

Keywords: ER stress, MitoQ, obese allergic asthma
Abstract
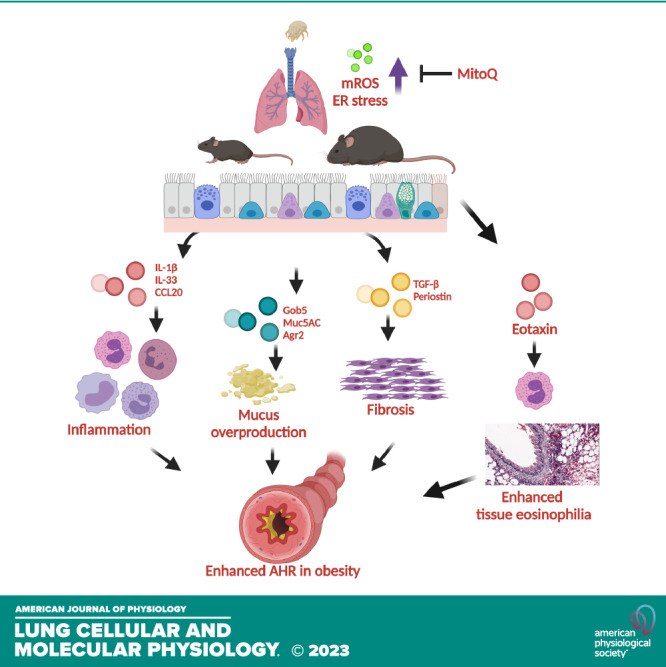
Obesity is associated with severe, difficult-to-control asthma, and increased airway oxidative stress. Mitochondrial reactive oxygen species (mROS) are an important source of oxidative stress in asthma, leading us to hypothesize that targeting mROS in obese allergic asthma might be an effective treatment. Using a mouse model of house dust mite (HDM)-induced allergic airway disease in mice fed a low- (LFD) or high-fat diet (HFD), and the mitochondrial antioxidant MitoQuinone (MitoQ), we investigated the effects of obesity and ROS on HDM-induced airway inflammation, remodeling, and airway hyperresponsiveness (AHR). Obese allergic mice showed increased lung tissue eotaxin, airway tissue eosinophilia, and AHR compared with lean allergic mice. MitoQ reduced airway inflammation, remodeling, and hyperreactivity in both lean and obese allergic mice, and tissue eosinophilia in obese-allergic mice. Similar effects were observed with decyl triphosphonium (dTPP+), the hydrophobic cationic moiety of MitoQ lacking ubiquinone. HDM-induced oxidative sulfenylation of proteins was increased particularly in HFD mice. Although only MitoQ reduced sulfenylation of proteins involved in protein folding in the endoplasmic reticulum (ER), ER stress was attenuated by both MitoQ and dTPP+ suggesting the anti-allergic effects of MitoQ are mediated in part by effects of its hydrophobic dTPP+ moiety reducing ER stress. In summary, oxidative signaling is an important mediator of allergic airway disease. MitoQ, likely through reducing protein oxidation and affecting the UPR pathway, might be effective for the treatment of asthma and specific features of obese asthma.
INTRODUCTION
Obesity is a major risk factor for asthma, with increasing body mass index (BMI) associated with a dose dependent increase in the incidence of asthma (1–3). Obese individuals with asthma tend to have more severe disease, which is less responsive to standard controller therapy (4–6). Understanding the mechanisms of obese asthma is important to develop treatments for the unmet needs of this patient population.
Increased reactive oxygen species (ROS) contributes to severe asthma in people with obesity (7). A potential source of this oxidative stress is increased mitochondrial ROS (mROS), which has been reported in obesity-related asthma and in obese mouse models of asthma (8–12). Mitochondrial dysfunction, changes in bioenergetics, and increased ROS production have been found in the airway epithelium of obese asthma patients (13).
MitoQ is a lipophilic cation-tagged ubiquinone molecule that accumulates in the mitochondria (Supplemental Fig. S1) (14). The safety of the drug has been established in human clinical trials (15), and it is available as an over-the-counter antioxidant supplement in the United States. MitoQ is effective against LPS-induced acute lung injury (16) and ROS-induced airway smooth muscle remodeling in mouse models of chronic obstructive pulmonary disease (COPD) (17). The functional significance of targeting mROS in obese relative to lean allergic asthma has not been reported. Therefore, our objective was to examine the effect of MitoQ on the pathogenesis of asthma in mouse models of lean and obese allergic asthma.
Here, using a diet-induced obesity (DIO) model of obese allergic asthma in mice, we show that MitoQ reduced airway inflammation, remodeling, and methacholine hyperreactivity; similar effects were seen in lean and obese mice, although obese allergen-challenged mice had increased lung tissue eosinophils as compared with bronchoalveolar lavage fluid (BALF) eosinophils, which decreased following MitoQ; dTTP+ (an inactive analog lacking the antioxidant quinone moiety; Supplemental Fig. S1) had many similar effects. However, dTPP+ increased mROS production as measured by peroxiredoxin 3 (PRX3) hyperoxidation when compared with MitoQ in response to HDM confirming some reports that dTPP+ can increase mROS in certain cell types (18, 19). Using a proteomic approach, we found HDM increased cysteine oxidation in proteins of the unfolded protein response (UPR) pathway, which was attenuated with MitoQ. The UPR is implicated in the pathogenesis of asthma (20–23). Collectively our results show that the anti-allergic effects of MitoQ in obese mice are likely mediated in part by the attenuation of the UPR signaling.
METHODS
Cell Culture
Primary normal human bronchial epithelial cells (pHBE cells) obtained from ATCC (PCS-300-010) were cultured in airway epithelial cell basal medium (ATCC PCS-300-030) supplemented with bronchial epithelial growth kit (ATCC PCS-300-040). On day 1 of culture, the cells were incubated for 2 h in basal media without the growth kit. HDM (25 µg/mL) based on protein weight (Lot No: 360924, Endotoxin content: 872.5 EU/vial, Greer laboratories) dissolved in phosphate buffered sale (PBS) was added to the treatment groups and the control cells were exposed to an equal volume of PBS. On day 2, the MitoQ (MQ) group was treated with MitoQ or decyl-triphenylphosphonium (dTPP+) dissolved in dimethyl sulfoxide (DMSO) and control groups were treated with equal volume of DMSO. On day 3, the cells were treated with 25 µg/mL HDM together with a second dose of MitoQ or dTPP+. Twenty-four hours later the supernatants were collected for cytokine measurements and the cells were harvested in a buffer containing 20 mM Tris·HCl (pH 7.5), 150 mM NaCl, 0.5% Nonidet P-40 (NP-40), 10% glycerol, protease inhibitor cocktail (Sigma-Aldrich, P8340), phosphatase inhibitor cocktails 1 and 2 (Sigma-Aldrich, P5726, P0044), 5 mM Catalase, and 10 mM N-ethylmalemide. Experiments were performed in triplicate and each data point is an average of three independent experiments performed on different days (Supplemental Fig. S4A).
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Vermont (IACUC Protocol No: PROTO202000069). For the MitoQ (MQ)/dTPP+ experiments, male mice maintained on normal chow for 16 wk were used. To induce obesity, male C57BL/6NJ mice were used for all experiments because they are more susceptible to diet-induced obesity. Four-week-old mice were randomly assigned to a high-fat diet with 60% kcal from fat (Research Diets D12492) or a low-fat diet with 10% kcal from fat (Research Diets D12450B) for 20 wk before experiments. For the experiments, mice were separated into control, HDM, and HDM + MitoQ groups. The HDM and HDM + MitoQ groups were sensitized on days 1 and 7 with 25 µg HDM in 50-µL PBS (protein weight, Lot No: 360924, Endotoxin content: 872.5 EU/vial, Greer laboratories) via intranasal delivery followed by HDM challenges on days 14–18. The HDM + MitoQ/dTPP+ groups were treated with 6 mg/kg MitoQ or equimolar dTPP+ dissolved in PBS and administered via intraperitoneal injections on days 17–19 after the HDM administration. The control mice were treated with PBS. BALF and lungs were harvested on day 20 to study acute responses and day 27 to study late inflammatory responses (Fig. 1A and Supplemental Fig. S3A).
Figure 1.

