

Abstract
Background:
In a previous study, ondansetron, a serotonin 5-HT3 receptor antagonist, reduced drinking intensity (drinks/drinking day [DPDD]) among European-ancestry (EA) participants with moderate-to-severe alcohol use disorder (AUD) and variants in genes encoding the serotonin transporter (SLC6A4), and 5-HT3A (HTR3A) and 5-HT3B (HTR3B) receptors. We tested whether 1) ondansetron reduces DPDD among individuals of either European or African ancestry (AA) and 2) that reductions in DPDD are greatest among ondansetron-treated individuals with population-specific combinations of genotypes at SLC6A4, HTR3A, and HTR3B.
Methods:
In this 16-week, double-blind, placebo-controlled, parallel-group clinical trial, adults with AUD were randomized to receive low-dose oral ondansetron (0.33 mg twice daily) or placebo stratified by “responsive” vs. “non-responsive” genotype defined using population-specific genotypes at the three genetic loci. Generalized estimating equation regression models and a modified intent-to-treat analysis were used to compare the treatment groups on the primary outcome—DPDD—and two secondary outcomes—heavy drinking days per week [HDD] and drinks per day [DPD] across 16 weeks of treatment.
Results:
Of 296 prospective participants screened, 95 (58 EA and 37 AA) were randomized and received at least one dose of study medication. In the modified intent-to-treat analysis, the ondansetron group averaged 0.40 more DPDD (p=0.51), 1.35 times as many HDD (p=0.16), and 1.06 times as many DPD (p=0.59) as the placebo group. There were no significant interactions with genotype. There were no study-related serious adverse events (AEs) and similar proportions of participants in the two treatment groups experienced AEs across organ systems.
Conclusions:
We found no evidence that low-dose oral ondansetron is beneficial in the treatment of AUD, thus failing to confirm prior study findings. However, the study was underpowered to identify medication by genotype interactions.
Keywords: ondansetron, alcohol use disorder, randomized control trial, serotonin, pharmacogenetics
INTRODUCTION
Alcohol use disorder (AUD) is a heterogeneous, chronic, relapsing condition characterized by both acute and chronic heavy drinking (Witkiewitz et al., 2019). Globally, AUD is the seventh most common, and in the United States the third most common, preventable cause of morbidity and mortality (National Institute on Alcohol Abuse and Alcoholism, 2019). In 2020, 11.0% of US individuals 18 or older (27.6 million people) had a past-year alcohol use disorder (Substance Abuse and Mental Health Services Administration, 2021).
Despite the substantial clinical and public health burden of AUD, there is no broadly effective treatment for the disorder. Medications currently approved by the US Food and Drug Administration have modest therapeutic effects and are not well tolerated by some patients (Witkiewitz et al., 2019; Kranzler and Soyka, 2018). The heterogeneity in treatment response is due, in part, to individual differences in the molecular pathophysiology of AUD. A personalized pharmacogenetic approach offers the prospect of greater efficacy and tolerability by matching participants with specific genetic variants to appropriate treatments to increase the probability of a positive treatment outcome while potentially reducing adverse events.
Variation in the SLC6A4 gene, which encodes the serotonin transporter (5-HTT) protein, offers a promising target for a personalized approach to AUD pharmacotherapy. The 5-HTT is the main regulator of synaptic serotonin levels that modulate physiologic mechanisms underlying drinking behaviors (Kelai et al., 2003; Boyce-Rustay et al., 2006). Several studies have shown that two polymorphisms in SLC6A4 (5-HTTLPR in the promotor region and rs1042173 in the 3’-UTR region) differentially alter 5-HTT mRNA expression levels in individuals with AUD. These findings were subsequently validated in vitro in cells exposed to alcohol (Seneviratne et al., 2009; Seneviratne and Johnson, 2012; Johnson et al., 2008). These allelic variants also influenced the treatment response to ondansetron in adults with AUD. In an 11-week, controlled clinical trial involving 283 alcohol-dependent adults of European ancestry (EA).Participants with the 5-HTTLPR:LL genotype, either alone or in combination with the 3′-UTR variant rs1042173:TT, who received ondansetron (4 μg/kg orally, twice daily) had a significantly greater reduction in heavy drinking than those receiving placebo (Johnson et al., 2011). In a human laboratory study with a naturalistic drinking component, treatment with ondansetron (0.5 mg/day orally) for three weeks significantly reduced drinking intensity, compared to the 7 days preceding treatment, in adults with AUD and the 5-HTTLPR:LL genotype (Kenna et al., 2014; Kenna et al., 2009).
Ondansetron is a specific antagonist at serotonin-3 (5HT3) receptors, which in pre-clinical models are upregulated by the direct effects of alcohol (Engleman et al., 2008; Shelton et al., 2004). Variation in SLC6A4 is thought to increase the availability of synaptic serotonin, stimulating post-synaptic 5HT3 receptors, an effect counteracted by ondansetron’s 5HT3 receptor blockade. A post hoc analysis of the study (Johnson et al., 2011) evaluated the effect of allelic variation in genes encoding the 5-HT3A and 5-HT3B receptors (HTR3A and HTR3B, respectively) on the response to treatment with ondansetron (Johnson et al., 2013). It showed that three HTR3A and HTR3B genotypes (from among 18 SNPs selected for study) were significantly associated with a positive treatment response to ondansetron among EA participants.
The present study sought to confirm this pharmacogenetic effect on ondansetron’s efficacy and address several limitations of the earlier post-hoc pharmacogenetic analysis. First, we sought to expand the study population to include African ancestry (AA) adults with AUD and recruit an independent sample of EA adults. Second, we prospectively stratified the randomization using predicted ondansetron “responsive” and “non-responsive” genotype subgroups in both populations. Because different, but overlapping, variants in HTR3A and HTR3B are potential moderators of ondansetron response in the two population groups, we used different genotype combinations to define responsive and non-responsive subgroups in AAs and EAs.
We hypothesized that ondansetron would reduce drinking intensity (i.e., drinks/drinking day), the measure on which it showed the greatest efficacy in a prior study (Johnson et al., 2011), among individuals with the population-specific combination of responsive genotypes.
MATERIALS AND METHODS
Study Design.
We conducted a 16-week, randomized, double-blind, placebo-controlled, 2-arm, parallel-group, phase 2 clinical trial to determine the efficacy of oral ondansetron (0.33 mg twice daily) in reducing drinking intensity among heavy drinking adults with AUD. The randomization was stratified on selected genotypes at the 5-HTTLPR, 5-HT3A and 5-HT3B receptor genes (SLC6A4, HTR3A, and HTR3B, respectively). All participants received a low-intensity psychosocial intervention (Brief Behavioral Compliance Enhancement Treatment [BBCET]) (Johnson et al., 2007) at each treatment visit. The study was conducted from August 2015 to March 31, 2020 (when funding ended), at the University of Pennsylvania (UPenn) in Philadelphia, PA, and the University of Maryland Baltimore (UMB) in Baltimore, MD. The study was approved by the institutional review board at each site and the conduct of the study was monitored by a single independent data and safety monitoring board.
Participants and Procedures.
EA or AA men and women aged 18 to 70 were recruited through print, broadcast, and internet advertisements, distribution of flyers at relevant clinical and non-clinical community sites, and by outreach to potentially eligible patients identified in medical record searches in the Philadelphia and Baltimore metropolitan areas. Interested participants were briefly screened by telephone after they gave oral consent and, if potentially eligible, were invited for in-person screening.
