

Summary
The killer cell immunoglobulin-like receptor (KIR) recognizes human leukocyte antigen (HLA) class I molecules and modulates the function of natural killer cells. Despite its role in immunity, the complex genomic structure has limited a deep understanding of the KIR genomic landscape. Here we conduct deep sequencing of 16 KIR genes in 1,173 individuals. We devise a bioinformatics pipeline incorporating copy number estimation and insertion or deletion (indel) calling for high-resolution KIR genotyping. We define 118 alleles in 13 genes and demonstrate a linkage disequilibrium structure within and across KIR centromeric and telomeric regions. We construct a KIR imputation reference panel (nreference = 689, imputation accuracy = 99.7%), apply it to biobank genotype (ntotal = 169,907), and perform phenome-wide association studies of 85 traits. We observe a dearth of genome-wide significant associations, even in immune traits implicated previously to be associated with KIR (the smallest p = 1.5 × 10−4). Our pipeline presents a broadly applicable framework to evaluate innate immunity in large-scale datasets.
Keywords: killer cell immunoglobulin-like receptor, KIR, target sequencing, next-generation sequencing, imputation, PheWAS, biobank, immunogenetics
Graphical abstract
Highlights
-
•
Deep sequencing of killer cell immunoglobulin-like receptor genes in 1,173 individuals
-
•
Novel computational algorithm to determine the highest-resolution KIR alleles
-
•
Construction of KIR reference panel for accurate and biobank-scale imputation
-
•
Comprehensive PheWAS on clinical phenotypes and KIR alleles
Sakaue et al. generated the largest deep sequencing dataset of the killer cell immunoglobulin-like receptor (KIR) genes, a complex region interacting with HLA to regulate human innate immunity. The novel bioinformatics pipeline determined 118 KIR alleles in 13 genes and enabled biobank-scale imputation of KIR alleles, followed by comprehensive PheWAS.
Introduction
An overwhelming amount of genomics data produced over the past decade has largely decoded how genetic variations between individuals can lead to phenotypic variations between individuals.1 Although high-throughput genotyping and sequencing technology with scalable computational methods has driven this achievement, we still lack comprehensive understanding of specific genome regions. One example is the major histocompatibility complex (MHC) region, which is characterized by high-level polymorphism and a population-specific complex linkage disequilibrium (LD) structure. Motivated by its pleiotropic associations with complex human traits, construction of a population-specific human leukocyte antigen (HLA) reference panel from next-generation sequencing (NGS) data and development of HLA imputation methods have successfully contributed to fine mapping of numerous disease-associated loci.2,3
The killer cell immunoglobulin-like receptor (KIR) locus, located in 19q13.4 and expressed mainly on the natural killer (NK) cell surface, is another example of a complex locus characterized by high allelic diversity and complex genomic structure. In particular, the KIR region is comprised of a combination of KIR genes, copy number variations, large deletions/duplications, and single-nucleotide variants (SNVs) within each KIR gene. As many as 1,110 KIR alleles have been registered to date.4 Importantly, this diversity is expected to be closely related to the function of KIR genes, which is to recognize specific HLA class I molecules and regulate the activity of NK cells. When variations are located in specific positions of specific KIR alleles, they can change the affinity between those KIRs and interacting HLA class I molecules presenting self- or pathogen-derived antigens, changing the inhibitory or activating cytotoxic signaling initiated by NK cells. Variation in the function of NK cells has been reported to cause associations of KIR alleles with immune-related (e.g., psoriasis),5,6 infectious (e.g., hepatitis C virus),7 and reproductive (e.g., preeclampsia) diseases.8 Furthermore, KIR alleles and their combination with HLA alleles are critically associated with clinical outcomes in allogenic hematopoietic stem cell transplantation or treatment outcomes in cancer immunotherapy.9,10 However, the statistical significance of these associations has been relatively weak, possibly because the associations have tended to be reported using the “candidate-allele approach,” focused only on KIR allelic diversity, as opposed to a genome-wide approach. This has been partly due to a lack of large sample size for deriving robust associations. A methodology to determine KIR alleles at a population scale is thus warranted.
The challenge of elucidating the genomic and phenotypic landscape of the KIR region is linked to its complex structure, which makes it difficult to accurately define the individual composition of the KIR haplotype and alleles, even with NGS data. The complexity stems from (1) a large number of SNVs and insertions or deletions (indels) within each KIR gene, (2) the tandem duplications of KIR genes, and (3) the structural arrangements consisting of exponential combinations of KIR gene content and alleles.11,12 Thus, it is difficult to unambiguously map short reads from conventional NGS technology (i.e., ∼150 bp) to the KIR region. Carefully designed target sequencing with relatively longer read lengths and deep coverage and a computational pipeline to disentangle the individual structures and alleles of the KIR region are warranted, which would contribute to the clinically applicable quality of KIR typing. Given that such a customized sequencing technology is not scalable to hundreds of thousands of individuals, a methodology to impute KIR alleles from the SNVs genotyped from other platforms (e.g., SNP microarray or whole-genome sequencing [WGS]) is needed. Other studies have begun to address these limitations,13,14 but there remains a need to understand and interpret KIR alleles in biobank-scale data and explore the comprehensive landscape of associations between KIR alleles and human complex traits. Although target individuals from the current KIR data have been biased toward Europeans, the extreme diversity of the HLA and KIR region suggests population-specific frequencies of these alleles. Deeper insights into the genomic maps of non-Europeans at these critical loci have the potential to provide new information regarding population-specific selection pressure and prevent ongoing disparities in genomics-informed health initiatives.15
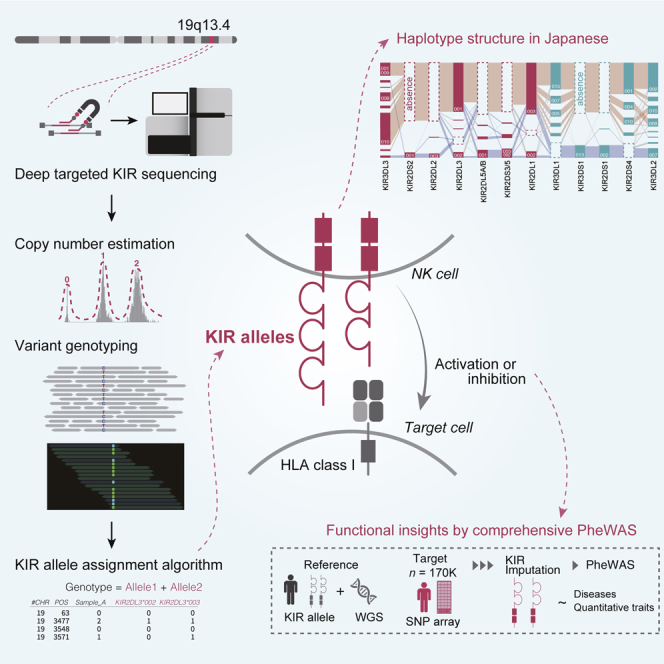
We conducted deep target sequencing of 16 KIR genes using a capture method with a read length of more than 300 bp in 1,173 individuals of Japanese ancestry (Figure 1A). We developed a custom bioinformatics pipeline focused on analysis of KIR loci. We incorporated SNV calling, indel calling, and copy number estimation using a machine learning framework, which enabled accurate determination of KIR gene content, copy numbers, and alleles at high resolution (Figure 1B). We used this catalog to (1) elucidate the frequency distribution, haplotype, and LD structure of the KIR region in the Japanese population and (2) construct a reference panel that can be used for KIR imputation by integrating with the WGS data (nreference = 689). With this reference panel, we implemented a KIR imputation method and applied it to biobank-scale genotype data of individuals of Japanese ancestry (ntotal = 169,907; Figure 1C). Finally, we performed a phenome-wide association study (PheWAS) of KIR alleles of 85 diverse complex traits to illustrate the comprehensive genomic and phenotypic landscape of the KIR region.
Figure 1.

Overview of this study
(A) Customized deep target sequencing of KIR genes was performed for 1,173 individuals.
(B) An integrative bioinformatics pipeline was devised to determine (i) the KIR gene content, (ii) copy number, and (iii) alleles. The coverage of aligned reads was used to determine the KIR gene content and copy number by kernel density estimation. The estimated copy number was used for ploidy-aware calling of SNVs and indels in the KIR region. Together with the public KIR allele database, the combination of SNVs and indels in the KIR region was used to determine the KIR alleles.
