

Keywords: carbon tetrachloride, hepatic stellate cells, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, transforming growth factor β1
Abstract
Estrogen signaling is protective against chronic liver diseases, although men and a subset of women are contraindicated for chronic treatment with 17β-estradiol (17β-E2) or combination hormone replacement therapies. We sought to determine if 17α-estradiol (17α-E2), a naturally occurring diastereomer of 17β-E2, could attenuate liver fibrosis. We evaluated the effects of 17α-E2 treatment on collagen synthesis and degradation rates using tracer-based labeling approaches in male mice subjected to carbon tetrachloride (CCl4)-induced liver fibrosis. We also assessed the effects of 17α-E2 on markers of hepatic stellate cell (HSC) activation, collagen cross-linking, collagen degradation, and liver macrophage content and polarity. We found that 17α-E2 significantly reduced collagen synthesis rates and increased collagen degradation rates, which was mirrored by declines in transforming growth factor β1 (TGF-β1) and lysyl oxidase-like 2 (LOXL2) protein content in liver. These improvements were associated with increased matrix metalloproteinase 2 (MMP2) activity and suppressed stearoyl-coenzyme A desaturase 1 (SCD1) protein levels, the latter of which has been linked to the resolution of liver fibrosis. We also found that 17α-E2 increased liver fetuin-A protein, a strong inhibitor of TGF-β1 signaling, and reduced proinflammatory macrophage activation and cytokines expression in the liver. We conclude that 17α-E2 reduces fibrotic burden by suppressing HSC activation and enhancing collagen degradation mechanisms. Future studies will be needed to determine if 17α-E2 acts directly in hepatocytes, HSCs, and/or immune cells to elicit these benefits.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in the world (1). Nearly 30% of the population in westernized countries have NAFLD and this number continues to increase due to rising cases of obesity and type 2 diabetes (T2D) (2). NAFLD encompasses a spectrum of disorders ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), which is characterized by lobular inflammation and hepatocyte ballooning in the presence or absence of fibrosis (3). NASH is a major risk factor for progression to cirrhosis and hepatocellular carcinoma (4) and represents the fastest-growing indication for liver transplantation in the United States over the past 2 decades (5). The severity of fibrosis in the setting of NASH was recently found to be the best predictor of mortality in patients with NAFLD (6).
Liver fibrosis manifests as an excessive accumulation of extracellular matrix (ECM) proteins, predominantly collagen, in response to chronic liver injury. Hepatic stellate cells (HSCs), a residential perisinusoidal mesenchymal cell type, maintain ECM homeostasis in the liver. During liver injury, quiescent HSCs transdifferentiate into an activated state and develop a profibrogenic phenotype that promotes fibrosis if unabated (7). Activated HSCs also produce transforming growth factor β1 (TGF-β1), which maintains HSC activation and is therefore the dominant mechanistic driver of liver fibrosis (7–9). The severity of liver fibrosis is partially controlled by the degree of collagen crosslinking because it increases resistance to proteolytic degradation. Collagen crosslinking is regulated by the activity of lysyl oxidase-like 2 (LOXL2) (10, 11) in liver. The degradation of liver collagen by matrix metalloproteinases (MMPs) also plays a critical role in the control of collagen accumulation. Both MMP2 and MMP9 degrade collagen in the liver, although it remains debated if either can be targeted to enhance collagen degradation (12–17).
Notably, the prevalence and severity of NAFLD, NASH, and liver fibrosis are greater in men than women (18–20) and men are twofold more likely to die from chronic liver disease (21). However, the prevalence and severity of NAFLD, NASH, and liver fibrosis become comparable between the sexes following menopause, particularly when hormone replacement therapies (HRT) are not initiated (22). Moreover, the duration of estrogen deficiency in postmenopausal women is associated with greater severity of liver fibrosis (23). These studies suggest that estrogen signaling is protective against liver disease onset and progression. Studies in rodents support the idea that estrogens, particularly 17β-estradiol (17β-E2), are protective against hepatic lipid deposition and fibrogenesis (24–28). However, chronic administration of 17β-E2 or combination HRTs is unrealistic in men due to increased stroke risk (29), prostate cancer development (30), and feminization (31). A subset of the female population also develops side effects with chronic HRT administration including headaches, nausea, and increased risk for breast cancer and cardiovascular events (32, 33). These observations, coupled with the fact that no medications are currently approved in the United States or Europe for the treatment of NAFLD, NASH, or liver fibrosis (34, 35), have led to the investigation of nonfeminizing estrogens for the treatment of liver diseases. Both naturally occurring and synthetic nonfeminizing estrogens have been studied in recent years for their ability to elicit health benefits (36, 37).
17α-Estradiol (17α-E2), a naturally occurring diastereomer of 17β-E2 with considerably less binding affinity for classical estrogen receptors (ERα and ERβ), was recently shown to extend lifespan of male mice in a dose-dependent manner (38, 39). We and others have since reported that 17α-E2 treatment reduces adiposity and improves a myriad of systemic metabolic parameters in obese and old male mice without inducing overt feminization (40–45). In liver, 17α-E2 dramatically reduces steatosis and age-related DNA damage, whereas also significantly improving hepatic insulin sensitivity in an ERα-dependent manner (40, 46). We also found that 17α-E2 attenuates several histological and transcriptional markers of liver fibrosis in models of diet-induced obesity (46). Given that 17α-E2 dramatically reduces adiposity, it remains unclear if the benefits of 17α-E2 on liver fibrosis are secondary responses to the reversal of metabolic sequela associated with obesity, or if 17α-E2 has direct effects on fibrogenesis-related mechanisms.
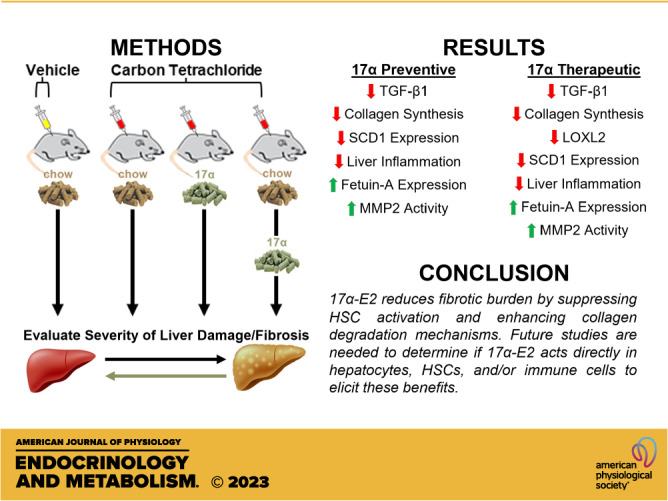
In the current study, we evaluated the effects of 17α-E2 treatment on collagen synthesis and degradation rates using novel tracer-based labeling approaches in male mice subjected to CCl4-induced liver fibrosis. We also assessed the effects of 17α-E2 on markers of HSC activation, collagen cross-linking, and collagen degradation. Finally, we also characterized the effects of 17α-E2 treatment of liver macrophage content and polarity due to their established role in exacerbating liver fibrosis (47, 48). We found that mice receiving 17α-E2 had significantly reduced collagen synthesis rates and greater collagen degradation rates, which was mirrored by TGF-β1 and LOXL2 protein content. These improvements were associated with increased MMP2 activity and suppressed stearoyl-coenyzme A desaturase 1 (SCD1) protein levels, the latter of which has been linked to fibrosis resolution (49, 50). We also found that 17α-E2 increased fetuin-A protein, a strong inhibitor of TGF-β1 signaling (51–55), and reduced macrophage activation and proinflammatory markers in the liver. We conclude that 17α-E2 acts in a multimodal fashion to reduce fibrotic burden in the liver.
METHODS
Animal Diets
TestDiet, a division of Purina Mills (Richmond, IN), prepared the diets for these studies. Chow-fed control animals received TestDiet 58YP (66.4% CHO, 20.5% PRO, 13.1% FAT), whereas mice treated with 17α-E2 received TestDiet 58YP that had been supplemented with 14.4 ppm of 17α-E2 (Steraloids, Newport, RI) during the manufacturing process.
Animal Experiments
Ten-week-old male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and were acclimatized to the Oklahoma City VA Health Care System vivarium for a period of 2 wk. Mice were individually housed with ISO cotton pad bedding, cardboard enrichment tubes, and nestlets at 22 ± 0.5°C on a 12:12-h light-dark cycle. Unless otherwise noted, all mice had ad libitum access to food and water throughout the experimental timeframe. At 12 wk of age, mice were randomized by body mass into one of five groups (Fig. 1): 1) short-term carbon tetrachloride (CCl4; n = 18): chow-fed, CCl4-treated twice weekly for 4 wk, 2) Vehicle (n = 15): chow-fed, vehicle-treated (olive oil) controls, 3) CCl4 (n = 15): chow-fed, CCl4-treated twice weekly for 8 wk, 4) CCl4 + 17α preventive (n = 18): chow + 17α-fed for 8 wk while simultaneously being CCl4-treated twice weekly for 8 wk, or 5) CCl4 + 17α therapeutic (n = 15): chow + 17α-fed for the final 4 wk while simultaneously being CCl4-treated twice weekly for 8 wk. CCl4 was administered as a 40% solution by intraperitoneal injection (1 µL/g body mass) on the first and fourth day of each week as previously described (56–58). We also administered deuterium oxide (D2O) in these studies so that collagen turnover rates could be evaluated. The Short-term CCl4 treatment group received 99% D2O intraperitoneal injections at day 0 to quickly enrich the body water pool (assumed to be 60% of body mass) to 5%, which was followed by 8% D2O in the drinking water to maintain a steady state of body water enrichment through the remainder of the labeling period (59). Mice in the Short-term CCl4 treatment group were euthanized at days 0, 4, 7, 14, 21, and 28 (n = 3/time point) over the 4-wk intervention period. This labeling strategy allowed us to determine how changes in collagen synthesis rates contribute to increases in collagen concentration over the first 4 wk of CCl4 administration. The Vehicle, CCl4, CCl4 + 17α preventive, and CCl4 + 17α therapeutic treatment groups were labeled for 28, 21, 14, 7, or 0 days (n = 3/time point/group) before the end of the 8-wk intervention. This labeling strategy allowed us to determine how CCl4 and 17α treatments impacted collagen synthesis and whether the treatments increased or decreased the pool of collagen that was resistant to turnover (i.e., fibrotic). At the conclusion of their respective interventional periods, mice were anesthetized with isoflurane and euthanized by exsanguination due to cardiac puncture. Blood was collected into EDTA-lined tubes, plasma was collected and frozen, and the mice were then perfused with ice-cold 1× PBS before tissues were excised, weighed, flash-frozen, and stored at −80°C unless otherwise noted. Following excision, small pieces of liver in the portal triad region were dissected and fixed in 4% PFA in preparation for paraffin- or cryo-embedding for future analyses. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the Oklahoma City VA Health Care System.
Figure 1.

