

Abstract
Objective:
To identify hallmark genes and biomolecular processes in aortitis using high-throughput gene expression profiling. Additionally, to provide a range of potentially new drug targets (genes) and therapeutics from a pharmacogenomic network analysis.
Methods:
Bulk RNA-seq was performed on surgically resected ascending aortic tissues from inflammatory (giant cell arteritis [GCA] with or without polymyalgia rheumatica [PMR] = 8; clinically isolated aortitis [CIA] = 17) and non-inflammatory (n = 25) aneurysms undergoing surgical aortic repair. Differentially expressed genes (DEGs) between the two patient groups were identified while controlling for clinical covariates. A protein-protein interaction model, drug-gene target information, and the DEGs were used to construct a pharmacogenomic network for identifying promising drug targets and potentially new treatment strategies in aortitis.
Results:
Overall, tissue gene expression patterns were the most associated with disease state than with any other clinical characteristic. We identified 159 and 93 genes that were significantly upregulated and downregulated, respectively, in inflammatory aortic aneurysms compared to non-inflammatory aortic aneurysms. We found that the upregulated genes were enriched in immune-related functions, whereas the downregulated genes were enriched in neuronal processes. Notably, gene expression profiles of inflammatory aortic aneurysms from patients with GCA were no different than those from patients with CIA. Finally, our pharmacogenomic network analysis identified genes that could potentially be targeted by immunosuppressive drugs currently approved for other inflammatory diseases.
Conclusion:
We performed the first global transcriptomics analysis in inflammatory aortic aneurysms from surgically resected aortic tissues. We identified signature genes and biomolecular processes, while finding that CIA may be a limited presentation of GCA. Moreover, our computational network analysis revealed potential novel strategies for pharmacologic interventions, and suggests future biomarker discovery directions for the precise diagnosis and treatment of aortitis.
Introduction
The etiology and pathogenic mechanisms leading to non-infectious inflammation of the aortic wall (aortitis) remain largely unknown. In the clinical practice, distinguishing patients with inflammatory aortic aneurysms from those with non-inflammatory aortic aneurysms may be difficult, since aortitis may be asymptomatic or associated with non-specific symptoms (1,2). Moreover, there are currently no laboratory diagnostic markers specifically for aortitis or for two of its most common underlying conditions, i.e., giant cell arteritis (GCA) and clinically isolated aortitis (CIA) (1,3,4). Therefore, the unmet need for patients with aortitis include the discovery of novel biomolecular features that stratify inflammatory and non-inflammatory aortic aneurysms, and thereby complement current diagnostic approaches and improve long-term treatment outcomes.
Previously, Quimson et al. investigated the histopathological and radiological differences between inflammatory and non-inflammatory aortic aneurysms in patients who underwent open aortic aneurysm repair (2). Their study uncovered five factors (i.e., age at the time of surgery, sex, absence of coronary artery disease, diameter of the aneurysms, arterial wall thickening) that were associated with aortitis. Moreover, the investigators found that, among patients who underwent open surgical repair of aortic aneurysms (inflammatory or non-inflammatory), elderly women with no history of coronary artery disease and aortic wall thickening were more likely to have histologic evidence of aortitis.
Despite the significance of previous observations by others, our understanding of aortitis can be advanced further by identifying disease-associated biomolecular processes using high-throughput technologies. To this point, genome-wide expression analyses with RNA-seq—which have yet to be performed in aortitis—provide a promising avenue for subsequent studies (5).
In this study, we performed for the first time global transcriptomic profiling using RNA-seq on surgically resected, inflammatory and non-inflammatory aortic aneurysms in order to reveal differences in their tissue gene expression. In all, our approach demonstrates the utility of bulk transcriptomic sequencing for the discovery of not only signature genes and cellular functions of aortitis, but also potentially novel therapeutic targets.
Materials and methods
Subject and aortic specimen identification
Subjects for which thoracic aortic replacement was performed between January 1, 2012 and December 31, 2019 were identified retrospectively through the use of current procedural terminology (CPT) coding. All aortic specimens had been previously reviewed by a vascular histopathologist. Inclusion criteria for aortitis samples was a description of “active giant cell aortitis” in the resected ascending aortic tissue. Charts were manually reviewed and patients with features suggestive of localized or systemic infection were excluded. Age- and sex-matched comparators were identified among patients with non-inflammatory aortic aneurysm resection during the same study period. Clinical parameters, including erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), historical use of glucocorticoids, glucocorticoid dose at time of surgery, presence of systemic symptoms, history of rheumatic disease, smoking history, and use of aspirin, statin and angiotensin converting enzyme/angiotensin II receptor blocker (ACE/ARB) medications, were abstracted for both inflammatory aortic aneurysm (aortitis) and non-inflammatory aortic aneurysm comparators. The clinical and demographic characteristics of the study participants are summarized in Table 1.
Table 1.
Clinical and demographic characteristics of study participants.
| Inflammatory aortic aneurysms (n = 25) | Non-inflammatory aortic aneurysms (n = 25) | |
|---|---|---|
| Sex (female/male) | 15/10 | 17/8 |
|
| ||
| Age (years) | ||
| Median [IQR] | 75.9 [70.5–78.0] | 72.1 [69.4–76.1] |
| Range (min–max) | 61.1–84.3 | 54.5–83.5 |
|
| ||
| ESR (mm/hr) | ||
| Median [IQR] | 6.0 [4.3–16.5] | 12.5 [7.8–25.3] |
| Range (min–max) | 0–25 | 2–43 |
| N/A (n) | 11 | 15 |
|
| ||
| CRP (mg/L) | ||
| Median [IQR] | 4.25 [3.0–5.7] | 2.9 [2.8–3.7] |
| Range (min–max) | 2.90–14.1 | 2.0–64.1 |
| N/A (n) | 11 | 17 |
|
| ||
| Treatment | ||
| Prednisone (n, %) | 1 (4.0%) | 0 (0.0 %) |
| Aspirin (n, %) | 15 (60.0%) | 12 (48.0%) |
| Statin (n, %) | 9 (36.0%) | 15 (60.0%) |
| ACE/ARB (n, %) | 11 (44.0%) | 18 (72.0%) |
|
| ||
| Smoking history (n) | ||
| Current | 6 | 3 |
| Former | 9 | 8 |
| Never | 10 | 14 |
|
| ||
| History of other rheumatic diseases | ||
| PMRα | 5 | 0 |
| GCAβ | 2 | 0 |
| GCA/PMRγ | 1 | 0 |
| Others (Iritis, Psoriasis, Gout) | 0 | 3 |
| None | 17 | 22 |
Polymyalgia rheumatic
Giant cell arteritis
Giant cell arteritis with polymyalgia rheumatica.
FFPE block sectioning and aortic tissue preparation
Formalin-fixed paraffin-embedded (FFPE) blocks containing ascending aortic aneurysm tissues were cut at 10 μm thick sections. Prior to cutting, the microtomes and workstations were cleaned to prevent DNase and RNase contamination. New blades were used between blocks. The floatation bath contained Milli-Q water (DNase- and RNase-free) and was cleaned between blocks. CitriSolv™ was used to remove paraffin. Tissues were then washed with absolute ethanol and dried with a heat block at 37°C.
