confirmed and segregated in additional family members by PCR for Sanger sequencing using established primer pairs and conditions [5]. For variants in the duplicated region of PKD1, LR-PCR was performed prior to exon-specific amplification using the previously described primer sets [6]. Regions of PKD1 and PKD2 that were poorly covered by the panel were further screened by LR-PCR methods in samples without a VOI identified by tNGS [16]. Large deletions in PKD1 or PKD2 were confirmed by established MLPA assays (MRC Holland, Amsterdam, The Netherlands) and evaluated in GeneMarker (SoftGenetics, State College, PA, USA). Additionally, a subset of samples was screened in full by the LR-PCR, Sanger and MLPA methodologies without prior panel analysis.
Variant pathogenicity and analysis
Variants were classified as VOIs if they were ultrarare (<10× for known dominant genes and <50× for recessive genes in gnomAD version 2.1.1) and predicted to alter either the protein sequence or splicing. VOIs were then evaluated for pathogenicity utilizing the previously defined criteria and classified into mutation strength groups [5].
Data analysis
Renal survival analysis was performed to study the relationship between PKD1 mutation type (truncating and non-truncating) and time to ESRD. Survival times were calculated as the time of onset of ESRD. Kaplan–Meier product-limit renal survival curves were constructed for patients with PKD1 truncating and non-truncating mutations using MATLAB and Statistics Toolbox, release 2012b (MathWorks, Natick, MA, USA). The mean survival time was calculated as the shortest survival time in which the estimated probability of renal survival was ≤0.5. The estimation is limited to the largest survival time if it is censored. The differences between both groups were evaluated for statistical significance using the logrank (Mantel–Cox) test ,where significance was established for P-values <.05.
Multivariable linear regression was conducted to assess associations between age, htTKV and eGFR while stratifying by PKD1 mutation status. Moreover, interaction terms between age and PKD1 mutation status were assessed. Sex-stratified analyses were conducted to evaluate the association between PKD1 mutation status and htTKV and eGFR. These models were followed by tests for interaction by including a product term (sex × PKD1 mutation) in the multivariable linear regression models. htTKV was modelled as log10 transformed variable, hence taking the antilog of the linear regression coefficients (β) yields an adjusted ratio of geometric means.
RESULTS
Study participants
A total of 126 ADPKD patients from 11 multiplex families and 25 singletons fulfilled the inclusion criteria and were enrolled in the study. The mean age of the cohort was 37.29 years (range 18–60) and 74/126 (58.7%) were males (Table 1). Of the recruited patients, 52/126 (41.3%) had liver cysts, confirmed by ultrasound, and 92/126 (73%) were diagnosed with hypertension (Table 1). Only 33/126 (26.2%) of the recruited patients were already at ESRD at the time of enrolment (Table 1).
Table 1:
Baseline characteristics of ADPKD patients based on their genetic diagnosis status.
| Characteristics | All patients (N = 126) | Solved (n = 112) | Unsolved (n = 14) | PKD1 truncating mutation (n = 74) | PKD1 non-truncating mutation (n = 36) |
|---|---|---|---|---|---|
| Age (years), mean (range) | 37.29 (18–60) | 38.32 (18–60) | 40 (26–56) | 37.93 (18–60) | 37.74 (18–58) |
| Male, n/N (%) | 74/126 (58.7) | 65/112 (58) | 9/14 (64.3) | 41/74 (55.4) | 21/36 (58.3) |
| Known liver cysts, n/N (%) | 52/126 (41.3) | 47/112 (42) | 5/14 (35.8) | 31/74 (41.9) | 15/36 (41.7%) |
| Known hypertension, n/N (%) | 92/126 (73) | 83/112 (74.1) | 9/14 (64.3) | 55/74 (74.3) | 25/36 (69.4) |
| Documented family history, n/N (%) | 112/126 (88.9) | 101/112 (90.2) | 11/14 (78.6) | 69/74 (93.2) | 30/36 (83.3) |
| Patients reaching ESRD, n/N (%) | 33/126 (26.2) | 33/112 (29.5) | 0/14 (0) | 20/74 (27) | 13/36 (36.1) |
ADPKD genetic diagnosis
DNA samples from the 126 PKD patients were analysed for PKD genetic diagnosis. From the enrolled families, 28/36 (77.8%) had a pathogenic mutation in PKD1 (NM_001009944) and 1/36 (2.8%) had a pathogenic variant in the IFT140 gene (NM_014714) resulting in a resolve rate of 29/36 (80.6%). The remaining 7/36 (19.4%) were unresolved genetically (Fig. 1A).
Figure 1:

Pathogenic variants linked to PKD in the cohort. (A) Genes linked to ADPKD in the presented cohort. (B) Details on the genetic modalities utilized for successful detection of ADPKD pathogenic variants. (C) An overview of types of PKD1 pathogenic variants.
Following the Mayo Clinic PKD pathogenicity guidelines, we report an overall molecular genetic diagnostic rate of 112/126 (88.9%) and 29/36 (80.6%) in families (Table 1). From the solved families, 19/29 (65.5%) were diagnosed using tNGS, 8/29 (27.6%) were diagnosed using LR-PCR and Sanger sequencing and 2/29 (6.9%) were diagnosed using MLPA (Fig. 1B).
Detected variants
A total of 29 pathogenic variants were detected in the resolved families and there were 9 variants of possible PKD relevance (Table 2). For the 28 detected PKD1 pathogenic variants, 7 (25%) were nonsense, 7 (25%) were missense, 6 (21.4%) were frameshift indel, 4 (14.3%) were large deletion, 2 (7.1%) were splicing variants and 2 (7.1%) were in-frame indel (Table 2 and Figs. 1C and 2). From a total of 29 ADPKD pathogenic variants detected, 20 (69%) were novel variants, while for the other possible relevant PKD variants, 4/9 (44.4%) were novel (Table 2). Conservation analysis of the detected variants is presented in (Supplementary Table 2).
