

Keywords: ARDS, COVID-19, malaria, red blood cell, sepsis
Abstract
The human immune system evolved in response to pathogens. Among these pathogens, malaria has proven to be one of the deadliest and has exerted the most potent selective pressures on its target cell, the red blood cell. Red blood cells have recently gained recognition for their immunomodulatory properties, yet how red cell adaptations contribute to the host response during critical illness remains understudied. This review will discuss how adaptations that may have been advantageous for host survival might influence immune responses in modern critical illness. We will highlight the current evidence for divergent host resilience arising from the adaptations to malaria and summarize how understanding evolutionary red cell adaptations to malaria may provide insight into the heterogeneity of the host response to critical illness, perhaps driving future precision medicine approaches to syndromes affecting the critically ill such as sepsis and acute respiratory distress syndrome (ARDS).
INTRODUCTION
Malaria has threatened human existence for millennia. It is uniquely positioned to be a driver of natural selection, given its propensity to infect children and cause fatality before reproductive age. In part, survival depends on red blood cell (RBC) mutations that confer innate resistance to the parasite (1–3). J.B.S. Haldane first noted in the 1940s that the geographical distribution of sickle cell disease, the most well-studied red blood cell disorder of the time, aligned with the tropical regions where malaria was endemic. He posited his since-termed “Malaria Hypothesis” that this overlap was no coincidence, but rather selection in those regions for heterozygotes who are more resistant to infection with the Plasmodium species that cause malaria (4). A.C. Allison lent credence to this hypothesis less than a decade later in a series of experiments that proved that the sickle cell polymorphism was balanced such that “the heterozygote has an advantage over either homozygote” (5). Since that time, malaria has become one of the most extensively studied and most elegant examples of an infectious force as the agent of natural selection.
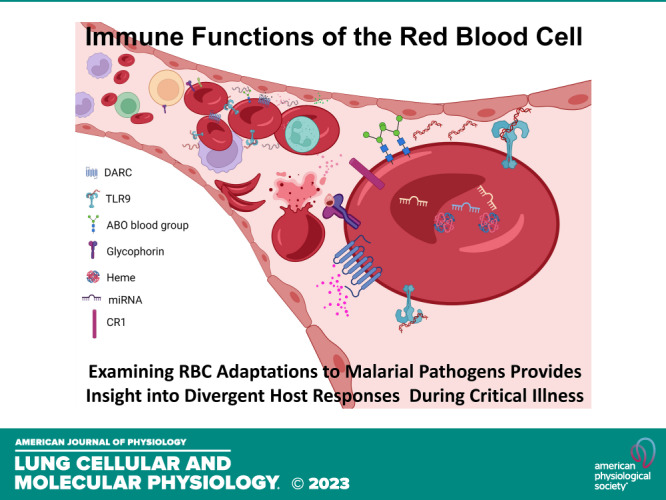
Malarial infection originates when female anopheles mosquitoes bite and inject plasmodium parasites (sporozoites) into the bloodstream. These sporozoites travel first to the liver where they undergo asexual multiplication, before emerging as merozoites, which invade and multiply in erythrocytes. Infected erythrocytes then burst, allowing the parasite to invade other neighboring erythrocytes (6). It comes as no surprise that the host target of malarial selection pressures is the same cell at which parasite sequestration and replication occurs—the red blood cell. Decades of work have been dedicated to investigating the link between malaria and known alterations in gas-exchanging properties of the RBC. Emerging evidence suggests further adaptations to virtually every component of the RBC, from membrane proteins to oxidative and glycolytic enzymes to the newly discovered expression of immune receptors on RBCs (Fig. 1; Table 1).
Figure 1.

Schematic overview of known and potential red blood cell adaptations to malaria. Known malaria adaptions include Duffy antigen receptor for chemokines (DARC), which modulates chemokine availability and neutrophil recruitment; hemoglobinopathies (sickle cell trait/disease and thalassemia); and enzymopathies (G6PD and pyruvate kinase deficiency), which can lead to hemolysis and subsequent free heme release, promoting inflammation and increase susceptibility to infection. Probable malarial adaptations include: ABO glycoproteins, which determine major ABO blood group, Toll-like receptor 9 (TLR9), which binds nucleic acids and mediates clearance of RBCs during inflammation, and miRNAs, which are abundant in RBCs but the functions of many remain undefined. G6PD, glucose-6-phosphate dehydrogenase; RBCs, red blood cells. [Image created with BioRender.com and published with permission.]
Table 1.
Non-gas exchanging functions of RBCs and hypothesized role in critical illness
| Hypothesized or Proven Adaptation | RBC Feature | Function | Adaptation to Malaria | Consequence to Critical Illness |
|---|---|---|---|---|
| Proven | Heme | Generates reactive oxygen species: activates TLR4 Amplifies cytokine production in the presence of LPS (7) | Hemoglobinopathies/thalassemia (8) | Increased susceptibility to nonmalarial infection (9, 10), anemia/transfusion dependence (11) |
| Duffy Antigen Receptor for Chemokines | Binds chemokines: maintains chemokine gradient (12) | Downregulation: decreased parasite entry (13, 14) | Increased susceptibility to ARDS (15, 16) | |
| Binds pathogens: entry point for P. vivax (13) | ||||
| Glucose-6-Phosphotase | Involved in pathway to consume reactive oxygen species (17) | Deficiency: increased intracellular oxidative stress decreases parasite survival (18) | COVID-19 severity (19) | |
| Pyruvate kinase | Catalyzes rate-limiting step in glycolysis to produce intracellular energy in nonmitochondrial cells (mature RBCs) (20) | Deficiency: decreased invasion by P. falciparum and increased phagocytosis of ring- and mature-stage-infected RBCs (20, 21) | Hemolytic anemia/transfusion dependency, functional asplenia (20) | |
| Hypothesized | Glycophorins | Binds pathogens: entry point for P. falciparum (22) | Dantu variant: structural variant in chromosome 4 region, which increases RBC surface tension and decreases merozoite invasion (23) | Potential predisposition to lysis/thrombosis |
| Complement Receptor 1 | Binds P. falciparum-induced ligand (24, 25) | Deficiency: increased cerebral malaria (26) | Increased RBC-bound complement fragments in COVID-19 and non-COVID-19 sepsis (27) | |
| Proinflammatory regulator abundant on RBCs (27) | Deficiency: reduced rosetting conferring protection (25) | |||
| Toll-like Receptor 9 | Binds nucleic acids (28, 29) | Deficiency/gene polymorphisms | Decreased capacity to bind mtDNA to ameliorate lung injury (28) | |
| Involved in P. yoeli infection (30, 31) | ||||
| ABO Glycoproteins | Surface proteins on RBCs responsible for determining ABO blood group (32) | Blood group O demonstrates different propensity to bind to Duffy binding-like regions of infected erythrocytes (33–36) | Blood group A may confer increased risk for ARDS in critically ill patients and associated with increased risk of AKI (37) |
AKI, acute kidney injury; ARDS, acute respiratory distress syndrome; COVID-19, coronavirus disease 2019; LPS, lipopolysaccharide; RBCs, red blood cells.
Critical illnesses such as sepsis and acute respiratory distress syndrome (ARDS) portend high morbidity and mortality with a multitude of mechanisms that collectively lead to profound inflammation, tissue injury, and ultimately organ failure. More and more, as we unveil these distinctive mechanisms, we start to recognize the diverse outcomes that stem from patient heterogeneity. For years, RBC dysfunction was perceived to accompany critical illness as a byproduct of excessive inflammation that resulted in RBC membrane distortion, oxidative disequilibrium, and insufficient oxygen delivery (38). However, recent studies have demonstrated that RBCs play an active role in modulating diverse immune functions critical for host defense, including chemokine regulation and nucleic acid scavenging. This long-overlooked erythrocyte contribution to host defense implores further understanding of its role in critical illness, namely, not as mere vessels for gas exchange but as active participants in immunoregulation. The coronavirus disease 2019 (COVID-19) pandemic has highlighted the remarkable heterogeneity of the host response to a specific pathogen. Given the importance of the host response to infection in mediating outcomes and treatment response, it is imperative that a comprehensive understanding of all potential modulators of the innate immune response is obtained. Here, we will review the emerging evidence for alterations in RBC immune function as a byproduct of the evolutionary response to malaria and will highlight some of the major advances over the past century in understanding RBC immunoregulatory biology. Equipped with this knowledge, we will be able to better understand how RBC immune function contributes to heterogeneous host responses in the face of pathogenic or sterile insults that underlie critical illnesses such as sepsis and ARDS.
