

Abstract
Tau-mediated neurodegeneration is a hallmark of Alzheimer’s disease. Primary tauopathies are characterized by pathological tau accumulation and neuronal and synaptic loss. ApoE-mediated neuroinflammation is involved in the progression of tau-mediated neurodegeneration, and emerging evidence suggests that the gut microbiota regulates neuroinflammation in an APOE-genotype dependent manner. However, evidence of a causal link between the microbiota and tau-mediated neurodegeneration is lacking. Here, we characterized a genetically engineered mouse model of tauopathy expressing human ApoE isoforms reared under germ-free conditions or after perturbation of their gut microbiota with antibiotics. Both these manipulations reduced gliosis, tau pathology, and neurodegeneration in a sex- and ApoE isoform-dependent manner. The findings reveal mechanistic and translationally relevant interrelationships between the microbiota, neuroinflammation, and tau-mediated neurodegeneration.
Graphical Abstract
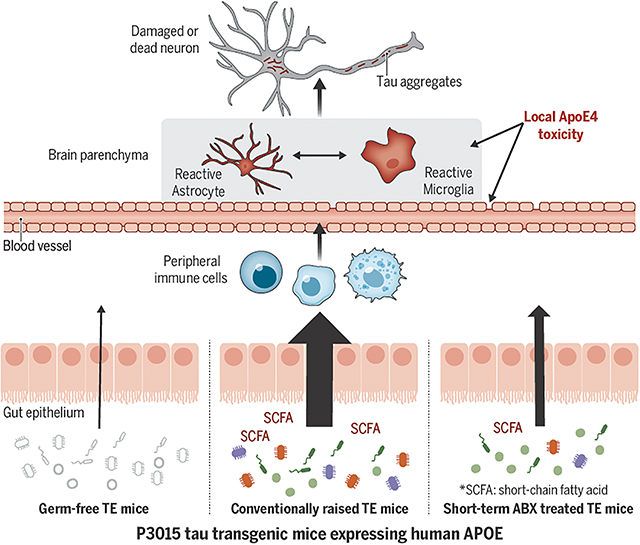
One-Sentence Summary:
Manipulation of gut microbiota attenuates brain atrophy in a genetically engineered mouse model of tau-mediated neurodegeneration.
Alzheimer’s disease (AD) is a fatal progressive neurodegenerative disease, characterized by early deposition of amyloid-β (Aβ) plaques, followed by pathological tau accumulation in the limbic system and neocortex; the latter is strongly linked to neuronal loss and brain atrophy (1). Evidence is mounting that there is a link between gut microbiota perturbations and Aβ deposition, potentially through effects on neuroinflammation and metabolic homeostasis (2–4). However, the contribution of the gut microbiota to tau-mediated neurodegeneration, which is strongly correlated with cognitive decline in AD and other tauopathies, has not been characterized. Furthermore, recent studies have reported that the configuration of the gut microbiota is differentially affected by APOE, the strongest genetic risk factor for AD and a known regulator of tau-mediated neurodegeneration (5, 6). Here, we used a mouse model of tauopathy with animals expressing different human ApoE isoforms to explore the hypothesis that the gut microbiota regulates tau-mediated neurodegeneration in an ApoE isoform-dependent manner.
TE4 germ-free mice are protected against tau-mediated neurodegeneration.
We began by rearing genetically engineered C57BL6J mice, containing a P301S human tau expressing transgene and a knocked-in human APOE4 gene (Tau/APOE4, abbreviated TE4) (7), under germ-free (GF) conditions. A second group of TE4 mice was exposed to microbes originating from their TE4 dams beginning at birth; these “conventionally-raised” mice were subsequently maintained under specified pathogen-free conditions in a mouse barrier facility. A third group of mice was reared under germ-free conditions until 12 weeks of age when they received an oral gavage of fecal microbiota sampled from 40-week-old, conventionally-raised TE4 mice (Ex-GF) (Fig. 1A). All mice were given a standard rodent chow rich in plant polysaccharides. We euthanized animals from all groups at 40 weeks of age – a time point when conventionally-raised TE4 mice display significantly greater tau-mediated neurodegeneration compared to (i) conventionally-raised P301S mice harboring other human APOE isoforms in their genomes, or (ii) conventionally-raised animals lacking both mouse and human APOE (7, 8). Unless otherwise indicated, all analyses were performed using biospecimens obtained from 40-week-old mice.
Fig. 1. TE4 germ-free (GF) mice are protected against tau-mediated neurodegeneration.
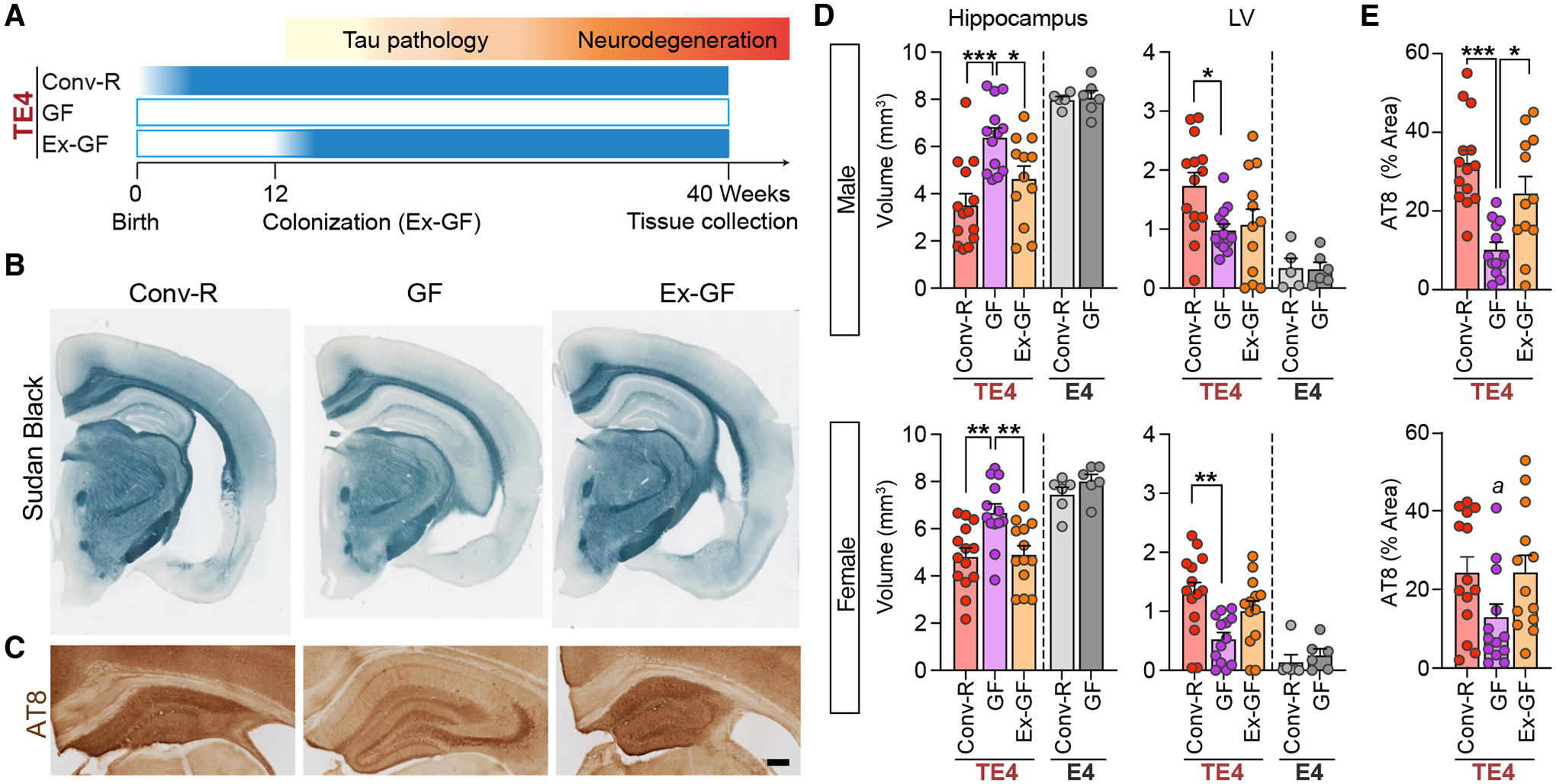
(A) Experimental design. TE4 mice were reared in a vivarium in a specified pathogen-free state (conventionally-raised; Conv-R; n=14/sex) or under germ-free conditions in gnotobiotic isolators (GF; n=13–14/sex) until they were euthanized at 40 weeks of age. A separate group of 12-week-old GF mice were colonized with fecal microbiota harvested from sex-matched 40-week-old conventionally-raised TE4 mice (Ex-GF; n=12/sex). (B) Representative images of 40-week-old male Conv-R, GF, and Ex-GF mouse brain sections stained with Sudan black. Scale bar, 1mm. (C) Representative images of p-tau staining (AT8) in the hippocampus of male mice. Scale bar, 250μm. (D) Volumes of the hippocampus (left) and lateral ventricles (LV; right) in male (top) and female (bottom) mice. E4 represents APOE4 knock-in mice that lack a P301S tau transgene (E) Percentage area covered by AT8 staining in sections prepared from the hippocampus of 40-week-old male (top) and female (bottom) TE4 mice. Data are presented as mean values ± SEM. Statistical significance was defined using a one-way ANOVA with Tukey’s post hoc test. *, p <0.05, **, p < 0.01, ***, p < 0.001. ‘a’ in (E) indicates that, in the post-hoc analysis, Tukey did not reveal significant differences, but LSD showed p < 0.05 (GF vs. Conv-R and Ex-GF). (see Table S1 for full statistical results).
Conventionally-raised (Conv-R) male and female TE4 mice displayed severe regional brain atrophy, manifested by hippocampal volume loss and lateral ventricle (LV) enlargement relative to E4 mice lacking the tau transgene (Fig. 1B,D). However, GF male and female TE4 mice had significant preservation of brain tissue compared to their conventionally-raised TE4 counterparts, as judged by hippocampal and LV sizes as well as by measurements of hippocampal neuronal layer thickness, a direct estimate of neuronal loss (Fig. S1C,D). Comparison of these areas of the brain in 12- and 40-week-old TE4 mice provided evidence that the protective effect produced by the GF state reflected a blockade of progression of neurodegeneration (Fig. S1E,F). We did not detect rescue from neurodegeneration in the entorhinal/piriform cortex of GF TE4 animals (Fig. S1G).
The GF rescue from neurodegeneration was reversed when animals were colonized with fecal microbial communities harvested from sex-matched conventionally-raised TE4 animals (Fig. S1H–J); 28 weeks after colonization, 40 week-old Ex-GF mice exhibited hippocampal and LV volumes and hippocampal neuronal layer thickness comparable to what was documented in Conv-R mice (Fig. 1B,D, S1D). Staining brain sections with a phospho-Tau antibody (AT8) revealed a marked decrease in tau phosphorylation in 40-week-old GF compared to 40-week-old Conv-R mice (Fig. 1C,E). In contrast, we did not observe any differences in hippocampal AT8 staining between 12-week-old GF and 12-week-old Conv-R mice (Fig. S1K,L). Thus, the microbiota greatly impacts the later appearance of tau-mediated neurodegeneration.