Table 2:
Details of ADPKD pathologic and possibly modifying variants.
| Pedigree | Gene | Ex/IVS | Protein variant | cDNA variant | Mutation type | Functional effect | Mayo Clinic designation | gnomAD1 | ADPKD Variant Database | dbSNP ID |
|---|---|---|---|---|---|---|---|---|---|---|
| Pathogenic variants | ||||||||||
| 1 | PKD1 | 5′UTR-Ex14 | p.Met1fs | c.(1-2kb)_(3295)del23.5kb | Large deletion | Truncating | DP | No | ||
| 2 | PKD1-TSC2 | 2–46 and 18–41 |
PKD1: p.Leu72fs* TSC2: p.Ala1614fs* |
Chr16: (2.121Mb)_(2.170Mb)del48.3kb-PKD1: c.(216)_(12909*)del30.9kb TSC2: c.(1840)_(5352)del17.4kb |
Large deletion | Truncating | DP | No | ||
| 3 | PKD1 | 5 | p.Glu226* | c.676G>T | Nonsense | Truncating | DP | No | ||
| 4 | PKD1 | 15 | p.Met1219fs | c.3656_6911del3256 | Large deletion | Truncating | DP | No | ||
| 5 | PKD1 | 15 | p.Arg1672fs | c.5014_5015delAG | Frameshift | Truncating | DP | Yes | rs1555455457 | |
| 6 | PKD1 | 15 | p.Gln2058Pro | c.6173A>C | Missense | Non-truncating | LP | 0 | No | |
| 7 | PKD1 | 15 | p.Arg2163* | c.6487C>T | Nonsense | Truncating | DP | Yes | ||
| 8 | PKD1 | 15 | p.Arg2220_Pro2224del5 | c.6657_6671del15 | InFrame D/I | Non-truncating | DP | Yes | ||
| 9 | PKD1 | 15 | p.Gln2243* | c.6727C>T | Nonsense | Truncating | DP | Yes | rs1567191609 | |
| 10 | PKD1 | 15 | p.Gly2278Arg | c.6832G>A | Missense | Non-truncating | LP | 0 | Yes | rs1555454145 |
| 11 | PKD1 | 15 | p.Trp2298Arg | c.6892T>G | Missense | Non-truncating | HLP | 0 | No | |
| 12 | PKD1 | IVS15 | p.Arg2306fs | c.6916-9G>A | Splice | Non-truncating | LP | 0 | Yes | |
| 13 | PKD1 | 17 | p.Cys2373* | c.7119C>A | Nonsense | Truncating | DP | Yes | ||
| 14 | PKD1 | 17 | p.Arg2404fs | c.7197_7209dup13 | Frameshift | Truncating | DP | No | ||
| 15 | PKD1 | 23 | p.Phe2806Ser | c.8417T>C | Missense | Non-truncating | LP | 0 | No | |
| 16 | PKD1 | IVS24 | p.Gly2983fs | c.8948+2dupT | Splice | Non-truncating | LP | No | ||
| 17 | PKD1 | 25 | p.Glu3020* | c.9058G>T | Nonsense | Truncating | DP | Yes | ||
| 18 | PKD1 | 25 | p.Leu3010Gln | c.9029T>A | Missense | Non-truncating | LP | 0 | No | rs750501225 |
| 19 | PKD1 | 26 | p.Pro3069fs | c.9205_9224del20insGACA | Frameshift | Truncating | DP | No | ||
| 20 | PKD1 | 36 | p.Ser3593Arg | c.10779C>G | Missense | Non-truncating | LP | 0 | No | |
| 21 | PKD1 | 39 | p.Tyr3734* | c.11202C>A | Nonsense | Truncating | DP | No | [54] | |
| 22 | PKD1 | 40 | p.Tyr3759fs | c.11274_11275delCT | Frameshift | Truncating | DP | No | rs1555446105 | |
| 23 | PKD1 | 40 | p.Asp3780_Asp3782del3 | c.11339_11347del9 | InFrame D/I | Non-truncating | HLP | No | ||
| 24 | PKD1 | 41 | p.Trp3842Arg | c.11524T>C | Missense | Non-truncating | HLP | 0x | Yes | rs1057518959 |
| 25 | PKD1 | IVS42-3′UTR | p.Val3905fs | c.11712+55_12909974del2.6kb | Large deletion | Truncating | DP | No | ||
| 26 | PKD1 | 43 | p.Gln3955fs | c.11863dupC | Frameshift | Truncating | DP | No | [43] | |
| 27 | PKD1 | 44 | p.Gln4005* | c.12013C>T | Nonsense | Truncating | DP | Yes | rs1567148587 | |
| 28 | PKD1 | 46 | p.Phe4219fs | c.12627_12655dup29 | Frameshift | Truncating | DP | No | ||
| 29 | IFT140 | 14 | p.Gly509? | c.1525-1G>A | Splice | Truncating | DP | NA | [16] | |
| Other possible relevant variants | ||||||||||
| 22 | PKD1 | 8 | p.Thr558Met | c.1673C>T | Missense | Non-truncating | LN | 8x | No | rs781572938 |
| Unsolved# | PKD1 | 15 | p.Arg1549Gln | 4646G>A | Missense | Non-truncating | LN | 2x | No | rs530555146 |
| 9 | PKD1 | 15 | p.His1769Tyr | c.5305C>T | Missense | Non-truncating | Mod | 5x | No | |
| Unsolved | PKD1 | 23 | p.Thr2756Ile | c.8267C>T | Missense | Non-truncating | VUS | 49x | No | rs141296093 |
| 22 | PKD1 | 27 | p.Arg3169Trp | c.9505C>T | Missense | Non-truncating | LN | 8x | No | rs765180455 |
| 21 | PKD1 | 36 | p.Lys3607Met | c.10820A>T | Missense | Non-truncating | VUS | 0x | No | |
| 4 | PKD2 | 15 | p.Met905Leu | c.2713A>C | Splice | Non-truncating | VUS | 77x | 2 | rs573871626 |
| Unsolved | IFT122 | 30 | p.Leu1257_Pro1263del | c.3769_3789+5del26 | Frameshift | Truncating | DP (rec) | 0x | No | |
| Unsolveda | COL4A1 | 30 | p.Pro739Leu | c.2216C>T | Missense | Non-truncating | VUS | 13x | No | |
aCo-inherited by the same patient.
dbSNP: Single Nucleotide Polymorphism Database; FS del: frameshift deletion; FS dup: frameshift duplication; IF del: in-frame deletion; DP: definitely pathogenic; LP: likely pathogenic; VUS: variant of uncertain significance; LN: likely neutral; Rec: recessive; HLP: highly likely pathogenic; Mod: moderate; Ex: exon; IVS: intervening sequence.