RNA purification, library preparation, and sequencing
Once dry, the tissues were added with buffer PDK and Proteinase K, and placed in a QIAcube for the RNA purification (extraction). On the instrument, the wash buffers used were RBC, RPE, and ethanol. RNA was then eluted in RNase-free water. The quality control of the total RNA was performed by the Qubit and Agilent 2100 Bioanalyzer (Agilent Technology). DV200 values (the percentage of RNA fragments > 200 nucleotides) were determined by 2100 expert software. Samples with DV200 values above 30% were used for library preparation. Libraries were prepared using the TruSeq RNA Exome Capture kit (Illumina, CA, USA) following the manufacturers’ protocol with minor modifications. Briefly, 500ng of FFPE RNA was used for synthesizing the first-strand cDNA at 42 °C, and the second strand cDNA was generated at 16 °C for one hour with a second-strand marking buffer. Double stranded cDNA was A-tailed, ligated with index adapters, and amplified over 15 cycles. The cDNA library was quantified using Qbit (Thermo Fisher Scientific, MA, USA) and Agilent TapeStation D1000 (Agilent, CA, USA), and 200ng of each library was pooled for exome enrichment and capture. The pooled library was amplified over 10 cycles after finishing the second enrichment. The final libraries were quantified by Agilent TapeStation D1000 and Qubit dsDNA BR Assay kit. Finally, the 101-bp, paired-end reads were sequenced on an Illumina HiSeq4000 platform. Importantly, samples of inflammatory and non-inflammatory aortic aneurysms were not sequenced separately, and thus negating the need for batch correction protocols.
Pre-processing and aligning RNA-seq data
FASTQC was used to estimate the quality of the generated paired-end reads (.fastq files). No files were reported to have a flag of poor sequence quality. Paired-end raw reads were trimmed by trimmomatic (v0.38) (6) with the following parameter: ILLUMINACLIP:TruSeq3-PE:2:30:10. Afterwards, STAR (v2.5.4b) (7) was used to align the trimmed paired-end reads on the human reference genome (hg38). RSEM (v1.3.1; --star-sjdboverhang 100) (8) was used to calculate transcripts per million (TPM) from the .bam files generated by STAR. Gene annotations for hg38 were retrieved from the UCSC genome browser.
Investigation of global transcriptome variance
Transcriptomes composed of log2-transformed TPM (with a pseudocount addition of 0.001) values of 26,475 genes from 50 samples (25 inflammatory aortic aneurysms and 25 non-inflammatory aortic aneurysms) were projected onto a principal component analysis (PCA) ordination plot. Agglomerative hierarchical clustering (Euclidean distance, complete-linkage) was performed on the gene expression profiles (n = 50) to observe clusters associated with clinical and demographic characteristics (i.e., inflammatory status of aortitis, ACE/ARB use, aspirin use, sex, history of other rheumatic diseases, smoking history, and statin use).
Identification of clinical covariates
A logistic linear regression model was used to identify clinical covariates associated with inflammatory and non-inflammatory aortic aneurysms. The inflammatory status (inflammatory or non-inflammatory) was used as the response variable, while the predictors (i.e., sex, smoking history, age, aspirin use, statin use, ACE/ARB use, history of other rheumatic diseases) were individually assessed. P-values were retrieved for the corresponding regression coefficient of the predictor variables. Predictors with the significance of P < 0.1 were considered as potential confounders and were adjusted for during the identification of differentially expressed genes (DEGs).
Identification of differentially expressed genes
DESeq2 (v1.26.0) (9) was used to identify DEGs between patient groups of (i) inflammatory (n = 25) and non-inflammatory (n = 25) aortic aneurysms, and (ii) aortitis with GCA/PMR (documented diagnosis or clinical features compatible with GCA and/or polymyalgia rheumatica [PMR]; n = 8) and CIA (n = 17; inflammatory aortitis without diagnosis of, or clinical features compatible with, either GCA or PMR). Of note, statin use, ACE/ARB use, and history of other rheumatic diseases were considered as potential confounders (P < 0.1, coefficient of the logistic regression model), and were adjusted for during the identification of upregulated (log2(fold-change) > 2, Benjamini-Hochberg (BH)-adjusted P < 0.01) and downregulated (log2(fold-change) < −2, BH-adjusted P < 0.01) DEGs. All DEG analysis results are summarized in Supplementary Table S1.
Functional annotation and gene-set enrichment analysis
The upregulated and downregulated DEGs were analyzed with PANTHER (v16.0) (10) and DAVID (v6.8) (11) for functional annotations and gene set enrichment analysis, respectively. PANTHER was used to obtain the genes’ protein class and biological pathway annotations; and DAVID was used to obtain statistically enriched Gene Ontology (GO) terms (12) from the following categories: Biological Process (GOTERM_BP_FAT), Cellular Component (GOTERM_CC_FAT), and Molecular Function (GOTERM_MF_FAT). GO terms with EASE scores (i.e., a P-value from a modified Fisher’s Exact test (11)) less than 0.05 were considered to be statistically enriched.
Construction of the pharmacogenomic network
A pharmacogenomic network was constructed with human protein-protein interaction (PPI) information from the STRING database (v11) (13) and drug-gene interaction (DGI) information from the Drug Gene Interaction Database (DGIdb) (v4.0) (14). First, a DEG-specific interactome was constructed by mapping the aforementioned upregulated and downregulated DEGs to the “high-confidence” (combined score > 0.7 in STRING) PPI network. From this high-confidence PPI network, the following nodes and edges were discarded during network construction: nodes (genes and proteins were considered as equals) that were not mapped by DEGs; edges (interaction between two different nodes) that do not connect two different DEGs. As a result, an interactome comprising 71 nodes (DEGs) and 122 edges was constructed (Supplementary Figure S1). From this, the largest connected component (LCC) of the interactome was identified, leaving 36 nodes and 92 edges. Finally, DGIdb was used to create a pharmacogenomic network by linking FDA-approved drugs that are known to target (directly or indirectly) any of the corresponding nodes of the LCC.
Results
Genome-wide expression profiles of inflammatory and non-inflammatory aortic aneurysms
Figure 1A illustrates our analysis strategy to identify differential gene expression signatures between inflammatory and non-inflammatory aortic aneurysms from 50 surgically resected aortic tissue samples. We first investigated whether the clinical characteristics (i.e., disease condition, ACE/ARB use, aspirin use, statin use, sex, history of other rheumatic diseases, smoking history) cluster according to the gene expression profiles acquired from the FFPE tissues (Materials and Methods). Using PCA (Figure 1B) and hierarchical clustering (Figure 1C), we observed that gene expression profiles of 26,475 genes were the most distinguishable based upon disease condition (inflammatory or non-inflammatory aortic aneurysm) compared to other clinical characteristics.
Figure 1. Data analysis pipeline and unsupervised clustering results on genome-wide expression (transcriptome) profiles of inflammatory and non-inflammatory aortic aneurysms.
