

Summary
The research of rare and devastating orphan diseases, such as idiopathic pulmonary fibrosis (IPF) has been limited by the rarity of the disease itself. The prognosis is poor—the prevalence of IPF is only approximately four times the incidence, limiting the recruitment of patients to trials and studies of the underlying biology. Global biobanking efforts can dramatically alter the future of IPF research. We describe a large-scale meta-analysis of IPF, with 8,492 patients and 1,355,819 population controls from 13 biobanks around the globe. Finally, we combine this meta-analysis with the largest available meta-analysis of IPF, reaching 11,160 patients and 1,364,410 population controls. We identify seven novel genome-wide significant loci, only one of which would have been identified if the analysis had been limited to European ancestry individuals. We observe notable pleiotropy across IPF susceptibility and severe COVID-19 infection and note an unexplained sex-heterogeneity effect at the strongest IPF locus MUC5B.
Keywords: idiopathic pulmonary fibrosis, cross-population analysis, GWAS, Global Biobank Meta-Analysis Initiative, ancestry, meta-analysis, fine-mapping, COVID-19, MUC5B
Graphical abstract
Highlights
-
•
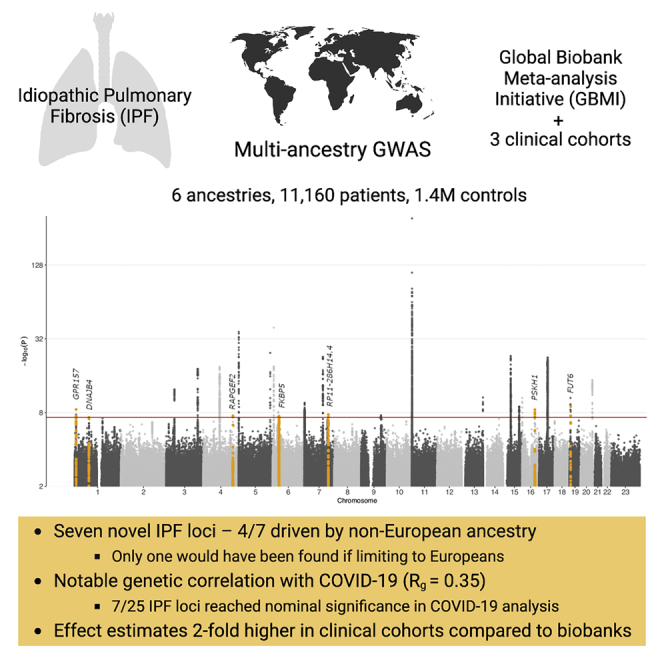
IPF meta-analysis across 6 ancestries with 11,160 cases and 1.4 M controls
-
•
Seven novel IPF loci—only one found if restricted to individuals of European ancestry
-
•
Genetic overlap with severe COVID-19: Rg ∼ 0.35, p = 0.001
-
•
Two-fold higher effect size estimates in clinical cohorts compared with biobanks
Partanen et al. present a multi-ancestry IPF meta-analysis with 11,160 cases and 1.4 M controls. They identify seven novel genome-wide significant loci, only one of which would have been identified if the analysis had been limited to Europeans. They also report notable pleiotropy across IPF susceptibility and severe COVID-19 infection.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive fibrotic disease of the lungs. It has no known etiology and pathogenesis, poor prognosis, and limited treatment options. The prevailing model of IPF pathogenesis suggests recurrent epithelial injury followed by aberrant repair and dysregulated interstitial matrix deposition with cell senescence playing an important role in promoting lung fibrosis.1
As IPF has, by definition, no identifiable cause, genome-wide approaches are especially attractive as they may provide insight into underlying causes, pathogenesis, and might potentially reveal novel therapeutic avenues. Genome-wide association studies (GWAS) of IPF have thus far reported at least 23 associated loci2, 3, 4, 5, 6, 7, 8, 9, 10, 11 highlighting genes involved in telomere maintenance,12 cell adhesion, airway clearance, and innate immunity. These studies have mainly been restricted to common variants in individuals of European descent, and have identified few associations to functional variants. The most recent and largest meta-analysis concluded that IPF is highly polygenic with a significant number of associated variants remaining to be identified.2 In addition, considerable genetic overlap between IPF and severe coronavirus disease 2019 (COVID-19) has been reported.13, 14, 15, 16
To further explore the genetics of IPF susceptibility, we performed the first multi-ancestry study on the genetics of IPF in six populations, altogether comprising a 4-fold increase in the number of patients compared with the largest IPF study to date, via meta-analysis of the Global Biobank Meta-Analysis Initiative (GBMI) with the most recent published IPF study.2 This allowed assessment of heterogeneity of effects over different ancestries, sex, and IPF diagnosis ascertainment between biobank and clinical cohort studies. With the increase in power, we were able to study population specific and rare variant effects. A notable fraction of the proposed loci have previously been associated with lung function. Fine-mapping in the Finnish population, making use of reduced allelic heterogeneity of a population isolate, identified a putative functional causal variant in the previously reported KIF15 locus. We also describe significant pleiotropy between IPF and COVID-19, beyond what is known to date. Finally, we suggest possible sex-based heterogeneity at MUC5B, the strongest genetic risk factor for IPF.
Results
Multi-ancestry meta-analysis reveals seven novel IPF loci
The GBMI IPF meta-analysis consisted of 8,492 cases and 1,355,819 controls representing 6 ancestries (Table 1). Out of 66.6 M variants included in analysis, 21.0 M were not present in the non-Finnish European (NFE) ancestry studies. While IPF prevalence and recruitment strategies varied greatly across contributing biobanks, the overall prevalence was 0.62% (Table S1). The GBMI IPF meta-analysis discovered 16 genome-wide significant loci, highlighting 2 potentially novel loci (Figure 1).
Table 1.
Ancestries in the GBMI IPF meta-analysis
| Ancestry | Biobanks | N IPF patients | N controls | Frac cases (%) |
|---|---|---|---|---|
| NFE | BioVU, CCPM, ESTBB, HUNT, MGB, MGI, UCLA, UKBB | 5,229 | 750,630 | 0.69 |
| FIN | FinnGen | 1,514 | 306,063 | 0.49 |
| EAS | BBJ, CKB, UCLA | 1,210 | 254,409 | 0.47 |
| AMR | BioMe, UCLA | 319 | 14,452 | 2.16 |
| AFR | BioMe, UCLA | 169 | 8,368 | 1.98 |
| SAS | GNH | 51 | 21,897 | 0.23 |
| 6∗ | 13∗ | 8,492∗ | 1,355,819∗ | 0.62∗ |
AFR, African/African American; AMR, Latino/admixed American; EAS, East Asian; FIN, Finnish; NFE, non-Finnish European; SAS, South Asian. Biobanks: BioME, BioVU, Colorado Center for Personalized Medicine Biobank (CCPM), Michigan Genome Initiative (MGI), UCLA Precision Health Biobank (UCLA), and Mass General Brigham (MGB) in America, BioBank Japan (BBJ) and China Kadoorie Biobank (CKB) in East Asia, and Genes & Health (GNH), Estonian Biobank (ESTBB), FinnGen project, Trøndelag Health Study (HUNT), and UK Biobank (UKBB) in Europe. Frac cases, fraction of cases in total sample. ∗Refers to values for GBMI as a whole.
Figure 1.

Genome-wide association results for IPF
Results from the joint meta-analysis are plotted in the top panel of the Miami plot and results from the GBMI meta-analysis in the bottom panel. Novel associations are highlighted in orange and annotated with the closest gene (index variant and variants in LD at r2 > 0.05 are highlighted, except for the PSKH1 signal for which variants within a 1 Mb window are highlighted due to missing LD information). Variants with p values ≤ 0.01 are plotted.
We then meta-analyzed the GBMI data with the largest IPF meta-analysis2 to date, later referred to as the Allen et al. study, including 10.8 M variants and increasing the number of cases and controls to 11,160 and 1,364,410, respectively. Altogether the joint meta-analysis identified 25 independent IPF-associated loci (Figure 1; Data S1; Table S2). We report genome-wide significant results at 14/23 previously reported loci, with a further 7/23 showing consistent direction of effects at varying levels of significance (Table S3). The linkage disequilibrium (LD) score regression intercept17 for the joint meta-analysis was not inflated (1.011) indicating independence of included studies. Quantile-quantile plots for meta-analyses are available in Figure S1.
Beyond confirming nearly all of the previous signals, we identified seven potentially novel loci (Table 2; Figure 1). One locus was driven by rs539683219 at 16q22.1, an intronic PSKH1 variant only found in the East Asian (EAS) population. Highlighting the importance of multi-ancestry analysis, four out of the seven novel loci were mostly driven by non-European ancestry, when assessed by highest minor allele frequency at population level within the meta-analysis. Minor allele frequency enrichment within the meta-analysis compared with NFEs was over 1.5-fold for these four index variants. Moreover, if only the European populations (including NFE and Finnish) were analyzed, only one of the seven loci reached genome-wide significance (Table S4).
Table 2.
Novel associations from the GBMI and joint IPF meta-analyses
| Variant | Rsid | Index variant gene | Most severe consequence | AF alt (%) | Max MAF pop | MAF enrichment vs NFE | OR [95% CI] | p value |
|---|---|---|---|---|---|---|---|---|
| 1_9107187_C_A | rs7549256 | GPR157 | intron | 64.16 | EAS | 1.5 | 0.91 [0.88–0.94] | 3.29E−09 |
| 1_77998184_T_C | rs4130548 | DNAJB4, GIPC2 | intron | 33.30 | NFE | 1 | 1.09 [1.06–1.13] | 4.91E−08 |
| 4_159892716_A_T | rs76537958 | RAPGEF2 | intergenic | 2.92 | AFR | 2.5 | 1.29 [1.18–1.42] | 2.94E−08 |
| 6_35707919_A_G | rs9380529 | FKBP5 | intron | 51.72 | NFE | 1 | 1.08 [1.05–1.12] | 3.33E−08 |
| 7_129095384_G_A | rs34288126 | RP11-286H14.4 | noncoding transcript exon | 12.70 | FIN | 1.1 | 1.13 [1.09–1.19] | 1.50E−08 |
| 16_67895674_TG_T | rs539683219 | PSKH1 | intron | 1.71 | EAS | Inf | 3.20 [2.17–4.70] | 3.52E−09 |
| 19_5840608_C_T | rs708686 | FUT6 | upstream gene | 31.47 | AMR | 1.6 | 1.11 [1.07–1.14] | 1.08E−09 |
Sample size weighted mean imputation info score ≥0.80 for all variants, index variant gene and most severe consequence information from variant effect predictor (VEP), variant = chrom_pos_ref_alt (GRCh38); AF alt = within the meta-analysis sample size weighted GRCh38 alternate allele frequency; max MAF population = population with highest minor allele frequency (MAF), MAF enrichment is calculated as highest MAF divided by MAF in NFE. Effects are given for the alt allele. AFR, African/African American; AMR, Latino/admixed American; EAS, East Asian; FIN, Finnish; NFE, non-Finnish European; SAS, South Asian. Inf, polymorphic in only one population. p value threshold for genome-wide significance pval < 5E−8.
