

Summary
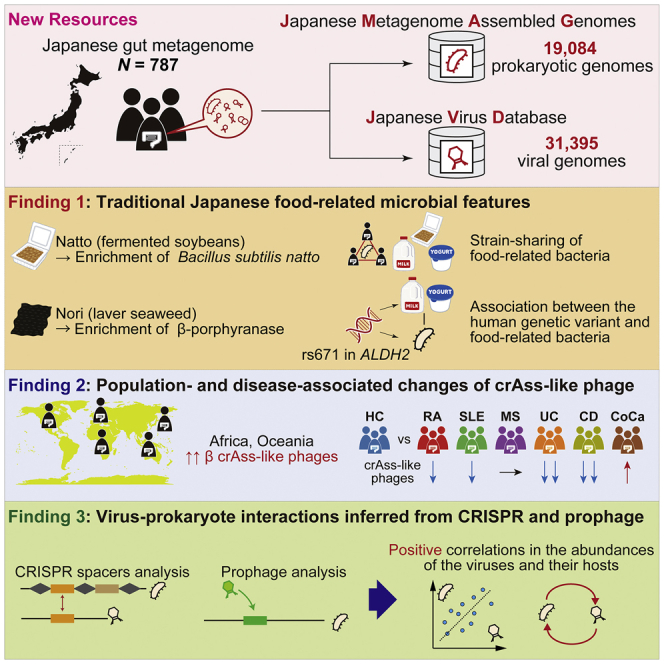
We reconstructed 19,084 prokaryotic and 31,395 viral genomes from 787 Japanese gut metagenomes as Japanese metagenome-assembled genomes (JMAG) and Japanese Virus Database (JVD), which are large microbial genome datasets for a single population. Population-specific enrichment of the Bacillus subtilis and β-porphyranase among the JMAG could derive from the Japanese traditional food natto (fermented soybeans) and nori (laver), respectively. Dairy-related Enterococcus_B lactis and Streptococcus thermophilus were nominally associated with the East Asian-specific missense variant rs671:G>A in ALDH2, which was associated with dairy consumption. Of the species-level viral genome clusters in the JVD, 62.9% were novel. The β crAss-like phage composition was low among the Japanese but relatively high among African and Oceanian peoples. Evaluations of the association between crAss-like phages and diseases showed significant disease-specific associations. Our large catalog of virus-host pairs identified the positive correlation between the abundance of the viruses and their hosts.
Keywords: metagenome-assembled genome, prokaryotic genome, viral genome, gut microbe, database
Graphical abstract
Highlights
-
•
Assembly of 19,084 prokaryotic and 31,395 viral genomes from Japanese gut metagenome
-
•
Traditional Japanese food-related features were observed in Japanese microbial genome
-
•
crAss-like phages were associated with populations and diseases
-
•
Abundances of bacteriophages and their hosts tended to be positively correlated
Tomofuji et al. reconstructed 19,084 prokaryotic and 31,395 viral genomes from Japanese gut metagenome shotgun sequencing data. They revealed the association between the gut microbiome and diet, populations, and diseases. Their genome catalog, JMAG and JVD, contributes to expanding the diversity of the microbial genome of a previously underrepresented population.
Introduction
The human microbiome is a complex microbial community inhabiting the human body. The largest community of the human microbiota resides within the gut and they interact with the host’s body via the immune system and metabolic reactions.1 Thus, understanding the human gut microbiome is important not only in terms of microbiology but also for medicine.
In gut microbiome studies, the genomic sequences of the individual microbes are important resources that by themselves reflect the diversity and function of the gut microbiome and also can be utilized as the reference genomes for quantification with metagenome shotgun sequencing (MSS) data. Therefore, great efforts have been spent on expanding the catalog of the gut microbe genomes. In addition to culturing efforts,2,3,4 genome assembly and binning from gut MSS data have greatly expanded the known diversity of the human gut microbiome.5,6,7 These efforts to recover metagenome-assembled genomes (MAGs) from large-scale human MSS data enabled us to survey the previously unknown part of the gut microbiome, especially for unculturable prokaryotes. Recently, several microbial genome databases, including MAG datasets, were integrated, and a Unified Human Gastrointestinal Genome (UHGG) collection comprised of 4,644 species-level genomes, which represented >200,000 non-redundant reference genomes, was released as the currently most comprehensive atlas of the human gut prokaryotes.8 However, current populational diversity of the prokaryotic genomes is still limited because the number of MAGs recovered from populations other than European, North American, and Chinese is relatively low. Therefore, reconstruction of the MAGs from currently underrepresented populations is warranted.
Although many of the gut microbiome studies have focused on the prokaryotes, viruses, mainly bacteriophages, are also highly abundant in the gut microbiome.9 Bacteriophages infect bacteria and regulate the bacteriome by either lysing their hosts or altering their physiological functions. In addition to the mediating effects, gut viruses are thought to directly interact with our body via the immune system.10,11 Various diseases, such as intestinal diseases12,13 and metabolic diseases,14,15 are associated with the gut virome. However, most of the human gut virome is still poorly characterized, partially because the traditional laboratory techniques, such as culturing, are typically low throughput and not applicable for some viruses. To overcome this problem, viral genomes have been recovered from the MSS data, and de novo assembly of the viral genomes greatly expanded the repertoire of the viral genomes and enable us to reveal a part of the gut virome.16,17 For example, crAss-like phages, one of the major components of the human gut viromes, were first discovered in 2014 by cross-assembly of the MSS data.18 Recently, a few studies recovered viral genomes from large-scale MSS data.19,20 However, the diversity of the gut viral genomes is still not saturated and the current populational diversity of the viral genomes remains limited, as is the case of the prokaryotes.
The Japanese have unique dietary culture and habits, which have resulted in the unique features of the gut microbiome, such as the enrichment of the enzymes degrading seaweed-derived polysaccharides,21 carbohydrate metabolism-related genes, and Actinobacteria, compared with other populations.22 However, most of the previous studies utilized reference bacterial genomes for phylogenetic analyses. Thus, the existence of the gut microbes that were not covered by the reference dataset have not been fully evaluated. In addition, previous analysis of the gut microbial genes lacked the link between the genes and their genome of origins, which hindered us from understanding the taxonomic features of the microbial genes. Also, few studies have focused on the Japanese gut virome,16 and there are only a small number of publicly available viral genomes recovered from the Japanese gut metagenome. Therefore, recovering MAGs and viral genomes from the Japanese gut metagenome is necessary for obtaining deep insights into the Japanese gut microbiome and complementing the microbial genome databases by increasing the populational diversity.
We recovered MAGs and viral genomes from the gut MSS data of 787 Japanese individuals.23,24,25,26,27 Utilizing these reconstructed microbial genomes, we evaluated the existence of the microbial taxa and genes that were specific to the Japanese, revealed the association of the crAss-like phages with the populations and diseases, and expanded the current knowledge of the virus-prokaryote interaction. The reconstructed microbial genomes and related information are available to the scientific community.
Results
Reconstruction of MAGs from the Japanese MSS data
To recover MAGs from the Japanese gut, we performed a single-sample metagenomic assembly and binning on 787 Japanese gut MSS data23,24,25,26,27,28 (Figure S1; Table S1). After the filtering based on the CheckM29 (>50% genome completeness, <5% contamination, and an estimated quality score >50; STAR Methods), we obtained 19,084 MAGs that met or exceeded the medium quality defined by “minimum information about a metagenome-assembled genome” standard30 (≥50% genome completeness and <10% contamination; Figures S2A–S2J; Table S2; Data S1), and we call this set of the MAGs the JMAG (Japanese metagenome-assembled genomes). We refer to the 11,917 MAGs with >90% genome completeness and <5% contamination as near-complete following the UHGG.8
The JMAG genomes were then clustered into 1,273 species-level clusters based on the average nucleotide identity (ANI). Although some of the species-level clusters had corresponding clusters in the UHGG (1,040 clusters composed of 18,734 MAGs), others did not (233 clusters composed of 350 MAGs). We assigned taxonomic information to the JMAG genomes with GTDB-tk31 and constructed a maximum-likelihood phylogenetic tree. Among the JMAG genomes presented in the UHGG, Firmicutes_A, Bacteroidota, and Actinobacteriota were frequent (Figure 1A). Among the JMAG genomes that did not present in the UHGG, the frequency of Actinobacteriota was higher than other MAGs, which reflected the high species-level diversity of the genus Collinsella (Figures 1A and 1B). To evaluate how representative the JMAG representative genomes were of Japanese gut microbial diversity, we mapped the gut MSS data against the 1,273 JMAG representative genomes. As for the Japanese gut MSS data, the mapping ratio to the 1,273 JMAG representative genomes was almost comparable with that of the 4,644 UHGG representative genomes despite the smaller number of the genomes in the JMAG than the UHGG (concordantly mapped read, 75.2% for the JMAG and 78.6% for the UHGG; overall mapped read, 82.6% for the JMAG and 86.4% for the UHGG; Figures S2K and S2L). Merging the 4,644 UHGG representative genomes and the 233 JMAG representative genomes that did not present in the UHGG only slightly improved the mapping ratio (concordantly mapped read, 79.1%; overall mapped read, 86.9%). Note that the differences in the mapping ratio between the JMAG and UHGG were larger in other populations than in Japan.
Figure 1.

Phylogenetic analysis of the JMAG genomes
(A) A pie chart illustrating the phylum-level phylogenetic composition of the JMAG, which had corresponding species-level clusters in the UHGG (top) or not (bottom). The phyla comprising <1% of each genome set are collapsed into “Other.”
(B) A maximum-likelihood phylogenetic tree reconstructed from 1,267 species-level representative bacterial MAGs. The color of the nodes represents whether the species-level clusters have corresponding clusters in the UHGG (navy) or not (magenta). An outer ring represents phylum-level taxonomic annotation. A bar plot depicted in the periphery of the tree represents the number of the MAGs belonging to the same cluster of the representative genomes.
(C) A scatterplot represents the number of the JMAG genomes in the species-level clusters (x axis) and non-Japanese-derived UHGG genomes belonging to the corresponding species-level clusters (y axis). The colors of the dots represent phylum-level taxonomy. The six species-level clusters that contained ≥10 JMAG genomes and ≤1 UHGG genome are represented by rhombus.
(D) A boxplot of the Bacillus subtilis abundances (RPKM) in the different populations. The boxplot indicates the median values (center lines) and IQRs (box edges), with the whiskers extending to the most extreme points within the range between (lower quantile − [1.5 × IQR]) and (upper quantile + [1.5 × IQR]).
(E) A non-metric multidimensional scaling plot of the Bacillus subtilis genomes. The colors of the dots represent the derivation of the genomes. The Bacillus subtilis natto and Bacillus subtilis 168 genomes in the GenBank are annotated and depicted in rhombus. IQR, interquartile range. See also Figures S1–S4, Table S2, and Data S1 and S2.
To evaluate whether the JMAG included the prokaryotic species that were underrepresented in the non-Japanese populations, we compared the number of the JMAG genomes in the species-level clusters and non-Japanese UHGG genomes belonging to the corresponding species-level clusters (Figure 1C). We found that six species-level clusters were enriched in the JMAG compared with the UHGG (≥10 JMAG genomes and ≤1 UHGG genome). MAGs in these species-level clusters, especially the unclassified Acutalibacteraceaem and Bacillus subtilis, had several carbohydrate active enzymes (CAZymes) that were specific to these species-level clusters among the JMAG (Figures S3A and S3B), suggesting that they might have unique metabolic functions in the Japanese gut microbiome. These CAZymes were underrepresented in the Unified Human Gastrointestinal Protein (UHGP) (Figure S3B), currently the largest gut microbiome protein database, and tend to be more abundant in Japanese than other populations (Figure S3C). Therefore, the JMAG captured a part of the gut microbial features that were underrepresented in the previous studies.
Bacillus subtilis was frequently seen in the JMAG (26 MAGs), while only an isolated genome was included in the UHGG. Bacillus subtilis was more frequent in the Japanese than other datasets of different populations, also in the read-based quantification approach (Figure 1D). To reveal the phylogenetic characteristics of the Bacillus subtilis genomes in the JMAG and UHGG, we retrieved 162 Bacillus subtilis genomes that were available in the GenBank for comparative analyses. Bacillus subtilis genomes in the JMAG were closely placed to Bacillus subtilis natto by ANI-based non-metric multidimensional scaling analysis (Figures 1E, S4A, and S4B; Data S2). Bacillus subtilis natto is a key component of a Japanese traditional fermented food natto. Thus, it was suggested that Bacillus subtilis in the JMAG was Bacillus subtilis natto and its frequent presence in the JMAG compared with the UHGG was the result of the Japanese unique diet.
Taxonomic and population annotation of the β-porphyranase
To gain functional insights into the reconstructed MAGs, we predicted 43,043,613 hypothetical proteins in the JMAG genomes and functionally annotated them. Most of the predicted proteins were covered by the eggNOG-mapper for the frequently reconstructed taxa, such as Firmicutes_A, Bacteroidota, and Actinobacteria, while they included a significant number of the functionary uncharacterized proteins (Figure S5A). Both the database coverage ratio and functional annotation ratio (ratio of the proteins that had any eggNOG-mapper hit and functionally characterized COG annotation, respectively; STAR Methods) for some taxa, such as Cyanobacteria and Verrucomicrobiota, were relatively low (Figure S5A). We found the phylum specificity of a part of the proteins. For example, GH92 (dbCAN2) and susD (Kyoto Encyclopedia of Genes and Genomes [KEGG] gene) were predominantly derived from the Bacteroidota (Figures S5B–S5E). We merged the predicted protein sequences of the JMAG to the UHGP and evaluated the overlap between the two datasets by clustering at 100%, 95%, 90%, and 50% sequence identities. Among the clusters that included the predicted proteins in the JMAG, 46.1%, 19.6%, 16.2%, and 9.5% were solely detected in the JMAG (Figure S5F).
