

PURPOSE
Pleuropulmonary blastoma (PPB) is the most common primary lung neoplasm of infancy and early childhood. Type II and type III PPB have historically been associated with a poor prognosis.
METHODS
Patients with known or suspected PPB were enrolled in the International PPB/DICER1 Registry. Medical records were abstracted with follow-up ascertained annually. All PPB diagnoses were confirmed by central pathology review. Beginning in 2007, the IVADo regimen (ifosfamide, vincristine, actinomycin-D, and doxorubicin) was recommended as a potential treatment regimen for children with type II and type III PPB. This regimen was compared with a historical control cohort.
RESULTS
From 1987 to 2021, 314 children with centrally confirmed type II and type III PPB who received upfront chemotherapy were enrolled; 132 children (75 with type II and 57 with type III) received IVADo chemotherapy. Adjusted analyses suggest improved overall survival for children treated with IVADo in comparison with historical controls with an estimated hazard ratio of 0.65 (95% CI, 0.39 to 1.08). Compared with localized disease, distant metastasis at diagnosis was associated with worse PPB event-free survival and overall survival with hazard ratio of 4.23 (95% CI, 2.42 to 7.38) and 4.69 (95% CI, 2.50 to 8.80), respectively.
CONCLUSION
The use of IVADo in children with type II and type III PPB resulted in similar-to-improved outcomes compared with historical controls. Inferior outcomes with metastatic disease suggest the need for novel therapies. This large cohort of uniformly treated children with advanced PPB serves as a benchmark for future multicenter therapeutic studies for this rare pediatric tumor.
INTRODUCTION
Pleuropulmonary blastoma (PPB) includes four main subtypes. Type I is a cystic lesion, type II is mixed cystic and solid, and type III is a solid multipatterned sarcoma. PPB is unique among pediatric solid tumors as it may progress from type I to type II to type III.1 Type Ir PPB is a cystic lesion lacking a primitive malignant component. Nearly all PPBs have biallelic DICER1 pathogenic mutations; more than 70% arise in the setting of germline pathogenic variants.2 In types II and III, the malignant cells are more likely to have acquired additional mutations, commonly involving TP53,3 and are associated with more aggressive clinical behavior, requiring more intensive treatment.
CONTEXT
Key Objective
Pleuropulmonary blastoma (PPB) is a rare but often aggressive pediatric tumor, diagnosed primarily in children age younger than 7 years. The International PPB/DICER1 Registry (ClinicalTrials.gov identifier: NCT03382158) was founded in 1987 to advance research for this rare tumor. This analysis examined outcomes for children with type II (mixed cystic and solid) and type III (solid) PPB treated with multiagent chemotherapy.
Knowledge Generated
Overall, 314 children with type II and type III PPB confirmed by central pathology review enrolled in the Registry. Of these, 132 were treated with a chemotherapy regimen consisting of ifosfamide, vincristine, actinomycin-D, and doxorubicin, determined by international consensus.
Relevance
This analysis reports treatment strategies and outcomes in the largest-ever cohort of children with advanced PPB treated in 204 centers in 47 countries. Future treatment regimens should optimize local control and address risk for CNS metastases. Ongoing international collaboration is encouraged.
Because PPBs histologically include components of rhabdomyosarcoma, agents known to have activity in sarcomas have generally been used.4-6 Some children have received additional treatment modalities including intracavitary cisplatin and high-dose chemotherapy with stem-cell rescue.7-10
In the largest published study of types II and III, the 5-year overall survival (OS) was 71% and 53%, respectively; this cohort received various chemotherapy regimens.11 Additionally, outcomes in smaller cohorts of patients with PPB have been described in the literature.4,5,12-20 The 5-year survival ranged from 45% to 75% for type II and 35%-67% for type III (Data Supplement, online only).
The International PPB (now PPB/DICER1) Registry was founded in 1987 to improve outcomes for children with this rare tumor. Initial findings included the familial nature21 and subtypes of PPB13 including the distinction between types I and Ir PPB.22 In 2009, Dr Hill and Registry colleagues described DICER1 as the main genetic factor underlying familial PPB. Key features of the Registry include free central pathology review, direct review of records with standardized abstraction, longitudinal follow-up since Registry inception, worldwide collaboration, and sustained philanthropic support.
The purpose of this study was to evaluate outcomes in the largest-ever cohort of children with type II and type III PPB receiving chemotherapy. Additionally, historically used chemotherapy regimens were compared with a standardized regimen recommended by an international consortium convened by the Registry and adapted from the study by Bisogno et al.6
METHODS
Individuals with suspected PPB were enrolled in the International PPB/DICER1 Registry. All study procedures were approved by the Children's Minnesota institutional review board and relevant human subjects committees (ClinicalTrials.gov identifier: NCT03382158). Written informed consent (including assent when applicable) was obtained. Medical records were abstracted. Eligibility for this analysis included type II or III PPB determined by central pathology review (L.P.D. or D.A.H.). Exclusion criteria included patients who did not receive chemotherapy at diagnosis and those who progressed to type II or III following type I PPB. Ascertainment of disease status was attempted annually.
