

Abstract
Warfarin, a commonly prescribed oral anticoagulant medication, is highly effective in treating deep vein thrombosis and pulmonary embolism. However, the clinical dosing of warfarin is complicated by high interindividual variability in drug exposure and response and its narrow therapeutic index. CYP2C9 genetic polymorphism and drug-drug interactions (DDIs) are substantial contributors to this high variability of warfarin pharmacokinetics (PK), among numerous factors. Building a physiology-based pharmacokinetic (PBPK) model for warfarin is not only critical for a mechanistic characterization of warfarin PK but also useful for investigating the complicated dose-exposure relationship of warfarin. Thus, the objective of this study was to develop a PBPK model for warfarin that integrates information regarding CYP2C9 genetic polymorphisms and their impact on DDIs. Generic PBPK models for both S- and R-warfarin, the two enantiomers of warfarin, were constructed in R with the mrgsolve package. As expected, a generic PBPK model structure did not adequately characterize the warfarin PK profile collected up to 15 days following the administration of a single oral dose of warfarin, especially for S-warfarin. However, following the integration of an empirical target-mediated drug disposition (TMDD) component, the PBPK-TMDD model well characterized the PK profiles collected for both S- and R-warfarin in subjects with different CYP2C9 genotypes. Following the integration of enzyme inhibition and induction effects, the PBPK-TMDD model also characterized the PK profiles of both S- and R-warfarin in various DDI settings. The developed mathematic framework may be useful in building algorithms to better inform the clinical dosing of warfarin.
SIGNIFICANCE STATEMENT
The present study found that a traditional physiology-based pharmacokinetic (PBPK) model cannot sufficiently characterize the pharmacokinetic profiles of warfarin enantiomers when warfarin is administered as a single dose, but a PBPK model with a target-mediated drug disposition mechanism can. After incorporating CYP2C9 genotypes and drug-drug interaction information, the developed model is anticipated to facilitate the understanding of warfarin disposition in subjects with different CYP2C9 genotypes in the absence and presence of both cytochrome P450 inhibitors and cytochrome P450 inducers.
Introduction
Warfarin is one of the most widely used oral anticoagulants worldwide (Barnes et al., 2015). Warfarin exhibits its pharmacological anticoagulation effects by inhibiting the vitamin K epoxide reductase to prevent the conversion of vitamin K epoxide to reduced vitamin K, which disrupts the vitamin K–dependent blood coagulation cascade (Goodman et al., 2011; Matalqah, 2019). Although warfarin is highly efficacious in reducing the risk of stroke in arterial fibrillation patients, the narrow therapeutic index and high interindividual variability in both drug exposure and response complicates its dosing (Jaffer and Bragg, 2003; Ufer, 2005; Flora et al., 2017). An inappropriate maintenance dose of warfarin resulting in drug exposure beyond the therapeutic window has been found to either compromise the therapeutic effects or introduce life-threatening bleeding risk (Kawai et al., 2014; Trusler, 2019).
Warfarin is administered as a racemic mixture. Although both S- and R-warfarin exhibit pharmacological activity, S-warfarin is suggested to be three- to sevenfold more active than R-warfarin (Flora et al., 2017; Udoamaka Ezuruike, 2019). The elimination of warfarin is primarily via cytochrome P450 (P450)-mediated metabolism, with negligible urinary excretion of the parent enantiomers (Ufer, 2005). Each warfarin enantiomer forms five monohydroxylated metabolites, namely 4’-, 6-, 7-, 8-, and 10-hydroxylated(OH) S- or R-warfarin, mediated by various P450s. S-warfarin is metabolized primarily by CYP2C9, whereas R-warfarin is metabolized by several P450 enzymes, including CYP1A2, CYP2C19, and CYP3A4, that have comparable contributions (Ufer, 2005; Flora et al., 2017; Pouncey et al., 2018).
CYP2C9 is subject to genetic polymorphism, which significantly influences warfarin exposure, particularly with respect to the S-7-OH metabolite, which is the primary elimination route for S-warfarin (Xue et al., 2017). CYP2C9 *2 (Arg144Cys) and *3 (Ile359Leu) variant alleles are associated with reduced metabolic activity and thus a higher exposure of the more pharmacologically active S-warfarin and an increase in the risk of dose-dependent adverse effects (Hamberg et al., 2007; Hamberg et al., 2010; Xue et al., 2017). More importantly, the frequency of the CYP2C9 *2 and *3 alleles can be as high as 15% in certain populations, such as Caucasians (Flora et al., 2017). Although the effect of these variants on warfarin exposure have been known for a long time, the impact of CYP2C9 *2 and *3 on the drug-drug interactions (DDIs) of warfarin has only recently been investigated. In addition, CYP2C9 *1B (-3089G>A and -2663delTG), a regulatory genetic polymorphism, may further complicate the dosing of warfarin in a DDI setting (Chaudhry et al., 2010).
Leveraging physiologic characteristics and the drug-related properties, physiology-based pharmacokinetic (PBPK) modeling is a valuable tool in model-informed drug development (Zhuang and Lu, 2016; Zhang et al., 2020). Importantly, the value of PBPK modeling in various drug development applications is gaining increasing acceptance by regulatory agency such as the US Food and Drug Administration in recent years (Grimstein et al., 2019). An important aspect of PBPK modeling in drug development applications is predicting clinical DDIs of drugs (Grimstein et al., 2019). A successful implementation of PBPK modeling is useful not only in gaining more mechanistic insights for the investigated products but also in supporting clinical decision making and regulatory submission (Alhadab and Brundage, 2020).
Although the clinical use of warfarin can be traced back to the 1950s, the impact of the CYP2C9 genotypes on the clinical DDIs of warfarin is poorly understood (Flora et al., 2017). Taking advantage of a single-dose warfarin clinical DDI study in healthy volunteers with various CYP2C9 genotypes and a target-mediated drug disposition (TMDD) model, our previous population pharmacokinetic (PK) analysis found that subjects with CYP2C9 *2 and *3 variants experience less reduction in S-warfarin clearance (CL) when warfarin is administered together with the P450 inhibitor fluconazole. In contrast, this population experienced a greater increase in S-warfarin CL when warfarin was administered together with the P450 inducer rifampin as compared with individuals possessing the wild-type genotype (CYP2C9*1/*1) (Cheng et al., 2022a). However, a more physiologically relevant PBPK model has not been developed to explain the CYP2C9 genotype-dependent DDIs of warfarin. In addition, it is unclear whether the TMDD mechanism used in our population PK analysis is needed in a PBPK model structure to explain the single-dose warfarin PK profiles collected up to 15 days following the drug administration. Thus, the objective of this study was to use a PBPK modeling approach to investigate the DDIs of warfarin in the presence of various CYP2C9 genotypes using the known P450 inhibitor fluconazole and P450 inducer rifampin.
Methods
Warfarin PBPK Model Structure
A diagram of the PBPK model is shown in Fig. 1, comprising 19 compartments in total. The PBPK model was compiled and implemented in R (version 3.6.3) using mrgsolve package (version 0.11.1) with mass balance differential equations (Elmokadem et al., 2019). The areas under the curve (AUCs) were calculated using R package PKPDmisc (version 3.0.0).
Fig. 1.

S- and R-warfarin PBPK model diagram. AD, adipose; BO, bone; BR, brain; Cp, plasma concentration of drug; Cvenous, venous blood concentration of drugs; DR, drug-receptor complex; HT, heart; MU, muscle; R, receptor; SK, skin; TH, thymus.
The full PBPK model structures of S- and R-warfarin were adapted from initial literature models with physiologic parameter values as shown in Table 1 (Peters, 2008). Standard weight-based allometric scaling coefficients of 0.75 and 1 were added on flow- and volume-based physiologic parameters, respectively (West et al., 1999; Anderson and Holford, 2009). The structure of the initial PBPK model incorporates 14 compartments representing various physiologic organs connected via arterial and venous blood flow components. In general, blood flows from the venous blood compartment and through the lungs into the arterial blood compartment, which further distributes blood into different organs. The physicochemical properties and blood- and plasma-binding related parameters were assumed to be the same between S- and R-warfarin, with values taken from the Sim-S-Warfarin compound file in Simcyp version 19 (Table 2) (Simcyp, 2020). Considering the relatively rapid and almost complete absorption of warfarin (Ufer, 2005), the advanced compartmental absorption and transit components of the original PBPK model (Peters, 2008) were not incorporated to reduce the model complexity. A simplified first-order absorption model was used instead, with drug administered into a gut lumen (GL) compartment. The organ distribution of S- and R-warfarin was assumed to follow a perfusion rate–limited manner into well stirred physiologic organs. The partition coefficients of S- and R-warfarin were assumed to be the same and were predicted using the Sim-S-Warfarin compound file with method 2 in Simcyp version 19 (Table 3) (Peters, 2008; Simcyp, 2020). The general form of the differential equation for a typical organ without elimination [brain, spleen (SP), pancreas (PA), stomach (ST), AD, adipose; BO, bone; BR, brain; HT, heart; MU, muscle; SK, skin; and TH, thymus] can be expressed using eq. 1:
 |
TABLE 1.