Further replication of the novel loci was attempted in two individual European ancestry cohorts (case count 792 and 664), where six loci were polymorphic and imputed at high quality (minimum imputation R2 = 0.98). Three of the six potentially novel findings replicated (at p value < 0.01 and with consistent direction of effects, Table S5).
Multiple novel variants associated with lung function and different organ manifestations
The Open Targets18 resource was used to assess previous findings for the proposed novel loci (Table S6).
Three of the seven novel loci have previously been implicated with lung function parameters forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC). First, at 6p21.31, the index variant rs9380529 in FKBP5 was in the 95% credible set of a FVC signal.19 The index variant for FVC (rs28435135, LD, r2 = 0.28) had a negative beta coefficient, i.e., decreasing vital capacity, consistent with higher risk of IPF suggested in the present study. In addition, rs9380529 was in LD (r2 = 0.64) with the lead variant (and included in the 95% credible set) for trunk and leg fat percentages in the UKB Neale v.2 analysis (http://www.nealelab.is/uk-biobank/). FKBP5 encodes a FK506-binding protein that has been shown to modulate the mTOR signaling pathway20 central in lung fibrinogenesis.21
Second, GPR157 at 1p36.22 has been associated with FEV1/FVC ratio,22 as the index variant rs7549256 from the current analysis was in LD (r2 = 0.55) with the reported index variant (no data on credible set or effect available). rs7549256 was also in LD (r2 = 0.64) with the index variant for insulin-like growth factor 1 levels.23
Third, no previous associations were found for the intergenic rs76537958 at 4q32.1, whereas variants in RAPGEF2 (LD with rs76537958: r2 < 0.1) have been associated with FVC in the UKB Neale v.2 analysis, as well as childhood and lifetime pneumonia.24 In addition, RAPGEF2 has been reported as a shared risk factor for both IPF and chronic obstructive pulmonary disease (COPD) in a network analysis.25
Regarding the four additional loci, rs4130548 at 1p31.1, has been implicated especially in body mass index, and was the index variant of the association signal.26 At 19p13.3, rs708686 upstream of FUT6 has been previously associated with carbohydrate antigen 19.9,27 blood protein levels (FUT3),28 and gallstones,28,29 among others. FUT6 affects fucosylation of mucins, including MUC5B, which may affect mucociliary clearance via changes in mucus viscoelasticity and pathogen binding.30 No previously reported associations were found for the non-coding transcript exon variant rs34288126 at 7q32.1, or the EAS-specific intronic rs539683219 in PSKH1 at 16q22.1. However, rs116906005, which is 190 kB downstream of the index variant in the PSKH1 locus (LD with rs539683219: r2 = 0.67 in EAS), has been previously associated with interstitial lung disease (ILD) in BioBank Japan.31
In contrast to other pulmonary diseases attributed to tobacco smoke exposure, such as COPD and lung cancer, no genome-wide significant association signal was seen in the CHRNA3/5 locus (OR[95% CI] = 1.05[1.02–1.08], p = 0.0034 for rs16969968), a known nicotine dependence locus.32
Expression levels and profiles across tissues and cell types for the genes at the novel loci, obtained from GTEx33,34 and IPF RNA sequencing studies,35, 36, 37 are displayed38 in Table S6, Figure S2, and Data S2. In summary, genes at five of the seven loci were expressed in the lung tissue (median TPM > 1, Table S6). Furthermore, genes at two loci (GIPC2 and FKBP5) have been reported to be differentially expressed in IPF lung compared with controls36 and, in addition to these, two more (GPR157 and RAPGEF2) in end-stage IPF37 (Table S6). Expression of the genes at the novel loci varied in level and tissue or cell-type specificity (Figure S2; Data S2). Of the genes differentially expressed in IPF, FKBP5, the gene with the highest expression level in lung, was expressed across different lung tissue cell types with highest expression levels among immune cells and fibroblasts, whereas GIPC2 was almost exclusively expressed in ciliated and endothelial/lymphatic cells.
Fine-mapping in the Finnish population suggests missense variant in KIF15 to be causal
To identify potential causal alleles within the identified loci, we performed fine-mapping of all identified loci in FinnGen, resulting in eight independent loci with suggested causal alleles (Table 3). None of the novel loci were successfully fine-mapped (i.e., had good quality credible sets with minimum LD between variants r2 0.25).
Table 3.
Fine-mapped good quality (minimum LD between variants r2 0.25) credible sets in FinnGen
| Locus | N variants in credible set | Credible set min LD (r2) | Highest PIP variant | rsid | Gene for most severe consequence | Most severe consequence | PIP |
|---|---|---|---|---|---|---|---|
| 3q26.2 | 35 | 0.91 | 3_169759718_A_G | rs12638862 | ACTRT3 | downstream gene | 0.080 |
| 3p21.31 | 22 | 0.48 | 3_44801967_G_T | rs138043992 | KIF15 | missense | 0.243 |
| 5p15.33 | 1 | 1.00 | 5_1272247_G_A | rs770066110 | TERT | stop gained | 1.000 |
| 5p15.33 | 1 | 1.00 | 5_1279370_T_C | rs776981958 | TERT | missense | 0.997 |
| 5q35.1 | 2 | 0.82 | 5_169588475_G_A | rs116483731 | SPDL1 | missense | 0.875 |
| 11p15.5 | 1 | 1.00 | 11_1219991_G_T | rs35705950 | MUC5B | upstream gene | 1.000 |
| 11p15.5 | 2 | 0.34 | 11_1468491_G_A | rs546531844 | MOB2 | downstream gene | 0.919 |
| 16p13.3 | 5 | 0.78 | 16_276685_G_A | rs184954013 | ARHGDIG | intron | 0.618 |
| 17q21.31 | 2,180 | 0.95 | 17_45753401_45753524del | rs1568002709 | CRHR1 | intron | 0.003 |
| 20q13.33 | 17 | 0.51 | 20_63582806_G_A | rs73315845 | GMEB2 | downstream gene | 0.077 |
Highest PIP variant = chrom_pos_ref_alt (GRCh38); PIP, posterior inclusion probability.
Fine-mapping suggested deleterious coding causal variants at three loci. In addition to the previously reported coding variants in TERT and SPDL1,4,6 fine-mapping identified a coding variant in KIF15 (predicted missense, rs138043992, AF = 0.29%, OR[95% CI] = 1.71[1.39–2.10], posterior inclusion probability (PIP) = 0.24, ENSP00000324020.4:p.Arg501Leu), enriched 2.6-fold in the Finnish population compared with non-Finnish non-Estonian Europeans (NFEEs) (gnomAD v2.1.1), and predicted as probably damaging by Polyphen and deleterious by SIFT.39,40
When a known locus near KANSL1/MAPT was assessed, the signal was fine-mapped to CRHR1 (intronic rs1568002709, AF = 11.7%, OR[95% CI] = 0.62[0.53–0.73], PIP = 0.003). There was, however, little resolution in the locus due to its exceptional LD structure41 (Data S1).
Fine-mapping further suggested two independent signals at 11p15.5; the well-established MUC5B and an independent signal downstream of MOB2. Causality of the MOB2 downstream variant (rs546531844, AF = 0.29%, 17 times enriched in Finns compared with NFEEs gnomAD v.2.1.1) was corroborated by two different fine-mapping methods. The "Sum of Single Effects" (SuSie) model42 suggested two 95% credible sets at the 11p15.5 locus. One of the credible sets included the MUC5B upstream gene variant rs35705950 (PIP = 1) and the other included two variants: a MOB2 downstream gene variant rs546531844 (PIP = 0.92) and a MUC6 missense variant rs148815783 (PIP = 0.072). Another fine-mapping method, FINEMAP,43,44 suggested four 95% credible sets (p = 0.55) at the locus. Three of the four credible sets consisted of one variant. Both the MUC5B upstream gene variant rs35705950 and the MOB2 downstream gene variant rs546531844 constructed their own one-variant credible sets (PIP = 1 and PIP = 0.96, respectively).
After conditioning the REGENIE GWAS on the MUC5B variant rs35705950 the MOB2 downstream gene variant rs546531844 association was no longer genome-wide significant and the effect size estimate was notably decreased (original beta = 1.35, original p = 1.33E–37, conditioned beta = 0.53, conditioned p = 1.41E−7). Imputation accuracy of the MOB2 variant was not optimal (INFO = 0.85) and LD between the MOB2 downstream variant and the MUC5B lead variant was low but not inexistent (r2 = 0.072, D’ = 0.766). Thus, the signal at MOB2 remains to be confirmed.
Pleiotropy of effects across COVID-19 severity
As considerable genetic overlap between IPF and severe COVID-19 caused by SARS-CoV-2 infection has been reported,13, 14, 15, 16,45 we assessed the shared genetic background of IPF and severe COVID-19 using the largest sample sizes available for both traits: the joint IPF meta-analysis reported here and the most recent COVID-19 Host Genetics Initiative results (data release 646). We discovered that, in addition to the four loci (MUC5B, DPP9, KANSL1/CRHR1, and ZKSCAN1)13, 14, 15, 16 previously associated with both IPF and COVID-19 hospitalization at a genome-wide level, three other genome-wide significant loci in the IPF meta-analysis passed the FDR-adjusted p value threshold of 0.05 in the COVID-19 scan (total 7/25, 28%; Figure 2; Table 4). As previously reported, the effect of MUC5B was reversed: the strong, established risk allele in IPF is clearly protective for severe COVID-19 (OR = 0.89, p = 1.2E−8). The ATP11A locus also demonstrated opposite effects for the two traits, while for the rest of the loci the direction of effects was shared. While genome-wide associations for both IPF and COVID-19 hospitalization have been reported separately in the 17q21.31 locus, we noted a shared signal at the locus with very high LD between the index variants (r2 = 0.97).
Figure 2.

IPF meta-analysis and COVID-19 hospitalization results
COVID-19 hospitalization results are shown in the top panel and IPF joint meta-analysis results in the bottom panel. Genome-wide significant IPF signals that reached FDR-adjusted p value < 0.05 in COVID-19 hospitalization scan are highlighted in yellow. Index variants in the IPF scan are plotted as diamonds in the COVID-19 results. Variants with p values ≤ 0.1 are plotted.
Table 4.