Overview of experimental design. Water bottles represent when 99% D2O bolus intraperitoneal injections were provided and 8% D2O was added to the drinking water. Yellow and red syringes represent when vehicle and CCl4 injections were performed, respectively. CCl4, carbon tetrachloride; D2O, deuterium oxide.
Plasma Alanine and Aspartate Transaminase Analyses
Plasma alanine aminotransferase (ALT) and aspartate aminotransaminase (AST) were evaluated using commercially available colorimetric kits from BioAssay Systems (Hayward, CA).
Liver Triglyceride Analyses
Liver samples (∼50 mg) were homogenized on ice for 60 s in 10× (v/w) RIPA Buffer (Cell Signaling, Danvers, MA) with protease and phosphatase inhibitors (Boston BioProducts, Boston, MA). Total lipids were extracted from 100 µL of homogenate using the Folch method (60). Samples were dried under nitrogen gas at room temperature before being resuspended in 100 µL of tert-butyl alcohol-methanol-Triton X-100 solution (3:1:1). Triglycerides (TG) were quantified spectrophotometrically using Free Glycerol and Triglyceride agents (Sigma Aldrich, St. Louis, MO) as previously described (61).
Liver Histology and Pathology Assessments
Liver samples were fixed in 4% PFA for 24 h, transferred to 1× PBS for 48 h, and then transferred to 70% EtOH until paraffin embedding occurred. H&E and Masson’s trichrome staining were performed by the OMRF Imaging and Histology Core Facility using established protocols. Images of H&E and trichrome stained slides were taken on an Olympus CX43 microscope and were evaluated by two clinical pathologists who were blinded to the treatment groups. NAFLD activity scores (NAS) and fibrosis staging were determined according to NASH Clinical Research Network standards (62, 63).
Liver TGF-β1 Quantification
Liver samples (∼100 mg) were homogenized on ice for 60 s in 10× (v/w) RIPA Buffer (Cell Signaling) with protease and phosphatase inhibitors (Boston BioProducts). The homogenates were spun at 17,000 rpm for 15 min at 4°C and the supernatant was collected. Total protein was then quantified using a Pierce bicinchoninic acid (BCA) kit (ThermoFisher Scientific, Waltham, MA). Supernatant (20 µL) was then diluted with TGF-β1 ELISA (Abcam, Waltham, MA) assay buffer (180 µL), which was then digested with 1 N HCl (20 µL) for 60 min at room temperature. The samples were then neutralized with 1 N NaOH (20 µL) and were then further diluted with ELISA assay buffer to a total volume of 1,200 µL (1:60 dilution). The samples were then evaluated according to the manufacturer’s instructions. TGF-β1 concentrations were normalized to total protein and expressed as nanogram/milligram protein.
Collagen Extraction and Isotopic Labeling Analyses
We performed collagen isolation and deuterium isotopic enrichment of collagen according to our previously published methods (59). Extracted collagen was derivatized for analysis of deuterium enrichment of alanine using Gas Chromatography-Mass Spectroscopy (7890 A GC-Agilent 5975 C MS, Agilent, Santa Clara, CA). To determine the precursor pool enrichment, plasma samples were prepared for analysis of deuterium enrichment on a liquid water isotope analyzer (Los Gatos Research, Los Gatos, CA). The precursor enrichment of alanine was then calculated by mass isotopomer distribution analysis. The deuterium enrichments of both the protein (product) and the precursor were used to calculate fraction new: Fraction new = Eproduct/Eprecursor, where the Eproduct is the enrichment (E) of protein-bound alanine and Eprecursor is the calculated maximum alanine enrichment from equilibration of the body water pool. The fraction of new data was then plotted across the time points and curves were fit to the data using one-phase associations. Two parameters of interest were then calculated from the curves using GraphPad Prism 9 and the one-phase association function. The software calculates rate parameter (k, 1/day), which reflects the protein synthetic rate, and plateau fraction new (p), which represents the proportion of the protein pool that is actively turning over (i.e., the dynamic protein pool), with 1.0 equal to 100% of the protein pool. Finally, rates of synthesis were multiplied by the concentration of collagen to calculate absolute rates of protein turnover.
Hydroxyproline Analyses
Powdered liver samples (∼200 mg) were evaluated for hydroxyproline content as previously described (64). In brief, samples were hydrolyzed in 500 µL of 6 M HCl at 105°C overnight. Samples were then vigorously vortexed and 10 µL of hydrolysate was mixed with 150 μL of isopropanol and 75 μL of acetate citrate buffer containing 1.4% chloramine‐T, were vigorously vortexed, and then left to oxide for 10 min at room temperature Samples were then mixed with 1 mL of a 3:13 solution of Ehrlich reagent [1.5 g of 4-(dimethylamino)benzaldehyde, 5 mL ethanol, 337-μL sulfuric acid] to isopropanol and incubated for 45 min at 55°C. Quantification was determined by extinction measurement of the resulting solution at 558 nm. A standard curve (0–1,000 μM, trans-4-hydroxy-l-proline) was included in each assay. Results are reported as microgram hydroxyproline/mg tissue.
Quantitative Real-Time PCR
Total RNA was extracted using TRIzol (Life Technologies, Carlsbad, CA) and RNA (2 µg) was reverse transcribed to cDNA with the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Real-time PCR was performed in a QuantStudio 12 K Flex Real-Time PCR System (ThermoFisher Scientific, Waltham, MA) using TaqMan Gene Expression Master Mix (ThermoFisher Scientific) and predesigned gene expression assays with FAM probes from Integrated DNA Technologies (Skokie, IL). Target gene expression was expressed as 2−ΔΔCT by the comparative CT method (65) and normalized to the expression of TATA-box binding protein (TBP).
Western Blot Analyses
Liver samples (∼100 mg) were homogenized on ice for 60 s in 10× (v/w) RIPA Buffer (Cell Signaling) with protease and phosphatase inhibitors (Boston BioProducts). The homogenates were spun at 17,000 rpm for 15 min at 4°C and the supernatant was collected. Total protein was then quantified using a Pierce BCA kit (ThermoFisher Scientific). Proteins were separated on Any kD Criterion TGX Stain-Free gels (Bio-Rad, Hercules, CA) at 75 V for 150 min in a running buffer (Cell Signaling). Protein was then transferred to 0.2 µm pore-size nitrocellulose membranes (Bio-Rad) at 75 V for 90 min on ice. All primary antibodies utilized have been commercially validated and include MMP-9 (Novus Biologicals, Centennial, CO; 1:1,000), MMP-2 (Cell Signaling; 1:1,000), TIMP-1 (Sino Biological, Houston, TX; 1:2,000), LOXL2 (Novus Biologicals; 1:2,000), PPARγ (Cell Signaling; 1:1,000), SCD-1 (Cell Signaling; 1:1,000), Fetuin-A (Abcam, 1:2,000), p65 (Cell Signaling; 1:1,000), phospho-p65 (serine536; Cell Signaling; 1:1,000), and GAPDH (Abcam; 1:2,500). Primary antibody detection was performed with IRDye 800CW Infrared Rabbit antibody (LI-COR Biotechnology, Lincoln, NE) at a concentration of 1:15,000. Only antibodies GAPDH was diluted in 5% dry milk (Cell Signaling) and all other antibodies were diluted in 5% BSA (Cell Signaling). Imaging was done on an Odyssey Fc Imaging System (LI-COR Biotechnology) with a 2-min exposure time at 800λ. Protein quantification was performed using Image Studio Software (LI-COR Biotechnology).
Gelatin Zymography Analyses
Activity of MMP-9 and MMP-2 were determined by gelatin zymography as described previously (17) with slight modifications. In brief, resolving gels were made with 4.6-mL sterile distilled water, 2.7-mL 30% acrylamide, 2.5-mL 1.5 M Tris (pH 8.8), 100-µL 10% SDS, 285-µL 8 mg/mL bovine skin type B gelatin, 6 µL TEMED, and 100-µL 10% APS to reach 8% acrylamide concentrations. Stacking gels were made with 3.4-mL sterile distilled water, 830-µL 30% acrylamide, 630-µL 1 M Tris (pH 6.8), 50-µL 10% SDS, 5-µL TEMED, and 50-µL 10% APS. Liver samples (∼100 mg) were homogenized on ice for 60 s in 10× (v/w) RIPA Buffer (Cell Signaling) with protease and phosphatase inhibitors (Boston BioProducts). The homogenates were spun at 17,000 rpm for 15 min at 4°C and the supernatant was collected. Total protein was then quantified using a Pierce BCA kit (ThermoFisher Scientific, Waltham, MA). Samples were prepared using nonreducing sample buffer and were loaded (20 μg/well) onto the gelatin gel along with recombinant MMP9 (Abcam) and MMP2 (BioLegend, San Diego, CA) as a standard control and run at 75 V for 150 min in running buffer (Cell Signaling). The gel was carefully removed and washed with Novex renaturing buffer (ThermoFisher Scientific) for 30 min at room temperature and was then washed with Novex developing buffer (ThermoFisher Scientific) for 30 min. Gels were then placed in Novex developing buffer and incubated for 16 h at 37°C. After incubation, gels were stained with Coomassie Brilliant Blue solution (0.5 g Brilliant Blue, 250 mL methanol, 100 mL acetic acid, 150 mL sterile distilled water) for 1 h, then washed with destaining solution (400 mL methanol, 100 mL acetic acid, and 500 mL sterile distilled water) until transparent bands were visualized in the blue background. Gels were then scanned and band intensity representing MMP activity was calculated using ImageJ densitometry software.
Immunofluorescence Analyses
Cryoembedded liver samples were sectioned (10 µm) and stained with primary antibodies against F4/80 (total macrophages; USBiological Life Sciences; 1:250), CD11c (M1: proinflammatory macrophages; Invitrogen; 1:300), CD206 (M2: anti-inflammatory macrophages; Cell Signaling; 1:500), and α smooth muscle actin (αSMA; activated HSCs; Novus; 1:500) as previously described (66). Secondary antibodies used include goat anti-Armenian hamster IgG Alexa Fluor 594 (Jackson ImmunoResearch Laboratories, West Grove, PA; 1:500), goat anti-chicken IgG Alexa Fluor 647 (Jackson ImmunoResearch Laboratories; 1:500), and goat anti-rabbit IgG Alexa Fluor 488 (Jackson ImmunoResearch Laboratories; 1:500). Sections were mounted in Prolong Diamond Mounting Medium with DAPI (Abcam) and images were acquired using a Leica 3 D Thunder scope from three nonintersecting fields per mouse. Intensity of fluorescence was measured as percent of total area using ImageJ after each image had its background intensity subtracted out.
Statistical Analyses
Results are presented as means ± SE with P values less than 0.05 considered significantly different. Analyses of differences between groups were performed by paired Student’s t test or one-way ANOVA with Tukey’s post hoc comparisons where appropriate using GraphPad Prism Software, Version 9.