Physiology parameter table
Values are extracted from literature (Peters, 2008).
| Tissue | Volume (mL, 70 kg Human) | Flow (mL/min, 70 kg Human) |
|---|---|---|
| Brain | 1450 | 700 |
| Hepatic artery | 302 | |
| Gut | 1650 | 1100 |
| Spleen | 192 | 77 |
| Pancreas | 77 | 133 |
| Stomach | 154 | 38 |
| Liver | 1690 | 1650 |
| Kidney | 280 | 1100 |
| Heart | 310 | 150 |
| Lung | 1172 | 5240 |
| Muscle | 35,000 | 750 |
| Adipose | 10,000 | 260 |
| Skin | 7800 | 300 |
| Bone | 4579 | 250 |
| Thymus | 29 | 80 |
| Arterial blood | 1698 | |
| Venous blood | 3396 |
TABLE 2.
Warfarin drug property–specific parameters
| S-Warfarin | R-Warfarin | Definitions | |
|---|---|---|---|
| MW | 308.3 (Simcyp, 2020) | Molecular weight | |
| Compound type | Monoprotic acid (Simcyp, 2020) | ||
| Log Po:w | 0.27 (Simcyp, 2020) | Logarithmic neutral species octanol:buffer partition coefficient | |
| pKa | 5.1 (Simcyp, 2020) | Acid dissociation constant | |
| CL (L/hour) | 0.260 (Cheng et al., 2022a) | 0.119 (Cheng et al., 2022a) | Total clearance |
| If *1B/*1B | × 0.885 (Cheng et al., 2022a) | Fractional multipliers of CL if other CYP2C9 genotypes | |
| If *1/*3 | × 0.607 (Cheng et al., 2022a) | ||
| If *2/*3 | × 0.277 (Cheng et al., 2022a) | ||
| If *3/*3 | × 0.215 (Cheng et al., 2022a) | ||
| CLR (L/hour) | 0.00369 (Cheng et al., 2022a) | 0.00436 (Cheng et al., 2022a) | Renal clearance |
| Kon (L/(µg*hour)) | 0.00494 (Cheng et al., 2022a) | 0.00137 (Cheng et al., 2022a) | Association rate constant |
| If *2/*3 | × 0.837 (Cheng et al., 2022a) | Fractional multipliers of Kon if other CYP2C9 genotypes | |
| If *3/*3 | × 0.518 (Cheng et al., 2022a) | ||
| Koff (/hour) | 0.0405 (Cheng et al., 2022a) | 0.0405 (Cheng et al., 2022a) | Dissociation rate constant |
| Rmax (µg/L) | 182 (Cheng et al., 2022a) | 188 (Cheng et al., 2022a) | Total receptor levels |
| If *1/*3 | × 0.479 (Cheng et al., 2022a) | Fractional multipliers of Rmax if other CYP2C9 genotypes | |
| If *2/*3 | × 2.51 (Cheng et al., 2022a) | × 0.506 (Cheng et al., 2022a) | |
| If *3/*3 | × 1.89 (Cheng et al., 2022a) | × 0.21 (Cheng et al., 2022a) | |
| Fa (%) | 100 (Simcyp, 2020) | Bioavailability | |
| Ka (/hour) | 1.85 (Simcyp, 2020) | Absorption rate constant | |
| Lag time (hour) | 0 (Simcyp, 2020) | Absorption lag time | |
| BP | 0.59 (Simcyp, 2020) | Blood-to-plasma ratio | |
| fup | 0.009 (Simcyp, 2020) | Fraction of unbound drug in plasma | |
TABLE 3.
Predicted S- and R-warfarin partition coefficients using method 2 (Rodgers et al.) in Simcyp version 19 (Simcyp, 2020)
| Kp Values | Sources | |
|---|---|---|
| Brain | 0.052 | Predicted |
| Gut | 0.162 | Predicted |
| Spleen | 0.101 | Predicted |
| Pancreas | 0.064 | Predicted |
| Stomach | 0.127 | Calculated as the average of nonadipose tissues |
| Liver | 0.090 | Predicted |
| Kidney | 0.134 | Predicted |
| Heart | 0.160 | Predicted |
| Lung | 0.215 | Predicted |
| Muscle | 0.038 | Predicted |
| Adipose | 0.040 | Predicted |
| Skin | 0.281 | Predicted |
| Bone | 0.103 | Predicted |
| Thymus | 0.127 | Calculated as the average of nonadipose tissues |
Kp, partition coefficient.
Vorgan is the organ volume, Qorgan is the tissue blood flow, Carterial is the drug concentration in arterial blood, Corgan is the drug concentration in each organ, Kporgan is the tissue partition coefficient defined as tissue-to-plasma concentration ratio of the drug, and BP is the blood-to-plasma concentration ratio of the drug. The equation for gut (GU) is also expressed as eq. 1 in general, except for an additional first-order absorption input from the GL compartment.
The equation for lung (LU) is expressed as eq. 2 as shown below:
 |
Elimination of S- and R-warfarin was incorporated for both liver and kidney. The CL parameter values were taken from our previous warfarin population PK study (Table 3) (Cheng et al., 2022a). The differential equation for liver (LI) is shown as eq. 3, assuming liver is receiving blood flow from the GU, SP, hepatic artery (HA), PA, and ST:
 |
CLint represents the hepatic intrinsic CL, and fup represents the free fraction in plasma.
Similarly, the differential equation for the other elimination organ, kidney (KI), is expressed as eq. 4:
 |
The hepatic CLint was calculated in a retrograde fashion from the CL terms using eq. 5 (Yang et al., 2007; Alhadab and Brundage, 2020):
 |
CLLI represents the hepatic CL, calculated as the difference between overall CL and renal CL (CLR) as shown in Table 3. Similarly, renal CL was used to derive the intrinsic clearance by the kidney (CLintKI) using the same method (eq. 6).
 |
An empirical TMDD mechanism assuming constant total receptor levels was further included in the venous blood compartment to account for saturable tissue binding of warfarin. The TMDD-related parameters were taken from our warfarin population PK study (Table 3) (Cheng et al., 2022a). The differential equations for the receptor compartment and the drug-receptor complex compartment are shown as eqs. 7 and 8:
 |
 |
R is the receptor concentration, and DR is the drug-receptor complex concentration. Kon and Koff are the association and dissociation rate constants. The initial condition for R was set as Total receptor levels, and the initial condition for DR was set as 0.
The differential equations for venous and arterial blood compartments are shown as eqs. 9 and 10.
 |
 |
In eq. 9, the blood flows from brain, kidney, liver, heart, muscle, adipose, skin, bone, and thymus are summed and are assumed as flowing to the venous blood compartment. V is an arbitrary volume term fixed at 1 L to convert a concentration into an amount. The plasma drug concentrations are predicted as Cvenous/BP for further analysis.
Multiplication factors were added on the absorption rate constants (MFka), tissue partition coefficients (MFkp), blood-to-plasma ratios (MFBP), free drug fractions in plasma (MFfup), association rate constant (MFkon), dissociation rate constant (MFkoff), and total receptor levels (MFRmax) for further parameter optimization, with initial values set at 1 (Peters, 2008; Alhadab and Brundage, 2020).
Clinical PK Data
The S- and R-warfarin plasma PK data used for developing our previous warfarin population PK model were used in this study for visualizing the PBPK model predictions (Cheng et al., 2022a). The warfarin PK data were collected in a clinical DDI study conducted with 29 healthy subjects with various CYP2C9 genotypes (CYP2C9 *1/*1, *1B/*1B, *1/*3, *2/*3, and *3/*3). Briefly, after enrollment, each subject went through three treatment periods. During treatment period 1, subjects were treated with a single 10-mg oral dose of racemic warfarin. Blood samples were collected up to 15 days postdose based on the subject’s CYP2C9 genotype, followed by a 7-day washout phase before entering the next treatment period. During treatment period 2, subjects were randomized to be treated with either 400 mg fluconazole or 300 mg rifampin once daily for 7 days, consecutively, to allow the concentration of each interacting drug reach steady state. On day 8, a single 10-mg oral dose of warfarin was administered in each subject, with blood samples collected following the same sampling scheme as treatment period 1, followed by another 7-day washout phase. During the sampling phase of period 2, interacting drugs were continuously administered with the same dosing regimens to maintain a steady-state concentration. The design of treatment period 3 was the same as treatment period 2, with subjects treated with the alternative interacting drug.