In 2007, the Registry began providing a roadmap for ifosfamide, vincristine, actinomycin-D, and doxorubicin (IVADo) chemotherapy. Treatment remained at the discretion of the treating physician. IVADo chemotherapy consists of ifosfamide 3 g/m2/dose IV on days 1 and 2 (6 g/m2/cycle) with MESNA, vincristine 1.5 mg/m2 IV on day 1 (maximum 2 mg), actinomycin-D 1.5 mg/m2 IV on day 1 (maximum 2 mg), and doxorubicin 30 mg/m2/dose IV on days 1 and 2 (60 mg/m2/cycle) for four 21-day cycles followed by IVA (ifosfamide, vincristine, and actinomycin-D) on day 1 for eight 21-day cycles. Dose adjustments were provided for patients age < 1 year. Suggested criteria for initiation of subsequent cycles included marrow recovery (absolute neutrophil count ≥ 750/μL and platelet count ≥ 75,000). Recommendations for local control were provided. If complete resection was not possible at diagnosis, resection was recommended at week 10 (or at week 19 if not achieved before). Focal radiotherapy was recommended between weeks 23 and 29 if complete resection was not achieved.
Patients who received at least one cycle of IVADo as first-line treatment were included in the IVADo cohort. Additionally, patients who received a maximum of one cycle of a different regimen followed by IVADo when central review results confirmed PPB were included in the IVADo cohort. Children who did not receive IVADo as initial chemotherapy were included in the historical cohort. Historical regimens were classified as vincristine, actinomycin-D and cyclophosphamide (VAC)–containing; anthracycline-containing; platinum- and anthracycline-containing; or other non-IVADo regimens that could not be further subcategorized.
Central surgical staging was completed (D.M.). Extent of disease at diagnosis was classified as local disease (confined to primary tumor site), locoregional disease (additional sites of disease within the chest, excluding type I/Ir, including lymph nodes), and distant metastasis (metastatic disease outside of the thorax with/without locoregional disease). Time-to-event (from start of chemotherapy) outcomes included OS and PPB event-free survival (EFS); the latter events were defined as recurrence, new metastasis, progression of type II or III PPB, or death. Subsequent neoplasms are expected within the context of a germline DICER1 variant and therefore were not defined as additional events.
Radiographic response was assessed using RECIST version 1.1.23 This subset analysis included children with measurable disease assessed by computed tomography (CT) scan following a diagnostic procedure who received IVADo as neoadjuvant chemotherapy and had a CT scan between 5 and 9 weeks following initiation of chemotherapy. Additionally, individuals were included if they fell outside of the time range but had cross-sectional imaging after the start of cycle 2 and before cycle 4. Partial response (PR) was defined as ≥ 30% decrease in sum of the diameters of the target lesions, and stable disease (SD) was defined as insufficient shrinkage and < 20% increase in sum of diameters of the largest target lesions.23 Original scans were centrally reviewed; if scans were unavailable, reports were used. When a distinction between cystic and solid components was available, only the solid component was included in the measurement.
Comparisons between type II and type III were made using Mann-Whitney, chi-square, and Fisher exact tests. Survival analyses were used for time-to-event outcomes. Kaplan-Meier curves were used to estimate PPB-EFS and OS on the basis of extent of disease at diagnosis and chemotherapy regimen. Unadjusted comparisons were assessed using log-rank tests. An adjusted analysis used stratified log-rank test after subclassifying patients using propensity score and PPB type.24 Balance between covariate distributions was assessed using standardized differences, with absolute difference of < 10% being considered well balanced.25 The propensity score model included age, sex, extent of disease at diagnosis, prechemotherapy degree of resection, and prechemotherapy radiotherapy use. Year of diagnosis could not be balanced, so an additional analysis included regression models to account for trends over time via linear associations (on the log hazard scale).26 When analyzing type II and III combined, regression adjustments for all covariates were included via Cox proportional hazards regression and IVADo was compared with specific historical chemotherapy regimens. Associations with individual prognostic factors and time-to-event outcomes were estimated using Cox proportion hazards model with stratification by PPB type and false-discovery rate to account for multiple comparisons.27
RESULTS
Of 582 cases of centrally reviewed PPB, 337 (58%) were type II or III and 314 (93%) individuals qualified for this analysis (Data Supplement). The overall cohort includes 173 (55%) children who were diagnosed with type II and 141 (45%) with type III. The children in the historical cohort were treated from 1973 to 2021 and children in the IVADo cohort were treated from 2007 to 2021.
Demographics are summarized in Table 1. DICER1-related neoplasms were observed before PPB in three patients. Concurrent types I and Ir were observed in eight children, suggestive of metachronous disease. DICER1 germline pathogenic variants were detected in 73% (85/116); tumor testing showed biallelic mutations in 91% (31/34).
TABLE 1.
Demographic, Clinical, Surgical, and Treatment Characteristics of Patients With Type II and III PPB

Locoregional spread was present at diagnosis in 7% (21/314), and distant metastatic disease was present at diagnosis in 6% (19/314) of patients. Locoregional spread and distant metastasis was more common in type III versus type II (P = .01). The most common sites of metastatic disease were bone and CNS. Type II tumors were typically smaller than type III (P = .0002).
Treatment
Forty-three percent (75/173) of children with type II and 40% (57/141) with type III received IVADo (Table 1). Interval from diagnosis to chemotherapy initiation was shorter for type III (median 0.3 months) than type II (median 0.5 months).
For type II, neoadjuvant chemotherapy was given following biopsy in 31% of patients. High-dose chemotherapy with stem-cell rescue was used (before any progression) in four cases. One patient with type II received intrapleural chemotherapy and one patient with type II received maintenance chemotherapy.
Compared with type II, more children with type III PPB received neoadjuvant chemotherapy (55%), consistent with the larger tumor size at diagnosis (P = .0001). High-dose chemotherapy with stem-cell rescue was used as part of primary treatment in 11 and maintenance chemotherapy was given to four patients with type III.