MitoQ attenuates acute responses to HDM: A: schematic for the acute HDM/MitoQ response. B–F: BAL total inflammatory cells, BAL macrophages, BAL neutrophils, BAL eosinophils, and BAL lymphocytes. G–K: analysis of lung tissue protein levels of innate cytokines IL-1β, IL-33, Eotaxin, CCL20, and mRNA levels of Th2 cytokine Il-4. P values <0.05 were regarded as discovery or statistically significant. Error bars ± SE. *Significant differences between PBS and HDM groups and number sign (#) indicates significant differences between HDM and HDM + MQ groups. HDM, house dust mite.
Tissue Staining
For Masson’s trichrome staining, paraffin-embedded lung tissue sections were deparaffinized followed by immersion in Bouin’s mordant solution for 1 h at 56°C. The sections were then stained in Weigert’s hematoxylin for 10 min, washed in running water, and stained with Biebrich scarlet acid fuchsin solution for 2 min. The slides were washed in running water again and immersed in phosphomolybdic/phosphotungstic acid solution for 10 min, and finally stained in aniline blue solution and 1% acetic acid for 5 min each and dehydrated and mounted using permaslip.
For Periodic acid-Schiff (PAS) staining, paraffin-embedded lung tissue sections were deparaffinized and rehydrated followed by incubation in 0.5% periodic acid for 5 min. After rinsing with distilled water, slides were immersed in Schiff’s reagent for 30 min followed by washing and staining with 0.55% potassium metabisulfite for 1 min, twice. The slides were washed in tap water and stained with hematoxylin counter stain for 10 min, then washed and dipped in 0.5% lithium carbonate for 10 s. The slides were then dehydrated and mounted using permaslip. The intensity of PAS and Massons’ trichrome staining was quantified by scoring of slides by three independent blinded investigators as described previously (24).
For eosinophil peroxidase (EPX) immunohistochemistry, paraffin-embedded tissue sections were deparaffinized and rehydrated, followed by antigen retrieval by boiling in sodium citrate buffer. After blocking with normal serum, sections were incubated with EPX antibody (1:200, Biorbyt, Cat. No. orb5168) overnight at 4°C. Sections were stained using the Vectastain ABC-alkaline phosphatase kit (Cat. No. AK-5200) and developed using the ImmPACT alkaline phosphatase substrate (SK-5105) following manufacturer’s protocol. EPX staining around large airways was quantified using Aperio imagescope software (Leica Biosystems).
Immunoprecipitation and Western Blotting
For the validation of GRP94 and PDIA3 sulfenylation, 250 μg total lung lysate prepared with RIPA buffer containing 5 mM catalase, 10 mM N-ethylmalemide, and DCP-BIO1 (dimedone tagged with biotin, Kerafast, EE0028) was incubated with streptavidin beads overnight at 4°C. After three washes with RIPA buffer to remove nonspecific interactions, the bound proteins were eluted by boiling equal volumes run on a 12% SDS-PAGE. Antibodies for GRP94 and PDIA3 [Cat No. ABISPA850F (Enzo Life Sciences Inc.) and LS-B9768-100 (LSBio) respectively] were used for detection. For detection of endoplasmic reticulum (ER) stress the following antibodies were used: phospho-IRE1 (Abcam, AB124945), phospho-EIF2α (Cell Signaling Technology, 9721S), ATF650 (Invitrogen, PA5-20216), phospho-PERK (Bioss antibodies; Cat No. BS-3330R), and CHOP (Novus Biologicals; NB600-1335). β-tubulin (Abcam, AB6046) was used as loading control. All the antibodies except CHOP was used at a 1:1,000 dilution in 2.5% bovine serum albumin (BSA). CHOP antibody was diluted to 1:500 in 2.5% BSA. Protein expression was quantified using ImageJ software (NIH, https://imagej.nih.goc/ii/).
ELISA
Lung tissue protein lysate was prepared using an extraction buffer containing 20 mM Tris·HCl (pH 7.5), 150 mM NaCl, 0.5% Nonidet P-40 (NP-40), 10% glycerol, protease inhibitor cocktail (Sigma-Aldrich, P8340), and phosphatase inhibitor cocktails 1 and 2 (Sigma-Aldrich, P5726, P0044). Eotaxin, IL-33, IL-1β, and CCL20 ELISAs (R&D systems) were performed on 50 µg total lung tissue protein lysate following the manufacturer’s protocol. Briefly, high-binding capacity ELISA plates were coated overnight at room temperature with capture antibody diluted in PBS. The plates were then blocked with 1% BSA and incubated with standards and samples overnight at 4°C. After washing, the plates were then incubated with the detection antibody followed by streptavidin-HRP and TMB detection. Absorbance was measured at 450 nm and 540 nm (background). For EPX ELISA, 50 µg of total lung lysate was used to measure tissue EPX following manufacturer’s protocol (Cusabio, Cat No. CSB-E15839m).
Lactate Dehydrogenase and Dead Cell Protease Activity Assays
BAL was incubated with lactate dehydrogenase (LDH) substrate and the reduction of NAD to NADH was detected colorimetrically at 450 nm following manufacturers’ protocol to measure LDH activity (Sigma-Aldrich; Cat No. MAK066). The AAF-aminoluciferin substrate was incubated with BALF and the luminescence generated by the breakdown product aminoluciferin was measured to assay dead cell protease activity (Promega; Cat No. G9290).
PRX3 Hyperoxidation Assay
Total lung protein lysate (250 µg) was incubated with 2 µg peroxiredoxin 3 (PRX3) antibody [mouse monoclonal AF17D2FF, Abfrontier (LF-MA0329)] overnight at 4°C on a shaker. Immunoprecipitation control sample was incubated with nonspecific mouse IgG. The protein-antibody complex was incubated with recombinant protein G agarose for 3 h at 4°C. The beads were washed three times with wash buffer containing 1% NP-40 and eluted by boiling in Laemmli sample buffer containing dithiothreitol (DTT). The samples were run in a 15% polyacrylamide gel and transferred to a polyvinylidene fluoride (PVDF) membrane, blocked using 5% BSA, and probed using peroxiredoxin-SO3 (PRXSO3 pan-PRX) antibody (Abfrontier, LF-PA0004). PRX3 antibody (Thermo scientific PA5-91918) was used for western blotting.
RNA Extraction and qRT-PCR
Total lung RNA was extracted using RNeasy mini kit (Qiagen) following manufacturers’ protocol. 1 µg of RNA was used for preparing cDNA (Promega) and qPCR was performed using iTaq SYBR green reagent (Biorad). The samples were run in duplicate for each gene and expression was normalized to the housekeeping genes Pp1ca and Ubc.
Flow Cytometry for Cell Counts
Bronchoalveolar lavage fluid (BAL) was collected from mice by injecting mice lungs with 1 mL PBS via tracheal cannula. Total cells were identified based on forward and side scatter parameters and immune cells were identified by hematoxylin and eosin staining (HEMA3 staining kit, Fisher) on cytospin preparations or by flow cytometry by the following cell surface markers: neutrophils (CD11c−, CD3/B220−, CCR3−), alveolar macrophages (CD11c+), eosinophils (CD11c−, CD3/B220−, CCR3+), and lymphocytes (CD11c−, CD3/B220+). Staining was performed after incubation with Fc block Trustain FcX (BioLegend 101320). Stained cells were analyzed using a Guava easycyte flow cytometer (Luminex corporation).
Pulldown of Cysteine-Sulfenylated Proteins and Identification Using LC-MS/MS
Sample preparation and trypsin digestion.
Total lung protein lysate (1 mg) extracted using a RIPA-based lysis buffer containing 5 mM catalase, 10 mM NEM and 10 mM DCP Bio-1 was used for binding to Neutravidin agarose beads overnight at 4°C. Following three washes with RIPA buffer to remove nonspecific interactions, the bound proteins were eluted by boiling. Equal volumes of the samples [Obese PBS (n = 5), Obese HDM (n = 5), Obese MitoQ (n = 5), Lean PBS (n = 4), Lean HDM (n = 4), Lean MitoQ (n = 4)] were run briefly onto the SDS-PAGE gels (1/3 of the whole length of the gel), which was then stained with silver. The gel pieces were minced to 1 mm3 cubes, combined, destained, and subjected to disulfide reduction and alkylation, and trypsin digestion protocols, as described previously (25).
Liquid chromatography-tandem mass spectrometry-based protein identification.
