

Abstract
Primary hematopoietic stem and progenitor cell (HSPC)-derived megakaryocytes are a valuable tool for translational research interrogating disease pathogenesis and developing new therapeutic avenues for patients with hematologic disorders including myeloproliferative neoplasms (MPNs). Thrombopoietin (TPO)-independent proliferation and megakaryocyte differentiation play a central role in the pathogenesis of essential thrombocythemia and myelofibrosis, two MPN subtypes that are characterized by increased numbers of bone marrow megakaryocytes and somatic mutations in either JAK2, CALR, or MPL. However, current culture strategies generally use healthy HSPCs for megakaryocyte production and are not optimized for the investigation of TPO-independent or TPO-hypersensitive growth and megakaryocyte-directed differentiation of primary patient–derived HSPCs. Here, we describe a detailed protocol covering all necessary steps for the isolation of CD34+ HSPCs from the peripheral blood of MPN patients and the subsequent TPO-independent differentiation into CD41+ megakaryocytes using both a collagen-based colony assay and a liquid culture assay. This protocol provides a novel, reproducible, and cost-effective approach for investigating megakaryocyte growth and differentiation properties from primary MPN patient cells that can be easily adapted for research on other megakaryocyte-related disorders.
This protocol was validated in: EMBO Rep (2022), DOI: 10.15252/embr.202152904
Graphical abstract
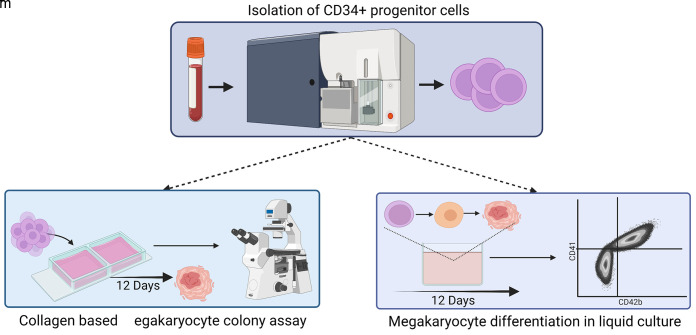
Schematic representation of the isolation of CD34+ progenitor cells and subsequent TPO-independent megakaryocyte differentiation
Keywords: Hematopoietic stem and progenitor cells, Megakaryocyte differentiation, Thrombopoietin, TPO-independent, Myeloproliferative neoplasm, Collagen, Interleukin-6, Interleukin-9
Background
Multipotent hematopoietic stem and progenitor cells (HSPCs) are not only responsible for life-long hematopoiesis but are also found to be the origin of many hematological malignancies (Bonnet and Dick, 1997; Jamieson et al., 2006; Woll et al., 2014; Reinisch et al., 2016). Myeloproliferative neoplasms (MPNs) are hematopoietic stem cell–derived diseases that are characterized by an aberrant proliferation of myeloid cells through constitutive activation of cytokine-signaling pathways (Tefferi and Pardanani, 2015; Spivak, 2017). In essential thrombocythemia (ET) and myelofibrosis (PMF), two common subtypes of MPNs, megakaryocyte lineage-biased differentiation and proliferation are hallmarks of disease pathogenesis (Vannucchi et al., 2013; Tefferi and Pardanani, 2019). Historically, thrombopoietin (TPO) was found to be a crucial growth factor in nearly all stages of megakaryocyte development with a profound effect on the survival, proliferation, and differentiation of committed HSPCs to the development of mature megakaryocytes (Debili et al., 1995; Choi et al., 1995; Broudy et al., 1995; Chen et al., 1995; Kojima et al., 1995). Thus, it is not surprising that the oncogenic drivers of MPNs were found to be activating mutations occurring in genes along the TPO signaling axis (MPL and JAK2), rendering cells TPO-independent and inducing increased megakaryopoiesis (Kralovics et al., 2005; Pikman et al., 2006; Woods et al., 2019). Additionally, mutations in the endoplasmic reticulum chaperone CALR, which has not been implicated in megakaryopoiesis to date, introduced structural changes, resulting in protein multimerization and binding to the TPO receptor (MPL). This binding results in an activation of downstream signaling in absence of TPO (Klampfl et al., 2013; Chachoua et al., 2016; Marty et al., 2016; Araki et al., 2016).
Despite recent advances in unveiling the oncogenic transformation of healthy HSPCs, detailed mechanisms of disease pathogenesis are still poorly understood. Current methods to investigate megakaryocyte-biased cellular transformation by MPN mutations either use TPO-dependent cell lines or costly HSPC differentiation assays that are not tailored to investigate TPO-independent cell growth (Lu et al., 2005; Araki et al., 2016; Elf et al., 2016; Pronier et al., 2018). Primary progenitor cells from patients with polycythemia vera, another MPN disease subtype, often form erythroid colonies in the absence of erythropoietin (Corre-Buscail et al., 2005), but assays to show TPO-independent growth or TPO-hypersensitive growth of ET- and PMF-derived cells are not well established. Current protocols for in vitro megakaryocyte differentiation focus on optimizing culture conditions to maximize the megakaryocyte output of healthy HSPCs, but do not consider MPN-related phenotypes. Here, we describe a reproducible and cost-effective step-by-step protocol for the isolation, culture, and TPO-independent megakaryocyte differentiation of primary MPN patient–derived CD34+ HSPCs to investigate disease-relevant characteristics. This approach uses fluorescence-activated cell sorting (FACS) to purify CD34+ HSPCs from the peripheral blood of MPN patients and an animal serum-free culture system for megakaryocyte differentiation in a clonogenic semi-solid media and collagen-based colony assay, as well as in liquid culture. This protocol facilitates future MPN research, providing an opportunity to test novel therapeutic interventions in a human pre-clinical setting (Tvorogov et al., 2022), but can be easily adapted to investigate other megakaryocyte-related diseases.