TE4 Germ-free mice exhibit reduced reactive gliosis.
While elevated levels of pathological p-tau may directly contribute to neuronal degeneration and death, there is strong evidence that reactive microglia and astrocytes are required for tau-mediated neurodegeneration (7, 9–11). Recent studies indicate that the gut microbiota contribute to glial functions (12–14), leading us to hypothesize that the microbiota may modulate tau-mediated neurodegeneration in our model by altering glial reactivity. Therefore, we first stained brain sections with markers for astrocytes and microglia (GFAP, Iba1, and CD68). Consistent with the amount of degeneration, expression of all these glial markers in the hippocampus was strongly reduced in GF compared to Conv-R male mice (Fig. 2A,B). Furthermore, analysis of glial morphology in the hippocampus (Fig. S2A–D) revealed that astrocytes in GF mice were larger with more branched processes compared to Conv-R mice. These morphological alterations of glial cells in GF mice were also observed at 12 weeks of age, when there is mild p-tau pathology but no evidence for neurodegeneration (Fig. S3).
Fig. 2. Germ-free TE4 mice exhibit reduced reactive gliosis.
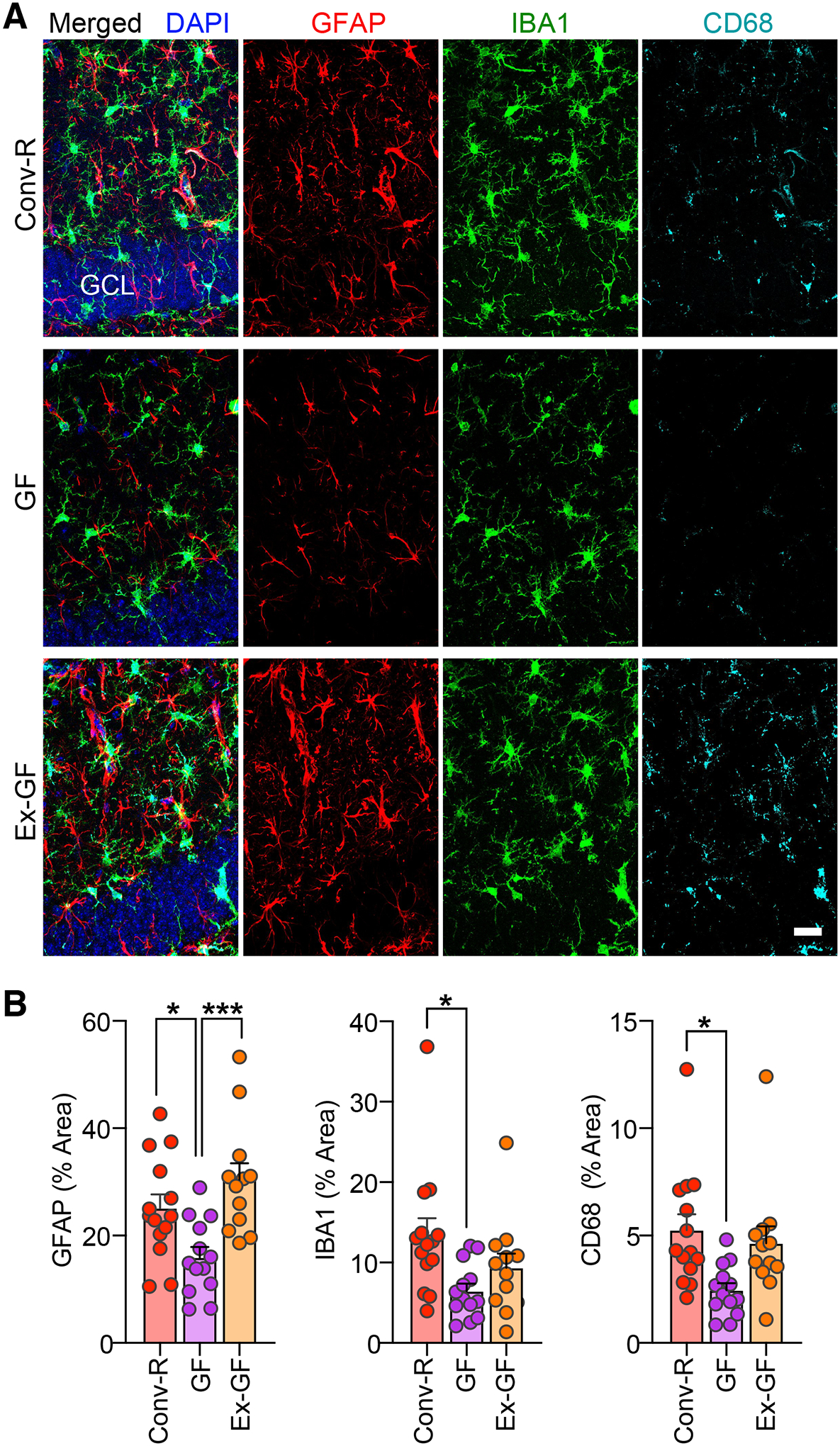
(A) Representative immunofluorescence images of hippocampal sections from 40-week-old male Conv-R, GF, and Ex-GF mice stained with antibodies to GFAP (red), Iba-1 (green), and CD68 (cyan), as well as DAPI (blue). Scale bar, 25 μm. GCL, granule cell layer. (B) Percent of the area of sections taken from the hippocampus covered by GFAP (left), Iba-1 (middle), CD68 (right) staining. Mean values ± SEM are shown. (n=12–14/group). Statistical significance was defined by one-way ANOVA with Tukey’s post-hoc test. *, p < 0.05, ***, p < 0.001. (See Table S1 for full statistical results).
Analysis of differential gene expression using nCounter (NanoString Technologies) showed that at 40 weeks of age, the GF state was associated with downregulation of genes related to “innate immune responses” (including sox9 and cx3cr1), “apoptosis” and “autophagy”, and upregulation of genes related to “neuronal activity” and “epigenetics” (Fig. S4A, Data S2). No significant differences in gene expression were detected when comparing Conv-R with Ex-GF mice (Data S3). Weighted Gene Co-expression Network Analysis using the Nanostring data identified five co-expression modules, two of which (modules Grey and Turquoise) exhibited significant correlation with either microbial colonization status or hippocampal size (Fig. S4B–D). Gene Ontology (GO) analysis demonstrated that module Grey was enriched for genes related to “epigenetics”, “T cell mediated immunity” (e.g., Foxp3 which is associated with Tregs), and “gliogenesis” (e.g., Tmem119 which is typically upregulated in “homeostatic” microglia). Module Turquoise was enriched for genes related to “cytokine production”, “cell death”, and “defense response” including APOE (Fig. S4E,F; Data S4). Analysis of eigengene (the first principal component of the expression matrix of the corresponding module) expression in module Grey and Turquoise by individual animal verified that increased and decreased expression, respectively, in these modules were tightly linked to the protective effect of the GF condition (Fig. S4G–J). Thus, GF conditions affect microglia and astrocyte reactivity or activation associated with tau-mediated neurodegeneration.
Antibiotic-induced gut microbiota perturbation protects against tau-mediated neurodegeneration.
To test whether the gut microbiota regulates tau-mediated neurodegeneration in an APOE isoform-dependent manner, we treated groups of conventionally-raised TE4 mice, P301S tau transgenic mice expressing human APOE3 (TE3) and P301S tau animals without APOE (TEKO) with an antibiotic cocktail (ABX) comprised of kanamycin, gentamicin, colistin, metronidazole, and vancomycin (4). Gavage with the antibiotic cocktail (or water in the case of controls) occurred daily from postnatal days 16–22. The fecal microbiota was serially sampled and animals were euthanized at 40 weeks of age (Fig. 3A). The short-term ABX treatment produced a marked, immediate decrease in the total number of viable bacteria; while this decrease in viability was transient, culture independent methods disclosed that the proportional representation of various bacterial taxa in the microbiota of ABX-treated groups remained different from controls throughout life (see Fig. S5A–E and below).
Fig. 3. Antibiotic-treatment and perturbation of the gut microbiota markedly protects against tau-mediated neurodegeneration in a sex- and APOE isoform-dependent manner.
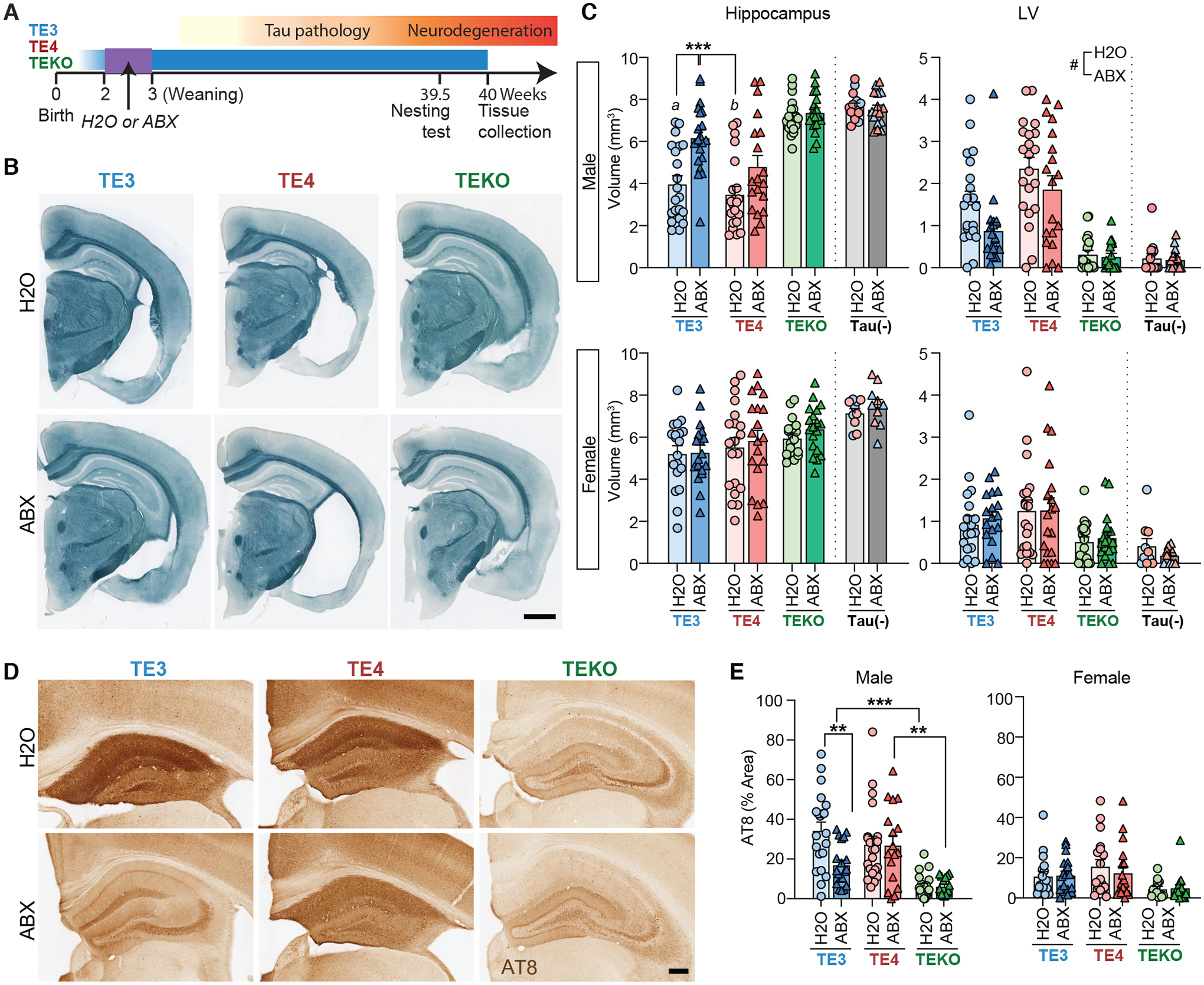
(A) Male and female TE3, TE4, and TEKO transgenic mice (n = 18–21/group) received a gastric gavage of a combination of antibiotics (ABX) from postnatal days 16–22. Controls were gavaged with water (H2O). Mice were euthanized at 40 weeks of age. (B) Representative images of male TE3/TE4/TEKO mouse brain sections stained with Sudan black. Scale bar, 1mm. (C) Volumes of the hippocampus (left) and lateral ventricles (right) in male (top) and female (bottom) animals. Tau (-) represents APOE3 (blue) or APOE4 (red) knock-in mice that lack a P301S tau transgene. (D) Representative images of p-tau staining (AT8) of hippocampal sections prepared from male mice. Scale bar, 250μm. (E) Percentage of the area covered by AT8 staining of hippocampal sections prepared from male (left) and female (right) mice. Mean values ± SEM are shown; Statistical significance defined by two-way ANOVA with Tukey’s post hoc test. *, p < 0.05, **, p < 0.01, ***, p < 0.001. Statistical significance of the main effects of treatments (H2O vs. ABX) were indicated as #, p < 0.05. In Fig. 2C (male hippocampus), ‘a’ indicates statistical significance compared to TEKO- H2O and ABX groups (p < 0.001). ‘b’ indicates statistical significance compared to TE3-ABX, TEKO-H2O and ABX (p < 0.001). (see Table S1 for full statistical results).