The tryptic peptides resuspended in 2.5% acetonitrile (ACN) and 2.5% formic acid (FA) in water and analyzed on the Q-Exactive Plus mass spectrometer coupled to an EASY-nLC 1200 system (Thermo Fisher Scientific). Samples were loaded onto a 100 μm × 320 mm capillary column packed with UChrom C18 (1.8 μm particle size, 120 A, Cat. No. PN-80001; Nanolcms, CA) at a flow rate of 300 nL/min. The column end was laser pulled to a ∼3 μm orifice and packed with minimal amounts of 5 µm Magic C18AQ before packing with the 1.8 μm particle size chromatographic materials. Peptides derived from proteins in DCP Bio-1 pull-downs were separated by a solvent system composed of solvent A: 100% water/0.1% FA and solvent B: 80% ACN/0.1% FA with a gradient of 0%–44% ACN/0.1% FA over 150 min, 44%–100% ACN/0.1% FA in 1 min and then 100% ACN/0.1% FA for 8 min, followed by an immediate return to 0% ACN/0.1% FA and a hold at 0% ACN/0.1% FA. Mass spectrometry data were acquired in a data-dependent “Top 10” acquisition mode with lock mass function activated (m/z 371.1012; use lock masses: best; lock mass injection: full MS), in which a survey scan from m/z 350–1,600 at 70,000 resolution (AGC target 1e6; max IT 100 ms; profile mode) was followed by 10 higher-energy collisional dissociation (HCD) tandem mass spectrometry (MS/MS) scans on the most abundant ions at 17,500 resolution (loop count = 10; AGC target 5e4; max IT 100 ms; centroid mode). MS/MS scans were acquired with an isolation width of 1.6 m/z and a normalized collisional energy of 26%. Dynamic exclusion was enabled (peptide match: preferred; exclude isotopes: on; underfill ratio: 1%; exclusion duration: 15 s). Minimum AGC target = 5 e2.
Database Searches
Raw files (.raw) were analyzed using Proteome Discoverer 2.4 (Thermo Fisher Scientific). Product ion spectra were searched using SEQUEST with the “Basic” Processing and Consensus workflows against a Uniprot Mus musculus protein database (UP000000589; downloaded March 3, 2020). Search Parameters were as follows: Full trypsin enzymatic activity; maximum allowed missed cleavages = 2, mass tolerance at 10 ppm and 0.02 Da for precursor ions and fragment ions, respectively; dynamic modification on methionine (oxidation: +15.995 Da), and static modification on cysteines (carbamidomethylation: +57.021 Da). Percolator was included in the workflow to limit the false discovery rate to less than or equal to 1%. For spectral counting analysis, the resulting msf result files were incorporated into Q + S 4.11 (Proteome Software, OR) with “prefiltered mode” and Protein Cluster Analysis for spectral counting analysis.
Measurement of AHR.
Mice were anesthetized by the intraperitoneal administration of sodium pentobarbital (90 mg/kg) followed by administration of pancuronium bromide, a paralyzing agent. The mice were then mechanically ventilated at 200 breaths/min using a Flexivent small animal ventilator (SCIREQ) via tracheal cannula. Aerosolized methacholine was used to induce airway hyperresponsiveness and Newtonian resistance (Rn), tissue damping (G), and tissue elastance (H) were measured every 10 s for 3 min (18 measurements each in total). The average of the 18 measurements was calculated excluding values whose coefficient of determination (COD) was less than 0.85 and plotted using Graphpad Prism 8 (Graphpad software Inc. CA).
Statistical Analysis
Experiments in mice were performed twice for each diet group and the data were pooled and analyzed by one or two-way analysis of variance (ANOVA), as appropriate and a two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli test to adjust for multiple comparisons. Data for all results are expressed as mean values ± SE. P values < 0.05 were regarded as discovery or statistically significant. The ROUT method was first used to identify outliers with a cutoff of false discovery rate Q = 1%, and they were removed from the analysis. All statistical analyses and plots were created using Graphpad Prism 8 (GraphPad Software Inc., CA). Detailed description of the statistical analysis is included in Supplemental Tables S1 and S2.
For mass spectrometry data analysis, the total spectral counts were statistically evaluated between “PBS,” “HDM,” and “MitoQ” treatment groups in both the lean and obese sample sets. Statistical tools in Scaffold Quantitative Analysis Mode were used for pairwise comparisons (two-tailed t test for PBS vs. HDM and HDM vs. MitoQ), with the following settings: No normalization was used; Min. Value = 0.5 (all spectral counts of 0 were replaced with a value of 0.5); and total spectra as quantitative method. Spectral counting reports were exported from Scaffold with “Show lower scoring peptides and <5% Probabilities” deactivated and Hide “Decoy.” The total spectral counts of all proteins (including protein cluster members) and the corresponding statistical analyses exported from Scaffold as “total spectrum counting reports” are uploaded to PRIDE. One of the cluster members was selected to represent the cluster and the associated spectral counts were derived from the peptides of all the members. For simplicity, only spectral counts of protein clusters were used to construct the volcano plots and heatmaps depicting differential sulfenylation of proteins between treatment groups in obese and lean sample sets. Volcano plots were created using fold-change values [log2 of the difference between the number of peptides of the two groups (PBS vs. HDM)] and –log10 of P values (PBS vs. HDM). Heat maps detailing significant increases in protein sulfenylation between HDM, and PBS groups were created by first scaling the total spectral counts in individual samples of the three treatment groups so that individual counts in all samples summed up to 100 (allowing fold-changes of proteins of varying abundances be represented on the same scale.) All volcano plots and heat maps were created using Graph Pad Prism 8 (GraphPad Software Inc., CA). The mass spectrometry/proteomics data, including raw (.raw), search (.msf), and analysis (.sf3) files, as well as MGF and mzIdentML files exported from Scaffold, have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Dataset identifier: PXD029715).
RESULTS
MitoQ Decreases HDM-Induced Inflammation
HFD increased total body and visceral adipose tissue weights compared with a low-fat diet. MitoQ did not affect total weight or visceral adiposity (Supplemental Fig. S2).
We investigated effects of HDM and MitoQ on inflammation using the regimen depicted in Fig. 1A. HDM increased BAL total cell counts (Fig. 1B), and neutrophils, eosinophils, and lymphocytes (Fig. 1, D–F), in both LFD and HFD mice. HDM did not affect BAL macrophages (Fig. 1C). Lymphocytes, but not total cells, macrophages, neutrophils or eosinophils, were significantly reduced by MitoQ (Fig. 1, B–F).
We investigated effects on lung tissue cytokines. Basal IL-1β was significantly higher in LFD than HFD mice (Fig. 1G). With HDM, levels of the IL-1 family members IL-33 and IL-1β were significantly higher in LFD than HFD mice (Fig. 1, G and H), whereas eotaxin (CCL11) was higher in HFD mice (Fig. 1I). MitoQ decreased eotaxin levels only in HFD mice (Fig. 1I). HDM-induced CCL20 protein and Il4 RNA were comparable between LFD and HFD mice (Fig. 1, J and K). MitoQ treatment significantly decreased IL-33, IL-1β, and CCL20 in both LFD and HFD mice, eotaxin in HFD-HDM mice, and Il4 RNA in LFD-HDM mice (Fig. 1, G–K). In summary, MitoQ alleviated many aspects of HDM-induced inflammation, with some notable differences between LFD and HFD mice.
Tissue Eosinophilia Is Increased in HDM-HFD Mice and Decreased by MitoQ
Eotaxin (CCL11), an eosinophil chemoattractant, was significantly higher in HDM-HFD mice (Fig. 1I); however, BAL eosinophil levels were similar in HDM-HFD and HDM-LFD mice (Fig. 1E). Peribronchiolar eosinophils and protein levels of eosinophil peroxidase (EPX) were increased in HDM-HFD (Fig. 2, A–C), and decreased by MitoQ (Fig. 2, A–C). In contrast, lung tissue IL-5, which induces eosinophil differentiation and migration, was elevated only in HDM-LFD mice, and unaffected by MitoQ (Fig. 2D). In summary, tissue eosinophils are increased in HDM-HFD mice and decreased by MitoQ; this is associated with changes in eotaxin but not IL-5.
Figure 2.

Tissue eosinophilia is elevated in HDM-challenged HFD mice and is attenuated with MitoQ treatment. A: representative images of EPX immunohistochemistry. Scale bars: 100 µm. B: quantification for the EPX staining. C: ELISA for EPX in the lung tissue. D: lung tissue IL-5 measured by ELISA. P values < 0.05 were regarded as discovery or statistically significant. Error bars ± SE. *Significant differences between PBS and HDM groups. #Significant differences between HDM and HDM + MQ groups. EPX, eosinophil peroxidase; HDM, house dust mite; HFD, high-fat diet.