KNOWN MALARIAL ADAPTATIONS AND THEIR RELEVANCE TO CRITICAL ILLNESS
The Erythrocyte Chemokine Receptor ACKR1—Duffy Antigen Receptor for Chemokines
Perhaps best known as the receptor by which Plasmodium vivax species gain entry into red blood cells, the Duffy Antigen Receptor for Chemokines (DARC) was initially reported as a chemokine binding protein on erythrocytes. Though unnamed and unidentified at that time, it was first described in a 1950 case report of a hemophilic patient who suffered a hemolytic transfusion reaction due to an antibody to an unknown substance on red blood cells (RBCs) (39). Subsequently, RBCs were demonstrated to carry a surface protein that binds the chemokine interleukin- 8 (IL-8), diminishing the concentration of soluble IL-8 in blood plasma samples and thereby dampening immune signaling (12). In 1993, Horuk et al. (13) united these two independent findings and showed that the Duffy antigen receptor, used by Plasmodium vivax for erythrocyte cell entry, was the same RBC receptor that bound IL-8. Since that time, DARC has been found to bind multiple inflammatory chemokines with high affinity (40, 41).
DARC belongs to a family of chemokine receptors that lack a G-coupled protein to mediate intracellular signaling, rendering the chemokine inaccessible to circulating immune cells (40–42). As such, DARC plays a significant immunoregulatory role by sequestering chemokines from immune cells such as neutrophils and acts as a reservoir for future release (43–46). In preclinical studies using lipopolysaccharide (LPS)-induced lung injury, erythrocyte DARC was found to dampen the local chemokine effects by soaking up chemokines in the lung microvasculature for release in the systemic circulation, preventing polymorphonuclear leukocyte infiltration into the alveolar space to cause lung injury (45).
It is thus of no surprise that RBC-derived cytokines play a role in transfusion-related lung injury. Lysis of transfused RBCs that contain chemokines can lead to profound lung inflammation and result in antibody negative-transfusion-related acute lung injury (TRALI) (47). Interestingly, prolonged RBC storage can result in decreased DARC expression on erythrocytes, thereby diminishing its chemokine-binding capacity (15). As such, these DARC-low erythrocytes are less capable of dampening local tissue inflammation following LPS-induced lung injury.
Consistent with the theory of malaria as a potent driver of human evolution, the DARC-null allele is present in as high as 90%–95% of sub-Saharan Africans in regions with the highest threat of P. vivax (14). In the United States, up to 68% of African Americans have the DARC-null allele (14). One observational study showed that patients with the Duffy-null phenotype with acute lung injury were at increased risk of mortality at 60 days, had increased number of days on mechanical ventilation, and had increased organ failure compared with the Duffy-positive counterparts (16). Of note, Duffy-positive African Americans had similar clinical outcomes compared with those of European descent. Though larger studies have yet to validate these findings, and plasma cytokines have yet to confirm the mechanism, these observations complement preclinical data denoting erythrocyte DARC as an essential player that tempers local tissue inflammation. Thus, the same adaptation that provides survival benefits to the threat of malaria infection may confer a higher risk of mortality when facing critical illness in an ICU setting, merely by altering the RBC’s ability to sequester and transport inflammatory chemokines.
Hemoglobinopathies
Hemoglobin has evolved to adapt to malarial pressures, rendering erythrocytes less hospitable to these invading parasites. However, under acute stress, hemoglobinopathies lead to hemolysis and subsequent release of chemokines and free heme into the circulation, promoting inflammation.
HbSS.
Of the hemoglobin adaptations to malaria-inducing pathogens, Hemoglobin SS (HbSS) is the most well-studied. HbSS arises from a single point mutation in the β-globin chain of the hemoglobin molecule. The resultant amino acid substitution of this mutation leads to polymerization of globin chains, which causes numerous and devastating sequelae of sickle cell disease (48). There have been countless studies confirming that the persistence of this hemoglobin mutation is due to the protective advantage against P. falciparum conferred in heterozygotes of the mutation (49).
The sequelae of sickle cell disease are known to impact host immunity indirectly via numerous vaso-occlusive mechanisms and splenectomy. Free heme released from hemolysis during vaso-occlusive crisis is extremely proinflammatory and can lead to endothelial damage (50). Sickled erythrocytes have also been shown to retain mitochondria, which can provide immunologically active mitochondria DNA (mtDNA) that ultimately leads to endothelial damage. Whether mitochondria retention is an advantageous adaption to malaria or a byproduct of abnormal erythroid development remains to be seen (51, 52). Nevertheless, mtDNA is elevated in patients with sickle cell disease and further increases during sickle cell crisis. Notably, mtDNA is also elevated in ARDS and is associated with increased mortality (53). It has long been appreciated that patients with sickle cell disease are at higher risk of severe disease and hospitalization from bacterial and viral infections and confer a higher risk for ICU admissions. Whether the level of mtDNA in patients with sickle cell disease correlates with a higher risk of acute chest syndrome or predisposition to severe ARDS has yet to be determined.
Despite long-held assumptions that the sickle cell trait is a silent protector against malaria, emerging evidence suggests that heterozygotes may have altered susceptibility to critical illness. Sickle cell trait is usually asymptomatic but in rare instances, severe dehydration or hypoxia can lead to erythrocyte sickling, vaso-occlusive crisis, and exertional rhabdomyolysis. Notably, a recent study found increased risk of mortality and acute kidney injury after SARS-CoV-2 infection in patients of African ancestry with sickle cell trait (54). Dedicated investigation into the impact of sickle cell trait on red blood cell immune function would help to better understand the potential consequences of this malarial adaptation on the host’s inflammatory responses.
Thalassemia.
Thalassemias are a heterogeneous group of red blood cell disorders in which the globin-chain units making up normal adult hemoglobin (α2β2) are reduced or absent. As a result, carriers of these mutations show microcytic anemia to varying degrees, and at least in the neonatal period, compensate with elevated levels of circulating fetal hemoglobin (γ4) (8). The prevalence of α- and β-thalassemia have similarly been shown to coincide with malaria-endemic regions, leading to hypotheses that these mutations also confer selective advantage against deadly malaria infections (55).
One proposed mechanism for that protection against malaria is increased susceptibility to P. vivax and P. falciparum during the neonatal period, likely due to increased erythrocytosis, acting as a natural vaccine and providing more enhanced protection in later life (56). Others have shown that parasitized α-thalassemic erythrocytes express higher levels of novel surface antigens (which are currently ill-defined) than wild-type parasitized erythrocytes, with a corresponding increase in antiparasitic antibody binding in vitro (57, 58). This enhanced detection and subsequent enhanced clearance of parasitized RBCs may explain how thalassemia protects against malaria.
Despite protection against malaria, thalassemia carriers are at increased infection risks due to several qualitative and functional defects in the immune system rendering them susceptible to pathogens such as Klebsiella pneumoniae and Yersinia enterocolitica infections (9). These defects range from defective T and B lymphocyte functions to altered chemotactic and phagocytotic processes to complement system deficiencies (9). Indirect consequences of chronic anemia also led to mortality and morbidity from increased exposure to transfusion-transmitted pathogens like Hepatitis C, chronic transfusion-related iron overload, and therapeutic splenectomies (11). Given this increased infection burden and mortality, this genetic adaptation to malaria has resulted in a less fit host if they live in regions with unlikely threat of malaria.