Radiotherapy was given before chemotherapy in eight patients. For patients with type II, 14% (24/173) received radiotherapy as part of primary treatment compared with 29% (41/141) of patients with type III (P = .0009).
Application of radiotherapy differed by extent of disease. In patients with type II PPB, 11% (17/159) with local versus 44% (4/9) with locoregional disease received radiotherapy. In type III PPB, 27% (31/115) with local and 42% (5/12) with locoregional disease received radiotherapy. See the Data Supplement for additional treatment characteristics by extent of disease at diagnosis.
As suggested by differences in tumor size, children with type II were more likely to attain gross total resection with negative margins than children with type III (51% v 35%).
Survival Outcomes
When assessing outcomes by extent of disease at diagnosis, distant metastasis had worse prognosis than local and locoregional disease. The 3-year PPB-EFS for type II was 69.3% (95% CI, 62.0 to 77.5) for local, 77.8% (95% CI, 54.9 to 100.0) for locoregional, and 20% (95% CI, 3.5 to 100.0) for distant metastatic disease (Table 2 and Fig 1A). Median time to event in the patients with distant metastasis was 9 months. The 3-year OS for type II was 82.8% (95% CI, 76.6 to 89.5) for local, 88.9% (95% CI, 70.6 to 100.0) for locoregional, and 40% (95% CI, 13.7 to 100.0) for distant metastatic disease (Fig 1B). Median time to death in the patients with distant metastasis was 19 months.
TABLE 2.
Outcome Data of Patients With Type II and Type III PPB by Local, Locoregional, and Distant Metastatic Disease

FIG 1.
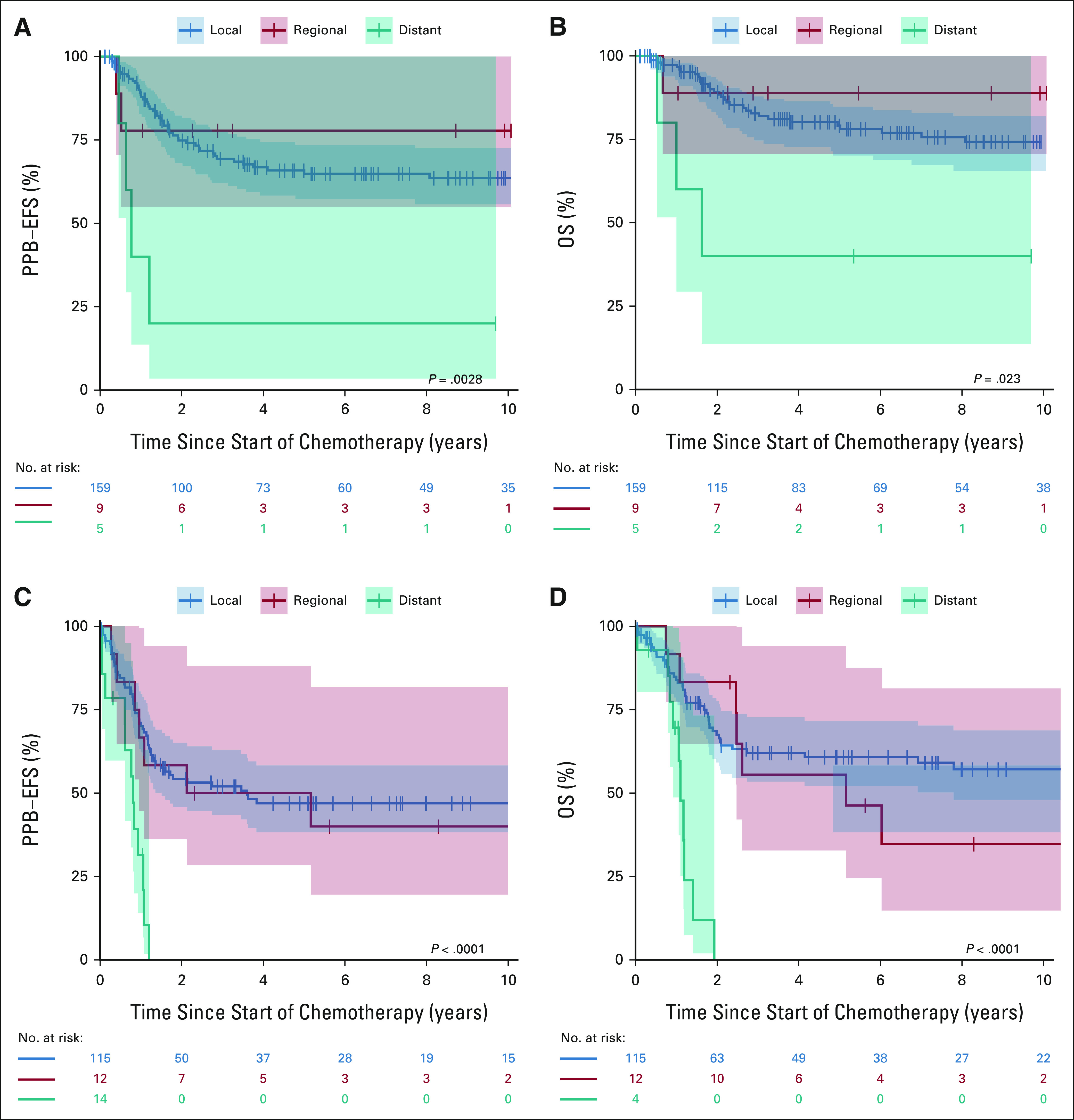
(A) PPB-EFS and (B) OS in patients with type II PPB by local disease (blue), locoregional disease (red), and distant metastasis (teal) at diagnosis. (C) PPB-EFS and (D) OS in patients with type III PPB by local disease (blue), locoregional disease (red), and distant metastasis (teal) at diagnosis. EFS, event-free survival; OS, overall survival; PPB, pleuropulmonary blastoma.