Figure 2:

The distribution of detected PKD1 variants. Variants highlighted in bold are pathogenic variants and those unhighlighted are the non-pathogenic variants with possible ADPKD relevance.
A case with the TSC2-PKD1 contiguous syndrome
Among the novel variants detected was a large deletion covering parts of PKD1 and TSC2 from PKD1 exon 2 to TSC2 exon 18. The full genomic location of the deletion is Chr16:(2.121Mb)(2.170Mb)del48.3kb. The TSC2 portion of the annotation is c.(1840)_(5352)del17.4kb(p.Ala1614fs*) and the PKD1 portion of the annotation is c.(216)_(12909*)del30.9kb(p.Leu72fs*) (Table 2 and Fig. 2). This contiguous gene syndrome deletion was detected in a 25-year-old male patient with early renal insufficiency (eGFR 9.8 ml/min/1.73 m2). Radiologic analysis revealed renal angiomyolipoma, pulmonary lymphangioleiomyomatosis and bilateral subependymal calcified nodules (Fig. 3A–E). The patient also showed multiple skin lesions including fibrous cephalic plaques, hypopigmented macules, angiofibroma and shagreen patches on the lower back.
Figure 3:

Diagnostic imaging of two rare PKD cases. (A) A CT scan of a patient with TSC2/PKD1 contiguous gene syndrome shows markedly enlarged kidneys with innumerable variable-sized cortical and parenchymal cysts, some showing scattered tiny mural calcifications. (B) Renal masses of mixed soft tissue and fat attenuation, suggestive of angiomyolipoma, are noted with a large amount of ascites. (C) A CT scan shows bilateral basal lung bronchiectatic changes along with atelectatic bands. (D and E) Small left hepatic lobe cysts were noted. (F and G) Multiple calcified subependymal nodules were noted. (H and I) A CT scan of the IFT140 patient shows enlarged kidneys by the presence of multiple variable-sized cortical and to lesser extent parenchymal renal cysts. (H) The largest cysts are the lower polar exophyting ones. (I) Small mural calcifications are seen at the right-sided lower polar cyst.
A case of co-inheritance of a PKD2 variant of uncertain significance (VUS) with a pathogenic PKD1 variant
In a rare case, genetic analysis revealed a 42-year-old singleton male co-inherited a large PKD1 deletion (c.3656_6911del3256) along with a PKD2 VUS, which may alter splicing (c.2713A>C), that was reported 77 times in gnomAD. The patient showed a more severe form of the disease, reaching ESRD at 39 years of age (Table 2 and Supplementary Table 2 and Supplementary Fig. 1).
IFT140 case of PKD
One pathogenic variant was detected in a gene other than PKD1. A splicing pathogenic variant at a conserved site was detected in IFT140 c.1525-1G>A with a truncating effect (Table 2 and Supplementary Table 1). This variant was detected in a 44-year-old female with normal eGFR and htTKV of 876 ml/m. The patient showed eight cysts in each kidney and no cysts in the liver (Fig. 3F–H).
Genetically unsolved PKD cases with inherited variants of PKD relevance
We detected four variants with possible PKD relevance in three unsolved cases (Table 2). In a 47-year-old female patient, which was not resolved genetically, a novel frameshifting variant (c.3769_3789+5del26) was detected in IFT122 (NM_052985), resulting in a frameshift mutation with a possible monoallelic pathogenic effect (Table 2). The patient had cysts in both kidneys with an htTKV of 539.5 cc/m and an eGFR of 78 ml/min/1.73 m2. The patient had no family history of PKD.
Genetic analysis also revealed co-inheritance of a missense variant (c.2216C>T, p.Pro739Leu) in COL4A1 (NM_001845) with uncertain significance with a missense non-truncating PKD1 variant (c.4646G>A, p.Arg1549Gln) (Table 2) in a 21-year-old female with normal kidney volume and function (Table 2). We also detected a PKD1 (c.8267C>T, p.Thr2756Ile) missense variant with uncertain significance in a 41-year-old male with enlarged kidneys (htTKV 2019.9 cc/m) and reduced kidney function (eGFR 42 ml/min/1.73 m2) (Table 2).
Clinical and renal survival analysis
We utilized htTKV and eGFR to determine the clinical course in patients with PKD1 truncating and non-truncating mutations and the unsolved cases. Figure 4 shows an association of age with eGFR and kidney volume (htTKV) according to PKD1 mutation status. Patients with PKD1 truncating (sex adjusted: β = −2.10, P < .001) and PKD1 non-truncating (sex adjusted: β = −1.95, P = .019) mutations demonstrated decreasing eGFR values as age increased (Fig. 4A). Moreover, there was no evidence for a difference in the association between age and eGFR according to PKD1 mutation type [P for interaction (age × PKD1 mutation) = .501]. Fig. 4B shows that htTKV increased as age increased among those with PKD1 truncating mutations (sex adjusted: β = 23.65, P < .001) and PKD1 non-truncating mutations (sex adjusted: β = 24.72, P = .034), with no indication for a difference in the magnitude of the association across mutation status [P for interaction (age × PKD1 mutation) = .817] (Supplementary Table 1).
