

Abstract
With the growing acceptance of the contribution of protein conformational ensembles to enzyme catalysis and regulation, research in the field of protein dynamics has shifted toward an understanding of the atomistic properties of protein dynamical networks and the mechanisms and time scales that control such behavior. A full description of an enzymatic reaction coordinate is expected to extend beyond the active site and to include site-specific networks that communicate with the protein/water interface. Advances in experimental tools for the spatial resolution of thermal activation pathways are being complemented by biophysical methods for visualizing dynamics in real time. An emerging multidimensional model integrates the impacts of bound substrate/effector on the distribution of protein substates that are in rapid equilibration near room temperature with reaction-specific protein embedded heat transfer conduits.
Introduction
It is now widely accepted that functions of proteins are intimately dependent on both their structural and dynamical properties [1–8]. While X-ray crystallography remains a powerful tool to view the 3-D arrangement of atoms within a protein, traditional X-ray techniques provide averaged positions of atoms in a protein, leading to minimal information regarding protein conformational dynamics [9,10]. Room temperature X-ray structural analyses have introduced the ability to infer dynamical networks from the detection of alternate protein side chain conformations [11–14] and recent advances in time-resolved X-ray crystallography using free electron lasers (XFEL) [15,16] have opened a window for the direct capture of dynamical information and the visualization of reaction intermediates. Proteins typically populate numerous states [17–23], due to their inherent fluctuations that promote changes in sidechain positioning, backbone rotations, large conformational changes and domain shifts. A major goal (and challenge!) is to discern the specific set of motions that influence biological function, which in the case of enzyme catalysis produce a combination of high regio-, stereo- and reaction-specificity with rate accelerations as high as 1030-fold [24]. Ongoing efforts in both computational and experimental methodology are rapidly advancing the “state of the art”, with sophisticated models for the physical origins of enzyme catalysis emerging that go beyond historical views such as “enhanced transition state binding” [25–27].
Can Differences in Protein Structure be Detected for ES vs ETS Complexes?
Over many decades, the mainstream explanation for the huge rate accelerations of enzyme catalyzed reactions was enhanced transition state stabilization, as proposed by Pauling [26,27]. The theory attributes the catalytic power of enzymes to a higher affinity of the protein catalyst for the activated complex/transition state in relation to the ground state. Though this theory has contributed tremendously to inhibitor design, it does not provide a biophysical understanding of the trajectory of enzyme catalysis. Reaction barrier crossings are expected to occur on the femtosecond time scale, leaving open the question of what physical processes control the activation event that transforms a thermodynamically stable ES (enzyme-substrate) complex to a state near or at the top of the reaction barrier and results in sub-millisecond to millisecond timescale processes. Efforts at distinguishing protein structural differences between ES and ETS (enzyme-transition state) complexes have been pursued via X-ray crystallography, hydrogen deuterium exchange (HDX) and molecular dynamics (MD) simulations.
Early on, Bruice and co-workers performed picosecond MD simulations on ES and ETS structures of Xanthobacter autotrophicus haloalkane dehalogenase. No major structural rearrangement was observed at this timescale with the exception of an additional hydrogen bond in the ETS structure compared to ES complex that would account for a lowering the activation barrier by ca. 2 kcal/mol [28]. In a second study, the same group pursued MD simulations of catechol O-methyltransferase (COMT) for 1 ns in the presence of catechol, catecholate or the transition state to distinguish active-site structure differences. The calculated root-mean-squared deviation and positional fluctuations of the active-site residues within 10 Å of the methyl group of S-adenosyl-L-methionine (AdoMet) for the catecholate and transition-state simulations were found to be similar. The authors suggested that the catalytic power of COMT in going from ES to ETS arises from the ability of the active site to arrange the ground state into tightly packed conformers [29]. This view is supported by secondary kinetic isotope effect measurements and extended GPU-based QM/MM calculations for the reaction of COMT with dopamine, indicating a ground state electrostatic compaction that is maintained through contact with a second sphere tyrosine that resides behind the methyl-bearing sulfur of AdoMet [30–34]. A recent application of temperature dependent hydrogen deuterium exchange (TDHDX, see below) to COMT has uncovered two orthogonal and potentially dynamical networks that connect protein-water surfaces to the primary reaction coordinate [33].
Utilizing another well studied enzyme system, Herschlag and co-workers collected room temperature X-ray crystallography data for the Pseudomonas putida ketosteroid isomerase (KSI). From careful examination of the structures of both substrate and TS analogs, the aggregate conformational ensembles indicate limited change in active site positioning (on the 1- to 1.5-Å scale, note that the crystal structure resolution is from 1.1 to 2.5 Å) throughout the KSI reaction cycle [35].
Recently, the TDHDX methodology has also been used to analyze the behavior of the TIM barrel enzyme adenosine deaminase in the presence of either a ground state or bis-substrate/tight binding inhibitor [36]. Evaluation of HDXMS across a range of temperatures for apo- and ligand bound complexes showed almost identical impacts on ΔH° for local protein flexibility from two ligands that differ by ca. 106-fold in net affinity [36]. It appears that either an ES- or ETS- like structure has undergone a similar global restructuring of enzyme, in preparation for optimal active site chemistry.
The lack of strong evidence for structural differences when comparing protein complexes of substrate or transition state analogs is in contrast to the evidence for protein preorganization [37]. Direct detection of the preorganization of enzyme active sites that involves, for example, large protein conformational changes or loop closure, is readily accessible from X-ray crystallography or SAXS (Small-angle X-ray scattering [38]) and provides the first level of understanding in the generation of active site geometries that are optimized for catalysis.
Delineating the Functional Importance of Conformational Landscapes and Ensembles.
Proteins in solvent are “wiggling and jiggling”. This property of constant flux produces a wide range of distributed substates referred to as the conformational landscape, that are treated as a continuum of thermodynamic states at equilibrium. Fluctuations within a protein permit jumps between ensembles over relatively low energy barriers and lead to rapid sampling of heterogeneous landscapes [39–45]. Transitions between distinct conformations on different timescales can be coupled to facilitate enzyme catalysis (Figure 1). Although conformationally heterogeneous states of proteins are implicated in many important biological processes, including protein folding, signal transduction, allosteric regulation, and enzyme substrate turnover, detailed knowledge of how the individual substates interconvert among each other and impact function is less clear. One important issue has been the mechanism whereby a small ligand, protein effector or substrate alters the protein conformational landscape. While the role of induced fit held in traction in enzymology for many years [46], it is increasingly accepted that related protein conformations exist in both ligand free and native protein states [40], undergoing a shift in their distribution upon interaction with substrate or effector.
Figure 1.