Among the proteins in the JMAG, we focused on β-porphyranase, which catalyzes the hydrolysis of the seaweed-derived polysaccharides, namely porphyran. A previous study identified β-porphyranase in the Phocaeicola plebeius (renamed from Bacteroides plebeius) genome and revealed that β-porphyranase was detectable in the Japanese gut but not in the European gut21 because the Japanese eat nori made from porphyra. However, its taxonomic origin and populational pattern were not fully evaluated because of the limited availability of the gut MSS data at that time. We identified the putative β-porphyranase sequences in the JMAG and UHGP, and all of them were placed close to the known β-porphyranase sequences in a maximum-likelihood phylogenetic tree, suggesting that our analysis successfully discriminated the β-porphyranase from other related proteins (Figure 2A). Among the β-porphyranase sequences in the JMAG, three sequences (JPN-Por1, JPN-Por-2, and JPN-Por-5) were also included in the UHGG (amino acid identity [AAI] > 99%), while the other five sequences were solely included in the JMAG (Figure S6A). We detected the 133 and 245 β-porphyranase sequences in the JMAG (ratio = 133/43,043,613 = 3.09 × 10−6) and UHGP (ratio = 245/625,255,473 = 3.92 × 10−7), respectively, suggesting that β-porphyranase was more frequent among the Japanese-derived gut prokaryotic genomes than those mainly derived from other populations. We evaluated the taxonomic origin of β-porphyranase and found that the majority of the taxonomy was Bacteroidota (78.9% in the JMAG and 93.9% in the UHGP), although Firmicutes_A-derived β-porphyranase proteins were also detected in both the JMAG and UHGP (18.8% and 5.3%, respectively; Figure 2B). At the genus level, we detected Phocaeicola and Bacteroides as the major origins of β-porphyranase both in the JMAG and UHGP. We also evaluated the populational pattern of β-porphyranase in the UHGP and found that most of the β-porphyranase sequences were derived from the Asian population (Figure 2C). As for the country-level annotation, although the ratio of the β-porphyranase sequences in the Chinese population was lower than the Japanese population (ratio = 177/125,294,874 = 1.41 × 10−6 for China and 13/2,048,327 = 6.35 × 10−6 for Japan), it was still higher than other country-level annotations, such as the US, Spain, and Denmark (ratio = 13/113,161,322 = 1.15 × 10−7, 11/43,819,760 = 2.51 × 10−7, 9/59,342,818 = 1.52 × 10−7, respectively; Figure 2C). β-porphyranase was more abundant in Japanese than other populations in the read-based quantification (Figures 2D and S6B). Thus, we replicated the high frequency of β-porphyranase in the Japanese gut metagenome, and newly revealed that β-porphyranase presented also in the gut metagenome of the Chinese population.
Figure 2.

Phylogenetic and interpopulational analysis of the β-porphyranase in the JMAG and UHGP
(A) A maximum-likelihood phylogenetic tree of the β-porphyranase sequences detected in the JMAG and UHGG. The β-porphyranase proteins and their related proteins (β-agarase and κ-carrageenase) were also utilized to reconstruct the phylogenetic tree. The colors and shapes of the nodes represent the derivation and name of the genes.
(B) Pie charts representing the phylogenetic composition of the MAGs, which are linked to the β-porphyranase proteins at the phylum (top) and genus level (bottom). The results for the JMAG (left) and UHGG (right) are described separately. The phyla comprising <1% of each genome set are collapsed into “Other.”
(C) Pie charts represent the origin of the β-porphyranase-linked MAGs at the region (top) and country (bottom) levels.
(D) A boxplot of the β-porphyranase abundances in the different populations. The boxplot indicates the median values (center lines) and IQRs (box edges), with the whiskers extending to the most extreme points within the range between (lower quantile − [1.5 × IQR]) and (upper quantile + [1.5 × IQR]). IQR, interquartile range. See also Figures S5 and S6.
Strains of food-associated bacteria were shared among the Japanese population
Utilizing the species-level representative genomes of the JMAG and the original MSS data, we evaluated the sharing of the prokaryotic strains among the Japanese by inStrain.32 We first performed per dataset analysis and found that strain sharing was reproducibly detected for 10 species in at least 3 datasets (Figure S7A) among the 1,273 species in the JMAG. As for these 10 species, we performed a strain-level comparison with all samples. We found that the majority of the individuals included in the analysis of the targeted species were involved in strain sharing for five species (Figures 3A and S7B), suggesting that strain sharing was relatively frequent for these species compared with the other species in the JMAG. Among the five species, Bacillus subtilis was considered to be derived from the Japanese traditional food natto as mentioned above. In addition, the other four species (Bifidobacterium animalis, Enterocossus_B lactis, Lactobacillus paracasei, and Streptococcus thermophilus) were reported to be associated with dairy products.33,34 Thus, it was suggested that food-related bacteria tended to be shared among the population at the strain level.
Figure 3.

Strain-level analysis and association tests with rs671 for food-related bacterial species
(A) Circulized arc diagrams representing the strain sharing among the subjects for the five food-related bacterial species. The nodes represent the individuals with the detection of each species of bacterium and the edges represent the sharing of the bacterial strains. The colors of the nodes represent the dataset of the individuals. Independent strain-sharing networks are depicted in different colors. The gray edges mean that the strain is shared only between a pair of individuals.
(B) Boxplots represent the abundances (mean coverage) of the five food-related bacterial species stratified by the rs671 genotypes. The boxplots indicate the median values (center lines) and IQRs (box edges), with the whiskers extending to the most extreme points within the range between (lower quantile − [1.5 × IQR]) and (upper quantile + [1.5 × IQR]). The “A” allele of the rs671 in ALDH2 had a negative association with natto consumption and a positive association with dairy consumption in the previous study.37 ∗p < 0.05; IQR, interquartile range. See also Figure S7 and Tables S3 and S4.
A missense variant rs671:G>A in ALDH2 is an East Asian-specific single-nucleotide polymorphism that is under the recent positive selection.35 The A allele of rs671 causes alcohol intolerance and has various pleiotropic associations with diseases, clinical biomarkers, and dietary habits.36,37 Since the consumption of natto and dairy was negatively and positively associated with the A allele of the rs671, respectively, we evaluated the association between the abundance of the five food-related bacterial species and the A allele of the rs671 (Figure 3B; Table S3; N = 546 in total). We found nominal associations for Enterococcus_B lactis (effect size = 0.270 and p = 0.034) and S. thermophilus (effect size = 0.122 and p = 0.048). Even when removing disease samples, the effect sizes for the Enterococcus_B lactis (effect size = 0.355 and p = 0.036) and S. thermophilus (effect size = 0.134 and p = 0.079) were consistent (Figure S7C). We performed a Mendelian randomization analysis38 to evaluate the effect of the dietary habits on the bacterial abundances and found that increased intake of dairy products could increase the abundances of the Enterococcus_B lactis (effect size = 2.385 and p = 0.034) and S. thermophilus (effect size = 1.077 and p = 0.047; Table S4).
Reconstruction of viral genomes from Japanese MSS data
We recovered viral genomes from the 787 Japanese MSS data (Figure S8). The viral genomes were extracted from the assembled contigs with VirSorter39 and VirFinder40 and subjected to CheckV.41 After CheckV, we retained the viral genomes that had ≥50% completeness and more viral genes than host genes. We obtained 31,395 viral genomes including 4,098 complete, 7,492 high-quality, and 19,805 medium-quality genomes (Table S5). We call this set of viral genomes as Japanese Virus Database (JVD). The 31,395 genomes were clustered into 12,213 clusters at ≥95% ANI, merged with the Gut Phage Database (GPD),19 Metagenomic Gut Virus (MGV),20 and taxonomic reference genomes (RefSeq and Yutin et al.42), and further clustered into 94,714 species-level viral operational taxonomic unit (vOTU) at ≥95% ANI. These species-level vOTUs were further clustered into 10,022 genus- and 2,577 family-level vOTUs based on the gene sharing ratio and AAI (STAR Methods; Tables S6 and S7). We assigned putative viral taxonomy to all the viral genomes based on the result of the clustering (Figure 4A). Siphoviridae (14.0%) and Myoviridae (9.3%) dominated the taxonomically annotated viruses, while crAss-like phages (2.6%) and Podoviridae (0.8%) also occupied a portion of the taxonomically annotated viral genomes. Salsmaviridae, a recently created viral family,43 also occupied a part of the taxonomically annotated viral genomes (0.7%).
Figure 4.

Reconstruction of the viral genomes and the comparison with the other databases
(A) A pie chart illustrating the family-level phylogenetic composition of the JVD. The families comprising <0.3% of the genomes are collapsed into “Other.”
(B) Venn diagrams represent the sharing of the vOTUs (left, family level; middle, genus level; right, species level) among the different databases. Reference genomes are composed of RefSeq and Yutin et al.42 (STAR Methods). See also Figures S8 and S9 and Tables S5, S6, and S7.
We evaluated the overlap between the JVD, previous studies (GPD and MGV), and reference genomes at the family, genus, and species levels. At the species level, the majority of the vOTUs that included the JVD genomes (62.9%) were not overlapped with the other databases (Figure 4B). Note that there was a relatively large overlap between the GPD and MGV because of the overlap of the original MSS dataset. In contrast, the majority of the family- and genus-level vOTUs were covered by the other databases (7.5% and 0.67% were novel, respectively).
We predicted and functionally annotated the protein sequences on the JVD viral genomes. The ratio of the proteins covered and functionally annotated by the current databases was lower for the crAss-like phages than the other viruses, possibly due to the relatively recent discovery and expansion of the crAss-like phage genomes (Figure S9A). Among the Virus Orthologous Groups44 and KEGG45 annotations of the JVD, typical viral proteins, such as the capsid proteins, terminase, and portal proteins, were observed as highly frequent proteins (Figures S9B and S9C). Among the KEGG pathways, virus-related pathways, such as DNA replication and homologous recombination, were frequently seen (Figure S9D). In addition, we could see the taxonomic tendency of the KEGG gene and pathways, such as the relatively high occurrence of dUTP pyrophosphatase and pyrimidine metabolism-related proteins in the crAss-like phage genomes. We also detected some auxiliary metabolic genes46 that potentially affect the metabolic function of their hosts (Figure S9E). Protein sequences were predicted also from the viral genomes in the GPD and MGV, merged with the JVD protein sequences, and clustered at 100%, 95%, 90%, and 50% amino acid sequence identity. Among the clusters that included the JVD proteins, 65.3%, 38.6%, 32.3%, and 19.4% were solely detected in the JVD, respectively (Figure S9F).
Interpopulational and case-control comparisons of the crAss-like phages
crAss-like phages were the bacteriophages that were reported to be abundant in the gut.18 Since it was discovered in 2014 by a cross-assembly of the human gut metagenome data,18 known diversity of the crAss-like phages has been expanded and now five subfamilies, namely αγ, β, δ, ε, and ζ, are recognized.42 We annotated the subfamily-level taxonomy to the crAss-like phage genomes based on the result of the genus-level vOTU clustering (Table S8). To validate the subfamily-level annotation, we made maximum-likelihood phylogenetic trees for the terminase (TerL), a marker protein of the crAss-like phages. The crAss-like phages belonging to the same subfamilies fell into the same clades, and those belonging to the same genus-level vOTU were placed closely (Figure 5A).
Figure 5.

Interpopulational and case-control comparisons of the crAss-like phages
(A) A maximum-likelihood phylogenetic tree reconstructed from the TerL proteins of the crAss-like phages. The color of the nodes represents the species-level clusters present only in the JVD (magenta), present only in the reference (green), or neither of the cases (navy). The outer rings represent the subfamily-level (inner) and genus-level (outer) taxonomic annotation of the crAss-like phages.
(B) A bar plot depicting the compositions of the subfamilies of the crAss-like phage genomes for the JVD and MGV. The genomes from the MGV are grouped according to their continental origin.
(C) A bar plot depicting the compositions of the subfamilies of the crAss-like phages calculated from the abundances (RPKM) in each group.
(D and E) Heatmaps represent the association of the crAss-like phages to the diseases (upper) and Shannon index (lower) at the subfamily (D) and genus (E) level, respectively. The colors indicate the Z score in each test. ∗p < 0.05. ∗∗p < 0.05/number of clades (per objective variables). ∗∗∗p < 0.05/number of tests across all diseases (only for association tests for diseases). See also Tables S8, S9, S10, and S11.
Then, we compared the subfamily-level composition of the crAss-like phage genomes among the various populational contexts. In the JVD, αγ followed by δ, ε, and ζ were frequent and β was minor among the crAss-like phage genomes. In the MGV, β crAss-like phages were also minor in Asia, Europe, and North America, as in the case of the JVD. In contrast, the composition of the β crAss-like phage genomes was significantly higher in Oceania and Africa than in Japan, Asia, Europe, and North America (Figure 5B; PFisher < 0.05/21 = 2.4 × 10−3). A relatively higher prevalence of the β crAss-like phages in Africa was also supported by the read-based quantification of the crAss-like phages (Figure 5C). Thus, it was suggested that the Japanese people’s subfamily-level composition of the crAss-like phages was mostly similar to populations such as Asian, European, and North American, and β crAss-like phages were associated with the African and Oceanian populations. These results might reflect the differences in dietary habits.
Although crAss-like phages were assumed to be a core component of the healthy gut virome, their association to diseases had not been fully evaluated. Therefore, we evaluated the association between the subfamily- and genus-level vOTU of the crAss-like phages and affection status of the diseases, namely rheumatoid arthritis (RA) (NCase = 113, NControl = 114), systemic lupus erythematosus (SLE) (NCase = 36, NControl = 205), multiple sclerosis (MS) (NCase = 30, NControl = 77), ulcerative colitis (UC) (NCase = 35, NControl = 40), Crohn disease (CD) (NCase = 39, NControl = 40), and colorectal cancer (CoCa) (NCase = 40, NControl = 39; Figures 5D and 5E; Tables S9, S10, and S11). The αγ, cluster_1743, cluster_1322, and cluster_655 crAss-like phages decreased at least nominally (p < 0.05) in both the RA and SLE patients. In MS patients, we could not detect any significant changes in the abundance of the crAss-like phages (p > 0.05). In patients with inflammatory bowel disease (IBD), namely UC and CD, most of the clades, including the αγ, cluster_1743, and cluster_655 decreased (p = 3.2 × 10−3 and 3.0 × 10−4 for αγ, p = 1.9 × 10−4 and 9.0 × 10−5 for cluster_1743, and p = 3.5 × 10−5 and 7.4 × 10−6 for cluster_655, respectively). In contrast, increases of some clades, such as the αγ crAss-like phages were observed in CoCa. Given that decreases of the diversity of the bacteria were reported for SLE,25 UC, and CD,47,48 but an increase was reported for CoCa,49 we hypothesized that crAss-like phages were associated with the diversity of the bacteria. We evaluated the association between the crAss-like phage clades and Shannon index, which is a measurement of the diversity of the bacteria, and found that most of the clades were positively associated with the Shannon index (Figures 5D and 5E; Tables S10 and S11).