Model Parameter Optimizations
The S- and R-warfarin PK profiles in CYP2C9 *1/*1 subjects treated with warfarin only were used for initial model optimization. The multiplication factors (MFka, MFkp, MFBP, and MFfup) in the PBPK model without TMDD and the multiplication factors (MFka, MFkp, MFBP, MFfup, MFkon, MFkoff, and MFRmax) in the PBPK model with TMDD were adjusted 0.1-, 0.25-, 0.5-, 1-, 2-, 4-, or 10-fold for the simulations. The resulting PBPK model predictions were overlaid with the S- and R-warfarin PK profiles in subjects with CYP2C9 *1/*1 treated with warfarin only to visualize the sensitivity of these multiplication factors on model predictions.
For the S-warfarin PK profiles in CYP2C9 *1/*1 subjects treated with warfarin only, MFkp and MFRmax were considered to be sensitive on model predictions and were selected to be further optimized. For R-warfarin PK profiles in CYP2C9 *1/*1 subjects treated with warfarin only, MFkp, MFRmax, and MFkon were considered to be sensitive on model predictions and were selected to be further optimized. Optimization was performed in R (version 3.6.3) using the Nelder-Mead method with the weighted least squared objective function (https://github.com/metrumresearchgroup/ub-cdse-2019/blob/master/content/tools_optimization_methods.md).
Following parameter optimization, the median predictions of the S- and R-warfarin PK profiles, in subjects with various CYP2C9 genotypes (*1/*1, *1B/*1B, *1/*3, *2/*3, and *3/*3) when warfarin is administered alone, were simulated and overlaid with the observations to visualize the model predictions.
PK Models for Interacting Drugs
Empirical PK models for fluconazole and rifampin were extracted from the literature and translated in R (version 3.6.3) using mrgsolve package (version 0.11.1) (Roos et al., 2008; Svensson et al., 2018). Briefly, the extracted fluconazole PK model is a one-compartment model with linear elimination, linear absorption, and the absorption lag time. The extracted PK model for rifampin is a one-compartment PK model with a nonlinear (Michaelis-Menten) elimination, a transit-compartment absorption process, a dose-dependent bioavailability component, and an enzyme turnover model to account for the autoinduction of rifampin. Both the fluconazole and the rifampin models were extracted with fixed and random effects. Fluconazole and rifampin PK profiles extracted from the literature were used for validating model predictions (Gross et al., 2001; Kumar et al., 2008; Wilkins et al., 2008; Seng et al., 2015).
Incorporating Drug-Drug Interactions into Warfarin PBPK Models
The hepatic intrinsic CL values for S- and R-warfarin were separated into five metabolic pathways (4’-, 6-, 7-, 8-, and 10-OH) pathways. The proportion of each metabolite as a function of the overall clearance was based on the results of our previous warfarin metabolites population PK modeling study (Cheng et al., 2022b). The metabolic elimination of S- and R-warfarin was assumed to be completely mediated by these five metabolic pathways. Thus, the metabolite proportions presented in the original study for each parent compound were summed and rescaled to 100% to calculate the new proportions of hepatic intrinsic CL mediated by the various metabolic pathways for use in this modeling analysis (Table 4).
TABLE 4.
The fractions of warfarin metabolic clearance by each metabolic pathway Fraction values are calculated based on literature (Cheng et al., 2022b). The metabolic fractions presented in the original study for each parent compound were summed and rescaled to 100% to calculate the new fractions of hepatic intrinsic CL mediated by the various metabolic pathways. The key assumption of this approach is to assume the metabolism of S- or R-warfarin is totally mediated by the respective five metabolic pathways.
| S-Warfarin | CYP2C9 *1/*1 | CYP2C9 *1B/*1B | CYP2C9 *1/*3 | CYP2C9 *2/*3 | CYP2C9 *3/*3 |
|---|---|---|---|---|---|
| 4’-OH (%) | 2.8 | 2.9 | 4.7 | 10.4 | 28.2 |
| 6-OH (%) | 19.8 | 16.8 | 20.1 | 17.6 | 20.6 |
| 7-OH (%) | 75.3 | 78.4 | 72.6 | 68.5 | 42.5 |
| 8-OH (%) | 1.5 | 1.2 | 1.7 | 1.9 | 4.8 |
| 10-OH (%) | 0.6 | 0.7 | 0.9 | 1.5 | 4.0 |
| R-Warfarin | CYP2C9 *1/*1 | CYP2C9 *1B/*1B | CYP2C9 *1/*3 | CYP2C9 *2/*3 | CYP2C9 *3/*3 |
| 4’-OH (%) | 2.8 | 3.3 | 1.8 | 1.8 | 1.8 |
| 6-OH (%) | 61.1 | 72.5 | 61.7 | 61.7 | 61.7 |
| 7-OH (%) | 9.8 | 11.7 | 9.9 | 9.9 | 9.9 |
| 8-OH (%) | 23.5 | 9.2 | 23.8 | 23.8 | 23.8 |
| 10-OH (%) | 2.8 | 3.3 | 2.8 | 2.8 | 2.8 |
The intrinsic hepatic CL of each metabolic pathway under the inhibitory effect of fluconazole was calculated using eq. 11:
 |
is the intrinsic hepatic CL of a particular metabolite pathway in the presence of the inhibitor fluconazole. Cfluc is the fluconazole plasma concentration predicted using an empirical PK model. Ki is the fluconazole inhibition constant.
The intrinsic hepatic CL of each metabolic pathway under the induction effect of rifampin was calculated using eq. 12.
 |
is the intrinsic hepatic CL of a particular metabolite pathway in the presence of the inducer rifampin. Crifa is the rifampin plasma concentration predicted using an empirical PK model. indmax and indC50 are the maximum fold increase in CLint that can occur following rifampin induction and the concentration of rifampin producing 50% of maximum induction of a particular metabolic pathway, respectively.
The overall hepatic intrinsic CL of each parent compound in the presence of fluconazole or rifampin was calculated as the summation of the hepatic intrinsic CL values of each metabolite pathway under inhibition or induction conditions.
The fluconazole and rifampin effects on CLR were included as multiplication factors based on a warfarin parent compound population PK analysis (Cheng et al., 2022a).
Population Simulations
Following the development of the S- and R- warfarin PBPK models and the validation of the fluconazole and rifampin empirical PK models, 30% interindividual variability was assumed as being log-normally distributed for the absorption rate constants, CL terms, TMDD-related parameters, and partition coefficients for performing the population simulations (Einolf et al., 2017). A virtual population with 500 subjects was simulated, with 100 subjects in each CYP2C9 genotype group (*1/*1, *1B/*1B, *1/*3, *2/*3, and *3/*3). The mean body weight of each genotype group was simulated based on the demographic information of the original study (Cheng et al., 2022a). Population-level simulations were performed using the dosing regimens of warfarin, fluconazole, and rifampin in the original study (Cheng et al., 2022a). To visualize the predictions, the medians and 5th and 95th percentiles of the simulated PK profiles at each time point were calculated and overlaid with the observations of either S- or R-warfarin PK profiles in subjects with different CYP2C9 genotypes under different cotreatments. The model codes for final S- and R-warfarin PBPK models, as well as the S- and R-warfarin PBPK models with the interacting drug components, are provided in Supplemental Materials.
Results
PBPK Model Structure
The PBPK model structure for S- and R-warfarin is shown in Fig. 1, with 14 physiologic organ compartments [LU, heart, brain, muscle, adipose, skin, SP, PA, LI, ST, GU, bone, KI, and thymus], venous and arterial blood compartments, a GL compartment for drug administration, an empirical receptor compartment, and an empirical drug-receptor complex compartment. Following the administration of drug in GL, drug is assumed to follow a first-order absorption (abs) into GU with a complete bioavailability and no delay (Table 2). LI and KI are assumed to be the organs of elimination. The empirical TMDD mechanism is arbitrarily embedded in the venous blood compartment.
To incorporate a drug-drug interaction mechanism, the hepatic CLint of S- and R-warfarin is separated into five metabolic pathways (4’-OH, 6-OH, 7-OH, 8-OH, and 10-OH). Fluconazole inhibition and rifampin induction effects were included in each metabolite pathway using the approach describe in Methods. The fluconazole and rifampin effects were also included as multiplication factors on the CLR. The inhibitory and induction parameters values used for simulations are displayed in Table 5 and Table 6, respectively.
TABLE 5.