At the in-person visit, after providing written informed consent, participants received a comprehensive medical and psychiatric evaluation to establish study eligibility. This included the Structured Clinical Interview for the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (SCID-IV) (First et al., 2002) administered by trained interviewers (except for the diagnosis of AUD, which was made using diagnostic criteria of the Diagnostic and Statistical Manual of Mental Disorders, 5th edition [DSM-5]), a standard medical history and physical examination, clinical laboratory tests (chemistry panel, CBC, urinalysis), 12-lead ECG, urine drug screen, urine pregnancy test (for women of reproductive potential), the Alcohol Use Disorders Identification Test (AUDIT), and an alcohol and other substance use history using the Time Line Follow-back method (TLFB) (Sobell et al., 1996) to cover the preceding 90 days.
Eligibility criteria
Inclusion criteria.
Participants were required to have a DSM-5 diagnosis of current alcohol use disorder (and an AUDIT score > 8 at the UMB site). Women had to consume ≥14 standard drinks/week and have ≥6 heavy drinking days (HDDs) [i.e., ≥4 standard drinks in a day (one standard drink = 14 g alcohol) and men had to consume ≥21 standard drinks/week and ≥6 HDDs, (≥5 standard drinks in a day) over the previous 28 days and be currently drinking during the 7 days prior to randomization. Participants had to report an intent to reduce or stop drinking, have a 6th grade or higher English language reading level, use adequate contraception (if a woman of reproductive potential); weigh < 110 kg; and self-identify as EA or AA. When ancestry was ambiguous, we used 24 genetic ancestry informative markers (AIMs) to determine ancestral proportions prior to randomization and included only participants with >75% European or >75% African ancestry (Johnson et al., 2011).
Exclusion criteria.
Participants were excluded if they reported >14 consecutive abstinent days in the 4 weeks preceding screening; had a Clinical Institute Withdrawal Assessment for Alcohol scale-revised (CIWA-Ar) score (Sullivan et al., 1989) >10 at the baseline visit, or any of the following current psychiatric disorders: schizophrenia or other psychotic disorder, bipolar disorder, severe major depressive disorder, post-traumatic stress disorder, panic disorder, eating disorder, or substance use disorder (other than alcohol, tobacco, or cannabis); current or recent (within 4 weeks prior to baseline) treatment with any medication likely to interact with ondansetron to produce adverse effects or with any investigational medicinal product within 30 days; current or recent participation in an AUD treatment program; have a score of >9 on the suicide risk assessment (module B) of the Mini International Psychiatric Interview (MINI) version 6.0 (Sheehan et al., 1998); have a clinically significant, unstable physical illness, body weight > 110 kg, a clinically significant abnormality on laboratory tests or ECG, a current or past history of clinically significant QT prolongation, or a first-degree biological relative with long QT syndrome.
Participants who met study eligibility criteria were randomized to receive treatment with either ondansetron or placebo using a block randomization procedure that balanced treatment groups within subgroups stratified by study site, race (EA or AA), sex, and genotype (Table 1). A pharmacist at the UMB Investigational Drug Service (IDS), who was not otherwise involved in the study, performed the randomization and assignment of eligible participants into treatment groups for both sites. All participants and other study staff were blinded to treatment condition.
Table 1:
Genotype groups used in ondansetron-responsive stratification for participant randomization
| Ancestry/Race | Gene | Ondansetron Responsive Genotypes | Ondansetron Non-Responsive Genotypes |
|---|---|---|---|
| European Ancestry | SLC6A4 | 5-HTTLPR:LL | 5-HTTLPR:LS/SS |
| rs25531:AA | rs25531:AG/GG | ||
| 5-HTTLPR + rs25531 = LALA | 5-HTTLPR + rs25531 = LGLA; LGLG; LAS; LGS; SS | ||
| rs1042173:TT | rs1042173:TG/GG | ||
| HTR3A | rs1150226: AG | rs1150226: AA/GG | |
| rs1176713: GG | rs1176713: GA/AA | ||
| HTR3B | rs17614942: AC | rs17614942: AA/CC | |
| African Ancestry | SLC6A4 | 5-HTTLPR:LL | 5-HTTLPR:LS/SS |
| rs25531:AA | rs25531:AG/GG | ||
| 5-HTTLPR + rs25531 = LALA | 5-HTTLPR + rs25531 = LGLA; LGLG; LAS; LGS; SS | ||
| rs1042173:TT | rs1042173:TG/GG | ||
| HTR3B | rs1176744:CC/CA | rs1176744:AA |
SLC6A4 – Serotonin transporter gene; HTR3A – gene encoding serotonin receptor subunit 3A; HTR3B – gene encoding serotonin receptor subunit 3B.
EA participants were stratified into two subgroups based on the presence of one or a combination of the ondansetron responsive genotypes (Table 1) as previously identified (Johnson et al., 2013) to achieve an equal probability of potentially responsive and non-responsive participants being assigned to receive ondansetron or placebo. Specifically, we used the same 5 genotypes tested in the preliminary study (Johnson et al., 2013) among EAs, and a 3-genotype combination in AAs that included the same SLC6A4 genotypes (5-HTTLPR:LL and rs1042173:TT), but a different, population-relevant HTR3B polymorphism. As the efficacy of ondansetron has not been specifically tested in predominantly AA populations stratified by HTR3A or HTR3B variants, we chose a missense polymorphism – rs1176744 in HTR3B – that has been associated with AUD in AAs and with various metabolic and substance use disorders (Hammer et al., 2009; Enoch et al., 2011; Kwon et al., 2019; Han et al., 2018; Horjales-Araujo et al., 2013). The prevalence of the minor rs1176744:C allele is highest among African-ancestry individuals (~40%)(Sherry et al., 2001).
AA participants were first stratified into two rs1176744 genotype subgroups: CC/CA or AA, with each subgroup having an equal probability of being assigned to receive ondansetron or placebo. Each rs1176744 genotype subgroup was then balanced by two additional polymorphisms: 5-HTTLPR and rs1042173. Based on these genotype combinations, there were 8 genotype strata (2 × 2 × 2 genotype combinations at the 3 loci) for AAs, and 2 genotype strata (responsive vs. non-responsive genotype combinations) for EAs. As described below, we reduced the multiple combinations among AAs to two strata—responsive and non-responsive genotype groups.
DNA extraction and genotyping procedures.
At the initial in-person screening visit, a peripheral venous whole blood sample was collected for DNA extraction and genetic analysis, which was conducted at the UMB Institute for Genome Sciences, according to previously established protocols (Johnson et al., 2011; Johnson et al., 2013). Genotyping the AIMs panel was conducted post hoc, except for participants whose self-reported ancestry was ambiguous (see Exclusion criteria above).
Following the baseline visit, participants entered a 16-week treatment period with weekly visits for the first 6 weeks, biweekly visits during weeks 7–12, and an end-point visit at week 16. A follow-up visit was conducted at week 20 to assess post-treatment safety and changes in drinking patterns. Assessments performed at each in-person visit are described below. The same study procedures were used at both sites.
Study medication.
Based on the weight-adjusted dose of ondansetron used in prior AUD clinical trials (4 μg/kg for individuals whose average weight was 82.5 kg) (Johnson et al., 2011; Johnson et al., 2000; Sellers et al., 1994), participants received ondansetron 0.33 mg or matching placebo capsules twice daily, formulated specifically for the study by the University of Maryland School of Pharmacy, Baltimore, MD. Each participant ingested an observed dose of ondansetron at the baseline visit (study Week 0) and the first weekly dose at study Weeks 4 and 8.
Study visits.
