

Abstract
Window-of-opportunity trials, during which patients receive short-duration pre-surgical therapies, provide a platform for understanding the therapies’ mechanisms of action, but will require a paradigm shift in trial design, specimen collection and analysis.
Cancer immunotherapy is a remarkable success story that has revolutionized cancer care across numerous tumor types1, and although most patients with cancer fail to respond to currently approved immunotherapies, there are nearly 5,000 novel agents in preclinical or clinical development that aim to improve outcomes2. However, progress in developing optimal cancer immunotherapies is impaired by the lack of a firm understanding of the mechanisms underlying the antitumor activity of these agents in humans, and of why there are such heterogeneous clinical responses. Without a clear understanding of how cancer immunotherapy agents modulate the immune system, the field cannot advance and patients will not see improved clinical outcomes. Although large clinical trials provide the best measures of clinical efficacy to support regulatory approvals, their size and design are not optimal for deep mechanistic studies that can yield predictive biomarkers of response, and by nature they are more onerous and take years to complete. There is therefore a need for smaller, shorter-duration, focused trials including extensive molecular and cellular characterizations to rapidly define mechanisms of drug response and resistance, enabling biomarker identification and more effective development and use of immunotherapies. Given the integral role of immune dysregulation in nearly all human disease, from Alzheimer’s to cardiovascular disease, a similar approach using focused trials with deep immune monitoring can broadly benefit clinical medicine as a whole.
Window trials
Neoadjuvant clinical trials in which patients receive brief pre-surgical therapeutic interventions, often termed window-of-opportunity or window trials, provide an invaluable opportunity to perform extensive molecular and cellular analysis that will elucidate how agents work in humans. Window trials are unique in that they provide large post-treatment tissue samples, as opposed to scant biopsy specimens, enabling the use of single-cell and multiplex technologies capable of defining the dynamic effects of therapy. As a result, window trials are morphing from a niche tool for clinical investigation to the centerpiece of an engine to drive discovery in translational immuno-oncology.
Window trial design facilitates understanding of the mechanisms of action of new therapies, as well as the heterogeneity of clinical response, after only a few weeks of preoperative therapy. These trials are not exploitative, but rather can be therapeutic3–5. Indeed, in contrast to the metastatic setting in which most clinical trials occur, surgical candidates with localized disease typically have smaller, less heterogeneous tumors and more intact immune systems than patients with metastatic disease who have progressed on standard cytotoxic therapy, potentially leading to more robust clinical responses6.
A paradigm shift focused on trial design endpoints and optimal biospecimen collection and analysis is required to accelerate the rational development of cancer immunotherapy. Optimal trial design in this window setting requires five key innovations in approach: multidisciplinary teams; novel statistical methods; maximum tissue access; definition of clinically meaningful response; and adaptive clinical trial designs.
Multidisciplinary teams
Window trials require multidisciplinary teams that harness expertise in surgery, pathology, radiology, interventional specialties, genomics, immunology and data science. These multidisciplinary teams, such as ours at Mount Sinai Hospital in New York established under the cooperative program TARGET (The Neoadjuvant Research Group to Evaluate Therapeutics), support trials that will yield relevant multidimensional datasets that can inform future clinical development of cancer immunotherapy, creating a virtuous cycle of bedside-to-bench-to-bedside-to-bench research (Fig. 1).
Fig. 1 |. A paradigm shift in clinical trials.