Volumetric analysis revealed that 40-week-old male mice in the H2O-treated control groups (TE3-H2O and TE4-H2O animals) had statistically significant hippocampal atrophy compared to mice lacking the APOE gene (TEKO) mice (Fig. 3B,C) (8). However, TE3-ABX, but not TE4-ABX mice, showed significant hippocampal preservation relative to controls. Enlargement of the LV and entorhinal/piriform cortical atrophy were also significantly attenuated by ABX treatment independent of APOE genotype, but the degree of ABX impact was higher in TE3 than TE4 mice in general (i.e., the log2-fold-changes by ABX in the LV size were −0.86 in TE3 and −0.34 in TE4 mice; the log2-fold-changes by ABX in the entorhinal/piriform cortex size were 0.57 in TE3 and 0.36 in TE4 mice; Fig. 2C, Fig. S5F). ABX treatment also prevented thinning of hippocampal neuronal cell layers independent of APOE genotype (Fig. S5G). These phenotypic effects of ABX treatment were seen in TE3 and TE4 males but not in females except in the case of the CA1 pyramidal layer which was slightly, albeit significantly, thicker in ABX-treated males and females (Fig. S5G). In concert with hippocampal brain atrophy, male TE3-ABX mice showed significantly lower AT8 staining relative to male TE3-H2O mice (Fig. 3D,E).
ABX treatment did not influence brain volume or early tau pathology in 12-week-old male TE3 mice (Fig. S5H–K). At 40 weeks of age, analysis of male cortical tissue also revealed that ABX-treatment reduced p-tau levels in the detergent soluble fraction (RIPA) and both p-Tau and human tau levels in the insoluble fraction (FA) across the three APOE genotype groups (Fig. S6). Furthermore, nest building behaviors, known to be sensitive to hippocampus damage and neurodegenerative disease (15), showed significant improvement in male TE3 and TE4 mice treated with ABX, which correlated with hippocampal volumes (Fig. S7)
Antibiotic-treatment alters astrocyte and microglial gene expression and morphological responses.
Single nucleus RNA-seq (snRNA-seq) of hippocampal tissue collected from Conv-R 40-week-old males that belonged to all three APOE genotype groups and were not exposed to antibiotics, identified 20 distinct clusters, which were categorized into excitatory/inhibitory neurons, astrocytes, microglia, oligodendrocytes, and oligodendrocyte progenitor cells (Fig. S8A,B). Cell proportion analysis showed that certain neuronal populations (e.g., exc1, exc2, and exc5) were reduced in the presence of tau pathology and expanded again with ABX-treatment, in agreement with the hippocampal volumetric data. The astrocyte population was reduced two-fold with ABX in the presence of tau pathology. The microglial population, which expanded ~7 fold (by proportion) with tau pathology, was reduced 2–3 fold with ABX-treatment.
The astrocyte cluster was re-scaled and re-clustered, revealing four subclusters (astro 0–3; Fig 4A and Fig. S8C). Re-clustering of the microglia cluster identified three subclusters (micro 0–2; Fig. 4H and Fig. S8D). Tau pathology resulted in a strong shift from astro0 to astro1 and micro0 to micro1. ABX treatment reversed these shifts in TE3 but not TE4 male mice, consistent with a stronger protective effect with ABX treatment in TE3 compared to TE4 male mice. Further differentially expressed gene (DEG) analysis between two clusters associated with pathological shifts revealed that the top upregulated pathways in astrocyte subcluster DEG (astro1 vs. astro0; Data S5) were associated with GO terms to “gliogenesis” and “cellular chemical homeostasis” (Fig. 4B) while the top upregulated pathways in the microglial subcluster DEG (micro1 vs. micro0; Data S6) were associated with GO terms related to “cell activation” and “small Gtpase-mediated signal transduction” (Fig. 4I). We used qPCR to verify upregulation of a subset of astro1 genes (e.g., Gfap, Vim) and micro1 genes (e.g., Arhgap25 and Itgax) (Fig. S9A,B). An alteration of glial gene expression was also observed in ABX treated TE3 mice at 12 weeks of age, well before neurodegeneration (Fig. S9C). Thus, the gut microbiota regulates expression of genes that are involved in reactive gliosis.
Fig. 4. Glial transcriptional and morphological responses to tau pathology are regulated by ABX-treatment.
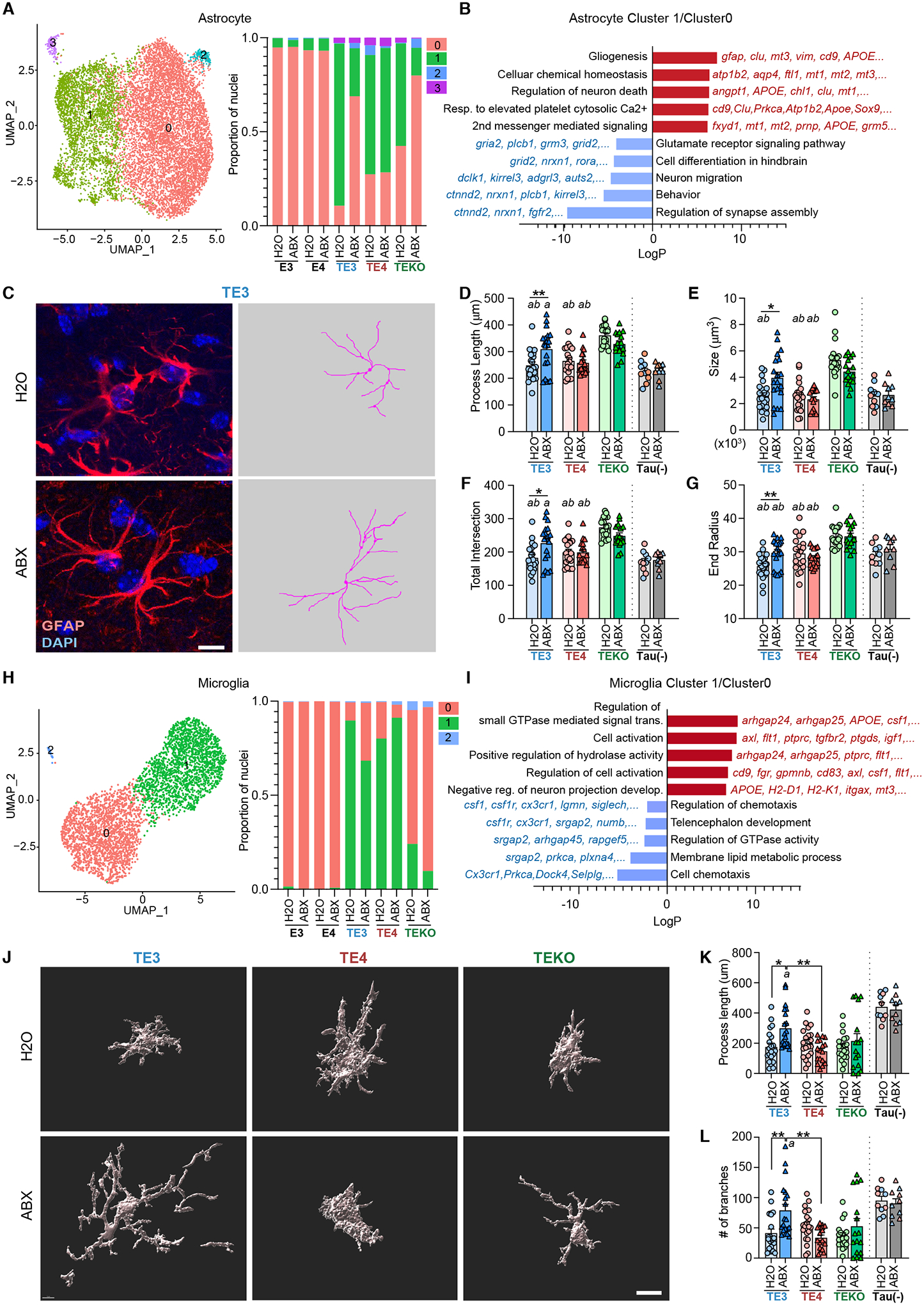
(A) The left panel shows UMAP plot of a re-clustered astrocyte population that identifies four distinguishable clusters (astrocyte cluster 0–3). The right panel shows relative frequency of all astrocyte clusters per genotype and treatment. (B) GO ‘Biological Process’ terms significantly enriched among differentially expressed genes (DEGs) (astrocyte cluster 1 vs.0). (C) Left side of panel; representative images of male TE3 mouse brain sections stained GFAP/DAPI. Right side of panel; traces of GFAP expression, generated using Simple Neurite Tracer (S7.), corresponding to the images on the left. Scale bar 10μm. (D-G) Morphometric analysis of astrocyte process lengths (D), size of GFAP+ astrocytes (from Convex Hull analysis) (E), total number of process intersections (F), and the end radius (from Sholl analysis) (G). (H) Left side of panel; UMAP plot of the re-clustered microglial population showing three distinguishable clusters (microglia clusters 0, 1, 2). Right side of panel; relative frequency (proportion of nuclei) of all microglia clusters per genotype and treatment group. (I) GO terms enriched in up-regulated and down-regulated DEGs (microglia cluster 1 vs. 0). (J) Imaris-based automatic reconstruction image of Iba1+ microglia. Scale bar 10μm. (K,L) Morphometric analysis of process lengths (K) and the number of branches (L) of Iba1+ microglial cells in male TE3, TE4, and TEKO mice. Tau(-) represents APOE3 (blue) or APOE4 (red) knock-in mice that lack a P301S transgene. Mean values ± SEM are presented with statistical significance defined by two-way ANOVA with Tukey’s post hoc test. *, p < 0.05, **, p < 0.01, ***, p < 0.001. ‘a’ and ‘b’ indicate statistical significance compared to TEKO-H2O (a) and TEKO-ABX (b, p <0.05). (see Table S1 for full statistical results).