Enzymopathies
G6PD deficiency.
Another well-studied RBC adaptation to malarial infection is glucose-6-phosphate dehydrogenase (G6PD) deficiency. G6PD is an enzyme in the pathway that consumes oxygen-free radicals within RBCs, thereby minimizing oxidative stress. This X-linked, inherited RBC disorder has increased prevalence in regions with the highest malaria burden, and the prevalent form of G6PD deficiency in Africa has been demonstrated to correlate with a 50% reduced risk of severe malaria (17). The hypothesized mechanism of protection is that the increase in oxidative stress seen in G6PD-deficient RBCs makes them a less hospitable host for malaria-causing parasites.
Although this adaptation offers protection from the potentially lethal pathogen, it comes at a cost. The oxidative stress leads to early RBC breakdown and, in situations of stress or exposure, widespread hemolysis, leading to free heme and chemokine release. Those with G6PD deficiency have also been shown to fare worse in the face of critical illness during COVID-19 infection. A retrospective chart review of hospitalized patients with COVID-19 pneumonia requiring supplemental oxygen found that the cohort with G6PD deficiency demonstrated prolonged hypoxia and a longer need for mechanical ventilation (19). Outside of COVD-19, no study specifically addressing whether G6PD deficiency confers a higher risk for ARDS mortality has been done.
Pyruvate kinase deficiency.
Pyruvate kinase (PK) deficiency has similarly been linked to resistance against P. falciparum. Pyruvate kinase catalyzes the rate-limiting step of glycolysis, a process that is crucial to energy production in nonmitochondrial cells like mature erythrocytes. In a study comparing in vitro P. falciparum inoculation of RBCs obtained from homozygous and heterozygous pyruvate kinase-deficient adults and controls, PK-deficient RBCs showed decreased invasion by P. falciparum (20). Furthermore, phagocytosis of ring-stage and mature-stage-infected erythrocytes was increased in samples from patients deficient in PK, indicating enhanced clearance of parasitized cells (20). Additional genotyping studies have indicated a global geographical codistribution between malaria and high frequency of PK deficiency (21).
Much like G6PD deficiency, this adaptation results in wide-ranging clinical consequences from mild hemolytic anemia to complete transfusion dependency. Though high-powered studies have yet to be performed to assess the susceptibility of individuals with PK deficiency to other pathogens and critical illnesses, the propensity toward anemia and the increased risk of functional asplenia are likely deleterious consequences accompanying the protection against P. falciparum.
PROBABLE ADAPTATIONS TO MALARIA
Beyond the RBC mutations that have been proven to provide malaria resistance to their host, there are numerous erythrocyte mutations that follow similar geographical patterns indicating similar evolutionary pressures and have shown some early associations with host resistance to infection.
ABO Blood Type
The ABO histo-blood group refers to a family of glycosyl-transferases encoded by the ABO gene that catalyze specific carbohydrate modifications on glycans and glycoproteins (32). These glycans are present on the surface of RBCs, platelets, endothelium, and epithelium. DNA sequencing has placed the development of the group O allele before human migration out of Africa, and it has been hypothesized that P. falciparum, which was active in Africa before the early migration of humans and was lethal before or during the human reproductive years, was the infectious disease driving the emergence the O allele component of ABO groups (59). This hypothesis is supported by the worldwide distribution of A and O blood groups, the ratio of which correlates with the global distribution of malaria (59).
Several studies have supported the hypothesis that group O is associated with improved outcomes of severe P. falciparum infection in both young adults and the fetuses of pregnant women with placental infections compared with group A (33–36). One study suggested that H antigen may be important for P. falciparum invasion, however, no other studies have addressed this hypothesis (60). The prevailing hypothesis is that the infected RBCs expressing the malarial encoded protein Pfemp contain Duffy binding-like regions that vary in cytoadhesion based on blood group, with group O being the least cytoadhesive and blood group A being the most cytoadhesive and demonstrating increased rosetting (59, 61, 62).
There is a growing body of work investigating ABO blood groups’ role following trauma and in critical illness (63). A series of studies demonstrated that the A group, and specifically the A1 allele, confers increased risk for ARDS in a cohort of critically ill patients (64). In a genome-wide association study of patients with COVID-19, blood group A was associated with a high risk of acquiring COVID-19, though this did not translate to higher rates of intubations in the severely ill patients with COVID-19 in a separate study (65, 66). The A group is also associated with acute kidney injury (AKI) risk in critically ill patients with trauma or severe sepsis (37). Recent evidence suggests that ABO expression on endothelium drives this association, which raises the important question of whether evolutionary adaptations to proteins on RBCs have bystander impacts on nonerythroid cells on which they are also expressed (64). Further work will be important in determining whether other RBC adaptations indirectly affect nonerythroid cells.
CR1
Complement receptor 1 (CR1), a regulatory component of one of the oldest arms of the innate immune system, the complement system, is more abundantly expressed in erythrocytes than anywhere else in the body (27).
The hypothesis that CR1 deficiency may be the result of selection pressures by malaria-causing species comes from cumulative research showing CR1 to be the host receptor for the P. falciparum-induced ligand, PfEMP1 (24, 26, 61). CR1 further participates in malaria pathogenicity by aiding in rosetting, the process by which parasitized erythrocytes grab on to neighboring erythrocytes to spread infection (25). Clinical studies of host susceptibility to malaria show conflicting evidence as to whether CR1 deficiency is beneficial or detrimental, likely attributable to the fact that there are several polymorphisms that alter receptor expression in different ways. One study showed that a CR1 polymorphism resulted in CR1 deficiency, which conferred protection against severe malaria by reducing rosetting of P. falciparum-infected cells (25). Other studies, however, showed that CR1 deficiency polymorphisms are associated with an increase in cerebral malaria (26). Supporting these findings another study demonstrated polymorphisms associated with higher erythrocyte CR1 were associated with protection from severe cerebral malaria (67). Given CR1’s role in dampening complement activation, one can speculate that perhaps the reduction in parasitic load is counterbalanced with increased inflammation leading to blood-brain barrier breakdown and parasitic access to the CNS.
A recent study suggested that increased function of the alternative complement pathway is associated with improved survival during critical illness (68). We have demonstrated that RBC-bound complement activation products are elevated in both COVID-19 and non-COVID sepsis (69). Yet, loss of CR1 density on erythrocytes in ICU patients with COVID-19 correlated with disease severity (27). Whether alterations in erythrocyte-specific CR1 expression drive sepsis or host responses in critical illness remains unknown. Further studies will be necessary to elucidate the role of red cell complement regulation in critical illness.
Toll-Like Receptor 9
Recognition of nucleic acids is a vital component of the innate immune system, which occurs through cytosolic and endosomal Toll-like receptors. Toll-like receptor 9 (TLR9) has been shown to mediate inflammatory responses to malarial DNA (70). TLR9 gene polymorphisms are associated with symptomatic malaria in Ghanaian children, and TLR9 has been shown to play an important role in the development of protective immunity to P. yoelli, especially in the regulation of pro- and anti-inflammatory cytokines (30, 71). TLR9 deficiency has also been shown to confer partial resistance to lethal infection from P. yoelli, likely due to malaria parasites requiring TLR9 signaling for immune evasion via regulatory T cells (31).
We have identified TLR9 presence on RBCs where it plays a role in scavenging cell-free DNA from the circulation during quiescent states (28). During inflammatory states, however, excess DNA binding (including mtDNA as well as CpG-DNA from pathogens) leads to RBC-TLR9-driven anemia and innate immune activation (29). RBCs loaded with TLR9-bound DNA effectively concentrate and deliver the CpG-containing DNA to tissue macrophages, which in turn phagocytose the RBCs, contributing to sepsis-induced anemia.