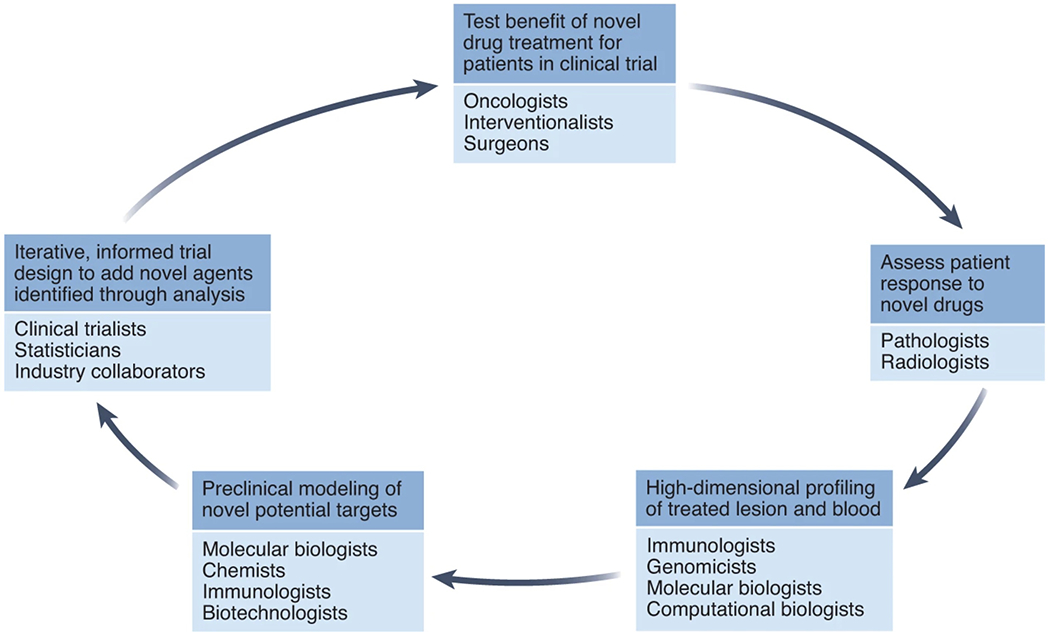
Large teams of physicians and scientists must collaborate to rapidly learn from trials, and inform subsequent trials, in order to optimize the learning potential that window trials provide.
Although immunotherapy trial design has historically been the realm of medical oncologists, leadership by surgical oncologists is key in the design and execution of window trials. Surgeon buy-in is critical not only to identify the optimal patient population and ensure recruitment, but also for collaboration with pathology and the research laboratory to facilitate rapid processing of tissue. Partnering with interventional radiologists, gastroenterologists and pulmonologists who employ state-of-the-art image guidance systems is also essential to obtain pretreatment biopsies that provide adequate viable tissue representative of the entire tumor, enabling sampling of multiple locations when significant intratumoral heterogeneity is evident on imaging.
Statistical models
Classically, biospecimen analysis has not been at the forefront of trial design but rather has been relegated to exploratory endpoints, often overlooked by statisticians and trialists alike. In a window trial design, optimal tissue analysis must be defined from the outset, requiring close collaboration between laboratory scientists, practitioners and statisticians.
The goal of all clinical trials is to enroll as few patients as possible to answer the question at hand while limiting exposure of patients to therapies that may be found ineffective or toxic. In addition, the immunophenotyping of pre- and post-treatment biospecimens, which makes window trials so valuable, is very expensive. Although the cohorts in most window trials may be small, the datasets are vast7. For example, analysis of a few dozen lung cancer lesions by CITE-seq (cellular indexing of transcriptomes and epitopes by sequencing) yielded single-cell proteomic and transcriptomic data from >500,000 individual cells, enabling the identification of predictive molecular signatures and revealing that the molecular programs and spatial distributions of tumor-infiltrating immune cells were much more similar across patients than between tumor and adjacent tissues within the same patient8, with identical immune molecular programs across different patient cohorts profiled with similar technologies8,9.
These data suggest that the immune system can respond to tumor cues in only a finite number of ways despite a myriad of tumor mutations, and although clinical responses may be heterogeneous, these findings provide statistical and rationale support for the use of small cohorts in exploratory window trials. The development of novel methods to calculate statistical power that integrate millions of data points captured per patient, from small patient numbers, is required to define minimal sample size for window trials.
Maximizing tissue access
Analysis of RNA and protein in immune and stromal cells isolated from pre- and post-treatment tumor specimens enables resolution of small subsets of immune cells integral to achieving a tumor-specific immune response. Such analysis is currently only possible on fresh tissue, necessitating immediate transport of resected specimens to pathology and rapid processing to ensure optimal tissue and single-cell viability of samples. Sampling multiple tumor regions, as well as adjacent tumor-free tissues, draining lymph nodes and peripheral blood, can define changes induced by brief therapeutic intervention, both within the tumor and systemically.