Conformational selection occurs in many biological processes including substrate binding, allosteric activation, directed evolution and thermal adaptation. Conformational ensemble redistribution has been detected in many systems (dihydrofolate reductase (DHFR) [50], Kemp eliminase [52,53], and tryptophan synthase [49]) using different techniques including X-ray crystallography, NMR, and computational simulations. For the above example, the impact of perturbation is shown to alter both the number of accessible protein sub-states and their relative populations (where green represents the most stable state).
The introduction of such conformation ensembles and selection not only deepens our understanding of how enzymes perform their unparalleled catalysis but is relevant to the fields of protein design and directed evolution. In many cases, directed evolution yields beneficial mutations at remote positions that would not have been predicted from simple structural inspection. Studies show that enhanced catalytic efficacy can be ascribed to a shift in the populations of conformational substates towards those that are more catalytically active and/or provide enhanced stabilization of catalytic intermediates [47–50]. The concept of rigidified active sites that are dependent on global protein conformational sampling was first apparent in a thermophilic alcohol dehydrogenase (ht-ADH), where the temperature dependence of the kinetic isotope was lost at elevated temperature, under conditions where enzyme had become more flexible and active [50]. Recent studies of directed evolution using a primitive form of dihydrofolate reductase (DHFR) illustrate this principle through the detection of kinetic evidence for a progressive restriction in substrate/cofactor positioning as enzyme variants arise with increased catalytic efficiency [50,51].
Broom et al. pursued room-temperature X-ray crystallography to study changes in the conformational ensemble during directed evolution for the designed Kemp eliminase [52]. Catalytic residues are seen to become increasingly rigidified through improved packing leading to an active site that is better pre-organized to favor productive positioning of the substrate. Given the intrinsic reactivity of the substrate of the Kemp eliminase, its overall rate enhancement is small relative to native enzymes. Nonetheless, the authors achieved a variant, labelled HG4, that is >700-fold more active than its’ predecessor. Crystallographic analysis of HG4 illustrated how mutations may shift the conformational ensemble toward an enrichment of catalytically competent substates [52]. The generation of new protein ensembles under the condition of directed evolution was also examined by Otten using the prototypic Kemp eliminase [53]. Through a combination of kinetic analyses, X-ray crystallography and NMR, these authors show how directed evolution gradually altered the conformational ensemble of the protein scaffold to shift to a narrow, highly active conformational ensemble and accelerate the catalytic efficiency by nearly nine orders of magnitude [53]. These experiments also uncover the complicating and off-pathway features that can arise in directed evolution, arguing for caution against overly simplistic molecular interpretations of its impact on enzyme function.
Is there a privileged path within the protein scaffold for the activation of enzyme active sites?
Historically, the protein matrix surrounding enzyme active sites was regarded primarily as a “scaffold” for the arrangement of active site catalytic residues that both generate a successful substrate binding site and provide catalytic rate enhancement. Many decades of active research in the fields of bioorganic and bioinorganic chemistry were focused on these active site elements, successfully producing small molecule mimics of the structural and catalytic elements of the active site [54,55], yet few models were able to approximate the enormous catalytic power of enzymes. In particular, the anisotropic topology of folded globular proteins points toward a role for catalytically privileged dynamical networks in the initiation of active site chemistry [56].
Approximately 20 years ago, Ranganathan et al. began this journey computationally, by analyzing the relatively small PDZ domain protein, comprised of 100 amino acids [57]. Their methodological approach used evolutionary data for protein families to define statistically based interactions between amino acid positions. In the case of the PDZ domain family, this analysis predicted a set of energetically coupled positions that included unexpected long-range interactions. Mutational studies confirmed the predictions, revealing sets of interacting residues that generate connected pathways through a protein fold and could serve as the basis for efficient energy conduction. Application of this method was subsequently extended to three structurally and functionally distinct protein families, detecting structural motifs for allosteric communication [58].
The experimental detection of intrinsic and long range thermally activated dynamical networks also began to emerge ca. 20 years ago through an analysis of HDX (using mass spectrometry for detection, HDX-MS) in the thermophilic alcohol dehydrogenase (ht-ADH) [59,60]. Analysis of networks in the well-studied DHFR have similarly implicated selected regions of protein that interact with bound substrates and extend out to the protein/solvent interface [61,62]. Dyer and co-workers recently tested the importance of site-specific protein solvent surfaces through an analysis of changes in DHFR activity via the photochemical activation of a surface attached gold nanoparticle [63].
The combination of experimental and computational studies of dynamic networks in enzyme is having an increasing impact on the origins of catalysis. For example, Bunzel et al. performed molecular dynamics simulations for the designed Kemp eliminase [64]. In their MD simulations (out to 5 microsec), a decreased fluctuation in the ETS ensemble was observed relative to ES that was concluded to reflect a network of residues that centers on the bound ligand and spans the protein. Two mutations introduced during evolution were found to be directly involved in the proposed network to tune the dynamic and packing properties for enhanced catalysis.
In a methodological development, Markin et al. designed a high-throughput microfluidic platform to introduce more than 1000 mutations (either a glycine or valine substitution) throughout the structure of the phosphate-irrepressible alkaline phosphatase of Flavobacterium and to correlate their impact with catalysis [65]. They observed impactful mutants that extend from the active site to the enzyme surface, providing a map of the underlying protein architecture in dictating catalysis. It was seen that positions that give catalytic effects tend to cluster, and the pattern is asymmetric and complex. Deleterious effects distribute throughout the enzyme with many of the largest effects distant from the active site.
Temperature dependent hydrogen deuterium exchange (TDHDX) as an emerging tool to uncover site specific thermal activation pathways.
Enzyme catalysis is, with few exceptions [66–68], thermally activated. While most discussions of enzyme effectiveness have been historically focused on their free energy of activation [69–72], emerging studies are beginning to address intrinsic differences in the way that enzymes undergo thermal activation relative to small molecules in condensed phase [73,74]. In this context, a set of experimental protocols has been developed that focuses on the TDHDX. This methodology can be readily applied to a wide range of enzymes, under conditions that are similar to those used for biochemical studies of enzyme function [74,75].
For HDX-MS studies in general, the course of deuteron incorporation into backbone amides is monitored as a function of time, undergoing time dependent quenching of protein samples and their subsequent digestion into small peptides that are selected to represent the entire protein sequence [76–78]. Single temperature HDX-MS is commonly used to investigate the extent of protein protection from bound ligands and/or protein/protein interactions, and also to study regions of proteins where no structural information is available (e.g., because of disordered structures that preclude positional X-ray structural data) or in protein-protein or protein-ligand interactions where structural effects propagate across long distances [79–81]. Though mapping deuteron uptake changes on a protein structure at a single temperature is extremely informative, these studies do not disclose the temperature dependent property of enzymes which, as noted above, is one of the key parameters underlying enzyme catalysis.