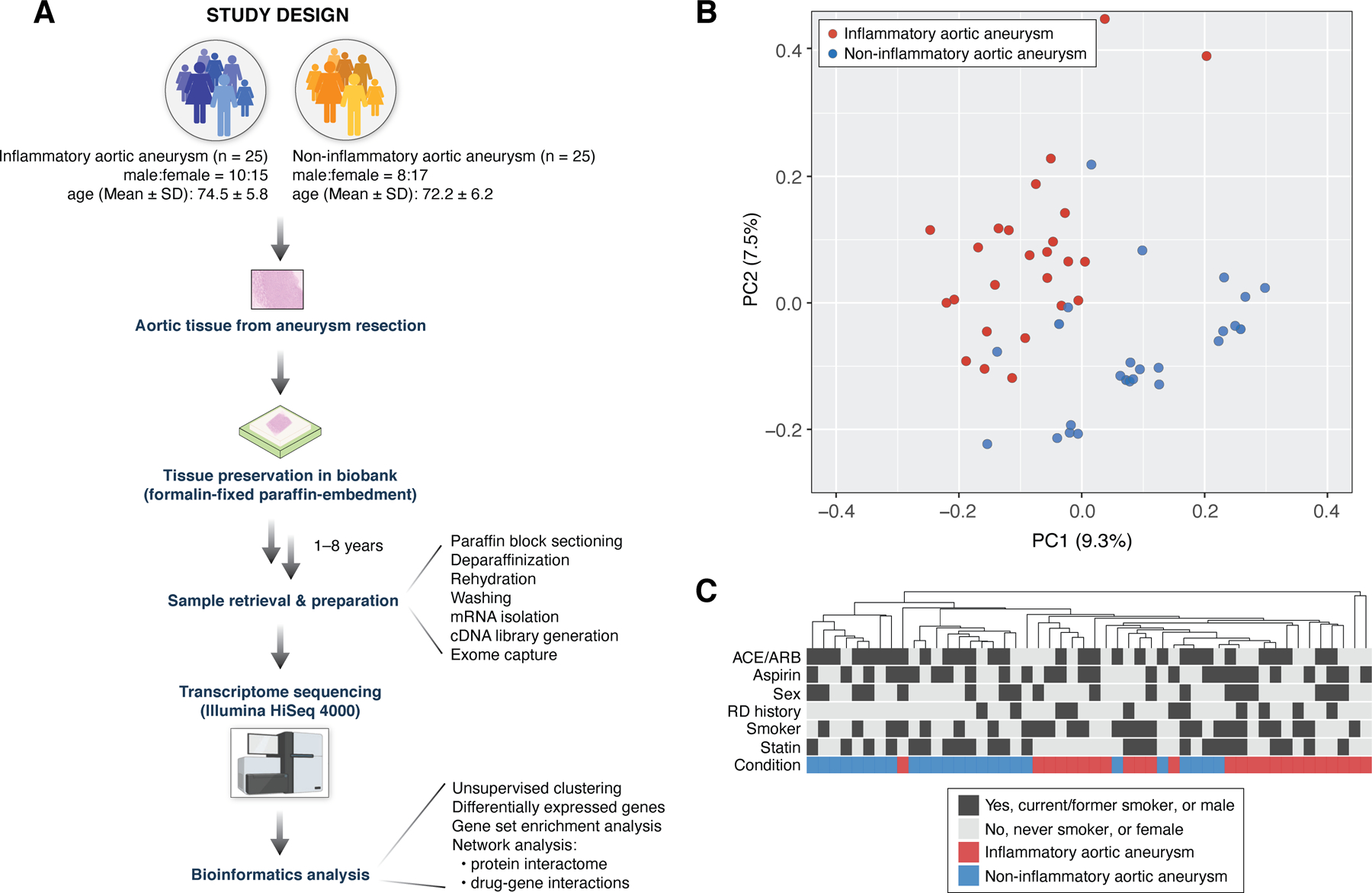
(A) Study design to investigate transcriptomic differences between inflammatory and non-inflammatory aortic aneurysms. (B) PCA on gene expression profiles (26,475 total genes) from 50 surgically resected ascending aortic tissue samples across the two patient groups (inflammatory aortic aneurysm, n = 25; non-inflammatory aneurysm, n = 25). (C) Hierarchical clustering on all 50 gene expression profiles shows that samples cluster together by disease condition (inflammatory/non-inflammatory aortic aneurysms) more so than by any other clinical characteristic (i.e., ACE/ARB use, aspirin use, sex, RD history, smoking history, and statin use). Heatmap of gene expression profiles is not shown due to space constraints. PCA: principal component analysis; ACE/ARB: angiotensin converting enzyme inhibitors/angiotensin receptor blockers; RD history: history of other rheumatic diseases.
Next, we examined whether there were any statistical associations between disease condition and other clinical characteristics to identify potential confounders in our study. Using logistic regression (Materials and Methods), we identified ACE/ARB use (P = 0.048), statin use (P = 0.093), and the history of other rheumatic diseases (P = 0.099) to be associated with disease condition. Henceforth, these clinical characteristics were considered as confounding variables while investigating the relationship between inflammatory/non-inflammatory aortic aneurysm and global gene expression. In contrast, aspirin use (P = 0.396), sex (P = 0.556), and smoking history (P = 0.260) were considered not to be associated with disease condition.
Identification of differentially expressed genes between inflammatory and non-inflammatory aortic aneurysms
We found transcriptomic differences between inflammatory and non-inflammatory aortic aneurysms while adjusting for the aforementioned clinical covariates. From 26,475 total genes, we identified 159 upregulated genes in inflammatory aortic aneurysms, including CXCL9, TEX28, CLEC5A, and OR8B2; and 93 downregulated genes, including PLD5, SFRP1, CARTPT, and FAR2P1 (Figure 2A). Among the 159 upregulated genes, 99 mapped onto PANTHER protein classes (Materials and Methods), including ‘Metabolite interconversion enzyme’ (24 of 99, 24.2%), ‘Defense/immunity protein’ (13 of 99, 13.1%) and ‘Intercellular signal molecule’ (12 of 99, 12.1%) (Figure 2B). Additionally, 29 upregulated genes mapped onto PANTHER biological pathways, of which ‘Inflammation mediated by chemokine and cytokine signaling pathway’ (6 of 29, 20.7%) was the most abundant annotation (Figure 2C). Alternatively, among the 93 downregulated genes, 50 genes mapped onto PANTHER protein classes, which include ‘Transporter’ (13 of 50, 26%), ‘Transmembrane signal receptor’ (7 of 50, 14%), and ‘Cell adhesion molecule’ (5 of 50, 10%) (Figure 2D). Finally, 56 of the downregulated genes mapped onto PANTHER biological pathways, of which ‘Wnt signaling pathway’ (6 of 56, 10.7%) was the most abundant (Figure 2E). P-values and fold-changes for all genes are listed in Supplementary Table S1. Full details of our functional classification results are summarized in Supplementary Tables S2–5.
Figure 2. Differentially expressed genes (DEGs) and their functional categories reveal transcriptomic signatures of inflammatory aortic aneurysms.
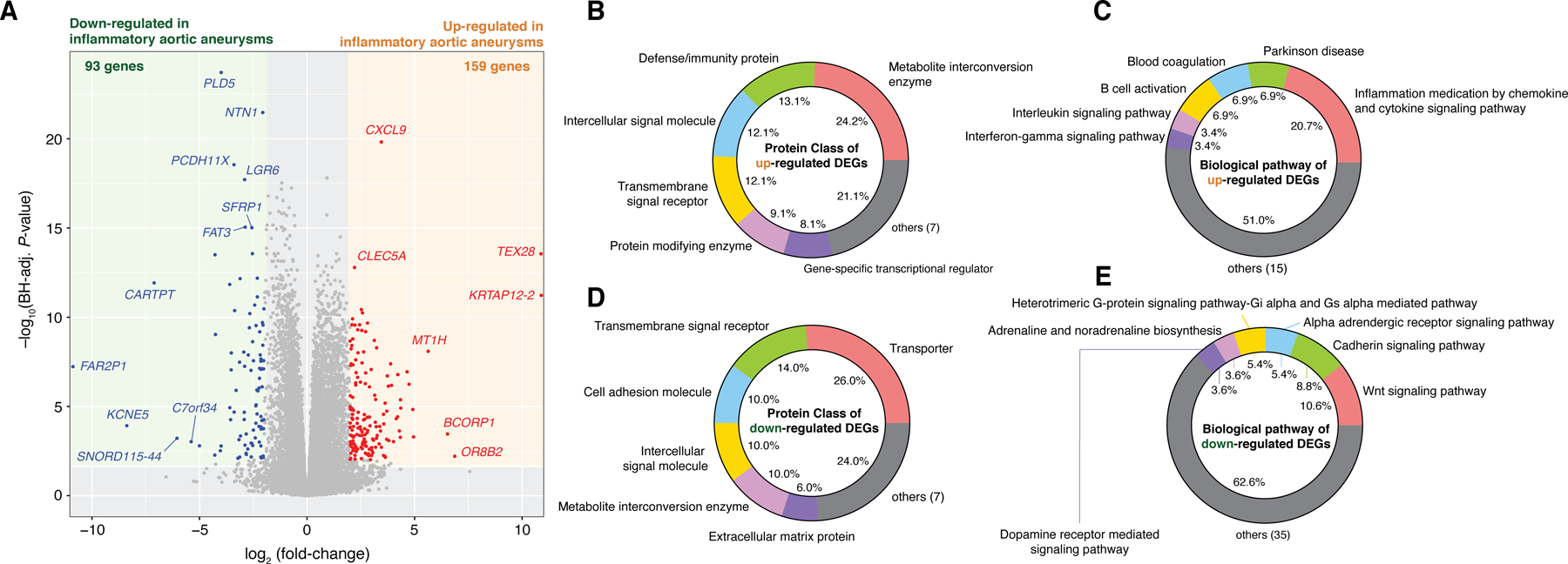
(A) A total of 159 and 93 genes were identified to be significantly upregulated and downregulated, respectively, in inflammatory aortic aneurysms (Benjamini-Hochberg adjusted P-value < 0.01 and |log2(fold-change in mean expression values)| > 2). (B) Protein classes and (C) biological pathways of the upregulated DEGs. (D) Protein classes and (E) biological pathways of the downregulated DEGs. DEGs and fold-changes were calculated by DEseq2 (v1.30.0) while controlling for ACE/ARB use, statin use, history of other RD. Functional classification of protein class and biological pathways was performed using the PANTHER Database (v16.0).