(C) With these KIR allele data and WGS data from 689 individuals, we implemented an in silico KIR imputation method. We imputed the KIR alleles in a large Japanese cohort (n = 169,907) to perform a PheWAS of the KIR alleles for 85 diverse complex traits.
Results
Deep target sequencing of KIR genes with a capture method
For 1,173 individuals of Japanese ancestry, we performed customized deep target KIR sequencing with a capture method (STAR Methods). We sequenced all 16 KIR genes including pseudogenes in the region:4 KIR3DL3, KIR2DS2, KIR2DL2, KIR2DL3, KIR2DL5A/B, KIR2DS3/5, KIR2DP1, KIR2DL1, KIR3DP1, KIR2DL4, KIR3DL1, KIR3DS1, KIR2DS1, KIR2DS4, KIR3DL2, and KIR3DX1. Of them, KIR3DL3, KIR3DP1, KIR2DL4, and KIR3DL2 are framework genes, which, in principle, are observed in every individual. The target KIR regions were designed to uniquely define each of the KIR genes despite their structural homology (Tables S1 and S2). We sequenced with relatively longer read lengths (350 bp and 250 bp for paired ends) and high depths (an average depth of 140.6 for the framework genes). The sequenced reads were mapped to the target contigs (STAR Methods). The relatively longer read design resulted in significantly better mapping quality and a larger fraction of uniquely mapped reads compared with simulated reads with a conventional read length of 150 bp, which were obtained by taking the first 150 bases of the original reads (paired t test, p < 1 × 10−323; Figure S1; STAR Methods). We then estimated the gene content and copy number of each KIR gene by quantifying the read depth. When determining the gene content (i.e., whether an individual has the gene) and the gene copy number (i.e., the number of gene copies), we took the ratio of the reads mapped to the gene to those mapped to KIR3DL3, which is one of the framework genes, and, in principle, all individuals should have two copies. To systematically assign the discrete number of gene copies to an individual, we performed unsupervised hard clustering based on the kernel density estimation of the read depth distribution within the study population (STAR Methods). Excluding KIR3DL3, KIR3DP1, KIR2DL4, KIR3DL2 (for which we assigned two copies to all individuals), and a pseudo-gene of KIR3DX1, we defined a gene set and copy number for the 11 KIR genes (Table S3). The variations in gene copy number were heterogeneous, according to each KIR gene in this Japanese cohort, and we confirmed that the range is mostly consistent with a previous study conducted in a Norwegian-German cohort.13
To validate the accuracy of the defined gene set, we genotyped 14 KIR genes by the polymerase chain reaction (PCR)-sequence-specific oligonucleotide (SSO) in 100 individuals with various sets of KIR genes to account for KIR diversity among the study population (STAR Methods). Using PCR-SSO, we determined gene content but could not determine the detailed KIR alleles. We observed almost perfect concordance of the gene content between target sequencing and PCR-SSO (mean concordance = 99.8%; Table S4), supporting the validity of our target capture method. There was no overrepresentation in the discordant results for a specific gene or a specific sample. To evaluate the strategy of using KIR3DL3 as a reference gene for copy number estimation, we determined the gene content by using (1) the coverage of the pseudo-gene of KIR3DX1, for which, in principle, all individuals should have two copies, and (2) the median coverage of all four framework genes as a reference. We confirmed that the gene copy numbers determined based on KIR3DL3 and those based on (1) or (2) were 99.9% and 99.3% concordant, respectively, which supported the robustness of our method.
Haplotypes of the KIR region have been classified into two large groups: group A haplotypes and group B haplotypes. Group B haplotypes are currently characterized by the presence of one or more of the following KIR genes within a haplotype: KIR2DL5, KIR2DS1, KIR2DS2, KIR2DS3, KIR2DS5, and KIR3DS1. Group A haplotypes are characterized by complete absence of these six genes.16,17 By using the KIR gene content data of the study population, we defined an individual haplotype combination that was A/A, A/B, or B/B. The frequencies of these haplotype combinations were 53%, 44%, and 3.4% in this study, respectively, which was consistent with the previous literature regarding Japanese individuals (n = 173; 58%, 40%, and 1.2%, respectively).18 Compared with worldwide populations, the prevalence of the group A haplotype in Japanese individuals was greater than in Europeans.19
To validate our analytic pipeline, we applied the same sequencing protocol and computational methods to samples of 24 Japanese individuals with the known KIR gene content and alleles using the previously established PCR-SSP typing method18 and to 52 individuals of International Histocompatibility Working Group (IHWG) cell lines with previously known KIR gene content, copy numbers, and alleles.11 As for the gene content, our pipeline showed 100% concordance with the Japanese samples (n = 24) and a mean concordance of 95.9% with those from the IHWG (n = 52; Table S5A). As for the gene copy numbers, we observed a mean concordance of 97.9% with samples from the IHWG (n = 52; Table S5B). Although we used a reference sequence of GRCh37/hg19 and an alternative haplotype of GL000209 as bait for the target capture method, this high concordance potentially suggests applicability of our sequencing and bioinformatics pipeline to diverse populations, with robustness to the variations in KIR alleles and polymorphisms.
Ploidy-aware genotyping and determination of the KIR alleles
To obtain high-resolution KIR alleles, accurate SNV and indel genotyping of the KIR genes is essential. Motivated by the validation result of our pipeline in accurately determining the KIR gene content and gene copy numbers, we performed ploidy-aware (i.e., copy-number-defined) genotyping of the KIR genes. For SNV calling, we used Genome Analysis Toolkit (GATK) with an option of ploidy, which was set as the estimated gene copy number of each KIR gene (STAR Methods). A small number of indels was critical for determining specific KIR alleles (e.g., a 22-bp insertion at position 6,833 in KIR2DS4 and a 1-bp deletion at position 9,608 in KIR2DL4). Because such indel calling by GATK did not have enough sensitivity, probably reflecting the highly polymorphic nature of this region, we used DeepVariant,20 which directly uses per-sample mapped reads in the deep-learning framework, to call indels (STAR Methods).
At the same time, we curated the nucleotide sequence data of KIR alleles that have been registered in the Immuno Polymorphism Database (IPD)-KIR database to date. We made a list of 706 SNVs and two indels residing within coding sequences of each KIR gene, which altogether discriminate a specific KIR allele from the others. We took the intersection of variants that were included in this list of variants and in a list of genotyped SNVs and indels of our dataset and utilized them to determine the KIR allele combination in our dataset. By obtaining all possible genotypes from the KIR alleles and matching them with the observed genotypes of these intersected variants, we defined 118 KIR alleles spanning 13 KIR genes. We investigated potentially novel allele combinations when the most probable candidate combination(s) of KIR alleles had at least one mismatched genotype. There were a few patterns of ambiguity (e.g., the difference between KIR3DL3∗001 and KIR3DL3∗009) for which it was technically difficult to uniquely select one allele over the other because of the paucity of variations within exons among the different KIR alleles.
To validate the accuracy of our pipeline when defining the KIR alleles, we again applied it to the Japanese dataset and IHWG dataset, where we had the previously established KIR allelic information based on PCR-SSP and pyrosequencing, respectively. We observed high concordance between the results from our method and the previously established alleles (the mean concordance was 98.6% and 94.6%, respectively; Table S6), which was comparable with the NGS-based determination of HLA alleles.2,21 We had a broader scope of KIR alleles than the previously released NGS-based pipeline11 (i.e., genotyping of KIR2DS1 and 2DS2), which is one of the advantages over the previous method.
LD and haplotype structure of the KIR region in Japanese individuals
Given the diversity and complex structure of the KIR region, elucidation of population-specific LD and haplotype structures would expand our understanding of this region. Historically, the KIR region is subdivided into a centromeric region and a telomeric region based on physical position. Between the centromeric and telomeric region exists a recombination hotspot, where reciprocal recombination is likely to occur to form new haplotypes by reassorting centromeric and telomeric gene content motifs.22 Although a strong LD within each of the regions has been reported, the LD across these regions has not been well described, especially for non-Europeans.23 By leveraging these high-resolution KIR gene content and allelic data from 1,173 Japanese individuals, we assessed the LD across the entire KIR region (Figure 2). The LD based on KIR gene content showed a moderate inter-region LD (for example, between KIR2DL5 [centromeric] and KIR3DL1 [telomeric]), as well as a strong intra-region LD (Figure 2A), which demonstrated the shared haplotypes stretching across the two regions. To elucidate the LD structure at a higher resolution, we calculated the allelic LD metric ε24, an entropy-based LD measurement index to quantify a pairwise LD between KIR genes by incorporating multiallelic information. We again observed intra- and inter-region LD across the KIR genes (Figure 2B), indicating that the KIR LD pattern is not only composed of the presence or absence of each KIR gene but also of the allelic diversity within and across the KIR genes.