COVID-19 pleiotropy
| Variant | rsid | Index variant gene | Most severe consequence | OR [95% CI] IPF | OR [95% CI] COVID-19 | p value IPF | p value COVID-19 |
|---|---|---|---|---|---|---|---|
| 1_77998184_T_C | rs4130548 | DNAJB4, GIPC2 | intron | 1.09 [1.06–1.13] | 1.04 [1.01–1.06] | 4.91E−08 | 2.28E−03 |
| 7_100020983_G_A | rs6963345 | ZKSCAN1 | intron | 1.16 [1.13–1.20] | 1.04 [1.02–1.06] | 1.27E−23 | 1.14E−03 |
| 11_1219991_G_T | rs35705950 | MUC5B | upstream gene | 2.76 [2.62–2.90] | 0.89 [0.86–0.93] | <1E−300 | 1.22E−08 |
| 13_112881427_C_T | rs12585036 | ATP11A | noncoding transcript exon | 0.89 [0.86–0.92] | 1.07 [1.04–1.09] | 2.35E−11 | 1.80E−06 |
| 16_276685_G_A | rs184954013 | ARHGDIG | intron | 1.87 [1.55–2.25] | 1.43 [1.1–1.86] | 1.30E−13 | 7.83E−03 |
| 17_46126154_C_T | rs113120855 | KANSL1 | intron | 0.80 [0.77–0.84] | 0.92 [0.89–0.95] | 8.92E−23 | 1.04E−08 |
| 19_4717660_A_G | rs12610495 | DPP9 | missense | 1.12 [1.08–1.15] | 1.11 [1.09–1.14] | 2.95E−11 | 6.09E−18 |
Results for genome-wide significant index variants in joint IPF meta-analysis with FDR-adjusted p value < 0.05 in COVID-19 hospitalization scan (n = 7/25, 28%). Variant = chr_pos_ref_alt (GRCh38), effects are given for the alt allele.
Secondly, 6 of the 17 loci from the COVID-19 hospitalization scan passed the FDR-adjusted p value threshold of 0.05 in the IPF meta-analysis (35%, Table S7), suggesting further shared etiology at CCHCR1, SLC22A31, and TAC4.
Formal colocalization analysis was not possible as COVID-19 results have not been fine-mapped. Genetic correlation between the traits, determined by LDSC,47 restricting to European samples for COVID-19 hospitalization and NFE samples for IPF, was 0.35 (95% CI 0.14–0.56, p = 0.001), somewhat higher compared with a previous estimate14 and with less uncertainty.
Pleiotropy of the IPF signals beyond COVID-19 hospitalization was explored by colocalization analysis in FinnGen, pointing to shared signals between IPF and osteoporosis, cancers, and hypothyroidism (Table S8). At 16p13.3 an intronic ARHGDIG variant (rs184954013, AF in Finns = 2.5%, AF in NFE = 0.29%) colocalized with the osteoporotic fracture signal (causal posterior agreement [CLPA] = 0.74) and also with any form of osteoporosis (CLPA = 0.58). As we have reported prior to this study,4 multiple malignancy-related colocalization signals were also detected (Table S8).
Sex-stratified analysis in biobanks suggests heterogenic effects at strongest IPF signal
Sex-stratified meta-analysis in the GBMI across six biobanks with results for both sexes identified a 1.6-fold larger effect for the strongest IPF-associated variant rs35705950 in the MUC5B locus in males (OR[95% CI] = 3.22[2.92–3.55], p = 1.0E−121) compared with females (OR[95% CI] = 2.04[1.82–2.29], p = 2.9E−34), Cochran’s Q p value for heterogeneity = 3.4E−9. To investigate whether the difference in effects was due to confounding by case ascertainment differences across contributing biobanks, we assessed the effect of rs35705950 in each contributing biobank in males and females, noting a weaker effect in females across biobanks (Figure S3; Table S9). The result was, however, not replicated in four clinical cohorts in sex-stratified analysis or sex interaction analysis (OR[95% CI]males = 4.81[4.37–5.30], OR[95% CI]females = 4.75[4.13–5.45] [Tables S10 and S11], and results in subsets of the FinnGen study [Table S12]). The lifetime incidence of IPF in the longitudinal FinnGen register follow-up stratified by sex and carrying a MUC5B risk allele is illustrated in Figure S4. MUC5B carrier status did not have an effect on the number of IPF deaths or lung transplants among IPF cases in FinnGen (Table S13).
The male-only meta-analysis identified two additional loci: 19q13.32 with index variant rs71338787 in EML2 and Xq28 with index variant rs5945238 in MPP1 (Table S14).
Heterogeneity assessment points to large effect of sample ascertainment
We observed heterogeneous effects across biobanks and ancestry at nearly half of the IPF genome-wide significant loci (11/25, 44%, FDR-adjusted Cochran’s Q p value < 0.05, mean heterogeneity index I2 = 0.62; Data S3; Table S2). Therefore, we explored whether there was a systematic difference between the effects observed in the latest IPF meta-analysis, involving carefully curated clinically defined IPF, and biobank defined IPF, generally obtained from ICD codes in electronic health records (EHR). Sample recruitment periods and time periods covered by the EHRs are available in Table S15. Limiting to samples of NFE descent and genome-wide significant loci in the meta-analysis, the effect size estimates were 2.1 times larger in the clinical cohort compared with the meta-analyzed biobank studies (Figure 3). Per each biobank, the median beta ratio (βGBMI/βAllen) varied from −0.04 to 0.62 (Figure S5).
Figure 3.

Effect size estimate comparison latest IPF meta-analysis versus GBMI
Scatter plot of absolute value of latest IPF meta-analysis beta against absolute value of meta-analyzed GBMI NFE beta with inverse variance weighted linear regression line (weights from Allen et al. study) and accompanying slope estimate. The analysis was performed within the non-Finnish European ancestry and variants included were genome-wide significant in the joint IPF meta-analysis.
To further study the effect of case ascertainment on effect size estimates, we divided the FinnGen study into three subsets based on diagnosis and original study cohort: a clinical IPF cohort (FinnishIPF,48 n cases = 205), other IPF patients (n = 1,366), and non-IPF ILD patients (n = 1,624) and compared effect size estimates from these cohorts with those of the latest IPF meta-analysis. Again, effect size estimates were 0.9, 1.4, and 2.5 times larger in the latest IPF meta-analysis compared with the Finnish IPF, other IPF, and non-IPF ILD cohorts (Figure S6), providing further evidence that effect sizes in highly ascertained IPF patients are substantially higher compared with patients identified from biobanks. We further explored the effect of defining IPF cases based on the slightly more inclusive PheCode definition used by most biobanks compared with a more rigorous definition (International Classification of Diseases [ICD]-10 code J84.1) in FinnGen and noted that effect size estimates were somewhat attenuated in the PheCode-based IPF cases (0.87-fold, 95% CI 0.83–0.91; Figure S7).
The large effect of case ascertainment on effect size estimates was corroborated by meta-regression results, where case ascertainment explained most of the observed heterogeneity (mean R2 = 55.8%), while ancestry, by comparison, explained very little (mean R2 = 6.2%). After adjusting for ancestry, all 11 loci with heterogeneous effects expressed evidence of remaining heterogeneity. To study the effect of case ascertainment, we compared the effect estimates of two clinical cohorts, the latest IPF meta-analysis and the FinnishIPF subcohort of FinnGen, to the estimates of the 13 GBMI biobanks (excluding the clinical subcohort from FinnGen). Having divided FinnGen into 2 subcohorts, 10 of the 25 loci expressed evidence of heterogeneity. However, after accounting for case ascertainment status (whether the studies were based on clinical cohorts or not), only 4 of these 10 loci expressed evidence of remaining heterogeneity.
Discussion
We conducted the first multi-ancestry meta-analysis of IPF increasing the number of cases over 4-fold compared with the latest IPF meta-analysis. Incorporating 11,160 patients from 6 ancestries allowed us to identify 7 novel loci associated with IPF susceptibility. Three of the identified loci, GPR157, FKBP5, and RAPGEF2 have been previously associated with lung function measurements. Furthermore, FKBP5 affects the mTOR signaling pathway,20 central in lung fibrinogenesis,21 and another novel locus, FUT6, affects mucin fucosylation potentially influencing mucociliary clearance.30 Genes at two of the novel loci (GIPC2, FKBP5) have been reported to be differentially expressed in IPF lung compared with controls.36
Highlighting the importance of multi-ancestry analysis, one of the novel loci (PSKH1) was only polymorphic in the EAS population and three additional index variants were enriched within the meta-analysis at least 1.5-fold in a non-European population compared with NFEs. Moreover, only one of the loci would have reached genome-wide significance had the analysis been restricted to European populations. The power boost of the multi-ancestry meta-analysis comes from an increase in both sample size and sample diversity, allowing identification of loci whose index variants are more frequent in other ancestries than European. In addition to boosting power, increasing the ancestral diversity of IPF genetic association studies allows analyzing a broader set of genetic variation, as nearly a third of the variants in GBMI were not present in the GBMI NFE studies. It also enables cross-validating new findings across biobanks and increases representation of understudied populations.
Fine-mapping in the Finnish population, enriched for deleterious low-frequency coding variants,49 suggested a predicted missense variant causal at the KIF15 locus. In future studies, after further increases in samples sourced from diverse ancestries, fine-mapping in multiple populations should be undertaken. Cross-ancestry fine-mapping, however, still has notable challenges to be resolved.50
COVID-19 severity and IPF share a notable proportion of their genetic background, as genetic correlation was estimated to be substantial (Rg ∼ 0.35), higher than previous estimates.14 Of the 25 IPF index variants, seven reached the FDR-adjusted nominal p value in the COVID-19 hospitalization scan (out of which three were genome-wide significant: CRHR1, in addition to the previously reported MUC5B and DPP9). Effects at these loci were mostly in the same direction for the two traits, but variants in MUC5B and ATP11A showed opposite directions of effects. The strongest IPF risk locus MUC5B confers protection from severe COVID-19 and has been associated with survival in IPF patients.51 Histologically, acute exacerbations of IPF present with diffuse alveolar damage, which is also present in severe COVID-19.52
Interestingly, we observed a sex-stratified effect in GBMI at MUC5B, the strongest common genetic risk factor for IPF, where the effect was 1.6 times larger in males compared with females. This result should, however, be considered with caution as it was not replicated in four clinical cohorts and may arise from confounding factors, such as ascertainment and age distribution differences in the sexes. These confounders should be investigated in future studies before the observed difference is inferred to represent a biological difference between the sexes. Yet, patient gender bias has been suggested to cause overdiagnosis of IPF in males and underdiagnosis in females,53 which would attenuate effect estimates in males and increase them in females. With relevance to misexpression of MUC5B in IPF, rs35705950 has been suggested to introduce a de novo binding site for HOXA9,54 a transcription factor differentially expressed by sex in whole blood55 and transcriptionally regulated by sex hormones.56,57 Also, FOXA2, a transcription factor binding 32 bp downstream of rs35705950 in the enhancer region 3 kB upstream of MUC5B, has a strong effect on MUC5B expression54 and has been shown essential for sexual dimorphism in liver cancer.58
The meta-analysis with contributing biobanks featuring a wide variety of sampling strategies enabled studying between study heterogeneity, which revealed that case ascertainment has a large effect on IPF effect size estimates. Nearly a half of the genome-wide significant IPF loci expressed evidence of heterogeneity, which was mainly due to differences in case ascertainment—whether the patients were recruited from a hospital’s pulmonary clinic or identified from health registries. Effect size estimates were more than 2-fold for clinical IPF cohorts compared with patients recruited from biobanks, in part due to the marginally more inclusive PheCode-based case definition used in the biobanks. For GWAS, however, the substantially larger number of patients available from biobanks benefits discovery even given the attenuated effect size estimates.