RESULTS
17α-E2 Treatment Attenuates Markers of Liver Injury
To investigate the effects of 17α-E2 on the development of liver fibrosis in mice, we administered CCl4 to induce hepatotoxic stress while also providing 17α-E2 for the entire duration of the 8-wk study (preventive group) or the final 4 wk of the study (therapeutic group). Consistent with our previous reports (40–43, 46, 67, 68), 17α-E2 treatment reduced body mass at the conclusion of the study in both the preventive and therapeutic groups (Fig. 2A). As expected, CCl4 administration resulted in liver damage as evidenced by increased circulating ALT and AST, which were significantly blunted in both the preventive and therapeutic groups following 17α-E2 treatment (Fig. 2, B and C). Liver mass and TG content were mildly increased by CCl4 administration (Fig. 2, D and E), which was prevented by 8 wk of 17α-E2 treatment but not 4 wk of therapeutic treatment (Fig. 2E). Pathological liver assessment confirmed liver damage, with CCl4 administration dramatically increasing the NASH Activity Score (NAS) and Brunt Fibrosis Score as compared to vehicle-treated controls (Fig. 2, F and G). Interestingly, 17α-E2 treatment only improved steatosis and inflammation in the preventive group, whereas both the preventive and therapeutic groups displayed declines in the severity of fibrosis following treatment (Fig. 2, F and G). Based on these observations, we subsequently evaluated the effects of 17α-E2 treatment on ECM homeostasis.
Figure 2.

17α-E2 treatment attenuates markers of liver injury. A: body mass at baseline (week 0; striped) and the conclusion (solid) of the 8-wk intervention (n = 15–19/group). Plasma ALT (n = 9–12/group; B), plasma AST (n = 6–14/group; C), liver mass (n = 15–19/group; D), liver TG [n = 11–17/group; E), and liver pathological NASH Activity and Brunt Fibrosis Scoring at the conclusion of the 8-wk intervention (n = 10–14/group; F). G: representative images of gross morphology, H&E stained (magnification = ×20; scale bar = 50 μm), and Masson’s trichrome stained (magnification = ×20; scale bar = 50 μm) liver at the conclusion of the 8-wk intervention. All data are shown as means ± SE and were analyzed by paired Student’s t test within treatment group (A) or one-way ANOVA with Tukey’s post hoc testing (B–F). We did not indicate statistical differences between vehicle and 17α-E2 treatment groups (preventive and therapeutic), or between 17α-E2 treatment groups (preventive and therapeutic), for purposes of visual clarity. *P < 0.05, **P < 0.01, ***P < 0.005. ALT, alanine aminotransferase; AST, aspartate aminotransaminase; NASH, nonalcoholic steatohepatitis; TG, triglycerides; 17α-E2, 17α-estradiol.
17α-E2 Treatment Suppresses Collagen Synthesis in the Liver
Similar to previous reports (69–72), CCl4 administration significantly increased hepatic TGF-β1 production (Fig. 3A) and suppressed cytochrome P450 2e1 (Cyp2e1) expression (Supplemental Fig. S1). Cyp2e1 mediates the hepatotoxicity of CCl4 by metabolizing it to the free radical trichloromethyl (69, 73). We also determined that CCl4 administration significantly increased hepatic αSMA production (Supplemental Fig. S2), which is highly expressed in activated HSCs (74). These observations indicate that HSCs had become activated and developed a profibrogenic phenotype. Interestingly, 17α-E2 treatment prevented and/or reversed liver αSMA and TGF-β1 production in the preventive and therapeutic groups, respectively (Supplemental Fig. S2; Fig. 3A). These observations suggest that 17α-E2 treatment likely reverses HSC activation and/or promotes apoptosis, which is known to occur during the resolution of liver fibrosis (75). Notably, Cyp2e1 expression was unchanged by 17α-E2 treatment, which suggests CCl4 metabolism is unchanged by 17α-E2. Using stable isotope labeling techniques, we evaluated liver collagen synthesis rates. In alignment with the increase in TGF-β1 production, CCl4 administration robustly increased the absolute rate of collagen synthesis by nearly threefold (Fig. 3B). The preventive and therapeutic 17α-E2 treatment groups displayed collagen synthesis rates that were half of the CCl4 treatment group, indicating a possible prevention of an increase or a renormalization of synthesis rates (Fig. 3B). Liver hydroxyproline, a marker of collagen content (76), increased following 4 (Supplemental Fig. S3) and 8 wk of CCl4 administration (Fig. 3C). 17α-E2 treatment significantly reduced hydroxyproline levels in the therapeutic group, but only induced a downward trend in the preventive group (Fig. 3C). By our stable isotope labeling approach, we were able to determine how much of the collagen pool was resistant to turnover (static), which we have previously shown to be indicative of a fibrotic phenotype (59). As anticipated, the percentage of collagen in the static pool was increased with CCl4 administration (Fig. 3D), as was the absolute amount of collagen in both the static and dynamic pools (Fig. 3E). Introducing 17α-E2 treatment at different phases of fibrosis development led to differential responses in collagen turnover dynamics. The preventive treatment group displayed a higher percentage of dynamic collagen compared with the CCl4 treatment group (Fig. 3D), but the overall difference in absolute collagen in both the static and dynamic pools was not found to be statistically different (Fig. 3E). Conversely, the therapeutic group displayed a higher percentage of collagen in the static pool compared with the CCl4 treatment group (Fig. 3D), although the absolute amount of collagen in the static pool was unchanged (Fig. 3E). This shorter duration of 17α-E2 treatment (4 wk) in the therapeutic group dramatically reduced the absolute amount of collagen in the dynamic pool, which was reflective of the overall lower collagen concentration (Fig. 3E). Collectively, these findings suggest that depending on the timing of administration, 17α-E2 treatment may slow the progression of fibrosis or resolve the collagen pool that is not cross linked. Therefore, we next sought to determine if 17α-E2 treatment alters the expression and/or activity of LOXL2 and MMPs.
Figure 3.

17α-E2 treatment suppresses collagen synthesis and fibrotic collagen deposition in liver. A: liver TGF-β1 at the conclusion of the 8-wk intervention (n = 13–17/group). B: collagen synthesis rates during the 8-wk intervention (n = 14–18/group). C: liver TGF-β1 hydroxyproline content at the conclusion of the 8-wk intervention (n = 14–19/group). Fractional percent (n = 15–18/group; D) and absolute content (n = 15–18/group; E) of static and dynamic collagen in liver at the conclusion of the 8-wk intervention. All data are shown as means ± SE and were analyzed by one-way ANOVA with Tukey’s post hoc testing. We did not indicate statistical differences between vehicle and 17α-E2 treatment groups (preventive and therapeutic), or between 17α-E2 treatment groups (preventive and therapeutic), for purposes of visual clarity. *P < 0.05, **P < 0.01, ***P < 0.005. TGF-β1, transforming growth factor β1; 17α-E2, 17α-estradiol.
17α-E2 Treatment Suppresses LOXL2 Expression and Increased MMP2 Activity
To determine if the declines in collagen synthesis following 17α-E2 treatment were due to changes in collagen crosslinking and/or degradation mechanisms, we first evaluated LOXL2 expression due to its key role in fibrotic matrix stabilization (11). It should also be noted that previous reports have established that the magnitude of LOXL2 expression is indicative of activity, and thus, an increase in crosslinked collagen fibrils (77). In alignment with previous reports (78, 79), CCl4 administration significantly increased LOXL2 expression at both the transcript and protein level (Fig. 4, A–C), indicating a higher level of collagen cross linking is occurring. 17α-E2 treatment did not alter Loxl2 transcription but did mildly suppress LOXL2 protein in the therapeutic group (Fig. 4, A–C), suggesting that a decline in collagen cross linking may be a minor mechanism by which 17α-E2 reduces liver fibrosis. We subsequently evaluated MMP expression and activity due to their critical roles in controlling collagen degradation. We focused most of our efforts on analyzing MMP2 and MMP9 because they control the final step in collagen degradation (7). We also evaluated the expression of tissue inhibitor matrix metalloproteinase 1 (TIMP1) due to its established role in inhibiting MMP activity (17). We found that CCl4 administration increased Mmp2 and Timp1 transcripts, but did not alter Mmp9 mRNA expression (Fig. 5, A–C). CCl4-mediated liver fibrosis was also associated with increased transcription of Mmp8, Mmp12, Mmp13, Mmp14, Mmp16, and Mmp19 (Supplemental Fig. S3). 17α-E2 treatment did not alter the transcription of any MMP we analyzed or Timp1 (Fig. 5, A–C; Supplemental Fig. S4). To our surprise, CCl4 administration did not alter MMP2 or MMP9 protein expression but did promote declines in TIMP1 protein (Fig. 5, D–G). 17α-E2 treatment mildly increased MMP2 protein expression, preferentially in the therapeutic group, but failed to modulate MMP9 or TIMP1 protein levels (Fig. 5, D–G). We next sought to determine if MMP2 or MMP9 activity was altered. As expected (12, 80–82), CCl4 administration dramatically increased MMP2 and MMP9 activity as determined by gelatin zymography (Fig. 5, H–J). Interestingly, 17α-E2 treatment further increased MMP2 activity in both the preventive and therapeutic groups (Fig. 5, H–J), indicating that declines in liver collagen by 17α-E2 are at least partially mediated through degradation mechanisms.
Figure 4.

17α-E2 treatment reduces the collagen cross-linking enzyme LOXL2 in liver. Liver Loxl2 mRNA (n = 9–12/group; A) and LOXL2 protein (n = 14/group; B) at the conclusion of the 8-wk intervention. C: representative immunoblots of liver LOXL2 and GAPDH at the conclusion of the 8-wk intervention. All data are shown as means ± SE and were analyzed by one-way ANOVA with Tukey’s post hoc testing. We did not indicate statistical differences between vehicle and 17α-E2 treatment groups (preventive and therapeutic), or between 17α-E2 treatment groups (preventive and therapeutic), for purposes of visual clarity. *P < 0.05, ***P < 0.005. LOXL2, lysyl oxidase-like 2; 17α-E2, 17α-estradiol.
Figure 5.

17α-E2 treatment increases collagen degradation by modulating MMP2 activity in liver. Liver Mmp2 mRNA (n = 9–12/group; A), Mmp9 mRNA (n = 9–12/group; B), Timp1 mRNA (n = 8–11/group; C), MMP2 protein (n = 12–17/group; D), MMP9 protein (n = 12–17/group; E), and TIMP1 protein (n = 12–17/group; F) at the conclusion of the 8-wk intervention. G: representative immunoblots of liver MMP2, MMP9, TIMP1, and GAPDH at the conclusion of the 8-wk intervention. Liver MMP2 (n = 10–14/group; H) and MMP9 (n = 11–14/group; I) enzymatic activity at the conclusion of the 8-wk intervention. J: representative images of liver gelatin zymography assessing MMP2 and MMP9 enzymatic activity. Signal intensity for each sample was normalized to the intensity of their corresponding positive control standard. All data are shown as means ± SE and were analyzed by one-way ANOVA with Tukey’s post hoc testing. We did not indicate statistical differences between vehicle and 17α-E2 treatment groups (preventive and therapeutic), or between 17α-E2 treatment groups (preventive and therapeutic), for purposes of visual clarity. *P < 0.05, **P < 0.01, ***P < 0.005. MMP, matrix metalloproteinase; TIMP1, tissue inhibitor matrix metalloproteinase 1; 17α-E2, 17α-estradiol.