Figure 2.

Linkage disequilibrium (LD) analysis of the KIR region
(A) Pairwise LD based on KIR gene content, which was measured as the r2 value of inter-gene copy number correlations. Dotted lines in white indicate the boundary between centromeric and telomeric regions.
(B) Pairwise LD based on KIR alleles, which was measured by the ε value of normalized entropy of the haplotype frequency. A higher ε value represents stronger LD. A dotted line in white indicates a boundary between the centromeric and telomeric regions of KIR.
To better understand the landscape of KIR diversity in the Japanese population, we applied (1) a dimensionality reduction method, t-distributed stochastic neighbor embedding (t-SNE),25 and (2) a haplotype visualization method, Disentangler,26,27 to the genotype and haplotype data of the KIR genes, respectively. First, we applied a non-linear dimensionality reduction method, t-SNE, to the KIR gene content (Figure 3A) and alleles (Figure 3B) to cluster the study population based on KIR diversity. This unsupervised clustering identified a major cluster, colored red in Figure 3A, that pointed to those who had two copies of a group A haplotype (A/A). On the other hand, the individuals in the second major cluster, colored blue in Figure 3A, had at least one copy of a specific group B haplotype that was characterized by (1) the presence of KIR3DS1 and KIR2DS1 and (2) the absence of KIR3DL1 and KIR2DS4. This structure was mostly preserved in the t-SNE application to the allelic data (Figure 3B). The large bottom cluster below the dotted line in Figure 3B mostly consisted of A/A haplotype individuals classified as the red cluster in Figure 3A but was more specifically characterized by complete absence of KIR2DL4∗00501, KIR3DS1∗01301, and KIR2DS1∗00201. To elucidate the KIR haplotype configuration, we used Disentangler, a graphics tool designed for visualizing high-dimensional haplotype configurations across multiallelic genetic markers such as HLA and KIR alleles. We observed the long-range haplotypes that corresponded to the group A and group B haplotypes (red and blue, respectively, in Figure 3C) in the Japanese population.
Figure 3.

The LD and haplotype structure of KIR region
(A) Unsupervised sample clustering by t-SNE based on KIR gene content. We manually categorized the plots into three clusters (red, blue, and pink). A/A, A/B, and B/B indicate the haplotype combination of the individuals in the cluster.
(B) Unsupervised sample clustering by t-SNE based on the KIR alleles. We annotated individuals with colors according to the clusters defined in (A). We segregated the points into top and bottom clusters by a dotted line.
(C) Visualization of haplotype structures of the KIR genes by Disentangler. The vertically stacked bars represent each of the KIR genes. A tile in a bar represents a KIR allele, and a segment connects two alleles on adjacent genes. Tiles with dotted frames indicate that the haplotype did not have the KIR gene. The height of each tile and the thickness of each segment correspond to the frequencies of the KIR allele and haplotype, respectively. The pink tiles are centromeric KIRs and the green tiles are telomeric KIRs. The representative haplotypes are colored manually in red (group A haplotypes) and in blue (group B haplotypes).
These analyses supported the historically defined categorization of the group A/B haplotype from the recently developed dimensionality reduction method and might suggest a sub-classification strategy based on the high-resolution allelic data.
Construction of a biobank-scale KIR imputation method by integrating high-resolution KIR alleles and WGS genotype
A biobank-scale PheWAS of the KIR region will provide comprehensive insights into how the diversity of KIR alleles affects a spectrum of human complex traits. To conduct PheWAS, an accurate and scalable method to impute KIR alleles from the existing SNV data (genotyped by SNP microarray or WGS) is warranted because it would be challenging to perform customized deep sequencing of the KIR region in hundreds of thousands of individuals. Given that the genotype data of current biobanks have been generated on different genotyping or sequencing platforms, it is also warranted to construct a KIR reference panel that includes as many variants in the KIR region as possible to encompass diverse potential scaffold variants. We integrated WGS data for some of the individuals in the study cohort (nreference = 689) with the high-resolution KIR gene copy number and allele to construct a reference panel for KIR imputation, which included 11 KIR gene copy numbers, 52 high-resolution (7-digit maximum) KIR alleles, and 151,350 SNVs on chromosome 19 after quality control (STAR Methods). The tagging SNPs in LD with KIR alleles are essential in an accurate imputation based on a hidden Markov model (HMM).28 We observed that the R2 values of LD between KIR alleles and the best tagging SNPs were always higher when including whole-genome-sequenced variants than when limiting the variants to those on the genotyping platforms (Figure S2). The higher R2 values in WGS suggested an advantage of including whole-genome variants in the reference panel. The imputation accuracy of the constructed KIR reference panel was empirically evaluated by simulating the SNPs on the genotyping array (Illumina OmniExpressExome) as an imputation scaffold and performing 10-fold cross validation (STAR Methods).29 We observed a high imputation accuracy for KIR gene copy numbers (mean concordance = 99.7%; Table S7A) and KIR alleles (mean concordance = 99.7%; Table S7B), which was comparable with that for HLA alleles.2,30
We next sought to investigate the imputation accuracy of the constructed KIR reference panel in other populations. We genotyped IHWG samples (n = 39) using the Illumina OmniExpressExome genotyping array. The principal-component analysis with 1000 Genomes Project individuals suggested that the IHWG samples are mostly of European ancestry. We imputed KIR alleles using this genotype as a scaffold and the KIR reference panel of Japanese (nreference = 689) and assessed the accuracy of imputation by comparing the imputed alleles with those from the previously defined KIR alleles based on pyrosequencing. Although the imputation pipeline generally worked, we observed a relatively low accuracy (mean concordance = 88.9%; Table S8) compared with imputation in Japanese individuals (Table S9; Figure S3), which underscores the necessity of constructing a population-specific KIR reference panel.
Last, we benchmarked our imputation pipeline by comparing it with the previously published KIR imputation method of KIR∗IMP.13 KIR∗IMP imputes KIR gene copy number and haplotypes but cannot impute KIR alleles, and the reference panel was constructed from individuals in the United Kingdom. We observed that the imputation accuracy of KIR copy number was comparable with or slightly better in our pipeline than in KIR∗IMP for the IHWG dataset (mean concordance = 90.1% [ours] versus 85.3% [KIR∗IMP]; Table S10A) and Japanese dataset (mean concordance = 94.6% [ours] versus 93.9% [KIR∗IMP]; Table S10B). We also had a larger scope of KIR genes whose gene content can be imputed.
PheWAS in 170,000 individuals revealed no significant association of the investigated KIR alleles with complex human traits
The high-resolution KIR reference panel enabled us to conduct a biobank-scale KIR allelic imputation, followed by comprehensive PheWAS in the KIR region. We imputed the KIR alleles in BioBank Japan genome-wide association study (GWAS) data (n = 164,540)31, 32, 33 and case-control cohort GWAS data of inflammatory bowel disease (n = 5,367)34,35 and associated the imputed KIR alleles with 85 human complex traits (40 diseases and 45 quantitative traits; Table S11). When conducting KIR imputation using these biobank-scale genotype data, we used minmac3 software instead of beagle because of computational scalability. We again confirmed the high imputation accuracy in this modified pipeline by splitting the cohort into two batches evenly, with a smaller number of individuals in the reference panel (n = 295; mean concordance = 93.9% for gene copy number and 96.1% for alleles; Table S9; STAR Methods). We imputed 24 common KIR genes and alleles with an estimated imputation accuracy of R2 > 0.5. PheWAS revealed no significant association of these alleles with these 85 traits after Bonferroni correction of multiple testing (smallest p = 1.5 × 10−4 in KIR3DL2∗010 for serum total protein level; number of tests = 24 markers × 85 traits = 2,040; significance threshold p = 0.05/2,040 = 2.5 × 10−5; Figure 4; Table S12). We could not robustly replicate the previously reported associations of the investigated KIR alleles with immune-related diseases (e.g., ulcerative colitis, Crohn’s disease,36 rheumatoid arthritis,37 type 1 diabetes,38 and HCV hepatitis;7 smallest p = 0.0063 in KIR3DL2∗001 for type 1 diabetes).
Figure 4.