GCA/PMR and CIA display no significant differences in tissue gene expression
GCA is typically associated with various clinical presentations, such as headache, temporal artery abnormalities, elevated markers of inflammation, and PMR (4,15). Meanwhile, patients with CIA are generally asymptomatic, and aortitis is often incidentally identified within histopathology (16). Interestingly, however, inflammatory aortic aneurysms of GCA and CIA are radiographically and histopathologically indistinguishable. Moreover, it remains unclear whether CIA is truly isolated to the aorta or represents a subclinical systemic vasculitis. As there has not been any investigation into the gene expression differences between GCA and CIA, we sought to compare transcriptomes of aortic tissue resections between these two clinical phenotypes. Strikingly, there were no significant differences in gene expression profiles between GCA/PMR and CIA (BH-adjusted P < 0.1) (Supplementary Table S6), indicating that CIA may be pathophysiologically closely related to GCA.
Functional enrichment of differentially expressed genes between inflammatory and non-inflammatory aortic aneurysms
Having identified DEGs and their protein class and biological pathway annotations, we next investigated whether the up/downregulated genes display statistically significant enrichment in GO terms (Materials and Methods). Among the 159 upregulated genes, we identified 228 enriched GO terms (Supplementary Table S7); the top 10 of which were all associated with immune response (Figure 3A), as can be expected given the nature of the disease under study. Interestingly, several GO terms were of the response to microbial agents (e.g., ‘Cellular response to interferon-gamma’, ‘Response to molecule of bacterial origin’, and ‘Defense response to bacterium’) (Supplementary Table S7).
Figure 3. Gene Ontology (GO) enrichment analysis reveals strong upregulation of immune response and downregulation of neuronal activity in inflammatory aortic aneurysms.
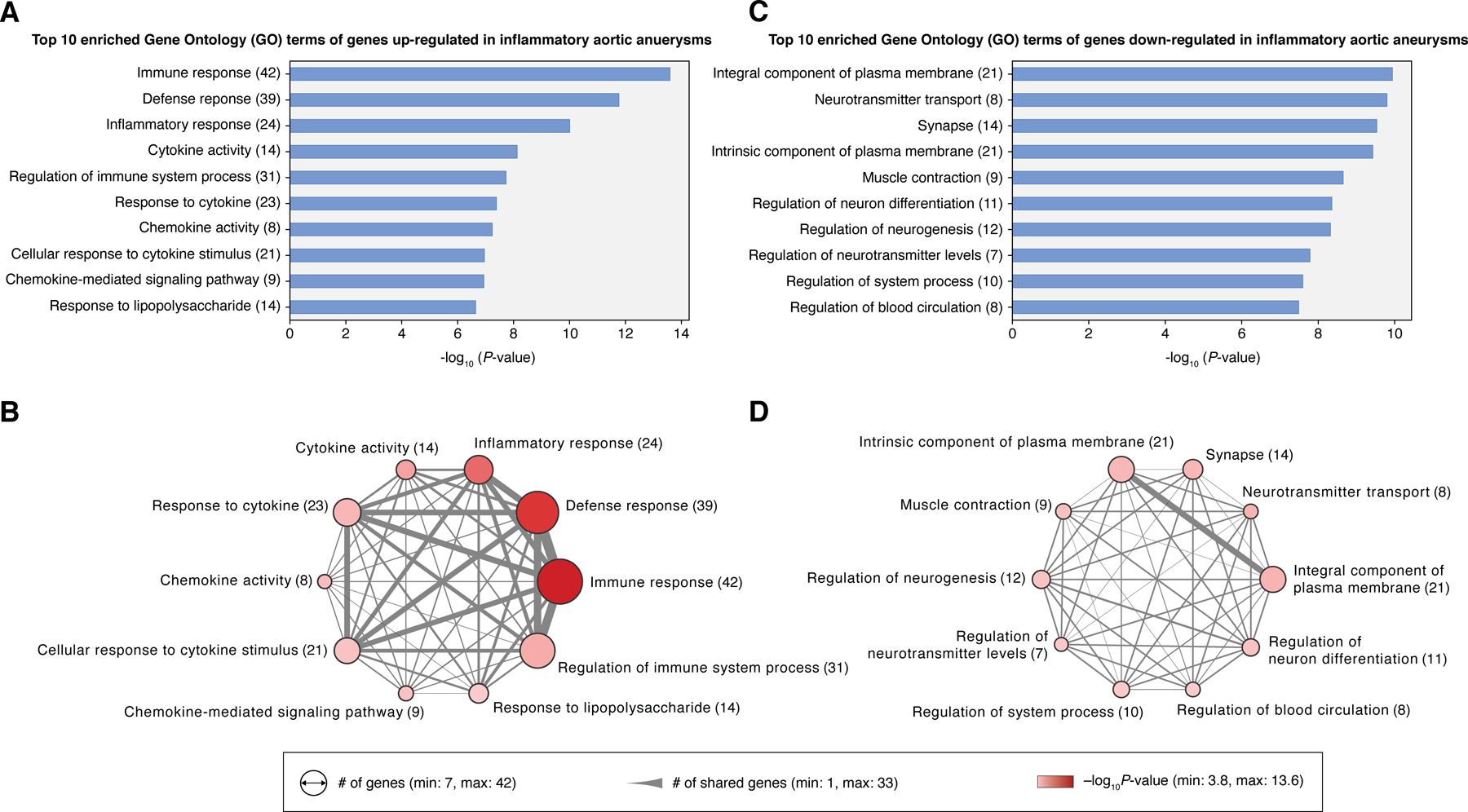
(A) Top 10 enriched GO terms of the upregulated DEGs (n = 159) and (B) their gene set similarities illustrated in a network diagram. (C) Top 10 enriched GO terms of the downregulated DEGs (n = 93) and (D) a network of their gene set similarities. Size of nodes in the network corresponds to the number of genes of each GO term. The width of the edge represents the number of genes common to both GO terms. Colors of nodes (pink to red) signify the statistical significance of GO enrichment. Gene set enrichment analysis on GO terms was performed in DAVID (v6.8).