In the pursuit of TDHDX, the EX-2 regime is the targeted regime, and this can generally be realized through a screening of experimental conditions and confirmed from the patterns of mass spectrometric analysis. The general formulation for EX-2 is: kHDX = Kop kexc where kexc represents the intrinsic rate constant for hydrogen deuterium exchange within a transiently unfolded region of protein and Kop represents the equilibrium constant for local protein unfolding. From studies at a single temperature one obtains ΔGobs = ΔG° + ΔG‡. The incorporation of temperature dependency into HDX allows a formal separation of the ΔHobs = ΔH° + ΔH‡. As ΔH‡ has a well-established and fairly constant value (ΔH‡~17 kcal/mol) through peptide studies [12,82], ΔH° becomes the dominant new parameter that reports on temperature dependent changes in flexibility driven by native protein dynamics [74]. The introduction of a perturbation to the native folded protein structure further simplifies interpretation, with an observed ΔΔHobs = ΔH°(variant) − ΔH°(WT) that essentially eliminates the contribution of ΔH‡ from analyses.
A general TDHDX approach in the Klinman lab is to target hydrophobic side chains for site specific mutagenesis, with the goal of generating modest protein packing defects that impact the rate and especially the ΔH‡ of catalysis. Using the enzyme soybean lipoxygenase (SLO) as a prototype, TDHDX experiments on wild type and mutant forms of SLO have uncovered site specific trends in the enthalpic barriers for HDX-MS within two distal and solvent-exposed loops that correlate well with the enthalpic barriers for catalysis [74]. The remarkable specificity of the inferred thermal conduit in SLO has been replicated and expanded using room temperature X-ray studies that compare changes in the position of amino acid side chains between WT and function-altering mutants [83].
The TIM barrel family of proteins represents ca. 10% of structurally characterized enzyme structures and uses a conserved scaffold to catalyze five out of six of the known classes of enzyme reaction [84]. The TIM barrel scaffold has also served as a primary starting point for de novo enzyme design [85]. The diversity of the evolved chemical reactivity within TIM barrel enzymes provides an excellent platform for testing whether evolution of active site chemistry may be linked to the emergence of positionally selective thermal conduits. Thus far, several types of TIM barrel enzyme classes have been studied by TDHDX: a murine adenosine deaminase (mADA) [86], cf. Figure 3, and yeast enolase [87], together with the partial TIM barrel enzyme COMT discussed above [33]. In the case of mADA, Phe61, one of four side chains that generate a hydrophobic wall behind bound substrate was targeted for mutagenesis by insertion of a series of hydrophobic aliphatic side chains. In the case of catalysis these mutations produce a linear increase in ΔH‡ that correlates with changes in ΔH° obtained from TDHDX. Two thermal networks that reach from opposing protein/solvent interfaces toward the active site zinc ion and substrate binding site have been identified [86]. In an analogous study, experiments were performed as a function of time, temperature and mutation on yeast enolase [87], uncovering a completely distinct and horseshoe shaped region where changes in protein flexibility from TDHDX correlate with changes in ΔH‡ for enzyme catalysis. Once again, these reach from protein/solvent interfaces toward either the active site base or the metal cofactors that surround and activate bound substrate. The resulting data strongly implicate a general presence of thermal energy transfer pathways that are embedded within an enzyme’s structure and are unique to each reaction catalyzed. We further propose that site specific thermal conduits result from co-evolutionary changes within the protein scaffold, to accompany the emergence of new active site structures and chemical reactivity.
Figure 3.
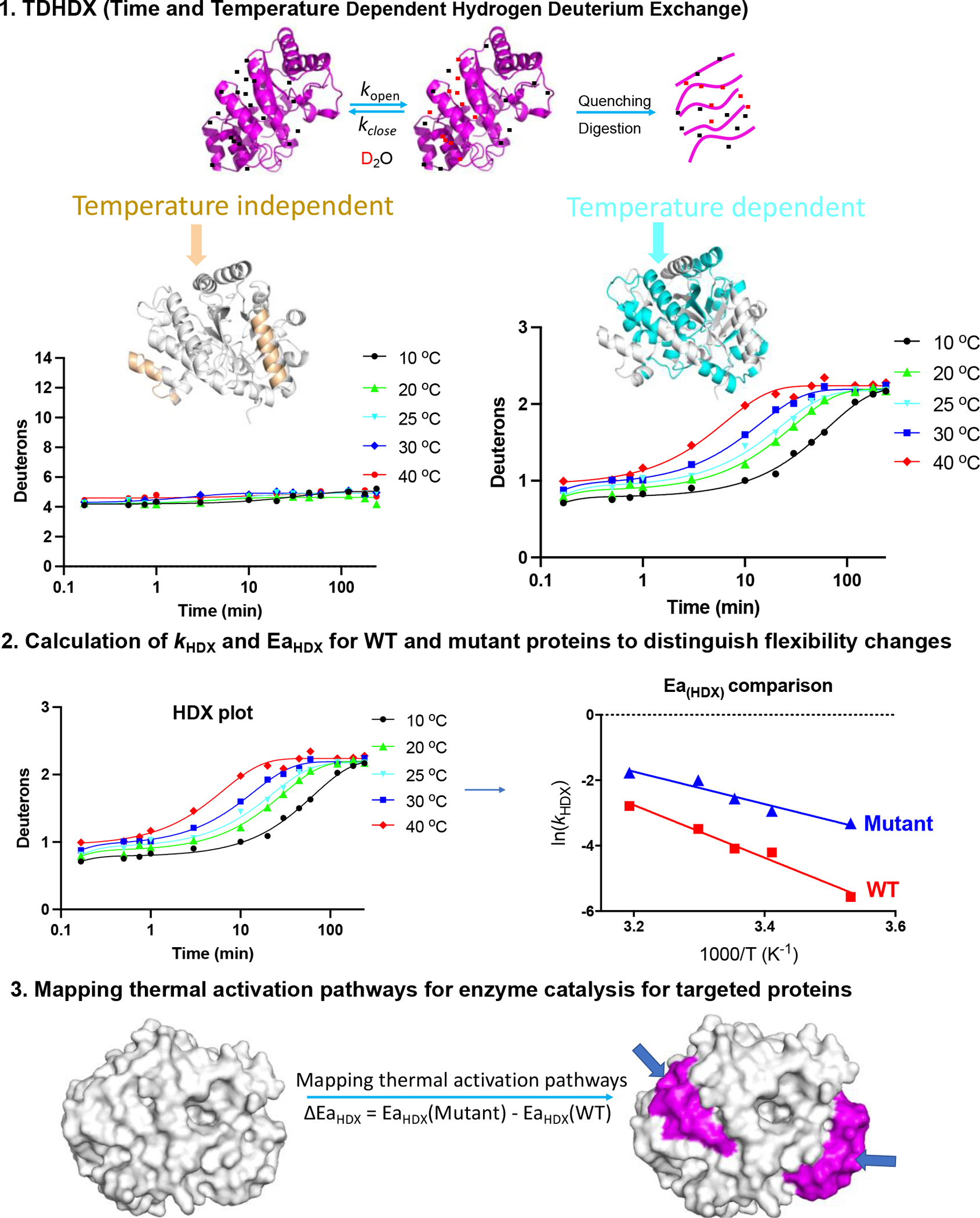
1. Procedures for uncovering thermal transfer pathways using temperature dependent hydrogen deuterium exchange coupled to mass spectrometry (TDHDX) using data from mADA as an example. (1) HDX samples are digested into small peptides and analyzed by mass spectrometry to detect deuteron incorporation. Most peptides will manifest at least 2 types of patterns: temperature independent plots (left panel, mapped in gold for the two peptides of this type in mADA) with the example shown undergoing rapid exchange for 4/14 amides and no further detectable change in deuteron uptake throughout the experimental period; and temperature dependent plots (right panel, mapped in cyan for the 10 peptides of this type in mADA) and where temperature dependency is apparent throughout the time course. 2. Peptides with apparent temperature dependent trends are fitted to a three-exponential equation (see Ref [86,87] for details) providing rate constants for the different regimes of HDX exchange. Using weighted average rate constants, Arrhenius plots can be generated to calculate the observed activation energy for HDX (EaHDX) for each peptide. Accordingly, corresponding parameters are analyzed for mutants of interest and compared to the WT protein behavior. 3. using ΔEaHDX as a proxy, regions within a protein that show functionally relevant changes in protein flexibility are mapped onto the protein structure, to reveal possible thermal energy transfer pathways for efficient enzyme catalysis. Emerging models for enzyme catalysis invoke a redistribution of protein substates upon binding of substrate (Figure 1) that acts in concert with efficient heat transfer via embedded thermal conduit (Figure 2) to enhance the probability of reaction barrier crossings.