Reactive gliosis is a complex process that involves changes in gene expression and morphological remodeling: in general, homeostatic glial cells display highly branched thin processes in the homeostatic state, but in activated cells, processes become shorter as they retract and become hypertrophic (16). To further assess reactive gliosis, we performed GFAP, Iba1, and CD68 immunostaining. Although the coverage area of the immunostaining did not reveal significant ABX effects (Fig. S10A), morphometric analysis revealed that ABX drove astrocytes and microglia to a more homeostatic-like morphological state (e.g. increased length of processes and size of cells) in male TE3 mice, but not in male TE4 and TEKO, female TE3, or in male 12-week-old TE3 mice (Fig. 4C–G, J–L, and Fig. S10B–K). Thus, ABX-induced perturbation of the microbiota protects against tau-mediated neurodegeneration most strongly in male TE3 mice and this effect is manifest by changes in multiple cell types, including astrocytes and microglia.
Antibiotic-treatment reshapes the bacterial communities and reduces SCFAs.
The microbiota perturbation induced by ABX was evidenced by (i) measurements of cecal weight compared to body weight (known to be markedly increased in GF compared to Conv-R animals and rapidly reduced in Ex-GF mice; Fig. S1A,B and Fig. S5D,E) and (ii) culture-independent analysis of the relative abundances of bacterial taxa in feces. The latter approach was based on sequencing PCR amplicons generated from variable region 4 of bacterial 16SrRNA genes (amplicon sequence variants or ASVs) and grouping these ASVs into taxonomic “bins”. Alterations in alpha and beta-diversity as well as the representation of phylum- and genus-level groups are summarized in Figures S11–13. Phylum- and genus-level changes in ABX-treated groups remained different from water-treated controls up to the time of euthanasia (Fig. S11D,E).
Linear Discriminant Analysis revealed that the members of the genera Helicobacter, Ruminococcus, and Butyricicoccus had lower relative abundance in the fecal microbiota of ABX-treated groups across male APOE isoform groups (Fig. 5A, Fig. S12A–C), but not in female TE3 mice (Fig. 5B, Fig. S13D). Recent studies suggest that the microbiota modulates glial activation through production of microbial metabolites such as short-chain fatty acids (SCFAs) (12, 13). Ruminococcus, and Butyricicoccus are associated with SCFA production. Also, the relative abundance of several bacterial family-level taxa known to produce SCFAs, such as Ruminococcaceae and Lachnospiraceae, were reduced with ABX across APOE genotypes (Fig. S12D).The representation of these taxa was greater in male compared to female members of the TE3-H2O treatment group (Fig. 5C) and lower in TE3 compared to TE4 mice (Fig. S13B,C). Gas chromatography-mass spectrometry of cecal samples disclosed that, consistent with our observation that SCFA-producing bacteria were reduced with ABX-treatment in males, acetate, propionate, and butyrate were significantly reduced across APOE genotypes in males, correlating with biomarkers of tau pathology (Fig. 4D,E, Fig. S13A), but not in females (Fig. S13E).
Fig. 5. Effects of ABX treatment on fecal microbiota composition and cecal levels of short chain fatty acids.
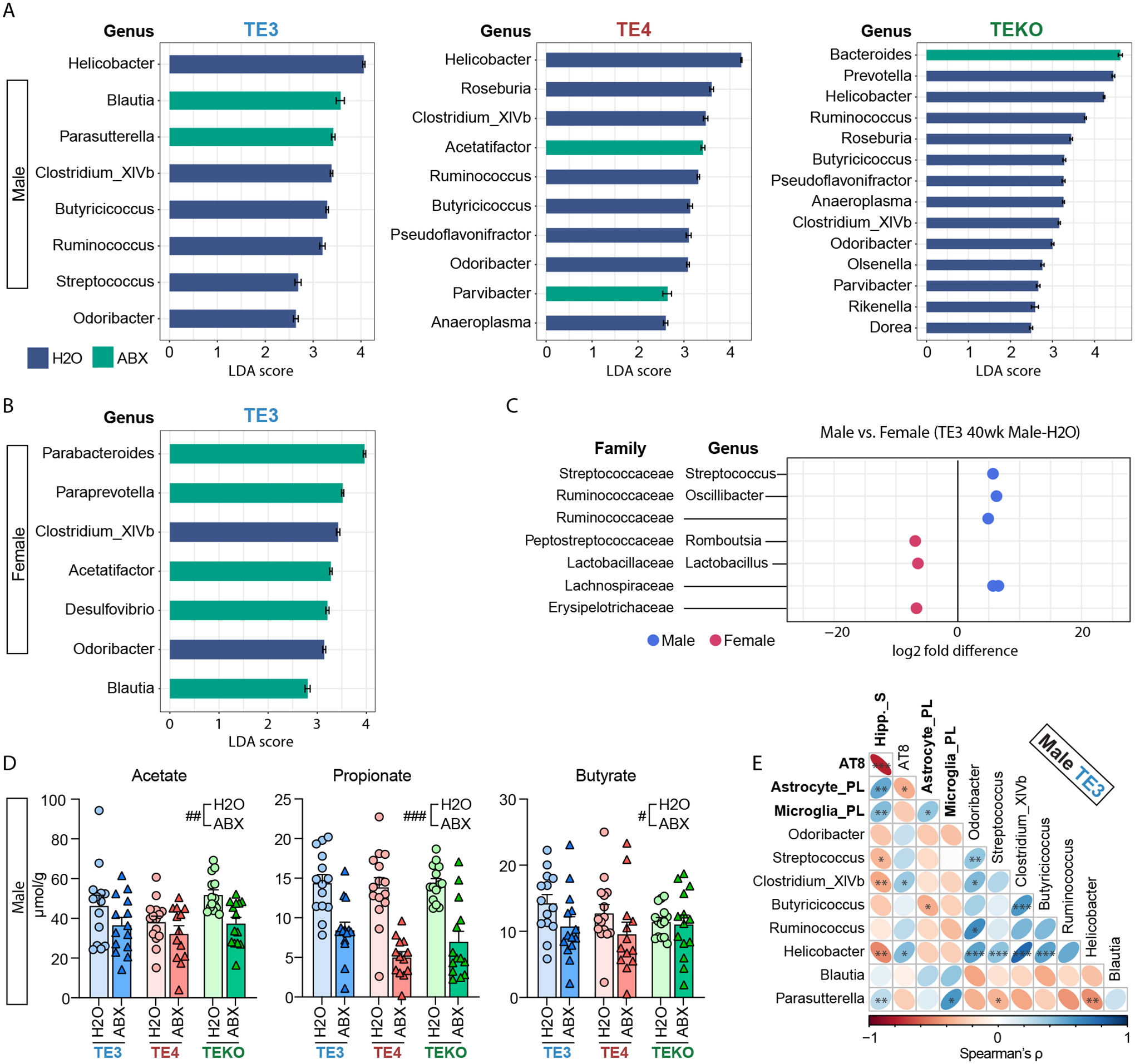
(A-B) Linear Discriminant Analysis (LDA) scores. Horizontal bars represent the LDA scores for each genus-level taxon in (A), male TE3, TE4, TEKO, and (B), female TE3 mice. Indigo and green bars represent taxon features with significantly higher representation in mice belonging to the control H2O and versus ABX treatment groups, respectively (LDA scores > 2). (C) Comparison of relative abundance of genera between 40-week-old male and female TE3-H2O mice. Family and genus assignments are shown. (D) Targeted GC-MS analysis of cecal short chain fatty acids in male 40-week-old TE3, TE4, and TEKO mice treated with ABX or H2O (n=14/group), tested by two-way ANOVA. Statistical significance of the main effects of treatments (H2O vs. ABX) were indicated as #, p < 0.05, ##, p < 0.01, ###, p < 0.001. (E) Correlogram showing the relationship between (i) eight genera identified as having differences in their relative abundance in male 40-week-old TE3 H2O vs ABX treated mice and (ii) biomarkers of tauopathy. These biomarkers (bold) include hippocampus size (Hipp._S; smaller value reflecting greater neurodegeneration), coverage of AT8 staining, process length of astrocytes (astrocyte_PL), and process length of microglial cells (microglia_PL). *, p < 0.05, **, p < 0.01, ***, p < 0.001. (see Table S1 for full statistical results).
ABX induced gut microbiota perturbation alters the peripheral immune system and the effects of SCFA on TE4 GF mice.
It is unclear if SCFA directly act on glial cells, because SCFA receptor-encoding genes (e.g., Ffar2) are not expressed in glial cells (12). However, we postulated that SCFA could affect other inflammatory mediators or immune cells that directly access the brain, or the immune milieu around the brain (i.e., the meninges). Indeed, we found reductions in meningeal NK and pDC cells in ABX-treated TE3 mice. Furthermore, γδ T and pDC cells were reduced in GF mice (Fig. S14A–C). These latter cells are known to release cytokines, such as IL-17, IFN type-I, and others, which may affect neuroinflammation in the brain (17–19).