Materials and Reagents
50 mL conical tubes (Corning, FalconTM, catalog number: 352098)
1.8 mL cryogenic tubes (Thermo Fisher Scientific, NuncTM, catalog number: 368632)
5 mL round bottom tubes (Corning, FalconTM, catalog number: 352054)
5 mL round bottom tubes with cell strainer cap (Corning, FalconTM, catalog number: 352235)
1.5 mL screw cap micro tube (Sarstedt, catalog number: 72692005)
1.5 mL safe-lock tubes (Eppendorf, catalog number: 0030123328)
Double chamber slides (Thermo Fisher Scientific, NuncTM, Lab-TekTM, catalog number: 177429)
100 mm culture dish (Corning, catalog number: 430591)
35 mm culture dish (Greiner Bio-One, catalog number: 627161)
48-well plate (tissue-culture treated) (Corning, CostarTM, catalog number: 3548)
3.5 mL transfer pipettes (Sarstedt, catalog number: 86.1171.001)
Parafilm (Bemis, catalog number: PM996)
Filter cards and spacers (Stemcell technologies, catalog number: 04911)
Phosphate buffered saline (PBS) (Gibco, catalog number: 10010015)
Lymphoprep (Stemcell technologies, catalog number: 07801)
RPMI 1640 (Sigma-Aldrich, catalog number: R8758)
IMDM (Pan-Biotech, catalog number: P04-20450)
Fetal bovine serum (FBS) (Pan-Biotech, catalog number: P30-19475)
Penicillin/streptomycin (Pan-Biotech, catalog number: P06-07100)
Dimethyl sulfoxide (DMSO) (WAK-Chemie, catalog number: WAK-DMSO-10)
DNase (Worthington, catalog number: LS002007)
MegacultTM-C collagen and medium with lipids, without cytokines (Stemcell technologies, catalog number: 04974)
Methanol (Merck, EMSURE®, catalog number: 106009)
Acetone (Sigma-Aldrich, catalog number: 320110)
Hydrochloric acid solution 6 M (HCl) (Merck, catalog number: 143007)
Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888)
Ammonium chloride (NH4Cl) (Sigma-Aldrich, catalog number: A9434)
Potassium bicarbonate (KHCO3) (Sigma-Aldrich, catalog number: 237205)
EDTA tetrasodium (Sigma-Aldrich, catalog number: 03699)
TRIS (Roth, PUFFERAN®, catalog number: 5429.1)
Tween 20 (Sigma-Aldrich, catalog number: P1379)
Evans Blue (Sigma-Aldrich, catalog number: E2129)
Serum-free expansion media II (SFEM II) (Stemcell technologies, catalog number: 09655) or StemPro 34 SFM + nutrient supplement (Thermo Fisher Scientific, catalog number: 10639011)
L-Glutamine (200 mM) (Sigma-Aldrich, catalog number: 59202C)
Recombinant human stem cell factor (SCF) (PeproTech, catalog number: 300-07
Recombinant human thrombopoietin (TPO) (PeproTech, catalog number: 300-18)
Recombinant human interleukin 3 (IL-3) (PeproTech, catalog number: 200-03)
Recombinant human interleukin 6 (IL-6) (PeproTech, catalog number: 200-06)
Recombinant human interleukin 9 (IL-9) (PeproTech, catalog number: 200-09)
Lipids cholesterol rich (Sigma-Aldrich, catalog number: L4646)
Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A7906)
EDTA (Thermo Fisher Scientific, catalog number: AM9260G, 0.5M solution)
FcR blocking reagent human (Miltenyi, catalog number: 130-059-901)
Mouse IgG1, κ isotype control (BioLegend, catalog number: 400101)
Mouse anti-human CD41 (BioLegend, catalog number: 303702)
ImmPRESS®-AP horse anti-mouse IgG polymer detection kit (Vector Laboratories, catalog number: MP-5402)
Alkaline phosphatase (AP) substrate kit, Vector® Red (Vector Laboratories, catalog number: SK-5100)
Mouse anti-human CD34 FITC (BD Biosciences, PharmingenTM, catalog number: 555821)
Mouse anti-human CD45 V500-C (BD Biosciences, HorizonTM, catalog number: 655873)
Mouse anti-human CD2 PE-Cy5 (BD Biosciences, PharmingenTM, catalog number: 555328)
Mouse anti-human CD3 PE-Cy5 (BD Biosciences, PharmingenTM, catalog number: 555334)
Mouse anti-human CD19 PE-Cy5 (BD Biosciences, PharmingenTM, catalog number: 555414)
Mouse anti-human CD14 PerCP-Cy5.5 (BD Biosciences, PharmingenTM, catalog number: 562692)
Mouse anti-human CD16 PE-Cy5 (BD Biosciences, PharmingenTM, catalog number: 555408)
Mouse anti-human CD56 PE-Cy5 (BD Biosciences, PharmingenTM, catalog number: 555517)
Mouse anti-human CD41 APC-Cy7 (BioLegend, catalog number: 303716)
Mouse anti-human CD42b APC (BioLegend, catalog number: 303912)
7-Amino-Actinomycin D (7-AAD) (BD Biosciences, PharmingenTM, catalog number: 559925)
Counting beads (Thermo Fisher Scientific, CountbrightTM, catalog number: C36950, LOT-specific concentration: 0.515 × 105 beads/50 µL)
RBC lysis buffer (see Recipes)
Freezing media (see Recipes)
Thawing media (see Recipes)
Staining buffer (SB) (see Recipes)
Antibody cocktail (see Recipes)
Megakaryocyte (Mk) colony media (see Recipes)
Megakaryocyte differentiation (Mk-diff) media (see Recipes)
Isotonic NaCl solution (0.15 M) (see Recipes)
TRIS buffer (0.5 M, pH 7.6) (see Recipes)
Wash buffer (see Recipes)
Substrate buffer (see Recipes)
Evans Blue solution (0.1%) (see Recipes)
Staining tray (see Recipes)
Equipment
Swing-bucket centrifuge (Eppendorf, model: 5810R)
Tabletop centrifuge (Eppendorf, model: 5427R)
Inverted light microscope (Zeiss, model: Primovert)
Upright light microscope (Olympus, model: BX51)
Fluorescence-activated cell sorter (BD Biosciences, model: Aria IIIu)
Flow cytometer (Beckman Coulter, model: Cytoflex S or BD Biosciences FACS CantoII)
Laminar flow workbench
Fume hood
Forceps
Scalpel
Software
FlowJo v10 (FlowJo LLC)
Prism 9 (GraphPad)
Procedure
-
Peripheral blood mononuclear cells (PBMC) collection and storage
Notes:
Collection of patient material has to be approved by a local ethics committee and every patient needs to give informed consent prior to material collection.