Dynamical Probes for the Temporal Resolution of Protein Motions.
The TDHDX methodology discussed above is analogous to the majority of biophysical probes that generate time averaged structural information. It is generally assumed that the source of the thermal activation of enzyme reactivity comes from collisions of bulk water with a regime of biological water that includes protein bound and highly structured waters [88]. With the increasing body of evidence for protein networks that provide high site selectivity with regard to thermal transfer from solvent to the components of the active site, many questions arise. In particular, the time scales and mechanisms for communication between bulk solvent and enzyme active sites become of paramount importance. The time scale for enzymatic reactions is millisecond, making observed rates 1012-fold slower than the actual (femtosecond) barrier crossings, leaving open a wide range of time dependent changes in protein structure that can be linked to catalytic activation.
In recent studies, Zaragoza et al. targeted the protein surface of SLO, through the introduction of site-specific cysteine residues that were modified through chemical attachment of a fluorescent molecule. Using Badan (6-bromoacetyl-2-dimethylaminonaphthalene) as the installed probe and focusing on time and temperature dependent Stokes shifts [89], initial experiments detected nanosecond transients that could be assigned to either bound water or free water undergoing rapid exchange with the bound water [88]. Quite significantly, incorporation of Badan at the primary loop identified by TDHDX for SLO leads to identical activation energies for the observed Stokes shifts and for catalytic turnover, while a control loop indicated no such correlation. Subsequently, five SLO variants within the network with different activation energies for catalysis were pursued, yielding an identity of Ea values for Stokes shifts to Ea values for active site catalysis [83]. These studies provide the first temporal resolution of nanosecond surface protein motions that reflect solvent-induced activation at a protein/water interface that is transmitted cooperatively over a distance of 15–30 Å to the buried active site [83]. Such behavior contrasts with the default role of distributed and rapidly equilibrating conformational landscapes as the dominant dynamical behavior impacting enzyme function (discussed above), leading to a new model for the essential role of protein dynamics in achieving optimal enzyme catalysis [83].
Integration of the dynamical properties of enzymes into de novo protein design.
Recent years have witnessed substantial progress in our ability to perform de novo design of proteins [90] and to introduce new functionalities into existing protein scaffolds [91,92]. Such protein design efforts have helped elucidate general principles of protein structure and test our understanding of the biophysical and functional mechanisms of naturally evolved proteins. However, computational protein design methods generally rely on the assumption of static conformations in order to handle the vast complexity of sequence space, generally providing relatively small overall rate accelerations. The success of design methods, especially in relation to functional proteins, is expected to benefit from explicit considerations of conformational heterogeneity and dynamics.
A common trend in evolutionary trajectories is that the resulting mutations are not necessarily found in or nearby the active site. Of particular interest to protein design, propagated effects can occur via long distance side chain reorganization. Jackson et al. analyzed interaction networks in the evolutionary trajectory of a phosphotriesterase from Pseudomonas diminuta, showing that while the atomic positions changed very little, the interaction network changed substantially. In particular, one loop gained more interactions, resulting in a loss of mobility, while removal of interactions to another loop caused increased mobility [93]. We note that while progress is being made, we remain a long way from being able to routinely improve protein function by manipulating conformational sampling. Going forward, a deeper understanding about enzyme catalysis is still required, especially in terms of how enzymes generate and utilize site selective networks that are in contact with solvent and transmit thermal activation to initiate active site chemistry.
Figure 2.