Fluconazole inhibitory parameters
| MW (g/mol) | 306.271 (PUBCHEM) |
|---|---|
| Ki (mg/L) | |
| 4’-OH-S | 8.88 (Brown et al., 2006; Damle et al., 2011) |
| 6-OH-S | 6.74 (Brown et al., 2006) |
| 7-OH-S | 6.74 (Brown et al., 2006) |
| 8-OH-S | 0.64 (Damle et al., 2011) |
| 10-OH-S | 19.3 (Brown et al., 2006; Damle et al., 2011) |
| 4’-OH-R | 8.88 (Brown et al., 2006; Damle et al., 2011) |
| 6-OH-R | 30.63 (Kunze et al., 1996) |
| 7-OH-R | 12.67 |
| 8-OH-R | 0.64 (Brown et al., 2006) |
| 10-OH-R | 19.3 (Brown et al., 2006; Damle et al., 2011) |
| Fluconazole effects on CLR (multiplication factor) | |
| S-warfarin | 0.847 (Cheng et al., 2022a) |
| R-warfarin | 0.752 (Cheng et al., 2022a) |
MW, molecular weight.
TABLE 6.
Rifampin induction parameters
indC50, concentration of rifampin producing 50% of maximum induction of a particular metabolic pathway; MW, molecular weight.
Model Parameter Optimizations
The model predictions of the S- and R-warfarin PK profiles, with and without a TMDD mechanism, in CYP2C9 *1/*1 subjects when warfarin is administered alone were overlaid with the observed values (Fig. 2). Inclusion of the TMDD mechanism substantially improved the model predictions for S-warfarin but only slightly improved the model predictions for R-warfarin.
Fig. 2.
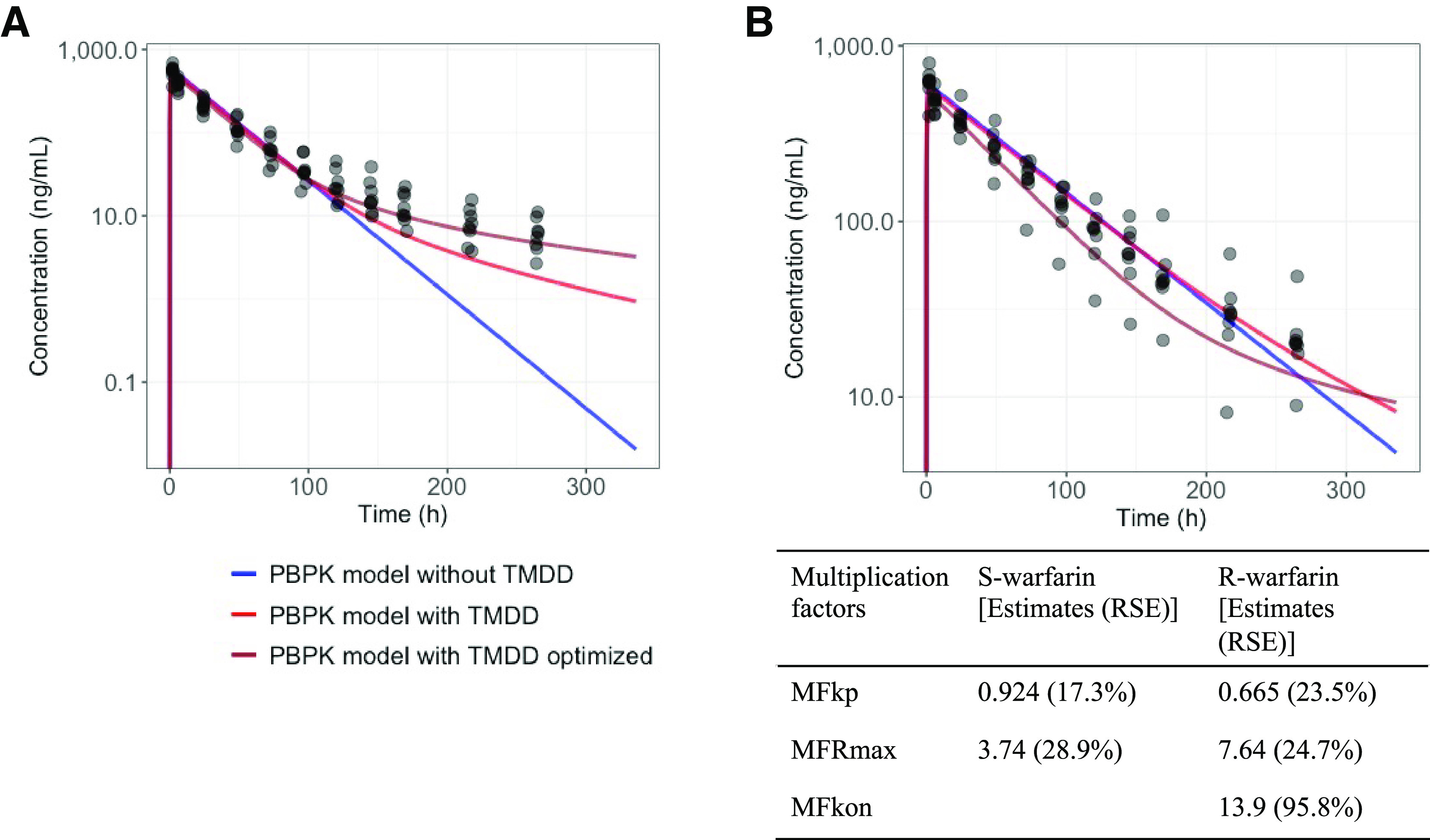
S-warfarin (A) and R-warfarin (B) PBPK model predictions in subjects with CYP2C9 *1/*1 when warfarin is administrated alone. Colors represent model predictions using a PBPK model without TMDD mechanism, with TMDD mechanism, and with TMDD mechanism following optimization. Table displays the multiplication factors for S- and R-warfarin following optimization. The multiplication factors are estimated based on the assumption that the volume of saturable binding target of warfarin is 1 L (arbitrarily fixed due to lack of relevant clinical information). RSE, relative standard errors.
Further sensitivity analyses were conducted on the multiplication factors, for both S- and R-warfarin PBPK models with and without the TMDD mechanism, to visualize the influence of each factor on the model predictions (Supplemental Figs. 1–4). MFka and MFfup had minimal influence on the model predictions for both the S- and R-warfarin PBPK models, with and without TMDD. For S-warfarin, MFkp, MFBP, and MFRmax substantially influenced the model predictions, whereas MFkon and MFkoff influenced the model predictions but to a lesser extent. For R-warfarin, MFkp, and MFBP substantially influenced the model predictions, whereas MFRmax, MFkon and MFkoff only slightly influenced the R-warfarin PK model predictions.
Sensitive parameters MFkp, MFRmax, and MFkon were selected for optimizations. Both optimizations converged successfully with the optimized values of the multiplication factors displayed in the table insert of Fig. 2. Optimization of the multiplication factors further improved the model predictions for both S- and R-warfarin PK profiles (Fig. 2).
The PBPK models including a TMDD mechanism that were achieved following the optimizations were expanded to incorporate subjects with various CYP2C9 genotypes when warfarin is administered alone. The predicted S-and R-warfarin PK profiles adequately characterized the observations (Fig. 3).
Fig. 3.

Optimized S-warfarin (A) and R-warfarin (B) PBPK model with TMDD mechanism predictions overlaid with observations in subjects with various CYP2C9 genotypes when warfarin is administered alone. Dots represent observations. Red lines represent median predictions.
Validation of the Fluconazole and Rifampin Model Predictions
Fluconazole and rifampin PK profiles were extracted from the literature. The simulations conducted with the empirical PK models were able to capture the literature-extracted PK profiles of fluconazole and rifampin (Supplemental Figs. 5 and 6). These models were incorporated into the optimized S- and R-warfarin PBPK models that included a TMDD mechanism and were used for predicting S- and R-warfarin PK profiles in both inhibition and induction DDI settings.
Population Simulations
Population simulations for the S- and R-warfarin PK profiles when warfarin is administered alone were conducted following the incorporation of interindividual variation. The optimized PBPK models that include a TMDD mechanism were able to adequately characterize the S- and R-warfarin PK profiles when warfarin was administered alone (Fig. 4). Following the incorporation of the fluconazole inhibition and the rifampin induction, the optimized PBPK models that included a TMDD mechanism were also able to characterize the S- and R-warfarin PK profiles in respective inhibition and induction DDI scenarios. (Figs. 5 and 6).
Fig. 4.