17α-E2 Treatment Suppresses Hepatic SCD1 Expression and Increases Fetuin-A Expression
To determine if the declines in collagen synthesis following 17α-E2 treatment were due to changes in mechanisms linked to the resolution of liver fibrosis, we evaluated peroxisome proliferator-activated receptor-γ (PPARγ), SCD1, and fetuin-A expression. Several reports have established that PPARγ actively inhibits HSC activation and that its expression and activity are reduced once HSC activation occurs (83–87). Moreover, mice with HSC-specific PPARγ disruption display defects in the resolution of liver fibrosis following the discontinuation of hepatotoxic treatments (88). We found that CCl4 administration did not alter Pparγ transcription or translation in whole liver, whereas 17α-E2 treatment increased Pparγ mRNA and protein in the preventive group only (Fig. 6, A and C). In contrast to PPARγ, CCl4 administration significantly increased Scd1 transcripts and protein (Fig 6, B and D). SCD1 is the rate-limiting enzyme that catalyzes the formation of monounsaturated fatty acids and its association with liver steatosis and fibrosis has been known for years (50, 89–92). 17α-E2 treatment significantly reduced Scd1 transcripts in the preventive group (Fig. 6B), which was mirrored by a robust downregulation of SCD1 protein in the preventive group (Fig. 6D). Interestingly, the therapeutic 17α-E2 treatment group also displayed a trending reduction in SCD1 protein level (Fig. 6D), suggesting that a longer treatment duration may prove beneficial when intervening in the context of established liver fibrosis. We also evaluated fetuin-A expression due to it being a strong inhibitor of TGF-β1 signaling (51–55). We found that CCl4 administration did not alter fetuin-A protein levels in the liver but that both the preventive and therapeutic 17α-E2 treatment groups displayed increased levels of fetuin-A (Fig. 7, A and B). Collectively, these observations suggest that 17α-E2 treatment modulates several pathways linked to the prevention and/or resolution of liver fibrosis.
Figure 6.

17α-E2 treatment beneficially modulates fibrosis resolution genes, PPARγ and SCD1, in liver. Liver Pparγ mRNA (n = 9–12/group; A), Scd1 mRNA (n = 9–12/group; B), PPARγ protein (n = 12–17/group; C), and SCD1 protein (n = 8–14/group; D) at the conclusion of the 8-wk intervention. E: representative immunoblots of liver PPARγ, SCD1, and GAPDH at the conclusion of the 8-wk intervention. All data are shown as means ± SE and were analyzed by one-way ANOVA with Tukey’s post hoc testing. We did not indicate statistical differences between vehicle and 17α-E2 treatment groups (preventive and therapeutic), or between 17α-E2 treatment groups (preventive and therapeutic), for purposes of visual clarity. *P < 0.05, **P < 0.01, ***P < 0.005. PPARγ, peroxisome proliferator-activated receptor-γ; SCD1, stearoyl-coenzyme A desaturase 1; 17α-E2, 17α-estradiol.
Figure 7.

17α-E2 treatment increases fetuin-A, a potent inhibitor of TGF-β1, in liver. A: liver fetuin-A protein at the conclusion of the 8-wk intervention (n = 12–17/group). B: representative immunoblots of liver fetuin-A and GAPDH at the conclusion of the 8-wk intervention. All data are shown as means ± SE and were analyzed by one-way ANOVA with Tukey’s post hoc testing. We did not indicate statistical differences between vehicle and 17α-E2 treatment groups (preventive and therapeutic), or between 17α-E2 treatment groups (preventive and therapeutic), for purposes of visual clarity. *P < 0.05, **P < 0.01. TGF-β1, transforming growth factor β1; 17α-E2, 17α-estradiol.
17α-E2 Treatment Suppresses Hepatic Inflammation by Altering Macrophage Content and Polarity
Given the established role of macrophages in the progression and resolution of liver fibrosis (47, 48), we sought to determine if the aforementioned improvements in liver injury with 17α-E2 treatment were associated with changes in macrophage content and polarity in liver. In alignment with previous reports (66, 93), CCl4 administration significantly increased total macrophage (F4/80) content in the liver by over twofold (Fig. 8, A and B), while also increasing M1 proinflammatory macrophage (CD11c) content by over 10-fold (Fig. 8, A and C). These changes were mirrored by increased transcription of F4/80, C-C motif chemokine ligand 5 (Ccl5), and C-X-C motif chemokine ligand 2 (Cxcl2; Supplemental Fig. S5, A–C), the latter two of which are chemokines linked to liver fibrosis severity (94, 95). Liver expression of cytokines, tumor necrosis factor-α (Tnfα), and interleukin 1 β (Il1β), were upregulated by CCl4 administration but only Tnfα was found to be significantly increased (Supplemental Fig. S5, D and E). These observations suggest that both resident and recruited macrophages were activated in response to CCl4 administration, which has been established previously (47, 48). 17α-E2 treatment reduced total macrophage and M1 proinflammatory macrophage content in the liver in both treatment groups (Fig. 8, A–C), although these reductions were only found to be significantly different in the preventive treatment group; suggesting that 17α-E2 treatment duration likely plays a major role in the suppression of proinflammatory macrophage activity. Interestingly, CCl4 administration also mildly increased M2 anti-inflammatory macrophage content in the liver (Fig. 8, A and D), which has previously been described as a compensatory response to severe liver injury (93, 96). 17α-E2 treatment further increased M2 anti-inflammatory macrophage content in the liver by nearly twofold in both the preventive and therapeutic groups (Fig. 8, A and D), indicating that 17α-E2 robustly alters liver macrophage polarity within a few weeks of treatment. 17α-E2 treatment also suppressed F4/80, Ccl5, Cxcl2, Tnfα, and Il1β mRNA to varying degrees in both the preventive and therapeutic groups (Supplemental Fig. S5, A–E). These observations suggest that the resolution of liver fibrosis with 17α-E2 treatment may be at least partially mediated through actions in immune cells. For additional confirmation that 17α-E2 reduced the liver proinflammatory milieu, we evaluated the activation of the NF-κB subunit p65, which is commonly upregulated in chronic liver injury and fibrosis (97, 98). We found that CCl4 administration increased liver p65 serine536 phosphorylation by over threefold (Fig. 8, E and F), which represents the phosphorylation site with the most potent inducible response to inflammatory stimuli (99). 17α-E2 completely reversed this induction in both the preventive and therapeutic treatment groups (Fig. 8, E and F), indicating that 17α-E2 can reverse liver inflammation within a few weeks of treatment. Collectively, these observations suggest that 17α-E2 treatment may attenuate liver fibrosis by suppressing proinflammatory responses in the liver.
Figure 8.

17α-E2 treatment attenuates macrophage infiltration and proinflammatory responses in liver. A: representative immunofluorescence images of F4/80 (total macrophages), CD11c (M1: proinflammatory macrophages), and CD206 (M2: anti-inflammatory macrophages) in liver at the conclusion of the 8-wk intervention (magnification = ×320; scale bar = 50 μm). Percentage of liver area for F4/80 (n = 5–7/group; B), CD11c (n = 5–7/group; C), and CD206 (n = 5–7/group; D) at the conclusion of the 8-wk intervention. E: ratio of liver phospho to total p65 at the conclusion of the 8-wk intervention (n = 12–16/group). F: representative immunoblots of liver phosphor p65, total p65, and GAPDH at the conclusion of the 8-wk intervention. All data are shown as means ± SE were analyzed by one-way ANOVA with Tukey’s post hoc testing. We did not indicate statistical differences between vehicle and 17α-E2 treatment groups (preventive and therapeutic), or between 17α-E2 treatment groups (preventive and therapeutic), for purposes of visual clarity. *P < 0.05, ***P < 0.005. 17α-E2, 17α-estradiol.
DISCUSSION
17α-E2 is a naturally occurring diastereomer of 17β-E2 that ameliorates metabolic dysfunction in obese and aged male mice (40–42, 46, 68), which we surmise underlies its lifespan-extending effects (38, 39). We have previously reported that 17α-E2 reverses hepatic steatosis and improves hepatic insulin sensitivity in an ERα-dependent manner (46). Given that hepatic steatosis and insulin resistance are directly linked to the onset of liver fibrosis (16, 35), this study aimed to determine if 17α-E2 treatment could beneficially modulate fibrogenesis-related mechanisms in the absence of obesity and hepatic steatosis. Herein, we clearly demonstrate that preventive and therapeutic 17α-E2 treatments attenuate or reverse the progression of liver fibrosis by modulating collagen turnover. Collectively, our data suggest that 17α-E2 likely acts in a multimodal fashion through several cell types to attenuate liver injury and fibrosis.
One of the most important findings from this study was the observation that 17α-E2 significantly suppressed collagen synthesis in the setting of chronic liver injury without hepatic steatosis. This indicates that 17α-E2 reduces fibrogenesis through mechanisms other than the reduction of lipotoxicity. One such potential mechanism is the prevention and/or reversal of HSC activation, which is supported by our data showing declines in liver TGF-β1 production and increased liver fetuin-A production following 17α-E2 treatment. Fetuin-A antagonizes TGF-β1 binding to its receptor and modulates downstream Smad pathway activity, thereby inhibiting HSC activation (52–54). Potential HSC inactivation by 17α-E2 directly, or through fetuin-A-mediated mechanisms, is further supported by the data generated through labeling of liver collagen, which revealed that 17α-E2 treatment predominantly reduced the dynamic pool of collagen in the liver. This finding suggests that collagen production by HSCs was attenuated and that the degree of collagen cross linking of LOXL2 was also reduced. This was particularly evident in the therapeutic treatment group that received 17α-E2 for the final 4 wk of the intervention period. Interestingly, the therapeutic treatment group did not show declines in the static pool of liver collagen, indicating that 4 wk of 17α-E2 treatment is insufficient to induce the unwinding and degradation of stabile collagen fibrils. This is not incredibly surprising given the resistance of stabile collagen fibrils to proteolytic degradation (100), coupled with the fact that hepatic injury due to CCl4 administration was ongoing during 17α-E2 treatment. Conversely, the preventive treatment group, which received 17α-E2 for the entire 8-wk intervention period, displayed trending reductions in both the dynamic and static collagen pools. It is unclear if this observation indicates that concomitant 17α-E2 treatment reduced the level of stabile collagen fibril accumulation, or increased unwinding and degradation of cross-linked collagen fibrils. We speculate that the longer duration of 17α-E2 treatment in the preventive group likely did both as evidenced by reduced collagen synthesis rates and increased MMP2 activity. Future studies will be needed to determine if 17α-E2 reduces liver collagen production by directly inducing apoptosis or inactivation of HSCs, through fetuin-A-related mechanisms, or if 17α-E2 attenuates hepatocyte stress and/or immune cell activation and proinflammatory responses, which in turn reduce HSC activation.