Virus-host interaction analysis with CRISPR, prophage, and co-abundance
CRISPR (clustered regularly interspaced short palindromic repeats) and CRISPR-associated (Cas) proteins comprise the CRISPR-Cas system, a prokaryotic adaptive immune system against predators such as bacteriophages.50 The CRISPR-Cas system intakes short fragments of the viral sequences as CRISPR spacers to efficiently eject the viruses during subsequent infections. Thus, CRISPR sequences in the prokaryotic genomes are evidence of previous infections by viruses. Utilizing the CRISPR sequences in the JMAG genomes, we predicted the virus-prokaryote interaction. We detected 296,915 spacers in total, and 147,354 (49.6%) matched and 149,561 (50.4%) did not match the viral sequences recovered from the gut metagenome (Figure S10A). We then evaluated the taxonomic composition of the linked MAGs and viral targets of the CRISPR spacers, which reflected the host ranges of the viruses (Figure S10A; Table S12). For example, the major host of the crAss-like phages was Bacteroidota, while several crAss-like phages infected Firmicutes_A, as expected from previous studies.20,42 We also searched the viral target sequences of the CRISPR spacers in the 286,997 UHGG genomes,20 and 59% of the pairs of species-level vOTU and prokaryotic genus conferred from the analysis on the JMAG were replicated by the UHGG (Figure 6A). We also evaluated the virus-prokaryote interaction inferred from the proviral sequences in the JVD genomes (Figure S10B; Table S13). We got additional implications, such as the lack of the proviral sequences of the crAss-like phages and Salasmaviridae. The lack of proviral sequences of crAss-like phages in the JVD could reflect the lack of lysogeny of the crAss-like phages, as previously suggested.20 As for Salasmaviridae, it was reported that Salasmaviridae follow a strict lytic life cycle with no evidence of lysogenic activity.51 Thus, our large-scale analysis supported the previous implication for the newly classified virus.
Figure 6.

Virus-prokaryote interaction analysis based on the CRISPR and abundances
(A) Number of the pairs of the species-level vOTU and prokaryotic genus detected in the JMAG and UHGG.
(B) A quantile-quantile plot of the p values from the virus-prokaryote association analysis stratified by whether the virus-prokaryote pairs are supported by the CRISPR spacers in the JMAG (magenta) or not (gray). The x axis indicates −log10(P) expected from the uniform distribution. The y axis indicates the observed −log10(P). The diagonal dashed line represents y = x, which corresponds to the null hypothesis.
(C) A density plot of the Z score from the virus-prokaryote association analysis stratified by whether the virus-prokaryote pairs are supported by the CRISPR spacers in the JMAG (magenta) or not (gray). The upper limit of the Z score is set at 50. The diagonal dashed line represents y = x, which corresponds to the null hypothesis. The vertical dashed lines indicate the mean of the Z score for each group of the virus-prokaryote pair.
(D) A scatterplot of the odds ratios for being the Japanese-derived viruses (y axis) and prokaryotes (x axis) for the virus-prokaryote pairs supported by the CRISPR in the JMAG. The size of the dots represents the number of spacers supporting the virus-prokaryote pairs. The horizontal and vertical dashed lines represent odds ratio = 0 for virus and prokaryote, respectively.
(E) Violin plots of the species-level vOTU 23245 (left) and Blautia sp001304935 (right) abundances (RPKM) in each group. The red center lines indicate the median values. See also Figure S10 and Tables S12 and S13.
Co-abundance analysis of the virus and prokaryote had been used for implicating virus-prokaryote interaction, but how much did it concordant to the result of the CRISPR-based and prophage-based analyses, which had not been well evaluated. Utilizing this large dataset, we evaluated the association between the abundances of viruses and prokaryotes stratified by the existence of supports from the CRISPR spacers in the JMAG (Figure S10C; Table S12). Inflation of the p values of the virus-prokaryote association tests was much more severe for the pairs supported by the CRISPR spacers than those without supports (Figure 6B). Z scores of the virus-prokaryote pairs supported by the CRISPR spacers were severely biased positively, suggesting that the abundances of the viruses and their putative hosts tended to be positively correlated (Figure 6C). We performed the same analysis for the CRISPR sequences in the UHGG and the prophages in the JMAG and replicated the results obtained from the CRISPR sequences in the JMAG (Figures S10D–S10G).
Then, we performed the inter-database comparison of species-level vOTUs (JVD and MGV) and prokaryotic genome clusters (JMAG and UHGG) and integrated the results of these analyses based on the results of the CRISPR spacers. We calculated the odds ratio of the Japanese-derived genomes for each species-level vOTU and prokaryotic genome cluster. We found the enrichment of the CRISPR-supported virus-bacteria pairs that had the same sign of the log odds ratios for being Japanese derived (Figure 6D; STAR Methods). The log fold changes between the abundances in Japanese and other populations also tended to have same the signs for viruses and prokaryotes linked by the CRISPR spacers (Figure S10H). Thus, it was suggested that interpopulational differences of the viruses and their host were positively associated. For example, species-level vOTU 23245, which was frequently recovered and relatively abundant among the Japanese gut metagenome, infected Blautia sp001304935, which was also frequently recovered and relatively abundant among the Japanese gut metagenome (Figure 6E).
Virus-bacterium interaction network for crAss-like phages
Based on the result of the CRISPR analysis, we constructed a virus-bacterium interaction network of crAss-like phages (Figure 7A). The bacterial genera belonging to phylum Bacteroidota, such as Parabacteroides, Prevotella, Bacteroides, and Phocaeicola were highly connected to the crAss-like phages (Figure 7B), suggesting that the major host of the crAss-like phage was Bacteroidota as reported previously.42 In addition, several Firmicutes were also present in the network. Although most of the crAss-like phage subfamilies infected various bacterial genera, ε crAss-like phages had strong preferences for the genus Parabacteroides.
Figure 7.

Network plot for the crAss-like phages and their predictive hosts
(A) A network plot of the CRISPR-based links (edges) between the species-level vOTU of the crAss-like phages (circle nodes) and bacterial genus (rhombus nodes). The color of the edges represents the derivation of the CRISPR spacers. The color of the circle nodes represents the subfamily level taxonomic annotations of the crAss-like phage genomes. The color of the rhombus nodes represents the genus level taxonomic annotations of the candidate hosts of the crAss-like phages. The magenta dashed box indicates the interaction between the ε crAss-like phages and Parabacteroides.
(B) A bar plot indicates the top 10 bacterial genera that have the highest number of the species-level crAss-like phage vOTU linked by the CRISPR spacers.
Discussion
In this study, we reconstructed the 19,084 MAGs and 31,395 viral genomes from the 787 Japanese gut MSS data. Utilizing these data, we performed a comparative analysis among databases, interpopulational and case-control comparisons of the crAss-like phages, and virus-prokaryote interaction analysis.
While a large part of the species-level diversity of the Japanese gut prokaryotes was covered by the UHGG catalog possibly due to the partially westernized dietary habits of the Japanese, some Japanese population-specific traditional diet-associated features of the gut microbiome, such as the presence of the Bacillus subtilis natto and enrichment of β-porphyranase, were identified. Natto is a Japanese traditional fermented food that is still widely consumed and expected as a potential probiotic food.52 Although a previous 16S rRNA sequencing study suggested the presence of the family Bacillacea in the Japanese gut,53 whether it was Bacillus subtilis natto was not confirmed due to the insufficient taxonomic resolution. Thus, our analysis suggested that the reconstruction of the MAG enabled us to evaluate Bacillus subtilis natto in the gut more accurately than 16S rRNA analysis and could be useful for future implementation of the probiotics.
β-Porphyranase is an enzyme that degrades seaweed-derived polysaccharides that are contained in the nori, a traditional Japanese food made from porphyra.21 In our analysis, we confirmed the enrichment of the β-porphyranase in the Japanese gut with a large Japanese dataset, which had not been available in the previous study.21 Although not as apparent as in the Japanese population, the frequency of β-porphyranase was relatively high in the Chinese population. Relative enrichment of β-porphyranase in the Chinese population could be because the Chinese population also eats porphyra as zicai or the long-standing traffic among East Asia.
Through strain-level analysis, we revealed that five strains of food-related bacterial species were reproducibly shared among the Japanese. A previous comparative analysis of gut-derived and food-derived MAGs revealed that the major source of several gut bacteria, including L. paracasei and S. thermophilus, was food.33 Since the bacterial strains used for making fermented food are often determined by the manufacture, sharing of the strain for food-associated bacteria was expected when the major sources of the bacteria were food. rs671:G>A in ALDH2 is the East Asian-specific missense variant that is associated with alcohol intolerance. We identified the positive association between the abundance of dairy-associated bacteria and A alleles of the rs671, which was also associated with high dairy consumption.38 This finding suggested that human genetic variants could affect the gut microbiome via dietary habits, while we could not completely reject the possibility of the opposite (i.e., the high abundance of dairy consumption led to higher dairy consumption). Although not available for our datasets, future analysis with dietary information will be beneficial for deepening the insights into this association.
We mined the viral genomes from the MSS data. Among the taxonomically annotated viruses, Siphoviridae, Myoviridae, crAss-like phage, and Podoviridae were relatively frequent, as previously reported.16,17,20,54 In addition, newly classified Salasmaviridae was also relatively frequent. As observed in the previous studies,19,20 JVD included a significant amount of taxonomically unknown viruses possibly due to the underrepresentation of human gut phages in the taxonomic reference database. In contrast to prokaryotic genomes, a large part of the species-level diversity of the JVD was not covered by previous studies, such as GPD and MGV. This could be because of the enormous species-level diversity of the viruses, while differences in the populations and viral genome detection methods could also contribute.
We identified virus-prokaryote interaction by CRISPR and prophage analysis. Of the CRISPR spacers, 49.6% matched the viral sequence data composed of the JVD and the current largest gut virus databases (i.e., MGV and GPD), and future expansion of the viral sequence database may contribute to the further identification of the virus-prokaryote interaction. The abundance of the viruses and prokaryotes linked by the CRISPR spacers or proviral sequences was correlated positively in the gut. The Piggyback-the-Winner model,55 in which phages take a lysogenic or pseudo-lysogenic cycle to “piggyback on” the success of their host rather than killing their host is supposed to be a major strategy for the gut virome.56,57 Given that the lytic activities of the phages could result in a loss of positive correlation between the phages and their hosts,58 our results could reflect the peaceful symbiosis as indicated in the Piggyback-the-Winner model. The interpopulational differences of the number of the recovered genome or read-based abundance had the same trend for the virus-prokaryote pairs supported by the CRISPR spacers. These results suggested that interpopulational differences of the viruses and their hosts were positively associated possibly because the abundances of the viruses and their hosts tended to be positively correlated.
At the subfamily level, the frequency of the recovery and read-based abundance of the β crAss-like phages were relatively high in the populations with the non-westernized dietary habits, such as African compared with populations with westernized dietary habits, including the Japanese. This result could reflect the impact of dietary habits on the crAss-like phages. In case-control comparisons of crAss-like phages, we revealed that several clades of the crAss-like phages decreased in RA, SLE, UC, and CD patients, but increased in CoCa patients. During the preparation of this manuscript, a study on Dutch cohorts reported decreases of the crAss-like phages in IBD.59 Thus, decreases of the crAss-like phages in IBD could be a general event observed in multiple populations rather than a population-specific event. The diversity of the gut bacteriome has been reported to be associated with various diseases and is often suggested as a marker for microbiome health.60 The positive association between the crAss-like phage abundances and bacterial diversity suggested that the abundance of the crAss-like phage could reflect the overall healthiness of the gut microbiome.
In virus-prokaryote interaction analysis, we could not find the proviral sequences of the crAss-like phages. Since the currently isolated two crAss-like phages (ΦcrAss001 and 002) neither possess lysogeny-associated genes nor can form stable lysogens,61,62 this result could reflect the unique life cycle of crAss-like phages. Virus-prokaryote interaction analysis based on the CRISPR sequences predicted that the major host of the crAss-like phages was Bacteroidota, consistent with the previous finding.42 Although most of the crAss-like phage subfamilies infected the various bacterial genus, ε crAss-like phages mostly exclusively infected the genus Parabacteroides. The limited host range might reflect relatively short evolutionary distances (length of the branches in phylogenetic trees) among the currently identified ε crAss-like phages.
In summary, we recovered the MAGs and viral genomes from the Japanese gut MSS data. Based on the recovered microbial genomes, we revealed the features of the Japanese gut metagenome, associations of the crAss-like phages to populations and diseases, and virus-prokaryote interactions. The reconstructed microbial genomes and related information are available at the National Bioscience Database Center (https://humandbs.biosciencedbc.jp). We believe that our dataset, which includes MAGs, viral genomes, and CRISPR spacers, will be a useful resource for future studies.
Limitation of the study
The JVD did not include viruses that were classified as RNA viruses or eukaryotic viruses because they were not efficiently detected by our pipeline due to the nature of the sequencing data and property of the virus detection pipeline. Future investigation on the other type of datasets such as meta-transcriptome data and further expansion of the reference databases will be beneficial to increase the known diversity of the gut virome.