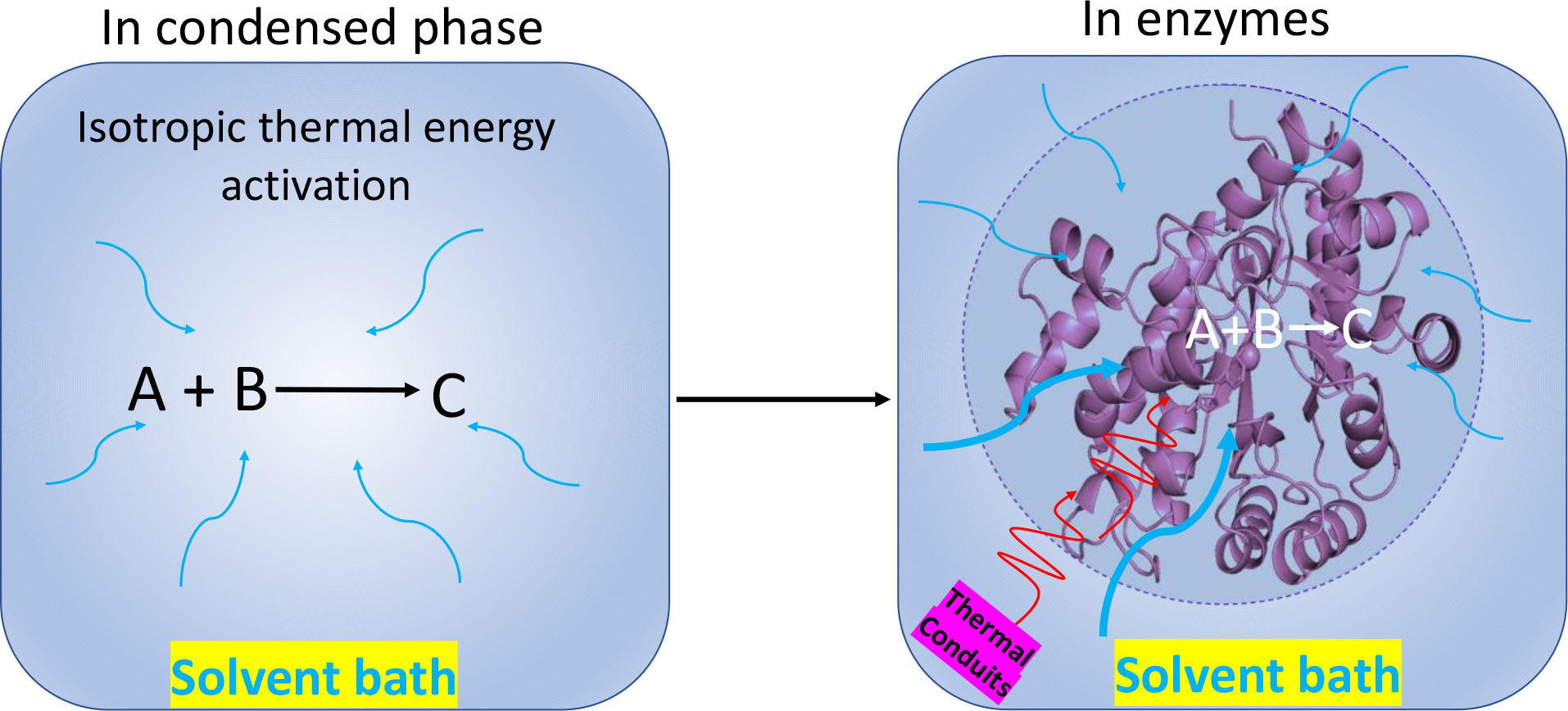
Collision theory states that for a chemical reaction to occur in condensed phase, the reacting particles must collide with one another. The rate of the reaction depends on the frequency and direction of collisions. For such temperature dependent reactions, thermal energy is expected from all directions in the solvent bath via isotropic thermal energy transfer (Left). As a comparison, enzymes are anisotropic structures, with active sites for chemical reactivity that are generally protected from direct collisions with solvent, implicating the protein scaffold as the basis for controlled heat transfer from the solvent bath to an enzyme active site (Right). Mechanistically, it is likely that enzymes will have evolved throughout time to construct privileged thermal conduits for efficient conduction of heat from the solvent to the active site that facilitate catalysis.
Acknowledgements
The authors thank the National Institutes of Health, United States for funding support (GM118117 to J.P.K.). We thank Professor Nigel Richards from Cardiff University for insightful discussions and comments.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Kohen A, Cannio R, Bartolucci S, Klinman JP: Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature 1999, 399:496–499. [DOI] [PubMed] [Google Scholar]
- 2.Knapp MJ, Rickert K, Klinman JP: Temperature-dependent isotope effects in soybean lipoxygenase-1: correlating hydrogen tunneling with protein dynamics. J Am Chem Soc 2002, 124:3865–3874. [DOI] [PubMed] [Google Scholar]
- 3.Benkovic SJ, Hammes-Schiffer S: A perspective on enzyme catalysis. Science 2003, 301:1196–1202. [DOI] [PubMed] [Google Scholar]
- 4.Tousignant A, Pelletier JN: Protein motions promote catalysis. Chemistry & biology 2004, 11:1037–1042. [DOI] [PubMed] [Google Scholar]
- 5.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, Kern D: A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450:913–916. [DOI] [PubMed] [Google Scholar]
- 6.Henzler-Wildman K, Kern D: Dynamic personalities of proteins. Nature 2007, 450:964–972. [DOI] [PubMed] [Google Scholar]
- 7.Klinman JP, Kohen A: Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu Rev Biochem 2013, 82:471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klinman JP: Dynamically achieved active site precision in enzyme catalysis. Acc Chem Res 2015, 48:449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DePristo MA, de Bakker PI, Blundell TL: Heterogeneity and inaccuracy in protein structures solved by X-ray crystallography. Structure 2004, 12:831–838. [DOI] [PubMed] [Google Scholar]
- 10.Kuzmanic A, Pannu NS, Zagrovic B: X-ray refinement significantly underestimates the level of microscopic heterogeneity in biomolecular crystals. Nat Commun 2014, 5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T: Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci U S A 2011, 108:16247–16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Den Bedem H, Bhabha G, Yang K, Wright PE, Fraser JS: Automated identification of functional dynamic contact networks from X-ray crystallography. Nat Methods 2013, 10:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y: A glimpse of structural biology through X-ray crystallography. Cell 2014, 159:995–1014. [DOI] [PubMed] [Google Scholar]
- 14.Keedy DA, Fraser JS, van den Bedem H: Exposing hidden alternative backbone conformations in X-ray crystallography using qFit. PLoS Comput Biol 2015, 11:e1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levantino M, Yorke BA, Monteiro DC, Cammarata M, Pearson AR: Using synchrotrons and XFELs for time-resolved X-ray crystallography and solution scattering experiments on biomolecules. Current opinion in structural biology 2015, 35:41–48. [DOI] [PubMed] [Google Scholar]
- 16.Šrajer V, Schmidt M: Watching proteins function with time-resolved x-ray crystallography. Journal of physics D: Applied physics 2017, 50:373001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vendruscolo M: Determination of conformationally heterogeneous states of proteins. Curr Opin Chem Biol 2007, 17:15–20. [DOI] [PubMed] [Google Scholar]
- 18.Ma B, Nussinov R: Enzyme dynamics point to stepwise conformational selection in catalysis. Curr Opin Chem Biol 2010, 14:652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyle N, Das RK, Pappu RV: A quantitative measure for protein conformational heterogeneity. J Chem Phys 2013, 139:09B607_601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramanathan A, Savol A, Burger V, Chennubhotla CS, Agarwal PK: Protein conformational populations and functionally relevant substates. Acc Chem Res 2014, 47:149–156. [DOI] [PubMed] [Google Scholar]
- 21.Keedy DA, Kenner LR, Warkentin M, Woldeyes RA, Hopkins JB, Thompson MC, Brewster AS, Van Benschoten AH, Baxter EL, Uervirojnangkoorn M, McPhillips SE, Song J, Alonso-Mori R, Holton JM, Weis WI, Brunger AT, Soltis SM, Lemke H, Gonzalez A, Sauter NK, Cohen AE, Bedem Hvd, E TR, Fraser JS: Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. Elife 2015, 4:e07574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agarwal PK, Doucet N, Chennubhotla C, Ramanathan A, Narayanan C: Conformational sub-states and populations in enzyme catalysis. Meth Enzymol 2016, 578:273–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrović D, Risso VA, Kamerlin SCL, Sanchez-Ruiz JM: Conformational dynamics and enzyme evolution. J R Soc Interface 2018, 15:20180330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipscomb WN: Acceleration of reactions by enzymes. Accounts of Chemical Research 1982, 15:232–238. [Google Scholar]
- 25.Wolfenden R, Snider MJ: The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res 2001, 34:938–945. [DOI] [PubMed] [Google Scholar]
- 26.Pauling L: Molecular architecture and biological reactions. Chem Eng News 1946, 24:1375–1377. [Google Scholar]
- 27.Pauling L: Nature of forces between large molecules of biological interest. Nature 1948, 161:707–709. [DOI] [PubMed] [Google Scholar]
- 28.•. Lightstone FC, Zheng Y-J, Bruice TC: Molecular dynamics simulations of ground and transition states for the Sn2 displacement of Cl-from 1, 2-dichloroethane at the active site of Xanthobacter autotrophicus haloalkane dehalogenase. J Am Chem Soc 1998, 120:5611–5621. MD simulation on COMT enzyme indicated that catalytic power of COMT in going from ES to ETS arises from the ability of the active site to arrange the ground state into tightly packed conformers.