Another interesting outcome from our study was the observation that 17α-E2 increased liver PPARγ in the absence of hepatic steatosis. The change in PPARγ, which was more robust in the preventive treatment group, has implications for the inactivation of HSCs and suppression of proinflammatory activity in monocytes and macrophages. Previous studies have established that PPARγ is highly expressed in quiescent HSCs and that its activity promotes apoptosis of activated HSCs and differentiation toward a quiescent phenotype (85, 101). PPARγ actions in monocytes and macrophages are similarly antifibrotic. PPARγ activation in M1 proinflammatory macrophages is anti-inflammatory by repressing NF-κB activity and the expression of downstream response genes (102). PPARγ activation in monocytes promotes their differentiation into M2 anti-inflammatory macrophages (103). Moreover, macrophage-specific deletion of PPARγ increases liver fibrosis and inflammation in mice subjected to CCl4 administration (84). Given that PPARγ activity in hepatocytes promotes steatosis (104), coupled with the fact that 17α-E2 reduces hepatic lipid deposition, we surmise that one of the mechanisms by which 17α-E2 attenuates liver fibrosis is by increasing PPARγ activity in HSCs and/or macrophages. It remains unclear if 17α-E2 directly modulates PPARγ activity in HSCs or monocytes/macrophages but cross talk between estrogen receptors (ERα and ERβ) and PPARγ has been documented previously (105–107), although most studies suggest antagonist interactions. We have previously reported that 17α-E2 can elicit genomic activity of ERβ (40), which is dominant to HSCs (108, 109), and ERα (40, 46), which is dominant to monocytes/macrophages (110, 111). However, it remains unresolved if the actions of 17α-E2 through ERα, and potentially ERβ, are through genomic or nongenomic mechanisms. Future studies will be needed to determine if PPARγ modulation by 17α-E2 is direct and if this occurs in HSCs, monocytes/macrophages, or both.
Our study also revealed that 17α-E2 treatment reduced liver SCD1 in the absence of hepatic steatosis. The suppression of SCD1 in hepatocytes is known to reduce steatosis (89, 91, 92) and 17β-E2 is reported to be a negative regulator of SCD1 expression (112–114). This suggests that declines in hepatic lipid accumulation with 17α-E2 treatment, which we also observed in the current study, is at least partially mediated through suppression of SCD1 in hepatocytes. What remains unclear is if 17α-E2 also directly suppresses SCD1 in HSCs, which was recently linked to an inhibition of fibrogenesis (50). As addressed earlier, HSCs express ERβ and 17α-E2 can elicit genomic activity through this receptor, therefore future studies should explore the possibility that 17α-E2 attenuates liver fibrosis through direct actions in HSCs.
Finally, our study revealed that 17α-E2 treatment elicited beneficial effects on liver macrophage content and polarity. Macrophages play a critical role in regulating the progression and resolution of liver fibrosis due to their plasticity and heterogeneity (47, 48, 115). Macrophage phenotypes, or polarization, are driven by the local microenvironment and are broadly categorized as M1 proinflammatory or M2 anti-inflammatory (48, 116), although several subtypes of both M1 and M2 polarization states have been described (117, 118). Our study clearly demonstrates that 17α-E2 treatment reduces CCl4-mediated increases in total and M1 proinflammatory macrophages in liver, in addition to dramatically suppressing NF-κB activity and the expression of downstream response genes. Furthermore, 17α-E2 treatment increased M2 anti-inflammatory macrophages in liver, which have recently been shown to protect hepatocytes against apoptosis and necroptosis (93, 96). Interestingly, 17β-E2 is widely reported to promote macrophage polarization toward an M2 anti-inflammatory state in an ERα-dependent manner (119–121). As alluded to above, we recently reported that 17α-E2 elicits beneficial metabolic effects through ERα (46), therefore we surmise that 17α-E2 directly modulates macrophage polarization through ERα. However, other mechanisms may also be contributing to the change in macrophage phenotype following 17α-E2 treatment, including the potential modulation by PPARγ and fetuin-A. PPARγ (102, 103) and fetuin-A (122–124) have been reported to suppress macrophage proinflammatory activity and promote M2 polarization and both were upregulated in liver by 17α-E2. At this juncture it remains unclear if 17α-E2 modulates PPARγ expression in macrophages, or if the rise in liver fetuin-A plays a mechanistic role in the shift toward M2 anti-inflammatory macrophage content following 17α-E2 treatment. Given the wealth of data indicating that ERα agonism in macrophages robustly alters the polarity state, we speculate that direct signaling of 17α-E2 through ERα in macrophages represents the dominant mechanism by which 17α-E2 alters proinflammatory outcomes. Future studies will be needed to determine if 17α-E2 directly modulates macrophage polarity to promote the resolution of liver fibrosis or if the changes in proinflammatory outcomes occur as secondary responses to 17α-E2 actions in hepatocytes and/or HSCs.
There are a few notable caveats to the current study that should be acknowledged. First, the primary limitation is that we are currently unable to determine if the benefits of 17α-E2 on liver fibrosis are largely mediated through a single cell type, hepatocytes, HSCs, or macrophages. Although our data suggest that 17α-E2 likely elicits benefits through direct actions in all three cell types, the current data set does not allow us to make definitive conclusions due to the significant amount of crosstalk between the aforementioned cell types in the setting of liver injury and fibrosis. Future studies utilizing hepatocyte-, HSC-, and macrophage-specific deletions of ERα or ERβ will provide tremendous insight into the effects of 17α-E2 on the prevention and/or resolution of liver fibrosis. Another potential avenue of investigation that remains unresolved is whether 17α-E2 acts predominantly through genomic or nongenomic mechanisms to elicit health benefits. We have previously reported that genomic actions of 17α-E2 and 17β-E2 through ERα are essentially identical in vitro (46), but additional studies evaluating nongenomic actions of 17α-E2 in hepatocytes and/or macrophages are still needed. Another minor limitation of our design and labeling approaches is that they did not allow us to determine if chronic 17α-E2 treatment increased the unwinding and degradation of static collagen, or if it prevents the cross linking of dynamic collagen, thereby reducing static collagen accumulation. Having a greater understanding of how 17α-E2 alters collagen synthesis and degradation mechanisms will provide insight into the translatability of 17α-E2 into humans with liver fibrosis. Finally, although CCl4 administration is an established model of liver fibrogenesis, it does not completely recapitulate the human NASH and liver fibrosis (35). Future studies utilizing a combination approach of western diet and CCl4 administration should be undertaken to determine if 17α-E2 also elicits benefits in a disease state that more closely resembles human NASH and liver fibrosis (125). We speculate that 17α-E2 will elicit even greater benefits in the model employing Western diet and CCL4 administration due to our previous work establishing that 17α-E2 reduces hepatic steatosis and insulin resistance (40, 46).
In summary, the data presented herein are the first to show that 17α-E2 reduces liver fibrosis by attenuating collagen synthesis and enhancing collagen degradation mechanisms. Our data suggests that 17α-E2 acts directly on hepatocytes, HSCs, and macrophages to attenuate liver fibrosis, although additional studies will be needed to confirm this suspicion. Our current study provides critical insight into how nonfeminizing estrogen compounds may have therapeutic potential for the treatment of chronic liver diseases.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental data associated with this article can be found at https://doi.org/10.6084/m9.figshare.21719378.v1.
GRANTS
This work was supported by the National Institutes of Health grants (R01 AG059430 to W.M.F., R56 AG067754 to B.F.M., and R01 AG070035 to M.B.S.) and the US Department of Veterans Affairs (I01 BX003906 and ISI BX004797 to W.M.F., Pilot Research Funding to M.B.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.A.M., B.F.M., and M.B.S. conceived and designed research; S.A.M., R.S., S.N.M., M.K., W.L., T.D.S., J.V.V.I., F.F.P.3rd., T.L., W.M.F., B.F.M., and M.B.S. performed experiments; S.A.M., R.S., S.N.M., M.K., W.L., T.D.S., J.V.V.I., F.F.P.3rd., T.L., W.M.F., B.F.M., and M.B.S. analyzed data; S.A.M., R.S., S.N.M., M.K., W.L., T.D.S., J.V.V.I., F.F.P.3rd., T.L., W.M.F., B.F.M., and M.B.S. interpreted results of experiments; S.A.M., R.S., and M.B.S. prepared figures; S.A.M. and M.B.S. drafted manuscript; S.A.M., R.S., S.N.M., M.K., W.L., T.D.S., J.V.V.I., F.F.P.3rd., T.L., W.M.F., B.F.M., and M.B.S. edited and revised manuscript; S.A.M., R.S., S.N.M., M.K., W.L., T.D.S., J.V.V.I., F.F.P.3rd., T.L., W.M.F., B.F.M., and M.B.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Catelyn Jones and Kennedy Gosnell for animal colony management and Claire Abbott for assistance with sample preparation.