Although the positively associated interpopulational differences were confirmed by the two different analyses (i.e., based on the number of the genomes and abundances) with the different outer datasets, batch/study effects were potential limitations of the current microbiome study focusing on the interpopulational differences. Ongoing efforts to collect and sequence stool samples from various populations in a unified framework will be promising.63
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Fecal samples | This study | N/A |
| Human DNA extracted from blood | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Tris-HCl | NIPPON GENE | Cat#316-90385 |
| SDS | Sigma Aldrich | Cat#28-3270 |
| EDTA | Nacalai Tesque | Cat#06894-14 |
| Phenol/chloroform/isoamyl alcohol | Nacalai Tesque | Cat#25970-56 |
| TE saturated phenol | Nacalai Tesque | Cat#26829-96 |
| Sodium acetate | Sigma Aldrich | Cat#28-1560 |
| Isopropanol | JUNSEI | Cat#67-63-0 |
| Ethanol | JUNSEI | Cat#64-19-5 |
| RNA later | Thermo Fisher Scientific | Cat#AM7021 |
| Critical commercial assays | ||
| KAPA Hyper Prep Kit | illumina | Cat#KK8504 |
| Glass beads (diameter 0.1 mm) | biospec | Cat#11079101 |
| Deposited data | ||
| Metagenome shotgun sequencing data | This study | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| Metagenome shotgun sequencing data | Kishikawa et al. 2020a23 | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| Metagenome shotgun sequencing data | Kishikawa et al. 2020b24 | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| Metagenome shotgun sequencing data | Tomofuji et al., 2021a25 | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| Metagenome shotgun sequencing data | Tomofuji et al., 2021b26 | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| Metagenome shotgun sequencing data | Otake et al., 202228 | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| Metagenome shotgun sequencing data | Yachida et al., 201927 | DDBJ Sequence Read Archive: DRA006684 |
| Metagenome shotgun sequencing data | Zhu et al. 202064 | Europea Nucleotide Archive: ERP111403 |
| Metagenome shotgun sequencing data | Dhakan et al. 201965 | Sequence Read Archive: SRP114847 |
| Metagenome shotgun sequencing data | Thomas et al. 201949 | Sequence Read Archive: SRP136711 |
| Metagenome shotgun sequencing data | Wirbel et al. 201966 | Europea Nucleotide Archive: ERP110064 |
| Metagenome shotgun sequencing data | Xie et al. 201667 | Europea Nucleotide Archive: ERP010700 |
| Metagenome shotgun sequencing data | Price et al. 201948 | Sequence Read Archive: SRP115494 |
| Metagenome shotgun sequencing data | Tett et al. 201968 | Sequence Read Archive: SRP168387 |
| Metagenome shotgun sequencing data | Tett et al. 201968 | Sequence Read Archive: SRP189832 |
| Metagenome shotgun sequencing data | Tett et al. 201968 | Sequence Read Archive: SRP189572 |
| RefSeq Virus | NCBI | https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/ |
| Bacillus subtilis genomes | NCBI GenBank | https://www.ncbi.nlm.nih.gov/genbank/ |
| crAss-like phage genomes | Yutin et al. 202142 | https://zenodo.org/record/4437596 |
| CRISPR spacers | Nayfach et al. 202120 | https://portal.nersc.gov/MGV |
| CRISPR spacers in JMAG genomes | This study | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| dbCAN HMMdb v10 | Zhang et al., 201869 | https://bcb.unl.edu/dbCAN2/index.php |
| GPD | Camarillo-Guerrero et al. 202119 | http://ftp.ebi.ac.uk/pub/databases/metagenomics/genome_sets/gut_phage_database/ |
| JMAG | This study | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| JVD | This study | National Bioscience Database Center (NBDC) Human Database: hum0197 |
| List of the AMGs | Kieft et al., 202046 | https://doi.org/10.1186/s40168-020-00867-0 |
| MGV | Nayfach et al. 202120 | https://portal.nersc.gov/MGV |
| Scripts for recovering and analyzing microbial genomes | This study |
https://doi.org/10.5281/zenodo.7053099 and https://github.com/ytomofuji |
| UHGG and UHGP | Almeida et al. 20218 | http://ftp.ebi.ac.uk/pub/databases/metagenomics/mgnify_genomes/ |
| VOG | Grazziotin et al., 201744 | https://vogdb.org |
| β-porphyranase sequences | Hehemann et al., 201021 | https://doi.org/10.1038/nature08937 |
| Bacillus subtilis genomes | NCBI GenBank | https://www.ncbi.nlm.nih.gov/genbank/ |
| Multiple sequence alignment files generated in this study (JMAG representative genomes, β-porphyranase, and TerL of crAss-like phages) | This study | https://doi.org/10.5281/zenodo.7053099 |
| Software and algorithms | ||
| Barrnap | https://github.com/tseemann/barrnap | https://github.com/tseemann/barrnap |
| bcl2fastq | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software/downloads.html |
| BMTagger | ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/70 | ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/ |
| bowtie2 | Langmead and Salzberg, 201271 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| CheckM | Parks et al., 201529 | https://github.com/Ecogenomics/CheckM |
| CheckV | Nayfach et al., 202141 | https://bitbucket.org/berkeleylab/checkv/ |
| CONCOCT | Alneberg et al., 201472 | https://github.com/BinPro/CONCOCT |
| coverM | Queensland University of Technology Microbiome Research Group | https://github.com/wwood/CoverM |
| DAS Tool | Sieber et al., 201873 | https://github.com/cmks/DAS_Tool |
| DIAMOND | Buchfink et al., 202174 | https://github.com/bbuchfink/diamond |
| dRep | Olm et al., 201775 | https://github.com/MrOlm/drep |
| eggNOG-mapper | Cantalapiedra et al. 202176 | https://github.com/eggnogdb/eggnog-mapper |
| EIGENSTRAT | Price et al., 200677 | https://www.hsph.harvard.edu/alkes-price/software/ |
| Ggraph | https://github.com/thomasp85/ggraph | https://github.com/thomasp85/ggraph |
| GTDB-tk | Chaumeil et al., 201931 | https://github.com/Ecogenomics/GTDBTk |
| Hmmer | http://hmmer.org/download.html | http://hmmer.org/download.html |
| inStrain | Olm et al., 202132 | https://github.com/MrOlm/instrain |
| Iqtree | Nguyen, L.-T et al., 201578 | http://www.iqtree.org |
| iTOL | Letunic & Bork, 201979 | https://itol.embl.de |
| MAFFT | Katoh & Standley, 201380 | https://mafft.cbrc.jp/alignment/software/ |
| Mash | Ondov et al., 201681 | https://github.com/marbl/Mash |
| MaxBin | Wu et al., 201682 | https://sourceforge.net/projects/maxbin/ |
| MCL | Enright et al., 200283 | http://micans.org/mcl/ |
| MetaBAT | Kang et al., 201984 | https://bitbucket.org/berkeleylab/metabat/src/master/ |
| MinCED | Bland et al., 200785 | https://github.com/ctSkennerton/minced |
| MMseqs2 | Steinegger & Söding, 201786 | https://github.com/soedinglab/MMseqs2 |
| MUMmer | Marçais et al., 201887 | https://github.com/mummer4/mummer |
| muscle | Edgar, 200488 | https://drive5.com/muscle/downloads_v3.htm |
| ncbi-blast-plus | Camacho et al., 200989 | https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastDocs&DOC_TYPE=Download |
| PLINK | Purcell et al., 200790 | https://www.cog-genomics.org/plink/ |
| PRINSEQ | Schmieder and Edwards, 201191 | http://prinseq.sourceforge.net/ |
| Prodigal | Hyatt et al., 201092 | https://github.com/hyattpd/Prodigal |
| Prokka | Seemann, 201493 | https://github.com/tseemann/prokka |
| Python | Python Software Foundation | https://www.python.org/downloads/release/python-376/ |
| R | The R Foundation for Statistical Computing | https://www.r-project.org |
| RefineM | Parks et al., 201794 | https://github.com/dparks1134/RefineM |
| Samtools | Li et al., 200995 | http://www.htslib.org/download/ |
| Script for clustering of the viral genomes | Nayfach et al., 202120 | https://github.com/snayfach/MGV |
| SPAdes | Prjibelski et al., 202096 | https://github.com/ablab/spades#sec5 |
| Trimmomatic | Bolger et al., 201497 | http://www.usadellab.org/cms/?page=trimmomatic |
| tRNAScan-SE | Chan et al., 202198 | http://lowelab.ucsc.edu/tRNAscan-SE/ |
| TwoSampleMR | Hemani et al., 201838 | https://mrcieu.github.io/TwoSampleMR/ |
| Vegan | https://github.com/vegandevs/vegan | https://github.com/vegandevs/vegan |
| VirFinder | Ren et al., 201740 | https://github.com/jessieren/VirFinder |
| VirSorter | Roux et al., 201539 | https://github.com/simroux/VirSorter |
| Custom codes used in this study | This study |
https://doi.org/10.5281/zenodo.7053099 and https://github.com/ytomofuji/JMAG_JVD |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yukinori Okada (yokada@sg.med.osaka-u.ac.jp).
Materials availability
The materials that support the findings of this study are available from the corresponding authors upon reasonable request. Please contact the lead contact for additional information.
Experimental model and subject details
Subject participation
818 Japanese gut metagenome sequencing data from 787 subjects were used in this study (Table S1). In addition, 432 gut metagenome sequencing data from various populations48,49,64,65,67,66,68 were used for the comparative analyses. Although most of the data was derived from previous studies,23,24,25,27,28 136 Japanese sequencing data (included healthy control [HC], Unruptured cerebral aneurysm [UA], Sub-arachnoid hemorrhage [SAH], and stroke [ST] subjects) was newly obtained in this study. The newly recruited HC subjects were enrolled at the Osaka University Graduate School of Medicine. Participants with UA and SAH were recruited from the Osaka University, Osaka Neurological Institution, Hanwa Memorial Hospital, and Iseikai Hospital as previously described.99 Participants with ST were recruited from the Osaka University.
Participants with extreme diets (e.g., strict vegetarians) were not included in the dataset. All subjects provided written informed consent before participation. Those who took antibiotics within a month were reported as the patients treated with antibiotics. The study protocol was approved by the ethics committees of Osaka University and related medical institutions.
Method details
Sample collection and DNA extraction
For the ST patients, fecal samples had been immediately frozen after production in an insulated container for storage at −20°C and subsequently stored at −80°C within 24 h after production. For the HCs, samples were stored at −80°C within 6 h after production. For the participants with UA, fecal samples were collected at home, immediately packed with frozen gel packs within insulated containers, and stored at −20°C. By the next day, the sample collection kits were returned by refrigerated shipping keeping at −20°C, and stored at −80°C until processing, as previously described.99 For the participants with SAH, the fecal samples were collected within 48 h following admission and before the induction of antibiotics to minimize changes in the gut microbial community, as previously described.99 Microbial DNA was extracted according to the previously described method.23 Briefly, 0.3 g glass beads (diameter: 0.1 mm) (BioSpec) and 500 μL EDTA-Tris-saturated phenol were added to the suspension, and the mixture was vortexed vigorously using a FastPrep-24 (MP Biomedicals) at 5.0 power level for 30 s. After centrifugation at 20,000 g for 5 min at 4°C, 400 μL of supernatant was collected. Subsequently, phenol-chloroform extraction was performed, and 250 μL of supernatant was subjected to isopropanol precipitation. Finally, DNAs were suspended in 100 μL EDTA-Tris buffer and stored at −20°C.
Whole-genome shotgun sequencing
A shotgun sequencing library was constructed using the KAPA Hyper Prep Kit (KAPA Biosystems), and 150-bp paired-end reads were generated on NovaSeq 6000. The sequence reads were converted to the FASTQ format using bcl2fastq (version 2.19).
Quality control of sequencing reads
We followed a series of steps to maximize the quality of the datasets. The main steps in the quality control process were as follows: (i) trimming of low-quality bases, (ii) identification and masking of human reads, and (iii) removal of duplicated reads. We marked duplicate reads using PRINSEQ-lite91 (version 0.20.4; -derep 1). We trimmed the raw reads to clip Illumina adapters and cut off low-quality bases at both ends using the Trimmomatic97 (version 0.39; parameters: ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10:8:true LEADING:20 TRAILING:20 SLIDINGWINDOW:3:15 MINLEN:60). We discarded reads less than 60 bp in length after trimming. Next, we performed duplicate removal by retaining only the longest read among the duplicates. When there were multiple reads with the same sequences and length, we randomly selected one of the reads. As a final quality control step, we aligned the quality-filtered reads to the human reference genome (hg38) using bowtie271 (version 2.3.5.1) with default parameters and BMTagger70 (version 3.101). We kept only the reads of which both paired ends failed to align in either tool.
Reconstruction of MAGs
The de novo assembly of the filtered paired-end reads into the contigs was conducted using SPAdes96 (version 3.13.0) with the ‘—meta’ option and the contigs longer than 2kbp were retained for subsequent binning. Then, filtered paired-end reads were mapped to the assembled contigs for quantifying the abundance of each contig with bowtie2 (version 2.3.5.1). Binning was performed per sample using three different tools with the default options; MaxBin82 (version 2.2.6), MetaBAT84 (version 2.12.1), and CONCOCT72 (version 1.0.0). DAS Tool73 (version 1.1.2) was used to integrate the results of the binning produced by the three tools. To refine the quality of the bins, we utilized RefineM94 (version 0.1.2) and filtered out scaffolds with the divergent genomic properties or incongruent taxonomic classification (based on Genome Taxonomy Database release 95). Then, we evaluated the quality of the MAGs with CheckM29 (version 1.0.12) using the ‘lineage_wf’ workflow to select only genomes that passed the following criteria; >50% genome completeness, <5% contamination, and an estimated quality score (completeness – 5 × contamination) > 50. After the filtering, we obtained 19,084 MAGs which were used for the subsequent analyses.
To evaluate the strain-level diversity of the MAGs, we mapped the filtered paired-end reads to the reconstructed MAGs with bowtie2 and calculated the average nucleotide diversity by inStrain32 (version 1.5.4) per sample. Evaluation of the average nucleotide diversity was performed per dataset because it was originally reported to be affected by the sequencing batches. Then, the read coverages of the reconstructed MAGs in originated samples were calculated by coverM (version 0.6.1). We searched for the presence of rRNAs in each MAG by barrnap (version 0.9) with the following parameters; --kingdom bac (for MAGs determined as bacteria by CheckM), --kingdom arc (for MAGs determined as archaea by CheckM), --reject 0.8, --evalue 1e-3. tRNAs of the standard 20 amino acids were identified by tRNAScan-SE98 (version 2.0.7) with the following parameters; -A (for MAGs determined as bacteria by CheckM), -B (for MAGs determined as archaea by CheckM).