Figure 4:

Relationship between eGFR and htTKV in the ADPKD cohort. (A) Plot of eGFR versus age for ADPKD patients with a truncating PKD1 mutation (red) and patients with a PKD1 non-truncating mutation (blue) shows a decreasing trend in both groups. (B) Plot of the htTKV in natural log scale versus age for ADPKD patients with a truncating PKD1 mutation (red) and patients with a PKD1 non-truncating mutation (blue) shows an increasing trend for both groups. (C) A plot shows a decreasing trend of eGFR versus age for the unsolved cases and PKD1 solved cases. (D) Plot of htTKV versus age shows a significant difference in the htTKV trend for PKD1 solved cases. Shaded areas indicate 95% confidence intervals.
Similarly, when testing the association between eGFR and age according to genetic status (solved and unsolved cases) (Fig. 4C), there was evidence for a decreasing trend in eGFR as age increased in those resolved PKD1 cases (sex adjusted: β = −1.99, P < .001) and in those unresolved cases (sex adjusted: β = −1.83, P = .014). There was no evidence for heterogeneity in these associations by PKD1 mutation status [P for interaction (age × PKD1 mutation) = .925]. Moreover, kidney volume showed an increasing trend as age increased among PKD1-resolved cases (sex adjusted: β = 24.22, P < .001), whereas there was no association among unresolved cases (sex adjusted: β = −9.16, P = .643; Fig. 4D). There was statistical evidence that the association between age and htTKV is different among PKD1 resolved and unresolved cases [P for interaction (age × PKD1 mutation) = .017] (Supplementary Table 1).
Kaplan–Meier renal survival analysis was performed to determine mean/median age to ESRD in PKD1 with truncated and non-truncated mutations (Fig. 5). Patients with PKD1 truncating mutations showed a shorter renal survival time {51.8 years [standard error (SE) 1.223]} compared with patients with non-truncating mutations [56.3 years (SE 0.918)], with statistical significance as indicate by the logrank test (P = .043) (Fig. 5 and Table 3). The overall mean age of ESRD, calculated by Kaplan– Meier renal survival analysis, for patients with PKD1 mutation was 53.9 years (SE 0.907), while the median was 55 years (SE 0.929) (Table 3).
Figure 5:
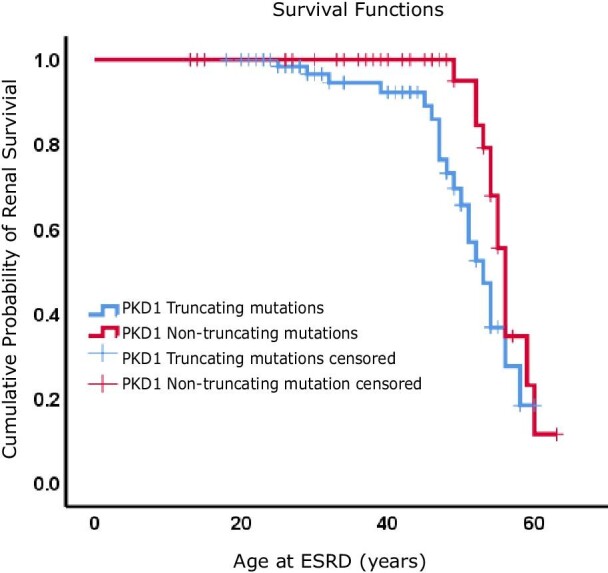
Kaplan–Meier renal survival analysis for ADPKD patients with PKD1 truncated and non-truncated mutations. Time to ESRD is plotted for each group.
Table 3:
Mean and median age of ESRD in patients with PKD1 mutations calculated by Kaplan–Meier analysis.
| Meana | Median | |||||||
|---|---|---|---|---|---|---|---|---|
| 95% confidence interval | 95% confidence interval | |||||||
| Group | Estimate | Standard error | Lower bound | Upper bound | Estimate | Standard error | Lower bound | Upper bound |
| PKD1 truncated mutation | 51.815 | 1.223 | 49.418 | 54.213 | 53 | 1.483 | 50.094 | 55.906 |
| PKD1 non-truncated mutation | 56.339 | 0.918 | 54.541 | 58.138 | 56 | 0.579 | 54.864 | 57.136 |
| Overall | 53.915 | 0.907 | 52.138 | 55.692 | 55 | 0.929 | 53.178 | 56.822 |
Estimation is limited to the longest survival time if it is censored.
DISCUSSION
This is the first report comprehensively describing the genetic landscape of ADPKD in Kuwait. We provide insights on the genotype–phenotype relationship in terms of disease severity evaluated by eGFR and htTKV. We recorded an overall molecular genetic diagnostic rate of 112/126 (88.9%) and 29/36 (80.6%) in families reporting 29 ADPKD pathogenic cases, of which 20/29 (69%) are novel and described for the first time. We highlight current limitations in the genetic diagnosis of ADPKD, as 19.4% of families were unsolved genetically.
Most pathogenic variants were detected in PKD1 (77.8%). Our results were supported by previous studies where PKD1 pathogenic variants were responsible for 67% and 77.7% of ADPKD cases in cohorts from Ireland and the USA, respectively [5, 27]. PKD1 exon 15 was the exon with highest number of pathogenic variants, but this result can be attributed to it genomic size (3620 bp) rather than being a mutation hot spot. In the PKD1 group, although the differences recorded in trends of eGFR decline and htTKV enlargement with age were not statistically significant between the PKD1 truncating and non-truncating groups, the more severe form of the disease in the PKD1 truncating group was demonstrated by the shorter time to ESRD, on average, when compared with patients with PKD1 non-truncating mutations (Fig. 4 and Table 3). At early stages of ADPKD, renal function remains stable and therefore eGFR may not be a reliable marker to monitor disease progression. On the other hand, a growing body of research has shown a progressive increase in TKV throughout the course of ADPKD, even at early disease stages where renal function is still intact, likely due to hyperfiltration of healthy nephrons [28–30]. Currently TKV is the preferred prognostic biomarker to monitor the progression of ADPKD and has been shown to predict the decline in eGFR [31]. It has been utilized as well as a surrogate marker for disease progression in ADPKD tolvaptan clinical trials [32].