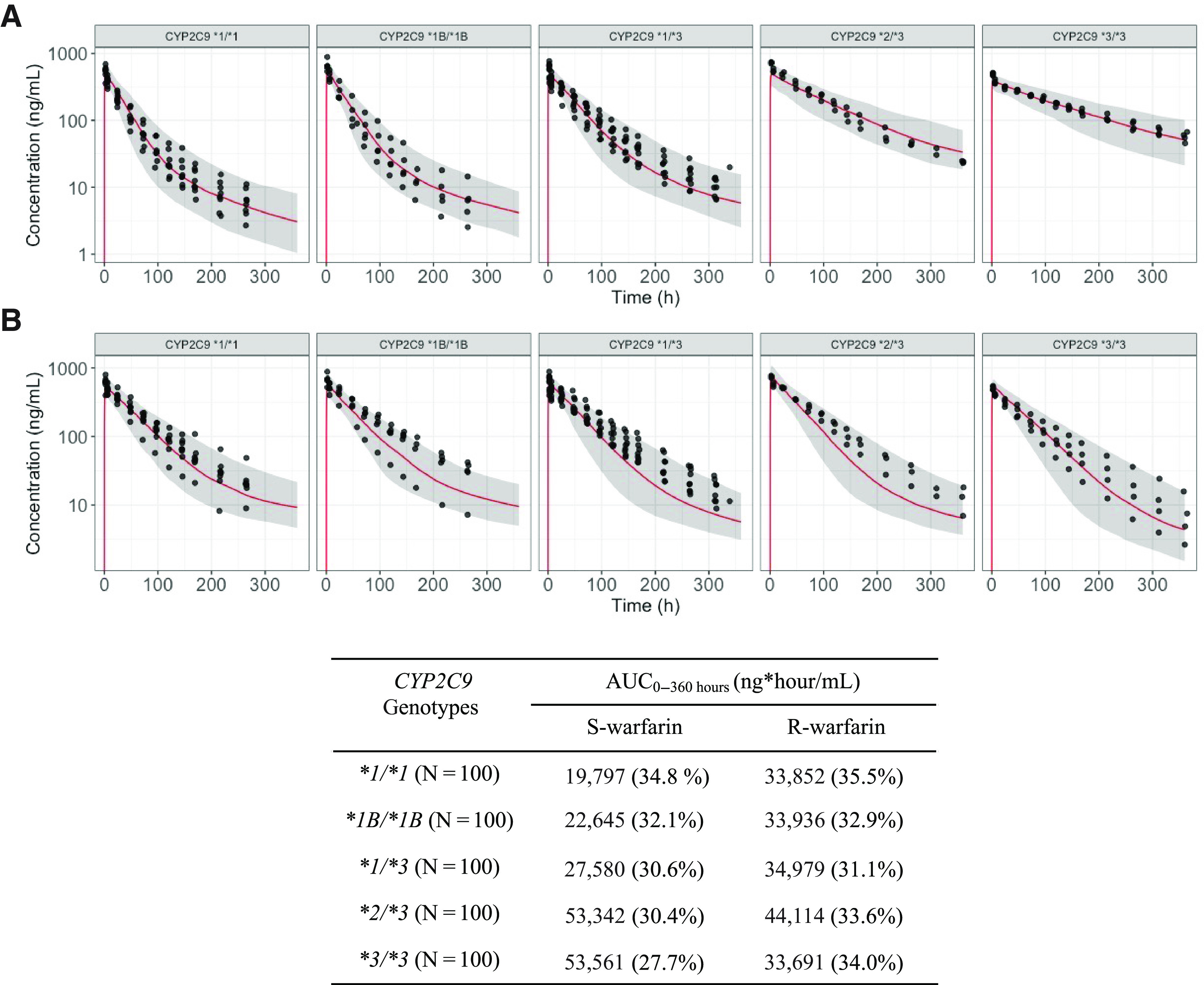
Optimized S-warfarin (A) and R-warfarin (B) PBPK model with TMDD mechanism population predictions overlaid with observations in subjects with various CYP2C9 genotypes when warfarin is administered alone. Dots represent observations. Red lines represent median predictions. Gray-shaded areas represent the area between 5th and 95th percentiles of the model predictions. Table shows the AUC0–360 hours of S- and R-warfarin by CYP2C9 genotypes. Values in table expressed as geometric means (coefficient of variations). AUC0–360, area under the curve from time 0 to 360 hours.
Fig. 5.
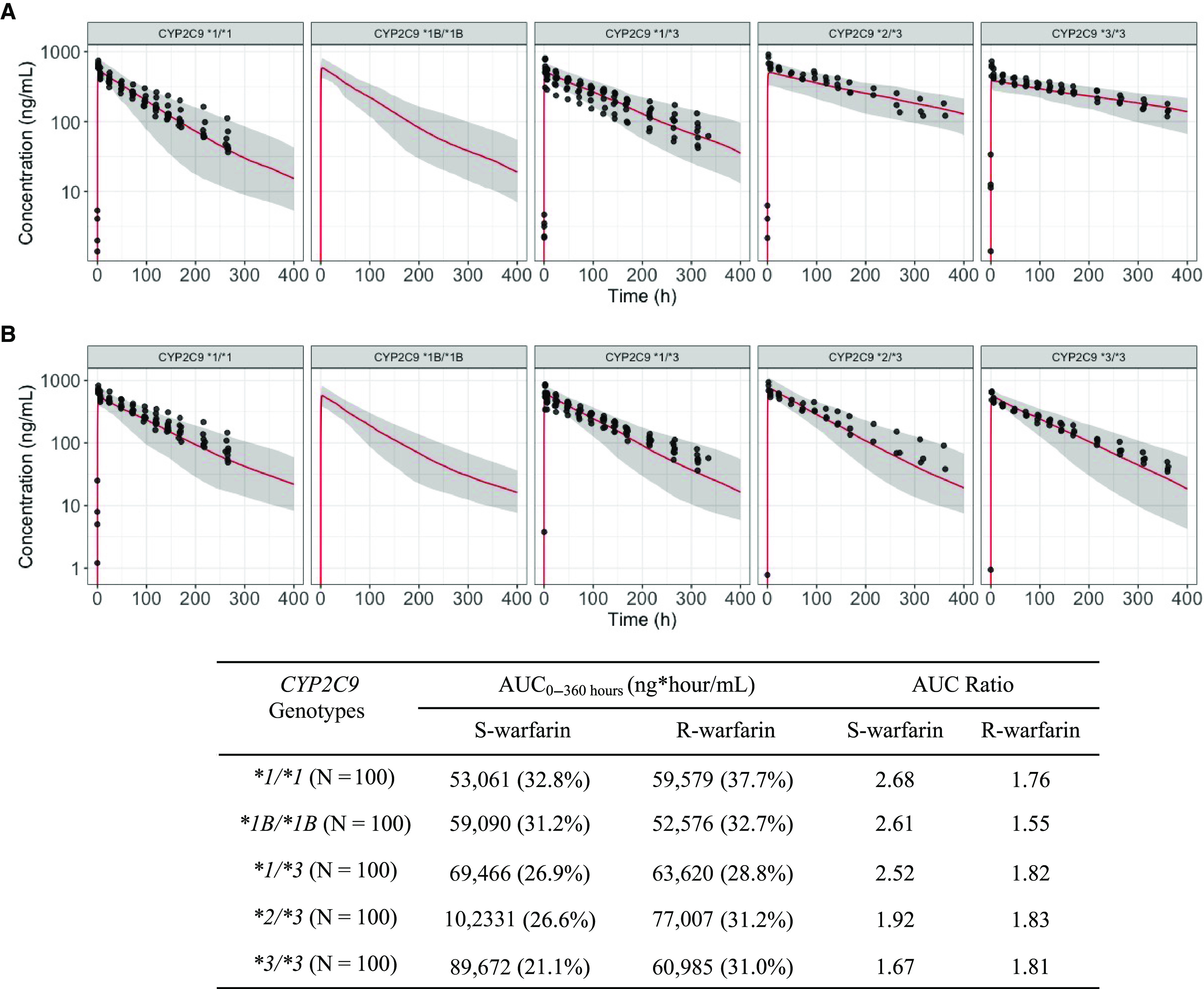
Optimized S-warfarin (A) and R-warfarin (B) PBPK model with TMDD mechanism population predictions overlaid with observations in subjects with various CYP2C9 genotypes when warfarin is administered together with fluconazole. Dots represent observations. Red lines represent median predictions. Gray-shaded areas represent the area between 5th and 95th percentiles of the model predictions. Table inset shows the AUC0–360 hours and AUC ratios of S- and R-warfarin by CYP2C9 genotypes when warfarin is coadministered with fluconazole. Values in table expressed as geometric means (coefficient of variations). AUC0–360, area under the curve from time 0 to 360 hours.
Fig. 6.