In comparison, 3-year PPB-EFS for type III was 52.0% (95% CI, 43.2 to 62.7) for local and 50.0% (95% CI, 28.4 to 88.0) for locoregional disease (Fig 1C). Importantly, all assessable patients with type III with distant metastases had a PPB event by 15 months. Median time to event was 44 months in children with locoregional disease and 10 months for children with distant metastasis. The 3-year OS for type III was 62.1% (95% CI, 53.2 to 72.5) for local and 55.6% (95% CI, 32.8 to 94.1) for locoregional disease (Fig 1D). All assessable type III patients with distant metastases at diagnosis died of disease by 2 years. Median time to death in children with locoregional disease was 62 months and 13 months for children with distant metastasis at diagnosis.
For type II PPB, progressive disease was observed in 52 patients compared with 59 patients with type III PPB. A total of 94 children died, with nearly all (94%) deaths related to PPB progression. The most frequent site of initial recurrent/progressive disease was the chest (local recurrence/progression) followed by CNS and bone.
Subsequent neoplasms, generally DICER1-related, were identified in 42 patients with a total of 54 neoplasms (Data Supplement).
Subtotal resection as best overall resection was associated with worse PPB-EFS and OS (Table 3). Additionally, tumor biopsy only without subsequent resection was also associated with inferior outcomes. This scenario represented six patients with type III PPB that progressed before resection. Importantly, tumor size > 10 cm and primary radiation therapy were not significantly associated with survival outcomes. Tumor size as a continuous variable was associated with worse OS but did not remain significant after multiple comparison correction. Pneumothorax demonstrated a protective association but did not remain significant after multiple comparison correction. Pneumothorax was more prevalent in type II and the protective association of pneumothorax was more pronounced in type II with a hazard ratio (HR) of 0.43 (95% CI, 0.23 to 0.81) versus III with a HR 0.74 (95% CI, 0.38 to 1.42).
TABLE 3.
Prognostic Factors and Corresponding HRs for PPB-EFS and OS for Patients With Type II and Type III Pleuropulmonary Blastoma (n = 314)

When considering patients treated with IVADo versus historical controls, the unadjusted HR was 0.94 (95% CI, 0.55 to 1.62) for EFS and 0.70 (95% CI, 0.34 to 1.44) for OS (Table 4). For type III PPB, the unadjusted HR was 0.74 (95% CI, 0.45 to 1.21) for EFS and 0.78 (95% CI, 0.45 to 1.36) for OS. The median follow-up was 36 months for the IVADo cohort and 56 months for the historical cohort. Outcome data by IVADo versus historic, Kaplan-Meier curves, and survival curves for future comparison studies are provided (Data Supplement).
TABLE 4.
Outcome Data Both Unadjusted and Adjusted for Covariates and Trends in Time

Stratification on the propensity score for IVADo versus historic cohorts led to improved balance on several factors including metastatic disease, degree of resection, and use of radiotherapy (Data Supplement). For type II, adjusted HR was 1.16 (95% CI, 0.64 to 2.09) for PPB-EFS and 0.93 (95% CI, 0.43 to 2.01) for OS (Table 4). For type III, adjusted HR was 0.52 (95% CI, 0.28 to 0.97) for PPB-EFS and 0.51 (95% CI, 0.26 to 1.01) for OS. When combining types II and III treated with IVADo versus historical regimens, the adjusted HR was 0.77 (95% CI, 0.51 to 1.18) for PPB-EFS and 0.65 (95% CI, 0.39 to 1.08) for OS. When adjusting for trends in time, the adjusted HR for type II and type III treated with IVADo versus historical cohort was 0.95 (95% CI, 0.57 to 1.59) for PPB-EFS and 0.98 (95% CI, 0.53 to 1.83) for OS.
Among specific chemotherapy regimens, non–anthracycline-containing regimens had inferior outcomes, although this was somewhat mitigated by trends in time (Data Supplement). When combining type II and type III PPB, the results suggested improved EFS and OS for IVADo, although mitigated when factoring in trends in time.
Radiographic Response
Figure 2A displays radiographic response to neoadjuvant IVADo in a subgroup of 29 children. Of these, 24 children (nine with type II and 15 with type III) had a CT scan performed within the defined time interval (Fig 2B). In type II, 44% (4/9) had PR and 56% (5/9) had SD. In type III, 53% (8/15) had PR and 47% (7/15) had SD, representing an overall response of 50% (95% CI, 31 to 69).
FIG 2.