At each treatment visit, participants had their breath alcohol concentration, body weight and vital signs measured. Blood samples for planned RNA expression studies were obtained at weeks 4, 8 and 12; urine screen for drugs of abuse at week 8, urine pregnancy test at weeks 4, 8, 12, and 16; and ECG at visit 5. At every visit, participants completed the Other Drug Use questionnaire, were queried regarding concomitant medication and treatment received since the last visit, reported study medication adherence for comparison with a count of returned medication and medication packaging, and were queried regarding adverse events.
Assessments
Measures of alcohol use.
The 10-item AUDIT (Bohn et al., 1995) was used at screening to assess hazardous/harmful alcohol consumption. The TLFB method (Sobell et al., 1996) was used to assess participants’ daily alcohol intake (in standard drinks) at each visit.
Primary efficacy outcome measure:
Weekly mean drinks per drinking day (DPDD) (adjusted for baseline value [as covariate] in the statistical analysis).
Secondary efficacy outcome measures:
Weekly number of drinking days (DD), weekly number of heavy drinking days (HDD), and weekly absence of heavy drinking days (No HDD).
Medication adherence.
Adherence to study medication was assessed at each treatment visit by self-report and by asking participants to return medication packaging and all unused study medication.
Adverse events.
Adverse events were assessed by open-ended questions at each visit.
Psychosocial counseling.
Manualized brief (lasting 10–15 minutes) individual counseling (BBCET) was delivered at each visit by a trained nurse or master’s-level clinician. BBCET uses a motivational enhancement approach to improve treatment adherence, especially for study medication (see Supplementary Material).
Follow-up visit.
An in-person or telephone follow-up visit was conducted at study Week 20, to assess the persistence of adverse events.
Other questionnaires and rating scales that were administered included the Clinical Institute Withdrawal Assessment Alcohol Scale Revised (CIWA-Ar) (Sullivan et al., 1989), MINI International Neuropsychiatric Interview (MINI 6.0) Suicide Risk Assessment Scale (Sheehan et al., 1998), Beck Depression Inventory (BDI) (Beck et al., 1993), and Short Index of Problems (SIP) (Alterman et al., 2009). These questionnaires and BBCET are described in Supplementary Table S1 and Supplementary Material.
Statistical Analysis
Data quality assessments.
Data collected at both sites were entered directly into a HIPAA-compliant, web-accessible electronic data capture and database system hosted and maintained by the UPenn Data Management Unit. Additional details are presented in Supplementary Material.
Assessment of genetic ancestry proportions for statistical modeling.
We estimated genetic ancestry proportions for each participant using the software program Structure (Pritchard et al., 2000), as detailed in Supplementary Material, and used the individual ancestry proportion estimates as covariates in all statistical models.
Power analysis.
The original protocol called for a sample size of 256, drawn equally from the two sites. Its primary aims were to examine drug-placebo differences among individuals with responsive genotypes. Due to delays in initiating the study imposed by the funding agency, we reduced the intended sample size to a total of 128 and dropped consideration of multiple individual genotypes, focusing instead on a binary responsive/non-responsive subgrouping.
In the planned sample of N=128, the primary goal of the study was to examine the ondansetron effect in the subsample of genotype-responsive participants. For a two-sided test at an alpha level of 5%, based on an expected sample size of 73 genotype-responsive participants (i.e., using the same genotype combination to stratify both the AA and EA groups) equally split between the ondansetron and placebo groups, and assuming 15–20% dropout and a within-participant correlation of about 0.6, the study had 84% power for an effect of Cohen’s d=0.59 in the genotype-responsive group. The group sizes for genotype-responsive participants achieved in the study were 34 in the ondansetron group and 39 in the placebo group, which yields 85% power for an effect of Cohen’s d=0.63 in the genotype responsive group. The study was also planned to examine the overall ondansetron effect, pooling over genotype. Based on an expected sample size of 64 in each of the ondansetron and placebo arms (i.e., N=128 over both sites), with the same assumptions as above, the study had 81% power to detect an effect of size of Cohen’s d=0.44 in the difference in the primary outcome measure (DPDD) between the two treatment groups (irrespective of genotype). In the study, we had samples sizes of 46 in the ondansetron group and 49 in the placebo group. With the same assumptions as above, this yielded 80% power for an effect of size of Cohen’s d=0.53. The study also planned to explore the difference between the ondansetron effects in the genotype-responsive and non-responsive participants but was not powered for the ondansetron-by-genotype interaction. For example, it provided 80% power for an interaction effect of Cohen’s d=0.8, e.g., an ondansetron effect of Cohen’s d=0.1 in the genotype non-responsive group and Cohen’s d=0.90 in the genotype-responsive group.
Outcome Analyses.
Outcome measures were analyzed using an intent-to-treat principle and included only randomized participants who received at least one dose of study medication. Randomized participants who did not receive a first dose of study medication had their randomization code re-assigned to the next qualified participant at that study site. One randomized participant at the UMB site started study medication and was later discovered to be ineligible. Their data were excluded from analysis.
Baseline Comparisons:
Descriptive statistics were calculated for continuous (mean, standard deviation) and categorical (counts, proportions) variables. We compared baseline characteristics across groups using t-tests for continuous variables and χ2 tests for categorical variables. Factorial models crossing treatment assignment with genotype group (responsive vs. non-responsive) were analyzed using a linear model for continuous variables and logistic regression for binary variables.
Of the 95 participants included in the analysis, 67 were from the UPenn site and 28 from the UMB site.
Comparisons over the 16-week treatment period:
The numbers of missed treatment visits and weeks with medication non-adherence (based on self-report) were compared using negative binomial models with robust standard errors. A week was defined as non-adherent if the participant reported less than seven days of medication use in that week. The analysis of drinking outcomes used generalized estimating equations models, with a normal distribution and identity link function for the DPDD variable, a Poisson distribution and log-link function for the HDD and DD variables, and a binomial distribution with logit link for the binary no-heavy-drinking-days variable. The independent variables of primary interest were two binary factors indicating treatment group (ondansetron vs. placebo), and genotype (responsive vs. non-responsive). To account for randomization strata, the models included factors for study site (UPenn vs UMB), sex (male vs. female), and genotype (responsive vs. non-responsive), with the baseline version of the response included to reduce error variance. To account for within-participant correlations among the weekly repeated measures, the models used a first-order autoregressive working correlation structure with robust standard errors. We first examined site-by-treatment effects, based on a model that included the site-by-treatment interaction, and report estimated ondansetron effects within each site. Next, to address the primary hypothesis concerning the overall ondansetron effect, we estimated the effects from a model with neither genotype-by-treatment nor site-by-treatment interaction terms. To address the primary hypothesis on the ondansetron effect within responsive participants, we included a genotype-by-treatment interaction term, and report estimates of the ondansetron effect within each of the responsive and non-responsive participants. We used pattern mixture models to examine the impact of missing data, with a binary variable indicating whether a participant completed the 16 weeks of treatment as a summary of missing data. Comparing results using the adjustment for genotype as a binary responsiveness variable vs. using ancestry scores yielded the same conclusions. We report results based on the binary responsiveness variable and include the approach involving ancestry scores in Supplementary Material.
Secondary analyses examined similar hypotheses for the number of heavy drinking days and number of drinking days, using the same analytic approach as described above.
Adverse Events:
We used the Medical Dictionary for Regulatory Activities (MedRA) (https://www.meddra.org/) to categorize all adverse events (AEs) by organ system.