Multiplex cytokine profiling revealed that plasma levels of MCP-3, IL-2R, BAFF, and Eotaxin were significantly reduced by ABX in mice (Fig. S15). The degree of reduction of Eotaxin, which can cross the blood brain barrier and activate glial cells, was greater in TE3 mice compared to those belonging to the other genotype groups. Gut microbiota can modulate systemic immunity including peripheral macrophages (20). ABX treatment of male TE3 mice significantly altered lung alveolar macrophage gene expression (Fig. S16). In turn, their cytokine release or interaction with adaptive immune cells could potentially impact brain pathology. The cytokine profiling data showed that some cytokines released by macrophages, such as IP-10, BAFF or that can stimulate macrophages such as MIP-1a were affected by ABX treatment. Brain border innate and infiltration of peripheral immune cells also may act on the tau pathology and neurodegeneration. Additional snRNA-seq analysis using hippocampal tissue revealed that the population of brain border and peripheral immune cells were increased in the tau mice at 40 weeks of age and were decreased by ABX-treatment. (Fig. S14D). While changes in these cytokines may be important to activate glial cells to drive tau-mediated neurodegeneration, the presence of ApoE in the brain is still necessary to drive tau-mediated neurodegeneration, because these cytokines in the plasma display high levels in TEKO mice that only display mild brain atrophy (Fig. 2C, S15B). Finally, to begin to test whether SCFA may be mediating the effects of GF conditions or ABX, we administered SCFA or control in drinking water to 10-week or 31-week-old TE4 GF mice for 5 or 4 weeks respectively. SCFA treatment altered alveolar macrophage gene expression in both the younger and older mice (Fig. S17). SCFA supplementation increased gliosis and p-tau pathology in the hippocampus of the older TE4 GF mice (Fig. S18) supporting the possibility that SCFA may be a key modulator of the microbiota effect on tau-mediated neurodegeneration.
Conclusion
Taken together, we have found that GF conditions are strongly neuroprotective against tau-mediated neurodegeneration in male and female TE4 mice. ABX treatment is also neuroprotective but its impact was limited to males, and greater in the presence of APOE3 than APOE4. Our results also suggest that APOE isoforms and sex differentially modulate the microbial response to ABX, resulting in significantly lower levels of SCFAs. The microbiota-associated metabolic changes are associated with altered peripheral cytokine responses, and strong changes in the innate immune response in the brain.
We hypothesize that the gut microbiota regulates the brain’s innate immune response to influence tau-mediated neurodegeneration in the brain. We speculate that in TE3 mice the ABX-induced microbiota perturbation that reduces peripheral immune activation may moderate the brain’s innate immune response, and eventually attenuate tau-mediated neurodegeneration. In contrast, in TE4 mice, the microbiota-associated peripheral immune changes were not altered as much by ABX-treatment. It is possible that the gain-of-function of APOE4 toxicity and the effects on the local innate immune response in the brain were stronger than that of APOE3, and ABX treatment ultimately failed to protect against tau-mediated neurodegeneration to the same degree. On the other hand, with the complete absence of the microbiota, even in TE4 mice, the depletion of microbially-produced metabolic and immune signals results in a strong reduction of the disease-associated astrocyte and microglial phenotypes, leading to a decrease in tau-mediated neurodegeneration (consistently, SCFA supplementation reversed the reduced gliosis and tau pathology in TE4 germ-free mice.
Further studies are needed to test these hypotheses and to gain greater understanding of how sex, microbiota manipulation, microbiota-linked metabolites, central or peripheral immune response, and the pathogenesis of neurodegeneration are related. A starting point for these follow-up analyses could involve gnotobiotic mice colonized with gut microbiota harvested from conventionally raised mice with different sex, age, and ApoE isoforms and from human donors (with or without tau-mediated neurodegeneration). It is possible that the TE4 GF mice colonized with different types of microbiota (e.g. harvested from wild-type mice, not necessarily from TE4-Conv mice) would reverse the GF rescue from neurodegeneration. It may be that the host genetic makeup in TE4 mice could reshape the effects of the transferred microbiota composition derived from a more general bacterial community and be sufficient to activate the metabolic-neuroinflammation axis. Alternatively, the sex, age, ApoE isoform status, and state of neurodegeneration may be critical in regard to the source from which the transferred microbiota are derived. Future studies are needed to sort out these possibilities. Nonetheless, these results raise the possibility that gut microbiota targeting may provide ways to prevent or treat progression of AD and primary tauopathies (21, 22).
Materials and Methods
Animals
All animal experiments were performed using protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University School of Medicine. All phenotyping and data analyses were performed by researchers who were completely blind to the experimental hypothesis and the treatments of mice.
P301S tau transgenic mice (PS19tg; Stock No. 008169, Jackson Laboratories) contain a transgene where expression of the human P301S tau mutant is driven by a mouse prion protein (Prnp) promoter. These animals had been backcrossed to C57BL/6 mice (Stock No. 027, Charles River) for more than 10 generations. P301S mice were crossed with human APOE knock-in mice where the endogenous mouse apoE gene was replaced by a human APOE gene flanked by LoxP sites (23) (APOE3flox/flox or APOE4flox/flox or ApoE knockout mice) to generate P301S∷APOE3/wt, APOE4/wt, or EKO/wt animals. These mice were then crossed to homozygous APOE3, APOE4, or EKO mice to generate P301S∷ApoE3/ApoE3, ApoE4/ApoE4, or EKO/EKO mice (abbreviated TE3, TE4, and TEKO, respectively). Sire and dams were randomly assigned from different litters to produce experimental animals. Dams that had received an antibiotic cocktail (see below) were euthanized after weaning their pups on postnatal day 23, and were not used for further breeding or any other experiments. At weaning (postnatal day23), pups were housed in same-sex, -genotype, -treatment groups (2–5 animals/cage). Conventionally-raised (Conv-R) mice were housed in a mouse barrier facility under specified pathogen-free conditions until they were euthanized at 40 weeks of age. All Conv-R mice in the antibiotic experiment were given the same type of food (#53WU, LabDiet, St. Louis, MO) ad libitum. All groups of animals were housed under the same conditions: same type of cage, bedding materials (#Bed-o’Cobs 1/8 BB40; the Andersons, Quakertown, PA), and nestlets (#NES7200; Ancare, Bellmore, NY) in the same room in the vivarium. A complete mouse cage consisted of a polycarbonate shoebox bottom equipped with a wire bar lid and a polycarbonate filter top containing a filter insert.
Conv-R TE4 mice were rederived as germ-free (GF) by embryo transfer. Embryos were harvested 1 day after mating, and transferred under sterile conditions to a pseudo-pregnant GF mother generated by mating to a vasectomized GF male. The transgenic GF descendants were intercrossed to produce TE4 GF mice. GF animals were reared in plastic flexible film gnotobiotic isolators (Class Biologically Clean Ltd., Madison, WI). GF status was verified by PCR of feces using universal bacterial 16S rRNA gene primers and by culturing fecal and skin swabs. The equivalent generation of Conv-R mice were used as controls.
GF mice were colonized with fecal microbiota samples collected from 40-week-old Conv-R TE4 mice to create Ex-GF animals. To do so, fecal samples from 4–5 mice/sex were pooled, homogenized in sterile reduced PBS containing 0.05% cysteine-HCL and 20% glycerol (5 pellets/10 mL) and stored in −80 °C until the time of the fecal microbiota transplantation. A 200 μL aliquot of the resulting suspension was thawed and administered to sex-matched 12-week-old GF mice by oral gavage on two occasions, separated by a 4-day interval. The Ex-GF mice were maintained in plastic flexible film gnotobiotic isolators (Class Biologically Clean Ltd., Madison, WI) (2–5 mice of the same sex/cage).
All Conv-R, GF, and Ex-GF mice (2–5 mice of the same sex/cage) were given the same type of autoclaved food (Teklab certified global 18% protein rodent diet; catalog number T2018SC.15) ad libitum. All animals were maintained under a strict light cycle (lights on at 0600 h and off at 1800 h). Fresh fecal pellets from each animal were collected directly in sterile 2 mL centrifuge tubes (Axygen; SCT-200-SS-R-S) and immediately stored in −80 °C until the time of DNA extraction. All fecal pellet samples were collected at between 1500h and 1700h to minimize circadian rhythm effects.
Antibiotic treatment
Pups assigned to the ABX treatment group were gavaged with 100 μL of an antibiotic cocktail that contained 4 mg/mL kanamycin (Sigma-Aldrich K4000), 0.35 mg/mL gentamicin (Sigma-Aldrich G1914); 8,500 U/mL colistin (Sigma-Aldrich C4461), 2.15 mg/mL metronidazole (Sigma-Aldrich M1547) and 0.45 mg/mL vancomycin (Sigma-Aldrich V2002) (prepared using autoclaved water). Gavage occurred daily from postnatal day 16 to 22 using animal feeding needles (Cadence; catalog number 7901). Control mice were gavaged with 100 μL of water (4, 24). After each gavage, mice were transferred to a new sterile cage to avoid contamination from accumulated feces in the old cages.
Nest-building behavior
A few days before euthanasia, group-housed mice were switched to individual housing (7, 25). Pre-weighed nestlets (2.5g; #NES7200, Ancare, Bellmore, NY) were introduced into each cage at ~1600h. The next morning at 1000h, the remaining nestlet was weighed. A 5-point scale was used to score the results: the scoring system is based on the percentage of nesting material remaining plus the shredding conditions: score = 1, nest shredding < 25%; 2, nest shredding 25%–50%; 3, nest shredding 50%–90%; 4, nest shredding > 90%, but nest was not compacted yet; 5, complete nest built (Fig. S7A).
Tissue collection
Mice received a lethal intraperitoneal injection of pentobarbital (200 mg/kg). Following perfusion with a solution of cold Dulbecco’s PBS containing 3 U/ml heparin, the brains were removed. The left hemisphere was fixed in 4% paraformaldehyde for 24 hours before being transferred to 30% sucrose and stored at 4 °C until sectioning. Brains were cut coronally into 50 μm-thick sections on a freezing sliding microtome (Leica SM1020R) and stored in cryoprotectant solution (0.2 M PBS, 15% sucrose, 33% ethylene glycol) at −20 °C until use. Right hemispheres were dissected, flash-frozen on dry ice, and stored at −80 °C for biochemical analyses.
Volumetric analysis
Volumetric analysis of the hippocampus, lateral ventricle (LV), and entorhinal/piriform (Ent/Piri) cortex was performed using a stereological method that involved assessing every sixth coronal brain section (300 μm between sections), starting rostrally from bregma −1.4 mm to bregma −3.1mm. All sections from each mouse were mounted on a glass slide. All mounted sections were completely dried, rinsed in water for 1 min and stained with 0.1% Sudan black in 70% ethanol at room temperature for 20 min, then washed in 70% ethanol for 1 min (three times). Sections were then washed in Milli-Q water three times and cover-slipped with Fluoromount. The stained sections were imaged with a NanoZoomer microscope (Hamamatsu); areas of interest were traced and measured in each section using the NDP viewer (Hamamatsu). Volume was calculated by the sum of area × 0.3 mm (7, 8). All staining and analysis of data obtained were performed by someone who was blind to the experimental hypothesis and the treatments of the animals being assessed.
Neuronal layer thickness measurement
Left hemi-brain sections from each mouse, corresponding approximately to bregma coordinates −1.7 and −2.7 mm, were mounted and stained in cresyl violet for 5 min at room temperature (7). Slices were sequentially dehydrated in 50%, 70%, 95% (three times) and 100% ethanol (twice) (1 min/treatment), then cleared in xylene for 4 min (twice), and cover-slipped in cytoseal60 (Thermo Fisher Scientific, catalog number 8310–16). Images were taken using Cytation 5 (Biotek) and analyzed with Gen5 Software (Biotek). Quantification of the thickness of the dentate granular cell layer and the CA1 pyramidal layer were measured by drawing a scale line that crossed the cell layers at two areas each section and obtaining the average value. All staining and analysis of data obtained were performed by individuals blind to the experimental hypothesis and the treatments of the animals being assessed.