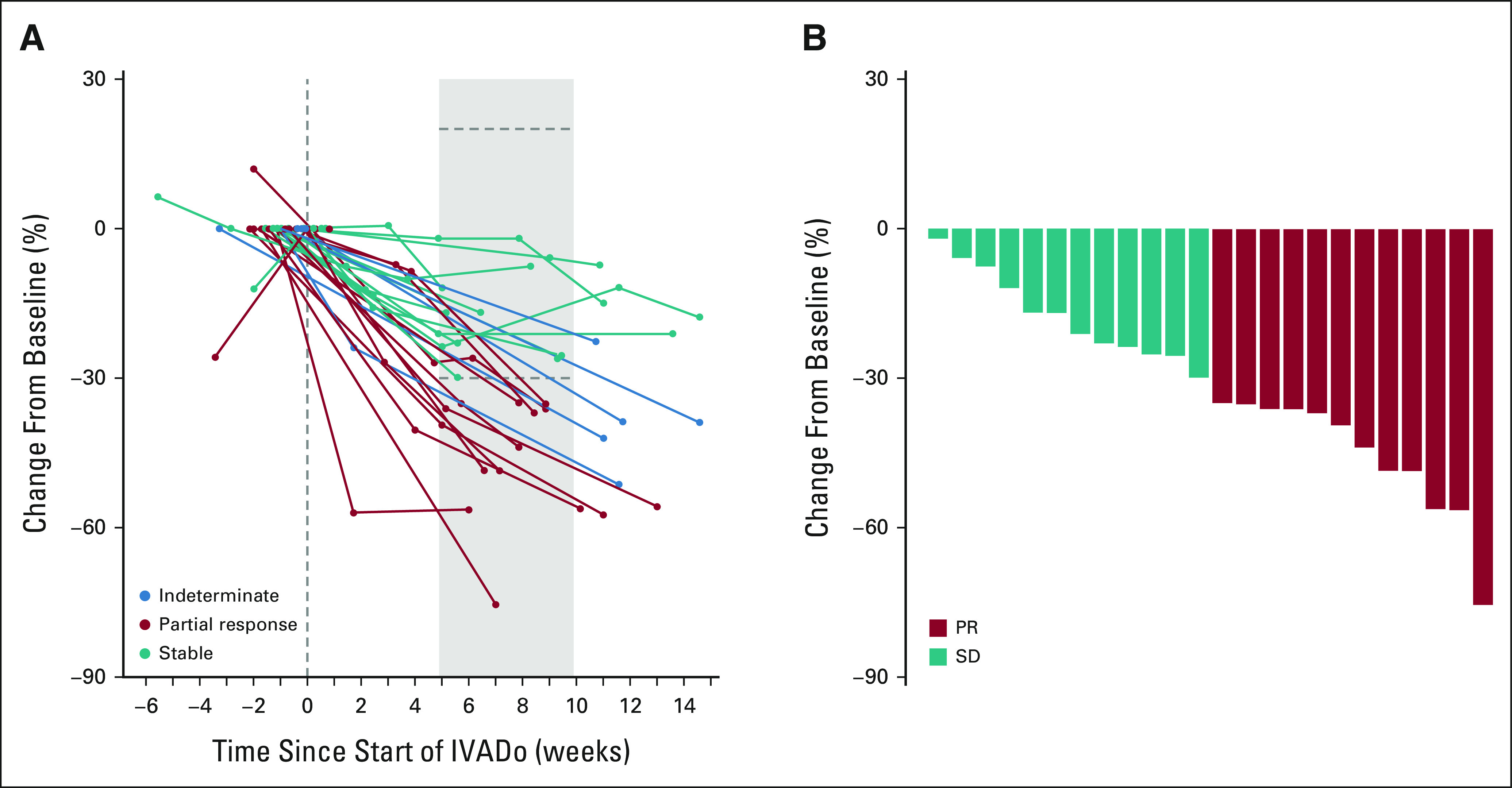
(A) Percent reduction of tumor size per RECIST criteria during treatment with neoadjuvant IVADo chemotherapy among patients with type II and type III PPB. Teal lines represent patients with SD, red lines represent patients with PR, and blue lines represent patients with scans falling outside the established assessment window. (B) Waterfall plot of best response during weeks 5-9 for patients receiving neoadjuvant IVADo chemotherapy, with teal representing SD and red representing PR per RECIST criteria. IVADO, ifosfamide, vincristine, actinomycin-D, and doxorubicin; PPB, pleuropulmonary blastoma; PR, partial response; SD, stable disease.
DISCUSSION
Our study evaluates outcomes in the largest-ever cohort of children with advanced PPB with detailed treatment and outcome data from 204 centers in 47 countries. This includes a cohort of 132 children from 103 centers in 24 countries treated with a uniform chemotherapy regimen, IVADo, developed through international consensus. This broad international effort provides several critical insights key to future treatment of children with PPB.
IVADo and historical cohorts were largely similar with respect to demographics, although children treated with IVADo were more likely to have metastatic disease at diagnosis and less likely to receive thoracic radiation therapy. When considering differences between the cohorts with an adjusted analysis (Table 4), on the basis of HRs, we estimate a 23% decrease in progressive disease and 35% decrease in death among children treated with IVADo compared with historical controls. These results were somewhat mitigated by trends in time. The results suggest effect sizes were modest because regimens used in the historical group include agents and dosages similar to IVADo. For example, 27% of children in the historical cohort received regimens incorporating doxorubicin; worst outcomes were observed in the VAC-only regimens (Data Supplement). In a prior study by Bisogno et al,12 patients with PPB who received doxorubicin fared better with a 5-year EFS of 70% versus 31.3% (P = .01).
Despite radiographic response to IVADo, R0 resection was achieved in only half of the children with type II and in only one third of the children with type III. These data demonstrate the need to optimize cytoreduction to facilitate local control.