Resolution of Airway Inflammation Is Enhanced by MitoQ
To determine effects of HDM and MitoQ on persistent inflammation, we performed a 28-day experiment (Supplemental Fig. S3A). BAL total cell counts, neutrophils, and eosinophils remained elevated in HDM-LFD mice (Supplemental Fig. S3, B, D, and E), whereas lymphocytes were significantly elevated in HDM-HFD mice (Supplemental Fig. S3F). MitoQ decreased BAL lymphocytes significantly only in HFD-mice (Supplemental Fig. S3F). Slightly higher levels of tissue eosinophils persisted in HDM-HFD compared with HDM-LFD mice; MitoQ enhanced resolution of inflammation in HDM-HFD mice (Supplemental Fig. S3G). In summary, MitoQ enhances resolution of eosinophilic and lymphocyte inflammation in obese allergic mice.
MitoQ Decreases Remodeling
MitoQ decreased HDM-induced goblet cell metaplasia detected by Periodic acid Schiff (PAS) staining only in HFD mice (Fig. 3, A and B). Genes associated with mucin production and secretion like Gob5 and Agr2 increase in asthma (26, 27). MitoQ decreased HDM-induced expression of Gob5 in both LFD and HFD mice (Fig. 3C) but of Agr2 only in HFD-HDM mice (Fig. 3D). MitoQ inhibited goblet cell metaplasia and markers of mucus production in response to HDM particularly in obese airway disease mice.
Figure 3.

Mucus production is increased by HDM and decreased by MitoQ. A: representative images of PAS staining. Scale bars: 100 µM. B: quantification of PAS staining. C and D: qRT-PCR of total lung RNA for genes involved in mucus production: Gob5 and Agr2. P values < 0.05 were regarded as discovery or statistically significant. Error bars ± SE. *Significant differences between PBS and HDM groups. #Significant differences between HDM and HDM + MQ groups. HDM, house dust mite; PAS, Periodic acid Schiff.
Massons’s trichrome (MT) staining revealed that HDM-induced collagen deposition and abundant immune cell infiltration in peribronchial and perivascular regions of both LFD and HFD mice (Fig. 4A), and this was decreased by MitoQ (Fig. 4B). BAL levels of the HDM-induced profibrotic growth factors TGF-β and periostin were also decreased by MitoQ (Fig. 4, C and D). HDM administration increases α-SMA gene expression significantly in both LFD and HFD mice, and this is inhibited by MitoQ (Supplemental Fig. S4). In summary, MitoQ treatment alleviates many features of HDM-induced airway remodeling in both lean and obese allergic asthma.
Figure 4.

HDM-induced fibrosis is reduced by MitoQ treatment. A: Masson’s Trichrome staining (MT) for collagen. Scale bars: 100 µM. B: MT staining quantification. C and D: BAL TGF-β and BAL periostin levels. P values < 0.05 were regarded as discovery or statistically significant. Error bars ± SE. *Significant differences between PBS and HDM groups. #Significant differences between HDM and HDM + MQ groups. HDM, house dust mite.
MitoQ Decreases Airway Hyperresponsiveness
Control HFD mice exhibited inherent airway hyperresponsiveness (AHR) to methacholine measured by central airway resistance (Rn; Fig. 5A). HDM increased Rn reactivity particularly in HDM-HFD mice (Fig. 5A). HDM increased AHR measured by tissue damping (G) and tissue elastance (H) in both LFD and HFD mice (Fig. 5, B and C). MitoQ decreased HDM-induced AHR particularly in Rn and G in both LFD and HFD mice (Fig. 5, A and B), whereas we did not see significant differences in H with MitoQ treatment, this might be related to slightly less physiological response to HDM compared with some other experiments (Supplemental Fig. S5C). In summary, obese allergic mice display increased Rn AHR in response to HDM, and AHR is decreased by MitoQ in both lean and obese mice.
Figure 5.

Analysis of methacholine-induced AHR in mice. A: central airway resistance (Rn) in response to methacholine. B: tissue resistance or damping G response to methacholine. C: tissue elastance H response to methacholine. P values < 0.05 were regarded as discovery or statistically significant. Error bars ± SE. *Significant differences between PBS and HDM groups. #Significant differences between HDM and HDM + MQ groups. AHR, airway hyperresponsiveness; HDM, house dust mite.
The Anti-Inflammatory Activity of MitoQ Is Mediated in Part by dTPP+
To understand the mechanism of action of MitoQ in vitro, we challenged pHBE cells with HDM, then compared effects of MitoQ with dTPP+ (Supplemental Fig. S5A). HDM increased CCL20 (Supplemental Fig. S5B); 100 nM MitoQ, but not dTPP+, significantly inhibited CCL20 production. At higher concentrations both MitoQ and dTTP+ inhibited CCL20 (Supplemental Fig. S5B). A similar response was found for IL-8 (Supplemental Fig. S5C).
To determine effects in vivo, we sensitized and challenged wild-type C56BL6/NJ mice with HDM and administered equimolar concentrations of intraperitoneal MitoQ or dTPP+ (Fig. 1A). The following HDM-induced responses were inhibited by both MitoQ and dTPP+: BAL total cells, neutrophils, and lymphocytes, but not eosinophils (Supplemental Fig. S5, D–H); lung tissue IL-33, CCL-20, and BAL periostin (Supplemental Fig. S5, I–K); AHR (Supplemental Fig. S6, A–C); and cytotoxicity measured by LDH and dead cell protease activity assays (Supplemental Fig. S6, D and E). The levels of caspase-9, an initiator caspase of the mitochondrial apoptosis pathway, increased with HDM treatment and decreased with both MitoQ and dTPP+, showing that drug treatments do not induce mitochondrial toxicity (Supplemental Fig. S6F).
To determine if both MitoQ and dTPP+ were affecting mROS, we measured hyperoxidation of peroxiredoxin 3 (PRX3, a mitochondrially localized peroxiredoxin): MitoQ decreased PRX3 hyperoxidation compared with HDM-dTPP+-treated mice (Supplemental Fig. S6, G and H), suggesting many of the anti-inflammatory effects of MitoQ in this model may be unrelated to effects on mROS.
MitoQ Reduces HDM-Induced Oxidation of Multiple Target Proteins
To investigate oxidative pathways affected by HDM, we measured proximal cysteine oxidation of proteins (28) using a dimedone based probe, DCP-Bio1, to purify the allergic airway “oxidome,” and then identified proteins using LC-MS/MS. In total, 1,151 proteins, belonging to 963 protein clusters, were identified. There were differences in protein oxidation between control LFD and HFD mice (Supplemental Fig. S7). A higher number of proteins were oxidized following HDM in HFD compared with LFD mice (Fig. 6, A and C). HDM-induced oxidation of 15 (HFD) and 5 (LFD) proteins was decreased by MitoQ, whereas 18 (HFD) and 4 (LFD) were unaffected by MitoQ (Fig. 6, A and C). As illustrated in the volcano plots (Fig. 6, B and D), there was an increase in global protein sulfenylation in the obese group versus lean HDM-treated groups.
Figure 6.

A and C: DCP Bio-1 pull-down samples from various conditions (PBS, HDM, HDM + MitoQ from lean and obese groups) were separated on SDS-PAGE. After in-gel digestion and mass spectrometry, protein abundances (reflecting sulfenylation) were analyzed by spectral counting and represented in volcano plots and heat maps. Log2 ratios (vs. PBS) of the total spectral counts for those proteins with increased sulfenylation in HDM (P < 0.05) are represented in heat maps, with protein accessions listed according to the corresponding average log2 fold-changes (from high to low). B and D: proteins with increased sulfenylation on HDM treatment and decreased with HDM + MitoQ treatment are highlighted in red in the volcano plot, whereas those with sulfenylation upregulated by HDM, but not subsequently diminished by MitoQ, are highlighted in green. Cutoffs of HDM/PBS at twofold (log2 −2 = −1 and log2 2 = 1) and P value at 0.05 (–log10 0.05 = 1.301) are indicated by dotted line(s) on the x- and y-axis, respectively. E: validation of ER-stress-related proteins: cysteine sulfenylation of GRP94 and PDIA3. HDM sample without DCP-Bio1 labeling was used as a negative control. F: comparison of cysteine sulfenylation of GRP94 and PDIA3 between MitoQ and dTPP+. G: detection of ER stress. Activation of ER stress through the IRE, ATF6, and PERK pathways was observed by detection of phospho-IRE1, ATF650, and phospho-EIF2α. β-Tubulin was used as the loading control. ER, endoplasmic reticulum; HDM, house dust mite.