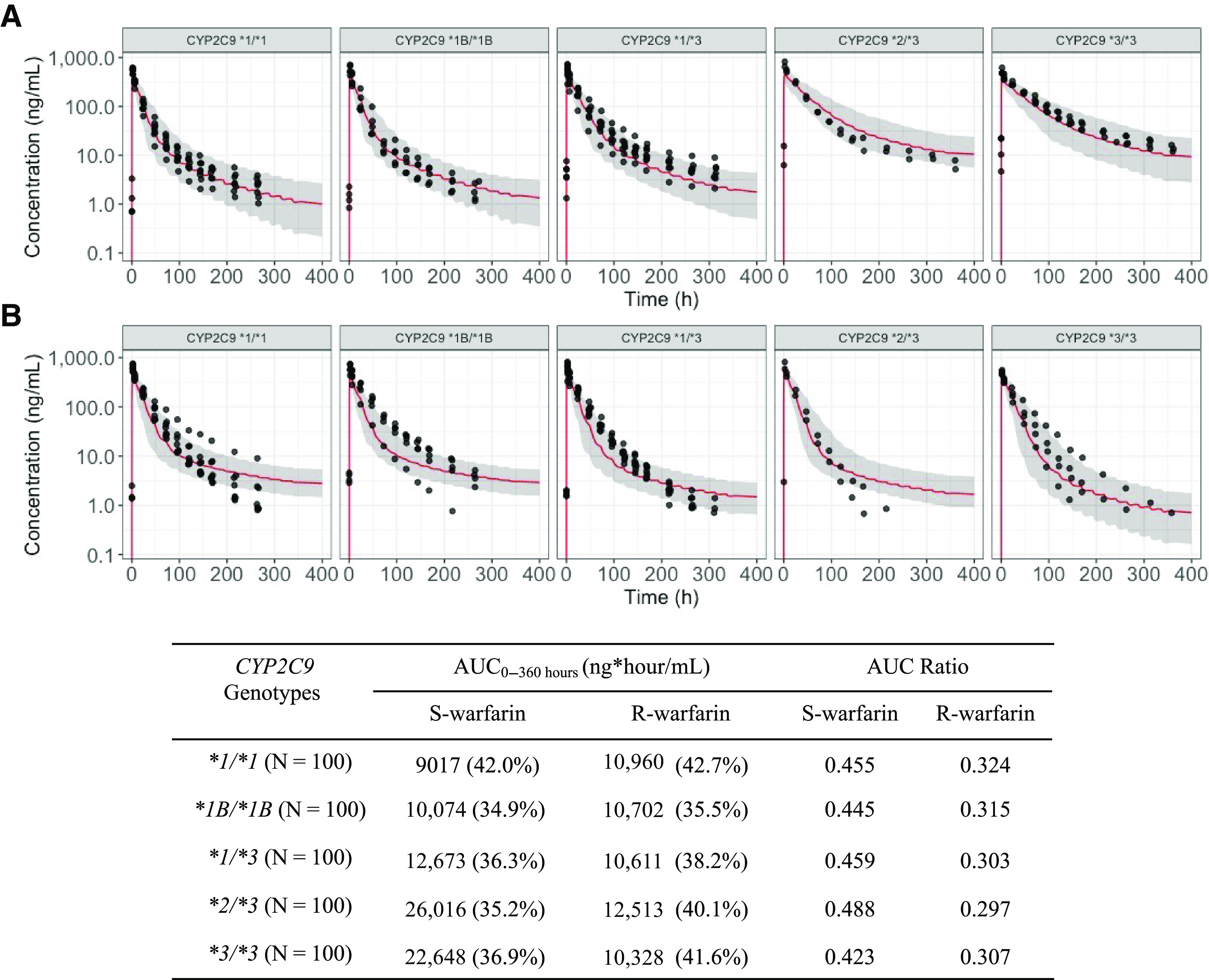
Optimized S-warfarin (A) and R-warfarin (B) PBPK model with TMDD mechanism population predictions overlaid with observations in subjects with various CYP2C9 genotypes when warfarin is administered together with rifampin. Dots represent observations. Red lines represent median predictions. Gray-shaded areas represent the area between 5th and 95th percentiles of the model predictions. Table inset shows the AUC0–360 hours and AUC ratios of S- and R-warfarin by CYP2C9 genotypes when warfarin is coadministered with rifampin. Values in table expressed as geometric means (coefficient of variations). AUC0–360, area under the curve from time 0 to 360 hours.
The areas under the curve from time 0 to 360 hours (AUC0–360) were calculated based on the population simulation for both S- and R- warfarin across various CYP2C9 genotypes, when warfarin is administered alone or together with fluconazole or rifampin (Figs. 4–6, table insets). The AUC ratios for S- and R-warfarin when warfarin is administered together with fluconazole are 1.67–2.68 and 1.55–1.83, respectively (Fig. 5, table inset). The AUC ratios for S- and R-warfarin when warfarin is administered together with rifampin are 0.423–0.488 and 0.297–0.324, respectively (Fig. 6, table inset).
Discussion
Leveraging information from the literature and available clinical PK data from our previous studies, the present study develops a PBPK model framework for each warfarin enantiomer. The developed PBPK model was able to capture the plasma PK profiles of each warfarin enantiomer collected up to 15 days following the administration of a single oral dose of warfarin in subjects with various CYP2C9 genotypes under different comedications. The developed PBPK models were able to characterize warfarin disposition in a more mechanistic manner and will be valuable for investigating the complicated dose-response relationship of warfarin.
Initially, a traditional PBPK model schematic was adapted from literature to predict warfarin PK profiles (Peters, 2008). However, we found that a traditional PBPK schematic fails to explain the warfarin enantiomer PK profiles (especially S-warfarin) when collected up to 11 days in CYP2C9 *1/*1 subjects following a single dose of warfarin administration, no matter how the model parameters were adjusted (Fig. 2; Supplemental Figs. 1 and 3). Interestingly, dose disproportionality of warfarin has been reported preclinically due to the presence of high-affinity and low-capacity binding sites of warfarin, which introduces the possibility of saturable tissue binding (Takada and Levy, 1979; Takada and Levy, 1980). Clinically, the saturable tissue binding of warfarin is observed as dose-dependent changes in the apparent volume of distributions (King et al., 1995). In fact, the term target-mediated drug disposition was first proposed by Dr. Gerhard Levy in 1994 on the basis of extensive preclinical PK research with small-molecule compounds like warfarin (Levy, 1994). Dr. Levy also proposed a TMDD model for warfarin to account for the observed PK nonlinearity in apparent volume of distribution observed clinically (Levy et al., 2003). Although the TMDD model is used frequently for modeling biologics, this model is gaining more attention recently to account for the saturable tissue or plasma binding observed in the PK profiles of small molecule compounds (Mager and Jusko, 2001; An et al., 2015; An, 2017; Bach et al., 2019). Our previous population PK analysis also suggested that a TMDD model was needed to characterize the warfarin PK profiles when collected up to 15 days following a single dose of warfarin after the administration of both a P450 inhibitor or a P450 inducer (Cheng et al., 2022a). In the current study, after including an empirical TMDD mechanism, we obtained a significant improvement in the PBPK model predictions of the S-warfarin PK profile. Further optimization of the PBPK model with a TMDD mechanism enabled the characterization of both the S- and R-warfarin PK profiles adequately in subjects with various CYP2C9 genotypes when warfarin was administered alone (Figs. 3 and 4).
CYP2C9 *2 and *3 variants are highly associated with the reduced metabolic activity of CYP2C9. Subjects possessing the CYP2C9 *2 and *3 variants experience higher S-warfarin exposure following warfarin administration and require lower warfarin maintenance doses (Dean, 2012). In subjects with the CYP2C9 *2 and *3 variants, non-CYP2C9–mediated elimination pathways occupy a higher proportion of overall S-warfarin elimination (Cheng et al., 2022b). A differential effect of fluconazole inhibition and rifampin induction on different metabolic pathways of S-warfarin was also noted, potentially explaining our observation of CYP2C9 genotype-dependent DDIs on S-warfarin PK in our previous population PK analysis (Cheng et al., 2022a). CYP2C9-mediated metabolic pathways constitute the largest proportion of S-warfarin elimination, yet elimination by CYP2C9 is reduced in subjects with *2 and *3 variants. Consequently, our findings suggest that the degree to which these individuals experience inhibition by fluconazole is lessened, resulting in a lower degree of drug inhibition interaction (genotype-dependent drug interactions). In contrast, because CYP2C9-mediated metabolic pathways are less inducible, the overall S-warfarin elimination in subjects with a lower proportion of CYP2C9-mediated elimination, such as subjects with *2 and *3 variants, are more susceptible to rifampin induction. Indeed, the Ki values (Table 5) reported in the literature suggest that CYP2C9-mediated S-warfarin metabolic pathways (6-OH-S and 7-OH-S) are inhibited to a greater extent compared with some of the other S-warfarin metabolic pathways, such as 10-OH-S. Furthermore, the indmax values we identified suggest that CYP2C9-mediated S-warfarin metabolic pathways are less inducible compared with other S-warfarin metabolic pathways mediated by other P450 enzymes (Table 6). More importantly, incorporating these inhibition- and induction-related parameters, our PBPK-TMDD model reflected the S-warfarin PK profiles when warfarin was administered with either fluconazole or rifampin (Figs. 5 and 6). Interestingly, a previous clinical DDI study conducted with flurbiprofen as the probe drug and fluconazole as the interacting drug showed that differential inhibition of CYP2C9- and non-CYP2C9–mediated pathways also resulted in CYP2C9 genotype-dependent DDIs (Kumar et al., 2008). The results of the present PBPK modeling study using S-warfarin as a probe drug are consistent with these previous study findings with flurbiprofen and fluconazole.