The top 10 GO terms shared a considerable number of genes which could be driving the robust immune signature (Figure 3B). In particular, we identified 19 genes common to the top 3 GO terms (i.e., ‘Immune response’, ‘Defense response’, ‘Inflammatory response’) enriched in the upregulated DEGs (Supplementary Figures 2A–C). Several of these genes were previously reported to be associated with autoimmune or inflammatory diseases. One example is IL23R, which encodes for the receptor of a key proinflammatory cytokine (IL-23) that stimulates the proliferation of Th17 cells in inflammatory diseases (17). IL23R is located upstream of the JAK-STAT signaling pathway, which has been implicated in the pathogenesis of several inflammatory and autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, and psoriasis (15). Similarly, we identified an upregulation of IL1A, which encodes for a proinflammatory cytokine (IL-1α) that can cause severe acute or chronic inflammation when dysregulated (18). The role of IL1A in human inflammatory aortic aneurysms is not fully understood; however, it has been shown in a mouse model that Il1a deficiency can be protective against the formation of Kawasaki disease-associated abdominal aortic aneurysm (19). In addition, we identified an upregulation of CCR6, which encodes for a receptor that can mediate the recruitment of immature/mature dendritic cells and other antigen-presenting cells (20). Immune cells that express CCR6 (such as CCR6+ T cells) have been reported to populate the wall infiltrate in GCA patients and can cause injury to vascular smooth muscle cells (21).
Meanwhile, from the 93 genes downregulated in inflammatory aortic aneurysm, we identified 206 enriched GO terms (Supplementary Table S8). The top 10 of these were mostly associated with neuronal activities (Figure 3C and Supplementary Figures 2D–F), which may possibly reflect an elevated presence of damaged neurons in the aorta resulting from sustained levels of inflammation. Notably, we observed that these top GO terms shared far fewer genes among each other than the top GO terms enriched in the upregulated genes (Figure 3D). Moreover, we only identified one common gene (ATP1A2) across the top 3 GO terms (i.e., ‘Integral component of plasma membrane’, ‘Neurotransmitter transport’, ‘Synapse’), which encodes an α2-subunit of the sodium/potassium pump primarily found in glial cells (22). To date, the role of ATP1A2 in inflammatory diseases remains unclear, although it has been suspected that this gene may be involved in neuroinflammatory processes (23).
Differential expression of genes targeted by aortitis treatment drugs
We next investigated known drug targets (i.e., genes) in aortitis. Specifically, we focused on the expression of genes known to be targeted by JAK inhibitors, interleukin-6 inhibitors, Th1/Th17 inhibitors, prednisone, and methotrexate (Figure 4). Of note, these drugs were not given to our study population except for prednisone (n = 1).
Figure 4. Transcriptomics analysis confirms higher expression of a subset of known target genes in aortitis.
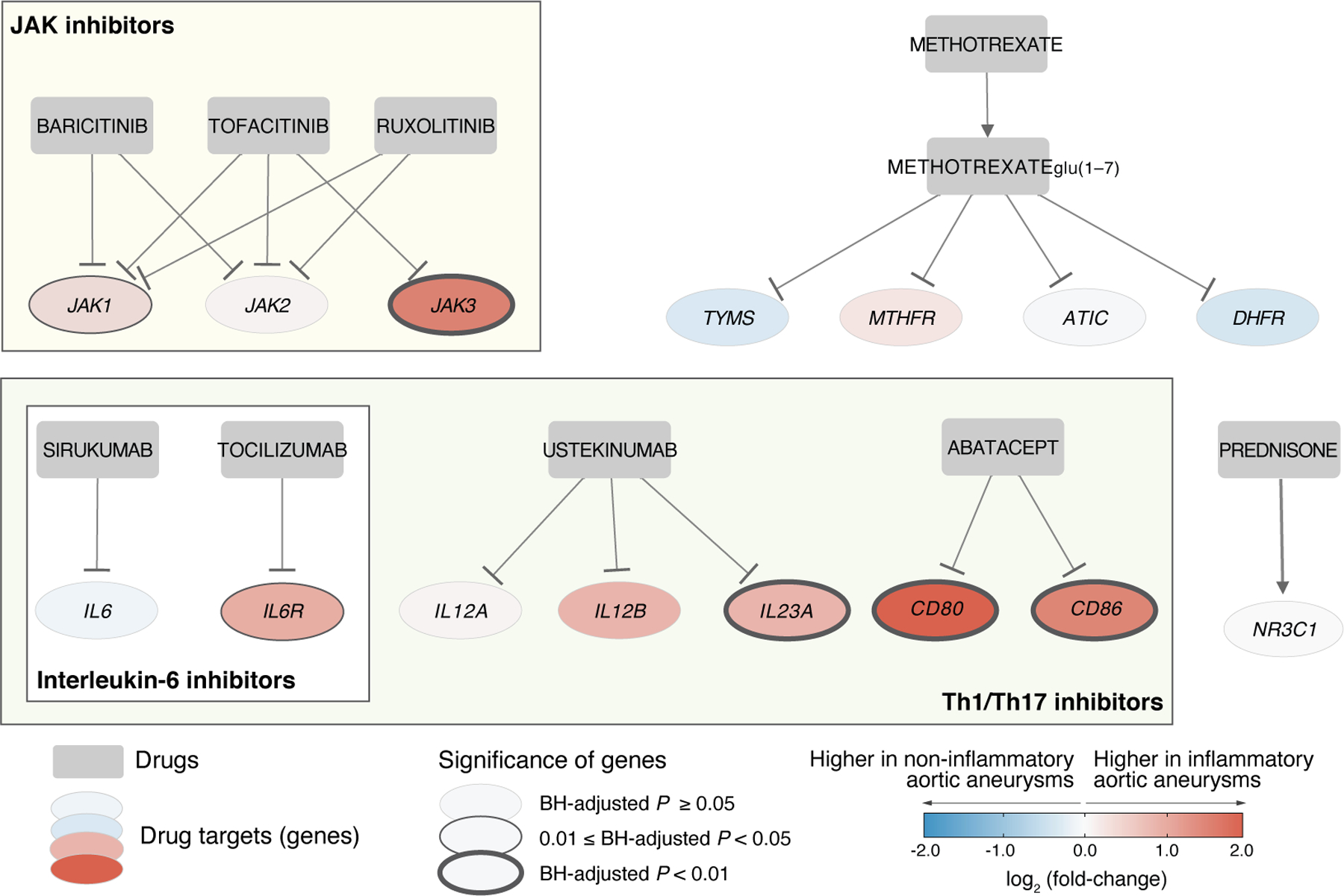
Among the known targets of aortitis immunosuppressive drugs (e.g., JAK inhibitors, IL6 inhibitors, Th1/Th17 inhibitors, prednisone, and methotrexate), six genes (JAK1, JAK3, IL6R, IL23A, CD80, and CD86) were found to display differential expression between inflammatory and non-inflammatory aortic aneurysms. Square and circle nodes indicate drug names and gene symbols, respectively. Edges with a hammerhead indicate inhibition. The color (blue to red) of each node represents gene expression fold-changes between inflammatory and non-inflammatory aortic aneurysms.
Baricitinib, tofacitinib, and ruxolitinib are JAK inhibitors that can target members of the JAK-STAT signaling pathway. Drugs that inhibit the JAK-STAT signaling pathway can potentially suppress vascular inflammation by reducing the activity of vascular dendritic cells and T cells (15,24–26). Interestingly, we identified JAK1 and JAK3 to be upregulated in inflammatory aortic aneurysms, which further supports the utility of JAK inhibitors for the treatment of aortitis. However, JAK2 was not identified to have differential expression between inflammatory and non-inflammatory aortic aneurysms.