A heatmap of PheWAS results of KIR alleles with 85 complex traits
Shown are the association results of KIR alleles (columns) with 85 complex traits (rows), demonstrated as −log10(P). The values of −log10(P) are colored according to the color scale at the top left. The traits are separated by dotted lines according to the phenotypic category. The abbreviations for the trait names are described in Table S11. Immune, immunological/allergic diseases; Inf, infectious diseases; CVD/metab, cardio/vascular/metabolic diseases; Malignancy, malignant diseases; Misc, miscellaneous diseases; Anth, anthropometric quantitative traits; BP, blood pressure-related quantitative traits; Metabolic, metabolic quantitative traits; Liver, liver-related quantitative traits; Other, other biochemical quantitative traits; Kidney, kidney-related quantitative traits, Hematological, hematological quantitative traits.
Considering the biological interplay between KIRs and HLA class I molecules, we assessed the interactive effect (i.e., epistasis) of a KIR allele and an HLA class I allele on these complex traits. We performed interaction analyses between the imputed KIR alleles and HLA class I alleles with a minor allele frequency of less than 0.05. We did not detect any significant interactive effects across the 85 traits after Bonferroni correction of multiple testing (smallest p = 3.4 × 10−5 in KIR3DL2∗007×HLA-C∗08:01 for serum potassium level; number of tests = 41,458; significance threshold p = 0.05/41,458 = 1.2 × 10−6; quantile-quantile (QQ) plot in Figure S4). We did not detect any evidence of epistasis reflecting the interaction between KIR alleles and HLA alleles within the Japanese population.
Discussion
We performed comprehensive target sequencing of KIR genes to uncover the genomic landscape of this region. We determined the highest-resolution KIR alleles in 1,173 individuals of Japanese ancestry. The KIR copy number and allele typing showed high concordance with previous studies in Japanese and European individuals, which suggests applicability of our pipeline to other global populations to increase the diversity in reference KIR alleles. The high-resolution map of KIR copy numbers and alleles disentangled the complex LD and haplotype structures. Strong LD was observed within the centromeric and telomeric regions and moderately spanned across the two regions beyond a recombination hotspot. A recently developed dimensionality reduction method elucidated the diversity of the KIR region. Although the gene-content-level analysis supported the historically defined haplotype categorization (i.e., groups A and B), the allele-level analysis suggested a classification strategy that could further divide individuals into subclusters. Construction of a reference panel of the high-resolution KIR alleles and the whole-genome variants, followed by an imputation of the biobank-scale GWAS data, showed the phenotypic associations of KIR genes with diverse human complex traits. Unexpectedly, the previously reported associations of KIR alleles with immune-related traits were not replicated at robust significance within the scope of our study, nor did we observe interactive effects between the KIR alleles and HLA alleles. However, we need to continue the effort to increase the sample size and population diversity in KIR association studies to validate our exploratory analyses. Given the high accuracy of our imputation method, application to other phenotypes, such as allogenic transplantation outcomes and treatment efficacy of cancer immunotherapy, is also warranted to uncover the biology and clinical importance of the KIR region.
We developed a new sequencing and bioinformatics pipeline to elucidate the KIR diversity of biobank-scale individuals, which provides a potential opportunity to be broadly applied in the clinical field.
Limitations of the study
Our study has potential limitations. First, although the optimized deep target sequencing technology and computational pipelines enabled us to create the high-resolution KIR catalog, there remains an upper limit when specifying the combination of KIR alleles without ambiguity. Although we implemented the function to flag potentially novel KIR allele combinations, detailed characterization of those alleles was beyond the scope of this study. One of the reasons was because we only included known exon variants in determining KIR alleles, considering the effect on the function of KIRs. An additional inclusion of intronic variants and novel variants would further fine-tune the KIR allele discovery with much less ambiguity. Second, our KIR sequencing data did not have phase information. We complemented this point with the computational pipelines characterized by (1) a matching algorithm with an exhaustive search for all possible combinations of genotypes in determining KIR alleles and (2) a computational phasing algorithm in the haplotype-based analysis. These pipelines enabled us to determine the high-resolution KIR alleles in most cases and to construct an imputation panel with high accuracy. Long-read sequencing technology that could address phase in the entire KIR region is needed to validate the accuracy of our results.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| The genotype data of BioBank Japan used in this study | Nagai et al. 201739 | Japanese Genotype-phenotype Archive (JGA) with accession code JGAS000114/JGAD000123 and JGAS000114/JGAD000220 which can be accessed through application at https://humandbs.biosciencedbc.jp/en/hum0014-latest |
| Deposited data | ||
| Individual-level KIR alleles and KIR imputation reference panel used in this study | This paper | the National Bioscience Database Center (NBDC) Human Database (https://humandbs.biosciencedbc.jp/en/) with the accession code hum0114 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| IPD-KIR | The KIR Nomenclature committee | https://www.ebi.ac.uk/ipd/kir/ |
| Experimental models: Cell lines | ||
| A cell line used for genotyping and constructing KIR reference in the Japanese population | the Japan Biological Informatics Consortium (JBIC) | https://bioresource.nibiohn.go.jp/psc/index.html |
| ECACC HLA typed collection | The International Histocompatibility Working Group (IHWG) | https://www.phe-culturecollections.org.uk/products/celllines/hlatyped/search.jsp |
| Software and algorithms | ||
| KIR genotyping algorithm | This paper |
https://github.com/saorisakaue/KIR_project https://doi.org/10.5281/zenodo.5908796 |
| BWA | Li et al. 200940 | http://bio-bwa.sourceforge.net |
| KernelDensity module | scikit-learn of python | https://scikit-learn.org/stable/modules/generated/sklearn.neighbors.KernelDensity.html |
| GATK | Van der Auwera & O'Connor. 202041 | https://software.broadinstitute.org/gatk/ |
| DeepVariant | Poplin et al. 201820 | https://github.com/google/deepvariant |
| blast | Altschul et al. 199042 | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| plink | Purcell et al. 200743 | https://www.cog-genomics.org/plink/ |
| eLD software | Okada 201824 | http://www.sg.med.osaka-u.ac.jp/tools.html |
| multicoreTSNE | python package | https://github.com/DmitryUlyanov/Multicore-TSNE |
| Disentangler software | Okada et al. 201144 | http://kumasakanatsuhiko.jp/projects/disentangler/ |
| Beagle | Browning and Browning. 200745 | https://faculty.washington.edu/browning/beagle/beagle.html |
| Minimac3 | Das et al. 201646 | https://genome.sph.umich.edu/wiki/Minimac3 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yukinori Okada (yokada@sg.med.osaka-u.ac.jp).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
To determine the high-resolution KIR gene content, copy number, and alleles in the Japanese population, we enrolled 1,173 individuals of Japanese ancestry, whose genomic DNA were obtained from Epstein-Barr virus transformed B-lymphoblast cell lines of unrelated Japanese individuals established by the Japan Biological Informatics Consortium (JBIC).29 Mean age of participants at recruitment was 47.4 years old, and 46.8% were female. Of them, 689 individuals for whom we conducted the whole-genome sequencing were enrolled in construction of the KIR imputation reference panel. In the PheWAS, 164,540 individuals were enrolled from BioBank Japan (BBJ) project, which is a hospital-based biobank that collaboratively collected DNA and serum samples from 12 medical institutions in Japan and recruited approximately 200,000 participants, mainly of Japanese ancestry, with the diagnosis of at least one of 47 diseases.39,47 Mean age of participants at recruitment was 63.0 years old, and 46.3% were female. Individuals who were identified as of non-Japanese origin either by self-reporting or by principal component analysis (PCA) were excluded, as described elsewhere.48 All the BBJ participants provided written informed consent as approved by the ethical committee of RIKEN Yokohama Institute and the Institute of Medical Science, the University of Tokyo. The sample information of the inflammatory bowel disease cohort of Japanese was extensively described elsewhere.34,35 This study was approved by the ethical committee of Osaka University Graduate School of Medicine.
Method details
Deep-target sequencing of KIR genes
We conducted a customized target sequencing of all KIR genes and pseudogenes in the region4 (i.e., KIR3DL3, KIR2DS2, KIR2DL2, KIR2DL3, KIR2DL5A/B, KIR2DS3/5, KIR2DP1, KIR2DL1, KIR3DP1, KIR2DL4, KIR3DL1, KIR3DS1, KIR2DS1, KIR2DS4, KIR3DL2, and KIR3DX1). The target sequence was extracted from the human reference genome of GRCh37/hg19 (which covers KIR3DX1, KIR3DL3, KIR2DL3, KIR2DP1, KIR2DL1, KIR3DP1, KIR2DL4, KIR3DL1, KIR2DS4, KIR3DL2) and alternative haplotype of GL000209 (which covers KIR2DL2, KIR2DL4, KIR3DS1, KIR2DL5A, KIR2DL5B, KIR2DS5, KIR2DS3, KIR2DS1, KIR2DS2; Table S1). A sequence capture method was performed by hybridization between DNA of an adapter-ligated library (KAPA Hyper Prep Kit, Roche, CA, USA) and a biotinylated DNA probe (SeqCap EZ choice kit, Roche, CA, USA), which was designed based on the target sequences of 16 KIR genes to uniquely discriminate each of the KIR genes, despite their structural homology. We described the exact target region design in Table S2 and Figure S5, and a total length of all target regions summed up to 182,016 bp. Paired-end sequence reads (350 bp read1 and 250 bp read2 in length) were obtained by using the MiSeq sequencer (illumina, CA, USA).