Analysis for the species-level representative MAGs
The 19,084 reconstructed MAGs were clustered into estimated species-level clusters (ANI ≥95%) by dRep75 (version 3.2.0) with the following parameters; -pa 0.9 -sa 0.95 -nc 0.30 -cm larger. Following score was calculated for each MAGs based on the output of CheckM and the genome with the highest score was selected as the representative genome for each species-level cluster; score = Completeness −5 × Contamination +0.5 × log10(N50). After the dereplication at the species level, we obtained 1,273 species-level clusters and representative genomes. We then annotated taxonomy to the species-level representative genomes with GTDB-tk31 (version 1.5.3) based on the Genome Taxonomy Database release 202. Taxonomy of the non-representative genomes was assigned according to the taxonomy of the representative genomes of their clusters. For the subsequent comparisons, the UHGG genomes were also subjected to taxonomic annotation because the reference database for GTDB-tk was updated from the version used in the original study.8
For each of the species-level representative genomes, we checked the existence of the same species-level clusters in the UHGG. First, we estimated the ANI between the 1,273 reconstructed MAGs and the 4,644 UHGG genomes by mash81 (version 2.3) with the sketch size 1000. Based on the result of mash, we extracted pairs of the genomes with mash-based ANI ≥90%. Then, we calculated ANI for the extracted pairs of the genomes with the dnadiff function of MUMmer87 (version 4.0.0.rc1). For each of the 1,273 MAGs, we assigned corresponding species-level genomes in the UHGG which had ANI ≥95%, aligned fraction ≥30%, and the highest ANI to the query MAGs.
Among the 1,273 species-level representative MAGs, we extracted 1,267 bacterial MAGs for the construction of a maximum-likelihood phylogenetic tree. A multiple sequencing alignment (MSA) of the core genes generated by GTDB-tk were subjected to the iqtree78 (version 2.1.2). ‘LG + F + R10’ was chosen as the best-fit model by the ModelFinder100 and constructed phylogenetic tree was visualized with iTOL79 (version 6).
Comparative analysis of Bacillus subtilis genomes
To characterize the reconstructed Bacillus subtilis MAGs (26 genomes), we performed a comparative analysis with the Bacillus subtilis genomes retrieved from the GenBank (162 genomes) and UHGG (1 genome). We calculated pair-wide ANI for all the pairs of the Bacillus subtilis genomes with the dnadiff function of MUMmer. Then, we performed hierarchical clustering by the hclust function in the R (version 4.0.1) with the ‘method = ”average”’ option. After clustering, we extracted a cluster which included all the Bacillus subtilis MAGs in the JMAG by cutree function in the R with the ‘k = 10’ option. Then, we performed NMDS of the extracted cluster.
To confirm that Bacillus subtilis MAGs in the JMAG were closely related to Bacillus subtilis natto, we checked the genetic variations of the degQ promoter and swrAA (yvzD) coding regions which were previously reported to be different between Bacillus subtilis natto and Bacillus subtilis 168101. We made MSAs of these genomic regions from the Bacillus subtilis natto genomes, Bacillus subtilis 168 genomes, and Bacillus subtilis genomes in JMAG by muscle88 (version 3.8.31).
For the comparison of the microbial abundances in boxplots, only the HC samples were used. Quality-controlled reads were down-sampled to 1,000,000 paired-ends reads to adjust the differences of the library sizes between the datasets. Then, the down-sampled reads were mapped to the reference genome of the reconstructed 1,273 MAGs with bowtie2 and the abundances were calculated as Reads Per Kilobase of exon per Million mapped reads (RPKM) by coverM.
Functional analysis of the MAGs
Protein-coding genes for each of the 19,084 MAGs were predicted with Prokka93 (version 1.14.6) with the specification of the kingdom annotated by CheckM. Predicted proteins were subjected to the eggNOG-mapper76 (version 2.1.2) for the annotation of Cluster of Orthologous Groups (COG) and KEGG and the calculation of the database coverage ratio and functional annotation ratio. The database coverage ratio was defined as the ratio of the protein sequences which were assigned with any eggNOG-mapper hits including unknown functions. The functional annotation ratio was defined as the ratio of the protein sequences which were assigned with COG annotations other than S (Function unknown) and R (General function prediction only). Annotation of the carbohydrate-active enzymes (CAZyme) was performed separately with the hmmscan function in hmmer (version 3.1b2) and dbCAN HMMdb v1069 was used as a reference hmm profile. E-values less than 1 × 10−18 were regarded as significant in the annotation of the CAZymes.
The predicted protein sequences on the MAGs were dereplicated with MMseqs286 (version 13.45111) with the following parameters; --cov-mode 1 -c 0.8 --kmer-per-seq 80 --min-seq-id 1. The Dereplicated set of the protein sequences were then merged with the UHGP-100 and subjected to further clustering with the following parameters; --cov-mode 1 -c 0.8 --kmer-per-seq 80. The ‘--min-seq-id’ option in the second clustering was set at 1, 0.95, 0.9, and 0.5 to dereplicate the protein sequences at 100%, 95%, 90%, and 50% amino acid sequence identity, respectively.
We identified the β-porphyranase sequences in the JMAG and UHGP. We first performed a blastp search with diamond74 (version 2.0.4) ‘--ultra-sensitive’ mode. The dereplicated protein sequences for the JMAG and UHGP were queried against the β-porphyranase sequences identified in the previous study21 and available in NCBI (PorA, PorB, PorC, PorD, and PorE). Since the β-porphyranase has high sequence similarity to other proteins such as β-agarase and κ-carrageenase, we set a relatively strict threshold for E-values (<1 × 10−40). In addition, we constructed a maximum-likelihood phylogenetic tree from the identified β-porphyranase sequences and other related proteins (i.e. β-porphyranase, β-agarase, and κ-carrageenase) published in the previous study21 for confirming that our pipeline discriminated β-porphyranase from other related proteins. First, we made an MSA with MAFFT80 (version 7.486) with the ‘--auto’ parameter. Then, we generated a phylogenetic tree by iqtree with the ‘VT + F + R4’ model which was chosen as the best-fit model by the ModelFinder and visualized it with iTOL. To profile the taxonomic and geographic features of the β-porphyranase among the JMAG and UHGP, we extracted all the protein sequences which belong to the protein clusters of the β-porphyranase. For the calculation of the AAI between the β-porphyranase sequences, we performed an all vs all blastp search with the default setting and pident was used as the AAI. For the read-based quantification of the β-porphyranase, we translated and mapped the 1,000,000 paired-ends reads against the non-redundant β-porphyranase sequences in the JMAG and UHGP (Figure S6A), using the ‘blastx’ function in the diamond. We extracted the blastx hits with ≥95% identity and E-value < 10−10. If the blastx had multiple hits, hits with the highest bitscore were selected. Abundance was calculated as a (total length of the alignment length of the query sequences)/(total sequencing length).
Analysis of the Japanese-specific species-level clusters
To identify Japanese-specific species-level clusters, we checked the (i) number of the JMAG genomes and (ii) number of the non-Japanese-derived MAGs contained in the corresponding UHGG clusters for all of the species-level clusters in the JMAG. The species-level clusters which contained ≥10 JMAG genomes and ≤1 UHGG genome were defined as the Japanese-specific species-level clusters. Based on the eggNOG-mapper annotation, we profiled the CAZyme profiles of the MAGs in these clusters. We extracted the CAZymes which satisfied (i) [within-cluster ratio of the MAGs which had the CAZymes] > 0.75, (ii) [within-cluster ratio of the MAGs which had the CAZymes] > 5 × [within-phylum ratio of the MAGs which had the CAZymes], and (iii) [within-cluster ratio of the MAGs which had the CAZymes] > 5 × [within-JMAG ratio of the MAGs which had the CAZymes]. We extracted the protein sequence clusters made by MMSeqs2 (dereplicated at 90% AAI) which included the extracted CAZymes. For the extracted protein sequence clusters, we checked the (number of the protein sequences from the Japanese-specific species-level cluster)/(number of the protein sequences in the JMAG) to evaluate the uniqueness of the CAZyme profiles of the Japanese-specific species-level clusters among the JMAG. We also checked the (number of the protein sequences in the JMAG)/(number of the protein sequences in the JMAG and UHGP) to evaluate the Japanese-specificity of the extracted CAZymes. For the read-based quantification of the CAZymes, we translated and mapped the 1,000,000 paired-ends reads against the extracted CAZyme sequences described in Figure S3A, using the ‘blastx’ function in the diamond. We extracted the blastx hits with ≥95% identity and E-value < 10−10. If the blastx had multiple hits, hits with the highest bitscore were selected. Abundance was calculated as a (total length of the alignment length of the query sequences)/(total sequencing length). Only the HC samples were used for the calculation of the mean abundances.
Strain-level analysis of the JMAG
Reference prokaryotic genomes composed of the 1,273 species-level representative JMAG genomes were indexed with bowtie2. Then, we mapped the quality-controlled sequencing reads to the reference genomes with bowtie2. The mapped-read data were converted to bam format by samtools95 (version 1.10) and individually subjected to the ‘profile’ function in inStrain with the ‘--database_mode’ option. Then, the results of the ‘profile’ function were merged with the ‘compare’ function in inStrain per dataset because merging the results of all the samples was not computationally scaled. We set a threshold for the population ANI (popANI; a metric introduced by Olm et al.32 to detect the strain-sharing) at ≥99.999% to define the sharing of the strain between two individuals according to the validation in the original study. As for the taxa for which strain sharing was detected in at least three datasets, the ‘compare’ function in inStrain was run with all the samples with the specification of the single taxa.
Association tests between food-related bacteria and rs671
We genotyped the 550 subjects using Infinium Asian Screening Array (Illumina, San Diego, CA, USA). This genotyping array was built using an East Asian reference panel including whole-genome sequences, which enabled effective genotyping in East Asian populations.
We applied stringent quality control filters to the genotyping dataset using PLINK90 (version 1.90b4.4) as described elsewhere.102 We confirmed that genotyping call rate was <0.98 for all the individuals. For pairs of closely related individuals (PI_HAT calculated by PLINK >0.185), we removed either of the related individuals. We confirmed that only the individuals of the estimated East Asian ancestry were included in this study, based on the principal component analysis with the samples of the HapMap project using EIGENSTRAT.77 After the quality control procedures, we obtained the genotype data of rs671 for 546 subjects (Table S3).
As for the five bacterial species which satisfied (number of the samples involved in the strain-sharing)/(number of the samples used for the analysis of the target species) ≥ 0.5 in the strain-sharing analysis, we obtained the abundances. Note that samples with the usage of antibiotics were not included in this analysis. Quality-controlled reads were mapped to the reference genome of the reconstructed 1,273 MAGs with bowtie2, and the mean coverages of each genome calculated by coverM genome function were divided by ‘total sequencing length/1,000,000,000’ and subjected to the log transformation.
We evaluated the association between the bacterial abundances and the genotypes of rs671 by linear regression analysis with the following formula; normalized abundance of the bacterial abundance ∼ rs671 genotype (dosage of the A allele) + age + sex + phenotype + dataset + total sequencing length. The significance of the associations was evaluated by Wald’s test for the effect size of the rs671 genotype. In the sub-analysis without disease samples, we performed linear regression analysis with the following formula; normalized abundance of the bacterial abundance ∼ rs671 genotype (dosage of the A allele) + age + sex + dataset + total sequencing length.
In the MR analysis for the five food-related bacteria, we used the result of the previous dietary habits GWAS in the Japanese population.37 Since dairy (milk and yoghurt) and natto had genome-wide association (p < 5 × 10−8) only with the rs671 (effect size = 0.113 and p = 6.4 × 10−18 for milk; effect size = 0.113 and p = 6.0 × 10−21 for yoghurt; effect size = −0.114 and p = 2.7 × 10−24 for natto), we performed Wald’s test as implemented in the TwoSampleMR package.38
Reconstruction of viral genomes
The assembled contigs longer than 5kbp were used for the detection of viral genomes by VirSorter39 (version 1.0.6) and VirFinder40 (version 1.1). VirSorter was performed using Viromes (–db 2) databases, and sequences sorted as viruses with the “most confident” prediction (category 1, 4) or “likely” prediction (category 2, 5) were extracted for further analysis. Contigs with the VirFinder score of ≥0.9 and p < 0.01 were also extracted for further analysis. We applied CheckV41 (software version 0.7.0, database version 1.0) to all the viral sequences to estimate the completeness of the viral genomes and remove the flanking host regions on the assembled prophages. Subsequently, we checked the number of the viral genes and host genes based on the CheckV annotations. We extracted 31,395 viral genomes of which genome completeness >50% and the number of viral genes > the number of host genes for further analyses.
Clustering and taxonomic annotation of the viral genomes
The 31,395 viral genomes were clustered into species-level vOTUs at the 95% ANI and 85% alignment fraction of the shorter sequence as previously described.20 We performed all vs all blast using the blastn function in the blast+89 (version 2.5.0) with the ’--max_target_seqs 10000′ option and the result were subjected to the greedy clustering with the previously published custom scripts.20 After the clustering, we obtained 12,213 species-level vOTUs. Same clustering procedures were performed for the viral genomes with completeness >50% in the GPD and MGV.
We extracted all the representative viral genomes from the JVD, GPD, and MGV and they were merged with the RefSeq viral genomes and previously published crAss-like phage genomes (taxonomic reference genomes) for subsequent clustering and taxonomic annotation. The merged viral genomes were clustered into species-level vOTU as described above and resulted in 94,714 species-level vOTUs. We extracted representative genomes from each of the species-level vOTUs and clustered them into family- and genus-level vOTUs based on the gene sharing ratio and AAI as previously described.20 The 94,714 viral genomes were subjected to prodigal92 (version 2.6.3) with the ‘-p meta’ option. Then all vs all blastp search by diamond was performed with the ‘--max_target_seqs 10000 --evalue 1e-5’ options. Then, pairwise gene-sharing and AAI were calculated for all the pairs of the viral genomes. For clustering, edges between viral genomes were filtered based on their minimum AAI and gene sharing ratio. We performed Markov clustering by MCL (version 14.137)83 using the following parameters and thresholds for gene sharing ratio and AAI; inflation factors: 1.1, 1.4, 2.0, 3.0, 4.0, 5.0, 6.0, 7.0; gene sharing ratio: 10, 15, 20, 30; AAI: 10, 15, 20, 25, 30, 40, 50, 60. We then selected the following filtering thresholds and MCL inflation factor that resulted in the highest accuracy103 for the family- and genus-level annotations of the RefSeq viral genomes and previously published crAss-like phage genomes; genus-level vOTU: ≥40% AAI, ≥30% gene sharing ratio, inflation factor = 2.0; family-level vOTU: ≥25% AAI, ≥10% gene sharing ratio, inflation factor = 1.4. In this setting, accuracies were 0.77 for the genus-level vOTU and 0.68 for the family-level vOTU (Table S6).