Perform all experimental steps in a sterile environment using a laminar flow workbench.
Alternatively, processed samples may be obtained from a biobank and used for subsequent analysis.
Dilute 15 mL of whole blood from MPN patients with 15 mL of PBS and slowly overlay on top of 15 mL Lymphoprep or similar density gradient reagents in a 50 mL conical tube.
Centrifuge at 800 × g at room temperature with brakes off for 30 min (excluding acceleration and deceleration times).
Carefully collect PBMCs from the whitish interphase layer between Lymphoprep (lower phase) and serum (upper phase) using a sterile 3.5 mL plastic transfer pipette. Avoid contamination of the layer with Lymphoprep.
Wash PBMCs by mixing with 40 mL of PBS and centrifuge at 350 × g at room temperature for 5 min.
-
Optional: If the cell pellet contains red blood cells (RBC), perform an RBC lysis to further purify PBMCs.
Suspend pellet in 10 mL of RBC lysis buffer (see Recipe 1) and incubate for 10 min at room temperature.
Wash PBMCs by mixing with 40 mL of PBS and centrifuge at 350 × g at room temperature for 5 min.
Resuspend pellet in 5 mL PBS and count the cells. Keep cells on ice during this step.
-
Take a small aliquot of cells (5 × 104) and determine the CD34+ cell fraction.
Resuspend cells in 50 µL staining buffer containing anti-human CD34 FITC antibody at a 1:25 dilution and incubate for 30 min at 4°C.
Wash cells by mixing with 1 mL of PBS and centrifuge at 350 × g at 4°C for 5 min.
Resuspend pellet in 100 µL staining buffer (see Recipe 2).
Add 2 µL of 7-AAD, incubate for 5 min, and acquire cells on flow cytometer.
Meanwhile, centrifuge the PBMCs at 350 × g at 4°C for 5 min, resuspend up to 5 × 106 cells in 1 mL of freezing media (see Recipe 3), and transfer to cryo tube for storage.
Freeze PBMCs in liquid nitrogen for long-term storage until required.
-
Primary human CD34+ cell isolation
Note: The CD34+ cell fraction observed in the peripheral blood of MPN patients is highly variable but generally much higher than normal healthy adults. We observed CD34+ HSPCs ranging from 0.2% to 20%. If this data is not available for your samples, we recommend thawing one vial of cells and determining the CD34+ cell frequency to estimate the number of PBMC vials needed.
Thaw frozen PBMCs in a water bath pre-heated to 37°C. The number of vials needed to get enough CD34+ cells for megakaryocyte differentiation is dependent on the predetermined frequency of CD34+ cells within total PBMCs.
Transfer cells from two vials of the same sample (1 × 107 cells) into one 50 mL tube and add 40 mL of thawing media (see Recipe 4) dropwise to the cells while gently swirling.
Incubate cells for 10 min at room temperature. The DNase in the thawing media will digest any DNA released from dead cells that would otherwise promote clumping of cells.
Centrifuge cells at 350 × g at room temperature for 5 min and remove supernatant.
Resuspend cell pellet in 135 µL of staining buffer (see Recipe 2).
Add 15 µL of FcR blocking reagent human, mix well, and incubate for 5 min at room temperature.
-
Add 40 µL of antibody cocktail (see Recipe 5), mix well, and incubate for 30 min at 4°C.
Notes:
Antibodies with different fluorescent conjugates can be used; however, lineage antibodies (CD2, CD3, CD19, CD14, CD16, CD56) should emit in the same channel as the viability dye for efficient exclusion.
At the initial setup of the experiment, unstained and single CD34 and CD45 antibodies as well as lineage antibody and 7-AAD stained cells should be prepared to correctly compensate all fluorophores and establish the population gates at the flow cytometer.
Wash cells once with 5 mL of staining buffer.
Centrifuge cells at 350 × g at 4°C for 5 min and remove supernatant.
Resuspend cells in 500 µL of staining buffer and filter the suspension through the cell strainer cap into a 5 mL round bottom tube.
Add 10 µL of 7-AAD and place tubes on ice prior to cell sorting.
-
Sort Lin- CD34+ CD45dim (Figure 1) hematopoietic stem and progenitor cells (HSPCs) into 1.5 mL screw cap tubes for downstream assays. Ideally, sort 8,000 CD34+ CD45dim HSPCs into a tube containing 400 µL of Mk colony media (see Recipe 6) and at least 100,000 CD34+ CD45dim HSPCs into a separate tube containing 200 µL Mk-diff media (see Recipe 7).