The estimated difference between the median age to ESRD in PKD1 truncating versus non-truncating groups (53 versus 56 years) was more narrow than other reports (55 versus 67 years) [15]. This can be influenced by many factors, including the high prevalence of comorbidities in Kuwait and, in particular, diabetes and obesity [33, 34]. Several reports shed light on the relationship between genotype and disease phenotype, where PKD1 truncating mutations were associated with a more severe form of the disease, in terms of onset to ESRD, when compared with PKD1 non-truncating mutations and PKD2 mutations [5, 11, 15, 35]. However, a recent study showed that 18% of ADPKD patients with PKD1 truncating mutations experienced a milder form of the disease [36]. Taken together, these results suggest possible involvement of genetic/environmental factors that, at present, are not fully understood.
One of the rare cases diagnosed was a 26-year-old male who had a novel large 48.3-kb deletion that covered PKD1 exons 2–46 and extended to exons 18–41 from the TSC2, which lies immediately adjacent to PKD1 (tail to tail) on chromosome 16p (Table 2). This patient showed early renal sufficiency that overlapped with a spectrum of tuberous sclerosis manifestations. The TSC2-PKD1 contiguous deletion syndrome is a rare condition reported for the first time in 1994 [37]. Since then, ≈70 cases have been reported in the literature [38–40]. Earlier clinical intervention, facilitated by earlier molecular diagnosis, is likely to improve disease management and outcomes.
Genetic analysis and mutation screening in PKD1 can be challenging technically due to multiple reasons. First, the gene is highly polymorphic and, as we have shown, mutations of different types can occur along the entire length of the gene without mutation hot spots (Fig. 2). Second, ADPKD-linked mutations are likely to be unique to a single family, as previous reports have shown that recurrent pathogenic variants are reported in only 30% of the total detected ADPKD mutations [41], which we also found in our cohort, as novel detected pathogenic mutations accounted for 72.4% of the ADPKD mutations reported. Third, and most importantly, exons 1–32 of PKD1, are duplicated six times and located in pseudo-regions 13–16 Mb proximal to the genuine PKD1, with a shared sequence homology of up to 97.8% [42]. This region of PKD1 represents a diagnostic challenge, and particularly to WES, as it has been reported that this technique has shown low sensitivity in the detection of mutations in this region of PKD1, which could limit its utility in clinical setups [43].
PKD2 is the second common gene responsible for ADPKD, as its accounts for 15% of the cases [5, 6]. However, in our cohort we did not detect any pathogenic variants in PKD2 that are linked to ADPKD (Fig. 1). Mutations in PKD2 are usually associated with a milder form of ADPKD, with a mean onset of ESRD of 79.7 years [13], so patients with PKD2-linked disease might not seek or present late to medical attention, which may explain their underrepresentation in our cohort [13, 44]. Referral bias cannot be excluded.
While the majority of ADPKD cases in our cohort were attributed to PKD1, we characterized one ADPKD case linked to a mutation in IFT140, the gene recently described as the third common ADPKD gene (Fig. 1 and Table 2) [16]. We could not perform comprehensive clinical analysis of the phenotype associated with IFT140 because we have one singleton in our cohort, but the patients showed a milder form of the disease as indicated by the normal kidney function and kidney volume. This agrees with the findings of a recent report that indicated monoallelic mutations in this gene are associated with a milder form of ADPKD even when compared with PKD2-linked disease [16]. These findings highlight the degree of complexity in ADPKD diagnosis and pathophysiology that is also reflected in the wide spectrum of disease manifestation. This phenotypic variability might be useful in guiding molecular testing.
Intrafamilial phenotypic variability has been estimated to occur in at least 12% of families with ADPKD, which provides a strong indication for the involvement of modifier genes [45]. We reported previously a novel PKD1 variant (c.5305C>T, p.His1769Tyr) demonstrating a disease-modifying role in trans with a truncating PKD1 mutation (c.6727C>T, p.Gln2243*) in a family with a history of ADPKD where patients who co-inherited the variant and mutation showed larger renal volumes and earlier renal insufficiency when compared with those who only carried the pathogenic mutation (Table 2) [46]. We further identified a total of nine variants, five of which were co-inherited with ADPKD pathogenic mutations in singleton cases (Table 2). The co-inheritance of certain variants in genes with ADPKD relevance can contribute to worse disease symptoms, as we reported in a case where a 42-year-old patient co-inherited a truncating large PKD1 deletion (c.3656_6911del3256) along with a VUS PKD2 splice site variant (c.2713A>C) (Table 2). The patient showed a more severe form of the disease demonstrated by an early onset of ESRD (39 years old). While understanding the exact prognostic effect of such genetic modifiers can help in improving disease management, it can be challenging, at times, and might require larger family-based cohort studies.
In our cohort, 19.4% of family cases were classified as unsolved, as no mutations linked to ADPKD were identified (Fig. 1), which is close to the 16% reported in Ireland [27]. Other studies reported 5–10% of unsolved cases in their PKD cohorts [15, 17, 47]. Despite the variable proportions between cohorts, which can be attributed to the design of the genetic testing pipeline, ADPKD genetically unsolved cases remain a diagnostic challenge that can limit prognostic application of genetic testing and affect any proposed family planning options. Some of these cases could be a result of genetic PKD1/PKD2 mosaicism, and conventional genetic analysis seems incapable of identifying such mutations [6]. The involvement of new genes other than the ones already linked to ADPKD is also a possibility. This genetic heterogeneity is also a contributor to the variable spectrum of ADPKD disease manifestations and severity. Our results indicated no apparent association between htTKV and age in our unresolved cases (Fig. 4D). This could be explained by the possible high degree of genetic heterogeneity of this particular group and subsequent variable disease manifestations.