REFERENCES
- 1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64: 73–84, 2016. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 2. Mantovani A, Byrne CD, Bonora E, Targher G. Nonalcoholic fatty liver disease and risk of incident type 2 diabetes: a meta-analysis. Diabetes Care 41: 372–382, 2018. doi: 10.2337/dc17-1902. [DOI] [PubMed] [Google Scholar]
- 3. Chen YY, Yeh MM. Non-alcoholic fatty liver disease: a review with clinical and pathological correlation. J Formos Med Assoc 120: 68–77, 2021. doi: 10.1016/j.jfma.2020.07.006. [DOI] [PubMed] [Google Scholar]
- 4. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43: S99–S112, 2006. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 5. Lazarus JV, Anstee QM, Hagström H, Cusi K, Cortez-Pinto H, Mark HE, Roden M, Tsochatzis EA, Wong VW, Younossi ZM, Zelber-Sagi S, Romero-Gómez M, Schattenberg JM. Defining comprehensive models of care for NAFLD. Nat Rev Gastroenterol Hepatol 18: 717–729, 2021. doi: 10.1038/s41575-021-00477-7. [DOI] [PubMed] [Google Scholar]
- 6. Hagstrom H, Nasr P, Ekstedt M, Hammar U, Stal P, Hultcrantz R, Kechagias S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol 67: 1265–1273, 2017. doi: 10.1016/j.jhep.2017.07.027. [DOI] [PubMed] [Google Scholar]
- 7. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158: 1913–1928, 2020. doi: 10.1053/j.gastro.2019.11.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: crosstalk with metabolism. JHEP Rep 1: 30–43, 2019. doi: 10.1016/j.jhepr.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 14: 397–411, 2017. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 10. Shao M, Ye Z, Qin Y, Wu T. Abnormal metabolic processes involved in the pathogenesis of non-alcoholic fatty liver disease (Review). Exp Ther Med 20: 26, 2020. doi: 10.3892/etm.2020.9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, Mikels-Vigdal A, Smith V, Schuppan D, Popov YV. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 66: 1697–1708, 2017. doi: 10.1136/gutjnl-2016-312473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geervliet E, Bansal R. Matrix metalloproteinases as potential biomarkers and therapeutic targets in liver diseases. Cells 9: 1212, 2020. doi: 10.3390/cells9051212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robert S, Gicquel T, Victoni T, Valenca S, Barreto E, Bailly-Maitre B, Boichot E, Lagente V. Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci Rep 36: e00360, 2016. doi: 10.1042/BSR20160107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang C, Zeisberg M, Mosterman B, Sudhakar A, Yerramalla U, Holthaus K, Xu L, Eng F, Afdhal N, Kalluri R. Liver fibrosis: insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology 124: 147–159, 2003. doi: 10.1053/gast.2003.50012. [DOI] [PubMed] [Google Scholar]
- 15. Friedman SL. Mac the knife? Macrophages—the double-edged sword of hepatic fibrosis. J Clin Invest 115: 29–32, 2005. doi: 10.1172/JCI23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov 19: 57–75, 2020. doi: 10.1038/s41573-019-0040-5. [DOI] [PubMed] [Google Scholar]
- 17. Lachowski D, Cortes E, Rice A, Pinato D, Rombouts K, Del Rio Hernandez A. Matrix stiffness modulates the activity of MMP-9 and TIMP-1 in hepatic stellate cells to perpetuate fibrosis. Sci Rep 9: 7299, 2019. doi: 10.1038/s41598-019-43759-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morán-Costoya A, Proenza AM, Gianotti M, Lladó I, Valle A. Sex differences in nonalcoholic fatty liver disease: estrogen influence on the liver-adipose tissue crosstalk. Antioxid Redox Signal 35: 753–774, 2021. doi: 10.1089/ars.2021.0044. [DOI] [PubMed] [Google Scholar]
- 19. Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, Abdelmalek MF, Suzuki A. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology 70: 1457–1469, 2019. doi: 10.1002/hep.30626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang JD, Abdelmalek MF, Pang H, Guy CD, Smith AD, Diehl AM, Suzuki A. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology 59: 1406–1414, 2014. doi: 10.1002/hep.26761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guy J, Peters MG. Liver disease in women: the influence of gender on epidemiology, natural history, and patient outcomes. Gastroenterol Hepatol (N Y) 9: 633–639, 2013. [PMC free article] [PubMed] [Google Scholar]
- 22. Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology 122: 1649–1657, 2002. doi: 10.1053/gast.2002.33573. [DOI] [PubMed] [Google Scholar]
- 23. Klair JS, Yang JD, Abdelmalek MF, Guy CD, Gill RM, Yates K, Unalp-Arida A, Lavine JE, Clark JM, Diehl AM, Suzuki A; Nonalcoholic Steatohepatitis Clinical Research Network. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology 64: 85–91, 2016. doi: 10.1002/hep.28514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu JW, Gong J, Chang XM, Luo JY, Dong L, Jia A, Xu GP. Effects of estradiol on liver estrogen receptor-alpha and its mRNA expression in hepatic fibrosis in rats. World J Gastroenterol 10: 250–254, 2004. doi: 10.3748/wjg.v10.i2.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu JW, Gong J, Chang XM, Luo JY, Dong L, Hao ZM, Jia A, Xu GP. Estrogen reduces CCL4- induced liver fibrosis in rats. World J Gastroenterol 8: 883–887, 2002. doi: 10.3748/wjg.v8.i5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Y, Wu L, Wang Y, Zhang M, Li L, Zhu D, Li X, Gu H, Zhang CY, Zen K. Protective role of estrogen-induced miRNA-29 expression in carbon tetrachloride-induced mouse liver injury. J Biol Chem 287: 14851–14862, 2012. doi: 10.1074/jbc.M111.314922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yasuda M, Shimizu I, Shiba M, Ito S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 29: 719–727, 1999. doi: 10.1002/hep.510290307. [DOI] [PubMed] [Google Scholar]
- 28. Hewitt KN, Pratis K, Jones ME, Simpson ER. Estrogen replacement reverses the hepatic steatosis phenotype in the male aromatase knockout mouse. Endocrinology 145: 1842–1848, 2004. doi: 10.1210/en.2003-1369. [DOI] [PubMed] [Google Scholar]
- 29. Abbott RD, Launer LJ, Rodriguez BL, Ross GW, Wilson PW, Masaki KH, Strozyk D, Curb JD, Yano K, Popper JS, Petrovitch H. Serum estradiol and risk of stroke in elderly men. Neurology 68: 563–568, 2007. doi: 10.1212/01.wnl.0000254473.88647.ca. [DOI] [PubMed] [Google Scholar]
- 30. Nelles JL, Hu WY, Prins GS. Estrogen action and prostate cancer. Expert Rev Endocrinol Metab 6: 437–451, 2011. doi: 10.1586/eem.11.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stratakis CA, Vottero A, Brodie A, Kirschner LS, DeAtkine D, Lu Q, Yue W, Mitsiades CS, Flor AW, Chrousos GP. The aromatase excess syndrome is associated with feminization of both sexes and autosomal dominant transmission of aberrant P450 aromatase gene transcription. J Clin Endocrinol Metab 83: 1348–1357, 1998. doi: 10.1210/jcem.83.4.4697. [DOI] [PubMed] [Google Scholar]
- 32. Howard BV, Rossouw JE. Estrogens and cardiovascular disease risk revisited: the women's health initiative. Curr Opin Lipidol 24: 493–499, 2013. doi: 10.1097/MOL.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dalal PK, Agarwal M. Postmenopausal syndrome. Indian J Psychiatry 57: S222–S232, 2015. doi: 10.4103/0019-5545.161483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Konerman MA, Jones JC, Harrison SA. Pharmacotherapy for NASH: current and emerging. J Hepatol 68: 362–375, 2018. [Erratum in J Hepatol 68: 1337, 2018]. doi: 10.1016/j.jhep.2017.10.015. [DOI] [PubMed] [Google Scholar]
- 35. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24: 908–922, 2018. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Engler-Chiurazzi EB, Covey DF, Simpkins JW. A novel mechanism of non-feminizing estrogens in neuroprotection. Exp Gerontol 94: 99–102, 2017. doi: 10.1016/j.exger.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Madak-Erdogan Z, Kim SH, Gong P, Zhao YC, Zhang H, Chambliss KL, Carlson KE, Mayne CG, Shaul PW, Korach KS, Katzenellenbogen JA, Katzenellenbogen BS. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci Signal 9: ra53, 2016. doi: 10.1126/scisignal.aad8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Strong R, Miller RA, Antebi A, Astle CM, Bogue M, Denzel MS, Fernandez E, Flurkey K, Hamilton KL, Lamming DW, Javors MA, de Magalhães JP, Martinez PA, McCord JM, Miller BF, Müller M, Nelson JF, Ndukum J, Rainger GE, Richardson A, Sabatini DM, Salmon AB, Simpkins JW, Steegenga WT, Nadon NL, Harrison DE. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 15: 872–884, 2016. doi: 10.1111/acel.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harrison DE, Strong R, Allison DB, Ames BN, Astle CM, Atamna H, Fernandez E, Flurkey K, Javors MA, Nadon NL, Nelson JF, Pletcher S, Simpkins JW, Smith D, Wilkinson JE, Miller RA. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging cell 13: 273–282, 2014. doi: 10.1111/acel.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stout MB, Steyn FJ, Jurczak MJ, Camporez JG, Zhu Y, Hawse JR, Jurk D, Palmer AK, Xu M, Pirtskhalava T, Evans GL, de Souza Santos R, Frank AP, White TA, Monroe DG, Singh RJ, Casaclang-Verzosa G, Miller JD, Clegg DJ, LeBrasseur NK, von Zglinicki T, Shulman GI, Tchkonia T, Kirkland JL. 17α-estradiol alleviates age-related metabolic and inflammatory dysfunction in male mice without inducing feminization. J Gerontol A Biol Sci Med Sci 72: 3–15, 2017. doi: 10.1093/gerona/glv309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steyn FJ, Ngo ST, Chen VP, Bailey-Downs LC, Xie TY, Ghadami M, Brimijoin S, Freeman WM, Rubinstein M, Low MJ, Stout MB. 17α-Estradiol acts through hypothalamic pro-opiomelanocortin expressing neurons to reduce feeding behavior. Aging Cell 17: e12703, 2018. doi: 10.1111/acel.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sidhom S, Schneider A, Fang Y, McFadden S, Darcy J, Sathiaseelan R, Palmer AK, Steyn FJ, Grillari J, Kopchick JJ, Bartke A, Siddiqi S, Masternak MM, Stout MB. 17α-Estradiol modulates IGF1 and hepatic gene expression in a sex-specific manner. J Gerontol A Biol Sci Med Sci 76: 778–785, 2021. doi: 10.1093/gerona/glaa215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Isola JVV, Veiga GB, de Brito CRC, Alvarado-Rincón JA, Garcia DN, Zanini BM, Hense JD, Vieira AD, Garratt M, Gasperin BG, Schneider A, Stout MB. 17α-estradiol does not adversely affect sperm parameters or fertility in male mice: implications for reproduction-longevity trade-offs. Geroscience 2022. doi: 10.1007/s11357-022-00601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garratt M, Bower B, Garcia GG, Miller RA. Sex differences in lifespan extension with acarbose and 17-α estradiol: gonadal hormones underlie male-specific improvements in glucose tolerance and mTORC2 signaling. Aging Cell 16: 1256–1266, 2017. doi: 10.1111/acel.12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garratt M, Lagerborg KA, Tsai YM, Galecki A, Jain M, Miller RA. Male lifespan extension with 17-α estradiol is linked to a sex-specific metabolomic response modulated by gonadal hormones in mice. Aging Cell 17: e12786, 2018. doi: 10.1111/acel.12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mann SN, Hadad N, Nelson Holte M, Rothman AR, Sathiaseelan R, Ali Mondal S, Agbaga MP, Unnikrishnan A, Subramaniam M, Hawse J, Huffman DM, Freeman WM, Stout MB. Health benefits attributed to 17α-estradiol, a lifespan-extending compound, are mediated through estrogen receptor alpha. eLife 9: e59616, 2020. doi: 10.7554/eLife.59616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, Schuppan D, Grønbæk H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 16: 145–159, 2019. doi: 10.1038/s41575-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 48. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol 60: 1090–1096, 2014. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 49. Iruarrizaga-Lejarreta M, Varela-Rey M, Fernández-Ramos D, Martínez-Arranz I, Delgado TC, Simon J, Juan VG, delaCruz-Villar L, Azkargorta M, Lavin JL, Mayo R, Van Liempd SM, Aurrekoetxea I, Buqué X, Cave DD, Peña A, Rodriguez-Cuesta J, Aransay AM, Elortza F, Falcón-Pérez JM, Aspichueta P, Hayardeny L, Noureddin M, Sanyal AJ, Alonso C, Anguita J, Martínez-Chantar ML, Lu SC, Mato JM. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun 1: 911–927, 2017. doi: 10.1002/hep4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bhattacharya D, Basta B, Mato JM, Craig A, Fernández-Ramos D, Lopitz-Otsoa F, Tsvirkun D, Hayardeny L, Chandar V, Schwartz RE, Villanueva A, Friedman SL. Aramchol downregulates stearoyl CoA-desaturase 1 in hepatic stellate cells to attenuate cellular fibrogenesis. JHEP Rep 3: 100237, 2021. doi: 10.1016/j.jhepr.2021.100237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rittenberg B, Partridge E, Baker G, Clokie C, Zohar R, Dennis JW, Tenenbaum HC. Regulation of BMP-induced ectopic bone formation by Ahsg. J Orthop Res 23: 653–662, 2005. doi: 10.1016/j.orthres.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 52. Verma-Gandhu M, Peterson MR, Peterson TC. Effect of fetuin, a TGFbeta antagonist and pentoxifylline, a cytokine antagonist on hepatic stellate cell function and fibrotic parameters in fibrosis. Eur J Pharmacol 572: 220–227, 2007. doi: 10.1016/j.ejphar.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 53. Zhou Y, Yang S, Zhang P. Effect of exogenous fetuin-A on TGF-β/Smad signaling in hepatic stellate cells. Biomed Res Int 2016: 8462615, 2016. doi: 10.1155/2016/8462615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Demetriou M, Binkert C, Sukhu B, Tenenbaum HC, Dennis JW. Fetuin/alpha2-HS glycoprotein is a transforming growth factor-beta type II receptor mimic and cytokine antagonist. J Biol Chem 271: 12755–12761, 1996. doi: 10.1074/jbc.271.22.12755. [DOI] [PubMed] [Google Scholar]
- 55. Swallow CJ, Partridge EA, Macmillan JC, Tajirian T, DiGuglielmo GM, Hay K, Szweras M, Jahnen-Dechent W, Wrana JL, Redston M, Gallinger S, Dennis JW. alpha2HS-glycoprotein, an antagonist of transforming growth factor beta in vivo, inhibits intestinal tumor progression. Cancer Res 64: 6402–6409, 2004. doi: 10.1158/0008-5472.CAN-04-1117. [DOI] [PubMed] [Google Scholar]
- 56. Bisht S, Khan MA, Bekhit M, Bai H, Cornish T, Mizuma M, Rudek MA, Zhao M, Maitra A, Ray B, Lahiri D, Maitra A, Anders RA. A polymeric nanoparticle formulation of curcumin (NanoCurc) ameliorates CCl4-induced hepatic injury and fibrosis through reduction of pro-inflammatory cytokines and stellate cell activation. Lab Invest 91: 1383–1395, 2011. doi: 10.1038/labinvest.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kovalovich K, DeAngelis RA, Li W, Furth EE, Ciliberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology 31: 149–159, 2000. doi: 10.1002/hep.510310123. [DOI] [PubMed] [Google Scholar]
- 58. Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 282: 23337–23347, 2007. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 59. Abbott CB, Lawrence MM, Kobak KA, Lopes EBP, Peelor FF 3rd, Donald EJ, Van Remmen H, Griffin TM, Miller BF. A novel stable isotope approach demonstrates surprising degree of age-related decline in skeletal muscle collagen proteostasis. Function (Oxf) 2: zqab028, 2021. doi: 10.1093/function/zqab028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: 497–509, 1957. [PubMed] [Google Scholar]
- 61. Stout MB, Liu LF, Belury MA. Hepatic steatosis by dietary-conjugated linoleic acid is accompanied by accumulation of diacylglycerol and increased membrane-associated protein kinase C ε in mice. Mol Nutr Food Res 55: 1010–1017, 2011. doi: 10.1002/mnfr.201000413. [DOI] [PubMed] [Google Scholar]
- 62. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149: 389–397.e10, 2015. doi: 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal; Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321, 2005. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 64. Smith LR, Hammers DW, Sweeney HL, Barton ER. Increased collagen cross-linking is a signature of dystrophin-deficient muscle. Muscle Nerve 54: 71–78, 2016. doi: 10.1002/mus.24998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 66. Itoh M, Suganami T, Kato H, Kanai S, Shirakawa I, Sakai T, Goto T, Asakawa M, Hidaka I, Sakugawa H, Ohnishi K, Komohara Y, Asano K, Sakaida I, Tanaka M, Ogawa Y. CD11c+ resident macrophages drive hepatocyte death-triggered liver fibrosis in a murine model of nonalcoholic steatohepatitis. JCI Insight 2: e92902, 2017. doi: 10.1172/jci.insight.92902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mann SN, Pitel KS, Nelson-Holte MH, Iwaniec UT, Turner RT, Sathiaseelan R, Kirkland JL, Schneider A, Morris KT, Malayannan S, Hawse JR, Stout MB. 17α-Estradiol prevents ovariectomy-mediated obesity and bone loss. Exp Gerontol 142: 111113, 2020. doi: 10.1016/j.exger.2020.111113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Miller BF, Pharaoh GA, Hamilton KL, Peelor FF, Kirkland JL, Freeman WM, Mann SN, Kinter M, Price JC, Stout MB. Short-term calorie restriction and 17α-estradiol administration elicit divergent effects on proteostatic processes and protein content in metabolically active tissues. J Gerontol A Biol Sci Med Sci 75: 849–857, 2020. doi: 10.1093/gerona/glz113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu J, Ma HY, Liang S, Sun M, Karin G, Koyama Y, Hu R, Quehenberger O, Davidson NO, Dennis EA, Kisseleva T, Brenner DA. The role of human cytochrome P450 2E1 in liver inflammation and fibrosis. Hepatol Commun 1: 1043–1057, 2017. doi: 10.1002/hep4.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, Haslett C, Simpson KJ, Sethi T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA 103: 5060–5065, 2006. doi: 10.1073/pnas.0511167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yao QY, Xu BL, Wang JY, Liu HC, Zhang SC, Tu CT. Inhibition by curcumin of multiple sites of the transforming growth factor-β1 signalling pathway ameliorates the progression of liver fibrosis induced by carbon tetrachloride in rats. BMC Complement Altern Med 12: 156, 2012. doi: 10.1186/1472-6882-12-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tuominen I, Fuqua BK, Pan C, Renaud N, Wroblewski K, Civelek M, Clerkin K, Asaryan A, Haroutunian SG, Loureiro J, Borawski J, Roma G, Knehr J, Carbone W, French S, Parks BW, Hui ST, Mehrabian M, Magyar C, Cantor RM, Ukomadu C, Lusis AJ, Beaven SW. The genetic architecture of carbon tetrachloride-induced liver fibrosis in mice. Cell Mol Gastroenterol Hepatol 11: 199–220, 2021. doi: 10.1016/j.jcmgh.2020.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wong FW, Chan WY, Lee SS. Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol Appl Pharmacol 153: 109–118, 1998. doi: 10.1006/taap.1998.8547. [DOI] [PubMed] [Google Scholar]
- 74. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 18: 151–166, 2021. doi: 10.1038/s41575-020-00372-7. [DOI] [PubMed] [Google Scholar]
- 75. Issa R, Williams E, Trim N, Kendall T, Arthur MJ, Reichen J, Benyon RC, Iredale JP. Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut 48: 548–557, 2001. doi: 10.1136/gut.48.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lee HS, Shun CT, Chiou LL, Chen CH, Huang GT, Sheu JC. Hydroxyproline content of needle biopsies as an objective measure of liver fibrosis: emphasis on sampling variability. J Gastroenterol Hepatol 20: 1109–1114, 2005. doi: 10.1111/j.1440-1746.2005.03901.x. [DOI] [PubMed] [Google Scholar]
- 77. Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, Barry V, Mikels-Vigdal A, Karpinski S, Kornyeyev D, Adamkewicz J, Feng X, Zhou Q, Shang C, Kumar P, Phan D, Kasner M, López B, Diez J, Wright KC, Kovacs RL, Chen PS, Quertermous T, Smith V, Yao L, Tschöpe C, Chang CP. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun 7: 13710, 2016. doi: 10.1038/ncomms13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chen HH, Waghorn PA, Wei L, Tapias LF, Schühle DT, Rotile NJ, Jones CM, Looby RJ, Zhao G, Elliott JM, Probst CK, Mino-Kenudson M, Lauwers GY, Tager AM, Tanabe KK, Lanuti M, Fuchs BC, Caravan P. Molecular imaging of oxidized collagen quantifies pulmonary and hepatic fibrogenesis. JCI Insight 2: e91506, 2017. doi: 10.1172/jci.insight.91506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Klepfish M, Gross T, Vugman M, Afratis NA, Havusha-Laufer S, Brazowski E, Solomonov I, Varol C, Sagi I. LOXL2 Inhibition paves the way for macrophage-mediated collagen degradation in liver fibrosis. Front Immunol 11: 480, 2020. doi: 10.3389/fimmu.2020.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Radbill BD, Gupta R, Ramirez MC, DiFeo A, Martignetti JA, Alvarez CE, Friedman SL, Narla G, Vrabie R, Bowles R, Saiman Y, Bansal MB. Loss of matrix metalloproteinase-2 amplifies murine toxin-induced liver fibrosis by upregulating collagen I expression. Dig Dis Sci 56: 406–416, 2011. doi: 10.1007/s10620-010-1296-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Prystupa A, Boguszewska-Czubara A, Bojarska-Junak A, Toruń-Jurkowska A, Roliński J, Załuska W. Activity of MMP-2, MMP-8 and MMP-9 in serum as a marker of progression of alcoholic liver disease in people from Lublin Region, eastern Poland. Ann Agric Environ Med 22: 325–328, 2015. doi: 10.5604/12321966.1152088. [DOI] [PubMed] [Google Scholar]
- 82. Onozuka I, Kakinuma S, Kamiya A, Miyoshi M, Sakamoto N, Kiyohashi K, Watanabe T, Funaoka Y, Ueyama M, Nakagawa M, Koshikawa N, Seiki M, Nakauchi H, Watanabe M. Cholestatic liver fibrosis and toxin-induced fibrosis are exacerbated in matrix metalloproteinase-2 deficient mice. Biochem Biophys Res Commun 406: 134–140, 2011. doi: 10.1016/j.bbrc.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 83. Hazra S, Xiong S, Wang J, Rippe RA, Krishna V, Chatterjee K, Tsukamoto H. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem 279: 11392–11401, 2004. doi: 10.1074/jbc.M310284200. [DOI] [PubMed] [Google Scholar]