Note: It may not be possible to sort such high numbers of cells for some of the samples. These experiments can be scaled depending on final numbers of cells obtained. Liquid culture differentiation assays have been carried out with as few as 1,000 cells/replicate.
Proceed with megakaryocyte colony formation assay (section C) and liquid culture differentiation (section D).
-
Differentiation and quantification of TPO-independent megakaryocyte progenitors in a collagen-based colony assay
Notes:
This assay can be used to analyze the ability of therapeutic drugs to interfere with the megakaryocyte differentiation of MPN patient–derived HSPCs.
This assay is designed to analyze samples using four replicates (two double chamber slides). To carry out more tests, it is possible to reduce the number of cells and volumes of all reagents by half and perform only duplicates.
The addition of 50 ng/mL TPO is required for optimal proliferation and megakaryocyte differentiation of healthy HSPCs. While addition of TPO is not required for CALRmut or MPLmut samples, in our experience, JAK2mut HSPCs will not grow in the complete absence of TPO. A low dose of TPO (5–10 ng/mL) may be required for JAK2V617F samples and should be compared to healthy controls to measure TPO hypersensitivity rather than factor-independence.
-
Mix sorted HSPCs in 1.7 mL of Mk colony media by pipetting (see Recipe 6).
If therapeutic compounds will be tested, they should be added to the Mk colony media prior to the cell suspension.
Label the double chamber slides appropriately with a pencil, as ink from standard laboratory markers or pens will become illegible during the fixation process.
-
Using a 2 mL serological pipette, carefully add 1.2 mL of cold collagen to the Mk colony media and mix by gently pipetting, avoiding any foaming of the media.
During this process, keep collagen on ice and prepare only one tube at a time to prevent solidification of collagen before seeding.
Dispense 0.75 mL of the cell suspension into each well of two double chamber slides (four replicates) and distribute the volume by circular motion.
Place each chamber slide along with a 35 mm dish filled with 3 mL of sterile water into a separate sterile 10 mm culture dish (Figure 2).
Close the 10 mm culture dish and place it into a humidified incubator at 37°C and 5% CO2 atmosphere for 12 days.
Check for colony formation using an inverted light microscope and proceed to fixation and staining if colonies are present (Figure 3).
Prepare 100 mL fixative (1:3 mixture of methanol and acetone) and place into a rectangular plastic or glass dish (at least 12 × 9 × 2 cm in size). Cover the dish with parafilm or an appropriate lid and place into a fume hood.
-
Remove one chamber slide at a time from the incubator and proceed immediately with disassembly as collagen becomes unstable at lower temperatures (Video 1).
Remove lid from the chamber slide.
Carefully lift the chamber starting at the labeled end of the slide.
Use a scalpel to cut the attachment points of the underlying rubber seal connecting it to the chamber (three in total).
Proceed to the other end of the slide until the chamber is disconnected. The rubber seal should still be attached to the slide.
Take caution to avoid moving or turning the two collagen pieces from each chamber. In case the collagen pieces have moved from the original position (e.g., moved out of the rubber seal boundaries), use a scalpel or forceps with smooth surface to carefully bring them back in place.
Use a scalpel to cut the rubber seal into 2–4 pieces (Video 2).
Carefully pull the pieces of rubber seal off the slide using forceps and avoid grabbing the collagen pieces.
Collagen pieces can be re-aligned again at this point before continuing to the fixation.
-
Remove any remaining culture media before fixation (Video 2).
Place a pre-cut spacer on the slide and make sure that collagen pieces are completely covered. Align the spacer to the slide prior to touching the collagen, as the collagen attaches to the spacer and allows no further adjustment afterwards.
Cover the spacer with a filter card and wait until it is fully soaked with culture media. Only apply slight pressure to the filter card if it does not soak the media on its own. Complete removal of culture media is important for proper fixation.
-
Remove the filter card and place the chamber slide with the spacer into the fixative. The whole slide should be submerged (Video 3).
Wait until the spacer starts floating. Then, grab one corner with a forceps and carefully pull it off the slide. The collagen should stay attached to the slide.
If the spacer does not float off on its own, gently rock the dish containing fixative to dislodge the spacer.
-
Fix slides for 20 min; then, remove them from the fixative and allow to air dry for 15 min.
Drying time can be extended to allow processing of all slides and perform subsequent staining of all slides in parallel.
If necessary, fixed slides can be stored at 2–8°C and stained within three days.
-
Proceed with immunocytochemical staining of colonies.
Prepare all buffers and reagents required for staining (see Recipes).
Place fixed and dried slides in horizontal position on a staining tray (either commercial or self-made (see Recipe 13, Figure 4). All subsequent steps are carried out at room temperature.
Rehydrate colonies by covering the whole slide with freshly made wash buffer (see Recipe 10) and incubate for 20 min at room temperature. Do not use a wash bottle as the pressure will cause the fixed collagen to dislodge.
Remove buffer by tilting the slide into a waste container. Ensure that collagen-embedded colonies are always covered with liquid in the following steps and do not dry out.
Block slides with six drops of 2.5% horse serum for 20 min at room temperature to prevent non-specific binding of antibodies. Other serums may be used as long as they match the species of the secondary antibody used.
Prepare 1:100 dilutions of mouse anti-human CD41 and mouse IgG1 control antibodies in wash buffer + 1% BSA.
Add 500 µL of CD41 antibody to the slides and incubate for 30 min at room temperature. At least one slide per staining batch should be incubated with mouse IgG1 control to assess potential unspecific staining of colonies.
Gently rinse the slides three times by covering the slide with wash buffer for 3 min and tilting off excessive liquid.