Immunohistochemistry
Left hemi-brain sections, corresponding approximately to bregma coordinates −1.5 and −1.8 mm, were used for immunohistochemistry. For AT8 staining, brain sections were washed in Tris-buffered saline (TBS) buffer three times followed by incubation in 0.3% hydrogen peroxide in TBS for 10 min at room temperature. After three washes in TBS, sections were blocked with 3% milk in 0.25% TBS-X (Triton X-100) for 30 min followed by incubation at 4 °C overnight with biotinylated AT8 antibody (Thermo Scientific, catalog number1020B, 1:500 solution). The next day, after washing 3 times with TBS, all sections were treated at room temperature for 60 min with reagents included in the VECTASTAIN Elite ABC-HRP Kit, followed by 3 washes in TBS. Finally, sections were developed and stained using ImmPACT DAB EqV Peroxidase Substrate. Slides were cover-slipped with cytoseal60 and scanned using a Nanozoomer microscope at 20X magnification. Images were extracted by using the NDP viewer and analyzed with ImageJ software (National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/). For immunofluorescence, sections were washed in TBS for 3 times (5 min/cycle). After washing, sections were blocked with a solution containing 3% BSA and 3% normal donkey serum in 0.25% TBS-X for 1 h at room temperature, followed by an overnight incubation at 4 °C with primary antibodies [mouse GFAP (EMD Millipore, MAB3402, 1:1,000), rabbit Iba1: Wako, (1:2000); rat CD68 (SeroTec, 1:500)]. The next day, after 3 times washes in TBS, slides were incubated with fluorescent-labeled secondary antibodies (Molecular Probes, 1:500) for one hour at room temperature. Sections were washed and incubated with 0.1% Sudan black solution in 70% ethanol for 10 min, washed once more, and mounted in ProLong Gold Antifade mounting medium (Molecular Probes, P36931). Images were obtained by using a Lecia Stellaris 5 confocal microscope and analyzed with ImageJ software.
Brain tissue sample processing for ELISA
Mouse posterior cortex was sequentially processed in (i) RAB buffer [100 mM MES, 1 mM EGTA, 0.5 mM MgSO4, 750 mM NaCl, 20 mM NaF, 1 mM Na3VO4, pH 7.0] supplemented with protease inhibitors (Complete, Roche) and phosphatase inhibitors (PhosSTOP, Roche); (ii) RIPA buffer [150 mM NaCl, 50 mM Tris, 0.5% deoxycholic acid, 1% Triton X-100, 0.1% SDS, 5 mM EDTA, 20 mM NaF, 1 mM Na3VO4, pH 8.0] supplemented by Complete and PhosSTOP, and (iii) 70% formic acid buffer as described (8). The tissue was then weighed and homogenized using a pestle (10 μL RAB buffer/1 mg tissue). After centrifugation at 50,000 × g for 20 min, the supernatant was taken as the RAB-soluble fraction, and the pellet was dissolved in RIPA buffer (10 μL buffer/ mg tissue) by sonication (1 min at 20% pulse, 1 s interval; Fisher scientific FB120, Pittsburgh, PA). After centrifugation at 50,000 × g for 20 min, the supernatant was taken as the RIPA-soluble fraction. The pellet was sonicated (1 min at 20% pulse, 1 s interval) in 70% formic acid (10 μL/1 mg tissue) and centrifuged at 50,000 × g for 20 min. The supernatant was taken as the formic-acid-soluble fraction. All fractions were stored in −80 °C until they were analyzed.
Sandwich ELISA
The levels of human total tau, phosphorylated tau and human ApoE were measured by sandwich ELISA and normalized to tissue weight. TAU-5 (mouse monoclonal, 20 μL/mL) was used as the coating antibody for the human tau ELISA, HJ14.5 (mouse monoclonal, 20 μL/mL) for the p-Tau ELISA and HJ15.3 (mouse monoclonal, 5 μL/mL) for the human ApoE ELISA. Biotinylated HT7 (mouse monoclonal, 200 ng/mL; Thermo Fisher) was used to detect antibodies for the human tau ELISA, biotinylated AT8 (mouse monoclonal, 300 ng/mL) for the p-Tau ELISA and biotinylated HJ15.7 (mouse monoclonal, 150 ng/mL) for the human ApoE ELISA.
Astrocyte Morphology Analysis
Z-stacks (20 μm) of DAPI- and GFAP- labeled immunofluorescence images were acquired on Lecia Stellaris 5 confocal microscope with a 40X objective and 1,024 X 1,024 pixel resolution. Simple Neurite Trace (SNT; ImageJ plug-in open-source tool) was used to reconstruct tri-dimensional arbors of GFAP-positive astrocytic main processes by semi-automatic tracing (26). For each mouse, three astrocytes were randomly selected in each section from two separate brain sections based on GFAP-stained structures enclosing a single DAPI-stained nucleus. The six astrocytes chosen from each mouse did not have processes that touched the edges of the field or were truncated. Fully traced astrocytes in SNT were used to obtain morphometric data; i.e., process length, end radius, the number of process branches, the total number of interactions between process branches and radius from Sholl analysis (27), and the volume occupied by the astrocytic process from Convex Hull analysis (28).
Microglia Morphology Analysis
Z-stacks (20 μm) of Iba1- labeled immunofluorescence images were acquired on an LSM 880 II Airyscan FAST confocal microscope (Zeiss) with a 60X objective, 1.8X zoom and 1,024 X 1,024 pixel resolution, For each mouse, a total of four z-stacks of the dentate gyrus region were taken from two separate brain sections. Microglia morphology analysis was performed on 3D images using Imaris 9.5 software (Bitplane). Morphology was analyzed using the Filament Tracer, with no loops allowed and spot detection mode to determine process start and end points per cell. Process reconstruction was made using the following custom settings: ‘detect new starting points’; ‘largest diameter 9.00 μm’, ‘seed points 2.00 μm’; ‘remove seed points around starting points’; ‘diameter of sphere regions: 15 μm’. All filament parameters were exported into separate Excel files and used for analyzing the number of process branches, process length, and process volume per cell. Image processing, three-dimensional reconstruction, and data analysis were performed in a blinded manner with regards to the experimental conditions.
Nanostring gene expression assay
RNA was isolated from mouse hippocampus using the RNeasy Mini Kit (QIAGEN, catalog number 74104). Quality control checks were performed on all samples to determine RNA concentration and integrity (RIN scores > 9.3). For the Nanostring gene expression assay, isolated RNA samples were processed by the Genome Technology Access Core at Washington University, using NanoString’s nCounter® Neuroinflammation panel (735 genes were detected out of 770 targeted genes). Background noise in the data was corrected to a thresholding count value of 20. Lane-by-lane technical variation was corrected by using the geometric median value of the positive-control set. Gene expression normalization was performed subsequently using the geNorm algorithm to select the optimal housekeeping genes (Mto1, Csnk2a2, Aars, Supt7l, Fam104a, Tbp, Ccdc127, Tada2b, Lars, Cnot10)
Differential gene expression was performed using nSolver 4.0 and the Advanced Analysis 2.0 plugin (NanoString). Fold-changes expression and p-values were calculated by linear regression analysis using negative binomial or log-linear models. P-values were corrected for multiple comparisons using the Benjamini-Hochberg method. Co-expression analysis was performed using the Weighted Gene Correlation Network Analysis (WGCNA) package in R (29). A soft thresholding power of 8 was selected to calculate the unsigned adjacency for normalized gene counts. Hierarchical clustering of the topological overlap matrix dissimilarity was used to produce a gene dendrogram. Gene modules were identified using a dynamic tree cut with a minimum module size of 20 genes. Eigengenes for each module were calculated, and correlations with dummy-coded APOE genotype or the size of the hippocampus as an index of neurodegeneration level were calculated. Gene Ontology (GO) enrichment analysis was performed using Metascape (30).
Single-nucleus RNA sequencing (SnRNA-seq) of frozen hippocampal tissue
Frozen hippocampus from n=5 mice of the same genotype and treatment were pooled as a single sample. Samples were selected based on those being closest to the mean values of hippocampal volumes. Tissue was homogenized using a Dounce homogenizer in 1 mL of lysis buffer [10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2; 0.005% NP40; and 0.2 U/mL RNase Inhibitor (prepared using nuclease-free water and chilled to 4 °C)] and incubated on ice for 15 min. A 30 mm MACS SmartStrainer was used to remove cell debris and large clumps, followed by centrifugation at 500 × g for 5 min at 4 °C. After carefully removing the supernatant, the pellet containing nuclei was resuspended with 5 mL of ‘nuclei wash and resuspension’ buffer (1% BSA plus 0.2 U/mL RNase Inhibitor in 1 X PBS). The cell debris removal step, centrifugation, and resuspension steps were repeated twice. Only 500 μL of nuclei wash and resuspension buffer was added in the last resuspension step. The resulting solution was mixed with 900 μL of Sucrose Cushion Buffer I [prepared by mixing 2.7 mL of Nuclei Pure 2M Sucrose Cushion Solution (MilliporeSigma, St. Louis) with 300 mL Nuclei Pure Sucrose Cushion Solution (MilliporeSigma, St. Louis) and then carefully layered to the top of 500 μl Sucrose Cushion Buffer I in a 2 mL eppendorf tube. Following centrifugation at 13,000 × g for 45 min at 4 °C, the nuclear pellet was resuspended in 500 μL nuclei wash and resuspension buffer. The concentration of nuclei was determined using a Countess instrument (Invitrogen) and DAPI staining. The concentration was adjusted to 1200 nuclei/mL using nuclei wash and resuspension buffer prior to snRNA-seq.
Isolated nuclei were used for droplet-based snRNA-seq using the Chromium Single Cell 3′ Reagent Kit (10x Genomics). Libraries were sequenced using a NovaSeq 6000 instrument (Illumina). Sample demultiplexing, barcode processing, and single-nuclei 3′ counting was performed using the Cell Ranger Single-Cell Software Suite (10x Genomics). Cell Ranger count was used to (i) align samples to a custom pre-mRNA reference package (mm10) containing the human APOE gene, (ii) quantify reads, and (iii) filter those reads with a quality score below 30. The Seurat v3 and SoupX R packages were used for subsequent analysis of the datasets (31–33). Contaminating cell-free RNA from each sample group was removed using SoupX. Nuclei with mitochondrial content >5% or total gene counts <200 or >5000 were removed (Seurat). For each group, the percent of mitochondria was regressed out as a nuisance variable, gene counts were normalized, and variable features identified using the SCTransform function in Seurat. The top 3000 variable genes were used to integrate experimental groups using the ‘PrepSCTIntegration’, ‘FindIntegrationAnchors’, and ‘IntegrateData’ commands in Seurat. Principal component analysis (PCA) was performed on the integrated dataset and the first 30 principal components were selected for downstream analysis using FindNeighbors. Clusters were identified using the FindClusters function with a granularity ranging from 0.1 to 1.2. Final clustering was performed using a resolution of 0.3. The first 30 principal components were passed into UMAP using the RunUMAP command with default parameters. Differential gene expression between each cell cluster and all other clusters was performed on SCT data to identify marker genes for each individual cell cluster. Clusters containing high mitochondrial genome content, or marker genes for more than one broad cell type (i.e., microglia and excitatory neurons) were removed and data were reclustered using the first 30 principal components and a resolution of 0.3. In total, 143,835 nuclei with a median UMI of 2,735 and median gene number of 1,664 across all 10 experimental groups were used in the final analysis.