RESULTS
The final study sample included 95 participants who received at least one dose of study medication (see CONSORT diagram [Figure 1], which shows the flow of study participants). Sociodemographic and clinical characteristics at baseline are presented in Table 2. The study sample consisted predominantly of middle-aged (mean=51.2 years, SD=2.08, range=24–70), married (57.6%), employed (81.2%), white (63%) males (71.2%) with a college education (mean=15.7 years of education, SD=3.25, range=8–28). Over the 3 months prior to the baseline visit, participants drank alcohol on a mean of 6.0 days per week (SD=1.4, range=0.7–7.0), drinking heavily on 5.1 days per week (SD=1.8, range=0.7–7.0). The lifetime prevalence of major depression was 21.5% and of any anxiety disorder 14.7%. There were no significant differences between the ondansetron and placebo groups on any baseline sociodemographic or clinical characteristics. The genotype frequencies were consistent with Hardy-Weinberg equilibrium (HWE) expectations for all polymorphisms tested in this study, as assessed by the software program Haploview (v. 4.0)(Barrett et al., 2005). For the 5 participants included in our analyses with ambiguous self-reported ancestry (UPenn=4; UMB=1), we inferred genetic ancestry using AIMs, which did not differ from self-reported ancestry for any of these individuals.
Figure 1:
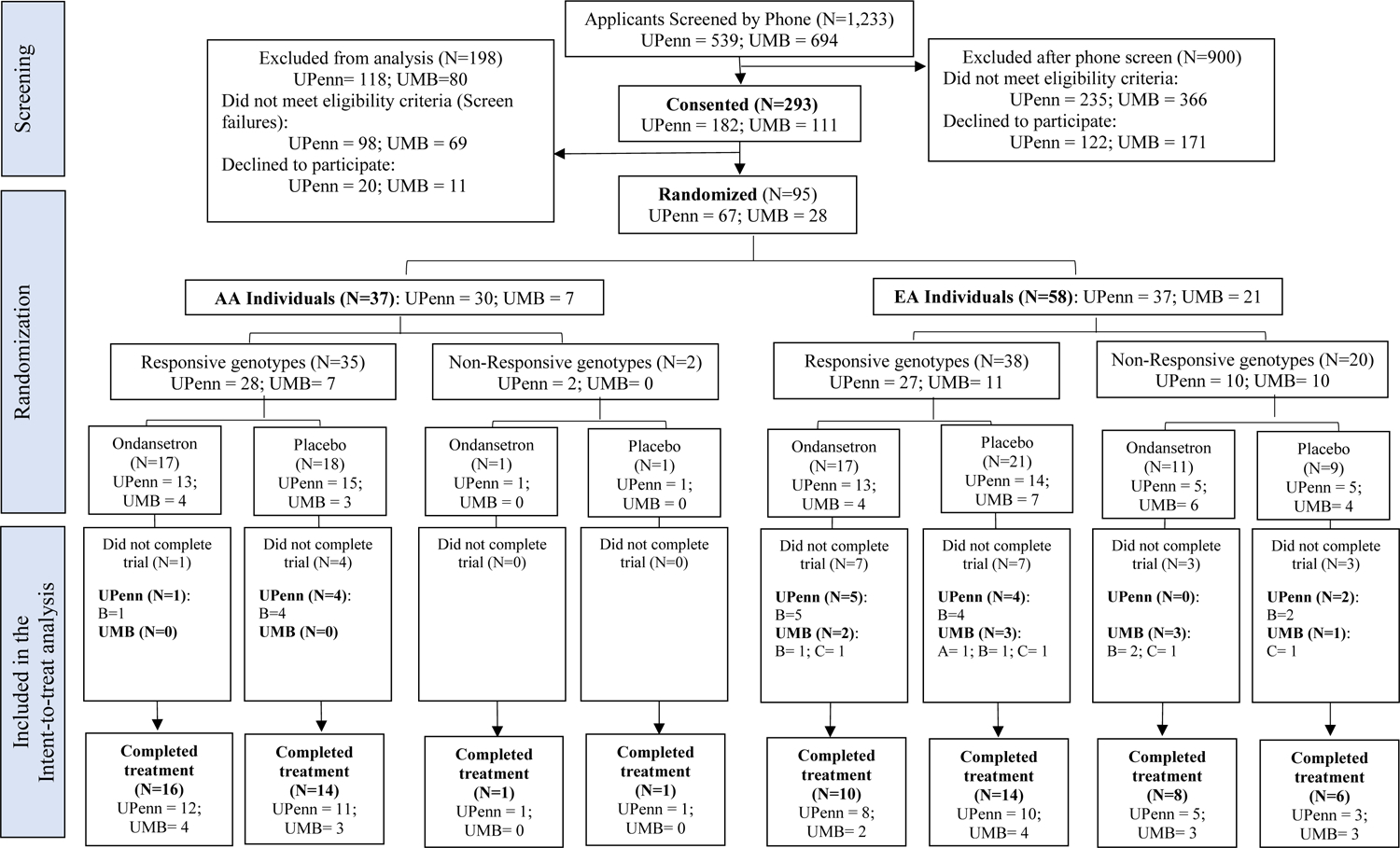
Consort flow diagram
A – Limiting adverse event; B – Lost to follow-up; C – Participant choice; D – Lack of efficacy; E – Other.
Table 2:
Baseline sociodemographic and clinical characteristics of randomized participants, by study site
| Demographics | Overall (N=95) N (% or SD) |
UPenn (N=67) N (% or SD) |
UMB (N=28) N (% or SD) |
P-value |
|---|---|---|---|---|
| Sex (Male) | 67 (70.1%) | 45 (67%) | 24 (86%) | 0.06 |
| Age (yr) | 51.5 (11.3) | 52.5 (11.1) | 49.0 (11.5) | 0.17 |
| Race (White) | 60 (63%) | 38 (56.7%) | 22 (78.6%) | 0.04 |
| Marital Status1 | 50 (53%) | 38 (57%) | 12 (43%) | 0.22 |
| Genotype (% Responsive) | 73 (77%) | 55 (82%) | 18 (64%) | 0.06 |
| Employment Status | 0.11 | |||
| Full-time | 49 (52%) | 30 (45%) | 19 (68%) | |
| Part-time | 14 (15%) | 12 (18%) | 2 (7%) | |
| Not Working | 32 (34%) | 25 (37%) | 7 (25%) | |
| Years of Education | 15.7 (3.2) | 15.9 (3.4) | 15.4 (3.0) | 0.55 |
| Annual Income Level | 0.60 | |||
| <$40,000 | 32 (34%) | 22 (33%) | 10 (36%) | |
| $40,000 - $79,999 | 17 (18%) | 11 (17%) | 6 (21%) | |
| $80,000 - $119,000 | 16 (17%) | 10 (15%) | 6 (21%) | |
| ≥$120,000 | 29 (31%) | 23 (35%) | 6 (21%) | |
| Clinical Measures | ||||
| AUDIT score | 20.2 (6.4) | 19.3 (6.2) | 22.4 (6.4) | 0.03 |
| Height (inches) | 68.0 (3.2) | 67.6 (2.9) | 69.1 (3.7) | 0.06 |
| Weight (pounds) | 183.6 (34.9) | 179.8 (31.4) | 192.5 (41.4) | 0.10 |
| BMI | 27.8 (4.5) | 27.6 (4.4) | 28.1 (4.9) | 0.65 |
| Lifetime Major Depression | 12 (13%) | 12 (17%) | 0 (0%) | 0.02 |
| Lifetime Anxiety Disorder | 10 (11%) | 9 (13%) | 1 (4%) | 0.15 |
| BDI Score | 5.2 (5.2) | 5.5 (5.2) | 4.6 (5.1) | 0.43 |
| Drinks per Drinking Day2 | 6.4 (2.8) | 6.0 (2.6) | 7.5 (3.1) | 0.01 |
| Pct. Drinking Days2 | 86.9 (18.5) | 85.6 (20.3) | 89.9 (13.5) | 0.31 |
| Pct. Heavy Drinking Days2 | 74.0 (18.3) | 69.9 (26.0) | 83.9 (20.5) | 0.01 |
| SIP Score2 | 13.4 (9.4) | 12.9 (9.3) | 14.5 (9.6) | 0.44 |
Married or cohabiting;
During the 90 days preceding the screening visit; SD-standard deviation; BDI-Beck Depression Inventory; SIP-Short Index of Problems
Treatment Completion.