In a CpG-induced lung injury model in mice, RBC-derived TLR9 binds CpG-DNA to ameliorate lung injury (28). Whether red cell TLR9 alters the host response in human lung injury remains to be seen, but if found, could indicate another mechanism through which RBC adaptation to malaria contributes to its immunomodulatory role to impact host response to critical illness.
miRNAs
RBCs contain abundant micro RNAs (miRNAs), which are noncoding RNAs that act as critical mediators of posttranscriptional regulation to enhance or suppress protein translation of its target. Despite the known abundance of miRNAs in RBCs, the specific roles of these miRNAs remain to be determined. Some have posited that erythrocytes may serve as repositories of miRNAs in the circulatory system, serving not only as markers of disease but as mediators of inflammation (72).
Studies have shown that miRNAs are important for host defense against intraerythrocytic pathogens like plasmodium species (73). At least two human miRNAs (miRNA-451 and miRNA-233) are more abundant in cells from individuals with sickle cell disease and sickle cell trait than normal red blood cells (74). The implication of this abundance in protection against malaria species was proven in in vitro experiments showing that miRNA-451 can be incorporated into P. falciparum mRNAs where it inhibits mRNA translation and parasite growth (74). When normal erythrocytes are manipulated to overexpress miRNA-451 and miRNA-223, these erythrocytes show reduced parasite growth. Conversely, blocking the expression of these miRNAs in erythrocytes from those with sickle cell disease and sickle cell trait led to diminished malaria resistance (74).
The numerous mechanisms by which miRNAs play a role in regulating immune cell function remain broad and under investigation. Both intracellular and extracellular miRNAs have been shown to mediate immune responses. Studies have found that tissue ischemia, like that seen in sepsis, causes release of cellular RNA, including miRNA, into the extracellular space. Once released, it may mediate local and systemic inflammatory responses via activation of pattern recognition receptors (75, 76). Alteration of circulating miRNA levels has even been linked to ARDS prognosis, with miRNA-126 showing the potential to predict 28-day mortality (77). miRNA-223, which is linked to increased resistance to P. falciparum as discussed earlier, also demonstrates a role in ameliorating acute lung injury (ALI) by decreasing IL-1b secretion and neutrophil recruitment (78, 79).
Glycophorin
Glycophorins (GYPs) are another surface protein on human erythrocytes, which exhibit diverse genetic variation. Human GYPs are determinants of the major MNS blood group and Gerbich blood group systems, some of which play a role as RBC receptors for P. falciparum invasion and other pathogens (22). GYPA, in particular, interacts with P. falciparum protein erythrocyte binding ligand 1 (EBL-1). Recent investigation has shown a large structural variant in the chromosome 4 region which gives rise to the Dantu glycophorin variant and is found predominantly in East African populations and has been associated with a 40% reduction in risk for severe malaria (23). This Dantu variant is believed to confer protection by increasing RBC surface tension to a high enough level that merozoite invasion rarely occurs (80).
The impact, if any, on critical illness survival for those with Dantu homozygosity remains unclear. However, it is tempting to speculate that mutations that alter membrane surface tension may predispose to RBC lysis or thrombosis thereby driving inflammatory dysregulation, especially in light of RBCs’ role in chemokine trafficking.
DISCUSSION
The need to better understand the role of RBC adaptions in host immunity is driven by the hypothesis that different populations may respond differently to modern protocols used in treating critical illness. Numerous studies have begun to address social determinants of health and other host factors that may contribute to the host response during critical illness, whereas factors such as RBC-mediated immunoregulation have been overlooked. As reviewed in this paper, we are only beginning to uncover the role RBCs play in mediating immune function. Beyond its gas exchange functions, this ubiquitous cell traverses the distance of the human body to transport chemokines, miRNA, cell-free DNA, and effectively alter, aid, or dampen the immune response. Selective pressures exerted over the millennia in malaria-endemic regions altered the makeup of erythrocytes in select individuals, impacting not only the cargo-carrying capacity but also the type of cargo it transports. Recognizing this previously overlooked compartment in critical illness (along with the selective pressures that alter the genetic makeup) adds another layer of complexity to the already heterogeneous population that makes up patients with ARDS and sepsis. Yet understanding it better may lend insight into why some protocolized therapies failed.
Take, for example, the multiple studies demonstrating that populations in malaria-endemic regions have not responded to protocols developed in cohorts from nonmalaria regions. Perhaps the most drastic example of this is the the Fluid Expansion as Supportive Therapy (FEAST) trial, in which fluid resuscitation strategies that have been widely endorsed by western-society pediatric life-support training programs were applied to children with severe febrile illness and impaired perfusion in Uganda, Kenya, and Tanzania, all regions in which centuries of endemic malaria has exerted its pressures (81). In contrast to original research performed on patients in nonendemic regions, the study found that excess mortality with fluid resuscitation was consistent across all groups, irrespective of physiological derangement or underlying microbial pathogen. A similar study in an adult population in Zambia in 2017 showed increased in-hospital mortality with early resuscitation with intravenous fluids and vasopressors compared with usual care (82). In the United States, a subgroup reanalysis of the Fluids and Catheters Treatment Trial (FACTT) trial, a study that initially concluded that a restrictive fluid resuscitation strategy, was superior to a liberal one in intubated patients with ARDS. Jolley et al. (83) found that although there was a decreased 1-yr mortality in black-identifying participants, the conservative fluid strategy actually led to a higher 1-yr mortality for white-identifying participants.
Despite limitations, this important observation that there is a heterogeneous treatment response calls for ongoing discussions regarding the roles of protocolized versus precision medicine. As Dellinger et al. (84) noted, although protocols are often borne out of necessity for quick action in resource- or time-limited settings, and often improve adherence to best practice standards, precision-based methods will likely be a critical component of future sepsis treatment.
The recent work that has been done in defining subgroups of patients with different host immune responses in sepsis lends credence to the benefits of precision medicine that could enhance well-designed protocols. These distinct phenotypes, derived from transcriptomic analysis of leukocytes in peripheral blood, have revealed a relatively immunosuppressed versus immunocompetent phenotype with differential response to steroid administration—the relatively immunocompetent group showing increased mortality with hydrocortisone use (85–87). Similarly, patients with acute respiratory distress syndrome (ARDS) have been stratified into two subgroups defined by levels of inflammatory biomarkers. Phenotype 1 (the hypoinflammatory subgroup) has lower mortality, more ventilator-free, and more organ failure-free days compared with the hyperinflammatory Phenotype 2 (88, 89). Much like the transcriptionally defined sepsis subgroups described earlier, the importance of stratifying patients in clinical trials by these phenotypes has been demonstrated by the use of steroids: Sinha et al. (90) recently demonstrated that steroids decreased mortality in Phenotype 2 patients with ARDS secondary to COVID-19, but not in Phenotype 1 patients. Emerging evidence demonstrating RBCs’ immunomodulatory roles coupled with millennia of selective evolutionary pressures on RBCs call for a reexamination of RBC heterogeneity as a variable that can modulate the host response to inflammation and infection. For example, one critical question would be how would subgroup analysis of the above studies segregate by RBC adaptations?
The current pandemic provides us with a unique opportunity to a single, ubiquitous pathogen through which we can gain insight into remarkably divergent host responses, including those involving RBC heterogeneity. Several of the RBC adaptations discussed in this review have shown significant differences in host response to coronavirus infection. A genome-wide association analysis of almost 2,000 patients with severe COVID-19 (defined by respiratory failure) found potential involvement of the ABO system in the severity of disease, with blood group A conferring higher risk (66). A large, multicenter retrospective chart review conducted from January 2020 to September 2020 of patients with COVID-19 found that, compared with Black-identifying individuals without sickle cell trait or sickle cell disease, Black-identifying individuals with sickle cell disease were at higher risk for hospitalization and the development of pneumonia, even when controlling for other preexisting conditions (91). RBCs obtained during the acute phase of infection from patients with moderate to severe COVID-19 pneumonia (defined as requiring supplemental oxygen support but not needing intensive care) were shown to induce endothelial injury via RBC-derived reactive oxygen species (ROS) and cytokines (92). In addition, a small but significant study found that, among hospitalized patients with COVID pneumonia, the group with G6PD deficiency was significantly more likely to have a worse P:F ratio, spend a longer period of time on mechanical ventilation, and have decreased hematocrit throughout their hospitalization (19). In a small retrospective study, the DARC polymorphism was suggested to be associated with COVID-19 severity infection though the study was underpowered to make definitive conclusions (93).