Using the clustering results, we performed taxonomic annotation of the viral genomes based on the taxonomic information of the reference genomes. First, if the viral genome was clustered into a species-level vOTU which contained taxonomic reference genomes, we annotated the taxonomic information which was concordant among the ≥75% of the taxonomic reference genomes and higher than genus-level. Second, the same procedures were repeated based on the genus-level vOTU information for unannotated genomes. Third, unannotated viral genomes clustered into a family-level vOTU which contains ≥2 taxonomic reference genomes, we annotated the taxonomic information which was concordant among the ≥75% of the taxonomic reference genomes and higher than family-level. After the taxonomic annotation procedures, we annotated family-level taxonomy for the 8,873 of 31,395 newly reconstructed viral genomes. The 9,167 GPD genomes and 43,817 MGV genomes were taxonomically annotated at the family level both in the original studies and this study. Although the recent taxonomic modification of the Podoviridae lowered the overall family-level taxonomic concordance between the original study and this study (73.1% for the GPD and 83.9% for the MGV), high family-level taxonomic concordance was observed for viruses that were not annotated as Podoviridae in the previous studies (95.7% for the GPD and 97.9% for the MGV).
Functional analysis of the viral genomes
Protein-coding genes for each of the viral genomes were predicted by prodigal with the ‘-p meta’ option. Predicted proteins were subjected to the eggNOG-mapper and hmmscan function in the hmmer against the VOG database44 (E-value < 1 × 10−5) for the annotation of the KEGG and VOG and calculation of the database coverage ratio and functional annotation ratio. The database coverage ratio was defined as the ratio of the protein sequences which were assigned with any eggNOG or VOG hits including unknown functions. The functional annotation ratio was defined as the ratio of the protein sequences which were assigned with either COG annotations other than S (Function unknown) and R (General function prediction only) or VOG annotations other than Xu (Function unknown). AMGs among the KEGG genes were defined based on the previously published manually curated list of the AMGs.46
Predicted protein sequences on the viral genomes from the JVD, MGV, and GPD were dereplicated with MMseqs2 with the following parameters; --cov-mode 1 -c 0.8 --kmer-per-seq 80 --min-seq-id 1. The Dereplicated set of the protein sequences from the JVD, MGV, and GPD were then merged and subjected to further clustering with the following parameters; --cov-mode 1 -c 0.8 --kmer-per-seq 80. The ‘--min-seq-id’ option in the second clustering was set at 1, 0.95, 0.9, and 0.5 to dereplicate the protein sequences at 100%, 95%, 90%, and 50% amino acid sequence identity, respectively.
Subfamily-level annotation of the crAss-like phages
We extracted the 1,378 putative crAss-like phage genomes which represented the species-level vOTUs. We annotate the subfamily of the crAss-like phage genomes based on the major annotation of the taxonomic reference genomes which were co-clustered into the same genus-level vOTUs. We checked the validity of the subfamily of crAss-like phages by constructing the maximum-likelihood phylogenetic trees for TerL, a previously reported marker gene.42,104 First, we identified the TerL from the predicted protein sequences on the crAss-like phage genomes by the hmmsearch function in hmmer against previously constructed hmm profiles.42 We then extracted the significant hits (E-value < 0.05) and constructed MSAs by MAFFT with the default parameter. The detection ratio of the TerL was 87.9%. We constructed phylogenetic trees from the MSAs by iqtree, and results were visualized by iTOL. ‘LG + R9’ model was selected as the best model by the ModelFinder.
Interpopulational comparisons of the crAss-like phages
For the interpopulational comparisons based on the number of the viral genomes, we utilized the viral genomes from the JVD and MGV. The geographical origin of the viral genomes from the MGV was defined in the original study. Fisher’s exact tests were performed for the contingency tables made from the following four numbers; (i) number of the β crAss-like phage genomes from the target population, (ii) number of the non-β crAss-like phage genomes from the target population, (iii) number of the β crAss-like phage genomes from the reference population, and (iv) number of the non-β crAss-like phage genomes from the reference population. We performed 21 tests in total.
For the comparison of the crAss-like phage abundances, only the HC samples were used. The 94,714 representative genomes of the species-level vOTUs were indexed with bowtie2. Quality-controlled reads were down-sampled to 1,000,000 paired-ends reads to adjust the differences of the library sizes between the datasets and mapped to the reference genome with bowtie2. Abundances were calculated as Reads Per Kilobase of exon per Million mapped reads (RPKM) by coverM and summed up for each subfamily and genus-level vOTU of the crAss-like phages. Then the compositions among the crAss-like phages were calculated for each sample and averaged over the samples from the same groups.
Case–control comparisons of the crAss-like phages
Samples with the usage of the antibiotics were removed from case–control comparisons (Table S9). Quality-controlled reads were mapped to the reference genome with bowtie2. Abundances were calculated as mean coverage of each viral genome by coverM genome function, divided by ‘total sequencing length/1,000,000,000’, and summed up for each subfamily and genus-level vOTU of the crAss-like phages. Only the clades which satisfied the following three criteria were retained and subjected to the log transformation for subsequent analyses; (i) detected in >20% of samples used for case–control comparison (ii) detected in both case and control samples (iii) adjusted mean coverage ≥0.001. We evaluated the association between the crAss-like phages and the diseases (RA, SLE, MS, UC, CD, and CoCa) by logistic regression analysis with the following formula; disease state ∼ crAss-like phage abundance + age + sex + dataset + total sequencing length. The significance of the associations was evaluated by Wald’s test for the effect size of the crAss-like phage abundance.
As for the tested clades, we also performed the association analyses with the Shannon index. Quality-controlled reads were down-sampled to 1,000,000 paired-ends reads to adjust the differences of the library sizes between the datasets. Then, the down-sampled reads were mapped to the reference genome of the reconstructed 1,273 MAGs with bowtie2 and the mean coverage matrix was calculated with coverM. The resulting matrix of the mean coverage was subjected to the diversity function in the R package vegan (version 2.5_6) to calculate the Shannon index. We evaluated the association between the crAss-like phages and Shannon index by linear regression analysis with the following formula; Shannon index ∼ crAss-like phage abundance + age + sex + phenotype + dataset + total sequencing length. The significance of the associations was evaluated by Wald’s test for the effect size of the crAss-like phage abundances.
Virus–prokaryote association analysis based on the CRISPR and prophages
We predicted the CRISPR sequences on the reconstructed MAGs with MinCED85 (version 0.4.2). Spacers within the predicted CRISPR sequences were queried against the viral contigs recovered from the gut metagenome data. Since the MGV and GPD have significant overlap, we performed MGV vs GPD blastn search and dereplicated at 100% ANI over 100% aligned fraction of the shorter sequences. Blast hits of the spacers with >95% ANI, end-to-end alignment, and spacer coverage >95% were retained for further analysis. For each spacer, we extracted all the blast hits with the highest bit-score, and if the phylum-, class-, order-, family-, genus-, or species-level taxonomy were consistent among more than a half of the blast subjects, taxonomic information of the target viruses was annotated to the spacer. If the taxonomy of the target of the spacer could not be determined, an “ambiguous” label was assigned. The same procedures were repeated for the CRISPR sequences identified within the UHGG in the previous study.20
In addition to the CRISPR sequences, we utilized prophage information to identify virus–prokaryote interaction. Among the proviral contigs determined by CheckV, we extracted contigs of which >50% were covered with host’s sequences.
Virus–prokaryote association analysis based on the abundance
Samples with the usage of antibiotics and insufficient clinical information were removed from this analysis as done in the association analysis between the crAss-like phages and Shannon index. Quality-controlled reads were mapped to the reference genomes of the 1,273 JMAG genomes, 4,644 UHGG genomes, and 94,714 representative genomes of the species-level vOTUs with bowtie2, respectively. The mean coverage of each genome was calculated by the coverM genome function and divided by ‘total sequencing length/1,000,000,000’.
We extracted the viruses and prokaryotes which were conferred to participate in the virus–prokaryote interaction from the JMAG CRISPR analysis, UHGG CRISPR analysis, and JMAG prophage analysis, respectively. Then, we retrieved the abundance of the viruses and prokaryotes which passed the following criteria; (i) detected in >20% of samples (ii) detected in all the datasets (iii) adjusted mean coverage ≥0.001. Abundances of the viruses and prokaryotes were subjected to the log transformation. We evaluated the association between the viruses and the prokaryotes by linear regression analysis with the following formula; prokaryotic abundance ∼ viral abundance + age + sex + phenotype + dataset + total sequencing length. The significance of the associations was evaluated by Wald’s test for the effect size of the viral abundance.
Comparison of the viral and prokaryotic numbers and abundances between Japanese and other populations
For each species-level vOTU, we counted the number of viruses derived from Japanese and other populations from the JVD and MGV. Then we defined the odds ratio for being Japanese-derived as follows; (number of the Japanese-derived viruses belonging to the vOTU/number of the other viruses belonging to the vOTU)/(number of the Japanese-derived viruses not belonging to the vOTU/number of the other viruses not belonging to the vOTU). For each species-level cluster of the JMAG genome which have the corresponding cluster in the UHGG, we merged the JMAG and UHGG clusters and defined the odds ratio for being Japanese-derived as follows; (number of the Japanese-derived bacterial genomes belonging to the cluster/number of the other bacterial genomes belonging to the cluster)/(number of the Japanese-derived bacterial genomes not belonging to the cluster/number of the other bacterial genomes not belonging to the cluster).
We extracted species-level vOTU–prokaryotic cluster pairs which were detected in the JMAG CRISPR analysis. Among the pairs, we retained only pairs whose species-level vOTU were included in both the JVD and MGV and the prokaryotic cluster was present in both the JMAG and UHGG. Then, we evaluated the enrichment of the virus–bacteria pairs which had same sign of the log odds ratios for being Japanese-derived based on the Fisher’s exact test.
We also checked the differences of the viruses-prokaryotes interaction by read count-based approach. We extracted species-level vOTU–prokaryotic cluster pairs which were detected in the JMAG CRISPR analysis. Quality-controlled reads were down-sampled to 1,000,000 paired-ends reads to adjust the differences of the library sizes between the datasets. Then, the down-sampled reads were mapped to the reference genomes and abundances were calculated as RPKM by coverM for the JMAG and viruses, respectively. For each comparison between Japan and other populations, only the pairs of which viruses and bacteria satisfied the following criteria were considered; (i) detected in >20% of samples (ii) detected in both of the populations (iii) RPKM ≥0.001. Fold changes between Japan and other populations were calculated for each species-level vOTU and prokaryotic cluster. Then, we evaluated the enrichment of the virus–bacteria pairs which had the same sign of the log fold-changes between the abundances in Japanese and other populations based on the Fisher’s exact test.
Network analysis of the crAss-like phages
We extracted all the CRISPR spacers in the JMAG and UHGG genomes which supported the link between the crAss-like phage species-level vOTUs and MAGs. Then, we counted the number of the spacers which supported the link between the crAss-like phage species-level vOTUs and bacterial genus for the JMAG and UHGG, respectively. We constructed a network plot with the ggraph package (version 2.0.4). We specified 'kk' as a layout option to place nodes based on the spring-based algorithm by Kamada and Kawai. The bar plot was based on the combined number of the spacers in the JMAG and UHGG.
Quantification and statistical analysis
Please refer to figure legends and method details for details of statistical analysis. Number of the samples used in the analyses are described in Tables S1, S3, and S9.
Acknowledgments
We would like to thank all the participants involved in this study. This research was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (19H01021 and 20K21834), the Japan Agency for Medical Research and Development (AMED; JP21km0405211, JP21ek0109413, JP21gm4010006, JP21km0405217, and JP21ek0410075), JST Moonshot R&D (JPMJMS2021 and JPMJMS2024), Takeda Science Foundation, Bioinformatics Initiative of Osaka University Graduate School of Medicine, and Grant Program for Next Generation Principal Investigators at Immunology Frontier Research Center (WPI-IFReC), Osaka University.
Author contributions
Y.T., T.K., and Y. Okada designed the study and conducted the data analysis. Y.T. and Y. Okada wrote the manuscript. Y.T., T.K., Y. Maeda, T.N., E.O., D.M., Y. Matsumoto, and S.N. conducted the experiments. Y.T., T.K., Y. Maeda, K.O., Y. Otake, S.K., T.N., T.O., E.O., M.K., M.T., N.O., K. Todo, K.Y., K.S., M. Yagita, A.H., H. Matsuoka, M. Yoshimura, S.O., S.S., and H. Iijima collected and managed the samples. H. Iijima, H. Inohara, H.K., T.T., H. Mochizuki, K. Takeda, A.K., and Y. Okada supervised the study.
Declaration of interests
The authors declare no competing interests.
Published: November 30, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100219.
Contributor Information
Yoshihiko Tomofuji, Email: ytomofuji@sg.med.osaka-u.ac.jp.
Yukinori Okada, Email: yokada@sg.med.osaka-u.ac.jp.
Supplemental information
We used metagenome shotgun sequencing data, which were generated in previous studies23,24,25,26,27,28,48,49,64,65,67,66,68 or this study. CD, Crohn disease; CoCa, colorectal cancer; HC, healthy control; MS, multiple sclerosis; NBDC, National Bioscience Database Center; RA, rheumatoid arthritis; SAH, sub-arachnoid hemorrhage; SLE, systemic lupus erythematosus; ST, stroke; UA, unruptured cerebral aneurysm; UC, ulcerative colitis.