- 84. Morán-Salvador E, Titos E, Rius B, González-Périz A, García-Alonso V, López-Vicario C, Miquel R, Barak Y, Arroyo V, Clària J. Cell-specific PPARγ deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J Hepatol 59: 1045–1053, 2013. doi: 10.1016/j.jhep.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 85. Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF Jr, Motomura K, Anania FA, Willson TM, Tsukamoto H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem 275: 35715–35722, 2000. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 86. Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, Ridolfi F, Trozzi L, Surrenti C, Casini A. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology 122: 1924–1940, 2002. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 87. Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, Bonacchi A, Caporale R, Laffi G, Pinzani M, Gentilini P. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology 119: 466–478, 2000. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- 88. Liu X, Xu J, Rosenthal S, Zhang LJ, McCubbin R, Meshgin N, Shang L, Koyama Y, Ma HY, Sharma S, Heinz S, Glass CK, Benner C, Brenner DA, Kisseleva T. Identification of lineage-specific transcription factors that prevent activation of hepatic stellate cells and promote fibrosis resolution. Gastroenterology 158: 1728–1744.e14, 2020. doi: 10.1053/j.gastro.2020.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X, Ntambi JM. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab 6: 484–496, 2007. doi: 10.1016/j.cmet.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 90. Lai KKY, Kweon SM, Chi F, Hwang E, Kabe Y, Higashiyama R, Qin L, Yan R, Wu RP, Lai K, Fujii N, French S, Xu J, Wang JY, Murali R, Mishra L, Lee JS, Ntambi JM, Tsukamoto H. Stearoyl-CoA desaturase promotes liver fibrosis and tumor development in mice via a Wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology 152: 1477–1491, 2017. doi: 10.1053/j.gastro.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhou Y, Zhong L, Yu S, Shen W, Cai C, Yu H. Inhibition of stearoyl-coenzyme A desaturase 1 ameliorates hepatic steatosis by inducing AMPK-mediated lipophagy. Aging (Albany NY) 12: 7350–7362, 2020. doi: 10.18632/aging.103082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dobrzyn P, Dobrzyn A, Miyazaki M, Cohen P, Asilmaz E, Hardie DG, Friedman JM, Ntambi JM. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc Natl Acad Sci USA 101: 6409–6414, 2004. doi: 10.1073/pnas.0401627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bai L, Liu X, Zheng Q, Kong M, Zhang X, Hu R, Lou J, Ren F, Chen Y, Zheng S, Liu S, Han YP, Duan Z, Pandol SJ. M2-like macrophages in the fibrotic liver protect mice against lethal insults through conferring apoptosis resistance to hepatocytes. Sci Rep 7: 10518, 2017. doi: 10.1038/s41598-017-11303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Li M, Sun X, Zhao J, Xia L, Li J, Xu M, Wang B, Guo H, Yu C, Gao Y, Wu H, Kong X, Xia Q. CCL5 deficiency promotes liver repair by improving inflammation resolution and liver regeneration through M2 macrophage polarization. Cell Mol Immunol 17: 753–764, 2020. doi: 10.1038/s41423-019-0279-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ge S, Yang W, Chen H, Yuan Q, Liu S, Zhao Y, Zhang J. MyD88 in macrophages enhances liver fibrosis by activation of NLRP3 inflammasome in HSCs. Int J Mol Sci 22: 12413, 2021. doi: 10.3390/ijms222212413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bai L, Kong M, Duan Z, Liu S, Zheng S, Chen Y. M2-like macrophages exert hepatoprotection in acute-on-chronic liver failure through inhibiting necroptosis-S100A9-necroinflammation axis. Cell Death Dis 12: 93, 2021. doi: 10.1038/s41419-020-03378-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Son G, Iimuro Y, Seki E, Hirano T, Kaneda Y, Fujimoto J. Selective inactivation of NF-kappaB in the liver using NF-kappaB decoy suppresses CCl4-induced liver injury and fibrosis. Am J Physiol Gastrointest Liver Physiol 293: G631–G639, 2007. doi: 10.1152/ajpgi.00185.2007. [DOI] [PubMed] [Google Scholar]
- 98. Toriumi K, Horikoshi Y, Yoshiyuki Osamura R, Yamamoto Y, Nakamura N, Takekoshi S. Carbon tetrachloride-induced hepatic injury through formation of oxidized diacylglycerol and activation of the PKC/NF-κB pathway. Lab Invest 93: 218–229, 2013. doi: 10.1038/labinvest.2012.145. [DOI] [PubMed] [Google Scholar]
- 99. Ghosh S, Karin M. Missing pieces in the NF-κB puzzle. Cell 109: S81–S96, 2002. doi: 10.1016/S0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 100. Liu SB, Ikenaga N, Peng ZW, Sverdlov DY, Greenstein A, Smith V, Schuppan D, Popov Y. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J 30: 1599–1609, 2016. doi: 10.1096/fj.14-268425. [DOI] [PubMed] [Google Scholar]
- 101. Bae MA, Rhee SD, Jung WH, Ahn JH, Song BJ, Cheon HG. Selective inhibition of activated stellate cells and protection from carbon tetrachloride-induced liver injury in rats by a new PPARγ agonist KR62776. Arch Pharm Res 33: 433–442, 2010. doi: 10.1007/s12272-010-0313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors: new targets for the pharmacological modulation of macrophage gene expression and function. Curr Opin Lipidol 14: 459–468, 2003. doi: 10.1097/00041433-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 103. Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab 6: 137–143, 2007. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 104. Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARγ1) overexpression. J Biol Chem 278: 498–505, 2003. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 105. Jeong S, Yoon M. 17β-Estradiol inhibition of PPARγ-induced adipogenesis and adipocyte-specific gene expression. Acta Pharmacol Sin 32: 230–238, 2011. doi: 10.1038/aps.2010.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chu R, van Hasselt A, Vlantis AC, Ng EK, Liu SY, Fan MD, Ng SK, Chan AB, Liu Z, Li XY, Chen GG. The cross-talk between estrogen receptor and peroxisome proliferator-activated receptor γ in thyroid cancer. Cancer 120: 142–153, 2014. doi: 10.1002/cncr.28383. [DOI] [PubMed] [Google Scholar]
- 107. Lin YM, Velmurugan BK, Yeh YL, Tu CC, Ho TJ, Lai TY, Tsai CH, Tsai FJ, Tsai CH, Huang CY. Activation of estrogen receptors with E2 downregulates peroxisome proliferator-activated receptor γ in hepatocellular carcinoma. Oncol Rep 30: 3027–3031, 2013. doi: 10.3892/or.2013.2793. [DOI] [PubMed] [Google Scholar]
- 108. Zhou Y, Shimizu I, Lu G, Itonaga M, Okamura Y, Shono M, Honda H, Inoue S, Muramatsu M, Ito S. Hepatic stellate cells contain the functional estrogen receptor beta but not the estrogen receptor alpha in male and female rats. Biochem Biophys Res Commun 286: 1059–1065, 2001. doi: 10.1006/bbrc.2001.5479. [DOI] [PubMed] [Google Scholar]
- 109. Zhang B, Zhang CG, Ji LH, Zhao G, Wu ZY. Estrogen receptor β selective agonist ameliorates liver cirrhosis in rats by inhibiting the activation and proliferation of hepatic stellate cells. J Gastroenterol Hepatol 33: 747–755, 2018. doi: 10.1111/jgh.13976. [DOI] [PubMed] [Google Scholar]
- 110. Murphy AJ, Guyre PM, Wira CR, Pioli PA. Estradiol regulates expression of estrogen receptor ERα46 in human macrophages. PLoS One 4: e5539, 2009. doi: 10.1371/journal.pone.0005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol 294: 63–69, 2015. doi: 10.1016/j.cellimm.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bryzgalova G, Lundholm L, Portwood N, Gustafsson JA, Khan A, Efendic S, Dahlman-Wright K. Mechanisms of antidiabetogenic and body weight-lowering effects of estrogen in high-fat diet-fed mice. Am J Physiol Endocrinol Physiol 295: E904–E912, 2008. doi: 10.1152/ajpendo.90248.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gao H, Bryzgalova G, Hedman E, Khan A, Efendic S, Gustafsson JA, Dahlman-Wright K. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol 20: 1287–1299, 2006. doi: 10.1210/me.2006-0012. [DOI] [PubMed] [Google Scholar]
- 114. Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, Dahlman-Wright K, Nilsson S, Gustafsson JA, Efendic S, Khan A. Evidence that oestrogen receptor-α plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia 49: 588–597, 2006. doi: 10.1007/s00125-005-0105-3. [DOI] [PubMed] [Google Scholar]
- 115. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115: 56–65, 2005. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Murray PJ. Macrophage polarization. Annu Rev Physiol 79: 541–566, 2017. doi: 10.1146/annurev-physiol-022516-034339. [DOI] [PubMed] [Google Scholar]
- 117. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science 356: 1026–1030, 2017. doi: 10.1126/science.aam7928. [DOI] [PubMed] [Google Scholar]
- 118. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20, 2014. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chakraborty B, Byemerwa J, Shepherd J, Haines CN, Baldi R, Gong W, Liu W, Mukherjee D, Artham S, Lim F, Bae Y, Brueckner O, Tavares K, Wardell SE, Hanks BA, Perou CM, Chang CY, McDonnell DP. Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J Clin Invest 131: e151347, 2021. doi: 10.1172/JCI151347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Campbell L, Emmerson E, Williams H, Saville CR, Krust A, Chambon P, Mace KA, Hardman MJ. Estrogen receptor-alpha promotes alternative macrophage activation during cutaneous repair. J Invest Dermatol 134: 2447–2457, 2014. doi: 10.1038/jid.2014.175. [DOI] [PubMed] [Google Scholar]
- 121. Pelekanou V, Kampa M, Kiagiadaki F, Deli A, Theodoropoulos P, Agrogiannis G, Patsouris E, Tsapis A, Castanas E, Notas G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J Leukoc Biol 99: 333–347, 2016. doi: 10.1189/jlb.3A0914-430RR. [DOI] [PubMed] [Google Scholar]
- 122. Dziegielewska KM, Andersen NA, Saunders NR. Modification of macrophage response to lipopolysaccharide by fetuin. Immunol Lett 60: 31–35, 1998. doi: 10.1016/s0165-2478(97)00126-0. [DOI] [PubMed] [Google Scholar]
- 123. Smith ER, Hanssen E, McMahon LP, Holt SG. Fetuin-A-containing calciprotein particles reduce mineral stress in the macrophage. PLoS One 8: e60904, 2013. doi: 10.1371/journal.pone.0060904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rudloff S, Janot M, Rodriguez S, Dessalle K, Jahnen-Dechent W, Huynh-Do U. Fetuin-A is a HIF target that safeguards tissue integrity during hypoxic stress. Nat Commun 12: 549, 2021. doi: 10.1038/s41467-020-20832-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y, Friedman SL. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 69: 385–395, 2018. doi: 10.1016/j.jhep.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data associated with this article can be found at https://doi.org/10.6084/m9.figshare.21719378.v1.
Data Availability Statement
Data will be made available upon reasonable request.