Despite the abovementioned studies, the role of RBC heterogeneity remains understudied in critical illness. The collection and biobanking of RBC samples combined with further translational studies will be necessary to elucidate the immunomodulatory functions of RBCs during critical illness. Given the accumulating evidence for RBC immune function and the recognition that host immune heterogeneity underlies differential severity to numerous pathogens, it is crucial that further investigations promoting precision medicine include large-scale exploration of RBC heterogeneity. By examining RBC heterogeneity through the most powerful evolutionary pressure exerted on them—malaria—we may continue to uncover previously unknown complexities of RBC immune function, and therefore host immune diversity. Moreover, given that these adaptations have been shaped by pressures that are unequal across geography, ancestry, and environment, we must account for genetic ancestry and environment as potential interacting or confounding factors in addition to the socioeconomic disparities already under investigation. Collectively, investigation of adaptations to malaria may provide a framework through which to better understand this missing piece of host immune diversity, in the current pandemic and beyond.
GRANTS
This work was supported in part by the NIH Grant R21 AI166813 and by the Penn Center for Precision Medicine at the University of Pennsylvania (to N.S.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D., L.W., and N.S.M. conceived and designed research; J.D., L.W., and N.S.M. prepared figures; J.D., L.W., and N.S.M. drafted manuscript; J.D., L.W., and N.S.M. edited and revised manuscript; J.D., L.W., and N.S.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Kaitlyn Eckart and Julia Yuen for administrative assistance. Graphical abstract image created with BioRender.com and published with permission.
REFERENCES
- 1. Miller LH, Mason SJ, Dvorak JA, McGinniss MH, Rothman IK. Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science 189: 561–563, 1975. doi: 10.1126/science.1145213. [DOI] [PubMed] [Google Scholar]
- 2. Miller LH, Mason SJ, Clyde DF, McGinniss MH. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N Engl J Med 295: 302–304, 1976. doi: 10.1056/NEJM197608052950602. [DOI] [PubMed] [Google Scholar]
- 3. Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, Rebelo S, Henri A, Beuzard Y, Soares MP. Sickle hemoglobin confers tolerance to plasmodium infection. Cell 145: 398–409, 2011. doi: 10.1016/j.cell.2011.03.049. [DOI] [PubMed] [Google Scholar]
- 4. Akide-Ndunge OB, Ayi K, Arese P. The Haldane malaria hypothesis: facts, artifacts, and a prophecy. Redox Rep 8: 311–316, 2003. doi: 10.1179/135100003225002952. [DOI] [PubMed] [Google Scholar]
- 5. Allison AC. Protection afforded by sickle-cell trait against subtertian malareal infection. Br Med J 1: 290–294, 1954. doi: 10.1136/bmj.1.4857.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mawson AR. The pathogenesis of malaria: a new perspective. Pathog Glob Health 107: 122–129, 2013. doi: 10.1179/2047773213Y.0000000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anderson HL, Brodsky IE, Mangalmurti NS. The evolving erythrocyte: red blood cells as modulators of innate immunity. J Immunol 201: 1343–1351, 2018. doi: 10.4049/jimmunol.1800565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shamoun M, Callaghan M. Thalassemia. In: Benign Hematologic Disorders in Children: A Clinical Guide, edited by Kamat DM, Frei-Jones M.. Cham: Springer International Publishing, 2021, p. 91–98. [Google Scholar]
- 9. Vento S, Cainelli F, Cesario F. Infections and thalassaemia. Lancet Infect Dis 6: 226–233, 2006. doi: 10.1016/S1473-3099(06)70437-6. [DOI] [PubMed] [Google Scholar]
- 10. Farmakis D, Giakoumis A, Polymeropoulos E, Aessopos A. Pathogenetic aspects of immune deficiency associated with beta-thalassemia. Med Sci Monit 9: RA19–RA22, 2003. [PubMed] [Google Scholar]
- 11. Ricerca BM, Di Girolamo A, Rund D. Infections in thalassemia and hemoglobinopathies: focus on therapy-related complications. Mediterr J Hematol Infect Dis 1: e2009028, 2009. doi: 10.4084/MJHID.2009.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darbonne WC, Rice GC, Mohler MA, Apple T, Hébert CA, Valente AJ, Baker JB. Red blood cells are a sink for interleukin 8, a leukocyte chemotaxin. J Clin Invest 88: 1362–1369, 1991. doi: 10.1172/JCI115442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Horuk R, Chitnis CE, Darbonne WC, Colby TJ, Rybicki A, Hadley TJ, Miller LH. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science 261: 1182–1184, 1993. doi: 10.1126/science.7689250. [DOI] [PubMed] [Google Scholar]
- 14. Howes RE, Patil AP, Piel FB, Nyangiri OA, Kabaria CW, Gething PW, Zimmerman PA, Barnadas C, Beall CM, Gebremedhin A, Ménard D, Williams TN, Weatherall DJ, Hay SI. The global distribution of the Duffy blood group. Nat Commun 2: 266, 2011. doi: 10.1038/ncomms1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mangalmurti NS, Xiong Z, Hulver M, Ranganathan M, Liu XH, Oriss T, Fitzpatrick M, Rubin M, Triulzi D, Choi A, Lee JS. Loss of red cell chemokine scavenging promotes transfusion-related lung inflammation. Blood 113: 1158–1166, 2009. doi: 10.1182/blood-2008-07-166264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kangelaris KN, Sapru A, Calfee CS, Liu KD, Pawlikowska L, Witte JS, Vittinghoff E, Zhuo H, Auerbach AD, Ziv E, Matthay MA, National Heart L, Blood Institute AN; National Heart, Lung, and Blood Institute ARDS Network. The association between a Darc gene polymorphism and clinical outcomes in African American patients with acute lung injury. Chest 141: 1160–1169, 2012. doi: 10.1378/chest.11-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luzzatto L, Ally M, Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Blood 136: 1225–1240, 2020. doi: 10.1182/blood.2019000944. [DOI] [PubMed] [Google Scholar]
- 18. Louicharoen C, Patin E, Paul R, Nuchprayoon I, Witoonpanich B, Peerapittayamongkol C, Casademont I, Sura T, Laird NM, Singhasivanon P, Quintana-Murci L, Sakuntabhai A. Positively selected G6PD-Mahidol mutation reduces Plasmodium vivax density in Southeast Asians. Science 326: 1546–1549, 2009. doi: 10.1126/science.1178849. [DOI] [PubMed] [Google Scholar]
- 19. Youssef JG, Zahiruddin F, Youssef G, Padmanabhan S, Ensor J, Pingali SR, Zu Y, Sahay S, Iyer SP. G6PD deficiency and severity of COVID19 pneumonia and acute respiratory distress syndrome: tip of the iceberg? Ann Hematol 100: 667–673, 2021. doi: 10.1007/s00277-021-04395-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ayi K, Min-Oo G, Serghides L, Crockett M, Kirby-Allen M, Quirt I, Gros P, Kain KC. Pyruvate kinase deficiency and malaria. N Engl J Med 358: 1805–1810, 2008. doi: 10.1056/NEJMoa072464. [DOI] [PubMed] [Google Scholar]