CHIL3, a macrophage marker previously reported to be oxidized in allergic asthma, was also identified in our study (29) (Supplemental Fig. S8). The ER-resident proteins GRP94 (Hsp90b1) and PDIA3, implicated in the unfolded protein response (UPR), were identified in oxidized form in both HFD and LFD mice. GRP78 (Hspa5), another ER-stress response protein, was identified only in the HDM-challenged HFD group (Fig. 6, A and C). We validated the oxidation of the ER-resident proteins and found that oxidation of GRP94 and PDIA3 increased with HDM administration and decreased with MitoQ treatment in both HFD and LFD mice. HDM without DCP-Bio1 labeling was used as a negative control (Fig. 6E). HDM-induced cysteine sulfenylation of the GRP94 was reduced only by MitoQ and not by dTPP+ (Fig. 6F and Supplemental Fig. S9). In summary, HDM induces protein oxidation of multiple cellular proteins including those in the UPR pathway, which can be inhibited by MitoQ. MitoQ, but not dTPP+, inhibited cysteine sulfenylation of the ER protein targets, suggesting effects on ER proteins are related to modulation by oxidation.
We then studied the effects on the canonical UPR transducers that can mediate features of allergic asthma (21, 22). Phospho-IRE1 and ATF6-50kD, activation markers of the IRE1 and ATF6 arms of the UPR pathway, were upregulated in both lean and obese allergic mice; phospho-PERK and phospho-EIF2α, involved in the PERK arm of the UPR, were upregulated only in HDM-HFD mice. MitoQ treatment attenuated the activation of the UPR transducers in both lean and obese allergic asthma (Fig. 6G and Supplemental Fig. S9), whereas dTPP+ reduced HDM-induced phosphor-IRE1 (IRE1 arm) and baseline CHOP (global downstream target of all the UPR pathways) (30) but not the ATF6 pathway (Supplemental Fig. S10). These data suggest that MitoQ attenuates HDM-induced UPR in response in part through its dTPP+ moiety that may be independent of quenching sulfenylation.
DISCUSSION
Obesity-related asthma is characterized by a severe, poorly controlled disease, which could be mediated by increased airway oxidative stress (8, 9, 31), as mitochondrial dysfunction and increased mROS are reported in obese asthma (13, 32–35). We found some differences between obese and lean animals challenged with HDM: lean animals had higher levels of IL-33 and IL1β in BAL, whereas obese animals had increased tissue eotaxin, tissue eosinophils, and airway reactivity measured by central airway resistance was elevated in obese control animals and further increased in obese-HDM compared with lean-HDM-challenged mice.
MitoQ consists of a lipophilic cation with a 10 carbon side chain (decyl-triphosphonium, dTTP+) tagged to a ubiquinone molecule (Supplemental Fig. S1); MitoQ accumulates in mitochondria to reduce mitochondrial ROS (14) and has been used to diminish mROS in clinical trials (36, 37). We found that MitoQ reduced inflammation, markers of mucus secretion and remodeling, and AHR in both lean and obese HDM-challenged mice and also reduced the increased lung tissue eosinophilia specific to obese allergic asthma. dTTP+ elicited many similar effects, despite increasing HDM-induced mROS; this suggest the efficacy of MitoQ in our model was not simply related to effects on mROS. In addition, MitoQ, through effects on cysteine oxidation and the UPR, may have efficacy in allergic airway disease and target the enhanced lung tissue eosinophilia of obese allergic asthma.
Existing data on mice models of obese allergic asthma report variable inflammatory responses (38–41). These differences could arise from the obesity model [ob/ob (Lepob) (41, 42) versus BALB/c (38) versus C57BL/6J (43) versus C57BL/6NJ (24)], asthma model [OVA (38, 41–43) versus HDM (24, 44)], and/or age of the mice (45). Here, using a 16-wk HFD C57BL/6NJ mice model, we showed that HDM-challenged lean animals develop a higher IL-1 and Th2 response, whereas eotaxin and tissue eosinophilia (but not airway lumen eosinophils) are increased in obese animals.
Higher levels of tissue, compared with airway, eosinophils have been found in obese people with asthma (46–48), and sputum eosinophils are lower in obese compared with lean people with asthma (49, 50). Using EPX as a marker we show that peribronchiolar and perivascular eosinophils are increased in obese allergic mice, similar to a report in an obese OVA model (42, 43). Tissue eosinophilia are potentially related to increased migration of eosinophils into the lungs from circulation (51), and perhaps decreased eosinophils in the BAL may reflect slower transit of eosinophils in obese mice. Epithelial transit of eosinophils might reflect resolution of inflammation (52); obese mice may have slower resolution of airway eosinophilic inflammation compared with lean mice (43, 53). Regardless of mechanism, distinction between tissue and airway eosinophilia is emerging as characteristic of obese asthma compared with lean asthmatics; we show this is related to higher levels of eotaxin and can be inhibited by MitoQ.
Obesity in mice is associated with increased and inherent airway hyperactivity (54), particularly in older mice (45). Here we show that 5-mo-old mice maintained on HFD for 16 wk develop inherent airway hyperreactivity to methacholine, which is further increased by allergen. To our knowledge, this is the first study to report that MitoQ reduces AHR in allergic airway disease, suggesting that the drug potentially has efficacy in lean and obese asthma.
Previous studies suggested that MitoQ may also induce effects through its dTPP+ backbone (55–57). In our experiments, we show that MitoQ at lower concentration was effective at reducing HDM-induced inflammatory cytokine production from pHBEs; at higher concentrations, dTPP+ functioned similarly to MitoQ. In mice treated with equimolar concentration of MitoQ or dTPP+, both reduce HDM-induced cellular inflammation, cytokine production and AHR. However, dTPP+ increased mROS production compared with MitoQ in response to HDM (previous reports suggest dTPP+ can increase mROS in certain cell types) (18, 19).
Although we did not anticipate the anti-inflammatory effects of dTTP, other studies have suggested that although systemic TPP might increase oxidative stress in the lungs (58), it can also have anti-inflammatory/proinflammatory properties (58, 59). The mechanisms of these effects are not clear, but dTPP+ can affect other mitochondrial functions such as calcium signaling (60) and ATP production (61). dTPP+ may also impart antioxidant effects by targeting mitochondrial function (62) that then affects other redox signaling pathways. Multiple mitochondrial pathways are likely involved in mediating allergic airway disease (63). MitoQ has also been shown to reduce mitochondrial DNA damage (64, 65) and induce HIF-1 destabilization and reduction in its transcriptional activity (66) in many models. Hence, the effects of dTTP+ and MitoQ on non-mROS pathways may potentially act to mitigate inflammation.
We next investigated oxidation of reactive cysteine residues in proteins by sulfenylation, as a key post-translational target of redox signaling. Oxidation status of many ER-resident proteins was increased by HDM and reduced only by MitoQ. The UPR is mediated through three pathways, namely, IRE1-XBP1 pathway, ATF6 pathway, and PERK-phospho-EIF2α-CHOP pathway (30). We and others have previously shown that ER stress and consequent UPR are elevated in mice models of allergic asthma and that inhibition of ER stress attenuates allergen-induced airway inflammation, AHR, and airway remodeling (20–23, 67–69). Furthermore, mitochondrial ROS is directly involved in elevated ER stress in allergic asthma (68). The mechanism by which mROS leads to protein oxidation and UPR in the ER is not well studied. However, recent evidence implicates both direct transfer via aquaporin channels as well as peroxiredoxin relay for the transfer of mROS to the ER providing a potential mechanism for mROS affecting ER protein oxidation and downstream ER stress.
Here, we show that MitoQ reduces the UPR via all three activation pathways whereas dTPP+ treatment reduces the IRE1 and PERK arms of the UPR. Phosphorylation of EIF2α is a nodal point of convergence for integrated stress response (ISR) pathways. The PERK-pEIF2α pathway is specifically upregulated in obese allergic mice and is reported to be also increased in certain cell types in obesity and hyperglycemia (70, 71). We speculate that this pathway might be important in the increased levels of eotaxin and eosinophils in lung tissue, though this would require further study. Detailed studies are needed to understand the mechanistic underpinnings of sulfenylation of ER proteins in obese asthma. Our data do suggest that sulfenylation of ER proteins and elevated ER stress are associated with obese allergic airway disease and can be targeted with MitoQ.