Among patients with advanced PPB, we confirmed that distant metastasis at diagnosis confers poor prognosis as was noted in prior studies. We did not observe a large survival difference between local and locoregional disease, although the number of patients with locoregional disease is small. Whether more intensive treatments such as additional surgery or radiation mitigated risk associated with locoregional disease is a subject of future analyses.
This analysis did not confirm the deleterious effect of size more than 10 cm noted on prior studies.11,16 When considering extent of resection, subtotal resection as best overall resection was associated with worse outcomes. Microscopic residual disease after best surgical resection did not reach statistical significance; however, the analysis did not consider the nonrandomized use of preoperative or postoperative radiotherapy, which may have mitigated the impact of microscopic residual disease. Thus, these results do not preclude administration of radiotherapy for residual disease or metastatic disease. Additional analyses specifically assessing the role and timing of radiation therapy are underway. Interestingly, pneumothorax at diagnosis demonstrated a protective association. We hypothesize this is in part due to earlier tumor detection in patients who presented with pneumothorax.
Children with advanced PPB and distant metastases represent an important group for whom treatment intensification should be considered. In this analysis, all children presenting with type III with distant metastatic disease experienced an event within 15 months of diagnosis.
Overall, more than a quarter of children with type II and nearly half of children with type III PPB will succumb to their cancer. Local recurrence within the thorax is the most common first failure event followed by CNS metastasis. Optimization of local control and integration of strategies to address risk for CNS disease is critical.
Prognosis for recurrent PPB remains poor.28 Novel strategies to prevent on-treatment and post-treatment recurrence are needed.
Another key finding is that children who underwent biopsy only, without subsequent tumor resection, fared especially poorly. This group includes individuals who progressed before resection. The magnitude of this result is likely because of a guarantee-time bias,29 but demonstrates that some children receiving a neoadjuvant strategy do not survive to undergo tumor resection.
The relationship between PPB and DICER1 biallelic mutations is well established.30 The majority of children with PPB have a pathogenic germline DICER1 variant. A subset of children in this analysis were diagnosed with a subsequent DICER1-related neoplasm, most of which were successfully resected and/or treated, highlighting the importance of genetic testing and tumor surveillance.31 Guidelines for surveillance and familial testing are available to facilitate the diagnosis of PPB and other DICER1-related neoplasms in their earliest, most curable forms.32 Given the natural history of PPB, attention to early diagnosis remains critical.
Strengths of this analysis include the largest-ever reported cohort of children with PPB, enrolled from 47 countries. All PPB diagnoses were confirmed by central pathology review, a key strength of this research because of the high rate of discordant diagnosis in rare tumors.33,34 Additionally, detailed data regarding surgical resection was relatively complete in this analysis with 86% having centrally reviewed surgical data.
Limitations to this study are inherent to the rarity of PPB and the retrospective nature of registry research. Surgical management and use and timing of radiotherapy remained at the discretion of the treating institution and therefore varied among patients. Data are missing for some variables, and the follow-up time for patients treated with IVADo is expectedly shorter than for historical controls. Although we have provided an adjustment for trends in time, we hypothesize improved ascertainment of progression events and both metastatic disease and locoregional disease at diagnosis in the IVADo cohort. Radiographic response data excluded children who did not undergo cross-sectional imaging in the designated interval; thus, children who died before these time points were not included in the analysis.
In conclusion, this study reports outcomes in the largest-ever cohort of children with advanced PPB. Novel approaches are needed to further improve outcomes and reduce risks of local and CNS recurrence; children with distant metastatic disease at diagnosis represent a particularly high-risk group. Importantly, the establishment of a large cohort of relatively uniformly treated children establishes feasibility for future prospective studies and provides a baseline for comparison with new therapeutic regimens. Finally, ongoing international collaboration is encouraged to optimize treatment for children with advanced PPB.
ACKNOWLEDGMENT
The authors wish to thank the many treating physicians, genetic counselors, patients, and families who collaboratively support the International PPB/DICER1 Registry as well as the Pine Tree Apple Classic Fund whose volunteers, tennis players, and donors have provided more than 35 years of continuous support for PPB Research. The authors gratefully acknowledge the contributions of Jason Albrecht to ongoing PPB research initiatives. The authors also wish to gratefully acknowledge the contributions of John R. Priest, Gretchen Williams, and Leslie Doros to PPB/DICER1 research.
Dave Watson
Research Funding: Merck (Inst)
Douglas R. Stewart
Employment: Genome Medical
Kelly N. Hagedorn
Consulting or Advisory Role: Abbott Laboratories, Boston Scientific, Saluda, Nevro
Junne Kamihara
Stock and Other Ownership Interests: PanTher Therapeutics, ROME Therapeutics, TellBio
Honoraria: Pfizer, NanoString Technologies, Foundation Medicine, Ikena Oncology
Consulting or Advisory Role: ROME Therapeutics, Tekla Capital Management, Foundation Medicine, NanoString Technologies, EMD Millipore, Pfizer
Research Funding: PureTech, Ribon Therapeutics, ACD Biotechne
Patents, Royalties, Other Intellectual Property: Patent on drug delivery device licensed to PanTher Therapeutics, Patents on Repeat RNA biomarkers and therapeutics licensed to Rome Therapeutics, Patents on Circulating Tumor Cell Biomarkers Licensed to TellBio, Inc
David B. Wilson
Research Funding: X4 Pharma, Novartis
Gianni Bisogno
Consulting or Advisory Role: Bayer
D. Ashley Hill
Employment: ResourcePath
Leadership: ResourcePath
Stock and Other Ownership Interests: ResourcePath
No other potential conflicts of interest were reported.