We present the first multi-ancestry meta-analysis of IPF to date with an over 4-fold increase in cases compared with the latest IPF meta-analysis. We discover multiple novel loci, the vast majority of which are driven by non-European populations and many of which have been linked to lung traits. We confirm and further describe the notable overlap of genetic determinants of IPF and severe COVID-19, calling for functional research. We describe a possible sex-dependent effect at the strongest IPF risk factor, the MUC5B locus, and demonstrate a 2-fold difference in effect size estimates derived from clinical cohorts as opposed to biobanks. To conclude, leveraging global multi-ancestry analysis further elucidates the genetic background of IPF by both revealing novel loci and providing increased resolution into previously identified ones.
Limitations of the study
Limitations of the study include between study heterogeneity, pointing to differing ascertainment between studies. However, this has limited impact on our novel findings, as only one of the novel loci showed evidence of heterogeneity (DNAJB4/GIPC2). Second, and with relevance to the former, the PheCode-based IPF case definition including other interstitial idiopathic pneumonias increased the risk of misclassification in biobanks and contributed to the observed heterogeneity. In addition, as recruitment of participants spanned over two decades, changes in diagnostic practices for IPF may introduce chronological bias. Third, even though samples representing four non-European ancestries were included, the sample was still dominated by participants of European ancestry. Furthermore, fine-mapping was successful for only a minority of the loci. Finally, the novel findings of this study require future functional studies to elucidate their possible biological effects.
Consortia
GBMI authors
Wei Zhou, Masahiro Kanai, Kuan-Han H. Wu, Humaira Rasheed, Kristin Tsuo, Jibril B Hirbo, Ying Wang, Arjun Bhattacharya, Huiling Zhao, Shinichi Namba, Ida Surakka, Brooke N. Wolford, Valeria Lo Faro, Esteban A. Lopera-Maya, Kristi Läll, Marie-Julie Favé, Sinéad B. Chapman, Juha Karjalainen, Mitja Kurki, Maasha Mutaamba, Juulia J. Partanen, Ben M. Brumpton, Sameer Chavan, Tzu-Ting Chen, Michelle Daya, Yi Ding, Yen-Chen A. Feng, Christopher R. Gignoux, Sarah E. Graham, Whitney E. Hornsby, Nathan Ingold, Ruth Johnson, Triin Laisk, Kuang Lin, Jun Lv, Iona Y. Millwood, Priit Palta, Anita Pandit, Michael H. Preuss, Unnur Thorsteinsdottir, Jasmina Uzunovic, Matthew Zawistowski, Xue Zhong, Archie Campbell, Kristy Crooks, Geertruida H. de Bock, Nicholas J. Douville, Sarah Finer, Lars G. Fritsche, Christopher J. Griffiths, Yu Guo, Karen A. Hunt, Takahiro Konuma, Riccardo E. Marioni, Jansonius Nomdo, Snehal Patil, Nicholas Rafaels, Anne Richmond, Jonathan A. Shortt, Peter Straub, Ran Tao, Brett Vanderwerff, Kathleen C. Barnes, Marike Boezen, Zhengming Chen, Chia-Yen Chen, Judy Cho, George Davey Smith, Hilary K. Finucane, Lude Franke, Eric R. Gamazon, Andrea Ganna, Tom R. Gaunt, Tian Ge, Hailiang Huang Jennifer Huffman Jukka T. Koskela, Clara Lajonchere, Matthew H. Law, Liming Li, Cecilia M. Lindgren, Ruth J.F. Loos, Stuart MacGregor, Koichi Matsuda, Catherine M. Olsen, David J. Porteous, Jordan A. Shavit, Harold Snieder, Richard C. Trembath, Judith M. Vonk, David Whiteman, Stephen J. Wicks, Cisca Wijmenga, John Wright, Jie Zheng, Xiang Zhou, Philip Awadalla, Michael Boehnke, Nancy J. Cox, Daniel H. Geschwind, Caroline Hayward, Kristian Hveem, Eimear E. Kenny, Yen-Feng Lin, Reedik Mägi, Hilary C. Martin, Sarah E. Medland, Yukinori Okada, Aarno V. Palotie, Bogdan Pasaniuc, Serena Sanna, Jordan W. Smoller, Kari Stefansson, David A. van Heel, Robin G. Walters, Sebastian Zöllner, BioBank Japan, BioMe, BioVU, Canadian Partnership for Tomorrow’s Health/Ontario Health Study, China Kadoorie Biobank Collaborative Group, Colorado Center for Personalized Medicine, deCODE Genetics, Estonian Biobank, FinnGen, Generation Scotland, Genes & Health, LifeLines, Mass General Brigham Biobank, Michigan Genomics Initiative, QIMR Berghofer Biobank, Taiwan Biobank, The HUNT Study, UCLA ATLAS Community Health Initiative, UK Biobank, Alicia R. Martin, Cristen J. Willer, Mark J. Daly, and Benjamin M. Neale
Author affiliations are available in the supplemental information section.
International IPF Genetics Consortium
Richard J. Allen, Helen L. Booth, William A. Fahy, Ian P. Hall, Simon P. Hart, Mike R. Hill, Nik Hirani, Richard B. Hubbard, R. Gisli Jenkins, Toby M. Maher, Robin J. McAnulty, Ann B. Millar, Philip L. Molyneaux, Vidya Navaratnam, Eunice Oballa, Helen Parfrey, Gauri Saini, Ian Sayers, Martin D. Tobin, Louise V. Wain Moira K. B. Whyte, Ayodeji Adegunsoye, Carlos Flores, Naftali Kaminski, Shwu-Fan Ma, Imre Noth, Justin M. Oldham, Mary E. Strek, Yingze Zhang, Tasha Fingerlin, David A. Schwartz, Beatriz Guillen-Guio, Maria Molina-Molina, Margaret Neighbors Xuting Sheng, Amy Stockwell, and Brian L. Yaspan.
Author affiliations are available in the supplemental information section.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Global Biobank Meta-Analysis Initiative (GBMI) genome-wide meta-analysis results | GBMI | https://www.globalbiobankmeta.org/resources |
| Allen et al. genome-wide meta-analysis results | Allen et al. (2020) | https://github.com/genomicsITER/PFgenetics |
| Software and algorithms | ||
| UCSC liftOver | https://genome.ucsc.edu/cgi-bin/hgLiftOver | |
| REGENIE | Mbatchou J. et al. (2021) | https://doi.org/10.1038/s41588-021-00870-7 |
| LD score regression | Bulik-Sullivan B. K. et al. (2015) | https://github.com/bulik/ldsc/ |
| PLINK 2.0 | Chang et al. (2015) | https://www.cog-genomics.org/plink/2.0/ |
| R statistical programming | https://www.r-project.org/ | |
| Other | ||
| GBMI custom scripts used for quality control, meta-analysis and summary | https://github.com/globalbiobankmeta | |
| FinnGen fine-mapping pipeline | https://github.com/FINNGEN/finemapping-pipeline | |
| Project code | This paper | https://doi.org/10.5281/zenodo.6993906 |
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead contact, Jukka Koskela (jukka.koskela@helsinki.fi).
Materials availability
This study did not generate new materials.
Experimental model and subject details
13 biobanks in Europe, Asia, and USA encompassing 6 ancestries contributed to the Global Biobank Meta-Analysis Initiative (GBMI) IPF meta-analysis, totaling 8,492 cases and 1,355,819 controls (Table 1, Table S1). Sample ancestry was determined by individual biobanks and successful determination was ensured by comparing projected principal components, calculated by each biobank for their samples based on marker loadings standardized for all GBMI biobanks, to the 1000 Genomes Project and the Human Genome Diversity Project (HGDP). Sample recruitment strategies differed between the biobanks (Table S1). The three clinical IPF cohorts of the latest IPF meta-analysis (Chicago, Colorado and UK), totaling 2,668 cases and 8,591 controls, all of European ancestry, are described elsewhere.2 The two cohorts used for replication (UUS and Genentech) were also used for replication in the latest IPF meta-analysis and are described elsewhere.2 There was sample overlap of 3,366 controls between the GBMI meta-analysis (UKBB) and the three clinical IPF cohorts (UK cohort) of the latest IPF meta-analysis, but there was no IPF case overlap. All analyses were limited to adults (age ≥18).
Method details
Phenotype definition and quality control
The GBMI phenotypic and genotypic quality control are described elsewhere.59 Analysis was performed in most biobanks for PheCode 502, constructed from health data available from each biobank (Table S16, phenotype definitions used in each biobank are in Table S15). IPF cases for the PheCode 502 were determined using the following International Classification of Diseases (ICD)-codes: ICD-9: 515, 515.0, ICD-10: J84.1, J84.10, J84.17, J84.8, J84.89. In the latest IPF meta-analysis, case definition was based on American Thoracic Society and European Respiratory Society guidelines, and quality control steps are described elsewhere.2
Meta-analysis
For GBMI, GWASs stratified by ancestry and sex were conducted in each biobank after standard sample-level and variant-level quality control. Thereafter, inverse-variance weighted fixed-effect meta-analyses were performed for all biobanks across all ancestries, all biobanks by each ancestry, and all biobanks by sex, detailed description elsewhere.59 Meta-analysis of the GBMI meta-analysis and the latest IPF meta-analysis, referred to as the joint meta-analysis, was likewise performed using the inverse-variance weighted fixed effects model in R (version 4.1). Prior to meta-analysis, the summary statistics of the latest IPF meta-analysis were lifted over from GRCh37 to GRCh38 using UCSC liftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Liftover results were verified by comparing the results to LiftOver results from Picard: of the 10,790,934 variants lifted over by UCSC liftOver 10,777,976 (99.9%) overlapped with LiftOver results from Picard, indicating high concordance in the results of the two methods.
All variants are reported based on the human genome reference sequence GRCh38. Only variants that were genome-wide significant in the joint meta-analysis were considered in downstream analyses. Genome-wide significant loci were determined by taking a 1 Mb region around each genome-wide significant variant and merging overlapping regions. The HLA region on chromosome 6 (GRCh38 chr6:28,510,120-33,480,577) was considered as one locus. Loci which did not include a previously reported IPF associated variant irrespective of the variant’s p value in the meta-analysis were considered novel.