- 29.Lau EY, Bruice TC: Comparison of the dynamics for ground-state and transition-state structures in the active site of catechol O-methyltransferase. J Am Chem Soc 2000, 122:7165–7171. [Google Scholar]
- 30.•. Zhang J, Klinman JP: Enzymatic methyl transfer: role of an active site residue in generating active site compaction that correlates with catalytic efficiency. J Am Chem Soc 2011, 133:17134–17137. Secondary isotope effects indicate active site compaction at the barrier crossing that are dependent on a tyrosine residue behind the methyl bearing sulfur of the AdoMet cofactor.
- 31.Zhang J, Kulik HJ, Martinez TJ, Klinman JP: Mediation of donor–acceptor distance in an enzymatic methyl transfer reaction. Proc Natl Acad Sci U S A 2015, 112:7954–7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kulik HJ, Zhang J, Klinman JP, Martínez TJ: How large should the QM region be in QM/MM calculations? The case of catechol O-methyltransferase. J Phys Chem B 2016, 120:11381–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.•. Zhang J, Balsbaugh JL, Gao S, Ahn NG, Klinman JP: Hydrogen deuterium exchange defines catalytically linked regions of protein flexibility in the catechol O-methyltransferase reaction. Proc Natl Acad Sci U S A 2020, 117:10797–10805. Using TDHDX-MS, this study uncovered two regions of the protein that could potentially provide thermal energy transfer from solvent to enzyme active site.
- 34.Czarnota S, Johannissen LO, Baxter NJ, Rummel F, Wilson AL, Cliff MJ, Levy CW, Scrutton NS, Waltho JP, Hay S: Equatorial active site compaction and electrostatic reorganization in catechol-O-methyltransferase. ACS catalysis 2019, 9:4394–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.•. Yabukarski F, Biel JT, Pinney MM, Doukov T, Powers AS, Fraser JS, Herschlag D: Assessment of enzyme active site positioning and tests of catalytic mechanisms through X-ray–derived conformational ensembles. Proc Natl Acad Sci U S A 2020, 117:33204–33215. Using room temperature X-ray crystallography data for the Pseudomonas putida ketosteroid isomerase (KSI), authors aimed to uncover the structural changes throughout the reaction cycle for KSI using protein complexes with substrate or transition state analogs; the aggregate conformational ensembles comparason indicated limited change in active site positioning (on the 1- to 1.5-Å scale, which is on the same level for the resolution of the crystal structure).
- 36.Gao S, Zhang W, Barrow SL, Iavarone AT, Klinman JP: Distinguishing the roles of substrate-induced changes to protein flexibility from embedded thermal activation networks in enzyme catalysis. J Biol Chem 2022, In Revision. [Google Scholar]
- 37.Warshel A: Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J Biol Chem 1998, 273:27035–27038. [DOI] [PubMed] [Google Scholar]
- 38.Petoukhov MV, Svergun DI: Applications of small-angle X-ray scattering to biomacromolecular solutions. Int J Biochem Cell Biol 2013, 45:429–437. [DOI] [PubMed] [Google Scholar]
- 39.Allison JR: Using simulation to interpret experimental data in terms of protein conformational ensembles. Curr Opin Struct Biol 2017, 43:79–87. [DOI] [PubMed] [Google Scholar]
- 40.••. Boehr DD, Nussinov R, Wright PE: The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 2009, 5:789–796. Conformational selection postulates that all protein conformations pre-exist, and ligand binding triggers the conformational selection to the most favored conformation via a population shift, redistributing the conformational states.