Additionally, the PBPK-TMDD model may be useful in informing the clinical use of warfarin. For instance, to reduce the risk of bleeding during surgery, warfarin treatment is typically discontinued about 5 days prior to surgery (Douketis et al., 2012). Following the inclusion of a TMDD mechanism, the model simulations based on the long half-life observed in our extended plasma sampling suggest that the pharmacologically more active S-warfarin may not be eliminated as fast as earlier literature would predict (Fig. 2A). Taking advantage of warfarin pharmacodynamic models published in the literature, it will be interesting to conduct simulations linking our PBPK-TMDD model together with a pharmacodynamic model to investigate the impact of this observed slower elimination of S-warfarin on the International Normalized Ratio and treatment outcomes following warfarin discontinuation.
Despite the potential uses of the warfarin PBPK-TMDD models, limitations exist in the current model structure. First, it is relatively empirical and arbitrary to embed the TMDD mechanism inside the venous blood compartment of a PBPK model structure to account for saturable tissue binding. A physiologically more relevant approach might be to incorporate the saturable tissue binding of warfarin into relevant organs, such as liver (Levy et al., 2003). However, lacking explicit clinical evidence about which organs exhibit saturable warfarin tissue binding, it was arbitrarily decided to embed the TMDD mechanism in the venous blood compartment of our PBPK model. Collecting additional information to inform an organ-specific TMDD mechanism clinically would be beneficial for future development of a more mechanistic PBPK model schematic for warfarin. In this regard, the PBPK model constructed in the present study can be easily adapted to incorporate an organ-specific TMDD mechanism, considering our model is developed using an open-source tool. Second, a significant assumption of the current PBPK model is that the hepatic CL of S- or R-warfarin is mediated completely by the five monohydroxylation pathways. Incorporating additional metabolic pathways of warfarin, such as ketone reduction and glucuronidation, might provide additional mechanistic insights to warfarin disposition but would require an even more extensive dataset (Ufer, 2005).
In summary, the present study found that a traditional PBPK model structure was inadequate to describe the PK profiles of warfarin enantiomers when collected up to 15 days following a single dose of warfarin. Instead, a PBPK model embedded with an empirical TMDD mechanism was able to characterize the single-dose warfarin PK profiles in subjects with clinically important CYP2C9 genotypes. Following the integration of fluconazole inhibition and rifampin induction, the developed PBPK-TMDD models were able to describe the S- and R-warfarin PK profiles under different cotreatments in subjects with various CYP2C9 genotypes. The developed PBPK models provide mechanistic insights regarding warfarin disposition and may also serve as a valuable tool to inform the clinical dosing of warfarin.
Acknowledgments
The authors would like to thank Dr. Kyle Baron for the insightful discussions and suggestions.
Abbreviations
- AUC
area under the curve
- CL
clearance
- DDI
drug-drug interaction
- DR
drug-receptor complex compartment
- GL
gut lumen
- GU
gut
- indmax
maximum fold increase in CLint that can occur following rifampin induction
- P450
cytochrome P450
- PBPK
physiology-based pharmacokinetic
- PK
pharmacokinetic
- R
receptor compartment
- TMDD
target-mediated drug disposition
Authorship Contributions
Participated in research design: Cheng, Flora, Rettie, Brundage, Tracy.
Participated in data collection: Flora, Rettie, Brundage, Tracy.
Performed data analysis: Cheng, Brundage.
Wrote or contributed to the writing of the manuscript: Cheng, Flora, Rettie, Brundage, Tracy.
Footnotes
This work was supported by National Institutes of Health Institute of General Medical Sciences [Grant R01-GM069753] (to T.S.T.) and [Grant R01-GM032165] (to T.S.T.)
The authors declare no conflict of interest.
1Current affiliation: Metrum Research Group, Tariffville, Connecticut.
2Current affiliation: GRYT Health Inc., Rochester, New York.
Cheng S., Flora D.R., Tracy T. S., Rettie A.E., Brundage R.C. A physiologically-based Pharmacokinetic (PBPK) Model embedded with a Target-Mediated Drug Disposition (TMDD) mechanism can characterize S-warfarin pharmacokinetic (PK) profiles in subjects with various CYP2C9 genotypes under different co-treatments. American Conference of Pharmacometrics (ACOP) 12.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Alhadab AA, Brundage RC (2020) Physiologically-Based Pharmacokinetic Model of Sertraline in Human to Predict Clinical Relevance of Concentrations at Target Tissues. Clin Pharmacol Ther 108:136–144. [DOI] [PubMed] [Google Scholar]
- An G (2017) Small-Molecule Compounds Exhibiting Target-Mediated Drug Disposition (TMDD): A Minireview. J Clin Pharmacol 57:137–150. [DOI] [PubMed] [Google Scholar]
- An G, Liu W, Dutta S (2015) Small-molecule compounds exhibiting target-mediated drug disposition - A case example of ABT-384. J Clin Pharmacol 55:1079–1085. [DOI] [PubMed] [Google Scholar]
- Anderson BJ, Holford NH (2009) Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24:25–36. [DOI] [PubMed] [Google Scholar]
- Bach T, Jiang Y, Zhang X, An G (2019) General Pharmacokinetic Features of Small-Molecule Compounds Exhibiting Target-Mediated Drug Disposition (TMDD): A Simulation-Based Study. J Clin Pharmacol 59:394–405. [DOI] [PubMed] [Google Scholar]
- Barnes GD, Lucas E, Alexander GC, Goldberger ZD (2015) National Trends in Ambulatory Oral Anticoagulant Use. Am J Med 128:1300–5.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HS, Galetin A, Hallifax D, and Houston JB (2006) Prediction of in vivo drug-drug interactions from in vitro data : factors affecting prototypic drug-drug interactions involving CYP2C9, CYP2D6 and CYP3A4. Clin Pharmacokinet 45:1035–1050. [DOI] [PubMed] [Google Scholar]
- Chaudhry ASUrban TJLamba JKBirnbaum AKRemmel RPSubramanian MStrom SYou JHKasperaviciute DCatarino CB, et al. (2010) CYP2C9*1B promoter polymorphisms, in linkage with CYP2C19*2, affect phenytoin autoinduction of clearance and maintenance dose. J Pharmacol Exp Ther 332:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Flora DR, Tracy TS, Rettie AE, Brundage RC (2022a) Pharmacokinetic Modeling of Warfarin I – Model-based Analysis of Warfarin Racemates with a Target Mediated Drug Disposition Model Reveals CYP2C9 Genotype-dependent Drug-drug Interactions of S-Warfarin. Drug Metab Dispos 50:1287–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Flora DR, Tracy TS, Rettie AE, Brundage RC (2022b) Pharmacokinetic Modeling of Warfarin II – Model-based Analysis of Warfarin Metabolites following Warfarin Administered either Alone or Together with Fluconazole or Rifampin. Drug Metab Dispos 50:1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damle B, Varma MV, Wood N (2011) Pharmacokinetics of voriconazole administered concomitantly with fluconazole and population-based simulation for sequential use. Antimicrob Agents Chemother 55:5172–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean L (2012) Warfarin Therapy and VKORC1 and CYP Genotype, in Medical Genetics Summaries (Pratt VM, Scott SA, Pirmohamed M, Esquivel B, Kane MS, Kattman BL, Malheiro AJ, eds) Bethesda, MD. [PubMed] [Google Scholar]
- Douketis JD, Spyropoulos AC, Spencer FA, Mayr M, Jaffer AK, Eckman MH, Dunn AS, Kunz R (2012) Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 141 (Suppl 2):e326S–e350S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einolf HJ, Lin W, Won CS, Wang L, Gu H, Chun DY, He H, Mangold JB (2017) Physiologically Based Pharmacokinetic Model Predictions of Panobinostat (LBH589) as a Victim and Perpetrator of Drug-Drug Interactions. Drug Metab Dispos 45:1304–1316. [DOI] [PubMed] [Google Scholar]