Cover slides with six drops of ImmPRESS®-AP secondary antibody and incubate for 30 min at room temperature.
Gently rinse the slides three times by covering the slide with wash buffer for 3 min and tilting off excessive liquid.
Prepare Vector® Red AP substrate. Add two drops of Reagent 1, Reagent 2 and Reagent 3 from the AP substrate kit to 5 mL of substrate buffer (see Recipe 11) and mix well.
Cover slides with 500 µL of AP substrate and incubate for 20 min at room temperature in the dark.
Gently rinse the slides three times by covering the slide with wash buffer for 3 min and tilting off excessive liquid.
Add 500 µL of 0.1% Evans Blue solution (see Recipe 12) and incubate for 5 min.
Carefully rinse slides with double-distilled water (ddH2O) until excessive blue dye is removed. Take caution, water stream might flush off the collagen. Let slides air dry before microscopy.
-
Count individual colonies using an upright light microscope (Figure 5).
CD41+ megakaryocyte colonies appear magenta and form colonies of variable size. Colonies can be categorized based on the number of containing cells: i) 5–20 cells; ii) 21–49 cells; and iii) > 50 cells. Large colonies arise from more primitive progenitors than small ones.
Accompanying myeloid colonies appear pale blue and are usually of small size.
In rare cases, mixed colonies containing cells of both types might occur.
How do you know the staining has worked? For each sample used, at least one well should be stained with the IgG control antibody instead of anti human-CD41 to allow for the detection of any non-specific binding. If all other slides which have been stained with the human CD41 antibody do not show signs of pink megakaryocyte colonies, but colonies were observed on the slide prior to fixation, this would indicate that something has gone wrong in the staining process and staining has failed. Additionally, we acknowledge that some patient samples will fail to differentiate or proliferate at all in culture, due to the inherent and unpredictable heterogeneity of primary patient material. In this instance, colonies may appear sparse or very small in size when analysed under a microscope prior to fixation and thus proceeding with the staining might not be reasonable.
-
Megakaryocyte-directed differentiation of HSPCs in liquid culture
Notes:
This assay can be used to analyze the ability of therapeutic drugs to interfere with the megakaryocyte differentiation of MPN patient–derived HSPCs.
TPO is not required for expansion and megakaryocyte differentiation of CALRmut or MPLmut MPN patient–derived HSPCs. In our experience, JAK2mut HSPCs will not grow in the complete absence of TPO. A low dose of TPO (5–10 ng/mL) may be required for JAK2V617F samples and should be compared to healthy controls to measure TPO hypersensitivity rather than factor-independence.
Make sure to pipette precise volumes in every step of the staining protocol to avoid any bias for the cell count via counting beads.
Centrifuge sorted CD34+ HSPCs at 350 × g for 10 min at 4°C and remove supernatant to remove all sheath fluid from the sorting process.
Resuspend pellet at 0.75 × 105–1.0 × 105 cells/mL in Mk-diff media (see Recipe 7).
-
Seed cells in triplicates into a 48-well plate (200–500 µL/well, 15–20,000 cells/well, ideally) for every experimental condition. Add your compound of interest or vehicle control (e.g., DMSO) to the cell suspension.
Note: If it is not possible to obtain such high numbers of CD34+ cells from samples, it is possible to scale down the number of cells and the volume of experimental setup. This experiment has been carried out with a minimum of 1,000 cells/replicate in a 96-well plate.
Fill all the perimeter wells with 1 mL of sterile water to prevent excessive evaporation of media.
Culture cells in a humidified incubator at 37°C and 5% CO2 atmosphere.
On days 4, 7, 10, and 12 mix the content of wells gently using a pipette and transfer 100 µL of cell suspension to a 1.5 mL microfuge tube.
Add 100 µL of fresh Mk-diff media into each well to replenish the culture.
Wash once with 1 mL of staining buffer (350 × g, room temperature, 5 min).
-
Resuspend cells in 50 µL of staining buffer containing human FcR blocking reagent and CD41 APC-Cy7 and CD42b APC antibodies (1:25 dilution each), and incubate for 20 min at 4°C.
Notes:
CD41 and CD42b antibodies can be used with different fluorescent conjugates to match flow cytometer configurations or additional fluorescent staining used in the experiment.
At the initial setup of the experiment, unstained and single antibody and 7-AAD stained cells should be prepared to correctly compensate fluorophores and set the population gates at the flow cytometer.
Wash once with 1 mL of staining buffer (350 × g, 4°C, 5 min).
Resuspend cell pellet in 133 µL of staining buffer, mix well, and transfer to a 5 mL round bottom FACS tube.
Add 2.0 µL of a viability dye (e.g., 7-AAD or DAPI) and 15 µL of counting beads resulting in 150 µL total volume. Vortex bottle of counting beads properly before use.
Incubate for 10 min at 4°C and measure on flow cytometer (Figure 6).
Calculate CD41+ CD42b- megakaryocyte progenitor or CD41+ CD42b+ mature megakaryocyte cell counts (see Data analysis).
Figure 1. Gating strategy for CD34+ cell purification via fluorescence-activated cell sorting (FACS).

(A) First, cell debris (top row, left) and remaining granulocytes (top row, middle) were excluded in SSC-A/FSC-A plots and selected single cells were gated in an FSC-A/FSC-H plot (top row, right). Next, HSPCs were pre-enriched by gating on lineage marker negative and live (7-AAD negative) cells (bottom row, right plot). HSPCs were further purified by gating on CD34+ CD45dim cells (bottom row, left plot). (B) Unstained (left plots) and single stained (center and right plots) cells were used to set up compensation controls and to identify the fluorescent threshold for accurate setup of population gates.