- 21. Machado P, Manco L, Gomes C, Mendes C, Fernandes N, Salome G, Sitoe L, Chibute S, Langa J, Ribeiro L, Miranda J, Cano J, Pinto J, Amorim A, do Rosario VE, Arez AP. Pyruvate kinase deficiency in sub-Saharan Africa: identification of a highly frequent missense mutation (G829A;Glu277Lys) and association with malaria. PLoS One 7: e47071, 2012. doi: 10.1371/journal.pone.0047071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Amuzu DS, Rockett KA, Leffler EM, Ansah F, Amoako N, Morang’a CM, Hubbart C, Rowlands K, Jeffreys AE, Amenga-Etego LN, Kwiatkowski DP, Awandare GA. High-throughput genotyping assays for identification of glycophorin B deletion variants in population studies. Exp Biol Med (Maywood) 246: 916–928, 2021. doi: 10.1177/1535370220968545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leffler EM, Band G, Busby GBJ, Kivinen K, Le QS, Clarke GM, et al. Resistance to malaria through structural variation of red blood cell invasion receptors. Science 356: eaam6393, 2017. doi: 10.1126/science.aam6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rowe JA, Moulds JM, Newbold CI, Miller LH. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature 388: 292–295, 1997. doi: 10.1038/40888. [DOI] [PubMed] [Google Scholar]
- 25. Cockburn IA, Mackinnon MJ, O'Donnell A, Allen SJ, Moulds JM, Baisor M, Bockarie M, Reeder JC, Rowe JA. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proc Natl Acad Sci USA 101: 272–277, 2004. doi: 10.1073/pnas.0305306101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gandhi M. Complement receptor 1 and the molecular pathogenesis of malaria. Indian J Hum Genet 13: 39–47, 2007. doi: 10.4103/0971-6866.34704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kisserli A, Schneider N, Audonnet S, Tabary T, Goury A, Cousson J, Mahmoudi R, Bani-Sadr F, Kanagaratnam L, Jolly D, Cohen JH. Acquired decrease of the C3b/C4b receptor (CR1, CD35) and increased C4d deposits on erythrocytes from ICU COVID-19 patients. Immunobiology 226: 152093, 2021. doi: 10.1016/j.imbio.2021.152093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hotz MJ, Qing D, Shashaty MGS, Zhang P, Faust H, Sondheimer N, Rivella S, Worthen GS, Mangalmurti NS. Red blood cells homeostatically bind mitochondrial DNA through TLR9 to maintain quiescence and to prevent lung injury. Am J Respir Crit Care Med 197: 470–480, 2018. doi: 10.1164/rccm.201706-1161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lam LKM, Murphy S, Kokkinaki D, Venosa A, Sherrill-Mix S, Casu C, Rivella S, Weiner A, Park J, Shin S, Vaughan AE, Hahn BH, Odom John AR, Meyer NJ, Hunter CA, Worthen GS, Mangalmurti NS. DNA binding to TLR9 expressed by red blood cells promotes innate immune activation and anemia. Sci Transl Med 13: eabj1008, 2021. doi: 10.1126/scitranslmed.abj1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gowda NM, Wu X, Gowda DC. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J Immunol 188: 5073–5085, 2012. doi: 10.4049/jimmunol.1102143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hisaeda H, Tetsutani K, Imai T, Moriya C, Tu L, Hamano S, Duan X, Chou B, Ishida H, Aramaki A, Shen J, Ishii KJ, Coban C, Akira S, Takeda K, Yasutomo K, Torii M, Himeno K. Malaria parasites require TLR9 signaling for immune evasion by activating regulatory T cells. J Immunol 180: 2496–2503, 2008. doi: 10.4049/jimmunol.180.4.2496. [DOI] [PubMed] [Google Scholar]
- 32. Dean L. Blood Groups and Red Cell Antigens. Bethesda, MD: National Center for Biotechnology Information (US), 2005. [Google Scholar]
- 33. Pathirana SL, Alles HK, Bandara S, Phone-Kyaw M, Perera MK, Wickremasinghe AR, Mendis KN, Handunnetti SM. ABO-blood-group types and protection against severe, Plasmodium falciparum malaria. Ann Trop Med Parasitol 99: 119–124, 2005. doi: 10.1179/136485905X19946. [DOI] [PubMed] [Google Scholar]
- 34. Loscertales MP, Brabin BJ. ABO phenotypes and malaria related outcomes in mothers and babies in The Gambia: a role for histo-blood groups in placental malaria? Malar J 5: 72, 2006. doi: 10.1186/1475-2875-5-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lell B, May J, Schmidt-Ott RJ, Lehman LG, Luckner D, Greve B, Matousek P, Schmid D, Herbich K, Mockenhaupt FP, Meyer CG, Bienzle U, Kremsner PG. The role of red blood cell polymorphisms in resistance and susceptibility to malaria. Clin Infect Dis 28: 794–799, 1999. doi: 10.1086/515193. [DOI] [PubMed] [Google Scholar]
- 36. Fischer PR, Boone P. Short report: severe malaria associated with blood group. Am J Trop Med Hyg 58: 122–123, 1998. doi: 10.4269/ajtmh.1998.58.122. [DOI] [PubMed] [Google Scholar]
- 37. Reilly JP, Anderson BJ, Mangalmurti NS, Nguyen TD, Holena DN, Wu Q, Nguyen ET, Reilly MP, Lanken PN, Christie JD, Meyer NJ, Shashaty MG. The ABO histo-blood group and AKI in critically ill patients with trauma or sepsis. Clin J Am Soc Nephrol 10: 1911–1920, 2015. doi: 10.2215/CJN.12201214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rogers S, Doctor A. Red blood cell dysfunction in critical illness. Crit Care Clin 36: 267–292, 2020. doi: 10.1016/j.ccc.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cutbush M, Mollison PL. The Duffy blood group system. Heredity (Edinb) 4: 383–389, 1950. doi: 10.1038/hdy.1950.31. [DOI] [PubMed] [Google Scholar]
- 40. Horuk R. The Duffy antigen receptor for chemokines DARC/ACKR1. Front Immunol 6: 279, 2015. doi: 10.3389/fimmu.2015.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tournamille C. Molecular basis and structure-activity relationships of the Duffy blood group antigens: chemokine and Plasmodium vivax receptors. Transfus Clin Biol 7: 497–509, 2000. doi: 10.1016/s1246-7820(00)80038-5. [DOI] [PubMed] [Google Scholar]
- 42. Tournamille C, Filipe A, Wasniowska K, Gane P, Lisowska E, Cartron J-P, Colin Y, Le Van Kim C. Structure–function analysis of the extracellular domains of the Duffy antigen/receptor for chemokines: characterization of antibody and chemokine binding sites. Br J Haematol 122: 1014–1023, 2003. doi: 10.1046/j.1365-2141.2003.04533.x. [DOI] [PubMed] [Google Scholar]
- 43. Rot A. Contribution of Duffy antigen to chemokine function. Cytokine Growth Factor Rev 16: 687–694, 2005. doi: 10.1016/j.cytogfr.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 44. Pruenster M, Mudde L, Bombosi P, Dimitrova S, Zsak M, Middleton J, Richmond A, Graham GJ, Segerer S, Nibbs RJ, Rot A. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat Immunol 10: 101–108, 2009. [Erratum in Nat Immunol 10: 223, 2009]. doi: 10.1038/ni.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee JS, Wurfel MM, Matute-Bello G, Frevert CW, Rosengart MR, Ranganathan M, Wong VW, Holden T, Sutlief S, Richmond A, Peiper S, Martin TR. The Duffy antigen modifies systemic and local tissue chemokine responses following lipopolysaccharide stimulation. J Immunol 177: 8086–8094, 2006. doi: 10.4049/jimmunol.177.11.8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fukuma N, Akimitsu N, Hamamoto H, Kusuhara H, Sugiyama Y, Sekimizu K. A role of the Duffy antigen for the maintenance of plasma chemokine concentrations. Biochem Biophys Res Commun 303: 137–139, 2003. doi: 10.1016/s0006-291x(03)00293-6. [DOI] [PubMed] [Google Scholar]