In summary, we have shown that MitoQ reduces allergen-induced inflammation, AHR, mucus hypersecretion, airway fibrosis, and protein oxidation in lean and obese mice. The effects of MitoQ due to its dTPP+ backbone are also mediated by attenuation of the UPR independent of effects on mROS, as we found many similar effects for both MitoQ and dTPP+. MitoQ treatment also reduces tissue eosinophilia, a specific feature of obese allergic asthma. MitoQ might be a viable therapy for treating allergic asthma and have efficacy on pathways particularly pertinent to obese asthma.
ETHICAL APPROVALS
Male C57BL/6NJ (The Jackson Laboratory) mice were used for all experiments as approved by the Institutional Animal Care and Use Committee of the University of Vermont (IACUC Protocol No: PROTO202000069).
DATA AVAILABILITY
The mass spectrometry/proteomics data, including raw (.raw), search (.msf), and analysis (.sf3) files, as well as MGF and mzIdentML files exported from Scaffold, have been deposited to the ProteomeXchange Consortiumvia via the PRIDE partner repository (Dataset identifier: PXD029715). Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S10 and Supplemental Tables S1 and S2: https://doi.org/10.6084/m9.figshare.21280395.
GRANTS
This work was supported in part by NIH Grants R01 HL136917 to A.E.D. and V.A. and Grants HL122383 and HL141364 to V.A. Y.-W.L., and C.G., and the Vermont Biomedical Research Network Proteomics Facility (RRID: SCR_018667) was supported by NIH Grant P20GM103449 (Vermont INBRE: Vermont Biomedical Research Network). This work was also supported by NIH Grants 1S10OD025030-01 (University of Vermont Microscopy imaging center) and T32HL076122 (NIH T32 grant).
DISCLOSURES
Y. Janssen-Heininger and V. Anathy hold patents: United States Patent No. 8,679,811, “Treatments Involving Glutaredoxins and Similar Agents”; United States Patent No. 8,877,447, “Detection of Glutathionylated Proteins”; United States Patent Nos. 9,907,828 and 10,688,150, “Treatments of oxidative stress conditions.” In the past, Y. Janssen-Heininger and V. Anathy have received consulting fees and laboratory contracts from Celdara Medical LLC, NH. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
R.C., A.E.D., and V.A. conceived and designed research; R.C., S.R.B., Z.F.M., J.W., S.C., C.G., A.K., N.C., I.M.B., C.R.M., N.D., M.A., and Y.-W.L. performed experiments; R.C. and Y.-W.L. analyzed data; R.C., Y.-W.L., A.E.D., and V.A. interpreted results of experiments; R.C. and Y.-W.L. prepared figures; R.C., A.E.D., and V.A. drafted manuscript; R.C., A.v.d.V., Y.J.-H., M.E.P., A.E.D., and V.A. edited and revised manuscript; R.C., S.R.B., Z.F.M., J.W., S.C., C.G., A.K., N.C., I.M.B., C.R.M., N.D., M.A., Y.-W.L., A.v.d.V., Y.J.-H., M.E.P., A.E.D., and V.A. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors express sincere thanks to Nirav Daphtary and Minara Aliyeva of the Vermont Lung Center for assistance with AHR experiments and analysis. The authors also thank Prof. Douglas Taatjes, Nicole Bufford, and Nicole DeLance of the University of Vermont Microscopy Imaging center for technical support. Graphical abstract image created with BioRender.com and published with permission.
REFERENCES
- 1. Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med 175: 661–666, 2007. doi: 10.1164/rccm.200611-1717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peters U, Dixon AE, Forno E. Obesity and asthma. J Allergy Clin Immunol 141: 1169–1179, 2018. doi: 10.1016/j.jaci.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rönmark E, Andersson C, Nyström L, Forsberg B, Järvholm B, Lundbäck B. Obesity increases the risk of incident asthma among adults. Eur Respir J 25: 282–288, 2005. doi: 10.1183/09031936.05.00054304. [DOI] [PubMed] [Google Scholar]
- 4. Schatz M, Hsu J-WY, Zeiger RS, Chen W, Dorenbaum A, Chipps BE, Haselkorn T. Phenotypes determined by cluster analysis in severe or difficult-to-treat asthma. J Allergy Clin Immunol 133: 1549–1556, 2014. doi: 10.1016/j.jaci.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 5. Vortmann M, Eisner MD. BMI and health status among adults with asthma. Obesity (Silver Spring) 16: 146–152, 2008. doi: 10.1038/oby.2007.7. [DOI] [PubMed] [Google Scholar]
- 6. Holguin F, Bleecker ER, Busse WW, Calhoun WJ, Castro M, Erzurum SC, Fitzpatrick AM, Gaston B, Israel E, Jarjour NN, Moore WC, Peters SP, Yonas M, Teague WG, Wenzel SE. Obesity and asthma: an association modified by age of asthma onset. J Allergy Clin Immunol 127: 1486–1493.e2, 2011. doi: 10.1016/j.jaci.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem 86: 715–748, 2017. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 8. Grasemann H, Holguin F. Oxidative stress and obesity-related asthma. Paediatr Respir Rev 37: 18–21, 2021. doi: 10.1016/j.prrv.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 9. Komakula S, Khatri S, Mermis J, Savill S, Haque S, Rojas M, Brown LAnn, Teague GW, Holguin F. Body mass index is associated with reduced exhaled nitric oxide and higher exhaled 8-isoprostanes in asthmatics. Respir Res 8: 32, 2007. doi: 10.1186/1465-9921-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernandez-Boyanapalli R, Goleva E, Kolakowski C, Min E, Day B, Leung DYM, Riches DWH, Bratton DL, Sutherland ER. Obesity impairs apoptotic cell clearance in asthma. J Allergy Clin Immunol 131: 1041, 2013. doi: 10.1016/j.jaci.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jaiswal AK, Makhija S, Stahr N, Sandey M, Suryawanshi A, Saxena A, Dagur PK, McCoy JP, Levine SJ, Mishra A. Dendritic cell-restricted progenitors contribute to obesity-associated airway inflammation via Adam17-p38 MAPK-dependent pathway. Front Immunol 11: 363, 2020. doi: 10.3389/fimmu.2020.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu X, Lin R, Zhao B, Guan R, Li T, Jin R. Correlation between oxidative stress and the NF-κB signaling pathway in the pulmonary tissues of obese asthmatic mice. Mol Med Rep 13: 1127–1134, 2016. doi: 10.3892/mmr.2015.4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Winnica D, Corey C, Mullett S, Reynolds M, Hill G, Wendell S, Que L, Holguin F, Shiva S. Bioenergetic differences in the airway epithelium of lean versus obese asthmatics are driven by nitric oxide and reflected in circulating platelets. Antioxid Redox Signal 31: 673–686, 2019. doi: 10.1089/ars.2018.7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murphy MP. Understanding and preventing mitochondrial oxidative damage. Biochem Soc Trans 44: 1219–1226, 2016. doi: 10.1042/BST20160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J 29: 4766–4771, 2015. doi: 10.1096/fj.15-275404. [DOI] [PubMed] [Google Scholar]
- 16. Cen M, Ouyang W, Zhang W, Yang L, Lin X, Dai M, Hu H, Tang H, Liu H, Xia J, Xu F. MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox Biol 41: 101936, 2021. doi: 10.1016/j.redox.2021.101936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, Bao W, Pavlidis S, Barnes PJ, Kanerva J, Bittner A, Rao N, Murphy MP, Kirkham PA, Chung KF, Adcock IM, COPDMAP. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 136: 769–780, 2015. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schibler J, Tomanek-Chalkley AM, Reedy JL, Zhan F, Spitz DR, Schultz MK, Goel A. Mitochondrial-targeted decyl-triphenylphosphonium enhances 2-deoxy-D-glucose mediated oxidative stress and clonogenic killing of multiple myeloma cells. PloS one 11: e0167323, 2016. doi: 10.1371/journal.pone.0167323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kloepping KC, Kraus AS, Hedlund DK, Gnade CM, Wagner BA, McCormick ML, Fath MA, Seol D, Lim T-H, Buettner GR, Goswami PC, Pigge FC, Spitz DR, Schultz MK. Triphenylphosphonium derivatives disrupt metabolism and inhibit melanoma growth in vivo when delivered via a thermosensitive hydrogel. PloS One 15: e0244540, 2020. doi: 10.1371/journal.pone.0244540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pathinayake PS, Waters DW, Nichol KS, Brown AC, Reid AT, Hsu AC-Y, Horvat JC, Wood LG, Baines KJ, Simpson JL, Gibson PG, Hansbro PM, Wark PAB. Endoplasmic reticulum-unfolded protein response signalling is altered in severe eosinophilic and neutrophilic asthma. Thorax 77: 443–451, 2022. doi: 10.1136/thoraxjnl-2020-215979. [DOI] [PubMed] [Google Scholar]