Fine-mapping
Fine-mapping was performed for the IPF GWAS in FinnGen release 7 using the "Sum of Single Effects" (SuSie) model42,60 and FINEMAP43,44 for corroborative analyses. Fine-mapping regions were defined by taking a 3 Mb window around each index variant in the joint IPF meta-analysis and merging overlapping regions. 95% credible sets (encompassing at least 95% of the probability of including the causal variant) were analyzed and the probability of variant causality was evaluated using the posterior inclusion probability (PIP). Conditional analysis in FinnGen was performed using REGENIE61 with dosages of the variant conditioned on as covariates alongside other covariates (age, sex, ten first principal components, and batch). Individual causal signals were explored in FinnGen using logistic regression with Firth correction implemented in the “logistf” R package.62
Phenome-wide lookup
To assess the shared effects of potentially novel loci, we considered associations with phenotypes in the Open Targets Genetics (OTG) obtained from the GWAS catalog. Linkage disequilibrium (LD) between variants was assessed using the LD pair tool63 (https://ldlink.nci.nih.gov/), restricting to the 1000 Genomes Project non-Finnish European sub-populations for variants polymorphic in non-Finnish Europeans and source population otherwise.
LD score regression intercept and genetic correlation
The LD score regression v.1.0.1 intercept17 was used to quantify the contribution of confounding biases to the IPF meta-analysis results. As this method depends on matching the LD structure of the analysis sample to a reference panel, the analysis was restricted to the NFE samples. LD score regression47 was also used to estimate the genetic correlation between IPF and COVID-19 hospitalization using samples of NFE and European ancestry for IPF and COVID-19, respectively. Pre-calculated LD scores from the 1000 Genomes European reference population were obtained online (https://data.broadinstitute.org/alkesgroup/LDSCORE/) and the analysis was conducted using the standard program settings for variant filtering (removal of non-HapMap3 SNPs, minor allele frequency of <1%, or allele mismatch with reference).
Colocalization
Colocalization analysis was performed in FinnGen release 7. Colocalization analysis was based on assessing agreement of the fine-mapped credible sets across two traits. Agreement was measured by causal posterior agreement (CLPA), calculated as the sum of minimum PIP between the two traits per variant in overlapping credible sets.
Sex-stratified and sex interaction analyses
We performed sex-stratified analysis in six biobanks in the GBMI. In FinnGen sex-stratified analyses were conducted in release 5 with 378 and 110524 female, and 650 and 86462 male IPF cases and controls, respectively. In addition, we performed sex-stratified and sex interaction analyses in four clinical cohorts (Colorado, UK, UUS, and Genentech) using the PLINK 2.0 software64 (https://www.cog-genomics.org/plink/2.0/). The analyses were not performed in the Chicago study for rs35705950 as the imputation quality (imputation R2) was less than 0.5. In the sex-stratified analysis the effect of the MUC5B variant rs35705950 on IPF case status was tested in males and females separately using logistic regression adjusting for the ten first principal components. The rs35705950-by-sex interaction analysis was performed using the following logistic regression model:
logit(P(Phenotypei)) = β0 + β1rs35705950i + β2Sexi + β3rs35705950i∗Sexi + β4PC1i +...+ β14PC10i + εi
where
Phenotypei is IPF status for individual i, rs35705950 is the dosage for rs35705950 (additive effect), Sex is binary coded, rs35705950∗Sex is the interaction term and PC1 to PC10 are the first ten standardised principal components.
The results from the four clinical cohorts were meta-analyzed using inverse-variance weighted fixed effect meta-analysis, implemented using the R package “metagen” (https://www.rdocumentation.org/packages/meta/versions/4.9-6/topics/metagen).
Heterogeneity evaluation
Heterogeneity of effect sizes across studies and between sexes was evaluated at each variant using Cochran’s Q p value and heterogeneity index. To study the contribution of different sample recruitment strategies on heterogeneity, effect size estimates of selected studies for genome-wide significant IPF loci were compared and an inverse-variance weighted linear regression line was fitted. To evaluate the extent to which sample recruitment and ancestry contributed to heterogeneity, we used meta-regression65 using the “meta” R package.66
Quantification and statistical analysis
Sample sizes and sample age and sex characteristics are available in Tables 1 and S1. All meta-analyses were performed using the inverse-variance weighted fixed effects model in R. The p value threshold for genome-wide significance was P < 5E-8. The p value for heterogeneity was calculated based on Cochran’s heterogeneity statistic.
Acknowledgments
We would like to thank Jaakko Kaprio, Juha Partanen, Sami Kilpinen, and Clara Benoit-Pilven from the University of Helsinki for providing insight on sections of the manuscript. This research used the SPECTRE High Performance Computing Facility at the University of Leicester.
This work was supported by the Doctoral Programme in Population Health, University of Helsinki (to J.J.P.); and The Finnish Medical Foundation (to J.J.P.); NHLBI (to J.O.); Wellcome Trust (221680/Z/20/Z to B.G.-G.); NHLBI (to D.A.S.), DoD (to D.A.S.); Spanish Ministry of Science and Innovation and Instituto de Salud Carlos III, co-financed by the European Regional Development Fund (ERDF) “A Way of Making Europe” from the European Union (EU) (RTC-2017-6471-1 and PI20/00876 to C.F.); Cabildo Insular de Tenerife (CGIEU0000219140 to C.F.); NIH (to I.N.), Medical Research Council – project grant (to R.G.J.); NIHR Research Professorship (to R.G.J.); GSK/Asthma + Lung UK Chair in Respiratory Research (C17-1) (to L.V.W.); Research Foundation of the Pulmonary Diseases HES (to R.K.); Jalmari and Rauha Ahokas Foundation (to R.K.); the Research Foundation of North Finland, Oulu, Finland, and a state subsidy of Oulu University Hospital (to R.K.); HUS State Research Funding (to M.M.); Academy of Finland (to J.T.K.). R.J.A. is an Action for Pulmonary Fibrosis Mike Bray Research Fellow. The work of the contributing biobanks for GBMI was supported by numerous grants from governmental and charitable bodies (see below). The research was partially supported by the National Institute for Health Research (NIHR) Leicester Biomedical Research Center; the views expressed are those of the author(s) and not necessarily those of the National Health Service (NHS), the NIHR, or the Department of Health. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The Graphical abstract was created with BioRender.com.
BioBank Japan Project: The BioBank Japan Project was supported by the Tailor-Made Medical Treatment program of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), the Japan Agency for Medical Research and Development (AMED). S.N. was supported by Takeda Science Foundation. Y.O. was supported by JSPS KAKENHI (19H01021 and 20K21834), and AMED (JP21km0405211, JP21ek0109413, JP21ek0410075, JP21gm4010006, and JP21km0405217), JST Moonshot R&D (JPMJMS2021 and JPMJMS2024), Takeda Science Foundation, and Bioinformatics Initiative of Osaka University Graduate School of Medicine, Osaka University.
BioMe - The Mount Sinai BioMe Biobank: The Mount Sinai BioMe Biobank has been supported by The Andrea and Charles Bronfman Philanthropies and in part by Federal funds from the NHLBI and NHGRI (U01HG00638001, U01HG007417, and X01HL134588). We thank all participants in the Mount Sinai Biobank. We also thank all our recruiters who have assisted and continue to assist in data collection and management and are grateful for the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai.
BioVU: The BioVU projects at Vanderbilt University Medical Center are supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH-funded Shared Instrumentation Grant nos. S10OD017985 and S10RR025141; CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/.
Colorado Center for Personalized Medicine (CCPM): CCPM would like to thank Richard Zane, Steve Hess, Sarah White, Emily Hearst, Emily Roberts, and the entire Health Data Compass team. CCPM was developed with support from UCHealth, Children’s Hospital Colorado, CU Medicine, CU Department of Medicine, and CU School of Medicine.
China Kadoorie Biobank collaborative group: The China Kadoorie Biobank collaborative group would like to thank the International Steering Committee: Junshi Chen, Zhengming Chen (PI), Robert Clarke, Rory Collins, Yu Guo, Liming Li (PI), Jun Lv, Richard Peto, Robin Walters, and Chen Wang. The China Kadoorie Biobank collaborative group would also like to thank the International Co-ordinating Centre, Oxford: Daniel Avery, Fiona Bragg, Derrick Bennett, Ruth Boxall, Ka Hung Chan, Yumei Chang, Yiping Chen, Zhengming Chen, Johnathan Clarke, Robert Clarke, Huaidong Du, Zammy Fairhurst-Hunter, Hannah Fry, Simon Gilbert, Alex Hacker, Parisa Hariri, Mike Hill, Michael Holmes, Pek Kei Im, Andri Iona, Maria Kakkoura, Christiana Kartsonaki, Rene Kerosi, Kuang Lin, Mohsen Mazidi, Iona Millwood, Qunhua Nie, Alfred Pozarickij, Paul Ryder, Sam Sansome, Dan Schmidt, Paul Sherliker, Rajani Sohoni, Becky Stevens, Iain Turnbull, Robin Walters, Lin Wang, Neil Wright, Ling Yang, Xiaoming Yang, and Pang Yao; and the National Co-ordinating Centre, Beijing: Yu Guo, Xiao Han, Can Hou, Chun Li, Chao Liu, Jun Lv, Pei Pei, and Canqing Yu. Regional coordinating centers would like to thank Guangxi Provincial CDC: Naying Chen, Duo Liu, and Zhenzhu Tang; Liuzhou CDC: Ningyu Chen, Qilian Jiang, Jian Lan, Mingqiang Li, Yun Liu, Fanwen Meng, Jinhuai Meng, Rong Pan, Yulu Qin, Ping Wang, Sisi Wang, Liuping Wei, and Liyuan Zhou; Gansu Provincial CDC: Caixia Dong, Pengfei Ge, Xiaolan Ren; Maiji CDC: Zhongxiao Li, Enke Mao, Tao Wang, Hui Zhang, and Xi Zhang; Hainan Provincial CDC: Jinyan Chen, Ximin Hu, and Xiaohuan Wang; Meilan CDC: Zhendong Guo, Huimei Li, Yilei Li, Min Weng, and Shukuan Wu; Heilongjiang Provincial CDC: Shichun Yan, Mingyuan Zou, and Xue Zhou; Nangang CDC: Ziyan Guo, Quan Kang, Yanjie Li, Bo Yu, and Qinai Xu; Henan Provincial CDC: Liang Chang, Lei Fan, Shixian Feng, Ding Zhang, and Gang Zhou; Huixian CDC: Yulian Gao, Tianyou He, Pan He, Chen Hu, Huarong Sun, and Xukui Zhang; Hunan Provincial CDC: Biyun Chen, Zhongxi Fu, Yuelong Huang, Huilin Liu, Qiaohua Xu, and Li Yin; Liuyang CDC: Huajun Long, Xin Xu, Hao Zhang, and Libo Zhang; Jiangsu Provincial CDC: Jian Su, Ran Tao, Ming Wu, Jie Yang, Jinyi Zhou, and Yonglin Zhou; Suzhou CDC: Yihe Hu, Yujie Hua, Jianrong Jin Fang Liu, Jingchao Liu, Yan Lu, Liangcai Ma, Aiyu Tang, and Jun Zhang; Qingdao CDC: Liang Cheng, Ranran Du, Ruqin Gao, Feifei Li, Shanpeng Li, Yongmei Liu, Feng Ning, Zengchang Pang, Xiaohui Sun, Xiaocao Tian, Shaojie Wang, Yaoming Zhai, and Hua Zhang; Licang CDC: Wei Hou, Silu Lv, and Junzheng Wang; Sichuan Provincial CDC: Xiaofang Chen, Xianping Wu, Ningmei Zhang, and Weiwei Zhou; Pengzhou CDC: Xiaofang Chen, Jianguo Li, Jiaqiu Liu, Guojin Luo, Qiang Sun, and Xunfu Zhong; Zhejiang Provincial CDC: Weiwei Gong, Ruying Hu, Hao Wang, Meng Wan, and Min Yu; and Tongxiang CDC: Lingli Chen, Qijun Gu, Dongxia Pan, Chunmei Wang, Kaixu Xie, and Xiaoyi Zhang. China Kadoorie Biobank gratefully acknowledges the participants, project staff, and the China National Centre for Disease Control and Prevention (CDC) and its regional offices. China’s National Health Insurance provides electronic linkage to all hospital treatment. Funding sources: baseline survey and first re-survey—Kadoorie Charitable Foundation, Hong Kong; long-term follow-up—UK Wellcome Trust (212946/Z/18/Z, 202922/Z/16/Z, 104085/Z/14/Z, 088158/Z/09/Z), National Natural Science Foundation of China (91843302), National Key Research and Development Program of China (2016YFC 0900500, 0900501, 0900504, 1303904); DNA extraction and genotyping – GlaxoSmithKline, UK Medical Research Council (MC-PC-13049, MC-PC-14135); core funding for the project to the Clinical Trial Service Unit and Epidemiological Studies Unit at Oxford University—British Heart Foundation (CH/1996001/9454), UK Medical Research Council (MC-UU-00017/1, MC-UU-12026/2, MC_U137686851), Cancer Research UK (C16077/A29186, C500/A16896).