The determination of KIR gene content and copy number
The sequenced reads were first mapped to the target genome sequence from the reference KIR sequences on human genome (GRCh37/hg19) and alternative haplotype of GL000209 (Table S1) using BWA (version 0.7.13)40 with default settings. We estimated the gene content and copy number of each KIR gene by quantifying the read depth, which was obtained by DiagnoseTargets module of GATK software (version3.6)41. We calculated a weighted average of read depth per gene, which was adjusted for the length of target regions within each KIR gene. To determine the gene content (i.e., the status whether an individual has the gene or not) and the gene copy number (i.e., the number of gene copies), we took the ratio of the reads mapped to the gene to those mapped to KIR3DL3, which is one of the framework genes and all individuals should have two copies in principle. For each of the 11 KIR genes other than the four frame work genes and a pseudo-gene of KIR3DX1, we collected this ratio across all samples, and computed a Gaussian kernel density estimate of the read depth distribution within the study population by KernelDensity module in python. To assign a discrete number of the gene copy number to an individual (e.g., 0,1,2,3,4), we performed unsupervised hard clustering by separating the kernel density- based distribution at the points of local minimum. The example of copy number assignment is shown in Figure S6. To assign the gene contents to an individual, we defined that an individual had the gene if the copy number of the gene equaled to or was more than one, and that the individual did not have the gene if the copy number of the gene was zero. We also performed the same analysis by using (i) the coverage of pseudogene of KIR3DX1 which also in principle all individuals have two copies, and (ii) the median coverage of all four framework genes as a reference for deriving the ratio, instead of KIR3DL3 for validation. We described the mean coverage of all four framework genes in Figure S7.
Assessment and comparison of mapping quality
To quantify the advantage of using relatively longer reads in KIR typing, we assessed and compared the mapping quality and the uniquely mapped rate between our strategy (i.e., 350/250 bp paired end) and conventional short reads (i.e., 150 bp paired end). To specifically compare the effect of read length while excluding the effects from sequences themselves, we obtained simulated short read sequences using the original read data. We used seqkit software to make a shortened fastq file with 150bp as the original fastq file by using “subset” function with “-r 1:150” option, thereby obtaining the first 150 bases of the original reads. Then, the obtained fastq files with shortened reads were mapped onto the reference using BWA software with the same settings as original protocol. For the mapping quality, we assessed the two datasets by CollectAlignmentSummaryMetrics function of Picard software (version 2.18.16). We collected the number of high-quality paired reads aligned to the reference sequence with a mapping quality of Q20 or higher over the total number of all-mapped paired reads. For the uniquely mapped rate, we counted the number of uniquely mapped reads using samtools (version 1.9). We then performed paired t-test of this metric (i.e., the fraction of high-quality reads and the fraction of uniquely mapped reads per individual) for statistical comparison.
The validation of KIR gene content by PCR-SSO method in selected subjects
We empirically confirmed the accuracy of the assigned KIR gene contents by evaluating the concordance with the PCR-SSO method for 14 KIR genes (GenoDive Pharma. Inc.). We selected 100 individuals from the cohort (n = 1,173) to maximize the diversity of KIR gene content combinations based on the results from our NGS-based pipeline, and performed the KIR typing by the PCR-SSO method in these individuals. Then, we investigated whether the gene content defined by our NGS-based pipeline was concordant with that by the PCR-SSO method for each of the 14 genes.
The ploidy-aware genotype calling of KIR region
To determine the KIR alleles at a high resolution, we next sought to identify the SNVs and indels in each of the KIR genes. Since we did not aim to determine the KIR alleles for the pseudogenes (i.e., KIR2DP1, KIR3DP1, and KIR3DX1), we performed genotyping of SNVs and indels in 13 KIR genes. For SNVs, we used HaplotypeCaller module of the GATK software with an option of ‘-ploidy’, which specified the gene copy number of each of the KIR genes. Since the individual ploidy could differ depending on the KIR genes, we conducted genotyping separately for each of the KIR genes. The output gVCFs were merged by GenotypeGVCFs module of the GATK software. For indels, it was technically difficult to perform the indel refinement in the KIR region by GATK, which resulted in few indels confidently called by the software. Since a 22bp insertion in KIR2DS4 and a 1 bp deletion in KIR2DL4 were critical in determining the alleles of these genes, we used DeepVariant software20 to complementarily call these indels. DeepVariant can detect SNPs and indels from a pileup image of the reference and read data around each candidate variant by using the deep learning model (the Inception), which achieved the high accuracy in the benchmarking for both SNPs and indels.49 We incorporated the results of the two indels from DeepVariant into a variant list for determining KIR alleles, together with SNV results from GATK.
The curation of nucleotide sequences of the KIR alleles from IPD reference
We collected the nucleotide sequence data of each of the KIR alleles that were registered to date at the IPD-KIR website. Since a complete genomic sequence data is not always available for all the KIR alleles, we curated the nucleotide sequence data, which is a fasta-based nucleotide coding sequences (CDS) of 887 KIR alleles. To obtain a list of the variants that discriminate each of the KIR alleles from the others, we ran the blast software42 to compare the CDS of one standard KIR allele (usually defined by the smallest allele nomenclature) with that of the investigated KIR allele for each of the 13 KIR genes. We thus extracted a list of 706 SNVs and 2 indels with the information of whether each KIR allele sequence has the variant or not. We finally converted the positional information of these variants from CDS-based to target sequence-based position.
The determination of the KIR alleles
We integrated a list of the genotyped variants from GATK and DeepVariant in our cohort with a list of the key variants in determining KIR alleles. We extracted the variants that are genotyped in our cohort and at the same time in the list of the key variants. Using these variants, we first made exhaustive patterns of all the possible genotypes from all the combinations of the KIR alleles at all the possible copy numbers of that gene. We then matched them with the observed genotype, together with the observed copy number of the gene. If there is no combination(s) of alleles which exactly matches with the observed genotype, we flagged them as potentially novel allele combinations, and also output the closest candidate combination of KIR alleles with a score quantifying the number of mismatches. We thus assigned the possible KIR allele combination(s) to each KIR gene in an individual. We finally merged the alleles into the lower digit when they are ambiguous at the higher digit (e.g., as we could not uniquely define one allele from KIR2DL3∗0020101, KIR2DL3∗0020102, or KIR2DL3∗0020103, these three were merged into KIR2DL3∗00201).
Assessment of LD structure of the KIR region
To evaluate the LD structure of the KIR region, we first assessed the LD pattern based on the KIR gene copy number. We note that the analyses here were not the haplotype-based but genotype-based. We re-coded the gene copy information of study individuals as a plink bed/bim/fam format, and calculated r2 value of LD by plink software.43 Next, we assessed pairwise LD based on the KIR alleles measured as the ε value, which utilizes differences of the normalized entropy of the haplotype frequency distributions between LD and the null hypothesis of linkage equilibrium (LE), using the eLD software.24 A higher ε value represents stronger LD.
Dimensionality reduction of samples based on KIR genes and alleles
We performed unsupervised clustering of the samples based on KIR gene contents and KIR alleles using a dimensionality reduction method of t-SNE. t-SNE is a non-linear dimensionality reduction method which converts similarities between data points to joint probabilities and minimizes the Kullback-Leibler divergence between the joint probabilities of the low-dimensional embedding and the high-dimensional data.50 We first applied t-SNE to the KIR gene content data using multicoreTSNE package of python software with default parameters. We manually categorized the plots into three clusters, which were colored in red, blue, and pink in Figure 3A. We next applied t-SNE to the KIR allele data. In Figure 3B, we annotated the individuals with colors according to the clusters defined in Figure 3A.