- 47. Vlaar AP, Juffermans NP. Transfusion-related acute lung injury: a clinical review. Lancet 382: 984–994, 2013. doi: 10.1016/S0140-6736(12)62197-7. [DOI] [PubMed] [Google Scholar]
- 48. Mangla A, Ehsan M, Agarwal N, Maruvada S. Sickle cell anemia. In: StatPearls. Treasure Island, FL: StatPearls Publishing, 2022. https://www.ncbi.nlm.nih.gov/books/NBK482164/. [PubMed] [Google Scholar]
- 49. Henrici RC, Sautter CL, Bond C, Opoka RO, Namazzi R, Datta D, Ware RE, Conroy AL, John CC. Decreased parasite burden and altered host response in children with sickle cell anemia and severe anemia with malaria. Blood Adv 5: 4710–4720, 2021. doi: 10.1182/bloodadvances.2021004704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc 68: 263–299, 2018. doi: 10.3233/CH-189012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tumburu L, Ghosh-Choudhary S, Seifuddin FT, Barbu EA, Yang S, Ahmad MM, Wilkins LHW, Tunc I, Sivakumar I, Nichols JS, Dagur PK, Yang S, Almeida LEF, Quezado ZMN, Combs CA, Lindberg E, Bleck CKE, Zhu J, Shet AS, Chung JH, Pirooznia M, Thein SL. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood 137: 3116–3126, 2021. [Erratum in Blood 140: 1327, 2022]. doi: 10.1182/blood.2020009063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Caielli S, Cardenas J, de Jesus AA, Baisch J, Walters L, Blanck JP, Balasubramanian P, Stagnar C, Ohouo M, Hong S, Nassi L, Stewart K, Fuller J, Gu J, Banchereau JF, Wright T, Goldbach-Mansky R, Pascual V. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell 184: 4464–4479.e19, 2021. doi: 10.1016/j.cell.2021.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Faust HE, Reilly JP, Anderson BJ, Ittner CAG, Forker CM, Zhang P, Weaver BA, Holena DN, Lanken PN, Christie JD, Meyer NJ, Mangalmurti NS, Shashaty MGS. Plasma mitochondrial DNA levels are associated with ARDS in trauma and sepsis patients. Chest 157: 67–76, 2020. doi: 10.1016/j.chest.2019.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Verma A, Huffman JE, Gao L, Minnier J, Wu W-C, Cho K, et al. Association of kidney comorbidities and acute kidney failure with unfavorable outcomes after COVID-19 in individuals with the sickle cell trait. JAMA Intern Med 182: 796–804, 2022. doi: 10.1001/jamainternmed.2022.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Flint J, Hill AV, Bowden DK, Oppenheimer SJ, Sill PR, Serjeantson SW, Bana-Koiri J, Bhatia K, Alpers MP, Boyce AJ, Weatherall DJ, Clegg JB. High frequencies of alpha-thalassaemia are the result of natural selection by malaria. Nature 321: 744–750, 1986. doi: 10.1038/321744a0. [DOI] [PubMed] [Google Scholar]
- 56. Weatherall DJ, Clegg JB. Genetic disorders of hemoglobin. Semin Hematol 36: 24–37, 1999. [PubMed] [Google Scholar]
- 57. Allen SJ, O'Donnell A, Alexander ND, Alpers MP, Peto TE, Clegg JB, Weatherall DJ. alpha+-Thalassemia protects children against disease caused by other infections as well as malaria. Proc Natl Acad Sci USA 94: 14736–14741, 1997. doi: 10.1073/pnas.94.26.14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Luzzi GA, Merry AH, Newbold CI, Marsh K, Pasvol G, Weatherall DJ. Surface antigen expression on Plasmodium falciparum-infected erythrocytes is modified in alpha- and beta-thalassemia. J Exp Med 173: 785–791, 1991. doi: 10.1084/jem.173.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cserti CM, Dzik WH. The ABO blood group system and Plasmodium falciparum malaria. Blood 110: 2250–2258, 2007. doi: 10.1182/blood-2007-03-077602. [DOI] [PubMed] [Google Scholar]
- 60. Pathak V, Colah R, Ghosh K. Correlation between 'H' blood group antigen and Plasmodium falciparum invasion. Ann Hematol 95: 1067–1075, 2016. doi: 10.1007/s00277-016-2663-5. [DOI] [PubMed] [Google Scholar]
- 61. Rowe JA, Handel IG, Thera MA, Deans AM, Lyke KE, Koné A, Diallo DA, Raza A, Kai O, Marsh K, Plowe CV, Doumbo OK, Moulds JM. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc Natl Acad Sci USA 104: 17471–17476, 2007. doi: 10.1073/pnas.0705390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen Q, Heddini A, Barragan A, Fernandez V, Pearce SF, Wahlgren M. The semiconserved head structure of Plasmodium falciparum erythrocyte membrane protein 1 mediates binding to multiple independent host receptors. J Exp Med 192: 1–10, 2000. doi: 10.1084/jem.192.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Reilly JP, Meyer NJ, Shashaty MG, Feng R, Lanken PN, Gallop R, Kaplan S, Herlim M, Oz NL, Hiciano I, Campbell A, Holena DN, Reilly MP, Christie JD. ABO blood type A is associated with increased risk of ARDS in whites following both major trauma and severe sepsis. Chest 145: 753–761, 2014. doi: 10.1378/chest.13-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Reilly JP, Meyer NJ, Shashaty MG, Anderson BJ, Ittner C, Dunn TG, Lim B, Forker C, Bonk MP, Kotloff E, Feng R, Cantu E, Mangalmurti NS, Calfee CS, Matthay MA, Mikacenic C, Walley KR, Russell J, Christiani DC, Wurfel MM, Lanken PN, Reilly MP, Christie JD. The ABO histo-blood group, endothelial activation, and acute respiratory distress syndrome risk in critical illness. J Clin Invest 131: e139700, 2021. doi: 10.1172/JCI139700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zietz M, Zucker J, Tatonetti NP. Associations between blood type and COVID-19 infection, intubation, and death. Nat Commun 11: 5761, 2020. doi: 10.1038/s41467-020-19623-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Severe Covid-19 GWAS Group, Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, et al. Genomewide association study of severe COVID-19 with respiratory failure. N Engl J Med 383: 1522–1534, 2020. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Teeranaipong P, Ohashi J, Patarapotikul J, Kimura R, Nuchnoi P, Hananantachai H, Naka I, Putaporntip C, Jongwutiwes S, Tokunaga K. A functional single-nucleotide polymorphism in the CR1 promoter region contributes to protection against cerebral malaria. J Infect Dis 198: 1880–1891, 2008. doi: 10.1086/593338. [DOI] [PubMed] [Google Scholar]
- 68. Bain W, Li H, van der Geest R, Moore SR, Olonisakin TF, Ahn B, Papke E, Moghbeli K, DeSensi R, Rapport S, Saul M, Hulver M, Xiong Z, Mallampalli RK, Ray P, Morris A, Ma L, Doi Y, Zhang Y, Kitsios GD, Kulkarni HS, McVerry BJ, Ferreira VP, Nouraie M, Lee JS. Increased alternative complement pathway function and improved survival during critical illness. Am J Respir Crit Care Med 202: 230–240, 2020. doi: 10.1164/rccm.201910-2083OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lam LKM, Reilly JP, Rux AH, Murphy SJ, Kuri-Cervantes L, Weisman AR, Ittner CAG, Pampena MB, Betts MR, Wherry EJ, Song W-C, Lambris JD, Meyer NJ, Cines DB, Mangalmurti NS. Erythrocytes identify complement activation in patients with COVID-19. Am J Physiol Lung Cell Mol Physiol 321: L485–L489, 2021. doi: 10.1152/ajplung.00231.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci USA 104: 1919–1924, 2007. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Omar AH, Yasunami M, Yamazaki A, Shibata H, Ofori MF, Akanmori BD, Shuaibu MN, Kikuchi M, Hirayama K. Toll-like receptor 9 (TLR9) polymorphism associated with symptomatic malaria: a cohort study. Malar J 11: 168, 2012. doi: 10.1186/1475-2875-11-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sun L, Yu Y, Niu B, Wang D. Red blood cells as potential repositories of MicroRNAs in the circulatory system. Front Genet 11: 442, 2020. doi: 10.3389/fgene.2020.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Duraisingh MT, Lodish HF. Sickle cell microRNAs inhibit the malaria parasite. Cell Host Microbe 12: 127–128, 2012. doi: 10.1016/j.chom.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. LaMonte G, Philip N, Reardon J, Lacsina JR, Majoros W, Chapman L, Thornburg CD, Telen MJ, Ohler U, Nicchitta CV, Haystead T, Chi JT. Translocation of sickle cell erythrocyte microRNAs into Plasmodium falciparum inhibits parasite translation and contributes to malaria resistance. Cell Host Microbe 12: 187–199, 2012. doi: 10.1016/j.chom.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zou L, He J, Gu L, Shahror RA, Li Y, Cao T, Wang S, Zhu J, Huang H, Chen F, Fan X, Wu J, Chao W. Brain innate immune response via miRNA-TLR7 sensing in polymicrobial sepsis. Brain Behav Immun 100: 10–24, 2022. doi: 10.1016/j.bbi.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xu J, Feng Y, Jeyaram A, Jay SM, Zou L, Chao W. Circulating plasma extracellular vesicles from septic mice induce inflammation via microRNA- and TLR7-dependent mechanisms. J Immunol 201: 3392–3400, 2018. doi: 10.4049/jimmunol.1801008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu X, Wu C, Gu W, Ji H, Zhu L. Serum exosomal MicroRNAs predict acute respiratory distress syndrome events in patients with severe community-acquired pneumonia. Biomed Res Int 2019: 3612020, 2019. doi: 10.1155/2019/3612020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 78. Feng Z, Qi S, Zhang Y, Qi Z, Yan L, Zhou J, He F, Li Q, Yang Y, Chen Q, Xiao S, Li Q, Chen Y, Zhang Y. Ly6G+ neutrophil-derived miR-223 inhibits the NLRP3 inflammasome in mitochondrial DAMP-induced acute lung injury. Cell Death Dis 8: e3170–e3170, 2017. doi: 10.1038/cddis.2017.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jiang Z-F, Zhang L, Shen J. MicroRNA: potential biomarker and target of therapy in acute lung injury. Hum Exp Toxicol 39: 1429–1442, 2020. doi: 10.1177/0960327120926254. [DOI] [PubMed] [Google Scholar]
- 80. Kariuki SN, Marin-Menendez A, Introini V, Ravenhill BJ, Lin YC, Macharia A, Makale J, Tendwa M, Nyamu W, Kotar J, Carrasquilla M, Rowe JA, Rockett K, Kwiatkowski D, Weekes MP, Cicuta P, Williams TN, Rayner JC. Red blood cell tension protects against severe malaria in the Dantu blood group. Nature 585: 579–583, 2020. doi: 10.1038/s41586-020-2726-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Maitland K, Kiguli S, Opoka RO, Engoru C, Olupot-Olupot P, Akech SO, Nyeko R, Mtove G, Reyburn H, Lang T, Brent B, Evans JA, Tibenderana JK, Crawley J, Russell EC, Levin M, Babiker AG, Gibb DM, Group FT; FEAST Trial Group. Mortality after fluid bolus in African children with severe infection. N Engl J Med 364: 2483–2495, 2011. doi: 10.1056/NEJMoa1101549. [DOI] [PubMed] [Google Scholar]
- 82. Andrews B, Semler MW, Muchemwa L, Kelly P, Lakhi S, Heimburger DC, Mabula C, Bwalya M, Bernard GR. Effect of an early resuscitation protocol on in-hospital mortality among adults with sepsis and hypotension: a randomized clinical trial. JAMA 318: 1233–1240, 2017. doi: 10.1001/jama.2017.10913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jolley SE, Hough CL, Clermont G, Hayden D, Hou S, Schoenfeld D, Smith NL, Thompson BT, Bernard GR, Angus DC, Investigators AN; ARDS Network Investigators. Relationship between race and the effect of fluids on long-term mortality after acute respiratory distress syndrome. secondary analysis of the National Heart, Lung, and Blood Institute Fluid and Catheter Treatment Trial. Ann Am Thorac Soc 14: 1443–1449, 2017. doi: 10.1513/AnnalsATS.201611-906OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dellinger RP, Levy MM, Schorr CA, Townsend SR. 50 years of sepsis investigation/enlightenment among adults-the long and winding road. Crit Care Med 49: 1606–1625, 2021. doi: 10.1097/CCM.0000000000005203. [DOI] [PubMed] [Google Scholar]
- 85. Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, Rautanen A, Gordon AC, Garrard C, Hill AV, Hinds CJ, Knight JC. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med 4: 259–271, 2016. doi: 10.1016/S2213-2600(16)00046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Antcliffe DB, Burnham KL, Al-Beidh F, Santhakumaran S, Brett SJ, Hinds CJ, Ashby D, Knight JC, Gordon AC. Transcriptomic signatures in sepsis and a differential response to steroids. From the VANISH randomized trial. Am J Respir Crit Care Med 199: 980–986, 2019. [Erratum in Am J Respir Crit Care Med 206: 1572–1573, 2022]. doi: 10.1164/rccm.201807-1419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wong HR, Hart KW, Lindsell CJ, Sweeney TE. External corroboration that corticosteroids may be harmful to septic shock endotype A patients. Crit Care Med 49: e98–e101, 2021. doi: 10.1097/CCM.0000000000004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, Calfee CS; ARDS Network. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med 195: 331–338, 2017. doi: 10.1164/rccm.201603-0645OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sinha P, Delucchi KL, Thompson BT, McAuley DF, Matthay MA, Calfee CS; NHLBI ARDS Network. Latent class analysis of ARDS subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med 44: 1859–1869, 2018. doi: 10.1007/s00134-018-5378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sinha P, Furfaro D, Cummings MJ, Abrams D, Delucchi K, Maddali MV, He J, Thompson A, Murn M, Fountain J, Rosen A, Robbins-Juarez SY, Adan MA, Satish T, Madhavan M, Gupta A, Lyashchenko AK, Agerstrand C, Yip NH, Burkart KM, Beitler JR, Baldwin MR, Calfee CS, Brodie D, O'Donnell MR. Latent class analysis reveals COVID-19-related acute respiratory distress syndrome subgroups with differential responses to corticosteroids. Am J Respir Crit Care Med 204: 1274–1285, 2021. doi: 10.1164/rccm.202105-1302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Singh A, Brandow AM, Panepinto JA. COVID-19 in individuals with sickle cell disease/trait compared with other Black individuals. Blood Adv 5: 1915–1921, 2021. [Erratum in Blood Adv 6: 2535, 2022]. doi: 10.1182/bloodadvances.2020003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mahdi A, Collado A, Tengbom J, Jiao T, Wodaje T, Johansson N, Farnebo F, Färnert A, Yang J, Lundberg Jon O, Zhou Z, Pernow J. Erythrocytes induce vascular dysfunction in COVID-19. JACC Basic Transl Sci 7: 193–204, 2022. doi: 10.1016/j.jacbts.2021.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Conrado MCAV, Dezan MR, Oliveira VB, Ziza KC, Fanciscani T, Rocha V, Mendrone-Júnior A, Dinardo CL. Association between FY*02N.01 and the severity of COVID-19: initial observations. Hematol Transfus Cell Ther 44: 213–217, 2022. doi: 10.1016/j.htct.2022.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]