There was no significant difference in 16-week study completion rate between the ondansetron group (89%, 41 participants) and the placebo group (78%, 38 participants) (χ2(1)=2.27, p=0.13, OR=2.27, 95% CI=(0.75, 7.46).
Self-reported Medication Adherence.
The placebo group averaged 5.87 days (SD=2.01, range 0.5–7) of self-reported medication ingestion per week, while the ondansetron group averaged 5.79 (SD=2.05, range 0–7). The medication groups were also comparable on the number of weeks of full adherence to treatment, defined as having taken medication on all 7 days in a week [ondansetron: mean=12.59, SD=4.67, range 0–16; placebo: mean=12.84, SD=4.54, range 1–16; χ2(1)=0.04, p=0.85). The interaction of genotype group with medication group [χ2(1) =0.20, p=0.65] and the main effect of genotype group on weeks of treatment received [χ2(1)=0.28, p=0.63] were not significant.
Ondansetron Effects: Interactions with Site and Overall
Primary outcome measure (DPDD):
The site-by-treatment effect was non-significant (GEE χ2(1)=3.14, p=0.08). At the UPenn site, the ondansetron group averaged 0.99 (SE=0.46, 95% CI=0.10–1.88, p=0.03) more DPDD per week than the placebo group. At the UMB site, the ondansetron group averaged 0.84 (SE=0.90, 95% CI=−0.93–2.62, p=0.35) fewer DPDD than the placebo group. Pooling across the two sites, the ondansetron group averaged 0.48 (SE=0.42, 95% CI=−0.35–1.31) more DPDD per week than the placebo group, which was not significant (GEE χ2(1)=1.27, p=0.26).
Secondary outcome measure (HDD):
The site-by-treatment effect was non-significant (GEE χ2(1)=2.11, p=0.15). At the UMB site, the mean number of HDD per week in the ondansetron group was 0.91 (SE=1.32, 95% CI=0.52–1.57, p=0.73) times the mean in the placebo group, while at the UPenn site the mean number of HDD per week in the ondansetron group was 1.45 (SE=1.16, 95% CI=1.08–1.57, p=0.02) times the mean in the placebo group. Pooling across the two sites, the mean number of HDD per week in the ondansetron group was 1.24 (SE=1.15, 95% CI=0.95–1.62, p=0.12) times the mean in the placebo group.
Secondary outcome measure (DD):
The site-by-treatment effect was non-significant (GEE χ2(1)=0.10, p=0.76). At the UMB site, the mean number of DD per week in the ondansetron group was 1.08 (SE=1.15, 95% CI=0.82–1.42, p=0.59) times the mean in the placebo group, while at the UPenn site mean number of DD per week in the ondansetron group was 1.03 (SE=1.08, 95% CI=0.88–1.20, p=0.75) times the mean in the placebo group. Pooling across the two sites, the mean number of DD per week in the ondansetron group was 1.04 (SE=1.07, 95% CI=0.91–1.19, p=0.59) times the mean in the placebo group.
Secondary outcome measure (No Heavy Drinking Days):
The site-by-treatment effect was non-significant (GEE χ2(1)=0.92, p=0.32). At the UMB site, the odds of No HDD in the ondansetron group were 0.92 times the corresponding odds in the placebo group (SE=1.75, 0.31–2.73, p=0.87), while at the UPenn site the odds of No HDD in the ondansetron group was 1.85 times the corresponding odds in the placebo group (SE=1.49, 0.88–3.98, p=0.13). Pooling across the two sites, the odds of No HDD in the ondansetron group was 1.49 times the corresponding odds in the placebo group (SE=1.39, 0.79–2.84, p=0.22).
Effects on the three outcome measures (i.e., DPDD, HDD, DD, and No HDD) were identical after adjustment for body weight at screening (see Supplementary Material).
Moderating Effect of Genotype Group on the Response to Ondansetron.
Table 3 shows the sociodemographic and clinical characteristics at baseline for the genotype-by-treatment groups. For the DPDD outcome variable, the treatment-by-genotype (responsive vs. non-responsive) interaction effect was not significant (GEE χ2(1)=0.04, p=0.85). In the responsive group, the ondansetron group averaged 0.43 (95% CI=(−0.48,1.35), p=0.35) more DPDD per week than the placebo group, compared to 0.64 (95% CI=(−1.26,2.55), p=0.51) in the non-responsive group. For the HDD outcome variable, the interaction was also non-significant (χ2(1)=0.23, df=1, p=0.63). In the responsive group, the mean number of HDD per week was 1.28 (95% CI=(0.96,1.72), p=0.09) times the mean in the placebo group, compared to 1.08 (95% CI=(0.58,2.00), p=0.80) in the non-responsive group. For the DD outcome variable, the interaction was also non-significant (χ2(1)=0.06, df=1, p=0.81). In the responsive group, the mean number of DD per week was 1.03 (95% CI=(0.88,1.20), p=0.71) times the mean in the placebo group, compared to 1.07 (95% CI=(0.78,1.48), p=0.66) in the non-responsive group. For the No Heavy Drinking Days response, the genotype by treatment effect was non-significant (GEE χ2(1)=0.61, p=0.43). In the responsive group the odds of No HDD in the ondansetron group was 1.29 times the corresponding odds in the placebo group (SE=1.46, 0.61–2.72, p=0.50), while in the non-responsive group the odds of No HDD in the ondansetron group was 2.37 times the corresponding odds in the placebo group (SE=1.92, 0.66–8.52, p=0.19).