Haplotype illustration of KIR region
We visualized the haplotype structures of the KIR genes by Disentangler software,44 which can visualize haplotype configurations across multiallelic genetic markers for which typical triangular heat maps with LD indices do not work. Disentangler internally applies a series of expectation-maximization algorithms to estimate the haplotype frequencies between adjacent markers, and it then uses this information to determine the order of the alleles for each marker, such that the number of crossing lines between adjacent markers is minimized. We used the re-formatted KIR alleles computationally phased by beagle software45 as an input for Disentangler. We colored the representative haplotypes in red (group A haplotypes) and in blue (group B haplotypes) in Figure 3C.
Whole-genome sequencing of selected individuals and construction of the reference panel for the KIR imputation
To construct a KIR imputation reference panel which can be applied to diverse genotyping arrays, we conducted WGS in selected samples whose KIR alleles are defined as described above (n = 689). In brief, we sequenced 295 samples using 2 × 150-bp paired-end reads on the HiSeq X platform with a mean depth of 16.7× and 394 samples using 2 × 150-bp paired-end reads on the NovaSeq platform with a mean depth of 16.1×, and processed the sequenced reads according to the standardized best-practice method proposed by GATK (ver.3.8-0). For QC, we additionally set exclusion filters for genotypes as follows: (1) DP <5, (2) GQ <20, or (3) DP >60 and GQ <95. We set these genotypes as missing and excluded variants with call rates <90% before variant quality score recalibration (VQSR). After performing VQSR, variants located in low-complexity regions (LCR), as defined by mdust software (“hs37d5-LCRs.20140224.bed”), were excluded. We then extracted variants located on chromosome 19, and merged them with the KIR gene content and allele data of the same 689 individuals, which we encoded as biallelic markers.
Imputation accuracy of the constructed reference panel was empirically evaluated by a cross validation approach. We randomly split the panel into 10 datasets (n = 56-83 [mean = 68.9] for each dataset). KIR alleles from one of the datasets were masked, and then imputed using the remaining nine datasets as an imputation reference using beagle software, which uses an HMM-based haplotype phasing and imputation algorithm. To evaluate the imputation accuracy in a realistic setting, we restricted the scaffold variants into those on the Illumina OmniExpressExome BeadChip array which we used in the real data of BioBank Japan, instead of using all the whole-genome-sequenced variants on chromosome 19. The concordances between the imputed and genotyped KIR allele dosages were calculated separately for each KIR gene content and each allele.
To evaluate the imputation accuracy in other populations than Japanese, we genotyped part of the IHWG samples (n = 40) using Illumina OmniExpressExome BeadChip. For sample QC, we excluded individuals with call rate < 98%. For variant QC, we removed (i) duplicated variants based on call rate, (ii) multi-mapped variants (iii) variants with call rate < 98%, (iv) variants with Hardy–Weinberg equilibrium P ≤ 1 × 10−10, and (v) variants with allele frequency difference from 1000 genomes project > 40%. After QC, we had 39 samples and 639,236 variants in total, of which 13,890 were on chromosome 19. We performed the principal component analysis to confirm the genetic ancestry of those samples (Figure S8). We phased 13,890 variants on chromosome 19 using shapeit2 software, and imputed KIR alleles by using the full reference panel of Japanese (nreference = 689) and minimac3 software.46 Here, we used minimac3 software instead of beagle for imputation to enable the direct comparison with the imputation conducted in BioBank Japan, which we will describe in the next section. The imputation accuracy was assessed by comparing the imputed allele dosages with those from the previously-defined KIR alleles based on pyrosequencing.
The benchmarking against KIR∗IMP software
To benchmark the imputation accuracy of our pipeline against the previously published imputation method, we performed the KIR gene copy number imputation by using KIR∗IMP software (http://imp.science.unimelb.edu.au/kir/documentation).13 We used phased haplotypes of (a) SNP genotype data (OmniExpressExome BeadChip array) from IHWG dataset and (b) mock GWAS genotype on the Illumina OmniExpressExome BeadChip array constructed from WGS of part of Japanese individuals in KIR reference panel. The haplotype phasing was performed using SHAPEIT2 software. We then uploaded the haplotypes to the software webserver, and obtained the result of imputed KIR gene copy numbers. The imputation accuracy was assessed by comparing the imputed copy number with those inferred from the NGS-based pipeline.
The KIR imputation in biobank-scale individuals and comprehensive PheWAS
Using the constructed high-resolution imputation reference panel, we imputed the KIR gene contents and alleles in the large-scale GWAS data of the BBJ individuals (n = 164,540)31,47,48 and inflammatory bowel disease (IBD) cohort (n = 5,367).34,35 The BBJ GWAS data was genotyped by the Illumina HumanOmniExpressExome BeadChip or a combination of the Illumina HumanOmniExpress and HumanExome BeadChips. We had previously confirmed that the genotype from these SNP arrays and that from the WGS were highly concordant (≥ 99.97%).51 The IBD cohort samples were genotyped by the ImmunoChip. Detailed characteristics of the GWAS data and the QC process were described elsewhere.34,35,48 While we used beagle software for KIR imputation in cross validation and in the IBD cohort, beagle software is currently not scalable to be applied to biobank-scale GWAS data such as BioBank Japan due to huge requirement of memory resources. Thus, we imputed the KIR genes and alleles using the standard genome-wide imputation softwares (i.e., Eagle [version 2.3] for haplotype phasing and minimac3 [version 2.0.1] for imputation). After the imputation, we applied post-imputation QC filtering of the variants (MAF ≥ 1% and imputation score Rsq ≥ 0.5). We again sought to benchmark this imputation strategy which used pre-phasing and minimac3 software instead of beagle, as well as in the meantime to perform benchmarking by using relatively independent dataset rather than ten-fold cross-validation approach. To this end, we created the new imputation reference panel using KIR alleles and WGS data of Hiseq X platform (n = 295), and on the other hand generated the mock GWAS dataset by including the variants on the Illumina OmniExpressExome BeadChip array from the WGS data of NovaSeq platform (n = 394). We then pre-phased the mock GWAS data using shapeit2 software52 (here we did not use Eagle2 for phasing since the sample size is small), and performed KIR imputation with this pre-phased genotype and the above mentioned reference panel from Hiseq X platform. We finally assessed the accuracy of imputation by calculating the concordances between the imputed and genotyped KIR allele dosages.
PheWAS was conducted to investigate the associations of the imputed KIR alleles with 85 human complex traits (23 diseases and 25 quantitative traits for BioBank Japan and 2 diseases [ulcerative colitis and Crohn’s disease] for the IBD cohort; details in Table S11). The diseases consisted of 4 major categories (immune/allergy [n = 7], cardiovascular and metabolic [n = 8], malignancy [n = 5], and other diseases [n = 5]). The quantitative traits consisted of 9 major categories (anthropometric [n = 3], blood pressure [n = 4], protein [n = 2], electrolyte [n = 4], metabolic [n = 6], liver-related [n = 6], other biochemical [n = 3], kidney-related [n = 4], hematological [n = 13]). Definition of the diseases and the process of patient registration are described elsewhere.47,53 For the controls in disease association studies, we used healthy participants in the IBD cohort, and used a mixed control group by excluding the subjects affected with the disease under investigation in BioBank Japan as described previously.2 Detailed processes of outlier exclusion, adjustment with clinical status, normalization methods of the quantitative traits are extensively described elsewhere.31,33 We evaluated the associations of the KIR alleles with the risk of the diseases using a logistic regression model, and with dosage effects on the normalized values of the quantitative traits using a linear regression model, using plink2 software. We assumed additive effects of the allele dosages on phenotypes in the regression models. We included age, sex, and the top genotype 20 PCs as covariates in the analysis of BioBank Japan, and included sex and the top genotype 20 PCs in the analysis of the IBD cohort to account for potential population stratification.
Quantification and statistical analysis
All the statistical methods, softwares, and a custom code used in this study are listed in the corresponding sections in the method details as well as the key resource table. The statistical significance was determined by properly accounting for multiple testing, by Bonferroni correction. All P values are two-sided.