The use of fresh tissues allows researchers to exploit multiple single-cell phenotyping tools. For many years, flow and mass cytometry have been used to characterize single-cell proteomic signatures10. Recently, single-cell sequencing platforms have led to the development of CITE-seq8, PIC-seq (single-cell RNA sequencing of physically interacting cells)11 and ATAC-seq (assay for transposase-accessible chromatin using sequencing)12, providing transcriptomic, proteomic and epigenetic data on individual and physically interacting cells. At the tissue level, multiplex imaging platforms enable characterization of the three-dimensional interface between immune cells, stroma and malignant cells13–16. New developments in spatial transcriptomics, such as the 10X Visium platform and Slide-seq, couple transcriptomic and spatial cellular data17–19.
Currently, owing to financial and technical demands, these deep-profiling techniques can be performed only in specialized laboratories. Biotechnologists are working to develop preservation protocols that do not compromise cell viability — preserving nucleic acid and protein content — while also optimizing next-generation spatial transcriptomics platforms to enable single-cell resolution of fixed tissues. The development of deep-profiling technologies tailored to fixed tumor lesions should permit window trials to include patients across many centers while allowing centralized analysis, thereby limiting batch effects and confounders based on machines or technicians.
Another significant change to the clinical pathway is the need for high-quality pretreatment biopsies, sometimes necessitating re-biopsy in patients who already carry a diagnosis. These pretreatment biopsies provide a baseline essential to interpreting the effect of treatment seen in the resected tumor, but require collaboration with the interventionalists who acquire the biopsies to minimize delay of treatment initiation and subsequent surgery.
Defining clinically meaningful response
As in most trials, tumor response is an important endpoint for window trials, though after only a few weeks of treatment robust pathological responses may not yet be present even in patients with robust antitumor immunity. Immunotherapy drugs work through the induction of a strong inflammatory response often associated with tumor-infiltrating innate immune cells and expansion of effector T cells that precedes tumor cell death. Defining parameters that incorporate both immune cell infiltration and tumor cell necrosis is therefore critical to assessing response to immunotherapy. Whereas there are validated pathological endpoints that correlate with survival in neoadjuvant chemotherapy trials, immunotherapy agents have disparate mechanisms of action, and there is the potential for priming systemic immune responses that may affect ultimate survival through elimination of micrometastatic disease, even if much of the primary tumor is still viable at time of resection. As such, assessments of response at the radiographic and histological level must be characterized in close collaboration between pathologists, immunologists and radiologists, and validated in subsequent larger trials to identify metrics that predict the ultimate goal, a benefit in overall survival.
Adaptive clinical trial models
An individual window trial not only provides information about the function of a novel agent, but also could generate biological data to inform subsequent trial design (Fig. 2). To harness biological data expeditiously, optimal window trials employ an adaptive-informed clinical trial design whereby small cohorts are treated and, based on the findings, new arms (or entirely new trials) are designed to investigate rational combinatorial approaches. In contrast to large trials with shallow profiling of pharmacodynamic effects, this small-cohort adaptive-design approach will enable rapid incorporation of emerging data into subsequent human subject cohorts, so as to move the field forward at the fastest pace possible.
Fig. 2 |. Iterative-design trials enable and inform rapid informative trials.