Table 3:
Baseline sociodemographic and clinical characteristics of randomized participants, by treatment group
| Demographics | Overall (N=95) N (% or SD) | Ondansetron (N=46) N (% or SD) | Placebo (N=49) N (% or SD) | P-value |
|---|---|---|---|---|
| Number of participants by study site (UPenn) | 67 (71%) | 32 (48%); | 35 (52%); | 0.84* |
| Sex (Male) | 67 (70.1%) | 34 (49%) | 35 (51%) | 0.79 |
| Age (yr) | 51.5 (11.3) | 52.9 (9.8) | 50.1 (12.4) | 0.23 |
| Race (White) | 60 (63%) | 28 (47%) | 32 (53%) | 0.65 |
| Marital Status1 | 50 (53%) | 19 (41%) | 31 (63%) | 0.03 |
| Genotype (% Responsive) | 73 (77%) | 34 (47%) | 39 (53%) | 0.51 |
| Employment Status | 0.55 | |||
| Full-time | 49 (52%) | 22 (48%) | 27 (55%) | |
| Part-time | 14 (15%) | 6 (13%) | 8 (16%) | |
| Not Working | 32 (34%) | 18 (39%) | 14 (29%) | |
| Years of Education | 15.7 (3.2) | 15.6 (3.5) | 15.8 (3.0) | 0.76 |
| Annual Income Level | 0.08 | |||
| <$40,000 | 32 (34%) | 18 (40%) | 14 (29%) | |
| $40,000 - $79,999 | 17 (18%) | 11 (24%) | 6 (12%) | |
| $80,000 - $119,000 | 16 (17%) | 4 (9%) | 12 (24%) | |
| ≥$120,000 | 29 (31%) | 12 (27%) | 17 (35%) | |
| Clinical Measures | ||||
| AUDIT | 20.2 (6.4) | 21.2 (6.7) | 19.3 (6.0) | 0.13 |
| Height (inches) | 68.0 (3.2) | 67.5 (3.2) | 68.5 (3.2) | 0.09 |
| Weight (pounds) | 183.6 (34.9) | 178.6 (35.7) | 188.2 (33.9) | 0.46 |
| BMI | 27.8 (4.5) | 27.4 (4.8) | 28.1 (4.3) | 0.62 |
| Lifetime Major Depression | 12 (13%) | 5 (11%) | 7 (14%) | 0.62 |
| Lifetime Anxiety Disorder | 10 (11%) | 7 (15%) | 3 (6%) | 0.15 |
| BDI Score | 5.2 (5.2) | 5.9 (5.5) | 4.7 (4.8) | 0.27 |
| Drinks per Drinking Day2 | 6.4 (2.8) | 6.5 (2.8) | 6.4 (2.9) | 0.80 |
| Pct. Drinking Days2 | 86.9 (18.5) | 86.3 (20.9) | 87.4 (16.2) | 0.77 |
| Pct. Heavy Drinking Days2 | 74.0 (18.3) | 75.8 (25.7) | 87.49 (16.2) | 0.52 |
| SIP Score2 | 13.4 (9.4) | 14.3 (10.2) | 12.5 (8.5) | 0.34 |
P-value for the comparison of study site by treatment; the other p-values refer to treatment analysis ignoring site; chi-square test for categorical variables and Kruskal-Wallis test for continuous variables
The results from models based on the ancestry scores are presented in Supplementary Material.
Observed Effect Sizes:
The targeted medication effect size among responsive genotype participants was Cohen’s d=0.6, corresponding to an average weekly difference of 0.6 standard deviations between the mean DPDD of the ondansetron and placebo groups, with the placebo group expected to show higher rates. In our data, the ondansetron effect ranged from d=−0.43 to 0.20, with an average of d=−0.19, so the effect size was smaller than that targeted, and in the opposite direction. The overall ondansetron effect, pooling responsive and non-responsive genotype individuals, was similar, with weekly effect sizes ranging from d= −0.48 to d=0.17, with an average of d=−0.22. The average weekly interaction effect, i.e., the difference between the weekly effect in responsive and non-responsive individuals, was d=0.12.
Integrity of medication blinding.
The medication blinding response was available for 23 of the 28 UMB participants (82%) and 60 of the 67 UPenn participants (90%). At the UMB site, 7 of 12 (58%) ondansetron participants thought that they were on the active medication, compared to 5 of 11 (45%) of placebo participants (chi-square(1)=0.38, p=0.54); at the UPenn site, 8 of 30 (27%) ondansetron participants thought that they were on the active medication, compared to 12 of 30 (40%) of placebo participants (chi-square(1)=1.20, p=0.27). Controlling for site by the Cochran-Mantel-Haenszel procedure showed no association between actual treatment group and perceived treatment group (Chi-square(1)=0.33, p=0.57)
Impact of Missing Data on the Primary Outcome Measure.
Available weekly TLFB data were the same in both groups: mean of 14.70 (SD=3.84) weeks in the ondansetron group vs. 13.88 (4.35) weeks in the placebo group (Kruskal-Wallis χ2(1) =1.93, p=0.17). TLFB data were available for 80 participants (84% of total study sample) for treatment week 16. Study completion rates did not differ significantly between the ondansetron group (89% [41 of 46]) and the placebo group (80% [38 of 49]) (χ2=1.62, df=1, odds ratio (OR)=2.10, 95% CI=(0.66,6.70), p=0.20) or between the UPenn site (88% [59 of 67]) and the UMB site (75% [21 of 28]) (χ2=2.53, df=1, OR=2.46, 95% CI=(0.79,7.62), p=0.11). A pattern mixture analysis using a binary indicator of complete data as a summary of missing data showed results very similar to those reported above. The interaction between treatment and completion status was not significant (χ2=0.01, df=1, p=0.93). The site-by-treatment effect was non-significant (GEE χ2(1)=2.78, p=0.10). At the UPenn site, the ondansetron group averaged 1.03 (SE=0.48, 95% CI=0.07–1.96, p=0.03) more DPDD per week than the placebo group. At the UMB site, the ondansetron group averaged 0.84 (SE=0.99, 95% CI= −1.09–2.79, p=0.39) fewer DPDD than the placebo group. Pooling across the two sites, the ondansetron group averaged 0.49 (SE=0.46, 95% CI=−0.40–1.39, p=0.28) more DPDD per week than the placebo group.
Adverse Events (AE).
There was no significant difference in the proportion of participants who reported at least 1 AE in the ondansetron group [40 of 46 (87%)] and the placebo group [40 of 49 (82%)] (χ2 (1)=0.51, p=0.48). Among participants reporting at least 1 AE, the median number of AEs in both medication groups was 2. Types of AEs that occurred in at least 10% of participants and the number of participants from each medication group that experienced the event are summarized in Supplementary Table S2. One participant (1.05%), who was in the placebo group, experienced a serious adverse event—atrial fibrillation—which was not related to study participation.
DISCUSSION
In this double-blind, randomized, placebo-controlled study of 95 heavy-drinking adults with AUD, we tested the efficacy of ondansetron in reducing alcohol use by applying a personalized medicine approach based on six genotypes in three serotonergic genes—SLC6A4, HTR3A, and HTR3B. The genetic variants were previously reported either to moderate the treatment effects of ondansetron in adults with AUD (Johnson et al., 2013; Seneviratne and Johnson, 2012; Johnson et al., 2011) and/or increase the risk of developing AUD (Seneviratne et al., 2013; Enoch et al., 2011). This is the first prospective, randomized treatment trial to examine ondansetron treatment responses moderated by variation in serotonergic genes in both AA and EA individuals with AUD.
Ondansetron had no significant effect on alcohol intake in the overall study population. However, treatment response varied by study site. The UPenn site enrolled a greater percentage of AA individuals (43.3%) than the UMB site (11.4%; p=0.04). At UPenn, ondansetron was associated with greater DPDD (p=0.03) and a trend towards greater HDD (p=0.08), effects that were not observed at UMB. These site differences may have resulted from the different ancestral distribution of participants across the two sites. Ondansetron has not been studied specifically as a treatment for AUD in AA populations (Cservenka et al., 2017) and AAs account for less than 25% of the participants in published phase II clinical studies of ondansetron (Brown et al., 2021; Johnson et al.; 2000, Skowronek et al., 2006) Another possible explanation for the site differences in ondansetron effects could be sample bias. The UMB site had substantially fewer participants who completed treatment than the UPenn site. Nonetheless, neither the pooled sample nor the individual-site data showed a beneficial effect of ondansetron in treating AUD, consistent with recent studies showing no efficacy of the medication in majority EA populations except when a personalized genomic approach was applied (Johnson et al., 2013).