The interleukin-6 (IL-6) inhibitors sirukumab and tocilizumab target the proinflammatory cytokine IL-6 and its receptor (IL-6R), respectively. This blocks the binding of IL-6 to IL-6R, and thereby reduces the recruitment of new macrophages (27). The upregulation of IL6R found in our study supports the use of tocilizumab for aortitis treatment (27). On the other hand, we did not identify differences in the expression of IL6 in resected tissues between inflammatory and non-inflammatory aortic aneurysms.
Ustekinumab and abatacept are Th1/Th17 inhibitors that suppress the activation of T cells by blocking proinflammatory cytokines (i.e., IL-12, IL-23) and Th1/Th17 membrane receptors (CD80, CD86). Our study identified genes that encode for the targets of abatacept (CD80 and CD86) to be upregulated in inflammatory aortic aneurysms. This finding is in line with the use of abatacept to reduce the activity of T cells by interrupting the communication between Th1/Th17 and antigen-presenting cells in aortitis (28). Likewise, among the proinflammatory cytokines targeted by ustekinumab, IL23A was found to be upregulated in inflammatory aortic aneurysms. Ustekinumab has been evaluated in patients with GCA and appears to be of limited benefit (29).
Lastly, the known gene targets of prednisone and methotrexate were not found to be differentially expressed in inflammatory aortic aneurysms. This result may help to confirm the relatively limited efficacy of these two drugs reported for aortitis (30,31).
Identification of potential drug targets through pharmacogenomic network analysis
Global transcriptomic profiling has been well demonstrated for drug target discovery (32), and several studies have coupled gene expression profiles with various systematic approaches for drug re-repurposing or biomarker discovery (32,33). Among various strategies for novel drug target identification, the network-based framework uses curated biological network topology (e.g., PPI network, metabolic network) to investigate the association across genes (or their products), diseases, and drugs (34). In this sense, we constructed a pharmacogenomic network composed of 36 genes and 92 interactions (representing the largest connected component; see Supplementary Figure 1 for the full interactome network) by integrating DEGs, a high-confidence PPI network, and drug-gene interactions (Materials and Methods). Our pharmacogenomic network identified 10 potentially new druggable gene targets in aortitis: BLK, CNR2, CR2, GZMB, IDO1, IFNG, IL1A, CXCL10, CXCL13, S1PR5 (Figure 5).
Figure 5. Pharmacogenomic network analysis uncovers the drug-gene interaction landscape in inflammatory aortic aneurysms.
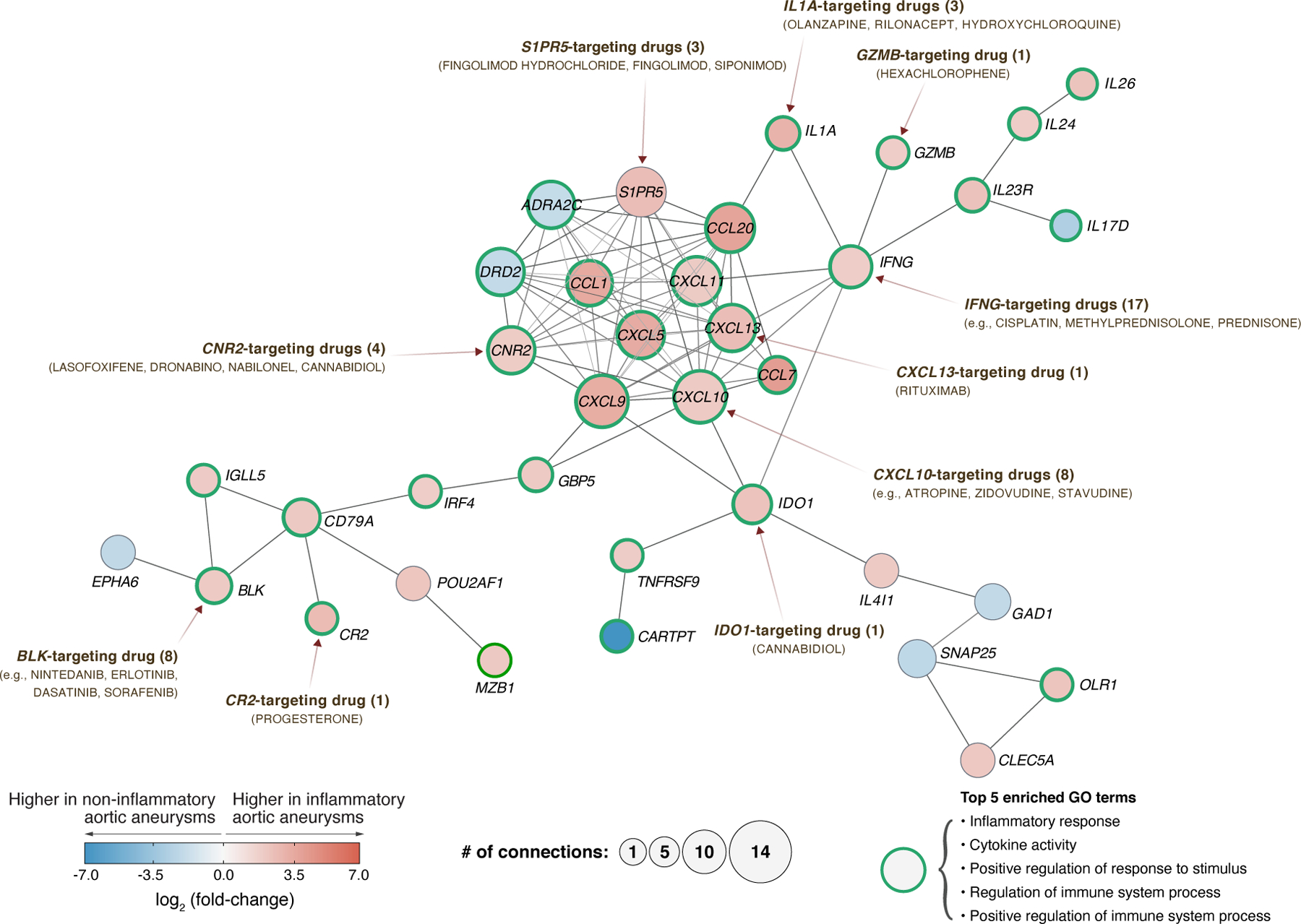
The largest connected component in the DEG-based protein-protein interaction network is composed of 36 genes (nodes) and 92 interactions (edges). Using pharmacological information, 10 of those 36 genes were identified as ‘druggable’ (both directly and indirectly) with FDA-approved, pharmaceutical drugs. In the pharmacogenomic network, 29 out of 36 genes are related to the top 5 enriched GO terms. Nodes with green borders represent genes that are related to the top 5 GO terms: Inflammatory response, Cytokine activity, Positive regulation of response to stimulus, Regulation of immune system process, and Positive regulation of immune system process. The color (blue to red) of each node represents gene expression differences between inflammatory and non-inflammatory aortic aneurysms. The size of each node represents its degree, i.e., number of connections to other nodes. GO enrichment analysis was performed on 36 genes using DAVID (v6.8). The information of drug-target genes were obtained from DGIdb (v4.0).