Acknowledgments
We thank all participants enrolled in this study. We thank Prof. Hisashi Arase for critical advice regarding the biological insights into the KIR genes. We would like to express our deepest gratitude to our co-author Prof. Hidetoshi Inoko, who passed away in January of 2022, for his invaluable contribution to creating a platform for performing KIR and HLA typing. This research was supported by JSPS KAKENHI grant 16H06279 (PAGS); the Tailor-Made Medical Treatment program (BioBank Japan Project) of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT); the Japan Agency for Medical Research and Development (AMED); MEXT KAKENHI (221S0002); and the Bioinformatics Initiative of Osaka University Graduate School of Medicine. S.S. was in part supported by The Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Kanae Foundation for the Promotion of Medical Science, the Astellas Foundation for Research on Metabolic Disorders, The JCR Grant for Promoting Basic Rheumatology, the Manabe Scholarship Grant for Allergic and Rheumatic Diseases, and the Uehara Memorial Foundation. Y.O. was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (19H01021 and 20K21834), AMED (JP20km0405211, JP20ek0109413, JP20ek0410075, JP20gm4010006, and 20km0405217), JST Moonshot R&D (JPMJMS2021 and JPMJMS2024), the Takeda Science Foundation, and the Bioinformatics Initiative of Osaka University Graduate School of Medicine, Osaka University. K.H. was supported by JSPS KAKENHI grant JP16H06502 “Neo-self.”
Author contributions
S.S. and Y.O. conceived and supervised the study. S.S., K.H., and Y.O. wrote the manuscript. S.S., J.H., T.N., Y.K., and Y.O. conducted the data analysis. K. Yamazaki, M.Y., N.Y., J.U., T.K., T. Matsui, S.M., Y.S., H.I., A.T., T. Morisaki, K.M., and K. Yamamoto managed samples and provided the data. S.S., J.H., K.H., H.N., M.Y., and I.I. conducted the experiments.
Declaration of interests
H.I. is a founder of GenoDive Pharma. Inc. J.H. is an employee of Teijin Pharma Limited.
Published: March 9, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100101.
Contributor Information
Saori Sakaue, Email: ssakaue@broadinstitute.org.
Yukinori Okada, Email: yokada@sg.med.osaka-u.ac.jp.
Supplemental information
Data and code availability
-
•
Individual-level KIR alleles and KIR imputation reference panel used in this study are deposited at the National Bioscience Database Center (NBDC) Human Database (https://humandbs.biosciencedbc.jp/en/) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The genomic DNA we used for KIR sequencing were obtained from a cell line established by the Japan Biological Informatics Consortium (JBIC), and can be purchased at https://bioresource.nibiohn.go.jp/psc/index.html. The International Histocompatibility Working Group (IHWG) cell lines were obtained from ECACC HLA typed collection (https://www.phe-culturecollections.org.uk/products/celllines/hlatyped/search.jsp). The genotype data of BBJ used in this study are available from the Japanese Genotype-phenotype Archive (JGA) through application at https://humandbs.biosciencedbc.jp/en/hum0014-latest. Accession numbers are listed in the key resources table. Other web resources used in this study are listed in the key resources table.
-
•
An original code used for determining ploidy (gene copy number) and allele genotype of KIR is available at https://github.com/saorisakaue/KIR_project with reference data curated from IPD-KIR allele database, both of which are publicly available (https://doi.org/10.5281/zenodo.5908796).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirata J., Hosomichi K., Sakaue S., Kanai M., Nakaoka H., Ishigaki K., Suzuki K., Akiyama M., Kishikawa T., Ogawa K., et al. Genetic and phenotypic landscape of the major histocompatibilty complex region in the Japanese population. Nat. Genet. 2019;51:470–480. doi: 10.1038/s41588-018-0336-0. [DOI] [PubMed] [Google Scholar]
- 3.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson J., Halliwell J.A., McWilliam H., Lopez R., Marsh S.G.E. IPD - the immuno polymorphism database. Nucleic Acids Res. 2013;41:1234–1240. doi: 10.1093/nar/gks1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holm S.J., Sakuraba K., Mallbris L., Wolk K., Ståhle M., Sánchez F.O. Distinct HLA-C/KIR genotype profile associates with guttate psoriasis. J. Invest. Dermatol. 2005;125:721–730. doi: 10.1111/j.0022-202X.2005.23879.x. [DOI] [PubMed] [Google Scholar]
- 6.Ahn R.S., Moslehi H., Martin M.P., Abad-Santos M., Bowcock A.M., Carrington M., Liao W. Inhibitory KIR3DL1 alleles are associated with psoriasis. Br. J. Dermatol. 2016;174:449–451. doi: 10.1111/bjd.14081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Re V., Caggiari L., De Zorzi M., Repetto O., Zignego A.L., Izzo F., Tornesello M.L., Buonaguro F.M., Mangia A., Sansonno D., et al. Genetic diversity of the KIR/HLA system and susceptibility to hepatitis C virus-related diseases. PLoS One. 2015;10:e0117420. doi: 10.1371/journal.pone.0117420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiby S.E., Walker J.J., O’Shaughnessy K.M., Redman C.W.G., Carrington M., Trowsdale J., Moffett A. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J. Exp. Med. 2004;200:957–965. doi: 10.1084/jem.20041214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mancusi A., Ruggeri L., Urbani E., Pierini A., Massei M.S., Carotti A., Terenzi A., Falzetti F., Tosti A., Topini F., et al. Haploidentical hematopoietic transplantation from KIR ligand-mismatched donors with activating KIRs reduces nonrelapse mortality. Blood. 2015;125:3173–3182. doi: 10.1182/blood-2014-09-599993. [DOI] [PubMed] [Google Scholar]
- 10.Trefny M.P., Rothschild S.I., Uhlenbrock F., Rieder D., Kasenda B., Stanczak M.A., Berner F., Kashyap A.S., Kaiser M., Herzig P., et al. A variant of a killer cell immunoglobulin-like receptor is associated with resistance to PD-1 blockade in lung cancer. Clin. Cancer Res. 2019;25:3026–3034. doi: 10.1158/1078-0432.CCR-18-3041. [DOI] [PubMed] [Google Scholar]
- 11.Norman P.J., Hollenbach J.A., Nemat-Gorgani N., Marin W.M., Norberg S.J., Ashouri E., Jayaraman J., Wroblewski E.E., Trowsdale J., Rajalingam R., et al. Defining KIR and HLA class I genotypes at highest resolution via high-throughput sequencing. Am. J. Hum. Genet. 2016;99:375–391. doi: 10.1016/j.ajhg.2016.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roe D., Kuang R. Accurate and efficient KIR gene and haplotype inference from genome sequencing reads with novel K-mer signatures. Front. Immunol. 2020;11:3102. doi: 10.3389/fimmu.2020.583013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vukcevic D., Traherne J.A., Næss S., Ellinghaus E., Kamatani Y., Dilthey A., Lathrop M., Karlsen T.H., Franke A., Moffatt M., et al. Imputation of KIR types from SNP variation data. Am. J. Hum. Genet. 2015;97:593–607. doi: 10.1016/j.ajhg.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ovsyannikova I.G., Schaid D.J., Larrabee B.R., Haralambieva I.H., Kennedy R.B., Poland G.A. A large population-based association study between HLA and KIR genotypes and measles vaccine antibody responses. PLoS One. 2017;12:e0171261. doi: 10.1371/journal.pone.0171261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin A.R., Kanai M., Kamatani Y., Okada Y., Neale B.M., Daly M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uhrberg M., Valiante N.M., Shum B.P., Shilling H.G., Lienert-Weidenbach K., Corliss B., Tyan D., Lanier L.L., Parham P. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7:753–763. doi: 10.1016/s1074-7613(00)80394-5. [DOI] [PubMed] [Google Scholar]
- 17.Hsu K.C., Chida S., Geraghty D.E., Dupont B. The killer cell immunoglobulin-like receptor (KIR) genomic region: gene-order, haplotypes and allelic polymorphism. Immunol. Rev. 2002;190:40–52. doi: 10.1034/j.1600-065x.2002.19004.x. [DOI] [PubMed] [Google Scholar]
- 18.Yawata M., Yawata N., Draghi M., Little A.-M., Partheniou F., Parham P. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J. Exp. Med. 2006;203:633–645. doi: 10.1084/jem.20051884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norman P.J., Stephens H.A.F., Verity D.H., Chandanayingyong D., Vaughan R.W. Distribution of natural killer cell immunoglobulin-like receptor sequences in three ethnic groups. Immunogenetics. 2001;52:195–205. doi: 10.1007/s002510000281. [DOI] [PubMed] [Google Scholar]