In contrast to the Johnson et al. (2013) study, we did not detect moderating (interaction) effects of selected genotypes on primary or secondary outcome measures. The responsive genotypes in our overall sample were present in 77% of the intent-to-treat population. Therefore, it is possible that the observed absence of a moderating effect was influenced by the unbalanced distribution of responsive vs. non-responsive genotypes across treatment cells, which could reduce statistical power in a small sample. Furthermore, the genetic variants used to identify potentially responsive subgroups that we utilized in this study were not identical to those of the Johnson et al. (2013) study, which included rs1042173:TT and 5HTTLPR:LL genotypes as one combined marker and categorized all individuals with only one of these two genotypes as non-responders, unless they were carriers for one or more of the remaining three markers tested in the EA genotype panel (i.e., rs1150226:AG and/or rs1176713:GG and/or rs17614942:AC). Here, we utilized each genotype as an individual marker and prospectively randomized participants into responsive vs. non-responsive groups. Second, we limited the AA genetic variants to only two of the five genotypes identified in the Johnson et al. (2013) study, plus an additional variant (rs1176744:AC/CC) not used in that study. Finally, the absence of a main effect of medication makes it more difficult to detect an interaction with genotype. We also conducted an exploratory analysis of moderating effects of the genotype combination tested in the Johnson et al. (2011) study (5HTTLPR:LL and rs1042173:TT as a single marker; 33% of the population) excluding rs11176744); the findings were similar to those with the responsive genotypes used in the present study (see Supplementary Material).
Despite these limitations, our study has several strengths. First, we had a higher completion rate (74.7%) for the 16-week treatment period than the approximately 50% completion rates reported in previous ondansetron clinical trials (Johnson et al., 2000; Johnson et al., 2011), which helped to protect against attrition bias. Second, the 16-week treatment period was 4 weeks longer than previous ondansetron trials for AUD treatment, providing a longer window for a possible treatment effect to emerge. Third, frequency of AEs were comparable in the ondansetron and placebo groups, so that, in contrast to other medications (e.g., topiramate) that cause high rates of adverse events (Johnson et al., 2007), unmasking of the blind is less likely to have occurred. This was supported by the responses provided by participants regarding the treatment to which they believed they were assigned. Lastly, participants were prospectively randomized into treatment groups stratified by ancestry-specific responsive vs. non-responsive genotype groups based on prior knowledge of their influence on ondansetron treatment effects or risk for developing AUD. This allowed a direct evaluation of the benefits of personalized treatment.
Although treatment response is partially genetically determined (Motsinger-Reif et al. 2013), it is a complex trait and thus influenced by multiple genetic variants of small effect (Manolio et al. 2009). Despite evidence that statistical power is enhanced in the study of treatment-relevant variants relative to disease-related ones (Maranville and Cox 2016), it may be unduly optimistic to expect that a single or even a handful of genetic variants could moderate treatment response, particularly for a pharmacodynamic effect. An alternative approach, such as the use of polygenic risk scores derived from genome-wide analyses of the response to the pharmacological treatment of AUD (Biernacka et al. 2021), is a promising alternative approach to advancing the precision medicine treatment of the disorder.
CONCLUSION
This 16-week, randomized, controlled trial showed no evidence that low-dose oral ondansetron is superior to an inactive placebo in treating AUD, either as a main effect or in interaction with a panel of genotypes. Due to a sample size that was smaller than planned, our trial was underpowered and may have thus failed to detect a moderating effect of previously proposed responsive and non-responsive genotype groups on the response to ondansetron. Larger clinical trials are needed to evaluate more conclusively the role of low-dose ondansetron in treating AUD.
Supplementary Material
Acknowledgment:
This study was funded by National Institute on Alcohol Abuse and Alcoholism grants R01 AA021164 (to HRK) and R01 AA021163 (to DAG). The funding agencies had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. The authors gratefully acknowledge the contributions to the conduct of the study made by the staff of the Penn Center for Studies of Addiction, particularly Gail Kaempf, APRN, and Laurie Downing, APRN, and the staff of the University of Maryland Baltimore Clinical Neurobehavioral Center.
Financial disclosures:
CS, DAG, KGL, CB, TP, KP have no disclosures to make. HRK is a member of advisory boards for Dicerna Pharmaceuticals, Sophrosyne Pharmaceuticals, and Enthion Pharmaceuticals; a consultant to Sobrera Pharmaceuticals; recipient of research funding and medication supplies for an investigator-initiated study from Alkermes; member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last three years by Alkermes, Dicerna, Ethypharm, Lundbeck, Mitsubishi, and Otsuka; and a holder of U.S. patent 10,900,082 titled: “Genotype-guided dosing of opioid agonists,” issued 26 January 2021.
Footnotes
ClinicalTrials.gov identifier (NCT number): NCT02354703
REFERENCES
- ALTERMAN AI, CACCIOLA JS, IVEY MA, HABING B & LYNCH KG 2009. Reliability and validity of the alcohol short index of problems and a newly constructed drug short index of problems. J Stud Alcohol Drugs, 70, 304–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRETT JC, FRY B, MALLER J & DALY MJ 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics, 21, 263–5. [DOI] [PubMed] [Google Scholar]
- BECK AT, STEER RA & BROWN GK 1993. Manual for Beck depression inventory-II, Psychological Corporation; San Antonio, TX. [Google Scholar]
- BOHN MJ, BABOR TF & KRANZLER HR 1995. The Alcohol Use Disorders Identification Test (AUDIT): validation of a screening instrument for use in medical settings. J Stud Alcohol, 56, 423–32. [DOI] [PubMed] [Google Scholar]
- BOYCE-RUSTAY JM, WIEDHOLZ LM, MILLSTEIN RA, CARROLL J, MURPHY DL, DAWS LC & HOLMES A 2006. Ethanol-related behaviors in serotonin transporter knockout mice. Alcohol Clin Exp Res, 30, 1957–65. [DOI] [PubMed] [Google Scholar]
- BROWN ES, MCARDLE M, PALKA J, BICE C, IVLEVA E, NAKAMURA A, MCNUTT M, PATEL Z, HOLMES T & TIPTON S 2021. A randomized, double-blind, placebo-controlled proof-of-concept study of ondansetron for bipolar and related disorders and alcohol use disorder. Eur Neuropsychopharmacol, 43, 92–101. [DOI] [PubMed] [Google Scholar]
- CSERVENKA A, YARDLEY MM & RAY LA 2017. Review: Pharmacogenetics of alcoholism treatment: Implications of ethnic diversity. Am J Addict, 26, 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGLEMAN EA, RODD ZA, BELL RL & MURPHY JM 2008. The role of 5-HT3 receptors in drug abuse and as a target for pharmacotherapy. CNS Neurol Disord Drug Targets, 7, 454–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENOCH MA, GORODETSKY E, HODGKINSON C, ROY A & GOLDMAN D 2011. Functional genetic variants that increase synaptic serotonin and 5-HT3 receptor sensitivity predict alcohol and drug dependence. Mol Psychiatry, 16, 1139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIRST M, GIBBON M, SPITZER R & WILLIAMS J 2002. User’s Guide for the Structured Clinical Interview for DSM-IV-TR Axis I Disorders – Research Version – (SCID-I for DSM-IV-TR, November 2002 Revision).