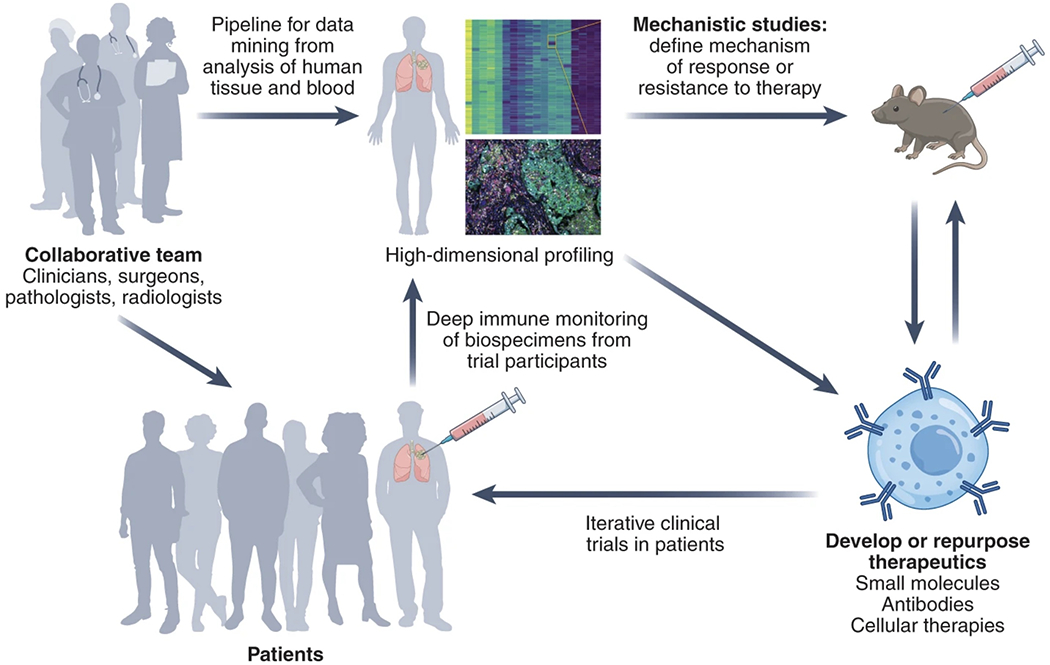
An in-depth analysis of treatment-naive tumors can characterize the dysregulation that limits the immune recognition and elimination of tumors; identify optimal models to study this dysregulation; model therapeutic interventions in preclinical models; and bring this understanding back to the clinic.
Beyond cancer
The main focus of window trials is in cancer therapeutics, but the utility of exhaustive immune phenotyping to understand novel agents is not limited to cancer. The central role of the immune system in most human diseases has increasingly become apparent, and this recognition has been accompanied by the development of immunomodulatory agents tackling these diseases treated by many distinct medical specialties. Additionally, there are numerous examples of agents in development in one setting being potentially repurposed when found effective at altering the outcome of another immune-related disease. For example, the interleukin-1β-depleting antibody canakinumab, initially developed to treat inflammasome-related illness, was subsequently used in a large trial to study its effect on cardiovascular disease and was then found in post-hoc analysis to affect the incidence of lung cancer20–22. Although these effects were only appreciated upon enrollment into a trial of over 10,000 patients, immune interrogation of novel agents early in their development can help clarify how these drugs work, how to use them more effectively and how to identify new and unanticipated indications (Fig. 3).
Fig. 3 |. Communication and collaboration across medical silos.
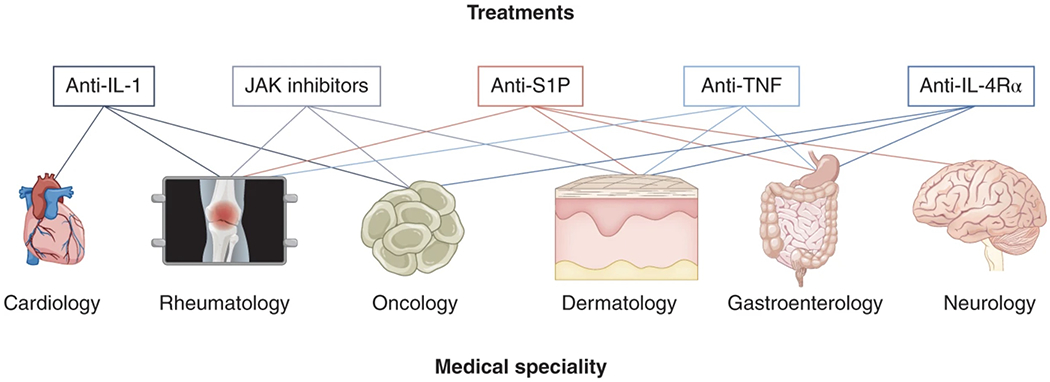
Immune dysregulation is at the heart of much of human pathology, and the repurposing of biologic therapies capable of disrupting the immunological milieu has potential applicability across many disease states that are the focus of disparate groups of physicians. Through the study of immunological programs in pathological conditions across disease states, and collaboration across silos, established biologic agents used in one setting may enable clinical advances in another. As an example, TNF blockade was initially developed in rheumatoid arthritis, and only a decade later became a standard therapy for inflammatory bowel disease (IBD); and IL-1 blockade, initially promising for inflammatory conditions, was later found to be effective in preventing cardiovascular disease and potentially cancer.