Estonian Biobank: Estonian Biobank research was supported by the European Union through Horizon 2020 research and innovation program under grant no. 810645 and through the European Regional Development Fund project no. MOBEC008, by the Estonian Research Council grant PUT (PRG1291, PRG687, and PRG184) and by the European Union through the European Regional Development Fund project no. MOBERA21 (ERA-CVD project DETECT ARRHYTHMIAS, GA no JTC2018-009), project no. 2014-2020.4.01.15-0012, and project no. 2014-2020.4.01.16-0125. Estonian Biobank would like to acknowledge Dr. Tõnu Esko, Dr. Lili Milani, Dr. Reedik Mägi, Dr. Mari Nelis, and Dr. Andres Metspalu, all from the Institute of Genomics, University of Tartu, Tartu, Estonia.
FinnGen: The FinnGen project is funded by two grants from Business Finland (HUS 4685/31/2016 and UH 4386/31/2016) and the following industry partners: AbbVie Inc., AstraZeneca UK Ltd, Biogen MA Inc., Bristol Myers Squibb (and Celgene Corporation & Celgene International II Sàrl), Genentech Inc., Merck Sharp & Dohme Corp, Pfizer Inc., GlaxoSmithKline Intellectual Property Development Ltd, Sanofi US Services Inc., Maze Therapeutics Inc., Janssen Biotech Inc., and Novartis AG. Following biobanks are acknowledged for delivering biobank samples to FinnGen: Auria Biobank (www.auria.fi/biopankki), THL Biobank (www.thl.fi/biobank), Helsinki Biobank (www.helsinginbiopankki.fi), Biobank Borealis of Northern Finland (https://www.ppshp.fi/Tutkimus-ja-opetus/Biopankki/Pages/Biobank-Borealis-briefly-in-English.aspx), Finnish Clinical Biobank Tampere (www.tays.fi/en-US/Research_and_development/Finnish_Clinical_Biobank_Tampere), Biobank of Eastern Finland (www.ita-suomenbiopankki.fi/en), Central Finland Biobank (www.ksshp.fi/fi-FI/Potilaalle/Biopankki), Finnish Red Cross Blood Service Biobank (www.veripalvelu.fi/verenluovutus/biopankkitoiminta) and Terveystalo Biobank (www.terveystalo.com/fi/Yritystietoa/Terveystalo-Biopankki/Biopankki/). All Finnish Biobanks are members of BBMRI.fi infrastructure (www.bbmri.fi). Finnish Biobank Cooperative-FINBB (https://finbb.fi/) is the coordinator of BBMRI-ERIC operations in Finland. The Finnish biobank data can be accessed through the Fingenious services (https://site.fingenious.fi/en/) managed by FINBB.
Genes and Health: Genes & Health is/has recently been core-funded by Wellcome (WT102627, WT210561), the Medical Research Council (UK) (M009017), Higher Education Funding Council for England Catalyst, Barts Charity (845/1796), Health Data Research UK (for London substantive site), and research delivery support from the NHS National Institute for Health Research Clinical Research Network (North Thames). Genes & Health is/has recently been funded by Alnylam Pharmaceuticals, Genomics PLC; and a Life Sciences Industry Consortium of Bristol-Myers Squibb Company, GlaxoSmithKline Research and Development Limited, Maze Therapeutics Inc., Merck Sharp & Dohme LLC, Novo Nordisk A/S, Pfizer Inc., Takeda Development Centre Americas Inc. We thank Social Action for Health, Centre of The Cell, members of our Community Advisory Group, and staff who have recruited and collected data from volunteers. We thank the NIHR National Biosample Centre (UK Biocentre), the Social Genetic & Developmental Psychiatry Centre (King’s College London), Wellcome Sanger Institute, and Broad Institute for sample processing, genotyping, sequencing, and variant annotation. We thank Barts Health NHS Trust, NHS Clinical Commissioning Groups (City and Hackney, Waltham Forest, Tower Hamlets, Newham, Redbridge, Havering, Barking, and Dagenham), East London NHS Foundation Trust, Bradford Teaching Hospitals NHS Foundation Trust, Public Health England (especially David Wyllie), Discovery Data Service/Endeavour Health Charitable Trust (especially David Stables)—for GDPR-compliant data sharing backed by individual written informed consent. Most of all, we thank all of the volunteers participating in Genes & Health. We would like to acknowledge the Genes & Health Research Team (in alphabetical order by surname): Shaheen Akhtar, Mohammad Anwar, Elena Arciero, Samina Ashraf, Gerome Breen, Raymond Chung, Charles J. Curtis, Maharun Chowdhury, Grainne Colligan, Panos Deloukas, Ceri Durham, Sarah Finer, Chris Griffiths, Qin Qin Huang, Matt Hurles, Karen A. Hunt, Shapna Hussain, Kamrul Islam, Ahsan Khan, Amara Khan, Cath Lavery, Sang Hyuck Lee, Robin Lerner, Daniel MacArthur, Bev MacLaughlin, Hilary Martin, Dan Mason, Shefa Miah, Bill Newman, Nishat Safa, Farah Tahmasebi, Richard C. Trembath, Bhavi Trivedi, David A. van Heel, and John Wright.
The HUNT Study: A special thanks to all the HUNT participants for donating their time, samples, and information to help others. The Trøndelag Health Study (HUNT) is a collaboration between HUNT Research Centre (Faculty of Medicine and Health Sciences, NTNU, Norwegian University of Science and Technology), Trøndelag County Council, Central Norway Regional Health Authority, and the Norwegian Institute of Public Health. The genotyping in HUNT was financed by the National Institutes of Health; University of Michigan; the Research Council of Norway; the Liaison Committee for Education, Research and Innovation in Central Norway; and the Joint Research Committee between St Olavs hospital and the Faculty of Medicine and Health Sciences, NTNU. The genetic investigations of the HUNT Study is a collaboration between researchers from the K.G. Jebsen Center for Genetic Epidemiology, NTNU and the University of Michigan Medical School and the University of Michigan School of Public Health. The K.G. Jebsen Center for Genetic Epidemiology is financed by Stiftelsen Kristian Gerhard Jebsen; Faculty of Medicine and Health Sciences, NTNU, Norway. We want to thank clinicians and other employees at Nord-Trøndelag Hospital Trust for their support and for contributing to data collection in this research project. We also acknowledge HUNT-MI Leadership: Kristian Hveem, Cristen Willer, Oddgeir Lingaas Holmen, Mike Boehnke, Goncalo Abecasis, Bjorn Olav Åsvold, and Ben Brumpton; Scientific Advisory Committee: Ele Zeggini, Mark Daly, and Bjørn Pasternak; HUNT Research Centre: Jørn Søberg Fenstad, Anne Jorunn Vikdal, and Marit Næss; HUNT Cloud: Oddgeir Lingaas Holmen, Sandor Zeestraten, and Tom Erik Røberg; data applications and registry linkages: Maiken E. Gabrielsen and Anne Heidi Skogholt; low-pass whole sequencing genome bioinformatics and statistical analysis: He Zhang, Hyun Min Kang, and Jin Chen; array genotyping: Sten Even Erlandsen and Vidar Beisvåg; GWAS bioinformatics, QC, imputation, and statistical analysis: Wei Zhou, Jonas Nielsen, Lars Fritsche, Hyun Min Kang, Oddgeir Holmen, Ben Brumpton, and Laurent Thomas; CNV calling: Ellen Schmidt and Ryan Mills; and statistical methods development for analyzing HUNT data: Wei Zhou and Shawn Lee. The K.G. Jebsen Centre for Genetic Epidemiology is financed by Stiftelsen Kristian Gerhard Jebsen. The genotyping in HUNT was financed by the National Institutes of Health; University of Michigan; the Research Council of Norway; Stiftelsen Kristian Gerhard Jebsen; the Liaison Committee for Education, Research and Innovation in Central Norway; and the Joint Research Committee between St Olav’s hospital and the Faculty of Medicine and Health Sciences, NTNU.
Mass General Brigham (MGB) Biobank: Samples, genomic data, and health information were obtained from the Mass General Brigham (MGB) Biobank, a biorepository of consented patients’ samples at Mass General Brigham (parent organization of Massachusetts General Hospital and Brigham and Women’s Hospital). We are grateful to all of the participants and clinical and research teams who made this work possible. Support for genotyping was provided through MGB Personalized Medicine. We would like to acknowledge MGB Biobank Leadership: Elizabeth W. Karlson, MD; Shawn N. Murphy, MD, PhD; Susan A Slaugenhaupt, PhD; Jordan W. Smoller, MD, ScD; and Scott T. Weiss, MD, MSc.
Michigan Genomics Initiative: The authors acknowledge the Michigan Genomics Initiative participants, Precision Health at the University of Michigan, the University of Michigan Medical School Central Biorepository, and the University of Michigan Advanced Genomics Core for providing data and specimen storage, management, processing, and distribution services, and the Center for Statistical Genetics in the Department of Biostatistics at the School of Public Health for genotype data curation, imputation, and management in support of the research reported in this publication.