- 21. Hoffman SM, Tully JE, Nolin JD, Lahue KG, Goldman DH, Daphtary N, Aliyeva M, Irvin CG, Dixon AE, Poynter ME, Anathy V. Endoplasmic reticulum stress mediates house dust mite-induced airway epithelial apoptosis and fibrosis. Respir Res 14: 141, 2013. doi: 10.1186/1465-9921-14-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoffman SM, Chapman DG, Lahue KG, Cahoon JM, Rattu GK, Daphtary N, Aliyeva M, Fortner KA, Erzurum SC, Comhair SAA, Woodruff PG, Bhakta N, Dixon AE, Irvin CG, Janssen-Heininger YMW, Poynter ME, Anathy V. Protein disulfide isomerase-endoplasmic reticulum resident protein 57 regulates allergen-induced airways inflammation, fibrosis, and hyperresponsiveness. J Allergy Clin Immunol 137: 822–832.e7, 2016. doi: 10.1016/j.jaci.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim SR, Kim DI, Kang MR, Lee KS, Park SY, Jeong JS, Lee YC. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor κB activation. J Allergy Clin Immunol 132: 1397–1408, 2013. doi: 10.1016/j.jaci.2013.08.041. [DOI] [PubMed] [Google Scholar]
- 24. Manuel AM, van de Wetering C, MacPherson M, Erickson C, Murray C, Aboushousha R, van der Velden J, Dixon AE, Poynter ME, Irvin CG, Taatjes DJ, van der Vliet A, Anathy V, Janssen-Heininger YMW. Dysregulation of pyruvate kinase M2 promotes inflammation in a mouse model of obese allergic asthma. Am J Respir Cell Mol Biol 64: 709–721, 2021. doi: 10.1165/rcmb.2020-0512OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hristova M, Veith C, Habibovic A, Lam Y-W, Deng B, Geiszt M, Janssen-Heininger YMW, van der Vliet A. Identification of DUOX1-dependent redox signaling through protein S-glutathionylation in airway epithelial cells. Redox Biol 2: 436–446, 2014. doi: 10.1016/j.redox.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schroeder BW, Verhaeghe C, Park S-W, Nguyenvu LT, Huang X, Zhen G, Erle DJ. AGR2 is induced in asthma and promotes allergen-induced mucin overproduction. Am J Respir Cell Mol Biol 47: 178–185, 2012. doi: 10.1165/rcmb.2011-0421OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Long AJ, Sypek JP, Askew R, Fish SC, Mason LE, Williams CMM, Goldman SJ. Gob-5 contributes to goblet cell hyperplasia and modulates pulmonary tissue inflammation. Am J Respir Cell Mol Biol 35: 357–365, 2006. doi: 10.1165/rcmb.2005-0451OC. [DOI] [PubMed] [Google Scholar]
- 28. Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev 113: 4633–4679, 2013. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang L, Wang M, Kang X, Boontheung P, Li N, Nel AE, Loo JA. Oxidative stress and asthma: proteome analysis of chitinase-like proteins and FIZZ1 in lung tissue and bronchoalveolar lavage fluid. J Proteome Res 8: 1631–1638, 2009. doi: 10.1021/pr800685h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Janssen-Heininger Y, Reynaert NL, van der Vliet A, Anathy V. Endoplasmic reticulum stress and glutathione therapeutics in chronic lung diseases. Redox Biol 33: 101516, 2020. doi: 10.1016/j.redox.2020.101516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holguin F, Fitzpatrick A. Obesity, asthma, and oxidative stress. J Appl Physiol (1985) 108: 754–759, 2010. doi: 10.1152/japplphysiol.00702.2009. [DOI] [PubMed] [Google Scholar]
- 32. Bhatraju NK, Agrawal A. Mitochondrial dysfunction linking obesity and asthma. Ann Am Thorac Soc 14, Suppl 5: S368–S373, 2017. doi: 10.1513/AnnalsATS.201701-042AW. [DOI] [PubMed] [Google Scholar]
- 33. Xu W, Ghosh S, Comhair SAA, Asosingh K, Janocha AJ, Mavrakis DA, Bennett CD, Gruca LL, Graham BB, Queisser KA, Kao CC, Wedes SH, Petrich JM, Tuder RM, Kalhan SC, Erzurum SC. Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J Clin Invest 126: 2465–2481, 2016. doi: 10.1172/JCI82925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nadeem A, Chhabra SK, Masood A, Raj HG. Increased oxidative stress and altered levels of antioxidants in asthma. J Allergy Clin Immunol 111: 72–78, 2003. doi: 10.1067/mai.2003.17. [DOI] [PubMed] [Google Scholar]
- 35. van der Vliet A, Janssen-Heininger YMW, Anathy V. Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol Aspects Med 63: 59–69, 2018. doi: 10.1016/j.mam.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RAJ, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int 30: 1019–1026, 2010. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 37. Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RAJ, Murphy MP, Taylor KM, Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord 25: 1670–1674, 2010. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 38. Silva FMC, Oliveira EE, Gouveia ACC, Brugiolo ASS, Alves CC, Correa JOA, Gameiro J, Mattes J, Teixeira HC, Ferreira AP. Obesity promotes prolonged ovalbumin-induced airway inflammation modulating T helper type 1 (Th1), Th2 and Th17 immune responses in BALB/c mice. Clin Exp Immunol 189: 47–59, 2017. doi: 10.1111/cei.12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ather JL, Chung M, Hoyt LR, Randall MJ, Georgsdottir A, Daphtary NA, Aliyeva MI, Suratt BT, Bates JHT, Irvin CG, Russell SR, Forgione PM, Dixon AE, Poynter ME. Weight loss decreases inherent and allergic methacholine hyperresponsiveness in mouse models of diet-induced obese asthma. Am J Respir Cell Mol Biol 55: 176–187, 2016. doi: 10.1165/rcmb.2016-0070OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park HJ, Lee J-H, Park YH, Han H, Sim DW, Park KH, Park J-W. Roflumilast ameliorates airway hyperresponsiveness caused by diet-induced obesity in a murine model. Am J Respir Cell Mol Biol 55: 82–91, 2016. doi: 10.1165/rcmb.2015-0345OC. [DOI] [PubMed] [Google Scholar]
- 41. Johnston RA, Zhu M, Rivera-Sanchez YM, Lu FL, Theman TA, Flynt L, Shore SA. Allergic airway responses in obese mice. Am J Respir Crit Care Med 176: 650–658, 2007. doi: 10.1164/rccm.200702-323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lintomen L, Calixto MC, Schenka A, Antunes E. Allergen-induced bone marrow eosinophilopoiesis and airways eosinophilic inflammation in leptin-deficient ob/ob mice. Obesity (Silver Spring) 20: 1959–1965, 2012. doi: 10.1038/oby.2012.93. [DOI] [PubMed] [Google Scholar]
- 43. Calixto MC, Lintomen L, Schenka A, Saad MJ, Zanesco A, Antunes E. Obesity enhances eosinophilic inflammation in a murine model of allergic asthma. Br J Pharmacol 159: 617–625, 2010. doi: 10.1111/j.1476-5381.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Diaz J, Warren L, Helfner L, Xue X, Chatterjee PK, Gupta M, Solanki MH, Esposito M, Bonagura V, Metz CN. Obesity shifts house dust mite-induced airway cellular infiltration from eosinophils to macrophages: effects of glucocorticoid treatment. Immunol Res 63: 197–208, 2015. doi: 10.1007/s12026-015-8717-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johnston RA, Theman TA, Lu FL, Terry RD, Williams ES, Shore SA. Diet-induced obesity causes innate airway hyperresponsiveness to methacholine and enhances ozone-induced pulmonary inflammation. J Appl Physiol (1985) 104: 1727–1735, 2008. doi: 10.1152/japplphysiol.00075.2008. [DOI] [PubMed] [Google Scholar]