- HAMMER C, KAPELLER J, ENDELE M, FISCHER C, HEBEBRAND J, HINNEY A, FRIEDEL S, GRATACOS M, ESTIVILL X, FICHTER M, FERNANDEZ-ARANDA F, EHRLICH S, RAPPOLD G & NIESLER B 2009. Functional variants of the serotonin receptor type 3A and B gene are associated with eating disorders. Pharmacogenet Genomics, 19, 790–9. [DOI] [PubMed] [Google Scholar]
- HAN H, LIU Q, YANG Z, WANG M, MA Y, CAO L, CUI W, YUAN W, PAYNE TJ, LI L & LI MD 2018. Association and cis-mQTL analysis of variants in serotonergic genes associated with nicotine dependence in Chinese Han smokers. Transl Psychiatry, 8, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HORJALES-ARAUJO E, DEMONTIS D, LUND EK, FINNERUP NB, BORGLUM AD, JENSEN TS, SVENSSON P & VASE L 2013. Polymorphism in serotonin receptor 3B is associated with pain catastrophizing. PLoS One, 8, e78889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON BA, AIT-DAOUD N, SENEVIRATNE C, ROACHE JD, JAVORS MA, WANG XQ, LIU L, PENBERTHY JK, DICLEMENTE CC & LI MD 2011. Pharmacogenetic approach at the serotonin transporter gene as a method of reducing the severity of alcohol drinking. Am J Psychiatry, 168, 265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON BA, JAVORS MA, ROACHE JD, SENEVIRATNE C, BERGESON SE, AIT-DAOUD N, DAWES MA & MA JZ 2008. Can serotonin transporter genotype predict serotonergic function, chronicity, and severity of drinking? Prog Neuropsychopharmacol Biol Psychiatry, 32, 209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON BA, ROACHE JD, JAVORS MA, DICLEMENTE CC, CLONINGER CR, PRIHODA TJ, BORDNICK PS, AIT-DAOUD N & HENSLER J 2000. Ondansetron for reduction of drinking among biologically predisposed alcoholic patients: A randomized controlled trial. JAMA, 284, 963–71. [DOI] [PubMed] [Google Scholar]
- JOHNSON BA, ROSENTHAL N, CAPECE JA, WIEGAND F, MAO L, BEYERS K, MCKAY A, AIT-DAOUD N, ANTON RF, CIRAULO DA, KRANZLER HR, MANN K, O’MALLEY SS, SWIFT RM, TOPIRAMATE FOR ALCOHOLISM ADVISORY, B. & TOPIRAMATE FOR ALCOHOLISM STUDY, G. 2007. Topiramate for treating alcohol dependence: a randomized controlled trial. JAMA, 298, 1641–51. [DOI] [PubMed] [Google Scholar]
- JOHNSON BA, SENEVIRATNE C, WANG XQ, AIT-DAOUD N & LI MD 2013. Determination of genotype combinations that can predict the outcome of the treatment of alcohol dependence using the 5-HT(3) antagonist ondansetron. Am J Psychiatry, 170, 1020–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KELAI S, AISSI F, LESCH KP, COHEN-SALMON C, HAMON M & LANFUMEY L 2003. Alcohol intake after serotonin transporter inactivation in mice. Alcohol Alcohol, 38, 386–9. [DOI] [PubMed] [Google Scholar]
- KENNA GA, ZYWIAK WH, MCGEARY JE, LEGGIO L, MCGEARY C, WANG S, GRENGA A & SWIFT RM 2009. A within-group design of nontreatment seeking 5-HTTLPR genotyped alcohol-dependent subjects receiving ondansetron and sertraline. Alcohol Clin Exp Res, 33, 315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNA GA, ZYWIAK WH, SWIFT RM, MCGEARY JE, CLIFFORD JS, SHOAFF JR, VUITTONET C, FRICCHIONE S, BRICKLEY M, BEAUCAGE K, HAASS-KOFFLER CL & LEGGIO L 2014. Ondansetron reduces naturalistic drinking in nontreatment-seeking alcohol-dependent individuals with the LL 5’-HTTLPR genotype: a laboratory study. Alcohol Clin Exp Res, 38, 1567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRANZLER HR & SOYKA M 2018. Diagnosis and Pharmacotherapy of Alcohol Use Disorder: A Review. JAMA, 320, 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWON YJ, HONG KW, PARK BJ & JUNG DH 2019. Serotonin receptor 3B polymorphisms are associated with type 2 diabetes: The Korean Genome and Epidemiology Study. Diabetes Res Clin Pract, 153, 76–85. [DOI] [PubMed] [Google Scholar]
- MERSHA TB & ABEBE T 2015. Self-reported race/ethnicity in the age of genomic research: its potential impact on understanding health disparities. Hum Genomics, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NATIONAL INSTITUTE ON ALCOHOL ABUSE AND ALCOHOLISM. Alcohol Facts and Statistics. https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/alcohol-facts-and-statistics. Accessed on August 4, 2022.
- PRITCHARD JK, STEPHENS M & DONNELLY P 2000. Inference of population structure using multilocus genotype data. Genetics, 155, 945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENEVIRATNE C, FRANKLIN J, BECKETT K, MA JZ, AIT-DAOUD N, PAYNE TJ, JOHNSON BA & LI MD 2013. Association, interaction, and replication analysis of genes encoding serotonin transporter and 5-HT3 receptor subunits A and B in alcohol dependence. Hum Genet, 132, 1165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENEVIRATNE C, HUANG W, AIT-DAOUD N, LI MD & JOHNSON BA 2009. Characterization of a functional polymorphism in the 3’ UTR of SLC6A4 and its association with drinking intensity. Alcohol Clin Exp Res, 33, 332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENEVIRATNE C & JOHNSON BA 2012. Serotonin transporter genomic biomarker for quantitative assessment of ondansetron treatment response in alcoholics. Front Psychiatry, 3, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEEHAN DV, LECRUBIER Y, SHEEHAN KH, AMORIM P, JANAVS J, WEILLER E, HERGUETA T, BAKER R & DUNBAR GC 1998. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry, 59 Suppl 20, 22–33;quiz 34–57. [PubMed] [Google Scholar]
- SHELTON KL, DUKAT M & ALLAN AM 2004. Effect of 5-HT3 receptor over-expression on the discriminative stimulus effects of ethanol. Alcohol Clin Exp Res, 28, 1161–71. [DOI] [PubMed] [Google Scholar]
- SHERRY ST, WARD MH, KHOLODOV M, BAKER J, PHAN L, SMIGIELSKI EM & SIROTKIN K 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res, 29, 308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SKOWRONEK MH, LAUCHT M, HOHM E, BECKER K & SCHMIDT MH 2006. Interaction between the dopamine D4 receptor and the serotonin transporter promoter polymorphisms in alcohol and tobacco use among 15-year-olds. Neurogenetics, 7, 239–46. [DOI] [PubMed] [Google Scholar]
- SOBELL LC, BROWN J, LEO GI & SOBELL MB 1996. The reliability of the Alcohol Timeline Followback when administered by telephone and by computer. Drug Alcohol Depend, 42, 49–54. [DOI] [PubMed] [Google Scholar]
- SUBSTANCE ABUSE AND MENTAL HEALTH SERVICES ADMINISTRATION 2021. Key substance use and mental health indicators in the United States: Results from the 2020 National Survey on Drug Use and Health. (HHS Publication No. PEP21-07-01-003, NSDUH Series H-56). Rockville, MD: Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration. Retrieved from https://www.samhsa.gov/data/ [Google Scholar]
- SULLIVAN JT, SYKORA K, SCHNEIDERMAN J, NARANJO CA & SELLERS EM 1989. Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict, 84, 1353–7. [DOI] [PubMed] [Google Scholar]
- WITKIEWITZ K, LITTEN RZ & LEGGIO L 2019. Advances in the science and treatment of alcohol use disorder. Sci Adv, 5, eaax4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.