UCLA ATLAS Community Health Initiative (UCLA): We gratefully acknowledge the resources provided by the Institute for Precision Health (IPH) and participating UCLA ATLAS Community Health Initiative patients. The UCLA ATLAS Community Health Initiative in collaboration with UCLA ATLAS Precision Health Biobank, is a program of IPH, which directs and supports the biobanking and genotyping of biospecimen samples from participating UCLA patients in collaboration with the David Geffen School of Medicine, UCLA CTSI, and UCLA Health. Members of the UCLA ATLAS Community Health Initiative include Ruth Johnson, Yi Ding, Vidhya Venkateswaran, Arjun Bhattacharya, Alec Chiu, Tommer Schwarz, Malika Freund, Lingyu Zhan, Kathryn S. Burch, Christa Caggiano, Brian Hill, Nadav Rakocz, Brunilda Balliu, Jae Hoon Sul, Noah Zaitlen, Valerie A. Arboleda, Eran Halperin, Sriram Sankararaman, Manish J. Butte, Clara Lajonchere, Daniel H. Geschwind, and Bogdan Pasaniuc, on behalf of the UCLA Precision Health Data Discovery Repository Working Group and UCLA Precision Health ATLAS Working Group.
UK BioBank: Access to data from the UK BioBank was obtained through application no. 31063. Principal investigators: Ben Neale, Claire Churchhouse. Project overview: “Methodological extensions to estimate genetic heritability and shared risk factors for phenotypes of the UK Biobank.” Website for Pan-UKBB results can be found here: https://pan.ukbb.broadinstitute.org/
Other G.D.S., T.R.G., and J.Z. are supported by a grant from the Medical Research Council for the Integrative Epidemiology Unit at the University of Bristol MC_UU_00011/1 & 4. J.Z. is supported by the Academy of Medical Sciences (AMS) Springboard Award, the Wellcome Trust, the Government Department of Business, Energy and Industrial Strategy (BEIS), the British Heart Foundation and Diabetes UK (SBF006\1117). J.Z. is funded by the Vice-Chancellor Fellowship from the University of Bristol. W.Z. was supported by the National Human Genome Research Institute of the National Institutes of Health under award no. T32HG010464.
ICDA: The authors would like to acknowledge the organizing committee of the International Common Disease Alliance for intellectual contributions on the set up of the GBMI as a nascent activity to the larger effort. We also thank them for the use of their slack platform. Website for ICDA can be found here: https://www.icda.bio/
The Hail Team and Data Management at the Stanley Center for Psychiatric Research: Hail is an open-source Python library that simplifies genomic data analysis in the cloud. It provides powerful, easy-to-use data science tools that can be used to interrogate biobank-scale genomic data and was used in the analysis of the data for this paper. We would especially like to thank Daniel King from the Hail team and Sam Bryant from the Stanley Center Data Management team for helping with the Google bucket set up and data sharing. The website for Hail can be found here: https://hail.is/.
Author contributions
Study design, J.J.P., P.H., W.Z., M.J.D., and J.T.K.; data collection/contribution, W.Z., J.M.O., N.J.C., J.B.H., D.A.S., T.E.F., C.F., I.N., B.L.Y., R.G.J., L.V.W., International IPF Genetics Consortium, GBMI, R.K., and M.M.; data analysis, J.J.P., P.H., W.Z., A.A.L., R.J.A., A.D.S., O.C.L., B.G.-G., and J.T.K.; writing, J.J.P., P.H., and J.T.K.; revision, J.J.P., P.H., M.A., R.K., M.J.D., and J.T.K. All authors provided critical input to interpretation of the data and have approved the final version of the manuscript.
Declaration of interests
A.S. and B.L.Y. are full-time employees of Genentech with stock and stock options in Roche. D.A.S. is the founder and chief scientific officer of Eleven P15, a company focused on the early diagnosis and treatment of IPF. T.E.F. is a consultant to Eleven P15. R.G.J. has received research funding from Astra Zeneca, Biogen, Galecto, GlaxoSmithKline, RedX, and Pliant; consulting fees from Bristol Myers Squibb, Daewoong, Veracyte, Resolution Therapeutics, RedX, and Pliant; payment for lectures, presentations, speakers bureaus, manuscript writing or educational events from Chiesi, Roche, PatientMPower, and AstraZeneca; payment for Participation on a Data Safety Monitoring Board or Advisory Board from Boehringer Ingelheim, Galapagos, and Vicore, had a Leadership or fiduciary role in other board, society, committee or advocacy group (unpaid) in NuMedii and Action for Pulmonary Fibrosis; and is a trustee for Action for Pulmonary Fibrosis. L.V.W. has received research funding from GSK and Orion Pharma and consultancy for Galapagos. M.J.D. is a founder of Maze Therapeutics. J.T.K. and M.J.D. are members of the Pfizer Finland FinnGen Advisory Board.
Inclusion and diversity
We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: October 12, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100181.
Contributor Information
Juulia J. Partanen, Email: juulia.partanen@helsinki.fi.
Jukka T. Koskela, Email: jukka.koskela@helsinki.fi.
Supplemental information
Data and code availability
Full joint meta-analysis summary statistics and GBMI meta-analysis results are available for downloading at https://www.globalbiobankmeta.org/resources and can be browsed at the PheWeb Browser http://results.globalbiobankmeta.org. Custom scripts used for quality control, meta-analysis and summary of the GBMI results are available at https://github.com/globalbiobankmeta. FinnGen fine-mapping pipeline scripts are available at https://github.com/FINNGEN/finemapping-pipeline. Original code generated within this project has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.6993906. Any additional information is available from the lead contact upon request.
References
- 1.Lederer D.J., Martinez F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 2.Allen R.J., Guillen-Guio B., Oldham J.M., Ma S.-F., Dressen A., Paynton M.L., Kraven L.M., Obeidat M., Li X., Ng M., et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2020;201:564–574. doi: 10.1164/rccm.201905-1017OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seibold M.A., Wise A.L., Speer M.C., Steele M.P., Brown K.K., Loyd J.E., Fingerlin T.E., Zhang W., Gudmundsson G., Groshong S.D., et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koskela J.T., Häppölä P., Liu A., Partanen J., Genovese G., Artomov M., Myllymäki M.N.M., Kanai M., Zhou W., Karjalainen J.M., et al. Genetic variant in SPDL1 reveals novel mechanism linking pulmonary fibrosis risk and cancer protection. bioRxiv. 2021 doi: 10.1101/2021.05.07.21255988. Preprint at. [DOI] [Google Scholar]
- 5.Ishigaki K., Akiyama M., Kanai M., Takahashi A., Kawakami E., Sugishita H., Sakaue S., Matoba N., Low S.-K., Okada Y., et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020;52:669–679. doi: 10.1038/s41588-020-0640-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dhindsa R.S., Mattsson J., Nag A., Wang Q., Wain L.V., Allen R., Wigmore E.M., Ibanez K., Vitsios D., Deevi S.V.V., et al. Identification of a missense variant in SPDL1 associated with idiopathic pulmonary fibrosis. Commun. Biol. 2021;4:392. doi: 10.1038/s42003-021-01910-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mushiroda T., Wattanapokayakit S., Takahashi A., Nukiwa T., Kudoh S., Ogura T., Taniguchi H., Kubo M., Kamatani N., Nakamura Y., Pirfenidone Clinical Study Group A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 2008;45:654–656. doi: 10.1136/jmg.2008.057356. [DOI] [PubMed] [Google Scholar]
- 8.Noth I., Zhang Y., Ma S.-F., Flores C., Barber M., Huang Y., Broderick S.M., Wade M.S., Hysi P., Scuirba J., et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir. Med. 2013;1:309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fingerlin T.E., Murphy E., Zhang W., Peljto A.L., Brown K.K., Steele M.P., Loyd J.E., Cosgrove G.P., Lynch D., Groshong S., et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013;45:613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen R.J., Porte J., Braybrooke R., Flores C., Fingerlin T.E., Oldham J.M., Guillen-Guio B., Ma S.-F., Okamoto T., John A.E., et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir. Med. 2017;5:869–880. doi: 10.1016/S2213-2600(17)30387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fingerlin T.E., Zhang W., Yang I.V., Ainsworth H.C., Russell P.H., Blumhagen R.Z., Schwarz M.I., Brown K.K., Steele M.P., Loyd J.E., et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet. 2016;17:74. doi: 10.1186/s12863-016-0377-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duckworth A., Gibbons M.A., Allen R.J., Almond H., Beaumont R.N., Wood A.R., Lunnon K., Lindsay M.A., Wain L.V., Tyrrell J., Scotton C.J. Telomere length and risk of idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease: a mendelian randomisation study. Lancet Respir. Med. 2021;9:285–294. doi: 10.1016/S2213-2600(20)30364-7. [DOI] [PubMed] [Google Scholar]
- 13.COVID-19 Host Genetics Initiative Mapping the human genetic architecture of COVID-19. Nature. 2021 doi: 10.1038/s41586-021-03767-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fadista J., Kraven L.M., Karjalainen J., Andrews S.J., Geller F., COVID-19 Host Genetics Initiative, Baillie J.K., Wain L.V., Jenkins R.G., Feenstra B. Shared genetic etiology between idiopathic pulmonary fibrosis and COVID-19 severity. EBioMedicine. 2021;65 doi: 10.1016/j.ebiom.2021.103277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.COVID-19 Host Genetics Initiative. Ganna A. Mapping the human genetic architecture of COVID-19: an update. medRxiv. 2021 doi: 10.1101/2021.11.08.21265944. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen, R.J., Guillen-Guio, B., Croot, E., Kraven, L.M., Moss, S., Stewart, I., Gisli Jenkins, R., and Wain, L.V. Genetic overlap between idiopathic pulmonary fibrosis and COVID−19. [DOI] [PMC free article] [PubMed]