Differential gene expression to identify marker genes was again performed using MAST (34) and broad cell types identified based on known cell-type specific markers. For subclustering analysis, nuclei from astrocyte or microglia clusters were extracted from the dataset, RNA counts were renormalized and the percentage of mitochondrial genes regressed out using the SCTransform command. PCA analysis was performed and the nuclei re-clustered. For astrocytes, in addition to regressing the percentage of mitochondrial genes, the percentage of Gh (growth hormone) and Prl (Prolactin) transcripts were also regressed out due to detection in the TEKO-H2O treatment group. The first 20 principal components were used, and clustering performed at a resolution of 0.15 (Data S5). For microglia, the first 10 PCs were used and clustering performed at a resolution of 0.1 (Data S6). Marker genes for subclusters were identified using MAST (Data S5–S6). The SCTransform function from the Seurat package was used to generate graphs from the data containing log-normalized values (35). Expression level represents the log-normalized values of gene counts. Gene counts for each cell were divided by the total counts for that cell and scaled before natural log transformation. Gene Ontology enrichment analysis was performed using Metascape (30).
Fluidigm Biomark HD Real-Time PCR
Hippocampus tissues were used for the gene expression analysis. Samples were selected based on the mean values of hippocampal volumes. RNA was exacted from frozen tissues using RNeasy Mini Kit (Qiagen) and converted to cDNA using the ‘high capacity RNA-to-cDNA kit’ from Thermo Fisher and the manufacturer’s instructions. Gene expression was carried out using Fluidigm Biomark HD Real-Time PCR System. Using Taqman primers, gene expression was quantitatively measured after normalization the housekeeping gene, Gapdh.
16S rRNA amplicon sequencing and analysis
For bacterial V4–16S rDNA amplicon sequencing, each frozen fecal sample was resuspended in 500 μl of a phenol:chloroform:isoamyl alcohol mixture (25:24:1), followed by addition of 710 μL of 2X Buffer A (200 mM NaCl, 200 mM Trizma base, 20 mM EDTA) and 20% SDS (500:210). The solution was shaken in a Mini-Beadbeater-96 (BioSpec Products) for 4 min with ~250 μl of 0.1 mm zirconia/silica beads and one 3.97 mm steel ball. After centrifugation at 3,220 × g for 4 min, 420 μL of the resulting aqueous phase was transferred to a well of a 96-well plate. A 100 μL aliquot of the crude extract was mixed with 400 μL of a mixture of Qiagen buffer PM and 3M NaOAc, pH5.5 (675:45), and the mixture was passed through a Qiagen QiaQuick 96 plate by centrifugation at 3,220 × g for 10 min, washed twice with 900 μL of Buffer PE by centrifugation, and finally eluted with 130 μL Buffer EB. Purified DNA was quantified using Invitrogen Quant-iT dsDNA BR kit and normalized to 2 ng/μL. Variable region 4 of the bacterial 16S rRNA gene was amplified by PCR using the following conditions: denaturation (94 °C for 2 min) followed by 26 cycles of 94 °C for 15 s, 50 °C for 30 s, and 68 °C for 30 s, followed by incubation at 68 °C for 2 min.
Sample-associated 16S rDNA amplicons were quantified, pooled and subjected to sequencing (Illumina MiSeq instrument, paired-end 250 nt reads). Reads were demultiplexed, trimmed to 200 nucleotides, and merged, followed by removal of chimeric sequences (DADA2 v. 1.13.0). Amplicon sequence variants (ASVs) were generated from demultiplexed paired-end reads with DADA2 and taxonomy was assigned based on the DADA2-formatted training dataset (36). Read quality control and the resolution of ASVs were performed with the dada2 R package (37, 38). ASVs that were not assigned to the kingdom Bacteria were filtered out. The remaining reads were assigned taxonomy using the Ribosomal Database Project (RDP trainset 16/release 11.5) 16S rRNA gene sequence database (38).
Analyses of alpha-diversity (richness, Faith’s phylogenetic diversity) and beta-diversity (weighted UniFrac distances) were performed using PhyloSeq and additional R packages (39). Taxa (ASVs) whose relative abundances differed significantly between sample groups were identified by performing pairwise comparisons using DESeq2 and MicrobiotaProcess packages (40). A correlation matrix was generated and plotted as ellipses using the corrplot package (41). R codes to generate 16S rRNA-related results and figures in this manuscript are available at GitHub: https://github.com/shandley/neurodegeneration_16S.
Gas Chromatography Mass Spectrometry (GC-MS) of short chain fatty acids
Short chain fatty acids (SCFA) were quantified by GC-MS using a previously described protocol (42). Cecal contents were weighed and placed in 2 mL glass screw cap vials. Ten microliters of a mixture of internal standards (20 mM of acetic acid-13C2,D, propionic acid-D 13 4 6, butyric acid- C4, lactic acid-3,3,3-D3, and succinic acid-13C4) was added to each vial, followed by 20 μL of 33% HCl and 1 mL diethyl ether. The solution was vortexed vigorously for 10 min. The two phases were separated by centrifugation (4,000 × g for 5 min). The upper organic layer was transferred into another vial and a second 1 ml diethyl ether extraction was performed. After combining the two ether extracts, a 60 μL aliquot was mixed with 20 μL N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) in a GC auto-sampler vial with a 200 μL glass insert, and the mixture was incubated for 2 h at room temperature. Samples were analyzed in a randomized order. Derivatized samples (1 μL) were injected with 15:1 split into an Agilent 7890A gas chromatography system coupled with 5975C mass spectrometer detector (Agilent). Analyses were carried on a HP-5MS capillary column (30 m × 0.25 mm, 0.25 μm film thickness, Agilent J & W Scientific) using electronic impact (70 eV) as ionization mode. Helium was used as a carrier gas at a constant flow rate of 1.26 mL/min, and the solvent delay time was set to 3.5 min. The column head pressure was 10 psi. The temperatures of injector, transfer line, and quadrupole were 270 °C, 280 °C, and 150 °C, respectively. The GC oven was programmed as follows; 45 °C held for 2.25 min, increased to 200 °C at a rate of 20 °C/min, increased to 300 °C at a rate of 100 °C/min, and finally held for 3 min. Quantification of SCFA was performed by isotope dilution GC-MS using selected ion monitoring (SIM). For SIM analysis, the m/z for native and labeled molecular peaks for SCFA quantified were 117 and 122 (acetate), 131 and 136 (propionate), 145 and 149 (butyrate), 261 and 264 (lactate), and 289 and 293 (succinate), respectively. Various concentrations of standards were spiked into control samples to prepare the calibration curves for quantification.
Flow cytometry
Mice were given a lethal dose of pentobarbital sodium (Fatal-Plus, Vortech) intraperitoneally and perfused through the heart with ice-cold PBS supplemented with 5U/mL heparin. The dural meninges and spleen were dissected and digested with 1.4 U/mL of Collagenase VIII (Sigma Aldrich, catalog number C2139) and 35 U/mL of DNAse I (Sigma Aldrich, catalog number DN25) for 20–30 min at 37 °C. Following the digestion step, the tissue was passed through 70 μm nylon mesh cell strainers (Fisher Scientific). Cells were then centrifuged at 340×g for 5 min or 450 × g for 4 min at 4°C, and then stained in PBS with Zombie NIR Fixable Viability Kit (1:500, BioLegend, catalog number 423105) for 15 min on ice. After one wash with FACS buffer (PBS with 2% fetal bovine serum or PBS with 1% bovine serum albumin, 2 mM EDTA, 25 mM HEPES) Fc-receptors were blocked with anti-CD16/32 (1:100, BioLegend, clone 93, catalog number 101302) for 15 min on ice. An equal volume of primary antibody mix was added, and cells were stained for 20 min on ice at 1:300 final dilution. Samples were then washed twice with FACS buffer and acquired on a spectral flow cytometer (Aurora, Cytek Biosciences). Data were unmixed using Spectral flow software (Cytek) and then gated and quantified using FlowJo v10.8.2 (Treestar).
The following antibodies were used for flow cytometry; anti-CD4 BUV395 (BD, GK1.5, catalog number 563790), anti-CD5 BUV496 (BD, 53–7.3, catalog number 741048), anti-CD27 BUV563 (BD, LG.3A10, catalog number 741275), anti-CD44 BUV615 (BD, IM7, catalog number 751414), anti-CD11c BUV737 (BioLegend, N418, catalog number 749039), anti-TCRb BUV805 (BD, H57–597, catalog number 748405), Ly6G BV421 (BioLegend, 1A8, catalog number 127628), anti-CD25 Pacific Blue (BioLegend, PC61, catalog number 102022), anti-CD19 BV480 (BD, 1D3, catalog number 566107), anti-I-A/I-E BV510 (BioLegend, M5/114.15.2, catalog number 107641), anti-CXCR3 (eBioscience, CXCR3–173, catalog number 63–1831), B220 BV650 (BD, RA3–6B2, catalog number 563893), anti-CD49a BV711 (BD, Ha31/8, catalog number 564863), anti-CD45 BV750 (BD, 30-F11, catalog number 746947), anti-Ly-6C AlexaFluor488 (BioLegend, HK1.4, catalog number 128022), anti-CD8a AlexaFluor532 (eBioscience, 53–6.7m catalog number 58–0081), anti-CD122 BrilliantBlue700 (BD, TM-b1, catalog number 742112), CD69 PE (Biolegend, H1.2F3, catalog number 104508), anti-CD186 (CXCR6) PE-Dazzle 594 (BioLegend, SA051D1, catalog number 151117), anti-CD127 PE-Cy5 (BioLegend, A7R34, catalog number 135016), anti-NK1.1 PE-Cy7 (eBioscience, PK136, catalog number 108714), anti-TCRg/d AlexaFluor647 (BioLegend,GL3, catalog number 118134), and anti-F4/80 AlexaFluor700 (BioLegend, BM8, catalog number 123130).