ADPKD genetic testing can improve the care of patients by guiding therapy, especially when integrated with other clinical parameters such as gender, hypertension and urological events to predict disease progression and aid personalized therapeutic decisions [48]. Genetic testing can also be useful to include/exclude patients for certain treatments, for example, excluding PKD1/2-negative ADPKD patients from tolvaptan treatment [49, 50].
Apart from conventional general CKD care measures, the current standard of care provided for ADPKD patients in Kuwait focuses mainly on managing the disease complications. While this may limit morbidity and mortality due to ADPKD-related complications, it does not delay disease progression. The current available treatment option for rapidly progressive ADPKD patients reaching ESRD in Kuwait is renal replacement therapy, which is placing a significant burden on ESRD care facilities. In a recent study on the dialysis population in Kuwait, PKD was shown to account for ≈2% of the total dialysis population [51]. However, unpublished data on renal transplants in Kuwait show that ADPKD cases accounted for 9.73% of total renal transplants performed between 2016 and 2019 (Supplementary Table 3). In 2018 the US Food and Drug Administration authorized use of the vasopressin V2 receptor antagonist tolvaptan as the first drug treatment for ADPKD [32]. Clinical trials showed that the use of tolvaptan reduced the growth of kidneys and slowed renal function decline in adult ADPKD patients at risk of rapid disease progression [32, 52, 53]. In Kuwait, tolvaptan use for ADPKD patients is due to start in 2022.
The presented study has some limitations. The relatively small cohort size and subsequent smaller subgroup sizes could restrict direct informative statistical comparisons. Some of the clinical data were self-reported by the recruited patients, thus the possibility of self-reporting bias cannot be excluded. There was also an underrepresentation of PKD2 cases, possibly caused by referral bias. Using ultrasonography to measure kidney volume is operator dependent and relies on geometric assumptions that could affect its accuracy when compared with CT or MRI.
CONCLUSION
The observed genetic heterogeneity of ADPKD along with the wide phenotypic spectrum reveal the level of complexity in disease pathophysiology. ADPKD genetic testing could improve the care of patients through improved disease prognostication, treatment guidance, support of clinical trials and genetic counselling. However, it is also important to overcome the hurdle of genetically unresolved ADPKD cases thorough improved genetic testing pipelines to ensure more effective implementation of ADPKD genetic testing in clinical setups.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the Ministry of Health in Kuwait and the management of Mubarak Al-Kabeer Hospital for supporting this work. We would also like to thank the Kuwait Foundation for the Advancement of Sciences (KFAS) for their unlimited support of clinical research in Kuwait. The study was reviewed and approved by the MOH Research Ethics Committee (reference 1139/2019) and the Joint Committee for the Protection of Human Subjects in Research of the Health Sciences Center at Kuwait University and the Kuwait Institute for Medical Specialization (reference VDR/JC/690). Written informed consent was obtained from all enrolled patients in accordance with the guidelines of the MOH Research Ethics Committee.
Contributor Information
Hamad Ali, Department of Medical Laboratory Sciences, Faculty of Allied Health Sciences, Health Sciences Center, Kuwait University, Jabriya, Kuwait; Department of Genetics and Bioinformatics, Dasman Diabetes Institute, Dasman, Kuwait.
Medhat Naim, Division of Nephrology, Mubarak Al-Kabeer Hospital, Ministry of Health, Jabriya, Kuwait.
Sarah R Senum, Division of Nephrology and Hypertension, Mayo Clinic, Rochester, MN, USA.
Ali AlSahow, Division of Nephrology, Al-Jahra Hospital, Ministry of Health, Al-Jahra, Kuwait.
Yousif Bahbahani, Division of Nephrology, Mubarak Al-Kabeer Hospital, Ministry of Health, Jabriya, Kuwait; Medical Division, Dasman Diabetes Institute, Dasman, Kuwait.
Mohamed Abu-Farha, Department of Biochemistry and Molecular Biology, Dasman Diabetes Institute, Dasman, Kuwait.
Jehad Abubaker, Department of Biochemistry and Molecular Biology, Dasman Diabetes Institute, Dasman, Kuwait.
Anwar Mohammad, Department of Biochemistry and Molecular Biology, Dasman Diabetes Institute, Dasman, Kuwait.
Adel Al-Hunayan, Department of Surgery, Faculty of Medicine, Health Sciences Center, Kuwait University, Jabriya, Kuwait.
Akram M Asbeutah, Department of Radiological Sciences, Faculty of Allied Health Sciences, Health Sciences Center, Kuwait University, Jabriya, Kuwait.
Mohamed Zayed, Department of Radiology, Mubarak Al-Kabeer Hospital, Ministry of Health, Jabriya, Kuwait.
Sriraman Devarajan, National Dasman Diabetes Biobank, Dasman Diabetes Institute, Dasman, Kuwait.
Naser Hussain, Division of Nephrology, Mubarak Al-Kabeer Hospital, Ministry of Health, Jabriya, Kuwait.
Sumi Elsa John, Department of Genetics and Bioinformatics, Dasman Diabetes Institute, Dasman, Kuwait.
Arshad Channanath, Department of Genetics and Bioinformatics, Dasman Diabetes Institute, Dasman, Kuwait.
Thangavel Alphonse Thanaraj, Department of Genetics and Bioinformatics, Dasman Diabetes Institute, Dasman, Kuwait.
Mohammad Al-Ali, Next Generation Sequencing Laboratory, Kuwait Medical Genetics Center, Ministry of Health, Sulaibikhat, Kuwait.
Mustafa AlMousawi, Department of Transplantation, Hamed Al Essa Organ Transplant Centre, Ministry of Health, Kuwait City, Kuwait.