- Elmokadem A, Riggs MM, Baron KT (2019) Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling With mrgsolve: A Hands-On Tutorial. CPT Pharmacometrics Syst Pharmacol 8:883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora DR, Rettie AE, Brundage RC, Tracy TS (2017) CYP2C9 Genotype-Dependent Warfarin Pharmacokinetics: Impact of CYP2C9 Genotype on R- and S-Warfarin and Their Oxidative Metabolites. J Clin Pharmacol 57:382–393. [DOI] [PubMed] [Google Scholar]
- Goodman LS, Brunton LL, Chabner B, Knollmann Br C (2011) Goodman & Gilman’s Pharmacological Basis of Therapeutics, McGraw-Hill, New York. [Google Scholar]
- Grimstein M, Yang Y, Zhang X, Grillo J, Huang SM, Zineh I, Wang Y (2019) Physiologically Based Pharmacokinetic Modeling in Regulatory Science: An Update From the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci 108:21–25. [DOI] [PubMed] [Google Scholar]
- Gross AS, McLachlan AJ, Minns I, Beal JB, Tett SE (2001) Simultaneous administration of a cocktail of markers to measure renal drug elimination pathways: absence of a pharmacokinetic interaction between fluconazole and sinistrin, p-aminohippuric acid and pindolol. Br J Clin Pharmacol 51:547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberg AK, Dahl ML, Barban M, Scordo MG, Wadelius M, Pengo V, Padrini R, Jonsson EN (2007) A PK-PD model for predicting the impact of age, CYP2C9, and VKORC1 genotype on individualization of warfarin therapy. Clin Pharmacol Ther 81:529–538. [DOI] [PubMed] [Google Scholar]
- Hamberg AK, Wadelius M, Lindh JD, Dahl ML, Padrini R, Deloukas P, Rane A, Jonsson EN (2010) A pharmacometric model describing the relationship between warfarin dose and INR response with respect to variations in CYP2C9, VKORC1, and age. Clin Pharmacol Ther 87:727–734. [DOI] [PubMed] [Google Scholar]
- Jaffer A, Bragg L (2003) Practical tips for warfarin dosing and monitoring. Cleve Clin J Med 70:361–371. [DOI] [PubMed] [Google Scholar]
- Kawai VKCunningham AVear SIVan Driest SLOginni AXu HJiang MLi CDenny JCShaffer C, et al. (2014) Genotype and risk of major bleeding during warfarin treatment. Pharmacogenomics 15:1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SY, Joslin MA, Raudibaugh K, Pieniaszek HJ Jr, Benedek IH (1995) Dose-dependent pharmacokinetics of warfarin in healthy volunteers. Pharm Res 12:1874–1877. [DOI] [PubMed] [Google Scholar]
- Krishna Machavaram LA Kim Crewe, Alice Ke, Oliver Hatley, Howard Burt, Iain Gardner, and Karen Rowland-Yeo (2017) Evaluation of In Vitro-In Vivo Extrapolation (IVIVE) of the induction potential for known CYP2C9 inducers, in International Society for the Study of Xenobiotics. [Google Scholar]
- Kumar V, Brundage RC, Oetting WS, Leppik IE, Tracy TS (2008) Differential genotype dependent inhibition of CYP2C9 in humans. Drug Metab Dispos 36:1242–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze KL, Wienkers LC, Thummel KE, Trager WF (1996) Warfarin-fluconazole. I. Inhibition of the human cytochrome P450-dependent metabolism of warfarin by fluconazole: in vitro studies. Drug Metab Dispos 24:414–421. [PubMed] [Google Scholar]
- Levy G (1994) Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther 56:248–252. [DOI] [PubMed] [Google Scholar]
- Levy G, Mager DE, Cheung WK, Jusko WJ (2003) Comparative pharmacokinetics of coumarin anticoagulants L: Physiologic modeling of S-warfarin in rats and pharmacologic target-mediated warfarin disposition in man. J Pharm Sci 92:985–994. [DOI] [PubMed] [Google Scholar]
- Mager DE, Jusko WJ (2001) General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 28:507–532. [DOI] [PubMed] [Google Scholar]
- Matalqah LMA (2019) Knowledge, Adherence, and Quality of Life among Warfarin Therapy Users, in Anticoagulation Drugs - the Current State of the Art (Kelleni M, ed) (2020). [Google Scholar]
- Pelletier RD, Lai WG, Wong YN (2013) Application of a substrate cocktail approach in the assessment of cytochrome P450 induction using cultured human hepatocytes. J Biomol Screen 18:199–210. [DOI] [PubMed] [Google Scholar]
- Peters SA (2008) Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin Pharmacokinet 47:261–275. [DOI] [PubMed] [Google Scholar]
- Pouncey DL, Hartman JH, Moore PC, Dillinger DJ, Dickerson KW, Sappington DR, Smith ES 3rd, Boysen G, Miller GP (2018) Novel isomeric metabolite profiles correlate with warfarin metabolism phenotype during maintenance dosing in a pilot study of 29 patients. Blood Coagul Fibrinolysis 29:602–612. [DOI] [PubMed] [Google Scholar]
- PUBCHEM Fluconazole compound summary, in, National Library of Medicine; National Center for Biotechnology Information, (https://pubchem.ncbi.nlm.nih.gov/compound/Fluconazole).
- PUBCHEM Rifampicin compound summary, in, National Library of Medicine; National Center for Biotechnology Information, (https://pubchem.ncbi.nlm.nih.gov/compound/Rifampicin).
- Roos JF, Kirkpatrick CM, Tett SE, McLachlan AJ, Duffull SB (2008) Development of a sufficient design for estimation of fluconazole pharmacokinetics in people with HIV infection. Br J Clin Pharmacol 66:455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seng KY, Hee KH, Soon GH, Chew N, Khoo SH, Lee LS (2015) Population pharmacokinetics of rifampicin and 25-deacetyl-rifampicin in healthy Asian adults. J Antimicrob Chemother 70:3298–3306. [DOI] [PubMed] [Google Scholar]
- Simcyp (2020) Simcyp Version 19 in, Certara, lnc.
- Svensson RJ, Aarnoutse RE, Diacon AH, Dawson R, Gillespie SH, Boeree MJ, Simonsson USH (2018) A Population Pharmacokinetic Model Incorporating Saturable Pharmacokinetics and Autoinduction for High Rifampicin Doses. Clin Pharmacol Ther 103:674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K, Levy G (1979) Comparative pharmacokinetics of coumarin anticoagulants XLIII: Concentration-dependent hepatic uptake of warfarin in rats. J Pharm Sci 68:1569–1571. [DOI] [PubMed] [Google Scholar]
- Takada K, Levy G (1980) Comparative pharmacokinetics of coumarin anticoagulants XLIV: Dose-dependent pharmacokinetics of warfarin in rats. J Pharm Sci 69:9–14. [DOI] [PubMed] [Google Scholar]
- Trusler MB (2019) Quality of warfarin management. Can Fam Physician 65:530. [PMC free article] [PubMed] [Google Scholar]
- Ezuruike U, Humphries H, Abduljalil K, Neuhoff S (2019) Use of a physiologically based pharmacokinetic–pharmacodynamic (PBPK-PD) model to guide dose adjustments to S-warfarin in different populations, in 20th Annual Simcyp Consortium Meeting. [Google Scholar]
- Ufer M (2005) Comparative pharmacokinetics of vitamin K antagonists: warfarin, phenprocoumon and acenocoumarol. Clin Pharmacokinet 44:1227–1246. [DOI] [PubMed] [Google Scholar]
- West GB, Brown JH, Enquist BJ (1999) The fourth dimension of life: fractal geometry and allometric scaling of organisms. Science 284:1677–1679. [DOI] [PubMed] [Google Scholar]
- Wilkins JJ, Savic RM, Karlsson MO, Langdon G, McIlleron H, Pillai G, Smith PJ, Simonsson US (2008) Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob Agents Chemother 52:2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Holford N, Ding XL, Shen ZY, Huang CR, Zhang H, Zhang JJ, Guo ZN, Xie C, Zhou L, et al. (2017) Theory-based pharmacokinetics and pharmacodynamics of S- and R-warfarin and effects on international normalized ratio: influence of body size, composition and genotype in cardiac surgery patients. Br J Clin Pharmacol 83:823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Costales C, Lazzaro S, Eatemadpour S, Kimoto E, Varma MV (2019) Physiologically-Based Pharmacokinetic Modeling Approach to Predict Rifampin-Mediated Intestinal P-Glycoprotein Induction. CPT Pharmacometrics Syst Pharmacol 8:634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jamei M, Yeo KR, Rostami-Hodjegan A, Tucker GT (2007) Misuse of the well-stirred model of hepatic drug clearance. Drug Metab Dispos 35:501–502. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yang Y, Grimstein M, Fan J, Grillo JA, Huang SM, Zhu H, Wang Y (2020) Application of PBPK Modeling and Simulation for Regulatory Decision Making and Its Impact on US Prescribing Information: An Update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J Clin Pharmacol 60 (Suppl 1):S160–S178. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Lu C (2016) PBPK modeling and simulation in drug research and development. Acta Pharm Sin B 6:430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]