- 17.Bulik-Sullivan B.K., Loh P.-R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium. Patterson N., Daly M.J., Price A.L., Neale B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghoussaini M., Mountjoy E., Carmona M., Peat G., Schmidt E.M., Hercules A., Fumis L., Miranda A., Carvalho-Silva D., Buniello A., et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 2021;49:D1311–D1320. doi: 10.1093/nar/gkaa840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shrine N., Guyatt A.L., Erzurumluoglu A.M., Jackson V.E., Hobbs B.D., Melbourne C.A., Batini C., Fawcett K.A., Song K., Sakornsakolpat P., et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat. Genet. 2019;51:481–493. doi: 10.1038/s41588-018-0321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hausch F., Kozany C., Theodoropoulou M., Fabian A.-K. FKBPs and the Akt/mTOR pathway. Cell Cycle. 2013;12:2366–2370. doi: 10.4161/cc.25508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodcock H.V., Eley J.D., Guillotin D., Platé M., Nanthakumar C.B., Martufi M., Peace S., Joberty G., Poeckel D., Good R.B., et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat. Commun. 2019;10:6. doi: 10.1038/s41467-018-07858-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kichaev G., Bhatia G., Loh P.-R., Gazal S., Burch K., Freund M.K., Schoech A., Pasaniuc B., Price A.L. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinnott-Armstrong N., Naqvi S., Rivas M., Pritchard J.K. GWAS of three molecular traits highlights core genes and pathways alongside a highly polygenic background. Elife. 2021;10:e58615. doi: 10.7554/eLife.58615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayden L.P., Cho M.H., McDonald M.-L.N., Crapo J.D., Beaty T.H., Silverman E.K., Hersh C.P., COPDGene Investigators ∗ Susceptibility to childhood pneumonia: a genome-wide analysis. Am. J. Respir. Cell Mol. Biol. 2017;56:20–28. doi: 10.1165/rcmb.2016-0101OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halu A., Liu S., Baek S.H., Hobbs B.D., Hunninghake G.M., Cho M.H., Silverman E.K., Sharma A. Exploring the cross-phenotype network region of disease modules reveals concordant and discordant pathways between chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Hum. Mol. Genet. 2019;28:2352–2364. doi: 10.1093/hmg/ddz069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Locke A.E., Kahali B., Berndt S.I., Justice A.E., Pers T.H., Day F.R., Powell C., Vedantam S., Buchkovich M.L., Yang J., et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olafsson S., Alexandersson K.F., Gizurarson J.G.K., Hauksdottir K., Gunnarsson O., Olafsson K., Gudmundsson J., Stacey S.N., Sveinbjornsson G., Saemundsdottir J., et al. Common and rare sequence variants influencing tumor biomarkers in blood. Cancer Epidemiol. Biomarkers Prev. 2020;29:225–235. doi: 10.1158/1055-9965.EPI-18-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emilsson V., Ilkov M., Lamb J.R., Finkel N., Gudmundsson E.F., Pitts R., Hoover H., Gudmundsdottir V., Horman S.R., Aspelund T., et al. Co-regulatory networks of human serum proteins link genetics to disease. Science. 2018;361:769–773. doi: 10.1126/science.aaq1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferkingstad E., Oddsson A., Gretarsdottir S., Benonisdottir S., Thorleifsson G., Deaton A.M., Jonsson S., Stefansson O.A., Norddahl G.L., Zink F., et al. Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat. Commun. 2018;9:5101. doi: 10.1038/s41467-018-07460-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janssen W.J., Stefanski A.L., Bochner B.S., Evans C.M. Control of lung defence by mucins and macrophages: ancient defence mechanisms with modern functions. Eur. Respir. J. 2016;48:1201–1214. doi: 10.1183/13993003.00120-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakaue S., Kanai M., Tanigawa Y., Karjalainen J., Kurki M., Koshiba S., Narita A., Konuma T., Yamamoto K., Akiyama M., et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021;53:1415–1424. doi: 10.1038/s41588-021-00931-x. [DOI] [PubMed] [Google Scholar]
- 32.Bierut L.J., Madden P.A.F., Breslau N., Johnson E.O., Hatsukami D., Pomerleau O.F., Swan G.E., Rutter J., Bertelsen S., Fox L., et al. Novel genes identified in a high density genome wide association study for nicotine dependence. Hum. Mol. Genet. 2007;16:24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eraslan G., Drokhlyansky E., Anand S., Subramanian A., Fiskin E., Slyper M., Wang J., Van Wittenberghe N., Rouhana J.M., Waldman J., et al. Single-nucleus cross-tissue molecular reference maps to decipher disease gene function. bioRxiv. 2021 doi: 10.1101/2021.07.19.452954. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reyfman P.A., Walter J.M., Joshi N., Anekalla K.R., McQuattie-Pimentel A.C., Chiu S., Fernandez R., Akbarpour M., Chen C.-I., Ren Z., et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang I.V., Coldren C.D., Leach S.M., Seibold M.A., Murphy E., Lin J., Rosen R., Neidermyer A.J., McKean D.F., Groshong S.D., et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68:1114–1121. doi: 10.1136/thoraxjnl-2012-202943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sivakumar P., Thompson J.R., Ammar R., Porteous M., McCoubrey C., Cantu E., 3rd, Ravi K., Zhang Y., Luo Y., Streltsov D., et al. RNA sequencing of transplant-stage idiopathic pulmonary fibrosis lung reveals unique pathway regulation. ERJ Open Res. 2019;5:00117–02019. doi: 10.1183/23120541.00117-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Speir M.L., Bhaduri A., Markov N.S., Moreno P., Nowakowski T.J., Papatheodorou I., Pollen A.A., Raney B.J., Seninge L., Kent W.J., Haeussler M. UCSC cell browser: visualize your single-cell data. Bioinformatics. 2021;37:4578–4580. doi: 10.1093/bioinformatics/btab503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng P.C., Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zody M.C., Jiang Z., Fung H.-C., Antonacci F., Hillier L.W., Cardone M.F., Graves T.A., Kidd J.M., Cheng Z., Abouelleil A., et al. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat. Genet. 2008;40:1076–1083. doi: 10.1038/ng.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang G., Sarkar A., Carbonetto P., Stephens M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. Roy. Stat. Soc. B. 2020;82:1273–1300. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benner C., Spencer C.C.A., Havulinna A.S., Salomaa V., Ripatti S., Pirinen M. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–1501. doi: 10.1093/bioinformatics/btw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benner C. Helsingin Yliopisto; 2019. FINEMAP : A Statistical Method for Identifying Causal Genetic Variants. [Google Scholar]
- 45.Wang L., Balmat T.J., Antonia A.L., Constantine F.J., Henao R., Burke T.W., Ingham A., McClain M.T., Tsalik E.L., Ko E.R., et al. An atlas connecting shared genetic architecture of human diseases and molecular phenotypes provides insight into COVID-19 susceptibility. Genome Med. 2021;13:83. doi: 10.1186/s13073-021-00904-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.COVID-19 Host Genetics Initiative A first update on mapping the human genetic architecture of COVID-19. Nature. 2022;608:E1–E10. doi: 10.1038/s41586-022-04826-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bulik-Sullivan B., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.-R., Perry J.R.B., Patterson N., Robinson E.B., et al. ReproGen Consortium. Psychiatric Genomics Consortium. Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3. Duncan L., ReproGen Consortium An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015;47:1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaunisto J., Salomaa E.-R., Hodgson U., Kaarteenaho R., Kankaanranta H., Koli K., Vahlberg T., Myllärniemi M. Demographics and survival of patients with idiopathic pulmonary fibrosis in the FinnishIPF registry. ERJ Open Res. 2019;5:00170–02018. doi: 10.1183/23120541.00170-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lim E.T., Würtz P., Havulinna A.S., Palta P., Tukiainen T., Rehnström K., Esko T., Mägi R., Inouye M., Lappalainen T., et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanai, M., Ulirsch, J.C., Karjalainen, J., Kurki, M., Karczewski, K.J., Fauman, E., Wang, Q.S., Jacobs, H., Aguet, F., Ardlie, K.G., et al. Insights from complex trait fine-mapping across diverse populations.
- 51.Dudbridge F., Allen R.J., Sheehan N.A., Schmidt A.F., Lee J.C., Jenkins R.G., Wain L.V., Hingorani A.D., Patel R.S. Adjustment for index event bias in genome-wide association studies of subsequent events. Nat. Commun. 2019;10:1561. doi: 10.1038/s41467-019-09381-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schaller T., Hirschbühl K., Burkhardt K., Braun G., Trepel M., Märkl B., Claus R. Postmortem examination of patients with COVID-19. JAMA. 2020;323:2518–2520. doi: 10.1001/jama.2020.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Assayag D., Morisset J., Johannson K.A., Wells A.U., Walsh S.L.F. Patient gender bias on the diagnosis of idiopathic pulmonary fibrosis. Thorax. 2020;75:407–412. doi: 10.1136/thoraxjnl-2019-213968. [DOI] [PubMed] [Google Scholar]
- 54.Helling B.A., Gerber A.N., Kadiyala V., Sasse S.K., Pedersen B.S., Sparks L., Nakano Y., Okamoto T., Evans C.M., Yang I.V., Schwartz D.A. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2017;57:91–99. doi: 10.1165/rcmb.2017-0046OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oliva M., Muñoz-Aguirre M., Kim-Hellmuth S., Wucher V., Gewirtz A.D.H., Cotter D.J., Parsana P., Kasela S., Balliu B., Viñuela A., et al. The impact of sex on gene expression across human tissues. Science. 2020;369:eaba3066. doi: 10.1126/science.aba3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang L., Pu Y., Hepps D., Danielpour D., Prins G.S. Posterior Hox gene expression and differential androgen regulation in the developing and adult rat prostate lobes. Endocrinology. 2007;148:1235–1245. doi: 10.1210/en.2006-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma L., Benson G.V., Lim H., Dey S.K., Maas R.L. Abdominal B(AbdB)HoxaGenes: regulation in adult uterus by estrogen and progesterone and repression in müllerian duct by the synthetic estrogen diethylstilbestrol (DES) Dev. Biol. 1998;197:141–154. doi: 10.1006/dbio.1998.8907. [DOI] [PubMed] [Google Scholar]
- 58.Li Z., Tuteja G., Schug J., Kaestner K.H. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell. 2012;148:72–83. doi: 10.1016/j.cell.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Global Biobank Meta-analysis Initiative. Zhou W. Global Biobank Meta-analysis Initiative: powering genetic discovery across human diseases. bioRxiv. 2021 doi: 10.1101/2021.11.19.21266436. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zou Y., Carbonetto P., Wang G., Stephens M. Fine-mapping from summary data with the “sum of Single effects” model. PLoS Genet. 2022;18:e1010299. doi: 10.1371/journal.pgen.1010299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mbatchou J., Barnard L., Backman J., Marcketta A., Kosmicki J.A., Ziyatdinov A., Benner C., O’Dushlaine C., Barber M., Boutkov B., et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 2021;53:1097–1103. doi: 10.1038/s41588-021-00870-7. [DOI] [PubMed] [Google Scholar]
- 62.Firth’s Bias-Reduced Logistic Regression [R package logistf version 1.24] (2020).
- 63.Machiela M.J., Chanock S.J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thompson S.G., Higgins J.P.T. How should meta-regression analyses be undertaken and interpreted? Stat. Med. 2002;21:1559–1573. doi: 10.1002/sim.1187. [DOI] [PubMed] [Google Scholar]
- 66.Balduzzi S., Rücker G., Schwarzer G. How to perform a meta-analysis with R: a practical tutorial. Evid. Base Ment. Health. 2019;22:153–160. doi: 10.1136/ebmental-2019-300117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full joint meta-analysis summary statistics and GBMI meta-analysis results are available for downloading at https://www.globalbiobankmeta.org/resources and can be browsed at the PheWeb Browser http://results.globalbiobankmeta.org. Custom scripts used for quality control, meta-analysis and summary of the GBMI results are available at https://github.com/globalbiobankmeta. FinnGen fine-mapping pipeline scripts are available at https://github.com/FINNGEN/finemapping-pipeline. Original code generated within this project has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.6993906. Any additional information is available from the lead contact upon request.