JMAG, Japanese metagenome-assembled genomes; MAG, metagenome-assembled genome; QS, quality score; rRNA, ribosomal RNA; tRNA, transfer RNA; UHGG, Unified Human Gastrointestinal Genome.
Alt, alternative; HC, healthy control; MS, multiple sclerosis; RA, rheumatoid arthritis; Ref, reference; SAH, sub-arachnoid hemorrhage; SD, standard deviation; SLE, systemic lupus erythematosus; UA, unruptured cerebral aneurysm.
SE, standard errorSE.
JVD, Japanese Virus Database; vOTU, viral operational taxonomic unit.
AAI, average amino acid identity; PPV, positive predictive value; vOTU, viral operational taxonomic unit.
vOTU, viral operational taxonomic unit.
vOTU, viral operational taxonomic unit.
CD, Crohn disease; CoCa, colorectal cancer; HC, healthy control; MS, multiple sclerosis; RA, rheumatoid arthritis; SAH, sub-arachnoid hemorrhage; SD, standard deviation; SLE, systemic lupus erythematosus; ST, stroke; UA, unruptured cerebral aneurysm; UC, ulcerative colitis.
CD, Crohn disease; CoCa, colorectal cancer; HC, healthy control; MS, multiple sclerosis; RA, rheumatoid arthritis; SE, standard error; SLE, systemic lupus erythematosus; UC, ulcerative colitis ∗p < 0.05, ∗∗p < 0.05/number of clades (per objective variables); ∗∗∗p < 0.05/number of tests across all diseases (only for association tests for diseases).
CD, Crohn disease; CoCa, colorectal cancer; HC, healthy control; MS, multiple sclerosis; RA, rheumatoid arthritis; SE, standard error; SLE, systemic lupus erythematosus; UC, ulcerative colitis ∗p < 0.05, ∗∗p < 0.05/number of clades (per objective variables); ∗∗∗p < 0.05/number of tests across all diseases (only for association tests for diseases).
CRISPR, clustered regularly interspaced short palindromic repeats; JMAG, Japanese metagenome-assembled genomes; MAG, metagenome-assembled genome; vOTU, viral operational taxonomic unit.
JMAG, Japanese metagenome-assembled genomes; MAG, metagenome-assembled genome; vOTU, viral operational taxonomic unit.
Sequences deposited as “Analysis” and “WGS” are available through the DDBJ search (https://ddbj.nig.ac.jp/search) and getentry (http://getentry.ddbj.nig.ac.jp/top-j.html), respectively.
Data and code availability
The JMAG genomes, JVD genomes, and CRISPR sequences are available in NBDC Human Database (http://humandbs.biosciencedbc.jp/) with the accession number of hum0197. The JMAG genomes, JVD genomes, and CRISPR sequences can also be downloaded from the DNA DataBank of Japan (DDBJ) with the accession numbers provided in Table S14. Detailed metadata for the JMAG and JVD genomes are provided as Tables S2 and S5, respectively. The MSS data are under the controlled access in NBDC Human Database (http://humandbs.biosciencedbc.jp/) with the accession number of hum0197 to protect the participants’ privacy. Applications from all the researchers who comply with the NBDC’s data terms of use are quickly assessed and accepted. Multiple sequence alignment files for the maximum-likelihood phylogenetic trees (representative JMAG genomes, β-porphyranase, and TerL of crAss-like phages) are available in Zenodo (https://doi.org/10.5281/zenodo.7053099). Codes used for the analysis and instructions for downloading JMAG genomes, JVD genomes, and CRISPR sequences from DDBJ are available in GitHub (https://github.com/ytomofuji/JMAG_JVD) and Zenodo (https://doi.org/10.5281/zenodo.7053099).
References
- 1.Holmes E., Li J.V., Marchesi J.R., Nicholson J.K. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012;16:559–564. doi: 10.1016/j.cmet.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Zou Y., Xue W., Luo G., Deng Z., Qin P., Guo R., Sun H., Xia Y., Liang S., Dai Y., et al. 1, 520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019;37:179–185. doi: 10.1038/s41587-018-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poyet M., Groussin M., Gibbons S.M., Avila-Pacheco J., Jiang X., Kearney S.M., Perrotta A.R., Berdy B., Zhao S., Lieberman T.D., et al. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat. Med. 2019;25:1442–1452. doi: 10.1038/s41591-019-0559-3. [DOI] [PubMed] [Google Scholar]
- 4.Forster S.C., Kumar N., Anonye B.O., Almeida A., Viciani E., Stares M.D., Dunn M., Mkandawire T.T., Zhu A., Shao Y., et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 2019;37:186–192. doi: 10.1038/s41587-018-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nayfach S., Shi Z.J., Seshadri R., Pollard K.S., Kyrpides N.C. New insights from uncultivated genomes of the global human gut microbiome. Nature. 2019;568:505–510. doi: 10.1038/s41586-019-1058-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P., et al. Extensive unexplored human microbiome diversity revealed by over 150, 000 genomes from metagenomes spanning age, geography, and lifestyle. Cell. 2019;176:649–662.e20. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Almeida A., Mitchell A.L., Boland M., Forster S.C., Gloor G.B., Tarkowska A., Lawley T.D., Finn R.D. A new genomic blueprint of the human gut microbiota. Nature. 2019;568:499–504. doi: 10.1038/s41586-019-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almeida A., Nayfach S., Boland M., Strozzi F., Beracochea M., Shi Z.J., Pollard K.S., Sakharova E., Parks D.H., Hugenholtz P., et al. A unified catalog of 204, 938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2021;39:105–114. doi: 10.1038/s41587-020-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shkoporov A.N., Hill C. Bacteriophages of the human gut: the “known unknown” of the microbiome. Cell Host Microbe. 2019;25:195–209. doi: 10.1016/j.chom.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 10.Keen E.C., Dantas G. Close encounters of three kinds: bacteriophages, commensal bacteria, and host immunity. Trends Microbiol. 2018;26:943–954. doi: 10.1016/j.tim.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guerin E., Hill C. Shining light on human gut bacteriophages. Front. Cell. Infect. Microbiol. 2020;10:481. doi: 10.3389/fcimb.2020.00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Norman J.M., Handley S.A., Baldridge M.T., Droit L., Liu C.Y., Keller B.C., Kambal A., Monaco C.L., Zhao G., Fleshner P., et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–460. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clooney A.G., Sutton T.D.S., Shkoporov A.N., Holohan R.K., Daly K.M., O’Regan O., Ryan F.J., Draper L.A., Plevy S.E., Ross R.P., Hill C. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe. 2019;26:764–778.e5. doi: 10.1016/j.chom.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Ma Y., You X., Mai G., Tokuyasu T., Liu C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome. 2018;6:24. doi: 10.1186/s40168-018-0410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao G., Vatanen T., Droit L., Park A., Kostic A.D., Poon T.W., Vlamakis H., Siljander H., Härkönen T., Hämäläinen A.M., et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA. 2017;114:E6166–E6175. doi: 10.1073/pnas.1706359114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujimoto K., Kimura Y., Shimohigoshi M., Satoh T., Sato S., Tremmel G., Uematsu M., Kawaguchi Y., Usui Y., Nakano Y., et al. Metagenome data on intestinal phage-bacteria associations aids the development of phage therapy against pathobionts. Cell Host Microbe. 2020;28:380–389.e9. doi: 10.1016/j.chom.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Gregory A.C., Zablocki O., Zayed A.A., Howell A., Bolduc B., Sullivan M.B. The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe. 2020;28:724–740.e8. doi: 10.1016/j.chom.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dutilh B.E., Cassman N., McNair K., Sanchez S.E., Silva G.G.Z., Boling L., Barr J.J., Speth D.R., Seguritan V., Aziz R.K., et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014;5:4498. doi: 10.1038/ncomms5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camarillo-Guerrero L.F., Almeida A., Rangel-Pineros G., Finn R.D., Lawley T.D. Massive expansion of human gut bacteriophage diversity. Cell. 2021;184:1098–1109.e9. doi: 10.1016/j.cell.2021.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nayfach S., Páez-Espino D., Call L., Low S.J., Sberro H., Ivanova N.N., Proal A.D., Fischbach M.A., Bhatt A.S., Hugenholtz P., Kyrpides N.C. Metagenomic compendium of 189, 680 DNA viruses from the human gut microbiome. Nat. Microbiol. 2021;6:960–970. doi: 10.1038/s41564-021-00928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hehemann J.-H., Correc G., Barbeyron T., Helbert W., Czjzek M., Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464:908–912. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- 22.Nishijima S., Suda W., Oshima K., Kim S.-W., Hirose Y., Morita H., Hattori M. The gut microbiome of healthy Japanese and its microbial and functional uniqueness. DNA Res. 2016;23:125–133. doi: 10.1093/dnares/dsw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kishikawa T., Maeda Y., Nii T., Motooka D., Matsumoto Y., Matsushita M., Matsuoka H., Yoshimura M., Kawada S., Teshigawara S., et al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population. Ann. Rheum. Dis. 2020;79:103–111. doi: 10.1136/annrheumdis-2019-215743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kishikawa T., Ogawa K., Motooka D., Hosokawa A., Kinoshita M., Suzuki K., Yamamoto K., Masuda T., Matsumoto Y., Nii T., et al. A metagenome-wide association study of gut microbiome in patients with multiple sclerosis revealed novel disease pathology. Front. Cell. Infect. Microbiol. 2020;10:585973. doi: 10.3389/fcimb.2020.585973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomofuji Y., Maeda Y., Oguro-Igashira E., Kishikawa T., Yamamoto K., Sonehara K., Motooka D., Matsumoto Y., Matsuoka H., Yoshimura M., et al. Metagenome-wide association study revealed disease-specific landscape of the gut microbiome of systemic lupus erythematosus in Japanese. Ann. Rheum. Dis. 2021;80:1575–1583. doi: 10.1136/annrheumdis-2021-220687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomofuji Y., Kishikawa T., Maeda Y., Ogawa K., Nii T., Okuno T., Oguro-Igashira E., Kinoshita M., Yamamoto K., Sonehara K., et al. Whole gut virome analysis of 476 Japanese revealed a link between phage and autoimmune disease. Ann. Rheum. Dis. 2022;81:278–288. doi: 10.1136/annrheumdis-2021-221267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yachida S., Mizutani S., Shiroma H., Shiba S., Nakajima T., Sakamoto T., Watanabe H., Masuda K., Nishimoto Y., Kubo M., et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019;25:968–976. doi: 10.1038/s41591-019-0458-7. [DOI] [PubMed] [Google Scholar]
- 28.Otake-Kasamoto Y., Kayama H., Kishikawa T., Shinzaki S., Tashiro T., Amano T., Tani M., Yoshihara T., Li B., Tani H., et al. Lysophosphatidylserines derived from microbiota in Crohn’s disease elicit pathological Th1 response. J. Exp. Med. 2022;219:e20211291. doi: 10.1084/jem.20211291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowers R.M., Kyrpides N.C., Stepanauskas R., Harmon-Smith M., Doud D., Reddy T.B.K., Schulz F., Jarett J., Rivers A.R., Eloe-Fadrosh E.A., et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017;35:725–731. doi: 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaumeil P.-A., Mussig A.J., Hugenholtz P., Parks D.H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–1927. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olm M.R., Crits-Christoph A., Bouma-Gregson K., Firek B.A., Morowitz M.J., Banfield J.F. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat. Biotechnol. 2021;39:727–736. doi: 10.1038/s41587-020-00797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasolli E., De Filippis F., Mauriello I.E., Cumbo F., Walsh A.M., Leech J., Cotter P.D., Segata N., Ercolini D. Large-scale genome-wide analysis links lactic acid bacteria from food with the gut microbiome. Nat. Commun. 2020;11:2610. doi: 10.1038/s41467-020-16438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morandi S., Cremonesi P., Povolo M., Brasca M. Enterococcus lactis sp. nov., from Italian raw milk cheeses. Int. J. Syst. Evol. Microbiol. 2012;62:1992–1996. doi: 10.1099/ijs.0.030825-0. [DOI] [PubMed] [Google Scholar]
- 35.Okada Y., Momozawa Y., Sakaue S., Kanai M., Ishigaki K., Akiyama M., Kishikawa T., Arai Y., Sasaki T., Kosaki K., et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 2018;9:1631. doi: 10.1038/s41467-018-03274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakaue S., Kanai M., Tanigawa Y., Karjalainen J., Kurki M., Koshiba S., Narita A., Konuma T., Yamamoto K., Akiyama M., et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021;53:1415–1424. doi: 10.1038/s41588-021-00931-x. [DOI] [PubMed] [Google Scholar]
- 37.Matoba N., Akiyama M., Ishigaki K., Kanai M., Takahashi A., Momozawa Y., Ikegawa S., Ikeda M., Iwata N., Hirata M., et al. GWAS of 165, 084 Japanese individuals identified nine loci associated with dietary habits. Nat. Hum. Behav. 2020;4:308–316. doi: 10.1038/s41562-019-0805-1. [DOI] [PubMed] [Google Scholar]
- 38.Hemani G., Zheng J., Elsworth B., Wade K.H., Haberland V., Baird D., Laurin C., Burgess S., Bowden J., Langdon R., et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roux S., Enault F., Hurwitz B.L., Sullivan M.B. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015;3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017;5:69. doi: 10.1186/s40168-017-0283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nayfach S., Camargo A.P., Schulz F., Eloe-Fadrosh E., Roux S., Kyrpides N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021;39:578–585. doi: 10.1038/s41587-020-00774-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yutin N., Benler S., Shmakov S.A., Wolf Y.I., Tolstoy I., Rayko M., Antipov D., Pevzner P.A., Koonin E.V. Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features. Nat. Commun. 2021;12:1044. doi: 10.1038/s41467-021-21350-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker P.J., Siddell S.G., Lefkowitz E.J., Mushegian A.R., Adriaenssens E.M., Alfenas-Zerbini P., Davison A.J., Dempsey D.M., Dutilh B.E., García M.L., et al. Changes to virus taxonomy and to the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2021) Arch. Virol. 2021;166:2633–2648. doi: 10.1007/s00705-021-05156-1. [DOI] [PubMed] [Google Scholar]