Near the hub of the pharmacogenomic network, we identified upregulated genes for cytokines (IL1A, INFG) and chemokines (CXCL5, CXCL9, CXCL10, CXCL11, CXCL13, CCL1, CCL7, CCL20) that can be targeted by drugs already known for the treatment of aortitis or other inflammatory diseases. For example, our results suggests that drugs targeting INFG (e.g., methylprednisolone, prednisone, cisplatin) and CXCL10 (e.g., atropine, zidovudine, atorvastatin) can be potentially used for the treatment of aortitis. Notably, IFN-γ (product of IFNG), which is produced by effector Th1 cells, is considered as one of the key cytokines involved in the pathogenesis of GCA (35). In addition, CXCL10, which is an interferon-stimulated gene that responds to different types of IFNs (i.e., IFN-α, IFN-β, IFN-γ, IFN-λ), encodes a chemokine that promotes the recruitment of CD8+ and Th1 cells (36). Of note, Corbera-Bellalta et al. demonstrated an ex vivo experiment in cultured GCA arteries showing reduced expression of CXCL9, CXCL10, and CXCL11 by blocking endogenous IFNγ with A6 abrogated STAT-1 phosphorylation (35). Furthermore, CXCL13, which is a chemokine that attracts B cells and contributes to the production of antibodies (37), may be potentially targeted with rituximab in aortitis. Rituximab is an anti-CD20 monoclonal antibody designed to target the surface of B cells and block interactions with effector T cells (38); preliminary studies have reported promising results for rituximab in GCA (39). In summary, our pharmacogenomic network analysis identified promising drug targets for potentially new treatment strategies in aortitis. We summarize all gene-gene and drug-gene interactions found within our pharmacogenomic network in Supplementary Table S9. In addition, a comprehensive summary of the enriched functions (GO terms) of all genes in the pharmacogenomic network is provided in Supplementary Table S10.
Discussion
In this study, we performed a genome-wide expression analysis to identify signature genes and biomolecular processes underlying aortitis. First, we compared gene expression profiles between inflammatory and non-inflammatory aortic aneurysms. Within inflammatory aortic aneurysms, we compared profiles between GCA/PMR and CIA. Notably, this study revealed for the first time that the transcriptomic signature of CIA is not different from that of aortitis related to GCA/PMR, suggesting that these two disease states largely share common pathophysiological mechanisms. Next, we investigated the functional annotations (i.e., protein class, biological pathway) and enriched GO terms of the DEGs. We found that the upregulated genes were generally enriched in immune processes, and in particular those in response to microbial agents. In contrast, the downregulated genes were enriched in neuronal processes. Finally, a pharmacogenomic network-based approach revealed a range of potentially new drug targets (genes) and therapeutics for the multimodal treatment of aortitis. The findings described herein motivate future research using multi-omics data or peripheral blood to investigate the wide landscape of biomolecular pathways and networks in aortitis, as well as advancing biomarker discovery (40), as we have demonstrated in another autoimmune disease (41,42).
Interestingly, a subset of the DEGs (BH-adjusted P < 0.05) were associated with receptors for pathogen-associated molecular patterns. It is yet unclear whether infectious agents play a causal role in blood vessel inflammation, and there has been no compelling evidence showing that abating aortitis symptoms are linked to the clearance of infections (43). Nevertheless, our study identified upregulation of viral ssRNA-specific endosomal pattern recognition receptors (TLR7, TLR8); other toll-like receptors (TLR1, TLR2, TLR4, TLR5, TLR6, TLR7, TLR8) and NOD-like receptors (NOD1, NOD2, NLRC4); pattern recognition receptor pathways that stimulate type 1 interferon production (MYD88, IRAK4, TRAF3, IKBKB, IRF5); members of the IFN-JAK-STAT pathway (IFNAR1, IFNAR2, IL10RB, IFNGR1, IFNGR2, JAK1, TYK2, STAT1, STAT2, IRF9); interferon regulatory factors (IRF1, IRF2, IRF4, IRF5, IRF8, IRF9); mature dendritic cell markers (CD80, CD83, CD86); and cell surface markers of plasmacytoid dendritic cells (CLEC4C, CCR7, LILRB4, NRP1), which are a subtype of dendritic cells that specifically sense viral RNA and DNA (44).
By providing a system-wide view of mechanistic gene (protein) interactions, our pharmacogenomic network analysis can facilitate the design of novel pharmacologic intervention strategies. For example, our analysis identified CD79A (immunoglobulin-associated alpha), which encodes a component of B cell antigen receptors, as a putative target in aortitis treatment. The product of CD79A is a highly reliable marker for B cells that is present on the cell surface throughout their life cycle (45). Although the efficacy of targeting B cells in aortitis has yet to be clearly and convincingly demonstrated, the role of B cells in aortitis is gradually being revealed (15). For example, van der Geest et al. reported changes in the distribution and homeostasis of B cells in GCA (46). Additionally, B cell-activating factor in GCA patients was reported to directly correlate with disease activity (47). In consideration of these findings, we can hypothesize that B cell suppressors may be beneficial for aortitis patients.
During the preparation of this manuscript, a transcriptomics study in large-vessel GCA was published by Vieira et al. (48). The investigators used microarray technology on aortic tissues from patients with GCA (n = 10) and controls (n = 9). Like our results above, upregulated gene sets for pathways involving interferons, JAK/STAT signaling, and proinflammatory cytokines and chemokines in GCA (compared to controls) were found. The authors too identified higher expression of members of the JAK/STAT signaling pathway (e.g., STAT1, STAT2) and type I-specific interferon response genes (e.g., EPSTI1) in inflamed aortic aneurysms in GCA. Therefore, our study using high-resolution transcriptome profiling by RNA-seq demonstrated favorable reproducibility of previous findings by others; and enabled novel insights including the discovery of other possible DEGs, that GCA and CIA are not different at the gene expression level, and new (albeit putative) targets of current aortitis treatment drugs (e.g., JAK inhibitors, Th1/Th17 inhibitors) and FDA-approved, off-label drugs.
We note a few limitations of this study. First, we acknowledge that 25 cases (inflammatory aortic aneurysms) and 25 controls (non-inflammatory aortic aneurysms) are a relatively small number of samples. Nevertheless, we were still able to identify a large number of statistically significant genes even after multiple hypothesis correction; this indicates that despite the limited sample sizes, global gene expression differences between inflammatory and non-inflammatory aortic aneurysms are sufficiently robust. Second, RNA-seq only allows us to observe the biomolecular processes within inflammatory aortic aneurysms at the gene expression level. Integrating our current findings with laboratory tests (e.g., RT-qPCR, immunohistochemistry staining) or other omics data, such as metabolomics, proteomics, and single-cell RNA-seq or mass cytometry (CyTOF), in future studies can confirm our results or elucidate additional details of the inherent biological processes in aortitis. Third, the current protein interactome network may not fully reflect the associations between the identified DEGs during the construction of the pharmacogenomic network. The stringent cutoff used to obtain the high-confidence network may lead to a loss in interactions between genes, and thereby missed opportunities to identify additional potential targets of known drugs. Nevertheless, the constructed pharmacogenomic network still identified genes that can be targeted by drugs conventionally prescribed for aortitis (e.g., prednisone) or other inflammatory diseases (e.g., rituximab). Last, our study used only FFPE samples from tissue biopsies, but did not investigate immune cells in circulation. Future immunophenotyping studies conducted with patient blood samples are needed to explore how the peripheral immune system is altered during disease onset and progression (49,50).
Supplementary Material
Acknowledgments
First and foremost, we thank our dear patients who volunteered for this study. We also thank the Mayo Clinic Genome Analysis Core staff members for making this work possible.
Funding
This work was supported in part by the Mayo Clinic Center for Individualized Medicine (to B.H and J.S.), Mayo Clinic Division of Rheumatology (M.J.K., C.M.W., and K.J.W.), and Mayo Clinic Benefactors.
Footnotes
Conflict of interest
All authors declare that they have no competing interests.
Ethics approval and consent to participate
This study was approved by the Mayo Clinic Institutional Review Board (no. 17-010612MJK) in accordance with the Declaration of Helsinki. All methods and procedures were performed in accordance with the Mayo Clinic Institutional Review Board guidelines and regulations.