- 41.Daniel RM, Dunn RV, Finney JL, Smith JC: The role of dynamics in enzyme activity. Annu Rev Biophys 2003, 32:69–92. [DOI] [PubMed] [Google Scholar]
- 42.Jamros MA, Oliveira LC, Whitford PC, Onuchic JN, Adams JA, Jennings PA: Substrate-specific reorganization of the conformational ensemble of CSK implicates novel modes of kinase function. 2012. [DOI] [PMC free article] [PubMed]
- 43.Kale S, Jordan F: Conformational ensemble modulates cooperativity in the rate-determining catalytic step in the E1 component of the Escherichia coli pyruvate dehydrogenase multienzyme complex. J Biol Chem 2009, 284:33122–33129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nashine VC, Hammes-Schiffer S, Benkovic SJ: Coupled motions in enzyme catalysis. Curr Opin Chem Biol 2010, 14:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei G, Xi W, Nussinov R, Ma B: Protein ensembles: how does nature harness thermodynamic fluctuations for life? The diverse functional roles of conformational ensembles in the cell. Chem Rev 2016, 116:6516–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koshland DE Jr: The key–lock theory and the induced fit theory. Angew Chem Int Ed 1995, 33:2375–2378. [Google Scholar]
- 47.Campbell E, Kaltenbach M, Correy GJ, Carr PD, Porebski BT, Livingstone EK, Afriat-Jurnou L, Buckle AM, Weik M, Hollfelder F, Tokuriki N, Jackson CJ: The role of protein dynamics in the evolution of new enzyme function. Nat Chem Biol 2016, 12:944–950. [DOI] [PubMed] [Google Scholar]
- 48.Hong N-S, Petrović D, Lee R, Gryn’ova G, Purg M, Saunders J, Bauer P, Carr PD, Lin C-Y, Mabbitt PD, Zhang W, Altamore T, Easton C, Coote ML, Kamerlin SCL, Jackson CJ: The evolution of multiple active site configurations in a designed enzyme. Nat commun 2018, 9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buller AR, Van Roye P, Cahn JK, Scheele RA, Herger M, Arnold FH: Directed evolution mimics allosteric activation by stepwise tuning of the conformational ensemble. J Am Chem Soc 2018, 140:7256–7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh P, Vandemeulebroucke A, Li J, Schulenburg C, Fortunato G, Kohen A, Hilvert D, Cheatum CM: Evolution of the Chemical Step in Enzyme Catalysis. ACS Catal 2021, 11:6726–6732. [Google Scholar]
- 51.Li J, Lin J, Kohen A, Singh P, Francis K, Cheatum CM: Evolution of Optimized Hydride Transfer Reaction and Overall Enzyme Turnover in Human Dihydrofolate Reductase. Biochemistry 2021, 60:3822–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.••. Broom A, Rakotoharisoa RV, Thompson MC, Zarifi N, Nguyen E, Mukhametzhanov N, Liu L, Fraser JS, Chica RA: Ensemble-based enzyme design can recapitulate the effects of laboratory directed evolution in silico. Nat Commun 2020, 11:1–10. A better pre-organized active site was detected for the Kemp eliminase what went through directed evolution. Crystallographic analysis illustrated how mutations may shift the conformational ensemble toward an enrichment of catalytically competent substates.
- 53.••. Otten R, Pádua RA, Bunzel HA, Nguyen V, Pitsawong W, Patterson M, Sui S, Perry SL, Cohen AE, Hilvert D, Kern D: How directed evolution reshapes the energy landscape in an enzyme to boost catalysis. Science 2020, 370:1442–1446. Directed evolution gradually altered the conformational ensemble of the protein scaffold to shift to a narrow, highly active conformational ensemble and accelerate the catalytic efficiency by nearly nine orders of magnitude for the designed Kemp eliminase.
- 54.Nothling MD, Xiao Z, Bhaskaran A, Blyth MT, Bennett CW, Coote ML, Connal LA: Synthetic catalysts inspired by hydrolytic enzymes. ACS Catal 2018, 9:168–187. [Google Scholar]
- 55.Raynal M, Ballester P, Vidal-Ferran A, Van Leeuwen PW: Supramolecular catalysis. Part 2: artificial enzyme mimics. Chem Soc Rev 2014, 43:1734–1787. [DOI] [PubMed] [Google Scholar]
- 56.••. Agarwal PK: A biophysical perspective on enzyme catalysis. Biochemistry 2018, 58:438–449. This perspective paper proposes networks of conserved residues that extend from the surface of the enzyme all the way to the active site, serving as pathways for energy transfer that enables thermodynamical coupling of the surrounding solvent with enzyme catalysis.
- 57.Lockless SW, Ranganathan R: Evolutionarily conserved pathways of energetic connectivity in protein families. Science 1999, 286:295–299. [DOI] [PubMed] [Google Scholar]
- 58.••. Süel GM, Lockless SW, Wall MA, Ranganathan R: Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat struct biol 2003, 10:59–69. This paper reports a set of energetically coupled positions that include unexpected long-range interactions in the PDZ domian protein, implicating the importance of networks of interactions along a long distance that could serve as a basis for efficient energy conduction.
- 59.•. Liang Z-X, Lee T, Resing KA, Ahn NG, Klinman JP: Thermal-activated protein mobility and its correlation with catalysis in thermophilic alcohol dehydrogenase. Proc Natl Acad Sci U S A 2004, 101:9556–9561. The first in depth study of HDX-MS as a funtion of temperature in an enzyme catalyzed reaction.
- 60.Nagel ZD, Cun S, Klinman JP: Identification of a long-range protein network that modulates active site dynamics in extremophilic alcohol dehydrogenases. J Biol Chem 2013, 288:14087–14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee J, Goodey NM: Catalytic contributions from remote regions of enzyme structure. Chem Rev 2011, 111:7595–7624. [DOI] [PubMed] [Google Scholar]
- 62.Singh P, Francis K, Kohen A: Network of remote and local protein dynamics in dihydrofolate reductase catalysis. ACS catal 2015, 5:3067–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.•. Kozlowski R, Zhao J, Dyer RB: Acceleration of catalysis in dihydrofolate reductase by transient, site-specific photothermal excitation. Proc Natl Acad Sci U S A 2021, 118. Using a gold nanoparticle attached to a specific site on the enzyme dihydrofolate reductase (DHFR), the authors were able to photothermally exicte specific ground-state conformations to accelerate catalysis.
- 64.•. Bunzel HA, Anderson J, Hilvert D, Arcus VL, van der Kamp MW, Mulholland AJ: Evolution of dynamical networks enhances catalysis in a designer enzyme. Nature Chemistry 2021, 13:1017–1022. Molecular dynamics on the designed Kemp eliminase detected two mutations introduced during evolution; these are found to be involved in a proposed network that tunes the dynamic and packing properties for enhanced catalysis.
- 65.•. Markin C, Mokhtari D, Sunden F, Appel M, Akiva E, Longwell S, Sabatti C, Herschlag D, Fordyce P: Revealing enzyme functional architecture via high-throughput microfluidic enzyme kinetics. Science 2021, 373:eabf8761. This study reported a designed high-throughput microfluidic platform to introduce more than 1000 mutations (either a glycine or valine substitution) throughout the structure of an alkaline phosphatase. Results showed that the pattern of mutations on impacting catalysis is asymmetric and complex.
- 66.Heyes DJ, Lakavath B, Hardman SJ, Sakuma M, Hedison TM, Scrutton NS: Photochemical mechanism of light-driven fatty acid photodecarboxylase. ACS catal 2020, 10:6691–6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sancar A: Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem Rev 2003, 103:2203–2238. [DOI] [PubMed] [Google Scholar]
- 68.Wang H, Lin S, Allen JP, Williams JC, Blankert S, Laser C, Woodbury NW: Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science 2007, 316:747–750. [DOI] [PubMed] [Google Scholar]
- 69.Abeles RH, Frey PA, Jencks WP: Biochemistry: Jones & Bartlett Pub; 1992. [Google Scholar]
- 70.Fersht A: Enzyme structure and mechanism. 1977.