Figure 2. Setup of a double chamber slide for the collagen-based megakaryocyte colony assay.

Double chamber slide seeded with CD34+ HSPCs along with a water-filled dish to maintain humidity during the 12-day culture.
Figure 3. Examples of megakaryocyte colonies prior to fixation and staining.

(A) Two dense, small colonies. (B) Medium sized colony. (C) Large colony. Scale bars represent 100 µm.
Video 1. Disassembly of double chamber slide prior to fixation.
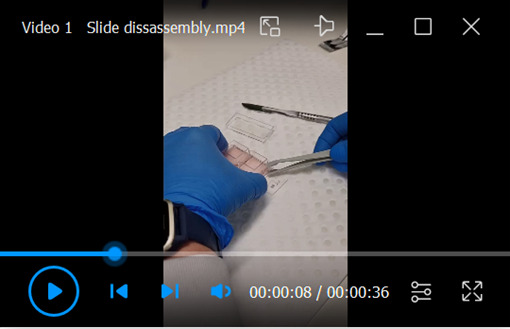
Removal of the chamber lid and detachment of the double chamber from the slide.
Video 2. Dehydration of the double chamber slide.
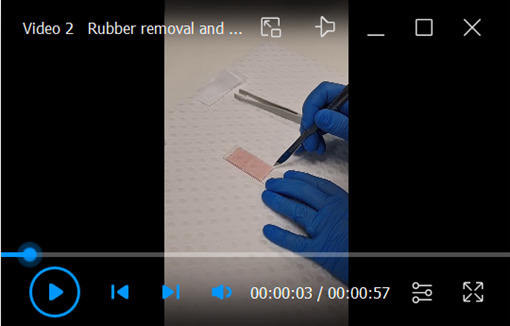
Removal of the rubber seal from the double chamber slide and dehydration of media from the collagen-embedded colonies.
Video 3. Fixation of the dehydrated slide.
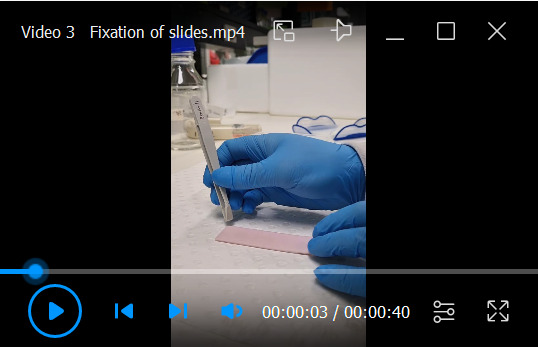
Fixation of collagen-embedded colonies on the dehydrated double chamber slide and removal of the spacer from the slide during the fixation process.
Figure 4. Preparation of equipment for staining of the collagen-based megakaryocyte colony assay.

(A) Example of a self-made staining tray using serological pipettes in a rectangular plastic dish. (B) Example of metal staining tray mounted over a sink.
Figure 5. Examples of megakaryocyte colonies upon fixation and staining.

(A) Small colony forming unit-megakaryocyte (CFU-Mk) (3–20 cells). (B) Medium sized CFU-Mk (21–49 cells). (C) Large CFU-Mk (>50 cells). (D) Large non-Mk colony near small CFU-Mk. (E) Two CFU-Mk colonies and one mixed CFU-Mk/non-Mk. (F) Non-Mk colony. Scale bars represent 100 µm. Colored arrows indicate colony types: red: Mk; blue: non-Mk; purple: mixed.
Figure 6. Gating strategy for evaluation of megakaryocyte differentiation via flow cytometry.

(A) Counting beads (high SSC-A, low FSC-A) and cells (excluding cell debris) were gated separately (top row, left plot). Beads were additionally gated in two independent fluorescent channels (e.g., ECD and Krome Orange) to exclude any contaminating cells in the initial beads gate (bottom row, left plot). After selecting cells based on morphology (top row, middle plot) and excluding cell doublets (top row, right plot) and dead cells (bottom row, right plot), megakaryocyte progenitors and mature megakaryocytes were evaluated by gating on CD41+ CD42b- and CD41+ CD42b+ cells, respectively (bottom row, center plot). (B) Unstained (left plots) and single stained (center and right plots) cells were used as compensation controls and for the identification of the fluorescent threshold for proper setup of population gates.
Data analysis
-
Megakaryocyte-directed differentiation in liquid culture
Note: We recommend performing flow cytometric data analysis using FlowJo, but software provided by cytometer manufacturers can be used as well.
Subset cell populations by applying the depicted gating strategy (Figure 6) to every sample.
Report the percentage of CD41+ CD42b- megakaryocyte progenitors and CD41+ CD42b+ mature megakaryocytes within live cells as an indication for the efficiency of cell differentiation.
Analyze the difference in cell differentiation between experimental conditions at each time point using two-way ANOVA (e.g., GraphPad Prism or similar software).
Extract the total numbers of events in the “pure beads,” the “CD41+ CD42b- megakaryocyte progenitors,” and the “CD41+ CD42b+ megakaryocytes” gate.
-
Calculate the number of CD41+ CD42b- megakaryocyte progenitors and CD41+ CD42b+ megakaryocytes that were present in each well of the liquid culture using the following equations:

Multiply the events acquired in either the “CD41+ CD42b- megakaryocyte progenitors” or the “CD41+ CD42b+ megakaryocytes” gate by the number of beads per 15 µL and by the total volume in the cell culture well. The bead concentration varies between batches and is stated on the label of each bottle. Divide the result by the events acquired in the “pure beads” gate and by the volume that was removed from the cell culture well (100 µL).