Availability of data and materials
Source codes and datasets that were used in this study are available at: https://github.com/jaeyunsung/Aortitis_2022.
References
- 1.Bossone E, Pluchinotta FR, Andreas M, Blanc P, Citro R, Limongelli G, et al. Aortitis. Vascul Pharmacol 2016;80:1–10. [DOI] [PubMed] [Google Scholar]
- 2.Quimson L, Mayer A, Capponi S, Rea B, Rhee RL. Comparison of aortitis versus noninflammatory aortic aneurysms among patients who undergo open aortic aneurysm repair. Arthritis rheumatol 2020;72:1154–1159. [DOI] [PubMed] [Google Scholar]
- 3.Ladich E, Yahagi K, Romero ME, Virmani R. Vascular diseases: aortitis, aortic aneurysms, and vascular calcification. Cardiovasc Pathol 2016;25:432–441. [DOI] [PubMed] [Google Scholar]
- 4.Gornik HL, Creager MA. Aortitis. Circulation 2008;117:3039–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet 2016;17:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 2019;47:D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiao X, Sherman BT, Huang DW, Stephens R, Baseler MW, Lane HC, et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012;28:1805–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anon. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 2021;49:D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, et al. DGIdb: mining the druggable genome. Nat Methods 2013;10:1209–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koster MJ, Warrington KJ. Giant cell arteritis: pathogenic mechanisms and new potential therapeutic targets. BMC rheumatol 2017;1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cinar I, Wang H, Stone JR. Clinically isolated aortitis: pitfalls, progress, and possibilities. Cardiovasc Pathol 2017;29:23–32. [DOI] [PubMed] [Google Scholar]
- 17.Duvallet E, Semerano L, Assier E, Falgarone G, Boissier M-C. Interleukin-23: a key cytokine in inflammatory diseases. Ann Med 2011;43:503–511. [DOI] [PubMed] [Google Scholar]
- 18.Di Paolo NC, Shayakhmetov DM. Interleukin 1α and the inflammatory process. Nat Immunol 2016;17:906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, et al. Role of Interleukin-1 Signaling in a Mouse Model of Kawasaki Disease-Associated Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol 2016;36:886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito T, Carson WF 4th, Cavassani KA, Connett JM, Kunkel SL. CCR6 as a mediator of immunity in the lung and gut. Exp Cell Res 2011;317:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng J, Ma-Krupa W, Gewirtz AT, Younge BR, Goronzy JJ, Weyand CM. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res 2009;104:488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGrail KM, Phillips JM, Sweadner KJ. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J Neurosci 1991;11:381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinoshita PF, Yshii LM, Orellana AMM, Paixão AG, Vasconcelos AR, Lima L de S, et al. Alpha 2 Na+,K+-ATPase silencing induces loss of inflammatory response and ouabain protection in glial cells. Sci Rep 2017;7:4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 2018;128:3041–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heine A, Held SAE, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, et al. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013;122:1192–1202. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation 2018;137:1934–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med 2017;377:317–328. [DOI] [PubMed] [Google Scholar]
- 28.Langford CA, Cuthbertson D, Ytterberg SR, Khalidi N, Monach PA, Carette S, et al. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis rheumatol 2017;69:837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matza MA, Fernandes AD, Stone JH, Unizony SH. Ustekinumab for the treatment of giant cell arteritis. Arthritis Care Res (Hoboken) 2021;73:893–897. [DOI] [PubMed] [Google Scholar]
- 30.Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002;46:1309–1318. [DOI] [PubMed] [Google Scholar]
- 31.Delecoeuillerie G, Joly P, Cohen de Lara A, Paolaggi JB. Polymyalgia rheumatica and temporal arteritis: a retrospective analysis of prognostic features and different corticosteroid regimens (11 year survey of 210 patients). Ann Rheum Dis 1988;47:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Kui L, Tang M, Li D, Wei K, Chen W, et al. High-throughput transcriptome profiling in drug and biomarker discovery. Front Genet 2020;11:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng Y, Wang Q, Wang T. Drug target protein-protein interaction networks: A systematic perspective. Biomed Res Int 2017;2017:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barabási A-L, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corbera-Bellalta M, Planas-Rigol E, Lozano E, Terrades-García N, Alba MA, Prieto-González S, et al. Blocking interferon γ reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis 2016;75:1177–1186. [DOI] [PubMed] [Google Scholar]
- 36.Peperzak V, Veraar EAM, Xiao Y, Babala N, Thiadens K, Brugmans M, et al. CD8+ T cells produce the chemokine CXCL10 in response to CD27/CD70 costimulation to promote generation of the CD8+ effector T cell pool. J Immunol 2013;191:3025–3036. [DOI] [PubMed] [Google Scholar]
- 37.Legler DF, Loetscher M, Roos RS, Clark-Lewis I, Baggiolini M, Moser B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J Exp Med 1998;187:655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor RP, Lindorfer MA. Drug Insight: the mechanism of action of rituximab in autoimmune disease—the immune complex decoy hypothesis. Nat Clin Pract Rheumatol 2007;3:86–95. [DOI] [PubMed] [Google Scholar]
- 39.Bhatia A Anti-CD20 monoclonal antibody (rituximab) as an adjunct in the treatment of giant cell arteritis. Annals of the Rheumatic Diseases 2005;64:1099–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sung J, Wang Y, Chandrasekaran S, Witten DM, Price ND. Molecular signatures from omics data: from chaos to consensus. Biotechnol J 2012;7:946–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hur B, Gupta VK, Huang H, Wright KA, Warrington KJ, Taneja V, et al. Plasma metabolomic profiling in patients with rheumatoid arthritis identifies biochemical features predictive of quantitative disease activity. Arthritis Res Ther 2021;23:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta VK, Cunningham KY, Hur B, Bakshi U, Huang H, Warrington KJ, et al. Gut microbial determinants of clinically important improvement in patients with rheumatoid arthritis. Genome Med 2021;13:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moiseev S, Novikov P, Smitienko I, Shchegoleva E. Giant cell arteritis, infections and biologics. Ann Rheum Dis 2017;76:e29. [DOI] [PubMed] [Google Scholar]
- 44.Li S, Wu J, Zhu S, Liu Y-J, Chen J. Disease-Associated Plasmacytoid Dendritic Cells. Front Immunol 2017;8:1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li R, Wang T, Bird S, Zou J, Dooley H, Secombes CJ. B cell receptor accessory molecule CD79α: Characterisation and expression analysis in a cartilaginous fish, the spiny dogfish (Squalus acanthias). Fish & Shellfish Immunology 2013;34:1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geest KSM van der, Abdulahad WH, Chalan P, Rutgers A, Horst G, Huitema MG, et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis rheumatol 2014;66:1927–1938. [DOI] [PubMed] [Google Scholar]
- 47.Geest KSM van der, Abdulahad WH, Rutgers A, Horst G, Bijzet J, Arends S, et al. Serum markers associated with disease activity in giant cell arteritis and polymyalgia rheumatica. Rheumatology 2015;54:1397–1402. [DOI] [PubMed] [Google Scholar]
- 48.Vieira M, Régnier P, Maciejewski-Duval A, Le Joncour A, Darasse-Jèze G, Rosenzwajg M, et al. Interferon signature in giant cell arteritis aortitis. J Autoimmun 2022;127:102796. [DOI] [PubMed] [Google Scholar]
- 49.Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang JS, Juran BD, Cunningham KY, Gupta VK, Son YM, Yang JD, et al. Single-cell mass cytometry on peripheral blood identifies immune cell subsets associated with primary biliary cholangitis. Sci Rep 2020;10:12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.