Fluorescence activated cell sorting (FACS) for lung macrophage isolation
Lung samples were harvested from the mice and chopped up with scissors into 1–2 mm sizes. The sliced lung samples were digested in digestion buffer containing 50U/mL DNase (Sigma), 100U/mL Hyaluronidase (Sigma), and 0.28U/mL Liberase (Roche) at 37°C for 45min. The mixtures were gently inverted a few times every 5–10 min. A final concentration of 10% FBS was used to stop the reaction and the samples were smashed through a 70 μm-diameter cell strainer. The cell suspensions were pelleted down by centrifugation at 456 × g for 5 min. Red blood cells were removed with 5 mL ACK buffer (150mM ammonium chloride, 10mM potassium bicarbonate, and .1mM EDTA) at room temperature for 2min. The reaction was stopped by adding 1 mL of FBS and cells were passed through a 70 μm strainer one more time. Cells were pelleted and ~5×106 cells were used for staining. Cell sorting was completed on a FACS AriaII. Staining was performed at 4 °C in the presence of Fc block (2.4G2; Leinco) in magnetic-activated cell-sorting (MACS) buffer (PBS + 0.5% BSA + 2mM EDTA). The following antibodies were used from Biolegend; BV510 anti-CD45 (30-F11), APC-Cy7 anti-CD11b (M1/70), Pacific-Blue anti-MHC-II (I-A/I-E) (M5/114.15.2), PercP-Cy5.5 anti-Ly6C/Ly6G (Gr-1) (RB6–8C5), APC anti-CD64 (X54–5/7.1). Antibodies obtained from Invitrogen included; PE-Cy7 anti-CD11c (N418), PE anti-SiglecF (1RNM44N). The alveolar macrophages were sorted as CD45+ SiglecF+ CD11c+ CD64+ CD11blo cells; Interstitial macrophages were sorted as CD45+ SiglecF− Gr1− CD11c− CD11b+ MHC-II+ CD64+ cells.
RNA Sequencing and analysis
RNA was extracted from FACS-sorted lung macrophages (described above) using the RNeasy Plus Micro Kit (QIAGEN, catalog number 74034). RNA samples were prepared according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina NovaSeq 6000. Basecalls and demultiplexing were performed with Illumina’s bcl2fastq2 software. RNA-seq reads were then aligned and quantitated to the Ensemble release 101 primary assembly with an Illumina DRAGEN Bio-IT on-premise server running version 3.9.3–8 software.
All gene counts were then imported into the R/Bioconductor package EdgeR and TMM normalization size factors were calculated to adjust for samples for differences in library size (43). Ribosomal genes and genes not expressed in the smallest group size minus one sample greater than one count-per-million were excluded from further analysis. The TMM size factors and the matrix of counts were then imported into the R/Bioconductor package Limma (44). Weighted likelihoods based on the observed mean-variance relationship of every gene and sample were then calculated for all samples and the count matrix was transformed to moderated log 2 counts-per-million with Limma’s voomWithQualityWeights (45). The performance of all genes was assessed with plots of the residual standard deviation of every gene to their average log-count with a robustly fitted trend line of the residuals. Differential expression analysis was then performed to analyze for differences between conditions; the results were filtered for only those genes with Benjamini-Hochberg false-discovery rate adjusted p-values less than or equal to 0.05. Gene Ontology enrichment analysis was performed using Metascape (30).
Quantification and statistical analysis
All data were expressed as mean values ± SEM. Statistical analysis was performed with JMP15 Pro (SAS Institute, Cary, NC, RRID:SCR_014242) and GraphPad Prism 9. Means between two groups were compared with a two-tailed, unpaired Student’s t-test. Comparisons of means from three groups with each other were analyzed with one-way ANOVA. Two-way ANOVAs were used to analyze between-subjects designs with two variable factors. Repeated-measures designs were analyzed using mixed-effects restricted maximum likelihood (REML) model. Tukey was used for post hoc pairwise comparisons. Fisher’s Exact test was used to analyze probability distributions. The strength of the linear relationship between two different variables was analyzed using Pearson’s or Spearman’s correlation. The null hypothesis was rejected at the p < 0.05 level. Statistical significance was taken as *, p < 0.05, **, p < 0.01, ***, p < 0.001. Statistical significance of the main effects without the interactions of two variables was indicated as #, p < 0.05, ##, p < 0.01, ###, p < 0.001. All statistical information is listed in Table S1.
Supplementary Material
Acknowledgments
We thank C. Nagler and her laboratory members at the University of Chicago for the discussion about the antibiotic treatment protocol; M. Colonna, Y. Shi, C. Wang, A. Cashikar, J. Long, N. Griffin, M. Cella, S. Wagoner, M. Hibberd, and K. de Lima at Washington University for technical discussions; J. Stanley, B. Boros, J. Serugo, S. Venkatesh, K. Mihindukulasuriya, L. Wang, for technical assistance; D. Bender at CHiiPs (Center for Human Immunology and Immunotherapy Programs at Washington University) for performing multiplex cytokine assay; C. Sawyer, C. Markovic, T. Sinnwell, E. Tycksen, and others at GTAC (Genome Technology Access Center at Washington University) for their contributions to generating genomic/transcriptomic datasets.
Funding:
Good Ventures (DMH), National Institutes of Health grant NS090934 (DMH).
Competing interests:
D.M.H: Co-founder, C2N Diagnostics LLC. Scientific advisory boards/consulting: Genentech, Denali, C2N Diagnostics, Cajal Neurosciences, and Alector. D.H.M. is an inventor on a patent licensed by Washington University to C2N Diagnostics on the therapeutic use of anti-tau antibodies and a patent licensed by Washington University to Eli Lilly on a humanized anti-Aβ antibody. The Holtzman lab receives research grants from the National Institutes of Health, Cure Alzheimer’s Fund, the Rainwater Foundation, the JPB Foundation, Good Ventures, Novartis, Eli Lilly and NextCure.
Footnotes
Data and materials availability:
snRNA-Seq sequencing datasets have been deposited in Gene Expression Omnibus under accession numbers GSE213446. 16s rDNA amplicon sequencing datasets have been deposited in ENA under accession numbers PRJEB55869.
References
- 1.Long J, Holtzman DM, Alzheimer Disease: An Update on Pathobiology and Treatment Strategies, Cell, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seo DO, Holtzman DM, Gut Microbiota: From the Forgotten Organ to a Potential Key Player in the Pathology of Alzheimer’s Disease., J Gerontol A Biol Sci Med Sci, 75, 1232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harach T et al. , Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota., Sci Rep, 7, 41802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dodiya HB et al. , Sex-specific effects of microbiome perturbations on cerebral Aβ amyloidosis and microglia phenotypes., J Exp Med, 216, 1542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parikh IJ et al. , Murine Gut Microbiome Association With APOE Alleles, Front Immunol, 11, 200 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran TTT et al. , genotype influences the gut microbiome structure and function in humans and mice: relevance for Alzheimer’s disease pathophysiology, FASEB j, 33, 8221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang C et al. , Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia., Neuron, 109, 1657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y et al. , ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy., Nature, 549, 523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y et al. , Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model., J Exp Med, 216, 2546 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mancuso R et al. , CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice., Brain, 142, 3243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mann CN et al. , Astrocytic a2-Na /K ATPase inhibition suppresses astrocyte reactivity and reduces neurodegeneration in a tauopathy mouse model, Sci Transl Med, 14, eabm4107 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erny D et al. , Host microbiota constantly control maturation and function of microglia in the CNS., Nat Neurosci, 18, 965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spichak S et al. , Microbially-derived short-chain fatty acids impact astrocyte gene expression in a sex-specific manner., Brain Behav Immun Health, 16, 100318 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampson TR et al. , Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease., Cell, 167, 1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jirkof P, Burrowing and nest building behavior as indicators of well-being in mice., J Neurosci Methods, 234, 139 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Vainchtein ID, Molofsky AV, Astrocytes and Microglia: In Sickness and in Health., Trends Neurosci, 43, 144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Marco Barros R, Fitzpatrick Z, Clatworthy MR, The gut-meningeal immune axis: Priming brain defense against the most likely invaders., J Exp Med, 219, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alves de Lima K et al. , Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons., Nat Immunol, 21, 1421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colonna M, Trinchieri G, Liu YJ, Plasmacytoid dendritic cells in immunity., Nat Immunol, 5, 1219 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Schuijt TJ et al. , The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia., Gut, 65, 575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y et al. , The Gut Microbiome as a Therapeutic Target for Cognitive Impairment., J Gerontol A Biol Sci Med Sci, 75, 1242 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo DO, Boros BD, Holtzman DM, The microbiome: A target for Alzheimer disease, Cell Res, 29, 779 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huynh TV et al. , Lack of hepatic apoE does not influence early Aβ deposition: observations from a new APOE knock-in model., Mol Neurodegener, 14, 37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stefka AT et al. , Commensal bacteria protect against food allergen sensitization., Proc Natl Acad Sci U S A, 111, 13145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deacon RM, Assessing nest building in mice., Nat Protoc, 1, 1117 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Tavares G et al. , Employing an open-source tool to assess astrocyte tridimensional structure, Brain Struct Funct, 222, 1989 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.SHOLL DA, Dendritic organization in the neurons of the visual and motor cortices of the cat., J Anat, 87, 387 (1953). [PMC free article] [PubMed] [Google Scholar]
- 28.Augusto-Oliveira M et al. , Plasticity of microglia., Biol Rev Camb Philos Soc, 97, 217 (2022). [DOI] [PubMed] [Google Scholar]
- 29.Langfelder P, Horvath S, WGCNA: an R package for weighted correlation network analysis., BMC Bioinformatics, 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y et al. , Metascape provides a biologist-oriented resource for the analysis of systems-level datasets., Nat Commun, 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species., Nat Biotechnol, 36, 411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stuart T et al. , Comprehensive Integration of Single-Cell Data., Cell, 177, 1888 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young MD, Behjati S, SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data., Gigascience, 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finak G et al. , MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data., Genome Biol, 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression., Genome Biol, 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Callahan BJ et al. , DADA2: High-resolution sample inference from Illumina amplicon data., Nat Methods, 13, 581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cole JR et al. , Ribosomal Database Project: data and tools for high throughput rRNA analysis., Nucleic Acids Res, 42, D633 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMurdie PJ, Holmes S, phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data., PLoS One, 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2., Genome Biol, 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu S, Yu G, MicrobiotaProcess: An R Package for Analysis, Visualization and Biomarker Discovery of Microbiome, Version 0.84, 3.12, (2021).
- 41.Wei T, Simko VR, R package “corrplot”: Visualization of a Correlation Matrix, WWW document, (2021).
- 42.Rey FE et al. , Metabolic niche of a prominent sulfate-reducing human gut bacterium., Proc Natl Acad Sci U S A, 110, 13582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data., Bioinformatics, 26, 139 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritchie ME et al. , limma powers differential expression analyses for RNA-sequencing and microarray studies., Nucleic Acids Res, 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu R et al. , Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses., Nucleic Acids Res, 43, e97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
snRNA-Seq sequencing datasets have been deposited in Gene Expression Omnibus under accession numbers GSE213446. 16s rDNA amplicon sequencing datasets have been deposited in ENA under accession numbers PRJEB55869.