Test for differences in cell numbers between experimental conditions at each time point using a two-way ANOVA.
Recipes
-
RBC lysis buffer
Prepare 10× stock solution. Dissolve 9.0 g of NH4Cl, 1.0 g of KHCO3, and 37 mg of EDTA tetrasodium in 100 mL ddH2O. Filter-sterilize buffer and store at 4°C. This buffer is stable for up to one year. Dilute 10× buffer with sterile ddH2O to get a 1× concentration prior to use.
-
Staining buffer (SB)
PBS
1% BSA
2 mM EDTA
-
Freezing media
RPMI 1640
20% FBS
1% penicillin/streptomycin
10% DMSO
-
Thawing media
RPMI 1640 or IMDM
10% FBS or 20% FBS
1% penicillin/streptomycin
20 U/mL DNase
-
Antibody cocktail
Note: This recipe is calculated to stain 1 × 107 PBMCs from one sample. Scale up for more cells per samples and for as many samples as required.
Mix 5 µL of each mouse anti-human fluorescence conjugated antibody:
CD34 FITC
CD45 V500-C
CD2 PE-Cy5
CD3 PE-Cy5
CD19 PE-Cy5
CD14 PerCP-Cy5.5
CD16 PE-Cy5
CD56 PE-Cy5
-
Megakaryocyte (Mk) colony media
Notes:
This recipe is calculated for one sample with four replicates. Scale up for as many samples or experimental conditions as required.
To prevent repeated freeze-thaw cycles, aliquot the MegacultTM-C medium with lipids into 15 mL tubes (1.7 mL medium each) at the first thawing and store aliquots at -20°C.
Prepare cytokines and potential therapeutic compounds at 8.25× concentration in IMDM with a final volume of 400 µL. Add IL-3 and IL-6 at a concentration of 82.5 ng/mL (final concentration in the assay will be 10 ng/mL). Finalize the colony media by adding 400 µL of cytokine mix into 1.7 mL of MegacultTM-C medium with lipids and mix well.
-
Megakaryocyte differentiation (Mk-diff) media
SFEM II or StemPro 34 + nutrient supplement and 2 mM L-Glutamine
20 ng/mL SCF
10 ng/mL IL-6
10 ng/mL IL-9
0.4% lipids cholesterol rich
-
Isotonic NaCl solution (0.15 M)
Dissolve 4.38 g of NaCl in 500 mL of ddH2O. Solution is stable for several months at room temperature.
-
TRIS buffer (0.5 M, pH 7.6)
Dissolve 3.03 g of TRIS in 50 mL of ddH2O. Adjust pH to 7.6 by adding HCl 6 M dropwise under constant swirling. Buffer is stable for one month at room temperature.
-
Wash buffer
Mix nine parts of isotonic NaCl solution with one part of TRIS buffer. Prepare wash buffer always fresh on day of use.
-
Substrate buffer
Dissolve 909 mg of TRIS in 50 mL of ddH2O. Adjust pH to 8.2–8.5 by adding HCl 6 M dropwise under constant swirling. Add 50 µL of Tween 20 and mix properly. Buffer is stable for one month at room temperature.
-
Evans Blue solution (0.1%)
Dissolve 50 mg of Evans Blue in 50 mL of ddH2O. Solution is stable for several months at room temperature.
-
Staining tray
Prepare a plastic dish of around 20 × 12 × 3 cm in size and cut two serological pipettes in length so they fit in the dish. Tape the serological pipettes 2 cm apart on the bottom of the dish using adhesive tape. Slides can rest on the pipettes for staining while excessive buffer and staining solutions are caught inside the dish.
Acknowledgments
This work was supported by the National Health and Medical Research Council Ideas Grant APP1182564 and APP2004288, and the Australian Cancer Research Foundation Discovery Accelerator, the Leukemia & Lymphoma Society Translational Research Program, the Hospital Research Fund, the Australian Medical Research Future Fund for Rare Diseases, Rare Cancers and Unmet Need, the Austrian Science Fund (grant numbers P32783 and I5021), the Austrian Society of Internal Medicine (Joseph Skoda Fellowship), the Austrian Society of Hematology and Oncology (Clinical Research Grant), and MEFOgraz. This protocol was adapted from previous studies (Cortin et al., 2009) and manufacturer recommendations (Stemcell technologies, MegacultTM-C Assays). Several figures were generated with BioRender with permissions to be published. We thank Suraiya Onnesha for assistance with video recording.
Competing interests
The authors have no competing interests to declare.