SUPPORT
The International PPB/DICER1 Registry is generously supported by the philanthropic efforts of the Pine Tree Apple Classic Fund (K.A.P.S., A.K.H., A.T.N., and Y.H.M.). The International PPB/DICER1 Registry is also supported by the Children's Minnesota Foundation (A.K.H. and A.T.N.), Mendon F. Schutt Foundation and Rein in Sarcoma (K.A.P.S., A.K.H., and A.T.N.). This analysis was supported by a grant from the Children's Minnesota Internal Research Grant Program (K.A.P.S. and A.K.H.) and supported by funding from National Institute of Health National Cancer Institute grants 1R37CA244940-01 (K.A.P.S. and A.K.H.), 2R01CA143167-06A1 (K.A.P.S., D.A.H., and Y.H.M.), 1R43CA228850 (K.A.P.S., A.K.H., and D.A.H.), and the Intramural Research Program of the Division of Cancer Epidemiology and Genetics of the National Cancer Institute, Rockville, MD (D.R.S.). A.T.N. received funding from the University of Minnesota Medical School Foundation for contributions to this research.
AUTHOR CONTRIBUTIONS
Conception and design: Kris Ann P. Schultz, Dave Watson, Arun A. Rangaswami, Carlos Rodriguez Galindo, Louis P. Dehner, D. Ashley Hill, Yoav H. Messinger
Provision of study materials or patients: Kris Ann P. Schultz, Douglas R. Stewart, Junne Kamihara, Sarah G. Mitchell, David B. Wilson, Katie Gettinger, Arun A. Rangaswami, Yoav H. Messinger
Collection and assembly of data: Kris Ann P. Schultz, Anne K. Harris, Alexander T. Nelson, Dave Watson, Doug Miniati, Douglas R. Stewart, Kelly N. Hagedorn, Sarah G. Mitchell, Katie Gettinger, Arun A. Rangaswami, Laura A. Harney, D. Ashley Hill, Yoav H. Messinger
Data analysis and interpretation: Kris Ann P. Schultz, Anne K. Harris, Alexander T. Nelson, Dave Watson, John T. Lucas Jr, Douglas R. Stewart, William Mize, Junne Kamihara, David B. Wilson, Arun A. Rangaswami, Carlos Rodriguez Galindo, Gianni Bisogno, Louis P. Dehner, D. Ashley Hill, Yoav H. Messinger
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Outcomes for Children With Type II and Type III Pleuropulmonary Blastoma Following Chemotherapy: A Report From the International PPB/DICER1 Registry
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Dave Watson
Research Funding: Merck (Inst)
Douglas R. Stewart
Employment: Genome Medical
Kelly N. Hagedorn
Consulting or Advisory Role: Abbott Laboratories, Boston Scientific, Saluda, Nevro
Junne Kamihara
Stock and Other Ownership Interests: PanTher Therapeutics, ROME Therapeutics, TellBio
Honoraria: Pfizer, NanoString Technologies, Foundation Medicine, Ikena Oncology
Consulting or Advisory Role: ROME Therapeutics, Tekla Capital Management, Foundation Medicine, NanoString Technologies, EMD Millipore, Pfizer
Research Funding: PureTech, Ribon Therapeutics, ACD Biotechne
Patents, Royalties, Other Intellectual Property: Patent on drug delivery device licensed to PanTher Therapeutics, Patents on Repeat RNA biomarkers and therapeutics licensed to Rome Therapeutics, Patents on Circulating Tumor Cell Biomarkers Licensed to TellBio, Inc
David B. Wilson
Research Funding: X4 Pharma, Novartis
Gianni Bisogno
Consulting or Advisory Role: Bayer
D. Ashley Hill
Employment: ResourcePath
Leadership: ResourcePath
Stock and Other Ownership Interests: ResourcePath
No other potential conflicts of interest were reported.
REFERENCES
- 1.Dehner LP, Watterson J, Priest J, Pleuropulmonary blastoma: A unique intrathoracic pulmonary neoplasm of childhood. Perspect Pediatr Pathol 18:214-226, 1995 [Google Scholar]
- 2.Brenneman M, Field A, Yang J, et al. : Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma/DICER1 syndrome: A unique variant of the two-hit tumor suppression model. F1000Res 4:214, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González IA, Mallinger P, Watson D, et al. : Expression of p53 is significantly associated with recurrence-free survival and overall survival in pleuropulmonary blastoma (PPB): A report from the International Pleuropulmonary Blastoma/DICER1 Registry. Mod Pathol 34:1104-1115, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Indolfi P, Bisogno G, Casale F, et al. : Prognostic factors in pleuro-pulmonary blastoma. Pediatr Blood Cancer 48:318-323, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Kirsch S, Leuschner I, Int-Veen C, et al. : Sixteen children with pleuropulmonary blastoma—Results of the German Cooperative Soft Tissue Sarcoma Group [abstract]. Sarcoma 9:88, 2005 [Google Scholar]
- 6.Bisogno G, Ferrari A, Bergeron C, et al. : The IVADo regimen—A pilot study with ifosfamide, vincristine, actinomycin D, and doxorubicin in children with metastatic soft tissue sarcoma: A pilot study of behalf of the European pediatric Soft tissue sarcoma Study Group. Cancer 103:1719-1724, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Kaneko H, Isogai K, Kondo M, et al. : Autologous peripheral blood stem cell transplantation in a patient with relapsed pleuropulmonary blastoma. J Pediatr Hematol Oncol 28:383-385, 2006 [DOI] [PubMed] [Google Scholar]