This exciting potential to repurpose drugs—particularly those that have been extensively studied and have well-characterized toxicity profiles—has raised awareness of the ways in which medical silos retard progress, and a discourse on how to encourage cross-medical-specialty collaboration has ensued23. At Mount Sinai, the INTERACT (Immunotherapy and Novel Target Research Across Clinical Teams) program was established to encourage collaboration and development of trials targeting shared immune pathways in different disease states while employing many of the same profiling technologies that are the backbone of the cancer-focused window trial programs in TARGET. For example, independent research teams characterized a type-2 immune milieu—most characteristic of atopic conditions such as eczema and asthma—in both lung cancer and keloid lesions24,25, and developed parallel clinical trials to treat these conditions with dupilumab, the IL-4Rα antibody, using biospecimen analysis pipelines optimized in TARGET.
Rapidly moving the field forward
Close partnerships between industry and academia, as well as mandated reporting of data from clinical trials, are needed to move the development of novel therapeutics forward at the fastest and safest pace possible. The analyses described come with great financial cost, but investment by industry and government alike in this approach would ensure that optimal therapies are pursued in the setting of phase 2/3 trials.
Window trials will enable the research community, across industry and academia, to stop the development of ineffective therapies, identify biomarkers to select optimal patient cohorts, understand mechanisms of resistance to inform combinatorial approaches and, ultimately, save money as well as reducing time commitment by physicians, scientists and patients alike. This human clinical trial paradigm should serve as a model for therapeutic development in cancer and beyond.
Acknowledgements
The TARGET and INTERACT teams comprise hundreds of physicians, researchers, coordinators and administrators, and the contributions of all these members is absolutely integral to the success of endeavors discussed here. We would like to acknowledge all these collaborators, as well as the support of S. Burakoff, R. Parsons, K. Goodman, F. R. Hirsch and D. Charney.
Competing interests
The authors have no competing interests in regards to the content of this commentary. They receive industry funding from partners to fund window studies, including Regeneron, Bristol Myers Squibb, Boehringer Ingelheim, Oncovir and Merck.
References
- 1.Sharma P. et al. Cancer Discov. 11, 838–857 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Upadhaya S, Hubbard-Lucey VM & Yu JX Nat. Rev. Drug Discov 19, 752 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Forde PM et al. N. Engl. J. Med 378, 1976–1986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang AC et al. Nat. Med 25, 454–461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uppaluri R. et al. Clin. Cancer Res 26, 5140–5152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinato DJ, Fessas P, Sapisochin G & Marron TU Hepatology 74, 483–490 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Stoeckius M. et al. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leader AM et al. Cancer Cell 39, I1594–1609.e12 (2021). [Google Scholar]
- 9.Lambrechts D. et al. Nat. Med 24, 1277–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Lavin Y. et al. Cell 169, 750–765.e717 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giladi A. et al. Nat. Biotechnol 38, 629–637 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Rose SA et al. Nat. Immunol 22, 914–927 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Remark R. et al. Sci. Immunol 1, aaf6925 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goltsev Y. et al. Cell 174, 968–981.e915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giesen C. et al. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Lin J-R et al. eLife 7, e31657 (2018).29993362 [Google Scholar]
- 17.Zhuang X. Nat. Methods 18, 18–22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merritt CR et al. Nat. Biotechnol 38, 586–599 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Ståhl PL et al. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Ridker PM et al. N. Engl. J. Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Ruperto N. et al. N. Engl. J. Med 367, 2396–2406 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Ridker PM et al. Lancet 390, 1833–1842 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Buckley CD et al. Nat. Immunol 22, 1344–1348 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Maier B. et al. Nature 580, 257–262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu J. et al. Front. Immunol 11, 597741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]