Ethics
Collection of peripheral blood from MPN patients at the Division of Hematology, Medical University of Graz was approved by the institutional review board (IRB approval: 30-464 ex 17/18) or requested from the South Australian Cancer Research Biobank (SACRB, CALHN approval: 12986). Blood was only collected from patients with informed consent.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1.Araki M., Yang Y., Masubuchi N., Hironaka Y., Takei H., Morishita S., Mizukami Y., Kan S., Shirane S., Edahiro Y., et al.(2016). Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 127(10): 1307-1316. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D. and Dick J. E.(1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3(7): 730-737. [DOI] [PubMed] [Google Scholar]
- 3.Broudy V. C., Lin N. L. and Kaushansky K.(1995). Thrombopoietin(c-mpl ligand) acts synergistically with erythropoietin, stem cell factor, and interleukin-11 to enhance murine megakaryocyte colony growth and increases megakaryocyte ploidy in vitro. Blood 85(7): 1719-1726. [PubMed] [Google Scholar]
- 4.Chachoua I., Pecquet C., El-Khoury M., Nivarthi H., Albu R. I., Marty C., Gryshkova V., Defour J. P., Vertenoeil G., Ngo A., et al.(2016). Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127(10): 1325-1335. [DOI] [PubMed] [Google Scholar]
- 5.Chen J., Herceg-Harjacek L., Groopman J. E. and Grabarek J.(1995). Regulation of platelet activation in vitro by the c-Mpl ligand, thrombopoietin. Blood 86(11): 4054-4062. [PubMed] [Google Scholar]
- 6.Choi E. S., Nichol J. L., Hokom M. M., Hornkohl A. C. and Hunt P.(1995). Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood 85(2): 402-413. [PubMed] [Google Scholar]
- 7.Corre-Buscail I., Pineau D., Boissinot M. and Hermouet S.(2005). Erythropoietin-independent erythroid colony formation by bone marrow progenitors exposed to interleukin-11 and interleukin-8. Exp Hematol 33(11): 1299-1308. [DOI] [PubMed] [Google Scholar]
- 8.Cortin V., Pineault N. and Garnier A.(2009). Ex vivo megakaryocyte expansion and platelet production from human cord blood stem cells. Methods Mol Biol 482: 109-126. [DOI] [PubMed] [Google Scholar]
- 9.Debili N., Wendling F., Katz A., Guichard J., Breton-Gorius J., Hunt P. and Vainchenker W.(1995). The Mpl-ligand or thrombopoietin or megakaryocyte growth and differentiative factor has both direct proliferative and differentiative activities on human megakaryocyte progenitors. Blood 86(7): 2516-2525. [PubMed] [Google Scholar]
- 10.Elf S., Abdelfattah N. S., Chen E., Perales-Paton J., Rosen E. A., Ko A., Peisker F., Florescu N., Giannini S., Wolach O., et al.(2016). Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov 6(4): 368-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jamieson C. H., Gotlib J., Durocher J. A., Chao M. P., Mariappan M. R., Lay M., Jones C., Zehnder J. L., Lilleberg S. L. and Weissman I. L.(2006). The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A 103(16): 6224-6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klampfl T., Gisslinger H., Harutyunyan A. S., Nivarthi H., Rumi E., Milosevic J. D., Them N. C., Berg T., Gisslinger B., Pietra D., et al.(2013). Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 369(25): 2379-2390. [DOI] [PubMed] [Google Scholar]
- 13.Kojima H., Hamazaki Y., Nagata Y., Todokoro K., Nagasawa T. and Abe T.(1995). Modulation of platelet activation in vitro by thrombopoietin. Thromb Haemost 74(6): 1541-1545. [PubMed] [Google Scholar]
- 14.Kralovics R., Passamonti F., Buser A. S., Teo S. S., Tiedt R., Passweg J. R., Tichelli A., Cazzola M. and Skoda R. C.(2005). A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352(17): 1779-1790. [DOI] [PubMed] [Google Scholar]
- 15.Lu X., Levine R., Tong W., Wernig G., Pikman Y., Zarnegar S., Gilliland D. G. and Lodish H.(2005). Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proceedings of the National Academy of Sciences 102(52): 18962-18967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marty C., Pecquet C., Nivarthi H., El-Khoury M., Chachoua I., Tulliez M., Villeval J. L., Raslova H., Kralovics R., Constantinescu S. N., et al.(2016). Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 127(10): 1317-1324. [DOI] [PubMed] [Google Scholar]
- 17.Pikman Y., Lee B. H., Mercher T., McDowell E., Ebert B. L., Gozo M., Cuker A., Wernig G., Moore S., Galinsky I., et al.(2006). MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 3(7): e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pronier E., Cifani P., Merlinsky T. R., Berman K. B., Somasundara A. V. H., Rampal R. K., LaCava J., Wei K. E., Pastore F., Maag J. L., et al.(2018). Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight 3(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinisch A., Thomas D., Corces M. R., Zhang X., Gratzinger D., Hong W. J., Schallmoser K., Strunk D. and Majeti R.(2016). A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med 22(7): 812-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spivak J. L.(2017). Myeloproliferative Neoplasms. N Engl J Med 376(22): 2168-2181. [DOI] [PubMed] [Google Scholar]
- 21.Tefferi A. and Pardanani A.(2015). Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol 1(1): 97-105. [DOI] [PubMed] [Google Scholar]
- 22.Tefferi A. and Pardanani A.(2019). Essential Thrombocythemia. N Engl J Med 381(22): 2135-2144. [DOI] [PubMed] [Google Scholar]
- 23.Tvorogov D., Thompson-Peach C. A. L., Foßelteder J., Dottore M., Stomski F., Onnesha S. A., Lim K., Moretti P. A. B., Pitson S. M., Ross D. M., et al.(2022). Targeting human CALR-mutated MPN progenitors with a neoepitope-directed monoclonal antibody. EMBO Reports 23(4): e52904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannucchi A. M., Lasho T., Guglielmelli P., Biamonte F., Pardanani A., Pereira A., Finke C., Score J., Gangat N. and Mannarelli C.(2013). Mutations and prognosis in primary myelofibrosis. Leukemia 27(9): 1861-1869. [DOI] [PubMed] [Google Scholar]
- 25.Woll P. S., Kjällquist U., Chowdhury O., Doolittle H., Wedge D. C., Thongjuea S., Erlandsson R., Ngara M., Anderson K., Deng Q., et al.(2014). Myelodysplastic Syndromes Are Propagated by Rare and Distinct Human Cancer Stem Cells In Vivo. Cancer cell 25(6): 794-808. [DOI] [PubMed] [Google Scholar]
- 26.Woods B., Chen W., Chiu S., Marinaccio C., Fu C., Gu L., Bulic M., Yang Q., Zouak A., Jia S., et al.(2019). Activation of JAK/STAT signaling in megakaryocytes sustains myeloproliferation in vivo. Clin Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]