Fahd Al-Mulla, Department of Genetics and Bioinformatics, Dasman Diabetes Institute, Dasman, Kuwait.
Peter C Harris, Division of Nephrology and Hypertension, Mayo Clinic, Rochester, MN, USA.
FUNDING
This work was funded by the KFAS research fund (PR17-13MM-07, to H.A.) and the National Institute of Diabetes and Digestive and Kidney Diseases (grant DK058816, to P.H.).
AUTHORS’ CONTRIBUTIONS
H.A. was responsible for conceptualization, data curation, funding acquisition, the investigation, project administration and writing the manuscript. M.N., A.AlSahow, Y.B., A.Al-Hunayan, N.H., M.Al-Ali and M.AlMousawi were responsible for patient recruitment and clinical analysis. S.R.S. was responsible for genotyping, data analysis and review and editing of the manuscript. M.A. and J.A. were responsible for resources and the investigation. A.M. was responsible for data analysis and visualization. A.M.A. and M.Z. were responsible for radiological analysis. S.D. was responsible for patient recruitment. S.E.J. and A.C. were responsible for genetic data analysis. T.A.T. was responsible for data interpretation. F.A. was responsible for supervision and data interpretation. P.C.H. was responsible for conceptualization, data analysis, the investigation, resources and review and editing of the manuscript.
DATA AVAILABILITY STATEMENT
The data underlying this article will be shared upon reasonable request to the corresponding author.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
REFERENCES
- 1. Suwabe T, Shukoor S, Chamberlain AMet al. Epidemiology of autosomal dominant polycystic kidney disease in Olmsted County. Clin J Am Soc Nephrol 2020;15:69–79. 10.2215/CJN.05900519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet 2019;393:919–35. 10.1016/S0140-6736(18)32782-X [DOI] [PubMed] [Google Scholar]
- 3. Bergmann C, Guay-Woodford LM, Harris PCet al. Polycystic kidney disease. Nat Rev Dis Primers 2018;4:50. 10.1038/s41572-018-0047-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luciano RL, Dahl NK. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol Dial Transplant 2014;29:247–54. 10.1093/ndt/gft437 [DOI] [PubMed] [Google Scholar]
- 5. Heyer CM, Sundsbak JL, Abebe KZet al. Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2016;27:2872–84. 10.1681/ASN.2015050583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hopp K, Cornec-Le Gall E, Senum SRet al. Detection and characterization of mosaicism in autosomal dominant polycystic kidney disease. Kidney Int 2020;97:370–82. 10.1016/j.kint.2019.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ong AC, Harris PC; A polycystin-centric view of cyst formation and disease: the polycystins revisited. Kidney Int 2015;88:699–710. 10.1038/ki.2015.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nauli SM, Alenghat FJ, Luo Yet al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003;33:129–37. 10.1038/ng1076 [DOI] [PubMed] [Google Scholar]
- 9. Su Q, Hu F, Ge Xet al. Structure of the human PKD1-PKD2 complex. Science 2018;361:eaat9819. 10.1126/science.aat9819 [DOI] [PubMed] [Google Scholar]
- 10. Hateboer N, Dijk MA v, Bogdanova Net al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet 1999;353:103–7. 10.1016/S0140-6736(98)03495-3 [DOI] [PubMed] [Google Scholar]
- 11. Harris PC, Hopp K. The mutation, a key determinant of phenotype in ADPKD. J Am Soc Nephrol 2013;24:868–70. 10.1681/ASN.2013040417 [DOI] [PubMed] [Google Scholar]
- 12. Cornec-Le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol 2018;29:13–23. 10.1681/ASN.2017050483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cornec-Le Gall E, Audrezet MP, Renaudineau Eet al. PKD2-related autosomal dominant polycystic kidney disease: prevalence, clinical presentation, mutation spectrum, and prognosis. Am J Kidney Dis 2017;70:476–85. 10.1053/j.ajkd.2017.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lavu S, Vaughan LE, Senum SRet al. The value of genotypic and imaging information to predict functional and structural outcomes in ADPKD. JCI Insight 2020;5:e138724. 10.1172/jci.insight.138724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cornec-Le Gall E, Audrezet MP, Chen JMet al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 2013;24:1006–13. 10.1681/ASN.2012070650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Senum SR, Li YSM, Benson KAet al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet 2022;109:136–56. 10.1016/j.ajhg.2021.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Porath B, Gainullin VG, Cornec-Le Gall Eet al. Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet 2016;98:1193–207. 10.1016/j.ajhg.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cornec-Le Gall E, Olson RJ, Besse Wet al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet 2018;102:832–44. 10.1016/j.ajhg.2018.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Besse W, Chang AR, Luo JZet al. ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol 2019;30:2091–102. 10.1681/ASN.2019030298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lanktree MB, Haghighi A, di Bari Iet al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol 2021;16:790–9. 10.2215/CJN.02320220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lanktree MB, Iliuta IA, Haghighi Aet al. Evolving role of genetic testing for the clinical management of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2019;34:1453–60. 10.1093/ndt/gfy261 [DOI] [PubMed] [Google Scholar]
- 22. Nestor JG, Marasa M, Milo-Rasouly Het al. Pilot study of return of genetic results to patients in adult nephrology. Clin J Am Soc Nephrol 2020;15:651–64. 10.2215/CJN.12481019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Snoek R, Stokman MF, Lichtenbelt KDet al. Preimplantation genetic testing for monogenic kidney disease. Clin J Am Soc Nephrol 2020;15:1279–86. 10.2215/CJN.03550320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. (PACI) PACI . Kuwaiti private households by size and gender and number of members. https://www.paci.gov.kw/ (20 April 2022, date last accessed). [Google Scholar]