- 46. Dixon AE, Poynter ME. Mechanisms of asthma in obesity. Pleiotropic aspects of obesity produce distinct asthma phenotypes. Am J Respir Cell Mol Biol 54: 601–608, 2016. doi: 10.1165/rcmb.2016-0017PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Desai D, Newby C, Symon FA, Haldar P, Shah S, Gupta S, Bafadhel M, Singapuri A, Siddiqui S, Woods J, Herath A, Anderson IK, Bradding P, Green R, Kulkarni N, Pavord I, Marshall RP, Sousa AR, May RD, Wardlaw AJ, Brightling CE. Elevated sputum interleukin-5 and submucosal eosinophilia in obese individuals with severe asthma. Am J Respir Crit Care Med 188: 657–663, 2013. doi: 10.1164/rccm.201208-1470OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van der Wiel E, Ten Hacken NH, van den Berge M, Timens W, Reddel HK, Postma DS. Eosinophilic inflammation in subjects with mild-to-moderate asthma with and without obesity: disparity between sputum and biopsies. Am J Respir Crit Care Med 189: 1281–1284, 2014. doi: 10.1164/rccm.201310-1841LE. [DOI] [PubMed] [Google Scholar]
- 49. van Veen IH, Ten Brinke A, Sterk PJ, Rabe KF, Bel EH. Airway inflammation in obese and nonobese patients with difficult-to-treat asthma. Allergy 63: 570–574, 2008. doi: 10.1111/j.1398-9995.2007.01597.x. [DOI] [PubMed] [Google Scholar]
- 50. Lessard A, Turcotte H, Cormier Y, Boulet LP. Obesity and asthma: a specific phenotype? Chest 134: 317–323, 2008. doi: 10.1378/chest.07-2959. [DOI] [PubMed] [Google Scholar]
- 51. Farahi N, Loutsios C, Tregay N, Wright AKA, Berair R, Lok LSC, Gillett D, Cullum I, Simmonds RP, Summers C, Wong A, Solanki CK, Buscombe J, Pang PH, Thavakumar A, Peters AM, Brightling CE, Condliffe AM, Chilvers ER. In vivo imaging reveals increased eosinophil uptake in the lungs of obese asthmatic patients. J Allergy Clin Immunol 142: 1659–1662.e8, 2018. doi: 10.1016/j.jaci.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Persson C, Uller L. Transepithelial exit of leucocytes: inflicting, reflecting or resolving airway inflammation? Thorax 65: 1111–1115, 2010. doi: 10.1136/thx.2009.133363. [DOI] [PubMed] [Google Scholar]
- 53. Uller L, Lloyd CM, Rydell-Törmänen K, Persson CG, Erjefält JS. Effects of steroid treatment on lung CC chemokines, apoptosis and transepithelial cell clearance during development and resolution of allergic airway inflammation. Clin Exp Allergy 36: 111–121, 2006. doi: 10.1111/j.1365-2222.2006.02396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim HY, Lee HJ, Chang Y-J, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 20: 54–61, 2014. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gottwald EM, Duss M, Bugarski M, Haenni D, Schuh CD, Landau EM, Hall AM. The targeted anti-oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue. Physiol Rep 6: e13667, 2018. doi: 10.14814/phy2.13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bond ST, Kim J, Calkin AC, Drew BG. The antioxidant moiety of MitoQ imparts minimal metabolic effects in adipose tissue of high fat fed mice. Front Physiol 10: 543, 2019. doi: 10.3389/fphys.2019.00543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang W, Cash N, Wen L, Szatmary P, Mukherjee R, Armstrong J, Chvanov M, Tepikin AV, Murphy MP, Sutton R, Criddle DN. Effects of the mitochondria-targeted antioxidant mitoquinone in murine acute pancreatitis. Mediators Inflamm 2015: 901780, 2015. doi: 10.1155/2015/901780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Powell RD, Goodenow DA, Christmas AB, McKillop IH, Evans SL. Effect of systemic triphenylphosphonium on organ function and oxidative stress. Am Surg 84: 36–42, 2018. doi: 10.1177/000313481808400119. [DOI] [PubMed] [Google Scholar]
- 59. Powell RD, Swet JH, Kennedy KL, Huynh TT, Murphy MP, Mckillop IH, Evans SL. MitoQ modulates oxidative stress and decreases inflammation following hemorrhage. J Trauma Acute Care Surg 78: 573–579, 2015. doi: 10.1097/TA.0000000000000533. [DOI] [PubMed] [Google Scholar]
- 60. Leo S, Szabadkai G, Rizzuto R. The mitochondrial antioxidants MitoE2 and MitoQ10 increase mitochondrial Ca2+ load upon cell stimulation by inhibiting Ca2+ efflux from the organelle. Ann NY Acad Sci 1147: 264–274, 2008. doi: 10.1196/annals.1427.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ozsvari B, Sotgia F, Lisanti MP. Exploiting mitochondrial targeting signal(s), TPP and bis-TPP, for eradicating cancer stem cells (CSCs). Aging (Albany NY) 10: 229–240, 2018. doi: 10.18632/aging.101384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Armstrong JA, Cash NJ, Morton JC, Tepikin AV, Sutton R, Criddle DN. Mitochondrial targeting of antioxidants alters pancreatic acinar cell bioenergetics and determines cell fate. Int J Mol Sci 20: 1700, 2019. doi: 10.3390/ijms20071700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Qian L, Mehrabi Nasab E, Athari SM, Athari SS. Mitochondria signaling pathways in allergic asthma. J Investig Med 70: 863–882, 2022. doi: 10.1136/jim-2021-002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hu Q, Ren J, Li G, Wu J, Wu X, Wang G, Gu G, Ren H, Hong Z, Li J. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis 9: 403, 2018. doi: 10.1038/s41419-018-0436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Williamson J, Hughes CM, Cobley JN, Davison GW. The mitochondria-targeted antioxidant MitoQ, attenuates exercise-induced mitochondrial DNA damage. Redox biology 36: 101673, 2020. doi: 10.1016/j.redox.2020.101673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sanjuán-Pla A, Cervera AM, Apostolova N, Garcia-Bou R, Víctor VM, Murphy MP, McCreath KJ. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1α. FEBS Lett 579: 2669–2674, 2005. doi: 10.1016/j.febslet.2005.03.088. [DOI] [PubMed] [Google Scholar]
- 67. Miller M, Tam AB, Cho JY, Doherty TA, Pham A, Khorram N, Rosenthal P, Mueller JL, Hoffman HM, Suzukawa M, Niwa M, Broide DH. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc Natl Acad Sci USA 109: 16648–16653, 2012. doi: 10.1073/pnas.1204151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee KS, Jeong JS, Kim SR, Cho SH, Kolliputi N, Ko YH, Lee KB, Park SC, Park HJ, Lee YC. Phosphoinositide 3-kinase-δ regulates fungus-induced allergic lung inflammation through endoplasmic reticulum stress. Thorax 71: 52–63, 2016. doi: 10.1136/thoraxjnl-2015-207096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nakada EM, Bhakta NR, Korwin-Mihavics BR, Kumar A, Chamberlain N, Bruno SR, Chapman DG, Hoffman SM, Daphtary N, Aliyeva M, Irvin CG, Dixon AE, Woodruff PG, Amin S, Poynter ME, Desai DH, Anathy V. Conjugated bile acids attenuate allergen-induced airway inflammation and hyperresponsiveness by inhibiting UPR transducers. JCI Insight 4: e98101, 2019. doi: 10.1172/jci.insight.98101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vega-Martín E, González-Blázquez R, Manzano-Lista FJ, Martín-Ramos M, García-Prieto CF, Viana M, Rubio MA, Calle-Pascual AL, Lionetti L, Somoza B, Fernández-Alfonso MS, Alcalá M, Gil-Ortega M. Impact of caloric restriction on AMPK and endoplasmic reticulum stress in peripheral tissues and circulating peripheral blood mononuclear cells from Zucker rats. J Nutr Biochem 78: 108342, 2020. doi: 10.1016/j.jnutbio.2020.108342. [DOI] [PubMed] [Google Scholar]
- 71. Escribano-Lopez I, Bañuls C, Diaz-Morales N, Iannantuoni F, Rovira-Llopis S, Gomis R, Rocha M, Hernandez-Mijares A, Murphy MP, Victor VM. The mitochondria-targeted antioxidant MitoQ modulates mitochondrial function and endoplasmic reticulum stress in pancreatic β cells exposed to hyperglycaemia. Cell Physiol Biochem 52: 186–197, 2019. doi: 10.33594/000000013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S10 and Supplemental Tables S1 and S2: https://doi.org/10.6084/m9.figshare.21280395.
Data Availability Statement
The mass spectrometry/proteomics data, including raw (.raw), search (.msf), and analysis (.sf3) files, as well as MGF and mzIdentML files exported from Scaffold, have been deposited to the ProteomeXchange Consortiumvia via the PRIDE partner repository (Dataset identifier: PXD029715). Data will be made available upon reasonable request.