- 8.de Castro CG Jr, de Almeida SG, Gregianin LJ, et al. : High-dose chemotherapy and autologous peripheral blood stem cell rescue in a patient with pleuropulmonary blastoma. J Pediatr Hematol Oncol 25:78-81, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Bekassy AN, Garwicz S, Wiebe T, et al. : Uncertain role of high dose chemotherapy with autologous stem cell support in pediatric pleuro-pulmonary blastoma (PPB). Med Pediatr Oncol 28:75-76, 1997 [DOI] [PubMed] [Google Scholar]
- 10.Katzenstein HM, Petricca S, Ricketts R, et al. : Intracavitary cisplatin therapy for pediatric malignancies. Pediatr Blood Cancer 55:452-456, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Messinger YH, Stewart DR, Priest JR, et al. : Pleuropulmonary blastoma (PPB): A report on 350 central pathology reviewed confirmed cases by the International PPB Registry. Cancer 121:276-285, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bisogno G, Brennan B, Orbach D, et al. : Treatment and prognostic factors in pleuropulmonary blastoma: An EXPeRT report. Eur J Cancer 50:178-184, 2014 [DOI] [PubMed] [Google Scholar]
- 13.Priest JR, McDermott MB, Bhatia S, et al. : Pleuropulmonary blastoma: A clinicopathologic study of 50 cases. Cancer 80:147-161, 1997 [PubMed] [Google Scholar]
- 14.Venkatramani R, Malogolowkin MH, Wang L, et al. : Pleuropulmonary blastoma: A single-institution experience. J Pediatr Hematol Oncol 34:e182-e185, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Zeng Q, Zhou C, He Y: Pleuropulmonary blastoma in children. Chin J Thorac Cardiovasc Surg 17:343-345, 2001 [Google Scholar]
- 16.Sparber-Sauer M, Seitz G, Kirsch S, et al. : The impact of local control in the treatment of type II/III pleuropulmonary blastoma. Experience of the Cooperative Weichteilsarkom Studiengruppe (CWS). J Surg Oncol 115:164-172, 2017 [DOI] [PubMed] [Google Scholar]
- 17.Christosova IR, Avramova BE, Drebov RS, et al. : Diagnosis and treatment of pleuropulmonary blastoma-single center experience. Pediatr Pulmonol 50:698-703, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Grigoletto V, Tagarelli A, Atzeni C, et al. : Pleuropulmonary blastoma: A report from the TREP (Tumori Rari in Età Pediatrica) Project. Tumori 106:126-132, 2020 [DOI] [PubMed] [Google Scholar]
- 19.Zhang N, Zeng Q, Ma X, et al. : Diagnosis and treatment of pleuropulmonary blastoma in children: A single-center report of 41 cases. J Pediatr Surg 55:1351-1355, 2020 [DOI] [PubMed] [Google Scholar]
- 20.Wang F, Zhuang L, Liang J, et al. : Treatment and prognosis of pleuropulmonary blastoma: A single-center report of 31 cases. Pediatr Pulmonol 57:2237-2243, 2022 [DOI] [PubMed] [Google Scholar]
- 21.Priest JR, Watterson J, Strong L, et al. : Pleuropulmonary blastoma: A marker for familial disease. J Pediatr 128:220-224, 1996 [DOI] [PubMed] [Google Scholar]
- 22.Hill DA, Jarzembowski JA, Priest JR, et al. : Type I pleuropulmonary blastoma: Pathology and biology study of 51 cases from the International Pleuropulmonary Blastoma Registry. Am J Surg Pathol 32:282-295, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Rubin DB: Estimating causal effects from large data sets using propensity scores. Ann Intern Med 127:757-763, 1997 [DOI] [PubMed] [Google Scholar]
- 25.Austin PC: Balance diagnostics for comparing the distribution of baseline covariates between treatment groups in propensity-score matched samples. Stat Med 28:3083-3107, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suissa S: Single-arm trials with historical controls: Study designs to avoid time-related biases. Epidemiology 32:94-100, 2021 [DOI] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y: Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57:289-300, 1995 [Google Scholar]
- 28.Schultz KAP, Williams GM, Stewart D, et al. : Association of recurrent or progressive p of form types II and III pleuropulmonary blastoma (PPB) with poor outcome: A report from the International PPB Registry. J Clin Oncol 33, 2015. (suppl 15; abstr 10014) [Google Scholar]
- 29.Giobbie-Hurder A, Gelber RD, Regan MM: Challenges of guarantee-time bias. J Clin Oncol 31:2963-2969, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill DA, Ivanovich J, Priest JR, et al. : DICER1 mutations in familial pleuropulmonary blastoma. Science 325:965, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez IA, Stewart DR, Schultz KAP, et al. : DICER1 tumor predisposition syndrome: An evolving story initiated with the pleuropulmonary blastoma. Mod Pathol 35:4-22, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultz KAP, Williams GM, Kamihara J, et al. : DICER1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res 24:2251-2261, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckardt MA, Graham DS, Singh AS, et al. : Pathologic discordance in sarcomas: Prospective comparison of external and sarcoma center pathologic diagnosis. J Clin Oncol 37, 2019. (suppl 15; abstr 11020) [Google Scholar]
- 34.Ray-Coquard I, Montesco MC, Coindre JM, et al. : Sarcoma: Concordance between initial diagnosis and centralized expert review in a population-based study within three European regions. Ann Oncol 23:2442-2449, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]