- 20.Poplin R., Chang P.C., Alexander D., Schwartz S., Colthurst T., Ku A., Newburger D., Dijamco J., Nguyen N., Afshar P.T., et al. A universal snp and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018;36:983. doi: 10.1038/nbt.4235. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y.-Y., Mimori T., Khor S.-S., Gervais O., Kawai Y., Hitomi Y., Tokunaga K., Nagasaki M. HLA-VBSeq v2: improved HLA calling accuracy with full-length Japanese class-I panel. Hum. Genome Var. 2019;6 doi: 10.1038/s41439-019-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parham P. MHC class I molecules and KIRS in human history, health and survival. Nat. Rev. Immunol. 2005;5:201–214. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- 23.Vierra-Green C., Roe D., Jayaraman J., Trowsdale J., Traherne J., Kuang R., Spellman S., Maiers M. Estimating KIR haplotype frequencies on a cohort of 10,000 individuals: a comprehensive study on population variations, typing resolutions, and reference haplotypes. PLoS One. 2016;11:e0163973. doi: 10.1371/journal.pone.0163973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okada Y. eLD: entropy-based linkage disequilibrium index between multiallelic sites. Hum. Genome Var. 2018;5:29. doi: 10.1038/s41439-018-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Der Maaten L. Accelerating t-SNE using tree-based algorithms. J. Machine Learn. Res. 2014:3221–3245. Volume 15 [Google Scholar]
- 26.Kumasaka N., Nakamura Y., Kamatani N. The textile plot: a new Linkage disequilibrium display of multiple-Single Nucleotide Polymorphism genotype data. PLoS One. 2010;5:e10207. doi: 10.1371/journal.pone.0010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada Y., Momozawa Y., Ashikawa K., Kanai M., Matsuda K., Kamatani Y., Takahashi A., Kubo M. Construction of a population-specific HLA imputation reference panel and its application to Graves’ disease risk in Japanese. Nat. Genet. 2015;47:798–802. doi: 10.1038/ng.3310. [DOI] [PubMed] [Google Scholar]
- 28.Browning B.L., Browning S.R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 2016;98:116–126. doi: 10.1016/j.ajhg.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirata J., Hirota T., Ozeki T., Kanai M., Sudo T., Tanaka T., Hizawa N., Nakagawa H., Sato S., Mushiroda T., et al. Variants at HLA-A, HLA-C, and HLA-DQB1 confer risk of psoriasis vulgaris in Japanese. J. Invest. Dermatol. 2018;138:542–548. doi: 10.1016/j.jid.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Raychaudhuri S., Sandor C., Stahl E.A., Freudenberg J., Lee H.S., Jia X., Alfredsson L., Padyukov L., Klareskog L., Worthington J., et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012;44:291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanai M., Akiyama M., Takahashi A., Matoba N., Momozawa Y., Ikeda M., Iwata N., Ikegawa S., Hirata M., Matsuda K., et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 2018;50:390–400. doi: 10.1038/s41588-018-0047-6. [DOI] [PubMed] [Google Scholar]
- 32.Ishigaki K., Akiyama M., Kanai M., Takahashi A., Kawakami E., Sugishita H., Sakaue S., Matoba N., Low S.K., Okada Y., et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020;52:669–679. doi: 10.1038/s41588-020-0640-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakaue S., Kanai M., Karjalainen J., Akiyama M., Kurki M., Matoba N., Takahashi A., Hirata M., Kubo M., Matsuda K., et al. Trans-biobank analysis with 676,000 individuals elucidates the association of polygenic risk scores of complex traits with human lifespan. Nat. Med. 2020;26:542–548. doi: 10.1038/s41591-020-0785-8. [DOI] [PubMed] [Google Scholar]
- 34.Gupta A., Juyal G., Sood A., Midha V., Yamazaki K., Vich Vila A., Esaki M., Matsui T., Takahashi A., Kubo M., et al. A cross-ethnic survey of CFB and SLC44A4, Indian ulcerative colitis GWAS hits, underscores their potential role in disease susceptibility. Eur. J. Hum. Genet. 2016;25:111–122. doi: 10.1038/ejhg.2016.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han B., Akiyama M., Kim K.K., Oh H., Choi H., Lee C.H., Jung S., Lee H.S., Kim E.E., Cook S., et al. Amino acid position 37 of HLA-DRβ1 affects susceptibility to Crohn’s disease in Asians. Hum. Mol. Genet. 2018;27:3901–3910. doi: 10.1093/hmg/ddy285. [DOI] [PubMed] [Google Scholar]
- 36.Saito H., Hirayama A., Umemura T., Joshita S., Mukawa K., Suga T., Tanaka E., Ota M. Association between KIR-HLA combination and ulcerative colitis and Crohn’s disease in a Japanese population. PLoS One. 2018;13:e0195778. doi: 10.1371/journal.pone.0195778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aghaei H., Mostafaei S., Aslani S., Jamshidi A., Mahmoudi M. Association study between KIR polymorphisms and rheumatoid arthritis disease: an updated meta-analysis. BMC Med. Genet. 2019;20:24. doi: 10.1186/s12881-019-0754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van der Slik A.R., Koeleman B.P.C., Verduijn W., Bruining G.J., Roep B.O., Giphart M.J. KIR in type 1 diabetes: disparate distribution of activating and inhibitory natural killer cell receptors in patients versus HLA-matched control subjects. Diabetes. 2003;52:2639–2642. doi: 10.2337/diabetes.52.10.2639. [DOI] [PubMed] [Google Scholar]
- 39.Nagai A., Hirata M., Kamatani Y., Muto K., Matsuda K., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Mushiroda T., et al. Overview of the BioBank Japan project: study design and profile. J. Epidemiol. 2017;27:S2–S8. doi: 10.1016/j.je.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van der Auwera G., O’Connor B., Safari an O.M.C. O’Reilly Media, Inc.; 2020. Genomics in the Cloud. [Google Scholar]
- 42.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 43.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okada Y., Yamazaki K., Umeno J., Takahashi A., Kumasaka N., Ashikawa K., Aoi T., Takazoe M., Matsui T., Hirano A., et al. HLA-Cw (∗)1202-B (∗)5201-DRB1 (∗)1502 haplotype increases risk for ulcerative colitis but reduces risk for Crohn’s disease. Gastroenterology. 2011;141:864–871.e1-5. doi: 10.1053/j.gastro.2011.05.048. [DOI] [PubMed] [Google Scholar]
- 45.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hirata M., Kamatani Y., Nagai A., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Kubo M., Muto K., Mushiroda T., et al. Cross-sectional analysis of BioBank Japan clinical data: a large cohort of 200,000 patients with 47 common diseases. J. Epidemiol. 2017;27:S9–S21. doi: 10.1016/j.je.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akiyama M., Ishigaki K., Sakaue S., Momozawa Y., Horikoshi M., Hirata M., Matsuda K., Ikegawa S., Takahashi A., Kanai M., et al. Characterizing rare and low-frequency height-associated variants in the Japanese population. Nat. Commun. 2019;10:4393. doi: 10.1038/s41467-019-12276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumaran M., Subramanian U., Devarajan B. Performance assessment of variant calling pipelines using human whole exome sequencing and simulated data. BMC Bioinformatics. 2019;20:342. doi: 10.1186/s12859-019-2928-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Maaten L., Hinton G. Visualizing data using t-SNE Laurens. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- 51.Okada Y., Momozawa Y., Sakaue S., Kanai M., Ishigaki K., Akiyama M., Kishikawa T., Arai Y., Sasaki T., Kosaki K., et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 2018;9:1631. doi: 10.1038/s41467-018-03274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delaneau O., Zagury J.F., Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 53.Sakaue S., Kanai M., Tanigawa Y., Karjalainen J., Kurki M., Koshiba S., Narita A., Konuma T., Yamamoto K., Akiyama M., et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021;53:1415–1424. doi: 10.1038/s41588-021-00931-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Individual-level KIR alleles and KIR imputation reference panel used in this study are deposited at the National Bioscience Database Center (NBDC) Human Database (https://humandbs.biosciencedbc.jp/en/) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The genomic DNA we used for KIR sequencing were obtained from a cell line established by the Japan Biological Informatics Consortium (JBIC), and can be purchased at https://bioresource.nibiohn.go.jp/psc/index.html. The International Histocompatibility Working Group (IHWG) cell lines were obtained from ECACC HLA typed collection (https://www.phe-culturecollections.org.uk/products/celllines/hlatyped/search.jsp). The genotype data of BBJ used in this study are available from the Japanese Genotype-phenotype Archive (JGA) through application at https://humandbs.biosciencedbc.jp/en/hum0014-latest. Accession numbers are listed in the key resources table. Other web resources used in this study are listed in the key resources table.
-
•
An original code used for determining ploidy (gene copy number) and allele genotype of KIR is available at https://github.com/saorisakaue/KIR_project with reference data curated from IPD-KIR allele database, both of which are publicly available (https://doi.org/10.5281/zenodo.5908796).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.