- 25. Pei Y, Obaji J, Dupuis Aet al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 2009;20:205–12. 10.1681/ASN.2008050507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levey AS, Stevens LA, Schmid CHet al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–12. 10.7326/0003-4819-150-9-200905050-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benson KA, Murray SL, Senum SRet al. The genetic landscape of polycystic kidney disease in Ireland. Eur J Hum Genet 2021;29:827–38. 10.1038/s41431-020-00806-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Gastel MDA, Edwards ME, Torres VEet al. Automatic measurement of kidney and liver volumes from MR images of patients affected by autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2019;30:1514–22. 10.1681/ASN.2018090902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grantham JJ, Torres VE, Chapman ABet al. Volume progression in polycystic kidney disease. N Engl J Med 2006;354:2122–30. 10.1056/NEJMoa054341 [DOI] [PubMed] [Google Scholar]
- 30. Chapman AB, Guay-Woodford LM, Grantham JJet al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003;64:1035–45. 10.1046/j.1523-1755.2003.00185.x [DOI] [PubMed] [Google Scholar]
- 31. Perrone RD, Mouksassi MS, Romero Ket al. Total kidney volume is a prognostic biomarker of renal function decline and progression to end-stage renal disease in patients with autosomal dominant polycystic kidney disease. Kidney Int Rep 2017;2:442–50. 10.1016/j.ekir.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Torres VE, Chapman AB, Devuyst Oet al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012;367:2407–18. 10.1056/NEJMoa1205511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alkandari A, Alarouj M, Elkum Net al. Adult diabetes and prediabetes prevalence in Kuwait: data from the cross-sectional Kuwait Diabetes Epidemiology Program. J Clin Med 2020;9:3420. 10.3390/jcm9113420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weiderpass E, Botteri E, Longenecker JCet al. The prevalence of overweight and obesity in an adult Kuwaiti population in 2014. Front Endocrinol (Lausanne) 2019;10:449. 10.3389/fendo.2019.00449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hwang YH, Conklin J, Chan Wet al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2016;27:1861–8. 10.1681/ASN.2015060648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lanktree MB, Guiard E, Akbari Pet al. Patients with protein-truncating PKD1 mutations and mild ADPKD. Clin J Am Soc Nephrol 2021;16:374–83. 10.2215/CJN.11100720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brook-Carter PT, Peral B, Ward CJet al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease—a contiguous gene syndrome. Nat Genet 1994;8:328–32. 10.1038/ng1294-328 [DOI] [PubMed] [Google Scholar]
- 38. Pan X, Yang C, Ma Set al. A case of TSC2-PKD1 contiguous deletion syndrome: clinical features and effective treatment for epilepsy. Int J Dev Neurosci 2021;81:191–9. 10.1002/jdn.10088 [DOI] [PubMed] [Google Scholar]
- 39. Osumi K, Suga K, Ono Aet al. Molecular diagnosis of an infant with TSC2/PKD1 contiguous gene syndrome. Hum Genome Var 2020;7:21. 10.1038/s41439-020-0108-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harris PC. The TSC2/PKD1 contiguous gene syndrome. Contrib Nephrol 1997;122:76–82. 10.1159/000059872 [DOI] [PubMed] [Google Scholar]
- 41. Rossetti S, Consugar MB, Chapman ABet al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007;18:2143–60. 10.1681/ASN.2006121387 [DOI] [PubMed] [Google Scholar]
- 42. Bogdanova N, Markoff A, Gerke Vet al. Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics 2001;74:333–41. 10.1006/geno.2001.6568 [DOI] [PubMed] [Google Scholar]
- 43. Ali H, Al-Mulla F, Hussain Net al. PKD1 duplicated regions limit clinical utility of whole exome sequencing for genetic diagnosis of autosomal dominant polycystic kidney disease. Sci Rep 2019;9:4141. 10.1038/s41598-019-40761-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Torra R, Badenas C, Perez-Oller Let al. Increased prevalence of polycystic kidney disease type 2 among elderly polycystic patients. Am J Kidney Dis 2000;36:728–34. 10.1053/ajkd.2000.17619 [DOI] [PubMed] [Google Scholar]
- 45. Lanktree MB, Guiard E, Li Wet al. Intrafamilial variability of ADPKD. Kidney Int Rep 2019;4:995–1003. 10.1016/j.ekir.2019.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ali H, Hussain N, Naim Met al. A novel PKD1 variant demonstrates a disease-modifying role in trans with a truncating PKD1 mutation in patients with autosomal dominant polycystic kidney disease. BMC Nephrol 2015;16:26. 10.1186/s12882-015-0015-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schrier RW, Abebe KZ, Perrone RDet al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014;371:2255–66. 10.1056/NEJMoa1402685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cornec-Le Gall E, Audrezet MP, Rousseau Aet al. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2016;27:942–51. 10.1681/ASN.2015010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schonauer R, Baatz S, Nemitz-Kliemchen Met al. Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet Med 2020;22:1374–83. 10.1038/s41436-020-0816-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sekine A, Hoshino J, Fujimaru Tet al. Genetics may predict effectiveness of tolvaptan in autosomal dominant polycystic kidney disease. Am J Nephrol 2020;51:745–51. 10.1159/000509817 [DOI] [PubMed] [Google Scholar]
- 51. AlSahow A, AlHelal B, Alyousef Aet al. Renal data from the Arab world: dialysis in Kuwait: 2013–2019. Saudi J Kidney Dis Transpl 2020;31:826–30. 10.4103/1319-2442.292317 [DOI] [PubMed] [Google Scholar]
- 52. Torres VE, Chapman AB, Devuyst Oet al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med 2017;377:1930–42. 10.1056/NEJMoa1710030 [DOI] [PubMed] [Google Scholar]
- 53. Torres VE, Chapman AB, Devuyst Oet al. Multicenter study of long-term safety of tolvaptan in later-stage autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2020;16:48–58. 10.2215/CJN.10250620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu D, Ma Y, Gu Xet al. Novel mutations in the PKD1 and PKD2 genes of Chinese patients with autosomal dominant polycystic kidney disease. Kidney Blood Press Res 2018;43:297–309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared upon reasonable request to the corresponding author.