- 71.Fersht A: Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding: Macmillan; 1999. [Google Scholar]
- 72.Jencks WP: Catalysis in chemistry and enzymology: Courier Corporation; 1987. [Google Scholar]
- 73.Winter SD, Jones HB, Răsădean DM, Crean RM, Danson MJ, Pantoş GD, Katona G, Prentice E, Arcus VL, van der Kamp MW, Pudney CR: Chemical mapping exposes the importance of active site interactions in governing the temperature dependence of enzyme turnover. ACS catal 2021, 11:14854–14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.••. Offenbacher AR, Hu S, Poss EM, Carr CA, Scouras AD, Prigozhin DM, Iavarone AT, Palla A, Alber T, Fraser JS, Klinman JP: Hydrogen–deuterium exchange of lipoxygenase uncovers a relationship between distal, solvent exposed protein motions and the thermal activation barrier for catalytic proton-coupled electron tunneling. ACS Cent. Sci 2017, 3:570–579. TDHDX experiments were conducted on wild type and mutant forms of soybean lipoxygenase (SLO). Results uncovered site specific trends in the enthalpic barriers for HDX-MS within two distal and solvent-exposed loops that correlate well with the enthalpic barriers for catalysis and serve as thermal transfer conduits.
- 75.Tajoddin NN, Konermann L: Analysis of temperature-dependent H/D exchange mass spectrometry experiments. Anal Chem 2020, 92:10058–10067. [DOI] [PubMed] [Google Scholar]
- 76.Konermann L, Pan J, Liu Y-H: Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chemi Soc Rev 2011, 40:1224–1234. [DOI] [PubMed] [Google Scholar]
- 77.Masson GR, Burke JE, Ahn NG, Anand GS, Borchers C, Brier S, Bou-Assaf GM, Engen JR, Englander SW, Faber J, Garlish R, Griffin PR, Gross ML, Guttman M, Hamuro Yoshitomo, Heck AJR, Houde D, Iacob RE, Jørgensen TJD, Kaltashov IA, Klinman JP, Konermann L, Man P, Mayne L, Pascal BD, Reichmann D, Skehel M, Snijder, Strutzenberg TS, Underbakke ES, Wagner C, Wales TE, Walters BT, Weis DD, Derek J. Wilson, Wintrode PL, Zhang Z, Zheng, Schriemer DC, Rand KD: Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat Methods 2019, 16:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.James EI, Murphree TA, Vorauer C, Engen JR, Guttman M: Advances in hydrogen/deuterium exchange mass spectrometry and the pursuit of challenging biological systems. Chem Rev 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deng B, Zhu S, Macklin AM, Xu J, Lento C, Sljoka A, Wilson DJ: Suppressing allostery in epitope mapping experiments using millisecond hydrogen/deuterium exchange mass spectrometry. In MAbs: Taylor & Francis: 2017:1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ramirez-Sarmiento CA, Komives EA: Hydrogen-deuterium exchange mass spectrometry reveals folding and allostery in protein-protein interactions. Methods 2018, 144:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ozohanics O, Ambrus A: Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes. Life 2020, 10:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bai Y, Milne JS, Mayne L, Englander SW: Primary structure effects on peptide group hydrogen exchange. Proteins: Struct Funct Bioinfo 1993, 17:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zaragoza JP, Offenbacher AR, Hu S, Gee CL, Firestein ZM, Minnetian N, Deng Z, Fan F, Iavarone AT, Klinman JP: Temporal and Spatial Resolution of a Protein Quake that Activates Hydrogen Tunneling in Soybean Lipoxygenase. bioRxiv 2022:2022.2003.2031.486235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wierenga R: The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS lett 2001, 492:193–198. [DOI] [PubMed] [Google Scholar]
- 85.Romero-Romero S, Kordes S, Michel F, Höcker B: Evolution, folding, and design of TIM barrels and related proteins. Curr Opin Struct Biol 2021, 68:94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.••. Gao S, Thompson EJ, Barrow SL, Zhang W, Iavarone AT, Klinman JP: Hydrogen–Deuterium Exchange within Adenosine Deaminase, a TIM Barrel Hydrolase, Identifies Networks for Thermal Activation of Catalysis. J Am Chem Soc 2020, 142:19936–19949. Using TDHDX on WT and mutant mADA, thermal transfter pathways were identified. Two primary thermal networks reach from opposing protein/solvent interfaces toward the active. Detailed analysis of the TDHDX was provided in this paper.
- 87.•. Thompson EJ, Paul A, Iavarone AT, Klinman JP: Identification of Thermal Conduits That Link the Protein–Water Interface to the Active Site Loop and Catalytic Base in Enolase. J Am Chem Soc 2021, 143:785–797. TDHDX on yeast enolase uncovered regions where changes in protein flexibility that correlate with changes in ΔH‡ for enzyme catalysis.
- 88.Nandi N, Bagchi B: Dielectric relaxation of biological water. J Phys Chem B 1997, 101:10954–10961. [Google Scholar]
- 89.••. Zaragoza JPT, Nguy A, Minnetian N, Deng Z, Iavarone AT, Offenbacher AR, Klinman JP: Detecting and characterizing the kinetic activation of thermal networks in proteins: thermal transfer from a distal, solvent-exposed loop to the active site in soybean lipoxygenase. J Phys Chem B 2019, 123:8662–8674. Bioconjugation of SLO and Badan (6-bromoacetyl-2-dimethylaminonaphthalene) provided a unique chance to study the temporal resolution solvation dynamics and its correlation with enyzme catalysis. Identical activation energies for stokes shifts and for catalysis were detected, implicating a cooperative and regional activation of the protein that is initiated at the protein surface.
- 90.Kaplan J, DeGrado W: De novo design of catalytic proteins. Proc Natl Acad Sci U S A 2004, 101:11566–11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ferrando J, Solomon LA: Recent progress using de novo design to study protein structure, design and binding interactions. Life 2021, 11:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cao L, Goreshnik I, Coventry B, Case JB, Miller L, Kozodoy L, Chen RE, Carter L, Walls AC, Park Y-J, Strauch E-M, Stewart L, Diamond MS, Veesler D, Baker D: De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370:426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.•. Jackson CJ, Foo J-L, Tokuriki N, Afriat L, Carr PD, Kim H-K, Schenk G, Tawfik DS, Ollis D: Conformational sampling, catalysis, and evolution of the bacterial phosphotriesterase. Proc Natl Acad Sci U S A 2009, 106:21631–21636. Interaction networks in the evolutionary trajectory of a phosphotriesterase from Pseudomonas diminuta showed that while the atomic positions changed very little, the interaction network changed substantially. Those interaction networks play an important role in enhanced catalysis.