- 44.Grazziotin A.L., Koonin E.V., Kristensen D.M. Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017;45:D491–D498. doi: 10.1093/nar/gkw975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanehisa M., Goto S. KEGG: Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kieft K., Zhou Z., Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome. 2020;8:90. doi: 10.1186/s40168-020-00867-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franzosa E.A., Sirota-Madi A., Avila-Pacheco J., Fornelos N., Haiser H.J., Reinker S., Vatanen T., Hall A.B., Mallick H., McIver L.J., et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019;4:293–305. doi: 10.1038/s41564-018-0306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., Andrews E., Ajami N.J., Bonham K.S., Brislawn C.J., et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas A.M., Manghi P., Asnicar F., Pasolli E., Armanini F., Zolfo M., Beghini F., Manara S., Karcher N., Pozzi C., et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019;25:667–678. doi: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deveau H., Garneau J.E., Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010;64:475–493. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- 51.Stanton C.R., Rice D.T.F., Beer M., Batinovic S., Petrovski S. Isolation and characterisation of the bundooravirus genus and phylogenetic investigation of the Salasmaviridae bacteriophages. Viruses. 2021;13:1557. doi: 10.3390/v13081557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang P., Gao X., Li Y., Wang S., Yu J., Wei Y. Bacillus natto regulates gut microbiota and adipose tissue accumulation in a high-fat diet mouse model of obesity. J. Funct.Foods. 2020;68:103923. doi: 10.1016/j.jff.2020.103923. [DOI] [Google Scholar]
- 53.Oki K., Toyama M., Banno T., Chonan O., Benno Y., Watanabe K. Comprehensive analysis of the fecal microbiota of healthy Japanese adults reveals a new bacterial lineage associated with a phenotype characterized by a high frequency of bowel movements and a lean body type. BMC Microbiol. 2016;16:284. doi: 10.1186/s12866-016-0898-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuo T., Sun Y., Wan Y., Yeoh Y.K., Zhang F., Cheung C.P., Chen N., Luo J., Wang W., Sung J.J.Y., et al. Human-gut-DNA virome variations across geography, ethnicity, and urbanization. Cell Host Microbe. 2020;28:741–751.e4. doi: 10.1016/j.chom.2020.08.005. [DOI] [PubMed] [Google Scholar]
- 55.Silveira C.B., Rohwer F.L. Piggyback-the-Winner in host-associated microbial communities. NPJ Biofilms Microbiomes. 2016;2:16010. doi: 10.1038/npjbiofilms.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reyes A., Haynes M., Hanson N., Angly F.E., Heath A.C., Rohwer F., Gordon J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moreno-Gallego J.L., Chou S.-P., Di Rienzi S.C., Goodrich J.K., Spector T.D., Bell J.T., Youngblut N.D., Hewson I., Reyes A., Ley R.E. Virome diversity correlates with intestinal microbiome diversity in adult monozygotic twins. Cell Host Microbe. 2019;25:261–272.e5. doi: 10.1016/j.chom.2019.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faruque S.M., Naser I.B., Islam M.J., Faruque A.S.G., Ghosh A.N., Nair G.B., Sack D.A., Mekalanos J.J. Seasonal epidemics of cholera inversely correlate with the prevalence of environmental cholera phages. Proc. Natl. Acad. Sci. USA. 2005;102:1702–1707. doi: 10.1073/pnas.0408992102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gulyaeva A., Garmaeva S., Ruigrok R.A.A.A., Wang D., Riksen N.P., Netea M.G., Wijmenga C., Weersma R.K., Fu J., Vila A.V., et al. Discovery, diversity, and functional associations of crAss-like phages in human gut metagenomes from four Dutch cohorts. Cell Rep. 2022;38:110204. doi: 10.1016/j.celrep.2021.110204. [DOI] [PubMed] [Google Scholar]
- 60.Mosca A., Leclerc M., Hugot J.P. Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Front. Microbiol. 2016;7:455. doi: 10.3389/fmicb.2016.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shkoporov A.N., Khokhlova E.V., Fitzgerald C.B., Stockdale S.R., Draper L.A., Ross R.P., Hill C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018;9:4781. doi: 10.1038/s41467-018-07225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guerin E., Shkoporov A.N., Stockdale S.R., Comas J.C., Khokhlova E.V., Clooney A.G., Daly K.M., Draper L.A., Stephens N., Scholz D., et al. Isolation and characterisation of ΦcrAss002, a crAss-like phage from the human gut that infects Bacteroides xylanisolvens. Microbiome. 2021;9:89. doi: 10.1186/s40168-021-01036-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rabesandratana T. Microbiome conservancy stores global fecal samples. Science. 2018;362:510–511. doi: 10.1126/science.362.6414.510. [DOI] [PubMed] [Google Scholar]
- 64.Zhu F., Ju Y., Wang W., Wang Q., Guo R., Ma Q., Sun Q., Fan Y., Xie Y., Yang Z., et al. Metagenome-wide association of gut microbiome features for schizophrenia. Nat. Commun. 2020;11:1612. doi: 10.1038/s41467-020-15457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dhakan D.B., Maji A., Sharma A.K., Saxena R., Pulikkan J., Grace T., Gomez A., Scaria J., Amato K.R., Sharma V.K. The unique composition of Indian gut microbiome, gene catalogue, and associated fecal metabolome deciphered using multi-omics approaches. GigaScience. 2019;8:giz004. doi: 10.1093/gigascience/giz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wirbel J., Pyl P.T., Kartal E., Zych K., Kashani A., Milanese A., Fleck J.S., Voigt A.Y., Palleja A., Ponnudurai R., et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019;25:679–689. doi: 10.1038/s41591-019-0406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie H., Guo R., Zhong H., Feng Q., Lan Z., Qin B., Ward K.J., Jackson M.A., Xia Y., Chen X., et al. Shotgun metagenomics of 250 adult twins reveals genetic and environmental impacts on the gut microbiome. Cell Syst. 2016;3:572–584.e3. doi: 10.1016/j.cels.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tett A., Huang K.D., Asnicar F., Fehlner-Peach H., Pasolli E., Karcher N., Armanini F., Manghi P., Bonham K., Zolfo M., et al. The Prevotella copri complex comprises four distinct clades underrepresented in westernized populations. Cell Host Microbe. 2019;26:666–679.e7. doi: 10.1016/j.chom.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., Busk P.K., Xu Y., Yin Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.BMTagger. ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/.
- 71.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alneberg J., Bjarnason B.S., de Bruijn I., Schirmer M., Quick J., Ijaz U.Z., Lahti L., Loman N.J., Andersson A.F., Quince C. Binning metagenomic contigs by coverage and composition. Nat. Methods. 2014;11:1144–1146. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 73.Sieber C.M.K., Probst A.J., Sharrar A., Thomas B.C., Hess M., Tringe S.G., Banfield J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018;3:836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buchfink B., Reuter K., Drost H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods. 2021;18:366–368. doi: 10.1038/s41592-021-01101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Olm M.R., Brown C.T., Brooks B., Banfield J.F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–2868. doi: 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cantalapiedra C.P., Hernández-Plaza A., Letunic I., Bork P., Huerta-Cepas J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021;38:5825–5829. doi: 10.1093/molbev/msab293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 78.Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Letunic I., Bork P. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17:132. doi: 10.1186/s13059-016-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu Y.-W., Simmons B.A., Singer S.W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 83.Enright A.J., Van Dongen S., Ouzounis C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kang D.D., Li F., Kirton E., Thomas A., Egan R., An H., Wang Z. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bland C., Ramsey T.L., Sabree F., Lowe M., Brown K., Kyrpides N.C., Hugenholtz P. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209. doi: 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Steinegger M., Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017;35:1026–1028. doi: 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 87.Marçais G., Delcher A.L., Phillippy A.M., Coston R., Salzberg S.L., Zimin A. MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 2018;14:e1005944. doi: 10.1371/journal.pcbi.1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hyatt D., Chen G.-L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 94.Parks D.H., Rinke C., Chuvochina M., Chaumeil P.-A., Woodcroft B.J., Evans P.N., Hugenholtz P., Tyson G.W. Recovery of nearly 8, 000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017;2:1533–1542. doi: 10.1038/s41564-017-0012-7. [DOI] [PubMed] [Google Scholar]
- 95.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Prjibelski A., Antipov D., Meleshko D., Lapidus A., Korobeynikov A. Using SPAdes de novo assembler. Curr. Protoc. Bioinformatics. 2020;70:e102. doi: 10.1002/cpbi.102. [DOI] [PubMed] [Google Scholar]
- 97.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chan P.P., Lin B.Y., Mak A.J., Lowe T.M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49:9077–9096. doi: 10.1093/nar/gkab688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kawabata S., Takagaki M., Nakamura H., Oki H., Motooka D., Nakamura S., Nishida T., Terada E., Izutsu N., Takenaka T., et al. Dysbiosis of gut microbiome is associated with rupture of cerebral aneurysms. Stroke. 2022;53:895–903. doi: 10.1161/STROKEAHA.121.034792. [DOI] [PubMed] [Google Scholar]
- 100.Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nishito Y., Osana Y., Hachiya T., Popendorf K., Toyoda A., Fujiyama A., Itaya M., Sakakibara Y. Whole genome assembly of a natto production strain Bacillus subtilis natto from very short read data. BMC Genomics. 2010;11:243. doi: 10.1186/1471-2164-11-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sakaue S., Yamaguchi E., Inoue Y., Takahashi M., Hirata J., Suzuki K., Ito S., Arai T., Hirose M., Tanino Y., et al. Genetic determinants of risk in autoimmune pulmonary alveolar proteinosis. Nat. Commun. 2021;12:1032. doi: 10.1038/s41467-021-21011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bin Jang H., Bolduc B., Zablocki O., Kuhn J.H., Roux S., Adriaenssens E.M., Brister J.R., Kropinski A.M., Krupovic M., Lavigne R., et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019;37:632–639. doi: 10.1038/s41587-019-0100-8. [DOI] [PubMed] [Google Scholar]
- 104.Yutin N., Makarova K.S., Gussow A.B., Krupovic M., Segall A., Edwards R.A., Koonin E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018;3:38–46. doi: 10.1038/s41564-017-0053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
We used metagenome shotgun sequencing data, which were generated in previous studies23,24,25,26,27,28,48,49,64,65,67,66,68 or this study. CD, Crohn disease; CoCa, colorectal cancer; HC, healthy control; MS, multiple sclerosis; NBDC, National Bioscience Database Center; RA, rheumatoid arthritis; SAH, sub-arachnoid hemorrhage; SLE, systemic lupus erythematosus; ST, stroke; UA, unruptured cerebral aneurysm; UC, ulcerative colitis.
JMAG, Japanese metagenome-assembled genomes; MAG, metagenome-assembled genome; QS, quality score; rRNA, ribosomal RNA; tRNA, transfer RNA; UHGG, Unified Human Gastrointestinal Genome.
Alt, alternative; HC, healthy control; MS, multiple sclerosis; RA, rheumatoid arthritis; Ref, reference; SAH, sub-arachnoid hemorrhage; SD, standard deviation; SLE, systemic lupus erythematosus; UA, unruptured cerebral aneurysm.
SE, standard errorSE.
JVD, Japanese Virus Database; vOTU, viral operational taxonomic unit.
AAI, average amino acid identity; PPV, positive predictive value; vOTU, viral operational taxonomic unit.
vOTU, viral operational taxonomic unit.
vOTU, viral operational taxonomic unit.
CD, Crohn disease; CoCa, colorectal cancer; HC, healthy control; MS, multiple sclerosis; RA, rheumatoid arthritis; SAH, sub-arachnoid hemorrhage; SD, standard deviation; SLE, systemic lupus erythematosus; ST, stroke; UA, unruptured cerebral aneurysm; UC, ulcerative colitis.
CD, Crohn disease; CoCa, colorectal cancer; HC, healthy control; MS, multiple sclerosis; RA, rheumatoid arthritis; SE, standard error; SLE, systemic lupus erythematosus; UC, ulcerative colitis ∗p < 0.05, ∗∗p < 0.05/number of clades (per objective variables); ∗∗∗p < 0.05/number of tests across all diseases (only for association tests for diseases).
CD, Crohn disease; CoCa, colorectal cancer; HC, healthy control; MS, multiple sclerosis; RA, rheumatoid arthritis; SE, standard error; SLE, systemic lupus erythematosus; UC, ulcerative colitis ∗p < 0.05, ∗∗p < 0.05/number of clades (per objective variables); ∗∗∗p < 0.05/number of tests across all diseases (only for association tests for diseases).
CRISPR, clustered regularly interspaced short palindromic repeats; JMAG, Japanese metagenome-assembled genomes; MAG, metagenome-assembled genome; vOTU, viral operational taxonomic unit.
JMAG, Japanese metagenome-assembled genomes; MAG, metagenome-assembled genome; vOTU, viral operational taxonomic unit.
Sequences deposited as “Analysis” and “WGS” are available through the DDBJ search (https://ddbj.nig.ac.jp/search) and getentry (http://getentry.ddbj.nig.ac.jp/top-j.html), respectively.
Data Availability Statement
The JMAG genomes, JVD genomes, and CRISPR sequences are available in NBDC Human Database (http://humandbs.biosciencedbc.jp/) with the accession number of hum0197. The JMAG genomes, JVD genomes, and CRISPR sequences can also be downloaded from the DNA DataBank of Japan (DDBJ) with the accession numbers provided in Table S14. Detailed metadata for the JMAG and JVD genomes are provided as Tables S2 and S5, respectively. The MSS data are under the controlled access in NBDC Human Database (http://humandbs.biosciencedbc.jp/) with the accession number of hum0197 to protect the participants’ privacy. Applications from all the researchers who comply with the NBDC’s data terms of use are quickly assessed and accepted. Multiple sequence alignment files for the maximum-likelihood phylogenetic trees (representative JMAG genomes, β-porphyranase, and TerL of crAss-like phages) are available in Zenodo (https://doi.org/10.5281/zenodo.7053099). Codes used for the analysis and instructions for downloading JMAG genomes, JVD genomes, and CRISPR sequences from DDBJ are available in GitHub (https://github.com/ytomofuji/JMAG_JVD) and Zenodo (https://doi.org/10.5281/zenodo.7053099).